 Jian Zhang1,2,3
Jian Zhang1,2,3 Mingxuan Zhao1,2,3Hongliang Zhang1,2,3Mengjia Han1,2,3Zhankun Tan1,2,3Peng Shang1,2,3*
Mingxuan Zhao1,2,3Hongliang Zhang1,2,3Mengjia Han1,2,3Zhankun Tan1,2,3Peng Shang1,2,3*- 1College of Animal Science, Xizang Agriculture and Animal Husbandry University, Linzhi, Xizang, China
- 2Key Laboratory of Tibetan Pig Genetic Improvement and Reproduction Engineering, Xizang Agriculture and Animal Husbandry University, Linzhi, Xizang, China
- 3Tibetan Pig Science and Technology Courtyard in Nyingchi, Key Laboratory of Tibetan Pig Genetic Improvement and Reproduction Engineering, Xizang Agriculture and Animal Husbandry University, Linzhi, Xizang, China
Weaned piglets are highly stress-vulnerable, with reduced immunity. Conventional use of antibiotics to prevent diarrhea and boost growth carries risks: bacterial resistance, drug residues, and intestinal flora imbalance, threatening food safety and public health. Meanwhile, demand for antibiotic-free livestock products is rising. Many countries have adopted policies restricting or banning antibiotics. Thus, developing safe, effective antibiotic alternatives is vital—to ensure piglet health, align with the industry’s green development, and meet market needs. This study experiment used a fecal microbiota transplantation method on weaned piglets and systematically evaluated the regulatory effects of fecal microbiota transplantation (FMT) on gut microbiota and host metabolism in weaned Tibetan piglets by integrating 16S rRNA sequencing and metabolomics, with experimental groups including a basal diet (Nor), lincomycin-supplemented (Ant), and FMT-supplemented (Fec) interventions. FMT significantly enhanced gut microbiota alpha diversity (Shannon index), enriching beneficial genera (e.g., Lactobacillus, Prevotella) and functional taxa (e.g., Eubacterium hallii group) to establish a core microbiota dominated by short-chain fatty acid producers and fiber-degrading bacteria, while antibiotics reduced Firmicutes abundance (p < 0.05) and promoted Proteobacteria proliferation. Metabolically, FMT activated tryptophan pathways (e.g., anti-inflammatory 5-hydroxyindole) and bile secretion (ko04976), whereas antibiotics suppressed amino acid metabolism (e.g., N-acetylglycine) and triggered oxidative stress (MAPK signaling). Notably, Streptococcus exhibited dual metabolic roles, positively correlating with phytoestrogens (R-equol) and negatively with biogenic amines (tyramine), highlighting its niche-specific regulatory potential. By reconstructing functional microbiota (e.g., Christensenellaceae) and metabolic networks (tryptophan/riboflavin pathways), FMT achieved comparable growth performance to antibiotics while mitigating dysbiosis and metabolic disturbances. These findings elucidate FMT’s mechanism in alleviating weaning stress through targeted enrichment of fiber-degrading and anti-inflammatory microbiota, coupled with metabolic synergy, thereby validating its feasibility as a non-antibiotic strategy and providing a theoretical framework for precision gut microbiota modulation in sustainable livestock production.
1 Introduction
Weaning represents one of the most critical physiological challenges in pig production, profoundly impacting piglet health and farm profitability (Liu et al., 2023; Perez-Palencia et al., 2022; Winters et al., 2023). During this phase, abrupt separation from the sow, a rapid dietary transition from nutrient-rich liquid milk to solid feed, and environmental stressors (e.g., regrouping, handler changes, and novel housing conditions) collectively induce multisystem stress (Camerlink et al., 2021; Kerschaver et al., 2023). Compounded by immature immune and digestive systems in piglets, these stressors synergistically contribute to the development of “post-weaning stress syndrome,” characterized by severe diarrhea, weight loss, growth retardation, reduced survival rates, and compromised disease resistance (Moeser et al., 2017; Dang et al., 2022). Such outcomes result in significant economic losses in intensive pig farming, underscoring the urgent need for effective antibiotic alternatives (Gomes et al., 2022; Xu et al., 2021; Ali et al., 2023). Emerging evidence suggests that fecal microbiota transplantation (FMT), which modulates gut microbiota composition, inhibits pathogenic microbes, and enhances feed intake, growth, and immune function, holds promise as a novel antibiotic substitute (Quanhang and Jian, 2019; Su et al., 2021).
FMT involves transferring fecal microbial communities from healthy donors to recipients to reshape gut microbiota. Initially applied in human medicine in 1958 by Eiseman and colleagues to treat pseudomembranous colitis, FMT has gained traction in veterinary research due to its potential to restore gut microbiota (Cold et al., 2021). Given the anatomical, physiological, and microbial similarities between pigs and humans, FMT strategies developed for humans may also benefit swine (Niederwerder et al., 2018). Studies in early-weaned piglets demonstrated that daily FMT administration significantly reduced diarrhea incidence, improved average daily gain (ADG), and enhanced intestinal barrier integrity and immune responses (Oladele et al., 2024; Mooyottu et al., 2025). While FMT is known to regulate host physiology via microbiota remodeling, its precise mechanisms require systematic investigation using multi-omics approaches.
16S rRNA sequencing enables precise tracking of microbial community dynamics post-FMT, such as sustained enrichment of Lactobacillus, highlighting FMT’s ability to selectively promote beneficial taxa (Ohara, 2019; Wang et al., 2021). Concurrently, metabolomics identifies downstream microbial–host interactions, including activation of tryptophan metabolism and accumulation of anti-inflammatory metabolites (e.g., 3-hydroxyanthranilic acid), elucidating how FMT enhances intestinal barrier function and nutrient utilization (Song et al., 2024; Zheng et al., 2022). Integrating these approaches not only confirms FMT’s structural optimization of gut microbiota but also links specific microbial taxa (e.g., Lactobacillus) to critical metabolic pathways, revealing a dual “microbiota remodeling–metabolic regulation” mechanism (Song et al., 2024). This strategy mitigates weaning stress with efficacy comparable to antibiotics while avoiding antimicrobial resistance risks, offering a comprehensive framework to decipher FMT’s biological mechanisms and advance targeted microbial therapies.
In this study, we systematically evaluated the effects of FMT gut microbiota in weaned Tibetan piglets by comparing fecal supernatant (Fec group), lincomycin (Ant group), and basal diet (Nor group) interventions. Using 16S rRNA sequencing and metabolomics, we aimed to quantify FMT’s impact on diarrhea reduction, feed efficiency, and ADG; characterize FMT-driven microbial community restructuring and its interaction with host metabolism; and elucidate the “microbiota–metabolite” axis underlying FMT-mediated intestinal health. Unlike previous single-omics studies, our integrated multi-omics approach not only validated FMT’s growth-promoting efficacy comparable to antibiotics but also uncovered its unique metabolic advantages, providing a theoretical basis for non-antibiotic microbial interventions. These findings offer scientific insights for improving health management in indigenous pig breeds and inform the development of precision nutritional strategies to optimize antibiotic alternatives in livestock production.
2 Materials and methods
2.1 Animal selection, experimental design, and diet setting
Animal experiments were conducted in strict accordance with the ethical requirements of the Laboratory Animal Ethics Committee of Tibet College of Agriculture and Animal Husbandry, China (License No. XZA-2005-012). The study was conducted in accordance with local laws and institutional requirements.
In the present study, 21 weaned Tibetan piglets (initial body weight 5.4 ± 0.2 kg) at 30 days of age were selected. These weaned Tibetan piglets are all from the Tibetan pig breeding base in Zengba Village, and the weaned piglets were clinically healthy, with normal feed intake and feces. They were tested to confirm that they were free of contagious diseases such as swine fever, blue ear disease, and other pathogens such as Salmonella and Escherichia coli. Furthermore, it was ascertained that the piglets had not been treated with any antibiotics or antimicrobial drugs in the past 30 days. The animals were divided into three experimental groups by randomization: the basal diet group (Nor), the lincomycin-treated group (Ant), and the fecal microbial extract-added group (Fec). The control group (Nor) was fed the basal diet, while the Fec and Ant groups were supplemented with 2,000 mL/kg of fecal bacterial extracts and 1,000 mg/kg of lincomycin hydrochloride, respectively, to the basal diet. The nutritional composition of the experimental diets is shown in Table 1 and was formulated according to the National Research Council (NRC) standards to meet or exceed the nutritional requirements for a 30-day trial period.
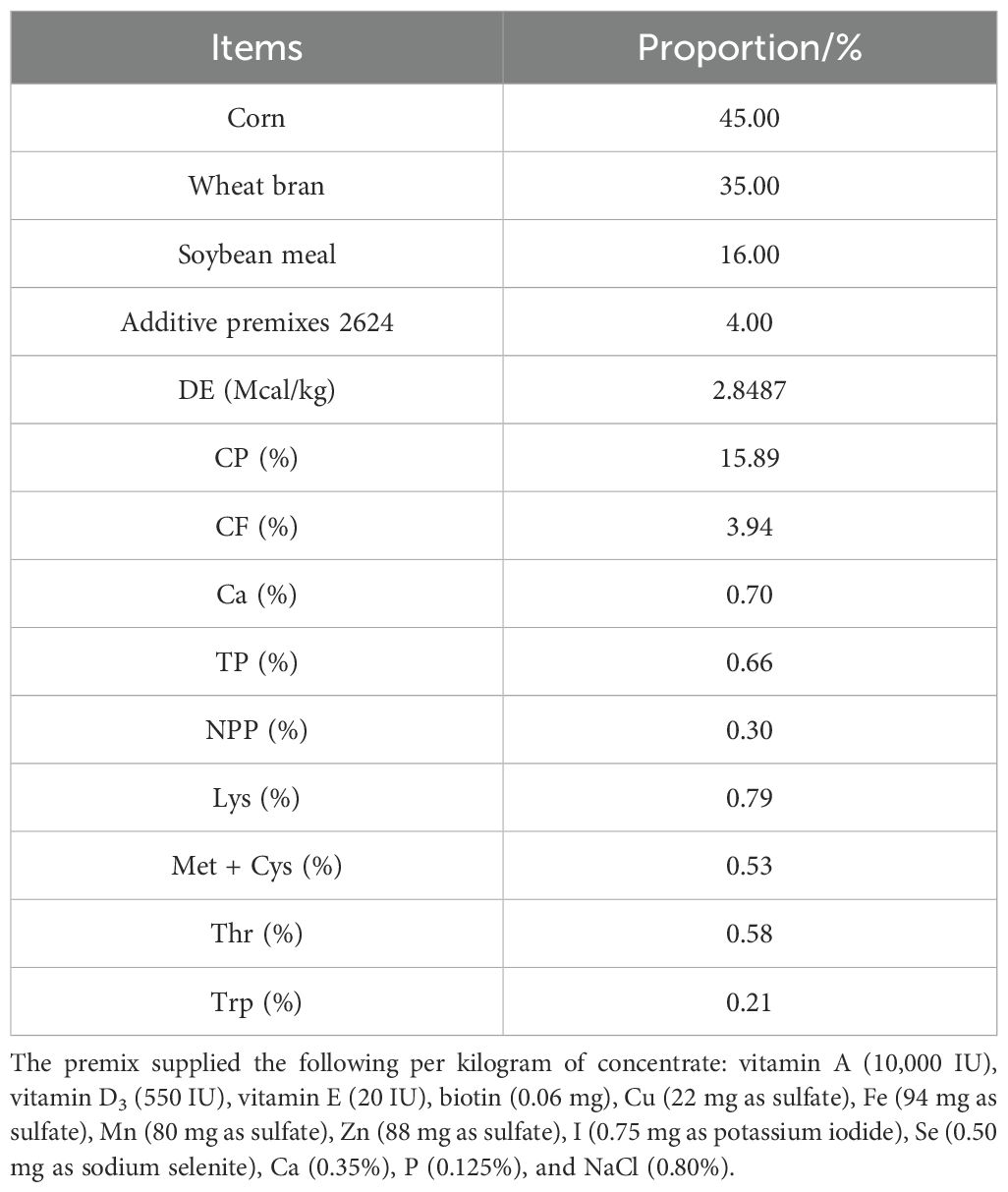
Table 1. Feed formulation and nutrient composition of diets.
