 David Antony Selby1*†
David Antony Selby1*† Rashika Jakhmola1,2†
Rashika Jakhmola1,2† Maximilian Sprang1,3†Gerrit Großmann1
Maximilian Sprang1,3†Gerrit Großmann1 Hind Raki1,4Niloofar Maani1,5Daria Pavliuk1,5
Hind Raki1,4Niloofar Maani1,5Daria Pavliuk1,5 Jan Ewald6Sebastian Vollmer1,5*
Jan Ewald6Sebastian Vollmer1,5*- 1Data Science and its Applications, German Research Center for Artificial Intelligence (DFKI), Kaiserslautern, Germany
- 2School of Computation, Information and Technology, Technical University of Munich, Munich, Germany
- 3Department of Dermatology, University Medical Center of the Johannes Gutenberg-University Mainz, Mainz, Germany
- 4College of Computing, University Mohammed VI Polytechnic, Ben Guerir, Morocco
- 5Department of Computer Science, University of Kaiserslautern–Landau (RPTU), Kaiserslautern, Germany
- 6Center for Scalable Data Analytics and Artificial Intelligence (ScaDS.AI) Dresden/Leipzig, Leipzig University, Leipzig, Germany
Background: Biomarker discovery and drug response prediction are central to personalized medicine, driving demand for predictive models that also offer biological insights. Biologically informed neural networks (BINNs), also referred to as visible neural networks (VNNs), have recently emerged as a solution to this goal. BINNs or VNNs are neural networks whose inter-layer connections are constrained based on prior knowledge from gene ontologies and pathway databases. These sparse models enhance interpretability by embedding prior knowledge into their architecture, ideally reducing the space of learnable functions to those that are biologically meaningful.
Methods: This systematic review-the first of its kind-identified 86 recent papers implementing BINNs/VNNs. We analyzed these papers to highlight key trends in architectural design, data sources and evaluation methodologies.
Results: Our analysis reveals a growing adoption of BINNs/VNNs. However, this growth is apparently juxtaposed with a lack of standardized, terminology, computational tools and benchmarks.
Conclusion: BINNs/VNNs represent a promising approach for integrating biological knowledge into predictive models for personalized medicine. Addressing the current deficiencies in standardization and tooling is important for widespread adoption and further progress in the field.
1 Introduction
High-throughput technologies have transformed biological research, enabling the collection of large-scale data from different molecular layers and leading to the emergence of multi-omics, an approach that combines information from diverse sources, such as genomics, transcriptomics, proteomics, and metabolomics (Sun and Hu, 2016). This allows researchers to analyse complex interactions and regulatory mechanisms that drive cellular function, disease progression and response to therapeutic interventions, with the potential to identify novel biomarkers and better understand how genetic and environmental factors influence disease (Subramanian et al., 2020).
However, multi-omics analysis is fraught with challenges due to high dimensional, heterogeneous data, requiring methods capable of modeling non-linear relationships across multiple processes. Modern machine learning (ML) techniques, such as deep learning, offer superior predictive capabilities at the expense of interpretability. In this context, even explainable AI (XAI) metrics may lack clear or robust biological interpretations (Hancox-Li, 2020; Slack et al., 2021; Wei et al., 2024).
To meet this demand, visible neural networks (VNNs), also known as biologically-informed neural networks (BINNs), have gained prominence (Selby et al., 2025). Unlike conventional neural networks (NNs), which learn relatively unconstrained functional approximations, VNNs incorporate prior knowledge directly into their architecture: previously “hidden” nodes map directly to entities such as genes or pathways, with inter-layer connections constrained by their ontology (see Figure 1).
