 Dan Wang1†
Dan Wang1† Zhenchen You1†
Zhenchen You1† Mengjie Yan1†Xihu Wang2Jianjun Ge2
Mengjie Yan1†Xihu Wang2Jianjun Ge2 Menghua Zhang1
Menghua Zhang1 Lei Xu1Shengchao Ma1Mingming Dong1Xixia Huang1*
Lei Xu1Shengchao Ma1Mingming Dong1Xixia Huang1*- 1College of Animal Science, Xinjiang Agricultural University, Urumqi, Xinjiang, China
- 2Xinjiang Hutubi Cattle Farm, Changji, Xinjiang, China
Suitable bedding provides cows with a clean and comfortable lying environment. However, with extended usage of bedding, microorganisms can be transferred to the mammary gland through the teat skin, adversely affecting the mammary glands of dairy cattle and altering the microbiota present in the milk. This study analyzed microorganisms in bedding composed of fermented cow manure, together with those in teat skin swabs and milk, and found that the bacterial richness and diversity of the bedding were maximal on the 14th day of use. The dominant bacterial phyla were Firmicutes, Actinobacteria, Proteobacteria, and Bacteroidetes, while the dominant genera were Corynebacterium_1, Bacillaceae_norank, Salinicoccus, and Dietzia. Analysis of the flora in the bedding over time showed that there was a gradual change toward moderately salinophilic genera. The composition of the teat-skin flora was highest on Day 21, with Firmicutes and Actinobacteria as the dominant phyla and Corynebacterium_1, Salinicoccus, Dietzia, Romboutsia, Nesterenkonia, and Turicibacter as the dominant genera. Correlation analysis of the teat-skin flora and somatic cell counts in the milk showed a positive association between the teat-skin flora composition and duration of bedding use, while changes in the abundance of mastitis-causing bacteria were positively correlated with somatic cell counts. Microbial abundance and diversity in the milk were highest on days 30 and 21, respectively, of bedding use. At the phylum level, Firmicutes, Proteobacteria, Bacteroidetes, and Actinobacteria dominated, while Staphylococcus, Pseudomonas, Lacto-bacillus, Enterococcus, Acinetobacter, and Bacteroides represented the core genera; correlation analysis further verified that mastitis-causing bacteria in milk were affected by the duration of bedding use and teat microorganisms, and were positively correlated with somatic cell counts. Thus, increased duration of bedding use resulted in changes in the microbial community structures of the bedding itself, the teat skin, and microorganisms in the milk, together with an increased number of mastitis-causing bacteria, considered to be related to the rise in the somatic cell count of the milk. Therefore, it is recommended that fermented cow manure bedding should be replaced in a cycle not exceeding 14 days.
1 Introduction
The welfare of dairy cows is essential to ensure good milk production and quality. Bedding is important for cows’ welfare, as they generally spend approximately 50 to 60% of the day resting to facilitate rumination and food digestion (Jensen et al., 2005), with adequate rest contributing to an estimated 24–28% increase in mammary-gland circulation, thus enhancing the feed conversion rate and increasing milk production (Metcalf et al., 1992; Cook and Nordlund, 2009). Beds are usually used for resting, and a dry, soft bed of appropriate size is important. However, poor bed design and bedding may lead to reduced milk production, as well as decreased reproduction (Rushen et al., 2007; Norring et al., 2008). Hard beds may also cause leg injuries and limping in cows (Tucker and Weary, 2004; Fregonesi et al., 2007; Li et al., 2021). Haley et al. (2001) compared the influence of concrete and cushioning on cow behavior, finding that 51% of cows preferred resting on soft bedding, while Norring et al. (2010) reported that cows rested for longer on rubber bedding than on concrete or sand bedding, and beds constructed of straw and rubber were preferred to those made of feces/straw and sand. Comfortable bedding constructed of cow manure may increase the times spent resting, as well as provide a means for using manure.
The skin of vertebrates constitutes the primary physical barrier between the animal and its external environment (Calamari et al., 2009; Cole and Hogan, 2016). The surface skin of the cow’s teats usually contains a large number of bacteria (Verdier-Metz et al., 2009; Braem et al., 2012; Monsallier et al., 2012), and when the cow is lying down, the teat will come into close contact with the bedding, resulting in the transfer of bacteria from the bedding to the skin and ultimately to the milk. Additionally, bacteria may be transferred from the milking equipment, the farm environment, soil, and forage, as well as being influenced by the stage of lactation (Vacheyrou et al., 2011; Esteban-Blanco et al., 2020). The quality and quantity of these teat-associated microbes varies in different farm environments (LeJeune and Rajala-Schultz, 2009) as well as with different types of bedding. Sorter et al. (2014) investigated 38 dairy farms and found that the number of bacteria in bedding made of feces was lower than that in other bedding types, while Zdanowicz et al. (2004) reported that the number of E. coli in sawdust bedding was twice that in sand and Rowbotham and Ruegg (2015) compared the effects of deep freshly laid sand bedding, treated and reused sand bedding, and cow manure bedding on the incidence of mastitis in primiparous Holstein cows, finding that the incidence of clinical mastitis in cows housed with freshly laid sand bedding was higher than that with the other two types of bedding (Rowbotham and Ruegg, 2016). Changing bedding made from manure on a daily basis reduces the total number of bacteria. Fermented cow manure, in particular, when used as bedding, is not favorable for the growth of microorganisms such as E. coli, and the growth of Streptococcus and Klebsiella is lower than in bedding made from fresh feces (Cole and Hogan, 2016). Therefore, variations in the types and functions of microbes in different bedding and teat skin may influence bacterial colonization of the mammary gland, potentially affecting the health of the organ. Microorganisms can enter the milk from a variety of sources, and once in the milk, they can have specific effects, such as promoting the fermentation of dairy products or causing milk spoilage (Montel et al., 2014). A study by Verdier-Metz et al. (2012) analyzed the microbial composition of milk and showed that the dominant bacteria included Firmicutes (76%), Actinobacteria (4.9%), Proteobacteria (17.8%), and Bacteroides (1.3%). The microbial composition of raw milk and cheese has been shown to differ, with previously unrecognized and diverse bacterial populations present in unpasteurized milk, mainly consisting of Lactococcus, Pseudomonas, and Leuconostoc. A number of anaerobic bacterial taxa were also detected, including Bacteroides, Fecalibacterium, Prevotella, and Streptococcus, all of which are commonly associated with the gut microbiota and may thus have entered the milk through fecal contamination. The management of pasture and the structure and composition of its specific microbiota have marked influences on the particular flavors of cheese (Hantsis-Zacharov and Halpern, 2007). Molecular biology and genomics can assist in analyzing the changes in the structure and composition of microbiota (Meng et al., 2019). Albino et al. (2017) analyzed the relationships between the somatic cell counts (SCCs) of milk, cow teat skin, bedding samples, and the number of bacteria in the milk and found that Streptococcus on teat skin were also present in both the milk and bedding.
The 16S rDNA gene encodes the small subunit rRNA of prokaryotic ribosomes, and it is the most commonly used indicator of bacterial phylogeny and taxonomic identification (Callahan et al., 2016). Sequencing of the 16S rDNA gene usually selects a specific variable region or several variable regions, and the structure and diversity of the microbiota in the samples can be analyzed using universal primers, providing information on the species composition and their relative abundance in the microbiota. At the same time, this technique can provide a comprehensive analysis of the microorganisms present in cow bedding, and the structure, composition, and diversity of those in the milk (Niu et al., 2022). The feeding environment of cows is the primary influence on the microbial composition of the milk. Fermentation of manure for use as bedding involves separation of the wet and dry manure, followed by fermentation before use; this not only solve the problem of manure removal and recycling on cattle farms but also addresses the issue of a source material for bedding, reducing the difficulty of manure treatment, and saving on costs (Callahan et al., 2016). The skin of the cow’s teat is critical for determining the microbial contents of the milk, while the microorganisms in the feces and bedding also have significant effects. This study, therefore, collected samples of manure used for cow bedding for different lengths of time, at the same time as collecting swabs of the teat skin and milk samples. The V3-V4 variable sequences of the 16s rDNA genes of microorganisms in these samples were sequenced on an Illumina MiSeq platform, to investigate the structure, composition, and diversity of the microbiota in the bedding, teats, and milk at different time points of bedding usage. The findings will provide a reference for cattle farms to determine the appropriate replacement cycle of the bedding and offer suggestions for pasture management.
2 Materials and methods
2.1 Sample collection
The study analyzed the bedding in barns housing adult cows in Hutubi, Xinjiang. The farm is located at E86.892848, N44.191178, located in a region of temperate continental arid and semi-arid climate with an average annual precipitation of 167 mm. The experiment was conducted in July, during which the monthly averages were: maximum temperature 32°C, minimum temperature 20°C, and relative humidity 20%. The farm cowshed was a loose pen-type cowshed, that used an automatic scraping board to clear the manure and used fermented cow manure as bedding. This farm maintained a herd of 1,500 Chinese Holstein. The standardized milking procedure, conducted three times daily, The milking procedure involves ensuring cow hygiene, pre-dipping with disinfectant, stripping three streams of milk per teat, wiping teats with disposable paper towels, attaching milking cups within 60 seconds, applying a post-milking iodine-based disinfectant to all teats. When newly laid, the bedding was dry, without odor and mildew. The thickness of the fermented cow manure bedding was 20–25 cm, and bedding was leveled and feces on the bedding were removed every morning.
The bedding samples were collected from the same barn on the 1st, 7th, 14th, 21st, and 30th days after the laying of the fermented cow manure bedding. Five bedding samples were randomly selected at each time point, with the cow occupying the entire bedding area. The “checkerboard” samples method, described in the “Technical Specification for Soil Environmental Monitoring in China” was used to collect the bedding samples (Food and Agriculture Organization of the United Nations, 2004, https://www.fao.org/faolex/results/details/en/c/LEX-FAOC193186/, Accessed June 1st 2019). Samples from every 5=five beds were mixed to form one replicate, 25 g of material was collected from each replicate, with 5 replicates, totaling 25 bedding samples; the samples were grouped as A1, A2, A3, A4, and A5. The samples were sealed in sterile sampling bags and were stored at -40°C for subsequent DNA extraction.
Random allocation eight single-parity cows in mid-lactation (90–144 days) with similar milk yields and SCC ≤ 200–000 cells/mL, no diagnosis of disease and treatment records, and with consistent levels of cleanliness of the limbs, feet, and udders were selected from the same barn. Swabs were taken from the teats of 8 selected cows on the 1st, 7th, 14th, 21st, and 30th days after the laying of the fermented cow manure bedding, and before milking and disinfection. Four skin swabs were collected from each cow using saline-soaked sterile cotton swabs, and the tips of the swabs from four milking areas of the same cow were placed in the same freezer tube and stored at -40°C.
After the collection of the skin samples, the teat was washed and dried with paper towel. The first 3 milk from each cow were discarded, after which 40 mL of milk was collected with sterile gloves into two sterile sample bottles. The milk samples were transported to the laboratory on dry ice; in the laboratory, the samples were thawed and centrifuged at 12000 rpm for 20 min to remove the upper layer of milk fat and supernatant. The precipitate was retained for DNA extraction. Meanwhile, 40 mL of fresh milk was collected on-site into sampling bottles containing bromonitol tablets for Dairy Herd Improvement (DHI) determination. Milk composition and somatic cell counts were simultaneously analyzed using a MilkScan™ FT+200 analyzer and Fossmatic™ FC5000 instrument, respectively (Foss, Denmark). DHI is a measurement and evaluation system that measures indicators such as milk production, milk fat percentage, milk protein percentage, lactose, and somatic cell count of lactating cows, combined with reproductive information such as parity and calving date of the herd. After analysis, it forms a report reflecting the information of the dairy farm’s diet nutrition, feeding management, reproductive management, sperm selection, disease prevention and control, and production performance. No ethical approval was required for this study. Through tripartite sampling (bedding-teat-milk) conducted at predetermined intervals, we systematically analyzed temporal microbial dynamics in fermented cattle bedding across distinct usage phases, while simultaneously characterizing associated microbial community structures.