2.2 Fecal suspension preparation
A total of 10 g of fresh feces was weighed, added with 40 mL of sterile PBS (0.1 mol/L, pH = 7.2) (feces-water ratio of 1:4), placed in a sterile beaker, stirred with a magnetic stirrer at a low speed (100–200 rpm) for 5 min, and mixed well to a paste. The suspension was passed through 2.0, 1.0, and 0.5 mm sterile stainless steel sieves to remove impurities such as crude fiber and fecal residue, and the filtrate was collected. The filtrate was transferred to a 50-mL sterile centrifuge tube and centrifuged at 200×g for 10 min at 4°C. The precipitate was discarded (residual impurities and host cells were removed), while the supernatant was retained. The supernatant was centrifuged at 3,000×g for 15 min at 4°C and discarded; the precipitate was retained. Twenty milliliters of sterile PBS was added to the precipitate, which was resuspended using a vortex shaker and then centrifuged at 3,000×g for 10 min at 4°C. This cycle was repeated twice to obtain the purified bacterial sludge. The precipitate was resuspended with sterile PBS solution, and 25% glycerol was added to prepare a fecal suspension. Sterile centrifuge tubes were dispensed and stored frozen at −80°C, and a 37°C water bath was used for 1 h before use. A fecal suspension with a bacterial load of 5 × 108 cfu/mL was prepared at the time of use.
2.3 Sample collection and processing
Fresh fecal samples were obtained from the piglets on days 0 and 30 of the experiment using rectal massage. Animals were fasted for 8 h prior to sampling in order to standardize intestinal contents while maintaining free access to water in order to reduce stress. This sampling method ensures aseptic operation and circumvents environmental contamination. The fecal samples were subjected to freeze-drying and pulverization (60 Hz, 30 s), and a 50-mg sample was placed in a centrifuge tube. Subsequently, 700 µL of precooled (−40°C) extract (methanol–water, 3:1 v/v, containing internal standard) was added, vortexed, and mixed for 30 s. The sample was then homogenized at 35 Hz for 4 min and sonicated in an ice-water bath for 5 min. The above homogenization–sonication steps were then repeated on three occasions. Subsequently, the samples were left to stand at 4°C in a homogenizer overnight. Following centrifugation at 4°C for 15 min at 12,000 rpm (centrifugal force 13,800×g, radius 8.6 cm), the resultant filtrate was filtered through a 0.22-µm microporous membrane and then diluted fivefold with the extract. The mixture was then vortexed for 30 s and combined into a quality control (QC) sample of 40 µL per sample. This sample was stored at −80°C for subsequent testing.
2.4 Liquid chromatography-mass spectrometry analysis
The target compounds were separated using a Waters UPLC column coupled with an EXIONLC system (SCIEX) ultra performance liquid chromatograph (UPLC). The acquisition and quantitative analysis of target compounds were performed using the SCIEX Analyst WorkStation software (v1.6.3). The raw mass spectrometry data were converted to TXT format by the MS converter software (v16.2.0.9), and then the peaks were extracted and annotated by using a self-developed R package in combination with a self-constructed database. Finally, orthogonal partial least squares discriminant analysis (OPLS-DA) was carried out. The validity of the model was evaluated by the R²Y and Q² parameters, and the metabolites were screened for differences based on the projected importance values (VIP > 1) of the variables analyzed by OPLS-DA between groups and the p-values of univariate tests. In order to enhance the metabolite coverage and detection efficacy, dual ionization modes of positive (POS) and negative (NEG) ions were utilized for metabolite detection, with the data from these two modes subsequently integrated for analysis using OPLS-DA.
Alpha diversity is defined as the abundance and isolation of species in a given habitat. The Chao1 and ACE indices were utilized to evaluate the species richness of the samples, with higher values denoting more significant diversity. The Simpson and Shannon indices were utilized to integrate species richness and evenness. The beta diversity analysis was employed to compare the species diversity among samples. This analysis reflected the distance relationship between samples and revealed the degree of differentiation of bacterial colonies. By quantifying the differences in colony composition among Nor, Ant, and Fec samples, the relative values of distances among samples were calculated, and the distance relationships were then visualized by downscaling the data. The principal coordinate analysis (PCoA) of the species composition of the three groups of samples was carried out by using the R software and QIIME software to characterize differences in species composition among samples based on the colony distance matrix.
2.5 Mass spectrometer parameters
LC-MS/MS analyses were performed using a SCIEX 6500 QTRAP + Triple Quadrupole Mass Spectrometer (equipped with an IonDriveTurbo VESI ion source) in multiple reaction monitoring (MRM) mode. The ion source parameters were configured as follows: ion spray voltage set to ±4,500 V, curtain gas pressure set to 35 psi, temperature was set to 400°C, ion source gas 1 and gas 2 pressures were set to 60 psi, and de-cluster voltage was set to ±100 V. These parameters were maintained consistently throughout the study.
2.6 Raw data preprocessing and quality control
The constructed libraries were then subjected to whole-genome sequencing on the Illumina NovaSeq high-throughput sequencing platform, employing a strategy that has been previously described in the literature as the “birdshot” approach. In order to evaluate the stability of the analysis system and to identify variables that demonstrate significant variation during the analysis process, it is imperative that all samples are incorporated within the QC sample category. During the instrumental analysis phase, QC samples were inserted at intervals of 8–10 samples to monitor system performance. In consideration of the multifaceted character of metabolomic data and the substantial interrelation between variables, conventional univariate analysis is challenging in facilitating expeditious and exhaustive extraction of pertinent information from the data. To address this challenge, OPLS-DA, a multivariate statistical methodology, was employed to perform dimensionality reduction and classification, thereby enabling the systematic analysis of the multidimensional data collected.
2.7 KEGG pathway analysis
The subsequent analysis of high-throughput sequencing data or metabolomics data yielded a list of differentially expressed genes (DEGs), differential flora, or differential metabolites. Subsequently, the metabolites were functionally annotated through the KEGG database (https://www.genome.jp/kegg/), and their corresponding KEGG Orthology (KO) numbers and the pathways to which they belong (e.g., metabolic pathways, signaling pathways) were obtained. The enrichment degree of metabolites in a specific pathway was analyzed using statistical methods, such as the hypergeometric test and Fisher’s exact test. Statistical methods (e.g., hypergeometric test, Fisher’s exact test) were used to analyze the degree of metabolite enrichment in a particular pathway and to determine whether the pathway was significantly associated with the study phenotype.
2.8 Statistical analysis
The study used the SPSS 18.0 software to implement Duncan’s multiple comparison test. The criteria for determining statistical significance were as follows: a p-value of less than 0.05 indicated a statistically significant difference between the groups; a p-value of less than 0.01 indicated a highly significant difference; and a p-value of greater than 0.05 indicated that the difference between the groups was not statistically significant.
3 Results
3.1 Quality control
In mass spectrometry-based metabolomics research, QC samples are essential for ensuring reliable, high-quality data. These samples were prepared by pooling equal aliquots of all experimental samples—including those from the Nor, Ant, and Fec groups—and were used to monitor stability across the entire workflow, from sample extraction and instrumental analysis to data acquisition. Though theoretically identical, QC samples often exhibited minor differences due to systematic errors introduced during these processes; smaller discrepancies indicated greater method stability and better data quality. To assess intra- and interbatch variations, QC samples were inserted at regular intervals throughout sequencing and metabolomics runs, helping verify the reliability and consistency of results. In this study, the dense clustering of QC samples in the PCA visualization (Figure 1A) further confirmed the robustness of our data.
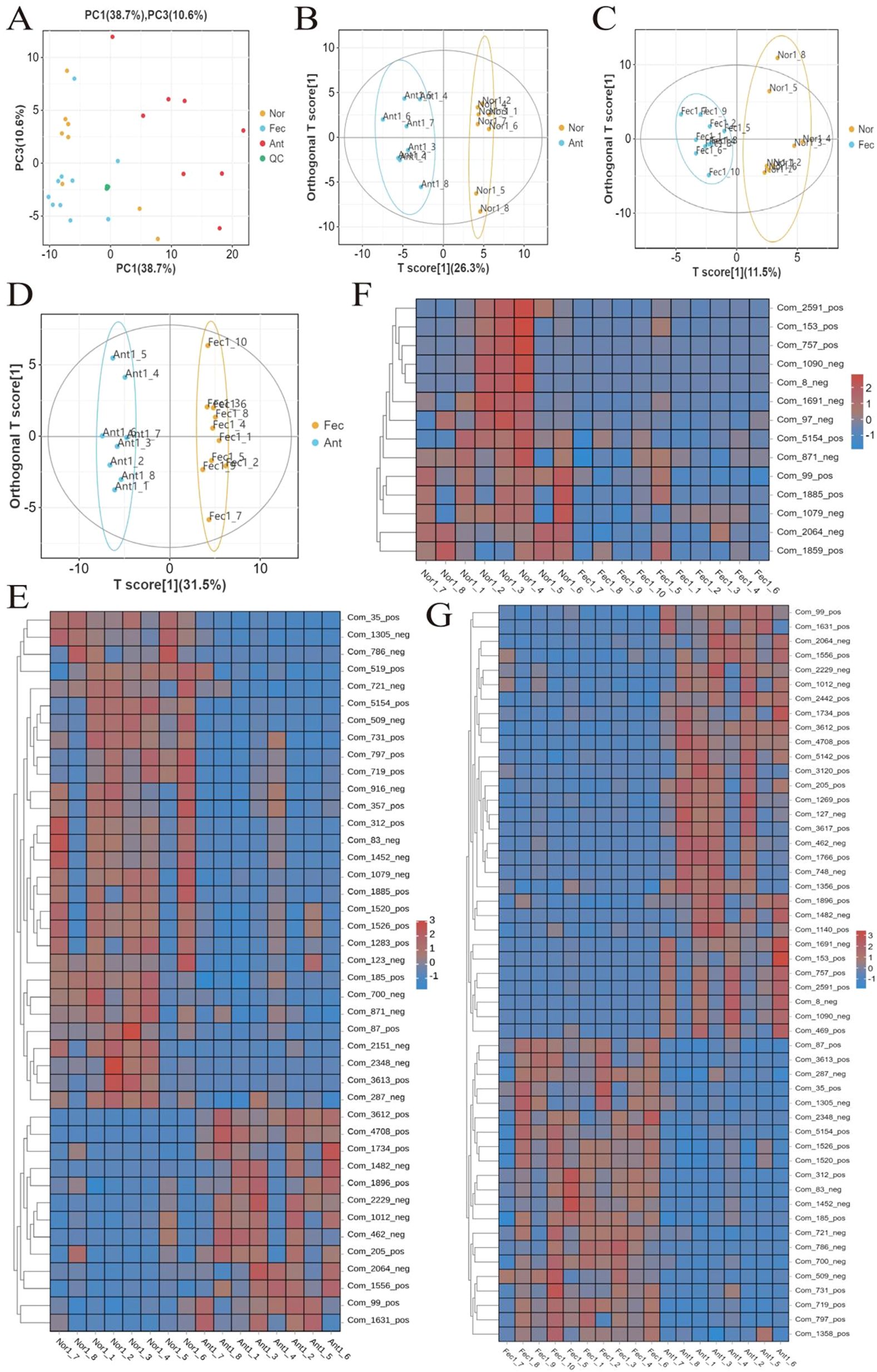
Figure 1. Quality assessment of metabolomics data and analysis of differential metabolites. (A) PCA analysis of QC samples showing tight clustering, indicating data reliability. (B–D) Validation of the OPLS-DA model (R2Y and Q2 values > 0.5) and permutation test results confirming model stability. (E–G) Bidirectional hierarchical clustering heatmap of samples and metabolites demonstrating gradient changes in differential metabolites across groups (red: upregulation; blue: downregulation).