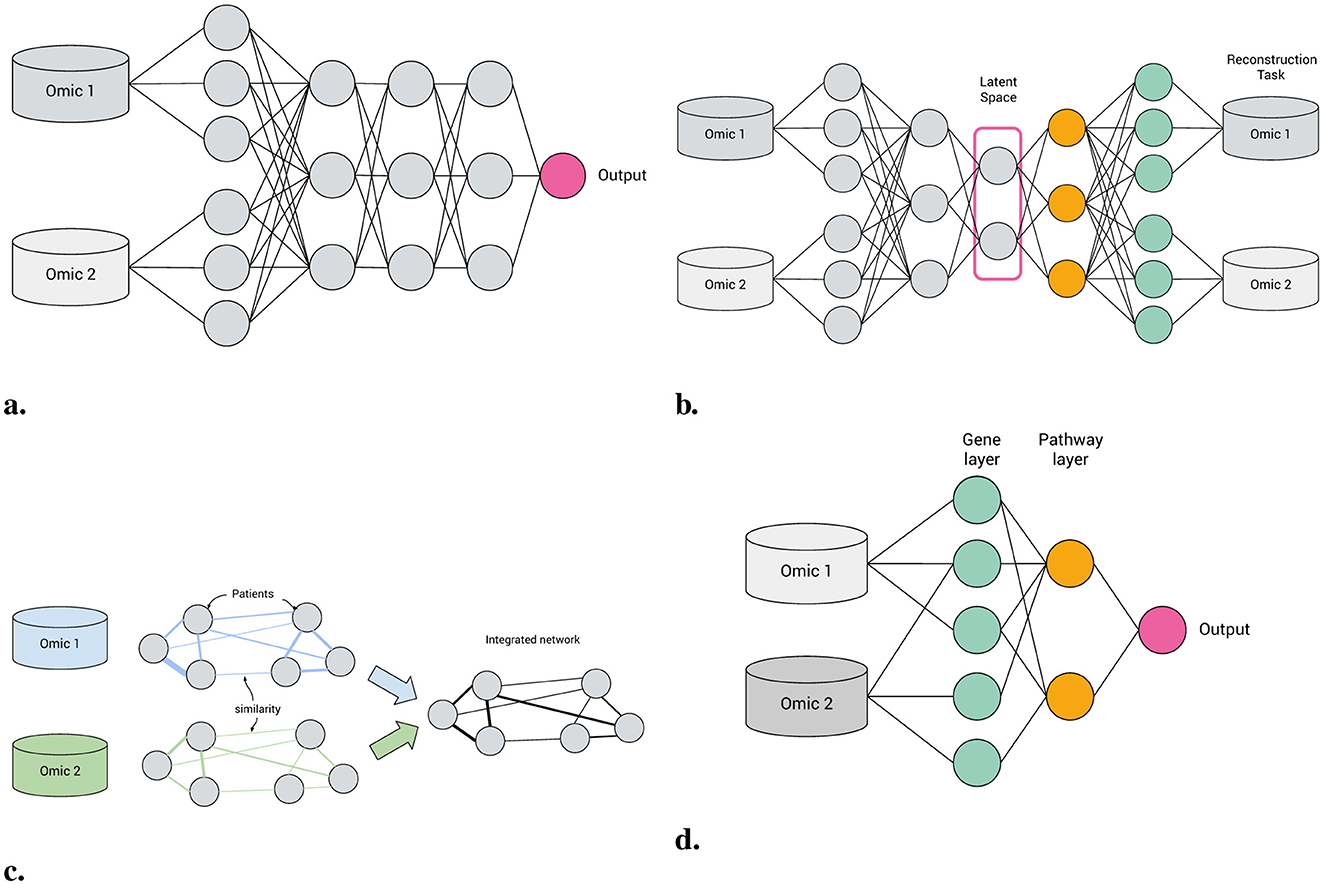
Figure 1. Machine learning approaches to multi-omics integration. (a) Feedforward neural network. (b) Autoencoder. (c) Patient similarity network. (d) Biologically-informed, visible neural network.
VNNs have the potential to enhance biomarker discovery and drug development by learning relationships between genes, pathways, drugs and disease phenotypes. Various VNN models have been proposed, but standard terminology and clear design practices—fostering scientific reproducibility, robustness and generalizability—do not yet exist.
Several existing survey papers offer a high-level overview of interpretable deep learning in omics, but without focussing on detailed comparisons of biologically-informed architectures. This review focusses specifically on VNNs, filling an important gap in the literature. We examine design considerations in constructing different VNN architectures and applying them to different tasks and data sources.
Our contributions are as follows: a taxonomy of BINN architectures, a critical appraisal of the dependencies, assumptions, data sources and tools involved in building BINNs/VNNs; and identification of several research gaps and future work. We consider three underlying research questions:
1. What advantages do BINNs offer over traditional ML models, and how is their relative performance typically benchmarked?
2. Are biological interpretations from VNNs robust to architectural design decisions and reproducible across studies?
3. Can VNNs uncover new scientific knowledge?
2 Background
A living cell is a complex system of interacting molecules, where metabolites and energy are used to form biomass. The cell's regulation is guided by the central dogma of biology: DNA encodes RNA, which, in turn, encodes proteins (Crick, 1970). RNA and proteins both play roles in metabolism, structure, replication, and other cellular processes, interacting with each other and with DNA. These interactions can regulate cellular functions, for example as recruiting transcription factors or opening chromatin in eukaryotes (Kadonaga, 1998). Gene interactions have been extensively studied, expanding our understanding of cellular interaction networks, or pathways (Yeger-Lotem et al., 2004). Pathways connect entities such as genes, proteins and metabolites with other tissue components, reflecting cellular functions from simple growth to complex immune response. Databases describing such molecular interactions are publicly available (see Section 5) representing decades of biomedical research into causal relationships.