2.2 DNA extraction
DNA extraction was performed using a Fast DNA Spin Kit for Soil (MP Biomedicals, Santa Ana, CA, USA), and the quality of the extracted DNA was checked by 2% agarose gel electrophoresis at 80 V for 30 min.
2.3 High-throughput sequencing
The V3-V4 universal primers 338F (5’-ACTCCTACGGGAGGCAGCAG-3’) and 806R (5’-GACTACHVGGGTWTCTAAT-3’) were used for PCR, and a barcoded tag of 8 nucleotide bases was added before the upstream sequence of the universal primers to form barcoded fusion primers. The PCR (Mastercycler, eppendof) reaction volume was 20 μL, containing 4 µL of 5×FastPfu Buffer, 2 µL of dNTPs (2.5 mM), 0.8 µL of 338F (5 µM), 0.8 µL of 806R (5 µM), 0.4 µLFastPfu polymerase, 0.2 µL of BSA, 10 ng of DNA, and made up to 20 µL with RNase-free dH2O up to 20 µL. The reaction conditions were 95 °C for 180 s, followed by 30 cycles of 95 °C for 30 s, 55 °C for 30 s, 45 °C for 30 s, and 72 °C for 600 s. The PCR products from the same samples were mixed together, and cut using AxyPrep DNA gel recovery kit (iGEM, Paris, France) to recover the PCR products which were then analyzed on 2% agarose gels. The results of the identification of the PCR products are shown in Supplementary Figure S1.
The library was constructed using TruSeqTM DNA Sample Prep Kit (Illumina, San Diego, CA, USA), quantified using the QuantiFluor™ -ST blue fluorescence quantification system (Promega, Madison, WI, USA) and PCR, and sequenced using MiSeq PE300 sequencing platform (Illumina).
2.4 Bioinformatics and statistical analysis
The original sequences were rejoined and screened for quality using QIIME software (Version 1.19). The primers were removed, and sequences with quality scores <20 or lengths <50 bp were removed. Sequences were clustered, checked for chimeras, and quality filtered with Q30≥90 using the USearch program to identify operational taxonomic units (OTUs) at the 97% similarity level.
Species annotation of representative OTU sequences with 97% similarity levels was performed using the RDP classifier Bayesian algorithm, and the community composition of each sample was assessed at each taxonomic level (kingdom, phylum, class, order, family, genus).
The structure and α diversity of the microbial communities in bedding samples collected at different time points were analyzed using one-way ANOVA in SPSS 22.0 (IBM Corp., Armonk, NY, USA), followed by the use of the least significant difference (LSD) to test the significance, with α diversity evaluated using theChao1, Shannon, Simpson, and ACE indices and R software used to plot the dilution curves.
The analysis of the intergroup differences in β diversity was performed by calculating Unifrac distances, followed by principal Co-ordinate analysis (PCoA) using multivariate statistical methods and permutational multivariate analysis of variance (PERMANOVA), to determine differences in the microbiota of bedding, teat skin, and milk at different time points.
Linear discriminant analysis effect size (LEfSe) was used to analyze the abundance of significantly different microbiota (e.g., biomarkers) at different time points after the laying of fermented cow manure bedding (e.g., between groups).
The workflow of the study is shown in Figure 1.
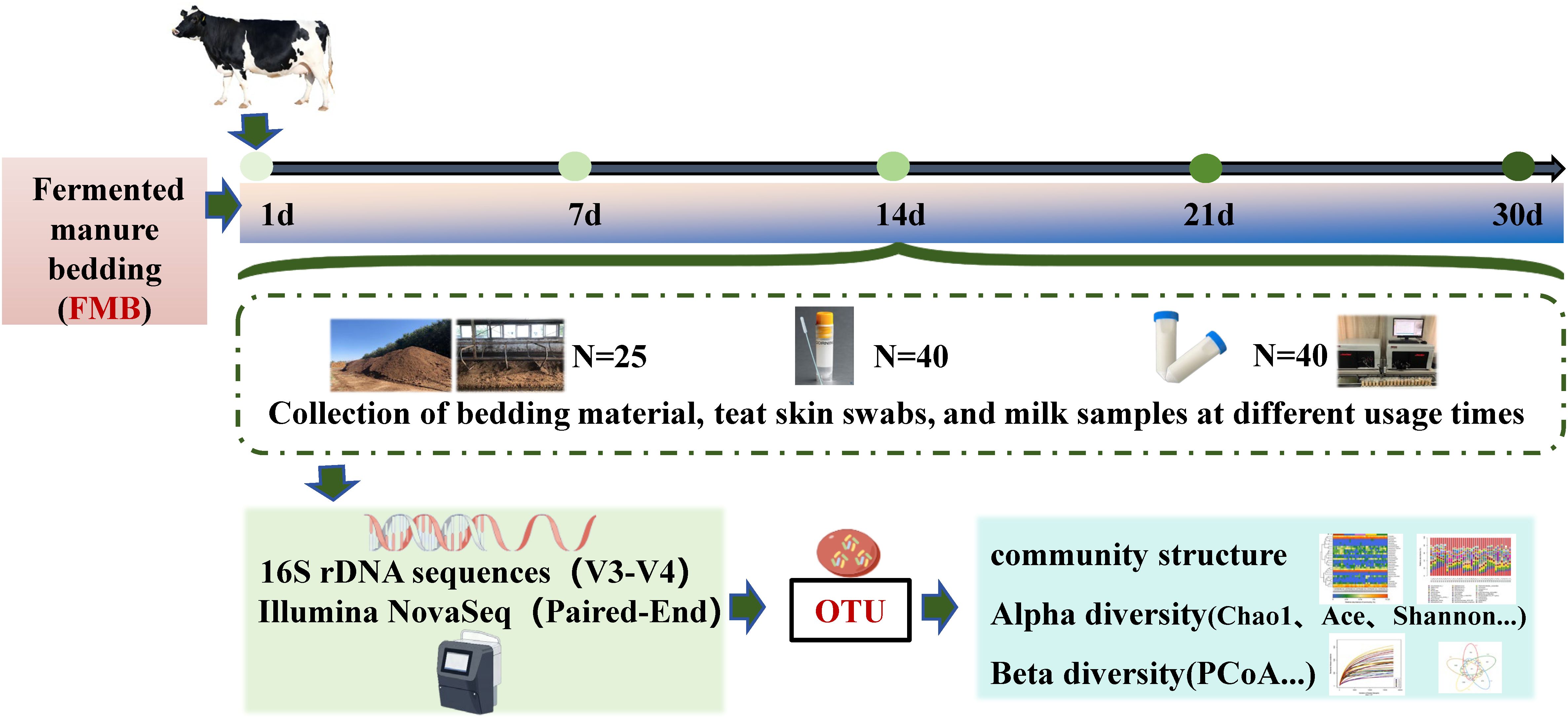
Figure 1. The workflow of the study.
3 Results
3.1 Diversity and structure of microbiota at different time points after the laying of fermented cow manure bedding
The sequencing of fermented cow manure bedding samples yielded a total of 2331 OTUs, 580 215–391 bases, and 1 311–650 sequences. The sample dilution curves were flat, and the sequencing depth was sufficient for diversity analysis (Figure 2A). The distribution of OTUs among the five collection time points of the fermented cow manure bedding (Figure 2B) showed that the number of OTUs shared among the five time points was 688, accounting for approximately 10% of the total number of OTUs. The number of unique OTUs in the fermented cow manure bedding was 59, 96, 144, 99, and 72 on the 1st (A1), 7th (A2), 14th (A3), 21st (A4), and 30th (A5) days, respectively. As the duration of bedding usage increased, the microbial components gradually increased, with the highest on day 14, after which both the structure and composition of the microbiota were observed to be reduced on the 21st and 30th days, but were nevertheless higher than those on the 1st and 7th days. The Chao1 and Shannon indices of the bedding were significantly higher after bedding use than those on day 1 (P<0.05), while the Chao1 index on day 14 was significantly higher than that on days 7 and 30 (P<0.05) (Figures 2C, D). As the duration of bedding usage increased, the abundance and diversity of the microbiota were found to be maximal on day 14. The OTU composition of different samples was analyzed to assess the differences between the samples. For example, the more similar the microbiota composition, the closer the distance reflected in thePCoA plots. The PCoA showed that although there was clustering of the microbiota from samples collected at different time points of bedding usage, some of the samples overlapped with each other, and the species composition and structure of the microbiota in the bedding at different stages showed similarities (Figure 2E). According to the results of multivariate permutation analysis of variance (Table 1), the microbiota structure on the 7th, 14th, 21st, and 30th day time points differed significantly from that on day 1 (P<0.05), and the composition of microbiota on day 30 was significantly different from that on the 7th and 14th days (P<0.05).
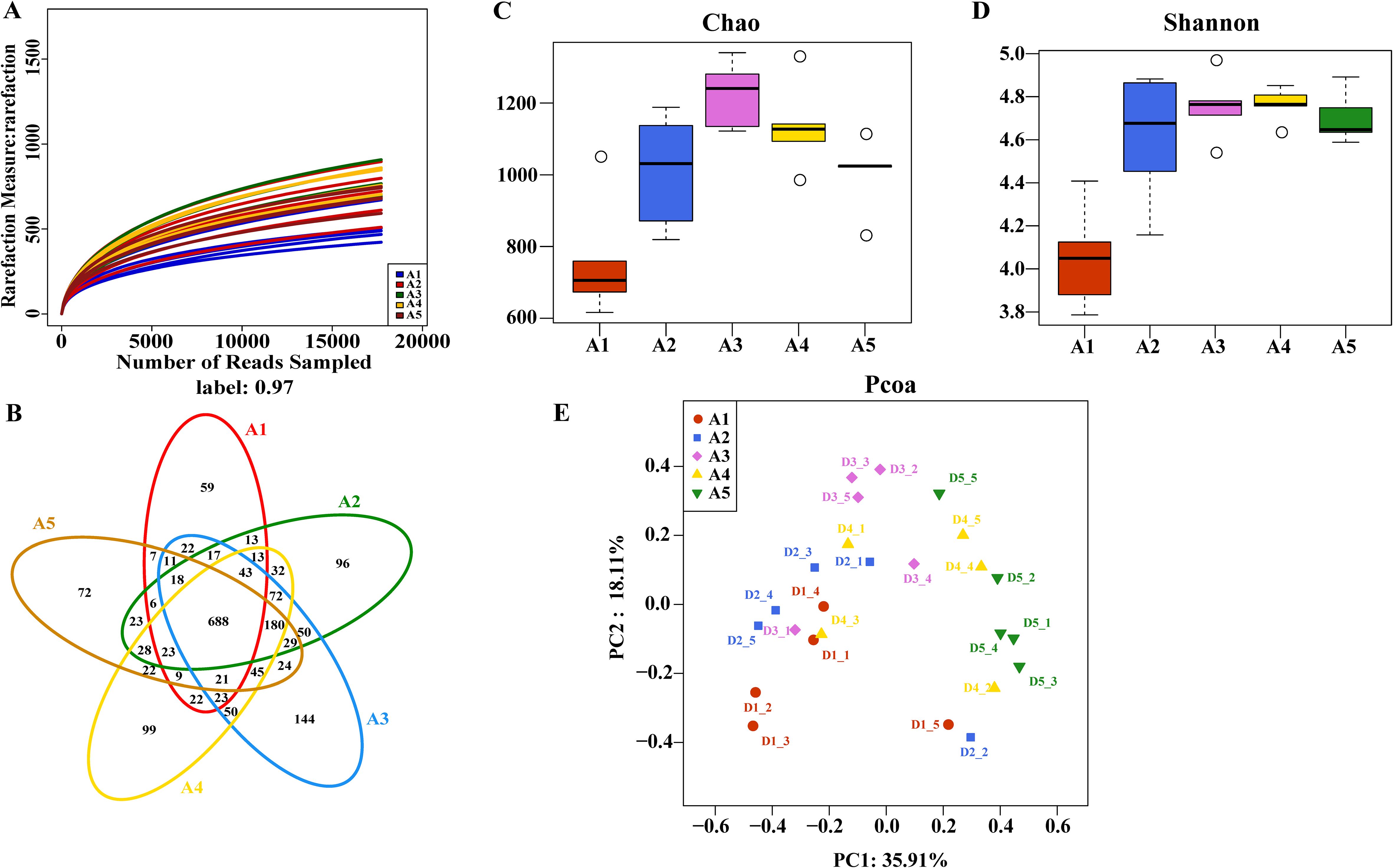
Figure 2. Characteristics of changes in microbial diversity of bedding material at different usage periods. (A) Sample rarefaction curves; (B) Distribution of microbial OTUs in fermented cow manure bedding at different time points; (C, D) Variations in microbial alpha diversity of fermented cow manure bedding samples at different time points; (E) Principal coordinate analysis of fermented cow manure bedding microbiota at different time points.