3.2 Multivariate statistical analysis
R2X, R2Y, and Q2 were commonly used metrics for this purpose. In general, R2Y and Q2 values greater than 0.5 indicated the reliability of the model (Table 2). The OPLS-DA model established in this experiment exhibited R2Y and Q2 values exceeding 0.5, thus signifying the stability, reliability, and statistical significance of the model (Figures 1B–D).
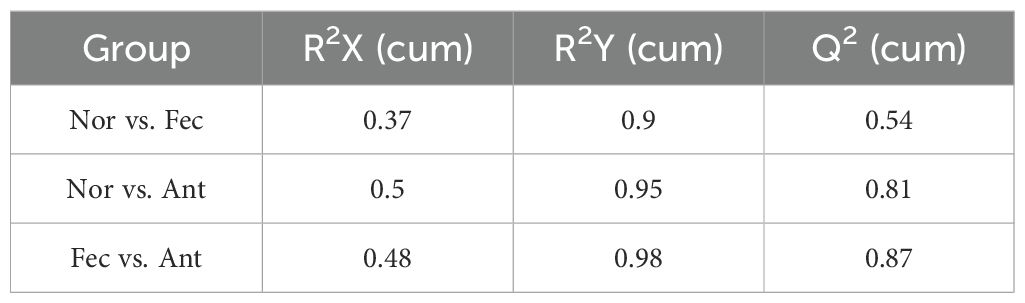
Table 2. The OPLS-DA model.
3.3 Screening for differential metabolites
Differential metabolites were identified based on three parameters: VIP, FC, and p-value. To be considered differential metabolites, the VIP value had to be greater than 1 and the p-value had to be less than 0.01. Furthermore, specific differential metabolites were selected based on the principle of p-value less than 0.01, VIP value greater than 1, and FC value greater than 1 or less than 1. These criteria were determined using VIP values obtained from statistical analyses and OPLS-DA.
At the 42nd day of the trial, in comparison with the control groups “Nor vs. Ant,” “Nor vs. Fec,” and “Fec vs. Ant,” a total of 20, 4, and 40 differential metabolites were identified, respectively, with 5, 0, and 23 being upregulated and 26, 4, and 17 being downregulated (Tables 3–5).
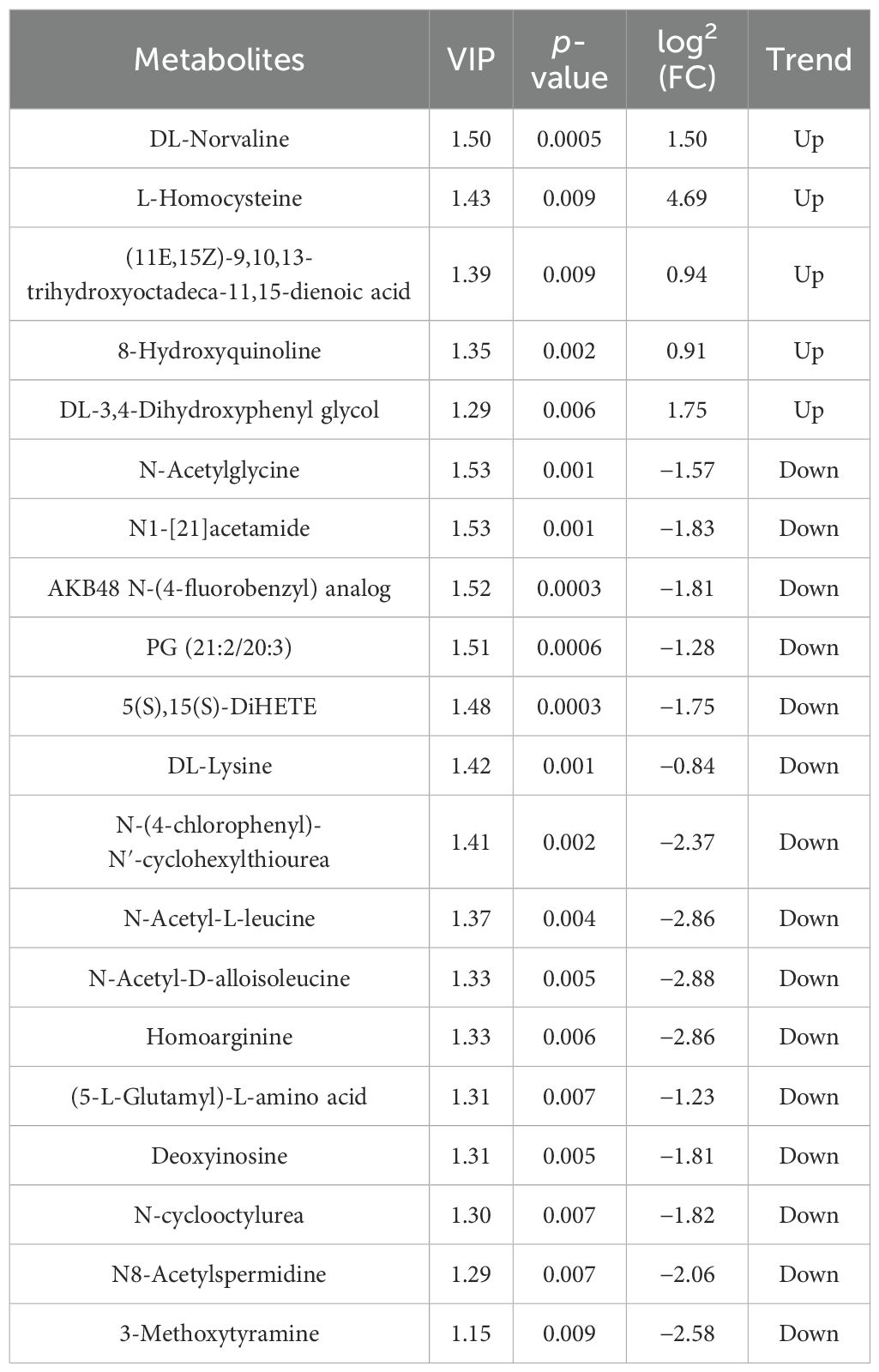
Table 3. Differential metabolites of Nor vs. Ant.
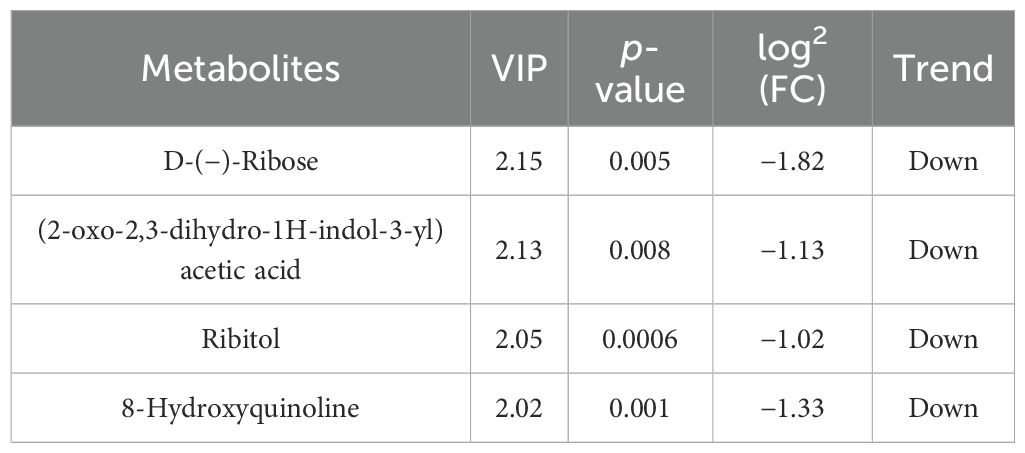
Table 4. Differential metabolites of Nor vs. Fec.