Multi-omics analysis uses data from heterogeneous sources to describe biological processes (Hasin et al., 2017; Subramanian et al., 2020). Modeling such sensitive and expensively collected data necessitates modern statistical methods that exploit existing domain knowledge to solve the “small data” problem (Rahnenführer et al., 2023). Biomarker discovery in single-omics data often relies on regularized linear models fit to a target of interest. Biomarkers are derived from signals of significant features, with enrichment analysis providing insights into their biological functions, or the features with largest coefficients identified as targets for validation in the laboratory (Ng et al., 2023).
Explainable AI (XAI) is a growing field of research that aims to produce methods to explain the reasoning behind ML models' predictions. Ante-hoc explainability refers to models that are intrinsically interpretable, whilst post-hoc methods are a way of gaining insights about “black box” model predictions in terms of their inputs (Retzlaff et al., 2024). Statistical models (or “data models”; Breiman, 2001), such as generalized linear models, are preferred when the main goal is inference: understanding relationships between variables that describe a natural process. On the other hand, when accurate prediction is a priority, especially when datasets are large and high-dimensional, black-box models (“algorithmic modelling”), are preferred, and capable of learning signals from data with intrinsic structure, such as images and sequences. However, larger samples and computational resources are necessary for such methods to outperform simpler models on tabular data (Grinsztajn et al., 2022).
To simplify the learning process, the search space can be constrained through provision of additional information from prior domain knowledge. Many taxonomies exist for these methods (Van Harmelen and Ten Teije, 2019; Karniadakis et al., 2021; Dash et al., 2022). Data augmentation involves generating additional data through preprocessing, e.g. varying contrast of an input image. Similarly, Yang et al. (2019) consider metabolite perturbations as input, but enrich this data through simulation. Another method involves crafting specialized loss functions for specific domains. For instance, Jia et al. (2019) uses a loss based on physical laws of energy distribution. Most relevant for this review is constraining the computational graph, creating an inductive bias in the neural network. Convolutional layers, for example, consider the dependence of neighboring pixels in an image, while neural ordinary differential equations (Chen R. T. et al., 2018) account for the structure of physical processes. VNNs also fall into this category and a taxonomy is given in Section 5.
Knowledge graphs (KGs) (Hogan et al., 2021) can integrate data from various sources, including scientific literature. Increasingly used in bioinformatics to encode and retrieve complex knowledge, KGs describe entities—including genes, proteins, pathways, diseases, and functional annotations—across multiple levels of organization (Yi et al., 2022). They are often built upon pathway databases, such as Reactome (Croft et al., 2011), Gene Ontology (The Gene Ontology Consortium et al., 2023), or KEGG (Kanehisa, 2000), that provide curated ontologies focussing on cellular processes, metabolism, interactions or signaling. These databases or networks can themselves be seen as “simple” KGs with restricted set of entities and relations. Biological KGs combined with network analysis methods offer the potential to discover or explain new relationships between drugs, genotypes and phenotypes (Chandak et al., 2023) and their embeddings have been employed in biomarker discovery (Galluzzo, 2022), multi-omics integration (Xiao et al., 2023) and drug-target discovery (Zahra et al., 2023); for example, GLUE is a KG-based architecture that integrates omics features based on regulatory interactions (Cao and Gao, 2022).
Constructing a NN based on such databases is a form of knowledge-intensive ML. Integrating the most general KGs may result in very large NNs that fail to learn relevant patterns. Prior knowledge integration is a universality–generalizability tradeoff: compared to a dense neural network, a VNN's function space is reduced, losing universality but generalizing better to the real-world context of the specific biological systems of interest (Miao, 2024). In most cases, this is achieved by leveraging pathway databases such as Gene Ontology, KEGG or Reactome to inform the design of hidden layers in the network, ensuring that (some or all of) the model's internal representations align with known entities and relationships. VNNs aim to emulate signaling processes, enhancing interpretability and biological relevance, thereby accurately modeling complex systems and facilitating discovery of novel insights. As Hanczar et al. (2020) observe, this approach aims to bridge the gap between data-driven ML models and mechanistic understanding, making it particularly valuable in fields like genomics and systems biology, where omics data lack the structure of imaging or text data commonly exploited by popular deep learning frameworks.