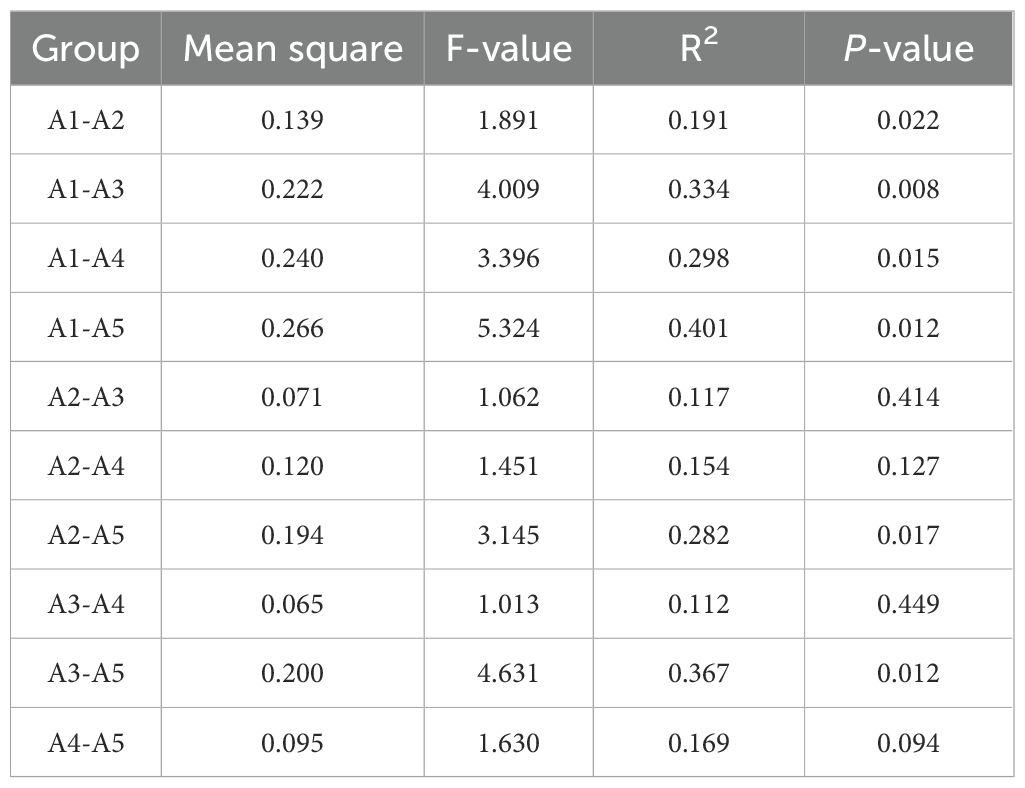
Table 1. Differences between groups of fermented cow manure bedding microbiota at different time points.
Based on the OTU annotation results, a total of seven dominant phyla with relative abundance > 1% were detected (Figure 3A), wherein Firmicutes showed the maximum relative abundance, which decreased as the duration of bedding usage increased. The abundance of Actinobacteria varied significantly, decreasing as the length of bedding usage increased, but with the abundance on day 30 exceeding that on day 1. The abundance of Proteobacteria and Bacteroidetes was markedly raised on days 21 and 30 relative to day 1 (P<0.05), while the abundance of Chloroflexi was reduced on the 7th and 14th days, and then continued to increase with an abundance on day 30 being significantly higher than that of the other groups (P<0.05). The abundance of Deinococcus-Thermus showed an increase followed by a decrease on the 7th day, while on day 30, its abundance was significantly higher than that of the other groups (P<0.05); the abundance of Halanaerobiaeota was the highest on the first day of use, and decreased continuously as the length of bedding usage extended (Figure 3B). At the genus level (Figure 3C), 23 genera were found with a relative abundance greater than 1%, of which the dominant genera were Corynebacterium_1, Bacillaceae_norank, Salinicoccus, and Dietzia. The abundance of Corynebacterium_1 was significantly higher than that of the other groups on day 1 (P<0.05), while the abundance of Salinicoccus increased with the bedding usage time, and on day 21, was significantly higher than that on day 7 (P<0.05). The abundance of Dietzia increased with the bedding usage time, and on day 30, it was significantly higher than that on day 7 (P<0.05), while the abundance of Staphylococcus was highest on day 21 (A4) and significantly higher than that on day 1 (P<0.05) (Figure 3D). Differential genera in the fermented cow manure bedding were analyzed using LEfSe (threshold set at 2), with a total of 2 phyla, 4 classes, 7 orders, 11 families, and 12 genera. At the genus level, the differential genera on day 1 (A1) of fermented cow manure bedding were Corynebacterium_1 and Planifilum, on day 7, the differential genera were Ruminococcaceae-UCG-005, Novibacillus, and Saccharomonospora, on day 14, the differential genus was Marinobacter, on day 21, the differential genera were Aliidiomarina and Marinospirillum, and on day 30, the differential genera compared to the other groups were Salinicoccus, Brachybacterium, Oceanobacter, and Ulvibacter (Figure 3E).
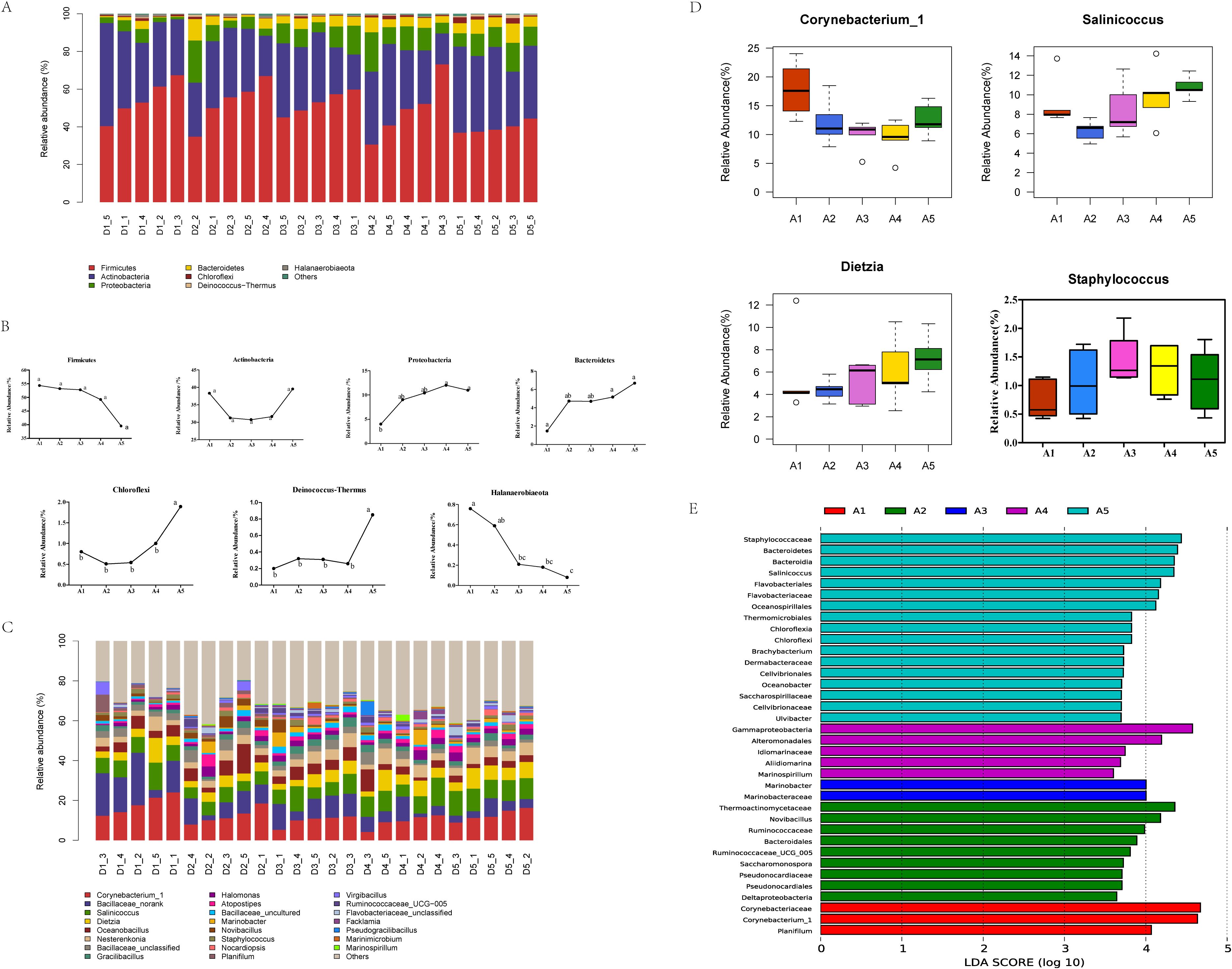
Figure 3. Analysis of the characteristics of microbial community structure changes in bedding material across different usage periods. (A) The distribution of fermented cow manure bedding microbiota at the phylum level at different time points; D1 represents day 1, D3 is day 7, D3 is day 14, D4 is day 21, and D5 is day 30; “_” indicates the sample number, with the same definitions applying below. (B) Variations in bacterial phyla in cow manure bedding at different time points. Different lowercase letters indicate significant differences (P<0.05), while the same lowercase letter indicates a non-significant difference (P>0.05). In the figure, a, b, c, d, e, f, and g represent Firmicutes, Actinobacteria, Proteobacteria, Bacteroidetes, Chloroflexi, Deinococcus−Thermus, and Halanaerobiaeota, respectively. (C) Variations in bacterial genera in cow manure bedding at different time points. (D) Changes in the abundance of genera in the microbiota of fermented cow manure bedding at different time points. (E) Map of the different microorganisms in fermented cow manure bedding at different time points.