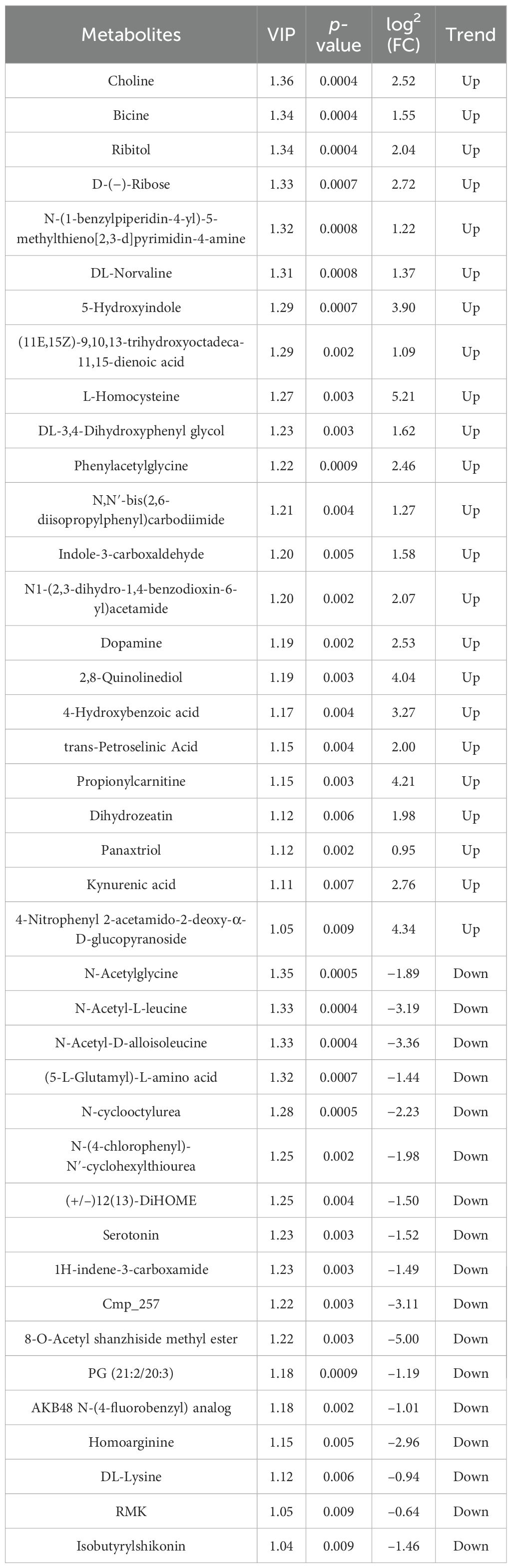
Table 5. Differential metabolites of Fec vs. Ant.
Based on untargeted metabolomics data, a bidirectional clustering heatmap of samples and metabolites was constructed using a hierarchical clustering algorithm. The heatmap revealed significant separation of the three groups of samples along the row direction, with tight clustering of intragroup samples. The color gradient (red: upregulation; blue: downregulation) further demonstrated gradient changes in differential metabolites across groups, suggesting their potential role in mediating phenotypic differences through the regulation of signaling pathways (Figures 1E–G).
3.4 KEGG analysis of differential metabolites and enrichment circle plot
KEGG enrichment bar plots and enrichment circle plots were generated to illustrate the metabolic pathways associated with different feed additives in the Nor and experimental groups. At the end of the experiment, compared with the Nor group (control group), the Ant group exhibited 15 differential metabolic pathways (Figure 2A), including tryptophan metabolism, benzoic acid family metabolism, serotonin receptor agonists, cholinergic synapse metabolism, and gap junction metabolism. The Fec group showed 15 metabolic pathways (Figure 2B), mainly including gap junction metabolism, riboflavin metabolism, and dopaminergic synapse. Additionally, 15 differential metabolic pathways were identified between the Fec and Ant groups (Figure 2C), primarily involving gap junction metabolism, tryptophan metabolism, CAMP signaling pathway, serotonin receptor agonists, and cholinergic synapse.
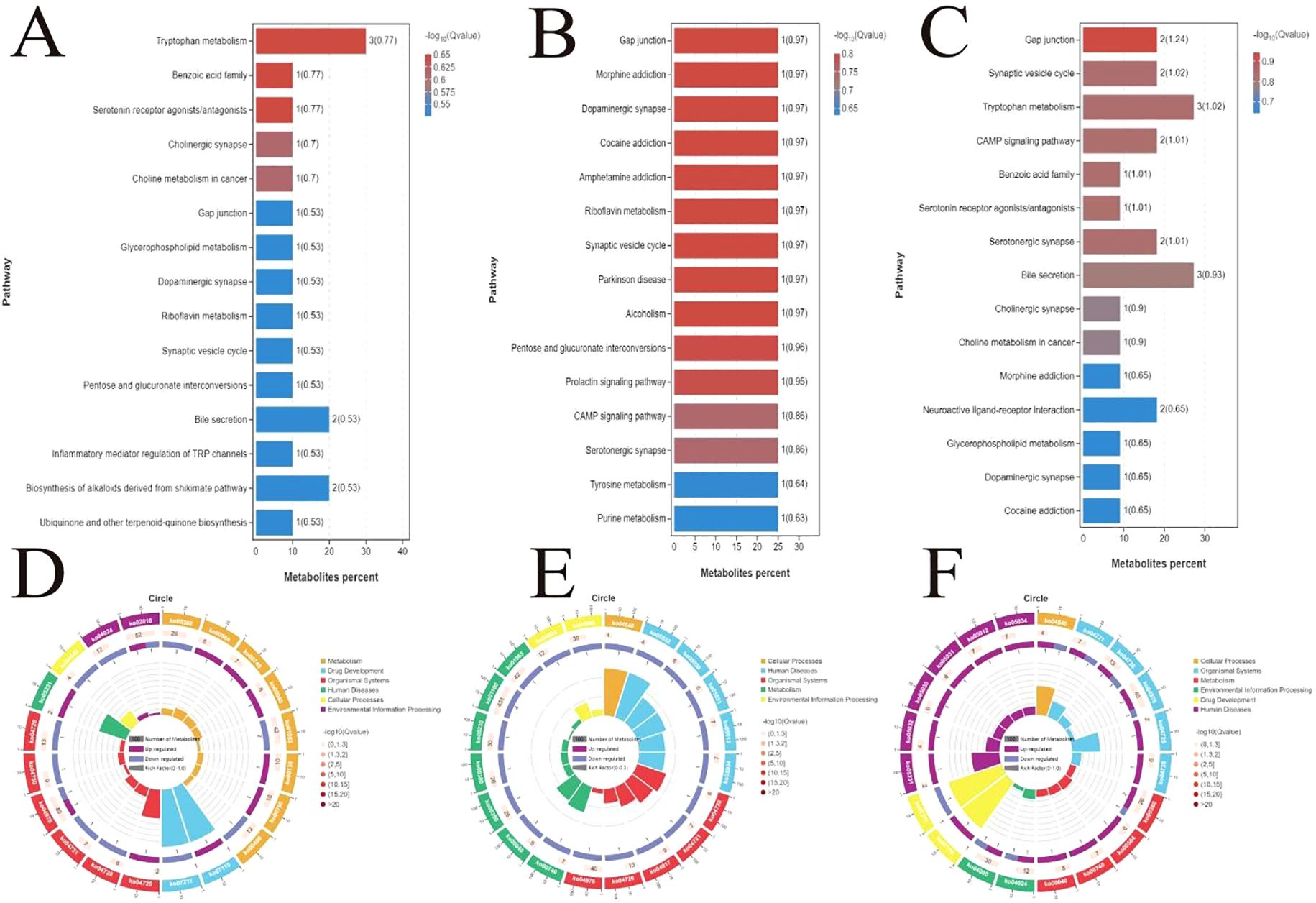
Figure 2. Screening of differential metabolic pathways across groups and KEGG enrichment visualization analysis. (A) Fifteen differential pathways between the Ant and Nor groups. (B) Fifteen differential pathways between the Fec group-specific pathways. (C) Fifteen differential pathways between the Fec and Ant groups. (D–F) KEGG enrichment circle plots of the top 20 enriched pathways.
The top 20 enriched pathways were selected to generate KEGG enrichment circle plots (Figures 2D–F). Based on the KEGG enrichment circle plot analysis, differentially abundant metabolites between the Nor and Ant groups were significantly enriched in metabolism (e.g., ABC transporters), signal transduction (e.g., MAPK signaling), and disease-related pathways (e.g., cancer pathways), suggesting that experimental interventions may mediate phenotypic differences through regulating efflux transport, cellular stress response, and metabolic reprogramming. The prominent enrichment of the ABC transporter pathway (ko02010) implies its potential role in drug resistance mechanisms, while activation of the MAPK pathway (ko00380) may correlate with inflammatory responses (Figure 2D). Differential metabolites between the Nor and Fec groups were significantly enriched in metabolism, organismal systems, and disease-related functional categories. The marked enrichment of metabolic pathways (ko01100) suggests potential involvement in energy metabolism reprogramming, such as enhanced glycolysis or TCA cycle activity, which may support cell proliferation or stress adaptation. The activation of bile secretion (ko04976) may correlate with abnormal lipid metabolism, including hypercholesterolemia or cholestasis. The enrichment of flavonoid biosynthesis (ko01063) likely reflects alterations in host–microbial co-metabolism products, exhibiting antioxidant effects. Additionally, the activation of neuroactive ligand–receptor interactions (ko04080) may involve neuroendocrine regulation, such as appetite modulation (Figure 2E). Differential metabolites between the Fec and Ant groups were significantly enriched in organismal systems, metabolism, and disease-related functional categories. The prominent enrichment of the bile secretion pathway (ko04976) suggests its potential involvement in lipid metabolism dysregulation or bile acid homeostasis imbalance, such as cholestasis or impaired cholesterol clearance. The activation of the neuro-metabolic hub (ko04080) may correlate with neuroendocrine regulation of metabolic adaptation, for instance, adrenergic signaling promoting gluconeogenesis or lipolysis during stress responses. Additionally, the significant enrichment of tryptophan metabolism (ko00380) implies its role in gut–brain axis signaling or immune modulation, exemplified by microbiota-derived indole metabolites modulating host inflammatory responses.
3.5 Analysis of fecal microbiota composition
Fecal samples from the Nor, Fec, and Ant groups were analyzed using 16S rRNA sequencing technology. The Chao1 (Figure 3A), Shannon (Figure 3B), and Simpson (Figure 3C) indices revealed that the Nor and Ant groups exhibited lower microbial diversity compared to the Fec group. PCoA based on weighted UniFrac distance demonstrated significant differences in beta diversity of the fecal microbiota among the Nor, Fec, and Ant groups (Figure 3D).