2.1 What's in a name?
“Visible neural network” (VNN) is one of myriad terms to describe a NN model whose hidden layers and connections are constrained by a pre-specified ontology; terminology is far from standardized, making literature search difficult. “Visible” emphasizes interpretability without restricting to biological applications: similarly, knowledge-primed (Fortelny and Bock, 2020), knowledge-guided (Lee and Kim, 2022; Hao et al., 2022) or knowledge-based neural networks (Ciallella et al., 2021), or ontology-based autoencoders (Doncevic and Herrmann, 2023; Joas et al., 2024) allow for hierarchical structures beyond biology. “BINNs” can also refer to biologically-informed neural-symbolic methods (Lagergren et al., 2020; Przedborski et al., 2021; Rodríguez et al., 2023), or biologically-inspired NNs found in connectomics (Klinger et al., 2021) or neuromorphic computing (Yamazaki et al., 2022). Here, “visibility” refers to the direct incorporation of biological pathway structures into the neural network architecture, offering intrinsic interpretability, such that VNNs sit within the broader category of BINNs.
3 Related work
VNNs are a growing sub-field at the intersection of deep learning-based multi-omics integration, interpretable ML methods for biology, and XAI more generally. Across these areas, various survey papers have been published that mention the concept of VNNs and BINNs, however, most do not discuss this area in detail. An overview of these review articles, including their scope and limitations, is given in Table 1.
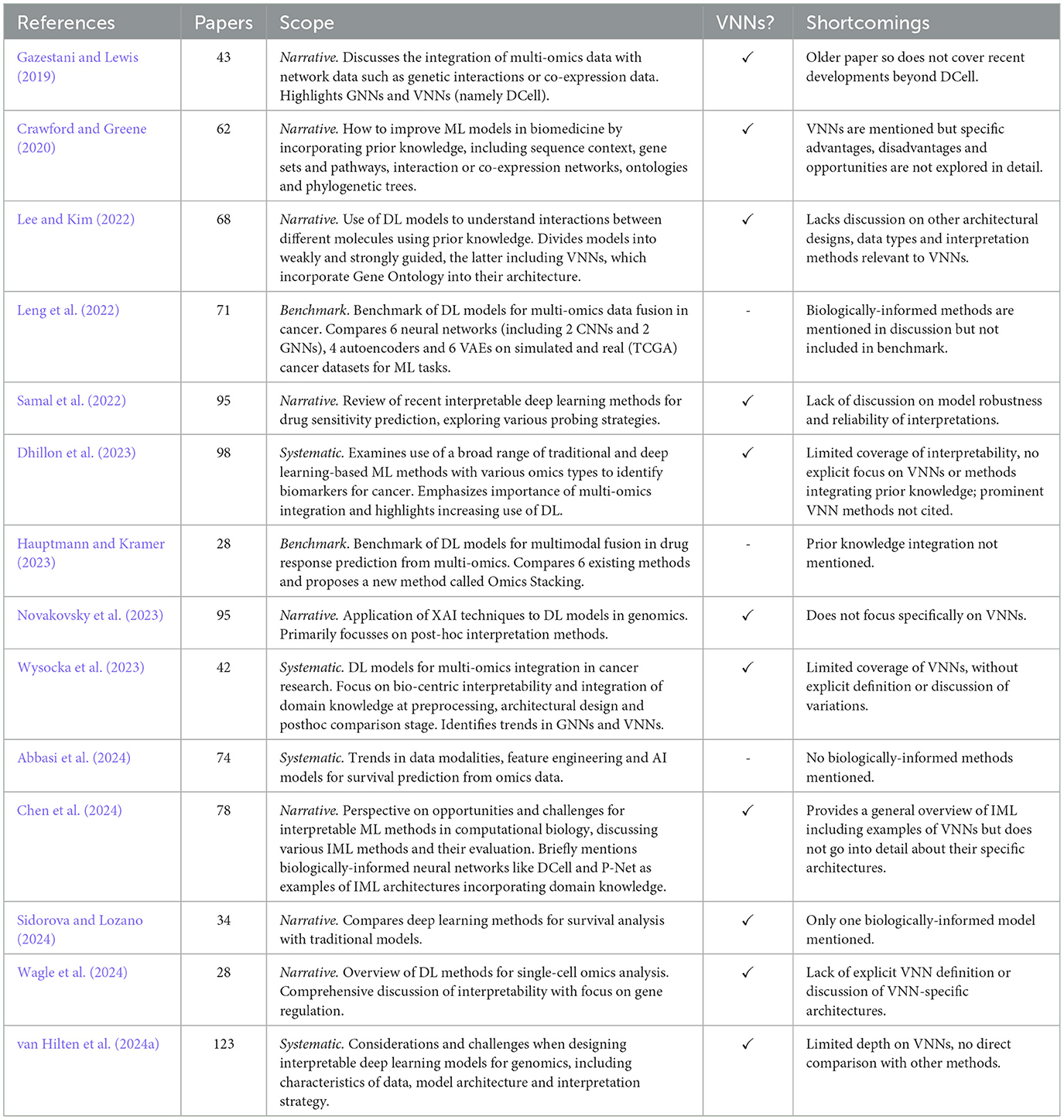
Table 1. Existing review papers on explainable deep learning for multi-omics.