3.2 Structural features of microbiota on cow teat skin at different time points of bedding usage
The sequencing results of teat skin swabs generated 3390 OTUs, 869 158–508 bases, and 1 986–008 sequences. Both the number of sequences and the species distribution gradually leveled off over time of bedding usage. The swab samples were found to have been sequenced at sufficient depth and the species distribution was even (Figures 4A, B). Analysis of the distribution of the swab OTUs among the five time points of bedding usage indicated that the number of shared OTUs was 947, accounting for approximately 28% of the total number of OTUs, while the numbers of unique OTUs were 86, 47, 254, 246, and 139 on the 1st (B1), 7th (B2), 14th (B3), 21st (B4), and 30th (B5) days, respectively. As the duration of bedding usage extended, the structure and composition of the microbiota on the cow teats were highest on days 14 and 21 (Figure 4C), with microbial abundance at its highest point on day 21, when it was significantly higher than on the days 1, 14, and 30 (P<0.05) and microbial diversity highest on day 14 when it was significantly higher than that on the other days (P<0.05) (Figures 4D, E). The PCoA of microbiota on cow teat skin samples at the different time points (Figure 4F) explained 18.1% and 10.9% of the total variance, respectively. The PCoA results showed that except for S1_7 in group B1, which was clearly separated from the other groups, the samples of the other groups were clustered at different time points with no significant separation. The results of multivariate permutation analysis of variance (Table 2) showed that the structure and abundance of microbiota on cow teat skin varied significantly among the different time points, suggesting that the duration of bedding usage was associated with significant changes in the structure and abundance of the microbiota on the cow teats.
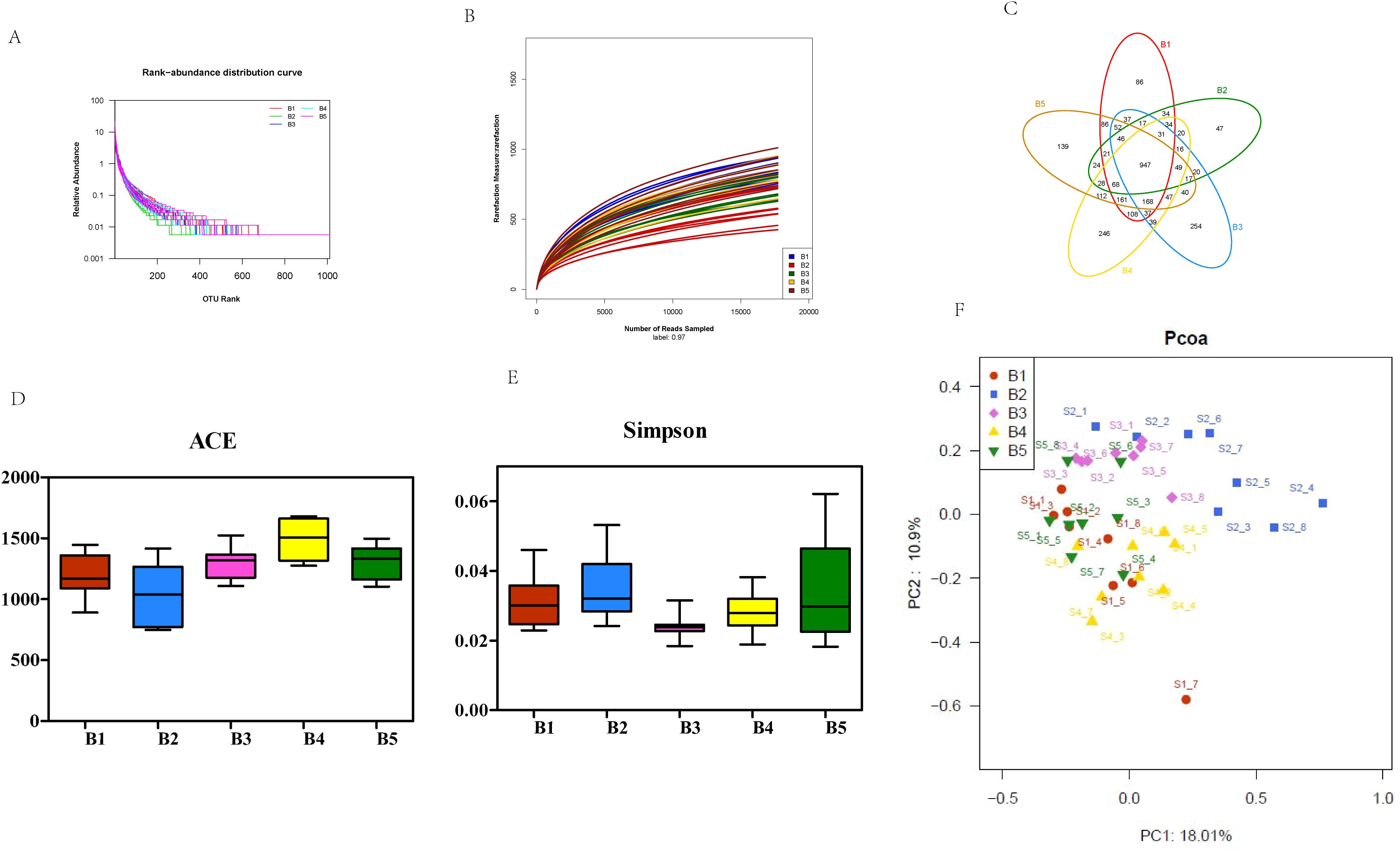
Figure 4. Characteristics of microbial diversity changes in teat skin swabs across different usage periods. (A) Sample Rank-Abundance curve. B1, day1; B2, day 7; B3, day 14; B4, day 21; B5, day 30. (B) Sample rarefaction curves. (C) Distribution of microbial OTUs in dairy cow teat skin samples at different time points. (D, E) Variations in the alpha diversity of cow teat skin microbiota at different time points. (F) PCoA of cow teat skin microbiota at different time points.
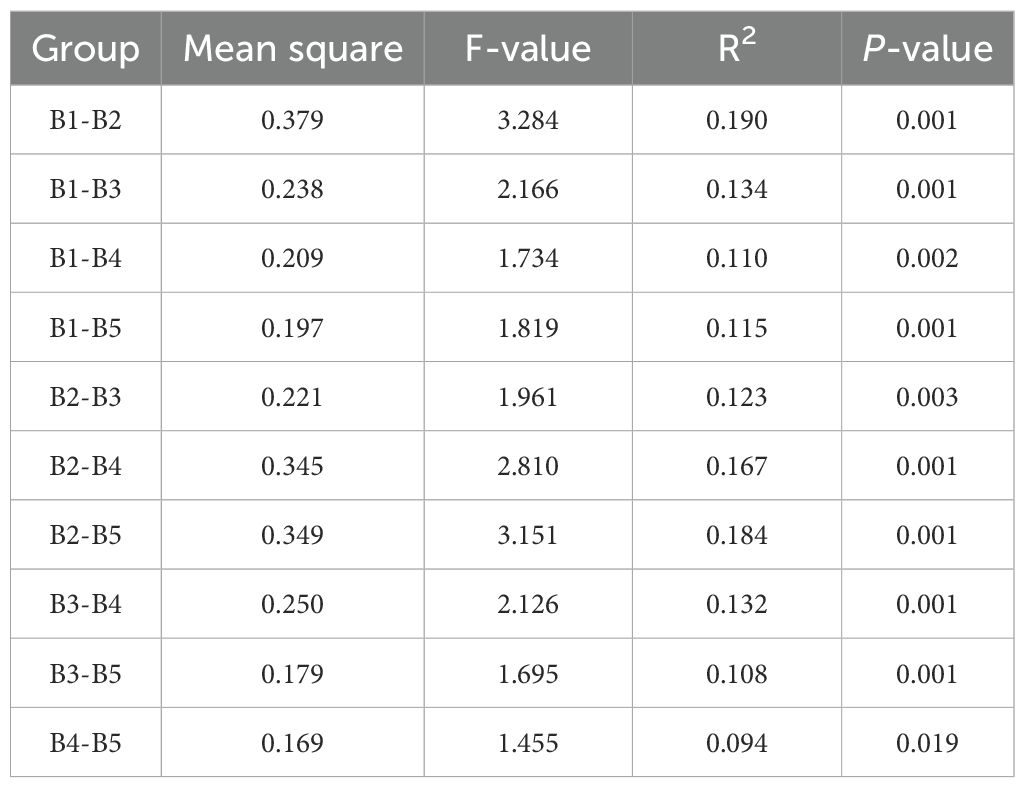
Table 2. Differences between cow teat skin microbiota at different time points.
The annotation results showed a total of 3390 OTUs representing 31 phyla, 64 classes, 175 orders, 331 families, and 840 genera. As shown in Figure 5A, the shared dominant phyla (relative abundance > 1%) by microbiota on cow teat skin samples at different time points included Firmicutes, Actinobacteria, Proteobacteria, Bacteroidetes, and Chloroflexi. The relative abundance of the five phyla differed at different time points in bedding usage (Figure 5B), with the abundance of Firmicutes being highest on days 1 and 14, and significantly higher than that on days 7 and 21 (P<0.05), while the abundance of Actinobacteria was highest on days 7 and 21, when it was significantly higher than that on day 14 (P<0.05). The abundance of Proteobacteria was highest on day 30, significantly higher than that on day 1 (P<0.05), and the abundance of Bacteroidetes was highest on days 21 and 30, although the difference in abundance among groups was not significant (P>0.05), the abundance of Chloroflexi was highest on day 21 when its abundance was significantly higher than that at other time points (P<0.05). As indicated, the microbial abundance in cow teat skin samples was affected by the length of bedding usage. Thirty-two microbial genera were detected in skin swabs with relative abundance > 1% at all time points of bedding usage (Figure 5C), and, in order of abundance, the dominant genera were Corynebacterium_1, Salinicoccus, Dietzia, Romboutsia, Nesterenkonia, and Turicibacter. The changes in genus abundance on cow teat skin at the different time points were compared with the SCC (Figures 5D, E), showing that as the duration of bedding usage increased, the abundance of Corynebacterium_1 and Paenibacillus decreased as the SCC increased, while the abundance of Dietzia, Staphylococcus, Escherichia-Shigella, and Streptococcus increased as the SCC increased. The abundance of various genera decreased in correspondence to increases in SCC, suggesting that microbiota on the skin of cow teats could affect the health of the mammary glands.