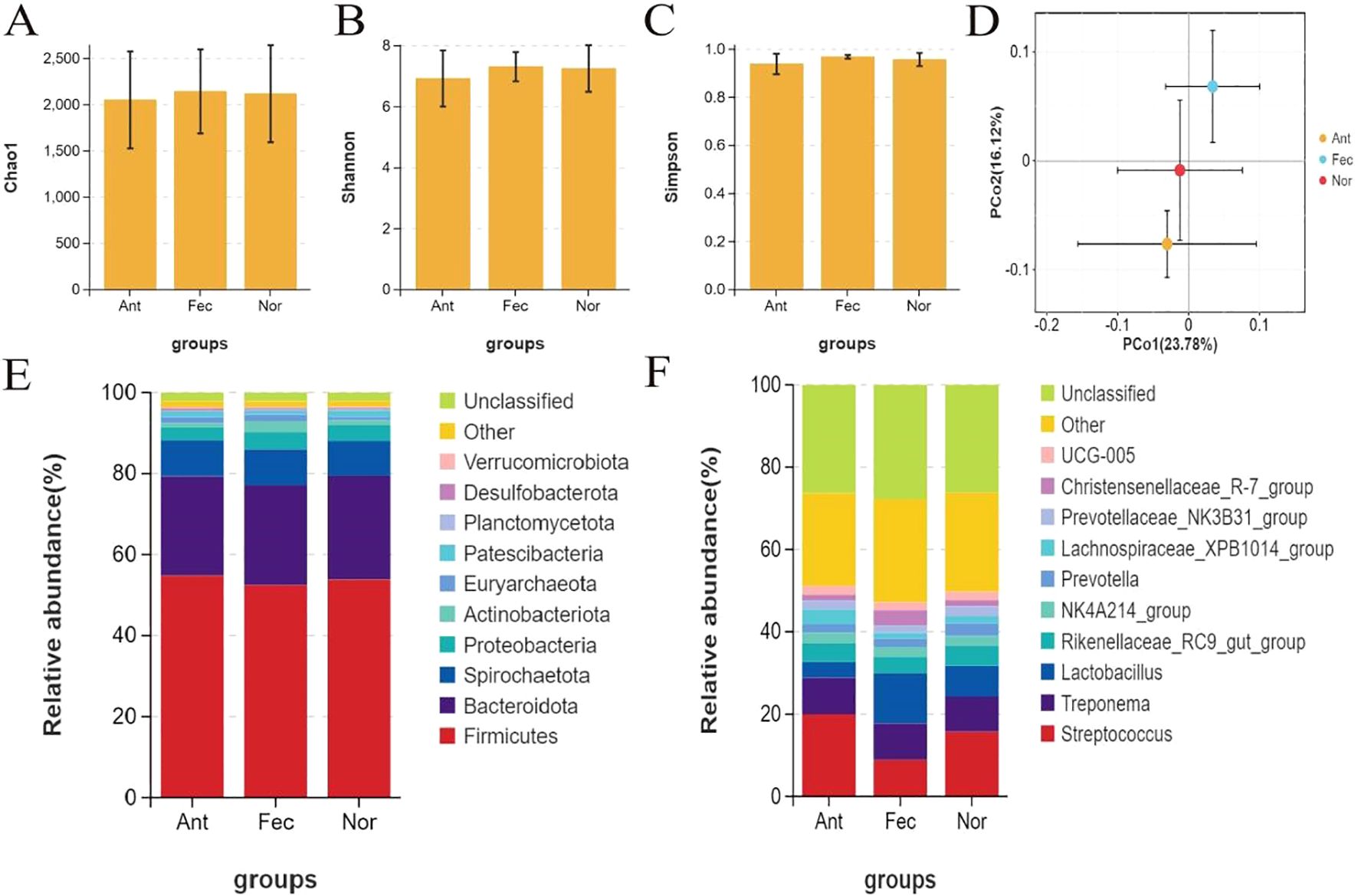
Figure 3. Analysis of fecal microbiota diversity and community structure across the three groups (A–C): alpha diversity indices (Chao1, Shannon, Simpson) showing lower microbial diversity in the Nor and Ant groups compared to the Fec group. (D) Principal coordinate analysis (PCoA) based on weighted UniFrac distance (beta diversity) revealing significant structural differences in microbial communities among the three groups. (E) Phylum-level community structure (shifting ratios of Firmicutes and Bacteroidota reflecting treatment effects). (F) Genus-level differences (probiotic genera enrichment in the Fec group, pathogenic genera proliferation in the Ant group, and homeostasis of core genera in the Nor group).
Analysis of the phylum-level microbial community structure in the Nor, Fec, and Ant groups via 16S rRNA sequencing revealed that Firmicutes and Bacteroidota were the dominant phyla, with changes in their ratios reflecting the regulatory effects of different treatment groups on the gut microbiota (Figure 3E). The increased abundance of Bacteroidota in the Fec group might be attributed to enhanced dietary fiber metabolism induced by fecal microbiota transplantation, while the significant reduction of Firmicutes accompanied by an increase in Proteobacteria in the Ant group suggests that antibiotics likely disrupted microbial balance and promoted pathogenic bacterial proliferation.
Analysis at the genus level via 16S rRNA sequencing revealed significant differences in gut microbiota among the Nor, Fec, and Ant groups (Figure 3F). The Fec group exhibited significant enrichment of probiotic genera (e.g., Lactobacillus and Prevotella) due to FMT, which may improve intestinal function by enhancing dietary fiber metabolism and short-chain fatty acid (SCFA) production. In the Ant group, a reduction in Firmicutes (e.g., Lactobacillus) and an increase in Proteobacteria (e.g., Streptococcus) were observed, potentially leading to elevated intestinal permeability and triggering endotoxemia, suggesting that microbial dysbiosis may induce inflammation or metabolic disorders. The stable abundance of core genera (e.g., Christensenellaceae_R-7_group, each accounting for ~8%) in the Nor group reflects the homeostatic maintenance function of the core microbiota.
3.6 Analysis of differences in the fecal microbiota
The Venn diagram of OTU distribution (Figure 4A) demonstrated distinct microbial community structures among the Nor (control), Fec (fecal microbiota transplantation), and Ant (antibiotic-treated) groups. A total of 2,284 OTUs were identified, with 81 OTUs shared across all groups, representing a conserved core microbiota. Unique OTUs were the highest in the Nor group (374), followed by Fec (145) and Ant (132), indicating reduced microbial diversity under antibiotic intervention. Pairwise overlaps revealed 919 OTUs shared between Fec and Nor, suggesting partial restoration of native microbiota through transplantation, while Ant exhibited limited overlap with both Fec (341 OTUs) and Nor (292 OTUs), consistent with antibiotic-induced suppression. Data derived from 16S rRNA sequencing (V3–V4 region) and analyzed via QIIME2 (v2021.4) highlighted the resilience of Nor’s baseline diversity and the functional recalibration achieved by fecal transplantation in the Fec group.
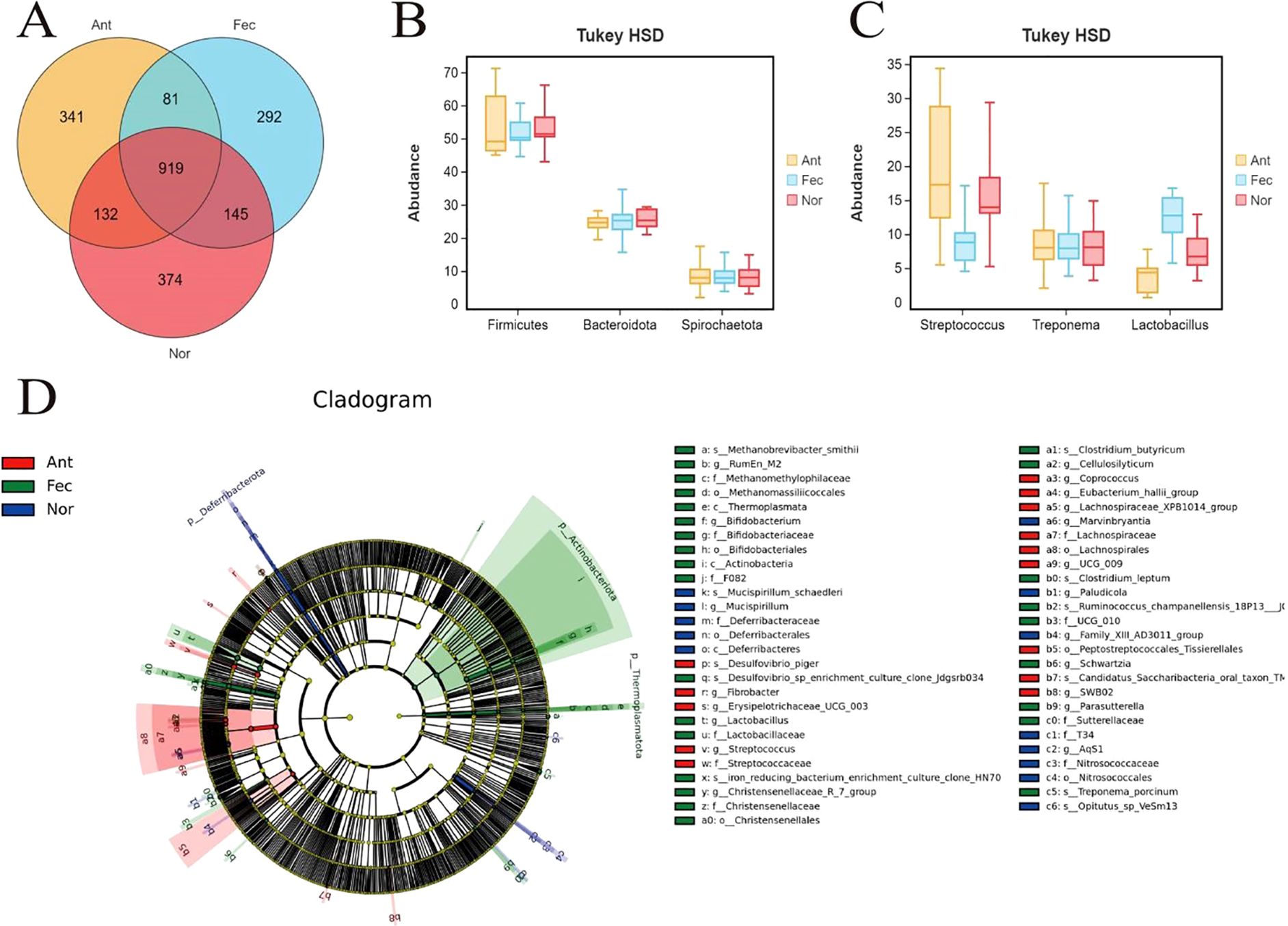
Figure 4. Multidimensional characterization of gut microbiota across the Nor, Fec, and Ant groups. (A) OTU diversity and core microbiota revealed by the Venn diagram. (B) Phylum-level abundance shifts via Tukey’s HSD test. (C) Genus-specific composition differences. (D) Metabolic functional enrichment identified by LEfSE analysis.
This study compared the differences in microbial abundance at the phylum level among the Nor, Fec, and Ant groups using Tukey’s HSD test (Tukey’s honestly significant difference) (Figure 4B). The Tukey’s HSD test is a post-hoc multiple comparison method applied after analysis of variance (ANOVA) to determine specific intergroup differences. The abundance of Firmicutes in the Ant group was significantly lower than in the Nor group (p < 0.05), possibly due to the selective inhibition of Gram-positive bacteria (as Firmicutes are predominantly Gram-positive) by antibiotics (e.g., vancomycin), leading to reduced microbial diversity. The abundance of Firmicutes in the Ant group was also significantly lower than in the Fec group (p < 0.01), suggesting that the inhibitory effect of antibiotics on Firmicutes outweighed the probiotic benefits of fecal microbiota transplantation. No significant difference was observed between the Nor and Fec groups, indicating that fecal microbiota transplantation did not markedly alter the baseline proportion of Firmicutes, likely due to their stable functional role in a healthy gut. The abundance of Bacteroidota in the Fec group was significantly higher than in the Nor group (p < 0.01), which might be attributed to the introduction of donor microbiota rich in Bacteroidota (e.g., Bacteroides succinogenes) via fecal microbiota transplantation, enhancing dietary fiber degradation capacity. The abundance of Bacteroidota in the Fec group was also significantly higher than in the Ant group (p < 0.01), reflecting the substantial suppression of Bacteroidota growth by antibiotics, potentially compromising mucus layer integrity. Antibiotic treatment (Ant group) significantly suppressed beneficial phyla (Firmicutes and Bacteroidota) while promoting the proliferation of potentially pathogenic phyla (Spirochaetota), which may impair host metabolic functions or trigger inflammatory responses. Fecal microbiota transplantation (Fec group) restored Bacteroidota abundance by introducing exogenous microbiota, likely enhancing fiber metabolism to increase SCFA production, lower intestinal pH, inhibit pathogen growth, and reduce opportunistic pathogen colonization (e.g., Spirochaetota) through competitive exclusion.