We consider three categories of reviews: narrative reviews, systematic reviews and quantitative benchmarks.
3.1 Narrative reviews
Gazestani and Lewis (2019) presented an early review of methods of ontology integration, including methods with hierarchical layers defined by gene ontology (i.e., VNNs), as well as network propagation models, a special case of GNNs. Crawford and Greene (2020) reviewed approaches for embedding biological structures into machine learning models, including sequence encodings, network embeddings and models constrained by ontological structures. Our review specifically narrows down to VNNs that use pathways and ontologies for structured input integration. Other relevant works include Novakovsky et al. (2023) on explainable AI in genetics, and Lee and Kim (2022), which distinguishes between “weak” (e.g., data-driven GNNs) and “strong” biological guidance (e.g., structured VNNs). Wagle et al. (2024) provided a review of interpretable deep learning models specifically for single-cell omics, highlighting the importance of model transparency in this context. Chen et al. (2024) provided a broad perspective on interpretable ML in computational biology. They mention biologically informed methods and categorize them, together with attention based algorithms, as “by-design” methods, which they see as naturally interpretable. Samal et al. (2022) reviewed interpretable deep learning models for drug sensitivity prediction, exploring various “probing” strategies for examining how input data is processed within the network, including some biologically-informed approaches. A brief and accessible high-level overview of BINNs is also given in Selby et al. (2025).
3.2 Systematic reviews
Wysocka et al. (2023) conducted a systematic review of BINNs. They derived a taxonomy of pre-processing, in-processing and post-hoc biological interpretability. Their review spans 42 publications up to 2022, sourced from PubMed and the Web of Science. Focussing on oncology, it covers a range of interpretability methods, including GNNs and post-hoc explainability measures for conventional deep learning models. About 10 models whose architecture is “explicitly defined” by domain knowledge are included. However, the relative merits of these architectures or their underlying assumptions are not critically evaluated. Dhillon et al. (2023) offered a more general systematic review of ML methods for multi-omics-based biomarker identification—again focussing on cancer—without a specific focus on biologically-informed models like VNNs. Abbasi et al. (2024) reviewed a large number of methods for survival analysis in multi-omics, using deep learning, traditional ML and statistical methods, but not any methods for knowledge integration. A recent systematic review paper by van Hilten et al. (2024a) includes 24 VNN papers, representing probably the most complete survey to date. They compare the popularity of VNNs with other methods, noting the sparsity of VNNs allows handling a greater number of input features than computationally intensive transformer models, though conventional NNs remain the most popular approach. However, the review does not distinguish different VNN architectures or discuss specific design decisions.
3.3 Benchmarks
Leng et al. (2022) performed a benchmark of deep learning models for multi-omics data fusion in cancer, comparing 6 NNs (including two CNNs and two GNNs), four autoencoders and six variational autoencoders on simulated and real cancer datasets for supervised and unsupervised learning tasks. However, their analysis did not cover any NNs with biologically-informed architectures. Similarly, Hauptmann and Kramer (2023) evaluated various deep learning-based multi-modal fusion approaches in drug response prediction and proposed a new method called Omics Stacking, but did not explore any VNNs. Other works extending specific methods include benchmarks of one or more VNN approaches (e.g., Esser-Skala and Fortelny, 2023): we discuss these in Section 5.
3.4 The gap we fill
In summary, most existing reviews either focus broadly on multi-omics ML approaches or are limited to specific contexts such as cancer or general interpretability methods. Our systematic review addresses this gap by focussing specifically on visible neural network architectures. We aim to provide a more detailed analysis of technical implementations, discussing the topic of robustness and reproducibility, covering the latest works in this area. Up-to-date coverage includes recent papers from 2024, focussing on applications beyond oncology and with an examination of ways in which VNNs have been both designed, implemented and evaluated, summarizing comparisons with traditional models and with each other.
4 Methods
Our aim was to compare architectural design considerations in BINN-like models and identify key contributions in the field. Thus we sought papers—for which the full text was available, to understand the model structure—using NN models informed by biological ontologies, applied to tabular (multi-)omics datasets.