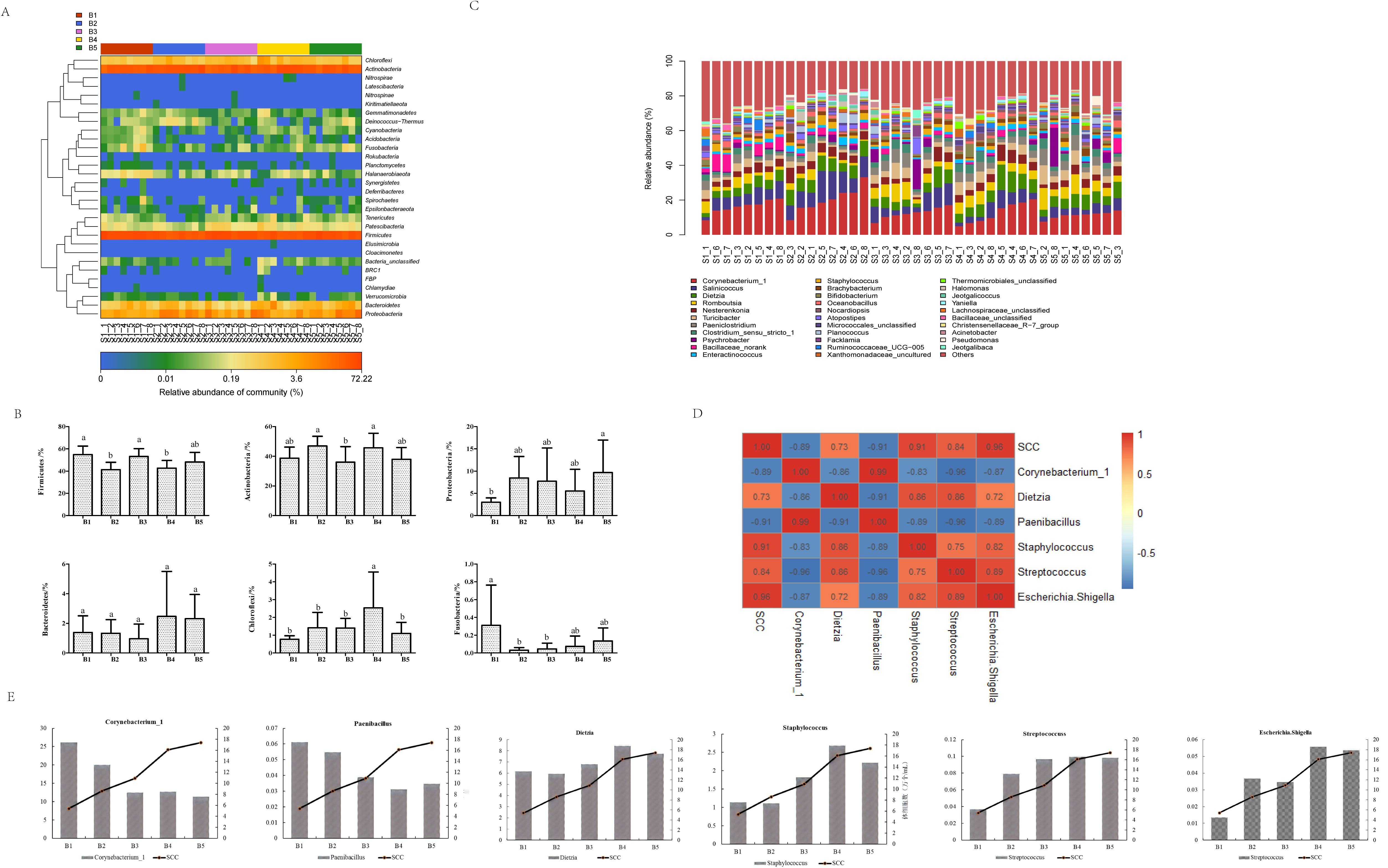
Figure 5. Characteristics of microbial community structure changes in dairy cow teat skin across different usage periods. (A) Distribution of microbial phyla on cow teat skin at different time points. S1, day 1; S2, day 7; S3, day 14; S4, day 21; S5, day 30; “_” indicates the sample number, the same below. Microbial community names are shown on the ordinate. (B) Changes in abundance of dominant bacteria in cow teat skin at different time points. (C) Distribution of cow teat skin microbiota at the genus level at different time points. (D) Correlation between teat microbiota and somatic cell counts in milk. (E) Trends in variation of microbial genera in teat skin and somatic cell counts.
3.3 Structural features of microbiota in milk at different time points of bedding usage
The sequencing results showed the presence of 8960 OTUs, 948 496–677 bases, and 2 129–466 sequences. The rank abundance curves (Figure 6A) and the dilution curves (Figure 6B) of the samples showed that the species composition was homogeneous, and the sequencing volume covered most microorganisms. The structure and composition of microbiota in the milk at different time points had common components, and there were also differences between different periods. The number of OTUs among the five time points was 811 (Figure 6C), accounting for 9% of the total number of OTUs. The numbers of unique OTUs in the milk were 755, 832, 870, 1,550, and 1,040 on the 1st (C1), 7th (C2), 14th (C3), 21st (C4), and 30th (C5) days of bedding usage, respectively. It can thus be seen that the composition of microbiota in milk peaked on day 21.
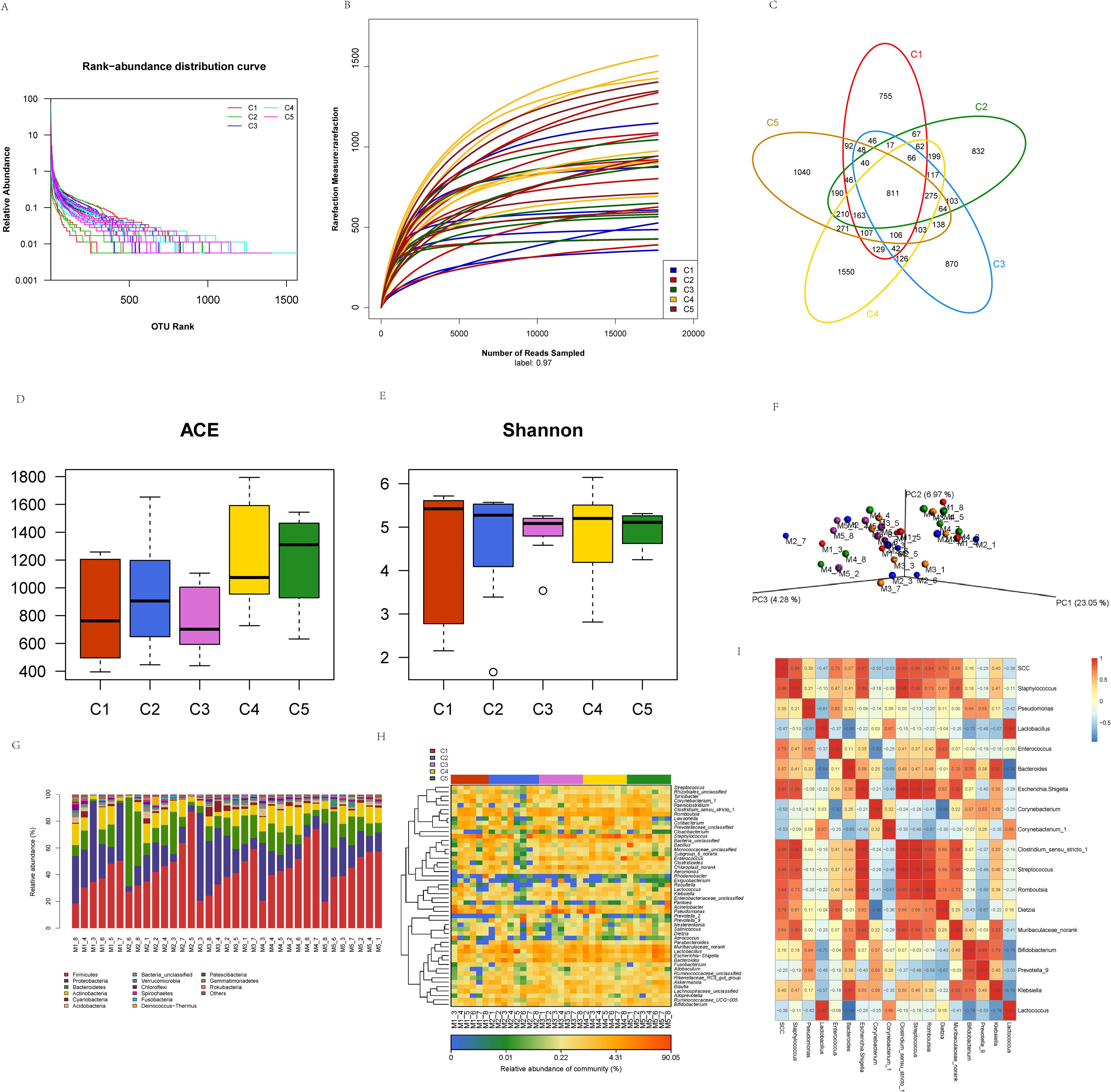
Figure 6. Characteristics of microbial diversity and community structure changes in milk across different usage periods. (A) Sample rank abundance curve. C1, day 1; C2, day 7; C3, day 14; C4, day 21; C5, day 30, the same below. (B) Sample rarefaction curves. (C) Distribution of microbial OTUs in milk at different time points. (D, E) Variations in alpha diversity in milk microbiota at different time points. (F) PCoA of milk microbiota at different time points. M1, day 1; M2, day 7’ M3, day 14; M4, day 21; M5, day 30; “_” indicates the sample number, the same below. (G) Distribution of milk microbiota at the phylum level at different time points. (H) Distribution of milk microbiota at the genus level at different time points. (I) Heatmap of milk microbes and somatic cell counts.
The ACE (Figure 6D), representing species richness, and Shannon (Figure 6E), representing species diversity, indices were determined. As the length of bedding usage increased, it was observed that microbial abundance in milk was highest on day 30, which was significantly higher than that on days 1, 7, and 14 (P<0.05). The microbial diversity was also highest on day 21, but the difference among the groups was not significant (P>0.05). All milk samples (5 time points) were analyzed by PCoA (Figure 6F), showing that the three principal components explained 23.05, 6.97, and 4.28% of the total variance, respectively. As indicated, there was no significant separation in microbial community structures and compositions among the different time points, and there was no significant difference in clustering, suggesting that the structure and composition of milk microbiota were similar among the different time points.
The microbial community structures in the milk at different time points of bedding usage varied, and the diversity and abundance of species also changed accordingly. At the phylum level (Figure 6G), the most common phyla were Firmicutes, Proteobacteria, and Bacteroidetes at the different time points. Statistical analysis of the relative abundance of the dominant phyla at different periods (Table 3) indicated that Firmicutes accounted for more than 30% of the phyla over the different time points, suggesting that Firmicutes was the most dominant phylum in the microbiota of milk samples. Proteobacteria was the second most dominant phylum in milk samples, with a larger proportion on day 1, while the abundance of Bacteroidetes decreased after day 7 following an initial increase, and Actinobacteria had a lower abundance than the other dominant phyla, although was more abundant on day 21. These results indicated that each sample had different distribution characteristics at the phylum level, and the four dominant phyla accounted for more than 80% of the total population. At different time points of bedding usage, the abundance of the top 50 genera in milk varied between different samples and time points (Figure 6H), with a greater abundance of Staphylococcus, Pseudomonas, Lactobacillus, Enterococcus, Acinetobacter, and Bacteroides. The abundance of Corynebacterium_1, Prevotella_9, Rhodanobacter, Raoultella, Lactococcus, Klebsiella, and Cloacibacterium, were also higher in some samples. The correlation between the milk microbes and SCC was investigated (Figure 6I), and the results revealed that SCC was negatively associated with the abundance of Lactococcus, Prevotella_9, Corynebacterium_1, Corynebacterium, and Lactobacillus, and positively related to the abundance of Staphylococcus, Klebsiella, Bifidobacterium, Muribaculaceae_norank, Dietzia, Romboutsia, Streptococcus, Clostridium_sensu_stricto_1, Escherichia. Shigella, Bacteroides, Enterococcus, and Pseudomonas.
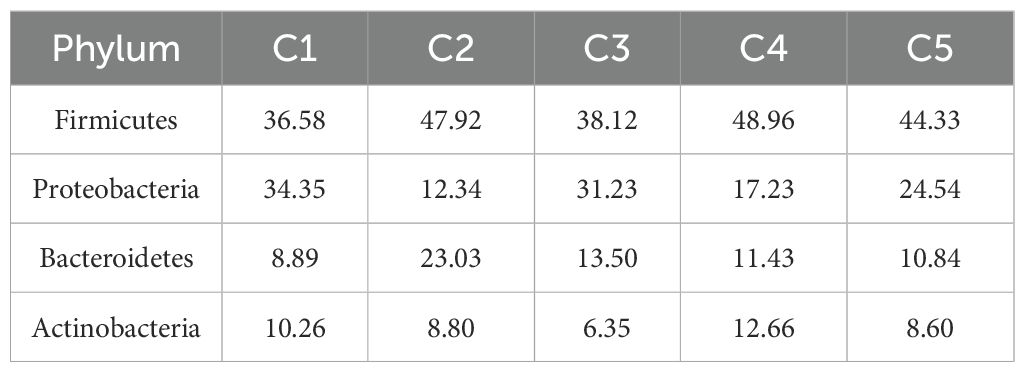
Table 3. The composition of milk microbial communities at the phylum level at different time points (%).