Through the Tukey’s HSD test analysis, significant differences were observed in the composition of the microbiota at the genus level among the three groups (Figure 4C). Specifically, the relative abundance of Streptococcus was significantly higher in the Nor group. In the Fec group, the abundance of Treponema was significantly increased, possibly related to fecal microbiota transplantation. In the Ant group, the abundance of Lactobacillus was significantly reduced, possibly due to antibiotic treatment or other external intervention factors. These differences not only reflect the ecological adaptability changes in the gut microbiota among the groups but may also have significant impacts on the health and survival of the host. Overall, the composition and abundance of the fecal microbiota exhibit distinct group-specific characteristics, revealing the important regulatory effects of environmental, dietary, or intervention factors on the gut microbiota.
LEfSE analysis revealed that the fecal microbiota of the Fec group exhibited significant advantages in metabolic functionality and gut homeostasis regulation (Figure 4D). Key enriched taxa included the Eubacterium hallii group (LDA = 4.5) and the Christensenellaceae_R-7_group (LDA = 4.0), which may enhance intestinal barrier integrity and mitigate inflammation through efficient SCFA synthesis and lipid metabolism modulation. Compared to the Ant group, the Fec group lacked pro-inflammatory taxa (e.g., Deferribacteraceae) and demonstrated markedly higher abundance of butyrate producers. Relative to the Nor group, the Fec group displayed a more balanced fiber-degrading capacity and substrate utilization efficiency, driven by synergistic interactions between Clostridium leptum (LDA = 3.5) and Fibrobacter. Additionally, the enrichment of Methanobrevibacter smithii (LDA = 2.8) suggested optimized hydrogen metabolism, supporting microbial ecological stability. Collectively, these features highlight the Fec group’s microbiota as a potential paradigm for metabolic health, anti-inflammatory activity, and functional diversity, aligning closely with an idealized gut microbial profile.
3.7 Functional analysis of the fecal microbiota
Comparative functional analysis of the gut microbiota among the Nor, Fec, and Ant groups using PICRUSt2 revealed distinct metabolic and adaptive responses (Figures 5A–E). The Ant group exhibited hyperactivation of carbohydrate and amino acid metabolism (KEGG units: 273,962.02 vs. 250,860.09 in the Nor group) alongside enhanced DNA replication and repair (129,789.82), indicative of compensatory survival strategies under antibiotic-induced stress, such as upregulated efflux pump activity and antioxidant synthesis. In contrast, the Fec group demonstrated partial metabolic recalibration (e.g., reduced carbohydrate metabolism compared to Ant) and enrichment of fiber-degrading functions (e.g., Bacteroidota), likely attributable to the introduction of exogenous microbiota. The Nor group maintained metabolic homeostasis with minimal pathogenic activity (e.g., infectious diseases: 4,717.11). Notably, while antibiotics suppressed pathogenic pathways (e.g., cancer-related functions: 0 in Ant), they disrupted ecological equilibrium. Conversely, FMT partially restored microbial functionality through enhanced SCFA synthesis and competitive exclusion of pathogens. These findings underscore the dual role of interventions: antibiotics as a double-edged sword and FMT as a partial restorative agent, emphasizing the need for balanced therapeutic strategies to preserve microbial resilience and host health.
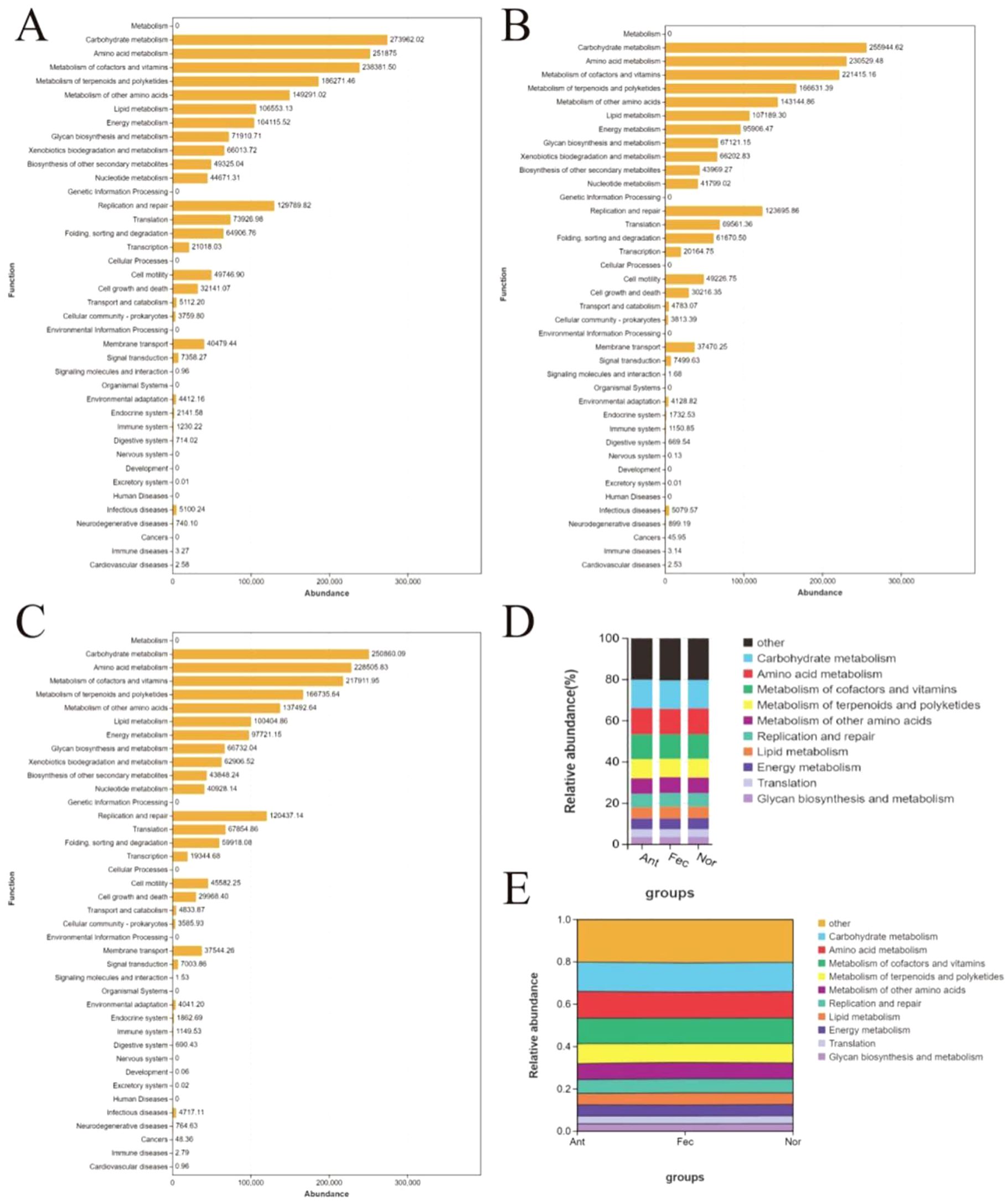
Figure 5. Functional profiling and dynamic shifts of gut microbiota in the Nor, Fec, and Ant groups via PICRUSt2 analysis. (A–C) Bar plot of core functional pathway abundance across groups. (D) Stacked Bar plot of functional pathway distribution across groups. (E) Sankey diagram of functional flux between microbial taxa and metabolic pathways.
3.8 Correlation between microbiota and metabolites
In this study, the correlation analysis results of metabolites and 16S rRNA are shown. As shown in Figures 6A, B, the analysis of correlations between metabolites and gut microbiota at the phylum level reveals that Firmicutes contributes to intestinal barrier protection and dietary utilization through tryptophan metabolites (e.g., the anti-inflammatory molecule indole-3-acrylic acid) and degradation of plant-derived compounds (e.g., caryophyllene oxide), while arecoline may exacerbate microbial dysbiosis by inhibiting its proliferation. Bacteroidota shows a positive correlation with secondary bile acids, suggesting its potential role in lipid metabolism regulation, whereas Proteobacteria is linked to oxidative stress markers, indicating inflammatory risks. Although the study uncovers functional modules of microbiota–metabolite interactions (anti-inflammatory and oxidative stress), experimental validation of causal relationships and optimization of statistical methods (e.g., multiple testing correction) are required to enhance reliability. Future efforts should integrate multi-omics data and clinical interventions to develop targeted microbial modulation strategies (e.g., probiotics or dietary interventions) for improving gut health and preventing metabolic diseases.