The systematic review was conducted following the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA; Page et al., 2021) guidelines to ensure transparency and rigor. The PRISMA flow diagram (Figure 2) outlines the process of study identification, screening, eligibility assessment and inclusion.
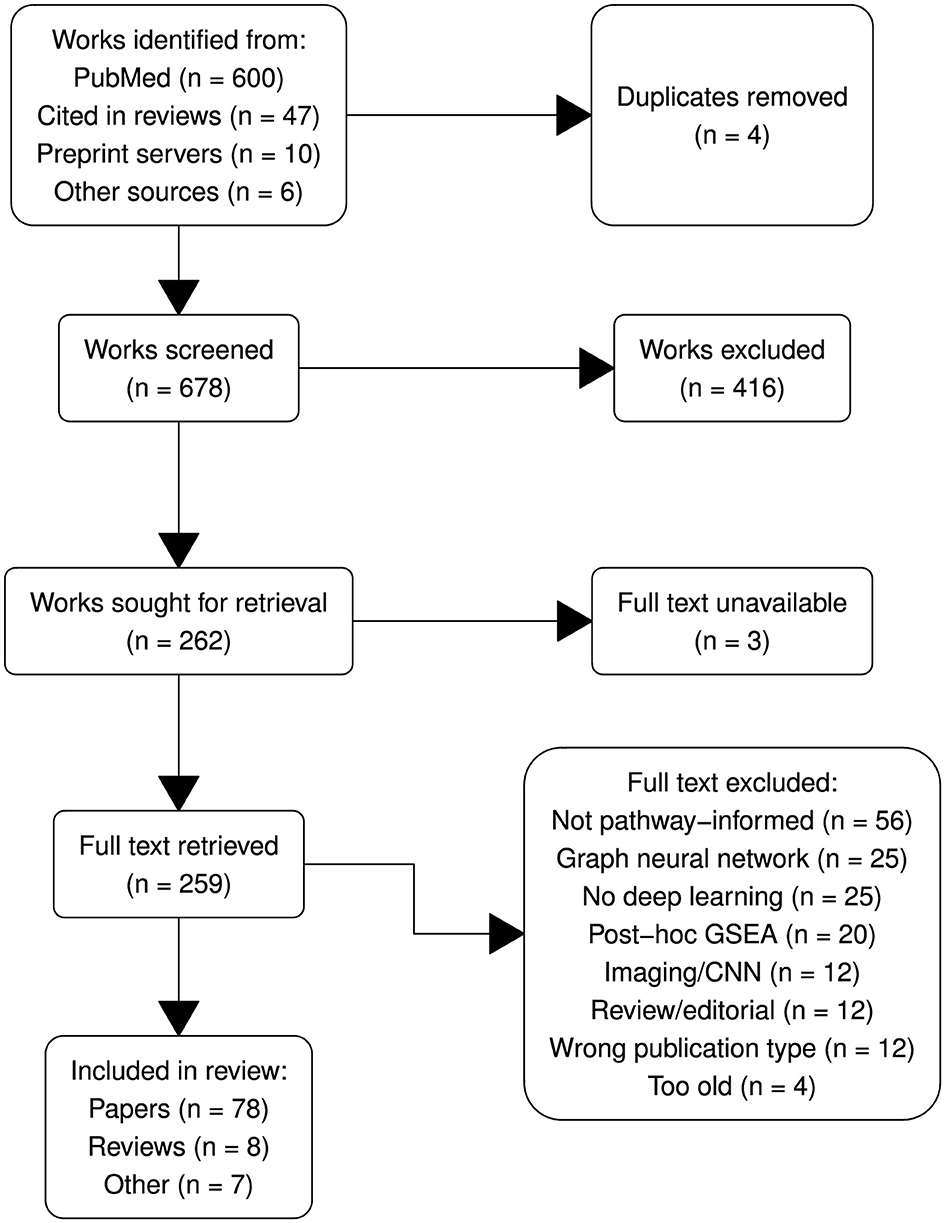
Figure 2. PRISMA diagram summarizing article selection.
4.1 Search strategy
We conducted a comprehensive search using PubMed to identify relevant papers. Unlike Wysocka et al. (2023) we chose not to restrict our search to works in oncology, instead focussing on biologically informed models in any application with the query multi-omics AND (deep learning OR computer science OR neural networks OR network analysis OR machine learning) AND (biologically informed), filtered to works published in the period 2018–2024: spanning from the earliest known BINN papers to the most recent publications at the time of retrieval.