Differences in the microbial community compositions among different groups of milk samples were analyzed by linear discriminant analysis. At the genus level (Figure 7A), the differential microorganisms in milk on day 1 were Gracilibacter and Limnobacter. Day 7 showed the highest number of differential microorganisms compared to other periods, with Lachnospiraceae_AC2044_group, Lachnoclostridium, Prevotella_2, Eubacterium_ruminantium_group, Lachnospira, Tyzzerella, Yaniella, Coprococcus_2, Agathobacter, Butyricicoccus, Finegoldia, Sutterella, and Candidatus_Solibacter. On day 14, the differential microorganisms were Allorhizobium_Neorhizobium_Pararhizobium_Rhizobium, Pseudomonas, RBG_16_58_14, and Dysgonomonas, while on day 21, the differential microorganisms were Bhargavaea, Kytococcus, Adhaeribacter, Phormidium_MBIC10003, and Pseudogracilibacillus, and on day 30, Thermoactinomyces and Ruminobacter were found. Overall, the differential microorganisms in milk varied according to the duration of bedding usage. We will advance to functional validation in the subsequent phase.
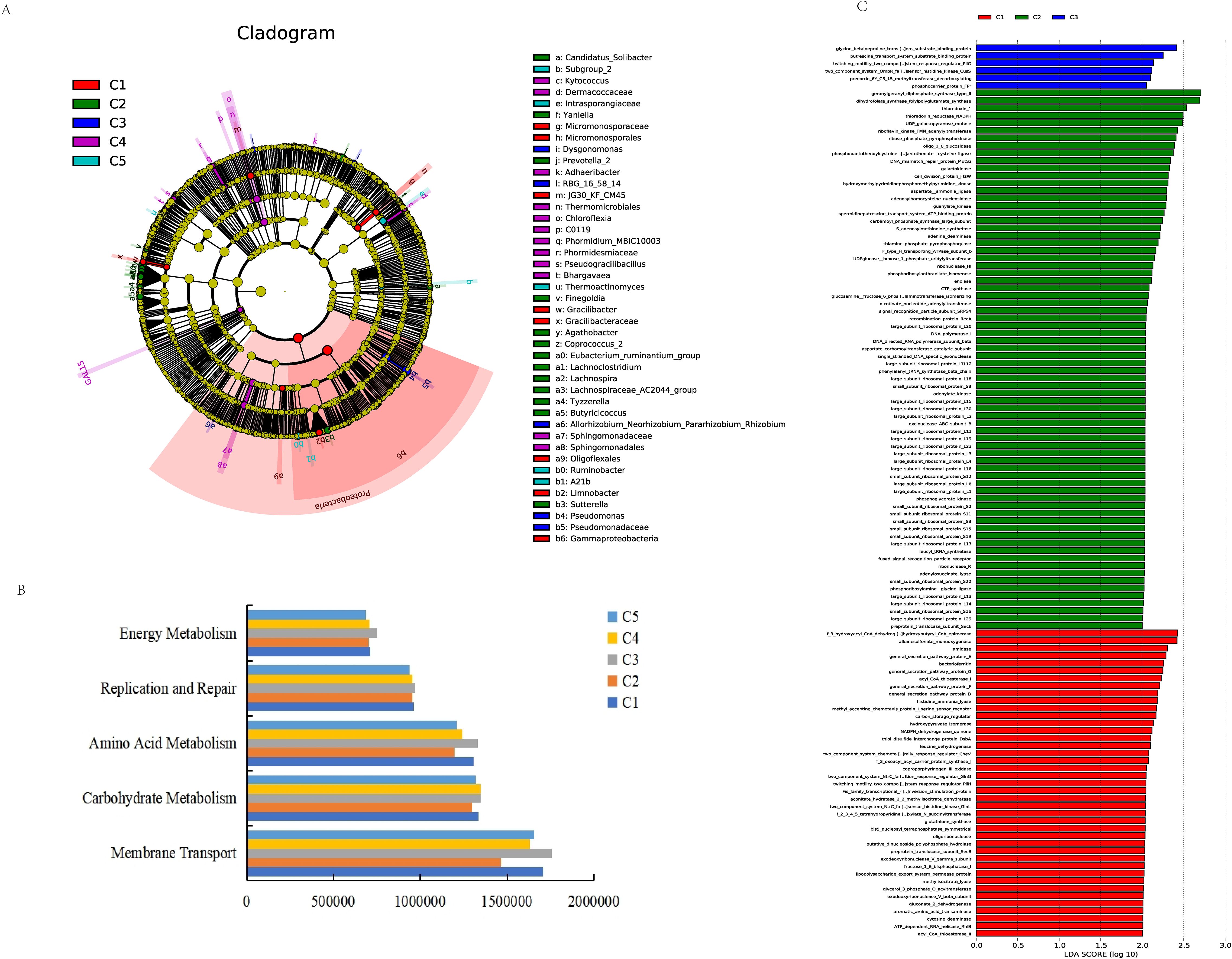
Figure 7. Changes in the functional profile of milk microbial communities across different usage periods. (A) Map showing the microorganisms present in milk at different time points. (B) The third-level KEGG analysis of milk samples at different time points. (C) KEGG pathway analysis of functional genes.
3.4 Functional analysis of milk microbiota at different time points
To evaluate the metabolic pathways associated with the milk microbiota at different time points of bedding usage, PICRUSt was used for functional prediction and KEGG annotation. This showed 41 metabolic pathways in the second level. A comparison of the metabolic pathways associated with microbiota in the bedding and teat skin samples identified five pathways in the three levels (Figure 7B), specifically associated with membrane transport, carbohydrate metabolism, amino acid metabolism, replication and repair, and energy metabolism. In the milk samples, these metabolic pathways were found to be more enriched on day 14, while the main functional genes of the third level were mainly enriched in the samples on days 1, 7, and 14 (Figure 7C). The pathways most enriched on days 1 and 7 were mostly related to protein secretion and DNA-associated proteins, and greater diversification in the enriched pathways was apparent as the bedding usage increased. The analysis indicated that the relative abundance of all metabolic pathways altered with the prolongation of bedding usage time.
4 Discussion
4.1 Microbial diversity at different time points of fermented cow manure bedding usage
The provision of clean, comfortable bedding is essential for the growth and welfare of dairy cattle. The use of manure for bedding can not only solve the problem of manure pollution on farms but also has advantages of resource utilization of manure and cost savings. After treatment, cow manure bedding is soft and comfortable, making it more convenient for subsequent manure treatment with sand bedding, resulting in less wear and reduced risk of pipeline clogging (Wu et al., 2021). Cow manure is also loose, does not clump easily, and is less costly and environmentally friendly compared to rubber bedding. Differences in the processes used for treating cow manure affect the community structures of its microbiota, and the number of bacteria in fermented cow manure is closely related to the degree of fermentation, fermentation time, and moisture content. The abundance and diversity of the microbiota in the fermented cow manure bedding were observed to increase as the duration of bedding usage increased, reaching its maximum abundance and diversity on the 14th day, with the community structures varying with the extent of bedding usage. The results of LEfSe analysis showed that the number of differential microorganisms in the bedding on day 30 was higher than that at the other time points, and that the majority were moderate Halocella. The microbiota composition was closely related to the saline alkaline environment of the region, and similar environmental conditions can lead to the development of microbiota with similar functions.
Firmicutes, Halanaerobiaeota, and Actinobacteria were found to be the dominant phyla, with their relative abundance varying according to the different bedding usage time points. The abundance of Firmicutes has been reported to be higher in the later stages of compost fermentation due to the thermotolerant and facultative anaerobic characteristics of the phylum (Tian et al., 2013; Zhang et al., 2014). The abundance of Firmicutes decreased as the bedding usage time increased, presumably because the fermented cow manure was in a loose state during usage, which is unfavorable to the growth of facultative anaerobic bacteria. These dominant phyla have different patterns of change in cow intestines during different growth periods, and remain relatively stable in the intestines in different growth periods (Dill-McFarland et al., 2017). The microbial composition of the fermented cow manure bedding resembled those of the gut microbiota and microbiota in fermented compost, varying in relative abundance according to the length of bedding usage. The abundance of Salinicoccus, Dietzia, and Staphylococcus tended to increase with prolongation of the bedding use duration. The comparison of differential microorganisms between different time points of bedding usage revealed that Corynebacterium_1, Ruminococcaceae_UCG_005, Novibacillus, and Planifilum were detected in the bedding, consistent with the findings of previous studies on the microbiota of the intestinal tract and during compost fermentation (Han et al., 2013; Yang et al., 2015; Song et al., 2017). All differential microorganisms were found to vary in abundance at the different bedding usage time points and tended to develop towards moderate Halocella. On the one hand, it may be due to the favorable conditions for microbial growth provided by the initially “clean” bedding, together with an accumulation of feces and urine during use, resulting in a differential microbial composition of intestinal, feces, and compost fermentation microorganisms at the beginning. On the other hand, the cattle farms in Xinjiang are located mostly in the Gobi area, characterized by a combination of salinity and desertification, intense salt accumulation, strong surface aggregation, and complex compositions of saline soils; thus, the microorganisms associated with the soil and water sources are mainly Halocella. With prolongation of bedding usage by the cow, the moderate Halocella gradually increased after prolonged exposure of the bedding to the environment.
4.2 Microbial diversity on cow teat skin in relation to different time points of bedding usage
From an ecological perspective, although each part of the cow’s body is characterized by a unique and specialized microbiome, the overall body can be considered a complex ecosystem (metacommunity), consisting of a variety of interrelated ecological microenvironments (Costello et al., 2009, Costello et al., 2012). In this study, the microbiota on teat skin had the highest number of OTUs on days 14 and 21, and their ACE and Simpson indices, representing abundance and diversity, respectively, increased with the prolongation of the bedding usage. The colonization and composition of microbiota on cow teat skin is not an independent process, as contact with the bedding and other parts of the cow’s environment alters the microbial compositions.
High-throughput sequencing and bioinformatics methods were used to analyze the influence of the different bedding usage time points on the structure of microbiota on cow teat skin, with the results showing that the dominant phyla of all samples at all five time points were Firmicutes, Actinobacteria, Proteobacteria, Bacteroidetes, and Chloroflexi. The abundance of Firmicutes and Actinobacteria accounted for more than 85% of the total abundance, which was consistent with the microbiota composition of Holstein cow teat skin reported by Braem et al. (2012). Firmicutes dominated all cow teat skin samples. Comparison with the dominant phyla of the fermented cow manure bedding showed that the dominant phyla of the teat skin samples were consistent with those in the bedding, and that the abundance of Proteobacteria, Bacteroidetes, and Chloroflexi reached a maximum on days 30 and 21, respectively, further demonstrating that microbial accumulation on cow teat skin was not an independent process, and that its abundance varied with the duration of bedding usage. Analysis of the teat skin microbiota at the genus level showed a predominance of Corynebacterium_1, Salinicoccus, Dietzia, Romboutsia, Nesterenkonia, and Turicibacter. These genera are found in a wide range of natural environments such as soils, deserts, and saline-alkaline zones, and even in the intestinal tracts of animals and composts of different materials. Based on the changes in microbiota composition at the phylum level, it was determined that the microbiota on the teat skin underwent significant community successional changes with extended bedding usage.