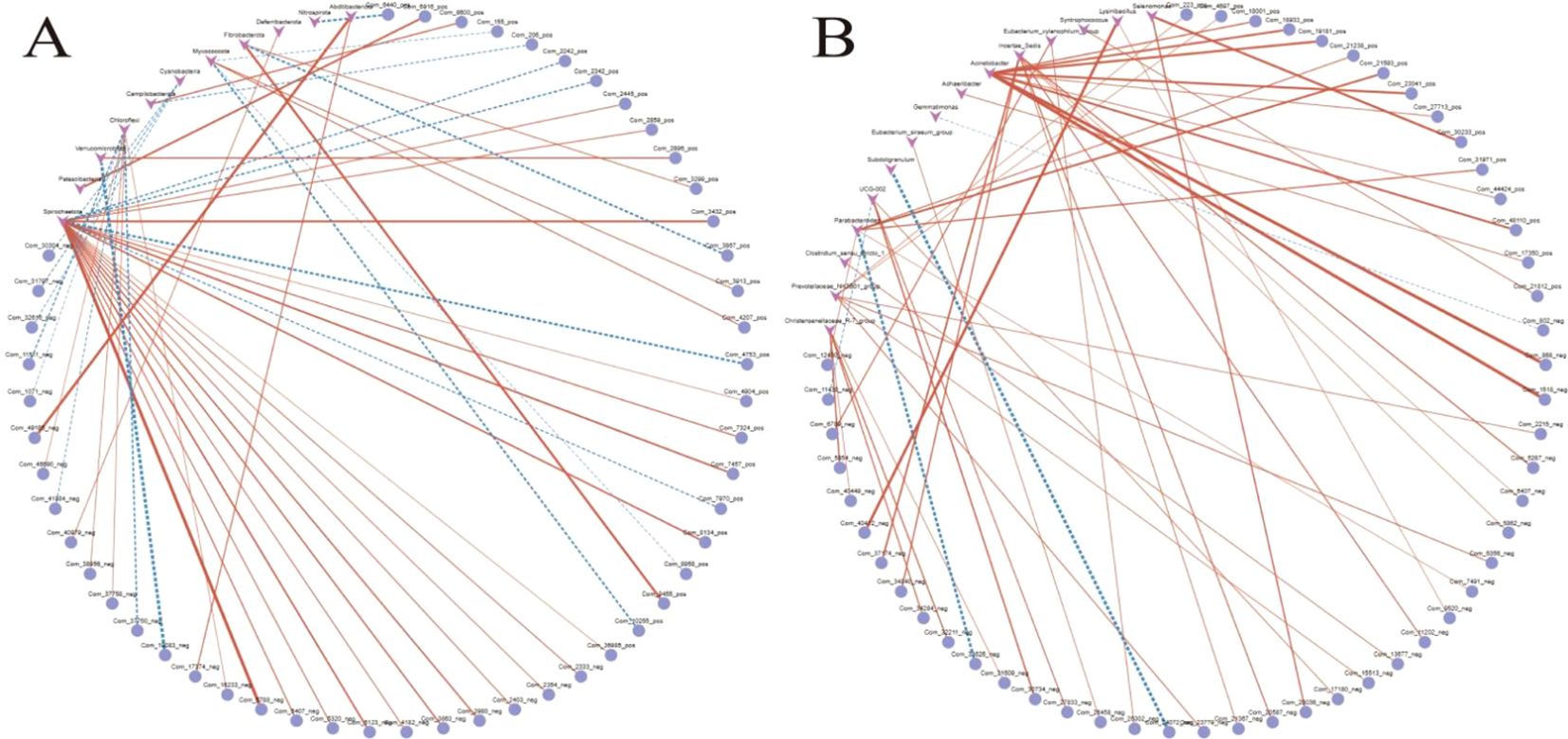
Figure 6. (A, B) Phylum-specific gut microbiota–metabolite associations linked to anti-inflammatory and oxidative stress pathways.
This study analyzed the correlations between Streptococcus and metabolites, revealing significant associations: positively correlated metabolites included (R)-equol (ρ = +0.583), a compound with estrogen-like activity; xanthurenic acid (ρ = +0.607), a tryptophan-derived metabolite; and panaxtriol (ρ = +0.533), a ginsenoside derivative, suggesting the potential roles of Streptococcus in host anti-inflammatory regulation, oxidative stress modulation, and plant-derived compound metabolism. In contrast, significant negative correlations with acetophenone (ρ = −0.647), tyramine (ρ = −0.649), and an unidentified metabolite RMK (ρ = −0.658) implied its involvement in metabolic balance regulation through degradation or inhibitory pathways. These findings highlight the dual functionality of Streptococcus in host-microbial metabolic interactions (promoting synthesis and substrate clearance).
4 Discussion
The findings of this study underscore the multifaceted role of FMT in mitigating weaning stress in piglets through targeted modulation of gut microbiota and host metabolic pathways. By integrating 16S rRNA sequencing and metabolomic analyses, we elucidated the mechanisms by which FMT restores microbial resilience and reprograms metabolic networks, offering a viable alternative to conventional antibiotic interventions. Weaning stress often leads to dramatic fluctuations in the structure of intestinal flora in piglets, while FMT constructs a more stable “ecological buffer system” for intestinal flora by enhancing microbial diversity (e.g., a significant increase in Shannon index). The enriched fiber-degrading taxa (e.g., Prevotella, Christensenaceae) can not only efficiently utilize feed fibers to generate SCFA but also inhibit the overproliferation of conditionally pathogenic bacteria (e.g., Aspergillus phylum) through a competitive exclusion mechanism, reducing intestinal inflammation triggered by bacterial imbalance, which is exactly what happens to piglets with diarrhea during weaning stress. Consistent with previous findings, these findings demonstrate the ability of FMT to reintroduce key taxa that are critical for fiber degradation and SCFA production (Sarah et al., 2024; Martinez-Gili et al., 2020). The enrichment of these taxa likely facilitates competitive exclusion of opportunistic pathogens, as evidenced by the suppression of Proteobacteria in the Fec group. In contrast, lincomycin-induced dysbiosis—marked by a 42% reduction in Firmicutes and expansion of Proteobacteria—can destabilize flora and make piglets more susceptible to stress—mirroring the outcomes of antibiotic overuse in human and veterinary medicine, where reduced microbial diversity correlates with increased susceptibility to enteric infections (Jukes et al., 2019; Gupta and Dey, 2023). These results reinforce the ecological principle that microbiota stability is pivotal for gut health, particularly during the vulnerable weaning transition (Chilloux et al., 2016; Arfken et al., 2019; Pluske et al., 2018).
The upregulation of anti-inflammatory indoles (e.g., 5-hydroxyindole) and SCFA precursors in the Fec group suggests a direct link between microbiota remodeling and host metabolic adaptation (Song et al., 2021; Martin-Gallausiaux et al., 2021; Deng et al., 2020). Indole derivatives, known activators of aryl hydrocarbon receptor (AhR) signaling, enhance intestinal barrier function by upregulating tight junction proteins, while SCFAs serve as both energy substrates for colonocytes and regulators of immune homeostasis (Lanza et al., 2022; Chen et al., 2021; Xu et al., 2023). In a similar manner, the neurometabolic hub (Ko04080) activates neuroendocrine regulation, which may be implicated in metabolic adaptation. This assists piglets in regulating energy metabolism during periods of stress. Furthermore, the predominance of the Fec group in these pathways serves to reduce the stress-exacerbating effects of antibiotics. Conversely, the depletion of amino acid metabolites (e.g., N-acetylglycine) and activation of MAPK signaling in the Ant group reflect a metabolic shift toward oxidative stress—a phenomenon previously linked to antibiotic-induced mitochondrial dysfunction (Muska and Mervyn, 2022; Kalghatgi et al., 2013). These findings highlight the divergent metabolic consequences of FMT vs. antibiotics: one promotes resilience through symbiotic metabolite production, while the other exacerbates stress via pathway disruption.
Firmicutes protects the intestinal barrier and promotes nutrient utilization through tryptophan metabolites (e.g., the anti-inflammatory molecule indole-3-acrylic acid) and degradation of plant-derived compounds; Bacteroidota is positively correlated with secondary bile acids, which may be involved in the regulation of lipid metabolism, and may contribute to the maintenance of stable energy metabolism and reduction of metabolic disorders induced by stress; and Proteobacteria is positively correlated with markers of oxidative stress. Proteobacteria is related to oxidative stress markers, suggesting that it may increase the risk of inflammation and exacerbate stress.
The dual metabolic roles of Streptococcus—synthesizing bioactive compounds ((R)-equol) while degrading harmful metabolites (tyramine)—provide novel insights into its ecological niche (Malik et al., 2020; Wang et al., 2024; van der Beek et al., 2019). While Streptococcus is often associated with opportunistic infections, our data suggest its commensal potential under FMT-mediated stabilization. The positive correlation between (R)-equol (a phytoestrogen with anti-inflammatory properties) and Streptococcus aligns with recent evidence of microbial estrogen metabolism influencing mucosal immunity (Baker et al., 2017; Park and Sohrabji, 2018). Conversely, its negative association with tyramine—a biogenic amine linked to intestinal hyperpermeability—implies a detoxification role (Lin et al., 2019). This duality challenges the conventional pathogen-centric view of Streptococcus and underscores the context-dependent nature of microbial functionality.
The activation of tryptophan metabolism and bile secretion pathways by FMT offers actionable targets for non-antibiotic interventions. Tryptophan metabolism plays a role in gut–brain axis signaling or immune regulation, colony-derived indole metabolites modulate host inflammatory responses, and differences in bile secretion pathways may affect lipid metabolism or bile acid homeostasis. For instance, precision probiotics combining Christensenellaceae and Lactobacillus could mimic FMT’s benefits, while dietary additives (e.g., prebiotic fibers) might selectively enrich SCFA producers (Çenesiz and Çiftci, 2020). Additionally, the Streptococcus–metabolite network identified here could inspire strain-specific therapies to modulate host inflammation or nutrient absorption.
In summary, this study delineates how FMT mitigates weaning stress by orchestrating microbial diversity and metabolic harmony. By contrasting FMT with antibiotic outcomes, we provide a mechanistic rationale for reducing antimicrobial reliance in livestock production. Future efforts should prioritize translating these insights into scalable interventions, ensuring both animal welfare and sustainable agriculture.
5 Conclusion
The above findings suggest that FMT has the ability to restore microbial stability and anti-inflammatory properties, as well as maintain intestinal barrier function. It can therefore be used as an effective, non-antibiotic strategy against weaning syndrome, providing an alternative to traditional antimicrobials. Causal validation via gnotobiotic models and clinical translation through precision microbial consortia or dietary interventions targeting Streptococcus–metabolite networks are critical next steps. This work establishes a mechanistic framework for optimizing livestock gut health through ecological modulation of host–microbiota dynamics.
Data availability statement
The data presented in the study are deposited in the NCBI repository, accession number PRJNA1306594.