However, non-standard terminology in this subfield means this query alone may not capture all relevant papers. For example, Ma et al. (2018) was one of the first papers to use the term “visible neural network,” but “multi-omics” or “omic” does not appear anywhere in the article, nor do phrases like “biologically informed” or “pathway.” Other relevant works may be published in conferences or periodicals not indexed by PubMed. Rather than contorting our search query to detect these (admittedly significant) edge cases, we opted to augment our main query with a Google Scholar search for “visible neural networks” as well as adding selected papers manually, including 47 works already included in existing reviews.
Periodically, our database of results has been updated by adding new articles reported as citing key review papers, according to Google Scholar. In this way we were able to include several very recently published works, e.g., Meirer et al. (2024); Liu P. et al. (2024).
4.2 Screening
After removing duplicates, a total of 678 unique records were screened by title and abstract using inclusion and exclusion criteria as defined above. The screening was performed by three reviewers—with backgrounds in statistics, computer science and bioinformatics—working independently. Disagreements were resolved by majority vote, following a discussion drawing on each reviewer's respective expertise.
4.3 Eligibility
Full-text versions of 262 articles were assessed for eligibility against the predefined criteria, which included methodological journal or conference papers proposing neural network approaches to model omics data that integrate prior biological knowledge into their network architecture. Studies were excluded at this stage if they did not involve NNs, if they were fully connected, non-biologically informed structures, or if biological interpretations only took the form of post-hoc gene-set enrichment analysis (GSEA). We focussed our attention on sparsely-connected feedforward neural networks (FFNNs) and autoencoders, while convolutional, attention or graph-based models were generally excluded, as were those designed solely for genomic/proteomic sequences or imaging data.
4.4 Inclusion
86 papers met the eligibility criteria and were included in the final analysis. These studies represent a variety of papers proposing or evaluating biologically-informed neural network architectures, as well as several relevant survey papers (see Section 3). We also highlight 7 “honorable mentions:” papers that do not strictly meet the inclusion criteria but are interesting examples of alternative approaches.
4.5 Reporting
The full list of included papers is given in Appendix Table A1. The PRISMA checklist is provided in the Supplementary materials.
4.6 Bibliometrics
Citation counts and relationships were retrieved from the CrossRef API using the R package rcrossref (Chamberlain et al., 2022) to construct a citation network between the papers (see Figure 3). We verified the existence of individual works in PubMed using the NCBI eUtils API

Figure 3. Bibliometric network of VNN papers, scaled by number of citations and shaded by year of publication.
5 Results
5.1 Overview
Our review examines the key advancements in visual neural networks (VNNs), focussing on taxonomy, architectural innovations, methods for evaluation and practical applications. The review identified major developments across diverse architectures—feed-forward neural networks, autoencoders and graph neural networks—evaluated on a variety of omics datasets and knowledgebases. A critical theme is the tradeoff between sparsity for interpretability and the robustness of explanations in complex biological datasets. We also explore how specific design decisions, such as data integration strategies and pathway representations, influence model performance.
We consider “strong” biological guidance (Lee and Kim, 2022) where models are constrained by prior knowledge, specifically the “in-processing” paradigm (Wysocka et al., 2023) where such information informs the model's internal architecture. By contrast, wrangling data into a graph structure and applying non-dedicated ML algorithms (such as GNNs) constitutes biologically-informed “data pre-processing” (Wysocka et al., 2023) and is not our focus here; similarly we do not dwell on post-hoc model explanations or enrichment analyses for non-biologically-informed models (e.g. Hanczar et al., 2020).
Post-2020, VNN research has grown significantly, with key works like DCell (Ma et al., 2018), DrugCell (Kuenzi et al., 2020), and P-Net (Elmarakeby et al., 2021) central to the citation network (Figure 3). P-Net has been reproduced by Pedersen et al. (2023) and built upon by Hao et al. (2018a); Hu et al. (2022); Hartman et al. (2023), applying the framework to other modalities and targets. Similarly, MOViDA (Ferraro et al., 2023) builds upon DrugCell, itself an extension of DCell. Approximately half of recent studies are uncited by prior reviews (see Figure 2), indicating untapped contributions. A full list of retrieved works and associated abbreviations is given in Appendix Table A1.
5.2 Taxonomy of architectures
Table 2 divides works according to their broad architecture: feed-forward neural networks (Figure 1d) and autoencoders (Figure 1b) constitute the majority of VNNs, but there also exist some GNNs and CNNs with apparent pathway-aware properties, and other notable approaches.