4.3 Effects of cow teat skin microbiota on SCC in milk
Based on the results of the high-throughput sequencing, the common bacteria present on the teat skin were compared with the milk SCC, showing that six genera showed marked abundance on day 21, among which Corynebacterium_1 was negatively correlated with the SCC. The effect of this bacterium on the milk microbiota requires further investigation. The abundance of Paenibacillus was negatively correlated with SCC, which is consistent with the findings of previous studies (Quigley et al., 2013; Oikonomou et al., 2014) and may contribute to the overall health of the cow. However, which Bacillus species are beneficial and their roles require further investigation. The abundance of Dietzia, Staphylococcus, Escherichia-Shigella, and Streptococcus increased with the duration of bedding usage and was positively correlated with SCC. These correlation results are consistent with the findings of Derakhshani et al. (2018). Dietzia can cause local and systemic infections in humans and animals (Koerner et al., 2009). We found that Dietzia on teat skin and SCC in the milk increased as the bedding usage increased. Competition exists between genera, and an increase or decrease in one microorganism can induce a change in the abundance of another microorganism. The development of mastitis is related to disordered microbiota, and the presence of a large number of symbiotic microbiota could modulate and reduce the susceptibility to mastitis. Thus, the effects of environmental factors on the health of cow mammary glands should be recognized.
4.4 Effects of fermented cow manure bedding usage on the diversity of microbiota in milk
Changes in the abundance and diversity of the milk microbiota occurred primarily on days 21 and 30, with the abundance reaching its highest level on day 21, although not changing significantly, consistent with the results of Cremonesi et al. (2018) who observed that the structure of microbiota in milk in the same species and under the same conditions was similar. We focused on a comprehensive investigation of the diversity, structure, and composition of the microbiota in milk throughout the entire usage cycle of fermented cow manure bedding. The dominant phyla in the milk were found to be Firmicutes, Proteobacteria, Bacteroidetes, and Actinobacteria, with the predominance of these phyla changing in correspondence with the length of bedding usage, consistent with the results of Pang et al. (2018) who evaluated milk microbiota using high-throughput sequencing. Greater abundance of Bacteroidetes was associated with lower humidity, while the abundance of Proteus was related to higher humidity. Therefore, variation in the abundance of the dominant milk phyla, as shown in the present study, was also influenced by environmental factors other than the duration of bedding usage.
At the genus level, the milk samples in the present study appeared to contain many of the same core genera as those reported in previous studies (Delbès et al., 2007; Rasolofo et al., 2010; Vacheyrou et al., 2011), including Staphylococcus, Pseudomonas, Lactobacillus, Enterococcus, Acinetobacter, and Bacteroides, all of which are frequently detected in healthy milk. Staphylococcus is a pathogen associated with mastitis, suggesting that cow teat skin is a source of microbes found in the milk.
The results of the high-throughput sequencing of milk samples from different time points, together with a comparison of the most common genera from cow teat skin and milk with SCC, indicated positive correlations between several genera and SCC. These were Streptococcus, Escherichia. Shigella, Klebsiella, Bacteroides, Enterococcus, Pseudomonas, and Dietzia. Of these, Escherichia-Shigella and Klebsiella are considered pathogens that cause cow mastitis, a disease resulting from pathogens in the digestive tract of the cow or its surrounding environment. Notably, Dietzia was found in the bedding, teat skin, and milk samples, and represented the genus with the highest abundance in both bedding and teat skin. Therefore, it can be assumed that mastitis-related bacterial pathogens colonize the mammary gland from the external environment, leading to increased SCC in the milk. Unlike mastitis-related bacterial pathogens, several microorganisms were negatively associated with SCC, and their presence laid the foundation for the microbiota balance in the milk; these included Lactococcus, Prevotella_9, Corynebacterium, and Lactobacillus. Corynebacterium was identified by Oultram et al. (2017) as a cow mastitis-associated pathogen, and an increase in other microorganisms may lead to a decline in its levels, creating a mutual inhibitory effect in the milk microbiota. Furthermore, the presence of a large number of symbiotic microbiota in milk could inhibit the growth of pathogenic bacteria, maintaining the milk microbiota in a relatively balanced state. Additionally, several individual samples were found to contain bacteria associated with cow intestines, compost fermentation, and the soil, demonstrating that the milk microbiota were derived not only from the teat skin of the cow but also from the intestine and environment. Therefore, it can be deduced that they also affect the composition of the microbiota in the mammary gland.
By comparing differential microorganisms in the milk at different time points of bedding usage, it was found that these microorganisms in milk were most abundant on day 7. The differential microorganisms found on day 1 showed a predominance of heat-tolerant anaerobes and aerobic heterotrophs compared with other time points, which may be related to the composting characteristics of fermented cow manure. On day 7, a predominance of rumen and gut microbiota was observed. On the 14th day, the differential bacteria were soil rhizobacteria and cow manure fermentation-associated bacteria. As the length of bedding usage increased, the differential microorganisms in the milk were found to be closer to the microorganisms observed in the soil on days 21 and 30. However, Kytococcus was found on day 21; these are a group of pathogenic bacteria that can cause pneumonia, bacteremia, and endocardial infection (Blennow et al., 2012). Additionally, the differential microorganisms in milk at time points showed a shift to microbiota such as initial-rumen or gut-soil.
PICRUSt was used primarily for analyzing high-throughput sequencing results for the prediction of the hypothetical metagenome and determining the potential functions of the microbiota in milk. The functional analysis of the predicted metagenome in this study showed that the most abundant functional categories included membrane transport pathways, carbohydrate metabolism, amino acid metabolism, replication and repair, and energy metabolism, consistent with the results of Lamendella et al. (2011), who examined the general metabolic functions (e.g., metabolism of amino acids, carbohydrates, and proteins) necessary for the survival of microorganisms, as well as with the results of Pannaraj et al. (2017) who evaluated the metagenomes of human milk. Lactose, the major carbohydrate in milk, is a potential carbon source for milk bacteria, and therefore carbohydrate metabolism is predicted to occur. Chen et al. (2018) also reported genes involved in replication and repair in milk in the intestines of piglets before weaning.
As the length of cow bedding usage increased, the abundance and diversity of the milk microbiota increased, reaching their highest levels on days 30 and 21, respectively. The dominant phyla in the milk microbiota were consistent with the bedding and teat skin results, with Staphylococcus, Pseudomonas, Lactobacillus, Enterococcus, Acinetobacter, and Bacteroides representing the core genera of the microbiota in milk. As the bedding usage duration increased, the content of mastitis-related bacterial pathogens and SCC in the milk increased, which further supported the results of the correlation between the teat microbiota and SCC. LEfSe analysis and the prediction of microbial functions showed that differential microorganisms in milk and their associated metabolic pathways altered with the duration of bedding usage. In this study, greater enrichment was found on day 14, and the metabolic pathways in the third level became more diversified as the duration of bedding usage increased, which may have been due to a combination of extended bedding usage, environmental factors, decreased host resistance, and other factors. The microenvironment of the cow mammary gland changed, resulting in a counter-environment for the growth of microbiota in milk, promoting an increase in pathways associated with substance transport and metabolism in the microorganisms, resulting in a wider range of metabolic pathways. It can thus be inferred that the duration of bedding usage affects the microbial functions of the second and third levels, and these functional differences provide further clues for the study of microbiota in milk and mammary gland health.
5 Conclusions
As the length of usage of fermented cow manure bedding increased, the abundance of mastitis-related bacterial pathogens (e.g., Staphylococcus, Escherichia, Klebsiella, Streptococcus, and Dietzia) in the milk and cow teat skin increased, together with increased SCC in the milk. The structures and metabolic functions of the microbiota associated with the bedding and milk changed as the length of bedding usage increased. Based on the abundance and diversity of microbiota in fermented cow manure bedding, teat skin, and milk samples, combined with the comprehensive analysis of the abundance of pathogenic bacteria, it is recommended that fermented cow manure bedding should be replaced in cycles of no more than 14 days.
Data availability statement
The data presented in this study are deposited in the NCBI database, accession number PRJNA1282870.
Ethics statement
The animal study was approved by Animal Care and Use Committee of Xinjiang Agricultural University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
DW: Methodology, Conceptualization, Writing – review & editing, Writing – original draft, Software, Visualization. ZY: Software, Conceptualization, Writing – original draft, Methodology. MY: Visualization, Conceptualization, Methodology, Writing – review & editing, Writing – original draft, Software. XW: Supervision, Investigation, Resources, Writing – review & editing. JG: Resources, Writing – review & editing, Investigation, Supervision. MZ: Supervision, Writing – review & editing, Data curation, Formal analysis. LX: Writing – review & editing, Investigation, Formal analysis, Data curation. SM: Software, Formal analysis, Writing – review & editing, Investigation. MD: Resources, Investigation, Formal analysis, Writing – review & editing, Supervision. XH: Supervision, Writing – review & editing, Project administration, Data curation, Methodology, Conceptualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by The National Natural Science Foundation of China (31860630). The funders played no role in study design, collection, analysis, data interpretation, manuscript writing, or decision to submit the manuscript for publication.