Ethics statement
The animal study was approved by the Laboratory Animal Ethics Committee of Tibet College of Agriculture and Animal Husbandry/Xizang Agricultural and Animal Husbandry University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
JZ: Investigation, Writing – original draft, Validation, Data curation, Conceptualization, Methodology, Supervision. MZ: Conceptualization, Validation, Writing – original draft. HZ: Writing – review & editing. MH: Writing – review & editing. ZT: Funding acquisition, Project administration, Writing – review & editing. PS: Funding acquisition, Writing – review & editing, Supervision, Project administration.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study received support from the Tibet Agriculture and Animal Husbandry College Talent Team Building Program (XZNMXYZFYC-2024-07); Science and Technology Projects of Xizang Autonomous Region, China (XZ202501ZY0147); National Natural Fund Program (32360843); Xizang Agriculture and Animal Husbandry University Doctoral Program in Forestry (Phase I) (538325001); and China Agricultural University-Tibet College of Agriculture and Animal Husbandry Joint Fund for Scientific Research Program (2023TC055).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ali M., Lee E. B., Hsu W., Suk K., Al S., Sayem J., et al. (2023). Probiotics and postbiotics as an alternative to antibiotics: an emphasis on pigs. Pathogens 12, 874. doi: 10.3390/pathogens12070874
Arfken A., Frey J., Ramsay T., and Summers K. L. (2019). 176 Characterizing the mycobiome in piglets during the weaning transition. J. Anim. Sci. 97, 177. doi: 10.1093/jas/skz258.364
Baker J. M., Al-Nakkash L., and Herbst-Kralovetz M. M. (2017). Estrogen–gut microbiome axis: physiological and clinical implications. Maturitas 103, 45–53. doi: 10.1016/j.maturitas.2017.06.025
Camerlink I., Proegger C., Kubala D., Galunder K., and Rault J. L. (2021). Keeping littermates together instead of social mixing benefits pig social behaviour and growth post-weaning. Appl. Anim. Behav. Sci. 235, 105230. doi: 10.1016/j.applanim.2021.105230
Çenesiz A. A. and Çiftci I. (2020). Modulatory effects of medium chain fatty acids in poultry nutrition and health. World’s Poultry Sci. J. 76, 234–248. doi: 10.1080/00439339.2020.1739595
Chen D. W., Chen C. M., Qu H. X., Ren C. Y., Yan X. T., Huang Y. J., et al. (2021). Screening of lactobacillus strains that enhance scfa uptake in intestinal epithelial cells. Eur. Food Res. Technol. 247, 1049–1060. doi: 10.1007/s00217-021-03686-1
Chilloux J., Neves A. L., Boulangé C. L., and Dumas M. E. (2016). The microbial-mammalian metabolic axis: a critical symbiotic relationship. Curr. Opin. Clin. Nutr. Metab. Care 19, 250. doi: 10.1097/MCO.0000000000000284
Cold F., Baunwall S. M. D., Dahlerup J. F., Petersen A. M., Hvas C. L., and Hansen L. H. (2021). Systematic review with meta-analysis: encapsulated faecal microbiota transplantation–evidence for clinical efficacy. Ther. Adv. Gastroenterol. 14, 569–580. doi: 10.1177/17562848211041004
Dang D. X., Liu Y., Chen N., and Kim I. H. (2022). Dietary supplementation of Aspergillus Niger-expressed glucose oxidase ameliorates weaning stress and improves growth performance in weaning pigs. J. Anim. Physiol. Anim. Nutr. 106, 258–265. doi: 10.1111/jpn.13576
Deng M., Qu F., Chen L., Liu C., Zhang M., Ren F., et al. (2020). Scfas alleviated steatosis and inflammation in mice with nash induced by mcd. J. Endocrinol. 245, 425–437. doi: 10.1530/JOE-20-0018
Gomes M. D. S., Alysson S., Valente Júnior Dante T., De O. L. L., Correia A. M., Sero N. V. L., et al. (2022). Effect of amino acid blend as alternative to antibiotics for growing pigs. J. Anim. Sci. 100, skac008. doi: 10.1093/jas/skac008
Gupta U. and Dey P. (2023). Rise of the guardians: gut microbial maneuvers in bacterial infections. Life Sci. 330, 121993. doi: 10.1016/j.lfs.2023.121993
Jukes C., Ijaz U., Buckley A., Spencer J., Irvine J., Candlish D., et al. (2019). Bile salt metabolism is not the only factor contributing to clostridioides (clostridium) difficile disease severity in the murine model of disease. Gut Microbes 11, 481–496. doi: 10.1080/19490976.2019.1678996
Kalghatgi S., Spina C. S., Costello J. C., Liesa M., Morones-Ramirez J. R., Slomovic S., et al. (2013). Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in mammalian cells. Sci. Trans. Med. 5, 192ra85. doi: 10.1126/scitranslmed.3006055
Kerschaver C. V., Turpin D., Michiels J., and Pluske J. (2023). Reducing weaning stress in piglets by pre-weaning socialization and gradual separation from the sow: a review. Animals 13, 1644. doi: 10.3390/ani13101644
Lanza M., Scuderi S. A., Filippone A., Casili G., Campolo M., Paterniti I., et al. (2022). The role of scfas on microbiota composition in a mouse model of ntg-induced migraine. FASEB J. 36 (1), 45–46. doi: 10.1096/fasebj.2022.36.S1.R4645
Lin T. H., Wang H. C., Cheng W. H., Hsu H. C., and Yeh M. L. (2019). Osteochondral tissue regeneration using a tyramine-modified bilayered PLGA scaffold combined with articular chondrocytes in a porcine model. Int. J. Mol. Sci. 20, 326. doi: 10.3390/ijms20020326
Liu S., Tao X., Deng B., Li Y., and Xu Z. (2023). Genome-wide analysis of long noncoding rnas in porcine intestine during weaning stress. Int. J. Mol. Sci. 24, 5343. doi: 10.3390/ijms24065343
Malik A., Kim Y. R., and Kim S. B. (2020). Genome mining of the genus streptacidiphilus for biosynthetic and biodegradation potential. Genes 11, 1166. doi: 10.3390/genes11101166
Martinez-Gili L., Mcdonald J. A. K., Liu Z., Kao D., Allegretti J. R., Monaghan T. M., et al. (2020). Understanding the mechanisms of efficacy of fecal microbiota transplant in treating recurrent clostridioides difficile infection and beyond: the contribution of gut microbial-derived metabolites. Gut Microbes 12, 1810531. doi: 10.1080/19490976.2020.1810531
Martin-Gallausiaux C., Marinelli L., Blottière H. M., Larraufie P., and Lapaque N. (2021). Scfa: mechanisms and functional importance in the gut. Proc. Nutr. Soc. 80, 37–49. doi: 10.1017/S0029665120006916
Moeser A. J., Pohl C. S., and Rajput M. (2017). Weaning stress and gastrointestinal barrier development: implications for lifelong gut health in pigs. Anim. Nutr. 3, 313–321. doi: 10.1016/j.aninu.2017.06.003
Mooyottu S., Muyyarikkandy M. S., Yousefi F., Li G., Sahin O., Burrough E., et al. (2025). Fecal microbiota transplantation modulates jejunal host-microbiota interface in weanling piglets. Microbiome 13, 45. doi: 10.1186/s40168-025-02042-9.DOI:10.1186/s40168-025-02042-9
Muska M. and Mervyn S. (2022). Do antibiotics cause mitochondrial and immune cell dysfunction? a literature review. J. Antimicrobial Chemotherapy 77, 1218–1227. doi: 10.1093/jac/dkac025
Niederwerder M. C., Constance L. A., Rowland R. R. R., Waseem A., Fernando S. C., Potter M. L., et al. (2018). Fecal microbiota transplantation is associated with reduced morbidity and mortality in porcine circovirus associated disease. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.01631
Ohara T. (2019). Identification of the microbial diversity after fecal microbiota transplantation therapy for chronic intractable constipation using 16s rRNA amplicon sequencing. PLoS One 14, e0214085. doi: 10.1371/journal.pone.0214085
Oladele P. O., Richert B. T., and Johnson T. A. (2024). 72 effect of in-feed fecal microbiota transplant on growth performance and gut microbiota dynamics of piglet subjected to post-weaning stress. J. Anim. Sci. 102, 47–48. doi: 10.1093/jas/skae102.056
Park M. J. and Sohrabji F. (2018). Abstract wp107: estrogen deficiency induces microbiota dysbiosis impacting stroke outcome in female rats. Stroke 49, AWP107–AWP107. doi: 10.1161/str.49.suppl_1.WP107
Perez-Palencia J. Y., Noah R., Haydon K. D., and Levesque C. L. (2022). Psi-3 effects of increasing dietary arginine supply on pig growth performance following weaning stress. J. Anim. Sci. 100, 197–198. doi: 10.1093/jas/skac064.333
Pluske J. R., Turpin D. L., and Kim J. C. (2018). Gastrointestinal tract (gut) health in the young pig. Anim. Nutr. 4, 187–196. doi: 10.1016/j.aninu.2017.12.004
Quanhang X. and Jian P. (2019). 350 the effect of early intervention with fecal microbiota transplantation combined c. butyricum and s. boulardii on weaning stress of piglets. J. Anim. Sci. 97, 94–95. doi: 10.1093/jas/skz258.195
Sarah A., Natalie H., Diptaraj C., Augusto S., Xiang Z., Sarah N., et al. (2024). Fecal transplant from long living ames dwarf mice alters the microbiome and biomarkers of liver health in control mice. Innovation Aging 8, 1141–1141. doi: 10.1093/geroni/igae098.3659
Song Q., Gao Y., Liu K., Tang Y., Man Y., and Wu H. (2024). Gut microbial and metabolomics profiles reveal the potential mechanism of fecal microbiota transplantation in modulating the progression of colitis-associated colorectal cancer in mice. J. Trans. Med. 22, 21. doi: 10.1186/s12967-024-05786-4
Song Y., Wang N., Chen L., and Fang L. (2021). Tr1 cells as a key regulator for maintaining immune homeostasis in transplantation. Front. Immunol. 12. doi: 10.3389/fimmu.2021.671579
Su Y., Li X., Li D., and Sun J. (2021). Fecal microbiota transplantation shows marked shifts in the multi-omic profiles of porcine post-weaning diarrhea. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.619460
van der Beek S. L., Zorzoli A., Çanak E., Chapman R. N., Lucas K., Meyer B. H., et al. (2019). Streptococcal dtdp-l-rhamnose biosynthesis enzymes: functional characterization and lead compound identification. Mol. Microbiol. 111, 951–964. doi: 10.1111/mmi.14197
Wang S., Chen H., Wen X., and Fan X. (2021). The efficacy of fecal microbiota transplantation in experimental autoimmune encephalomyelitis: transcriptome and gut microbiota profiling. J. Immunol. Res. 2021, 4400428. doi: 10.1155/2021/4400428
Wang H., Fan Q., Wang Y., Yi L., and Wang Y. (2024). Multi-omics analysis reveals genes and metabolites involved in streptococcus suis biofilm formation. BMC Microbiol. 24, 297. doi: 10.1186/s12866-024-03448-5
Winters J. F. M., Kobek-Kjeldager C., Foldager L., Tecles F., and Pedersen L. J. (2023). Stress responses in pigs postweaning: effect of heavier hybrid and weaning intact litters. Appl. Anim. Behav. Sci. 269, 106106. doi: 10.1016/j.applanim.2023.106106
Xu B., Fu J., Zhu L., Li Z., Jin M., and Wang Y. (2021). Overall assessment of antibiotic substitutes for pigs: a set of meta-analyses. J. Anim. Sci. Biotechnol. 12, 3. doi: 10.1186/s40104-020-00534-2
Xu B., Qin W., Chen Y., Tang Y., Zhou S., Huang J., et al. (2023). Multiomics analysis reveals gut microbiotaovary axis contributed to the follicular development difference between meishan and landrace×yorkshire sows. J. Anim. Husbandry Biotechnol. 14, 1954–1968. doi: 10.1186/s40104-023-00865-w
Keywords: 16S rRNA, metabolomics, weaned Tibetan piglets, growth performance, gut microbiota
Citation: Zhang J, Zhao M, Zhang H, Han M, Tan Z and Shang P (2025) Fecal microbiota transplantation alleviates weaning stress in Tibetan piglets by modulating gut microbiota–metabolite interactions. Front. Anim. Sci. 6:1634097. doi: 10.3389/fanim.2025.1634097
Received: 23 May 2025; Accepted: 21 July 2025;
Published: 26 August 2025.
Edited by:
Yong Su, Nanjing Agricultural University, ChinaReviewed by:
Liang Zhao, Washington State University, United StatesLong Pan, Nanjing Agricultural University, China
Copyright © 2025 Zhang, Zhao, Zhang, Han, Tan and Shang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Shang, bmVtb3NocG1oQDEyNi5jb20=