Acknowledgments
We would like to acknowledge and thank two breeding herds for their support and help, they are, Xinjinag Hutubi Cattle Farm, Xinjiang Tianshan Animal Husbandry and Bioengineering Co. We would like to thank the College of Animal Science, Xinjiang Agricultural University. At the same time, We would also like to thank every member who participated in the collection and collation of samples and data, sample extraction, and amplification.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fanim.2025.1614022/full#supplementary-material
References
Albino R. L., Taraba J. L., Marcondes M. I., Eckelkamp E. A., and Bewley J. M. (2017). Comparison of bacterial populations in bedding material, on teat ends, and in milk of cows housed in compost bedded pack barns. Anim. Prod. Sci. 58, 1686. doi: 10.1071/AN16308
Blennow O., Westling K., Fröding I., and Özenci V. (2012). Pneumonia and bacteremia due to kytococcus schroeteri. J. Clin. Microbiol. 50, 522–524. doi: 10.1128/JCM.01245-11
Braem G., De Vliegher S., Verbist B., Heyndrickx M., Leroy F., and De Vuyst L. (2012). Culture-independent exploration of the teat apex microbiota of dairy cows reveals a wide bacterial species diversity. Vet. Microbiol. 157, 383–390. doi: 10.1016/j.vetmic.2011.12.031
Calamari L., Calegari F., and Stefanini L. (2009). Effect of different free stall surfaces on behavioral, productive and metabolic parameters in dairy cows. Appl. Anim. Behav. Sci. 120, 9–17. doi: 10.1016/j.applanim.2009.05.013
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., and Holmes S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chen X., Xu J., Ren E., Su Y., and Zhu W. (2018). Co-occurrence of early gut colonization in neonatal piglets with microbiota in the maternal and surrounding delivery environments. Anaerobe 49, 30–40. doi: 10.1016/j.anaerobe.2017.12.002
Cole K. J. and Hogan J. S. (2016). Short communication: Environmental mastitis pathogen counts in free stalls bedded with composted and fresh recycled manure solids. J. Dairy Sci. 99, 1501–1505. doi: 10.3168/jds.2015-10238
Cook N. B. and Nordlund K. V. (2009). The influence of the environment on dairy cow behavior, claw health and herd lameness dynamics. Vet. J. 179, 360–369. doi: 10.1016/j.tvjl.2007.09.016
Costello E. K., Lauber C. L., Hamady M., Fierer N., Gordon J. I., and Knight R. (2009). Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697. doi: 10.1126/science.1177486
Costello E. K., Stagaman K., Dethlefsen L., Bohannan B. J. M., and Relman D. A. (2012). The application of ecological theory toward an understanding of the human microbiome. Science 336, 1255–1262. doi: 10.1126/science.1224203
Cremonesi P., Ceccarani C., Curone G., Severgnini M., Pollera C., Bronzo V., et al. (2018). Milk microbiome diversity and bacterial group prevalence in a comparison between healthy Holstein Friesian and Rendena cows. PloS One 13, 0205054. doi: 10.1371/journal.pone.0205054
Delbès C., Ali-Mandjee L., and Montel M.-C. (2007). Monitoring bacterial communities in raw milk and cheese by culture-dependent and -independent 16S rRNA gene-based analyses. Appl. Environ. Microbiol. 73, 1882–1891. doi: 10.1128/AEM.01716-06
Derakhshani H., Fehr K. B., Sepehri S., Francoz D., De Buck J., Barkema H. W., et al. (2018). Invited review: Microbiota of the bovine udder: Contributing factors and potential implications for udder health and mastitis susceptibility. J. Dairy Sci. 101, 10605–10625. doi: 10.3168/jds.2018-14860
Dill-McFarland K. A., Breaker J. D., and Suen G. (2017). Microbial succession in the gastrointestinal tract of dairy cows from 2 weeks to first lactation. Sci. Rep. 7, 40864. doi: 10.1038/srep40864
Esteban-Blanco C., Gutiérrez-Gil B., Marina H., Pelayo R., Suárez-Vega A., Acedo A., et al. (2020). The milk microbiota of the spanish churra sheep breed: new insights into the complexity of the milk microbiome of dairy species. Animals 10, 1463. doi: 10.3390/ani10091463
Fregonesi J. A., Veira D. M., Von Keyserlingk M. A. G., and Weary D. M. (2007). Effects of bedding quality on lying behavior of dairy cows. J. Dairy Sci. 90, 5468–5472. doi: 10.3168/jds.2007-0494
Haley D. B., De Passillé A. M., and Rushen J. (2001). Assessing cow comfort: effects of two floor types and two tie stall designs on the behavior of lactating dairy cows. Appl. Anim. Behav. Sci. 71, 105–117. doi: 10.1016/S0168-1591(00)00175-1
Han S.-I., Lee J.-C., Lee H.-J., and Whang K.-S. (2013). Planifilum composti sp. nov., a thermophile isolated from compost. Int. J. Syst. Evol. Microbiol. 63, 4557–4561. doi: 10.1099/ijs.0.053199-0
Hantsis-Zacharov E. and Halpern M. (2007). Culturable psychrotrophic bacterial communities in raw milk and their proteolytic and lipolytic traits. Appl. Environ. Microbiol. 73, 7162–7168. doi: 10.1128/AEM.00866-07
Jensen M. B., Pedersen L. J., and Munksgaard L. (2005). The effect of reward duration on demand functions for rest in dairy heifers and lying requirements as measured by demand functions. Appl. Anim. Behav. Sci. 90, 207–217. doi: 10.1016/j.applanim.2004.08.006
Koerner R. J., Goodfellow M., and Jones A. L. (2009). The genus Dietzia: a new home for some known and emerging opportunist pathogens. FEMS Immunol. Med. Microbiol. 55, 296–305. doi: 10.1111/j.1574-695X.2008.00513.x
Lamendella R., Santo Domingo J. W., Ghosh S., Martinson J., and Oerther D. B. (2011). Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol. 11, 103. doi: 10.1186/1471-2180-11-103
LeJeune J. T. and Rajala-Schultz P. J. (2009). Unpasteurized milk: a continued public health threat. Clin. Infect. Dis. 48, 93–100. doi: 10.1086/595007
Li H., Wang X., Wu Y., Zhang D., Xu H., Xu H., et al. (2021). Relationships among bedding materials, bedding bacterial composition and lameness in dairy cows. Anim. Biosci. 34, 1559–1568. doi: 10.5713/ajas.20.0565
Meng Q., Yang W., Men M., Bello A., Xu X., Xu B., et al. (2019). Microbial community succession and response to environmental variables during cow manure and corn straw composting. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.00529
Metcalf J. A., Roberts S. J., and Sutton J. D. (1992). Variations in blood flow to and from the bovine mammary gland measured using transit time ultrasound and dye dilution. Res. Vet. Sci. 53, 59–63. doi: 10.1016/0034-5288(92)90085-G
Monsallier F., Verdier-Metz I., Agabriel C., Martin B., and Montel M.-C. (2012). Variability of microbial teat skin flora in relation to farming practices and individual dairy cow characteristics. Dairy Sci. Technol. 92, 265–278. doi: 10.1007/s13594-012-0064-7
Montel M.-C., Buchin S., Mallet A., Delbes-Paus C., Vuitton D. A., Desmasures N., et al. (2014). Traditional cheeses: Rich and diverse microbiota with associated benefits. Int. J. Food Microbiol. 177, 136–154. doi: 10.1016/j.ijfoodmicro.2014.02.019
Niu K., Chao C., Zhang X., An Z., Zhou J., and Yang L. (2022). Effects of different microbial agents on bedding treatment of ectopic fermentation of buffalo manure. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.1080650
Norring M., Manninen E., De Passillé A. M., Rushen J., and Saloniemi H. (2010). Preferences of dairy cows for three stall surface materials with small amounts of bedding. J. Dairy Sci. 93, 70–74. doi: 10.3168/jds.2009-2164
Norring M., Manninen E., de Passillé A. M., Rushen J., Munksgaard L., and Saloniemi H. (2008). Effects of sand and straw bedding on the lying behavior, cleanliness, and hoof and hock injuries of dairy cows. J. Dairy Sci. 91, 570–576. doi: 10.3168/jds.2007-0452
Oikonomou G., Bicalho M. L., Meira E., Rossi R. E., Foditsch C., MaChado V. S., et al. (2014). Microbiota of cow’s milk; distinguishing healthy, sub-clinically and clinically diseased quarters. PloS One 9, e85904. doi: 10.1371/journal.pone.0085904
Oultram J. W. H., Ganda E. K., Boulding S. C., Bicalho R. C., and Oikonomou G. (2017). A metataxonomic approach could be considered for cattle clinical mastitis diagnostics. Front. Vet. Sci. 4. doi: 10.3389/fvets.2017.00036
Pang M., Xie X., Bao H., Sun L., He T., Zhao H., et al. (2018). Insights into the bovine milk microbiota in dairy farms with different incidence rates of subclinical mastitis. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.02379
Pannaraj P. S., Li F., Cerini C., Bender J. M., Yang S., Rollie A., et al. (2017). Association between breast milk bacterial communities and establishment and development of the infant gut microbiome. JAMA Pediatr. 171, 647. doi: 10.1001/jamapediatrics.2017.0378
Quigley L., O’Sullivan O., Stanton C., Beresford T. P., Ross R. P., Fitzgerald G. F., et al. (2013). The complex microbiota of raw milk. FEMS Microbiol. Rev. 37, 664–698. doi: 10.1111/1574-6976.12030
Rasolofo E. A., St-Gelais D., LaPointe G., and Roy D. (2010). Molecular analysis of bacterial population structure and dynamics during cold storage of untreated and treated milk. Int. J. Food Microbiol. 138, 108–118. doi: 10.1016/j.ijfoodmicro.2010.01.008
Rowbotham R. F. and Ruegg P. L. (2015). Association of bedding types with management practices and indicators of milk quality on larger wisconsin dairy farms. J. Dairy Sci. 98, 7865–7885. doi: 10.3168/jds.2015-9866
Rowbotham R. F. and Ruegg P. L. (2016). Bacterial counts on teat skin and in new sand, recycled sand, and recycled manure solids used as bedding in freestalls. J. Dairy Sci. 99, 6594–6608. doi: 10.3168/jds.2015-10674
Rushen J., Haley D., and De Passillé A. M. (2007). Effect of softer flooring in tie stalls on resting behavior and leg injuries of lactating cows. J. Dairy Sci. 90, 3647–3651. doi: 10.3168/jds.2006-463
Song W., Wang X., Gu J., Zhang S., Yin Y., Li Y., et al. (2017). Effects of different swine manure to wheat straw ratios on antibiotic resistance genes and the microbial community structure during anaerobic digestion. Bioresource Technol. 231, 1–8. doi: 10.1016/j.biortech.2017.01.054
Sorter D. E., Kester H. J., and Hogan J. S. (2014). Short communication: bacterial counts in recycled manure solids bedding replaced daily or deep packed in freestalls. J. Dairy Sci. 97, 2965–2968. doi: 10.3168/jds.2013-7814
Tian W., Sun Q., Xu D., Zhang Z., Chen D., Li C., et al. (2013). Succession of bacterial communities during composting process as detected by 16S rRNA clone libraries analysis. Int. Biodeter. Biodegr 78, 58–66. doi: 10.1016/j.ibiod.2012.12.008
Tucker C. B. and Weary D. M. (2004). Bedding on geotextile mattresses: how much is needed to improve cow comfort? J. Dairy Sci. 87, 2889–2895. doi: 10.3168/jds.S0022-0302(04)73419-0
Vacheyrou M., Normand A.-C., Guyot P., Cassagne C., Piarroux R., and Bouton Y. (2011). Cultivable microbial communities in raw cow milk and potential transfers from stabl es of sixteen french farms. Int. J. Food Microbiol. 146, 253–262. doi: 10.1016/j.ijfoodmicro.2011.02.033
Verdier-Metz I., Gagne G., Bornes S., Monsallier F., Veisseire P., Delbès-Paus C., et al. (2012). Cow teat skin, a potential source of diverse microbial populations for cheese production. Appl. Environ. Microbiol. 78, 326–333. doi: 10.1128/AEM.06229-11
Verdier-Metz I., Michel V., Delbès C., and Montel M.-C. (2009). Do milking practices influence the bacterial diversity of raw milk? Food Microbiol. 26, 305–310. doi: 10.1016/j.fm.2008.12.005
Wu H., Wang Y., Dong L., Hu H., Meng L., Liu H., et al. (2021). Microbial characteristics and safety of dairy manure ComPosting for reuse as dairy bedding. Biology 10, 13. doi: 10.3390/biology10010013
Yang G., Chen J., and Zhou S. (2015). Novibacillus thermophilus gen. nov., sp. nov., a gram-staining-negative and moderately thermophilic member of the family thermoactinomycetaceae. Int. J. Syst. Evol. Microbiol. 65, 2591–2597. doi: 10.1099/ijs.0.000306
Zdanowicz M., Shelford J. A., Tucker C. B., Weary D. M., and M. a. G. (2004). Bacterial populations on teat ends of dairy cows housed in free stalls and bedded with either sand or sawdust. J. Dairy Sci. 87, 1694–1701. doi: 10.3168/jds.S0022-0302(04)73322-6
Keywords: fermented manure bedding, teat skin, milk, 16S, somatic cell count
Citation: Wang D, You Z, Yan M, Wang X, Ge J, Zhang M, Xu L, Ma S, Dong M and Huang X (2025) Dynamics of microbial diversity in cow bedding composed of fermented manure and used for different times and their effects on milk quality. Front. Anim. Sci. 6:1614022. doi: 10.3389/fanim.2025.1614022
Received: 18 April 2025; Accepted: 09 June 2025;
Published: 09 July 2025.
Edited by:
Mohan Mondal, ICAR-National Dairy Research Institute, Eastern Regional Station, Kalyani, IndiaReviewed by:
Medhavi Sudarshan, Patliputra University, IndiaAtakan Koç, Aydin Adnan Menderes University, Türkiye
Copyright © 2025 Wang, You, Yan, Wang, Ge, Zhang, Xu, Ma, Dong and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xixia Huang, YXUtaHVhbmd4aXhpYUAxNjMuY29t
†These authors have contributed equally to the work