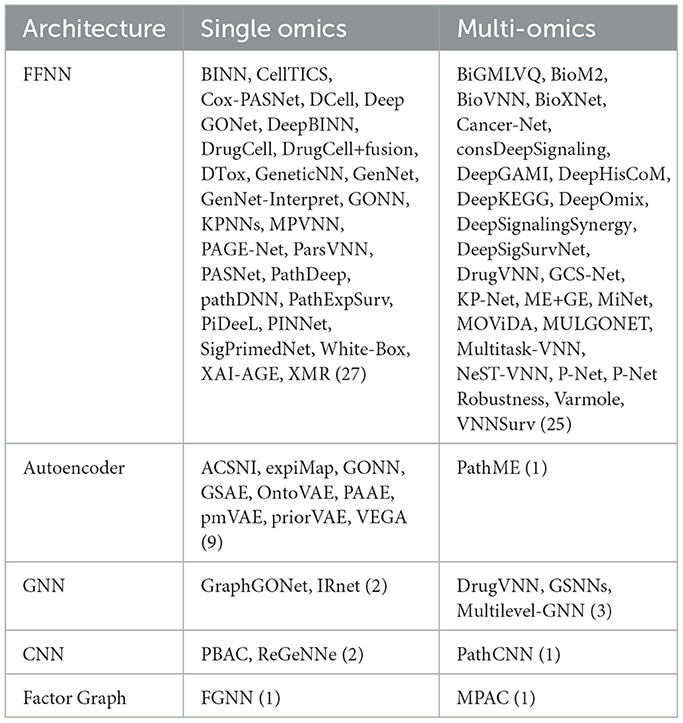
Table 2. Architectures used in included papers (numbers of papers in parentheses).
A VNN is a neural network model with at least one layer of structural sparsity based on connections not directly observable in the main data. Hence, patient similarity networks (Pai and Bader, 2018, Figure 1c) are not VNNs, because their inter-node connections are derived from the data.
Figure 4 gives a prototypical illustration, with various features of VNN models, described in detail later in this section and tabulated in Table 3. From left to right [input(s) to output] the model's abstraction becomes progressively more complex as later levels in the neural network correspond to higher tiers in the pathway hierarchy. Such a model can be constructed from a FFNN, with numbers of hidden layers and nodes within each layer corresponding to the desired gene and pathway levels. Initially densely connected, a masking matrix removes inter-layer connections that do not correspond to known biological relations.
• Feed-forward networks A “standard” NN for supervised learning, FFNNs may be applied to classification, regression or survival analysis tasks. Feed-forward VNNs may have deep structures with multiple layers of nested pathways, such as DCell (Ma et al., 2018) and P-Net (Elmarakeby et al., 2021); however the simplest networks, e.g., Cox-PASNet (Hao et al., 2018b) include just one pathway layer.
• Autoencoders Autoencoders are a class of deep learning models for unsupervised learning and consist of three components: an encoder, a latent space and a decoder (see Figure 1b). Input features are encoded through one or more hidden layers into a “bottleneck” layer representing a low-dimensional latent space; a decoder, typically mirroring the architecture of the encoder, then reconstructs the input features from this space. Some of the earliest BINNs are autoencoders, e.g., GSAE (Chen H.-I. H. et al., 2018). A typically used extension of this idea are variational autoencoders (VAE) which are probabilistic and generative models learning a multivariate distribution (e.g., Gaussian) as latent space, enabling latent space disentanglement and compactness. Examples include VEGA (Seninge et al., 2021) and ExpiMap (Lotfollahi et al., 2023); other models come in both AE and VAE variants, e.g., PAAE/PAVAE (Avelar et al., 2023), or FFNN and AE variants, e.g. GONN/GOAE (Peng et al., 2019). Liu B. et al. (2024) proposed “pathway-informed priors” where knowledge is integrated into the VAE loss term.
• Convolutional neural networks CNNs have been widely adopted in the analysis of structured datasets like images and sequences. Applying CNNs to multi-omics data is possible by combining with other data modalities such as sequences (DeepGo; Kulmanov et al., 2018) or by synthesizing “pathway images” from tabular data (PathCNN; Oh et al., 2021).
• Graph neural networks GNNs can directly leverage the topological structure of biological data, making them particularly well suited for tasks where the relationships between entities need to be explored. For example, GraphGONet (Bourgeais et al., 2022) uses the structure of the Gene Ontology graph directly; Yan et al. (2024) used a GNN to embed genes, followed by a “pathway aggregation block” resembling a FFNN, to obtain pathway-level features. As GNNs operate primarily through learned graph representations, they do not inherently enforce predefined biological hierarchies, which can make them less suitable for applications where pathway-level interpretability and strict adherence to known biological relationships are necessary.
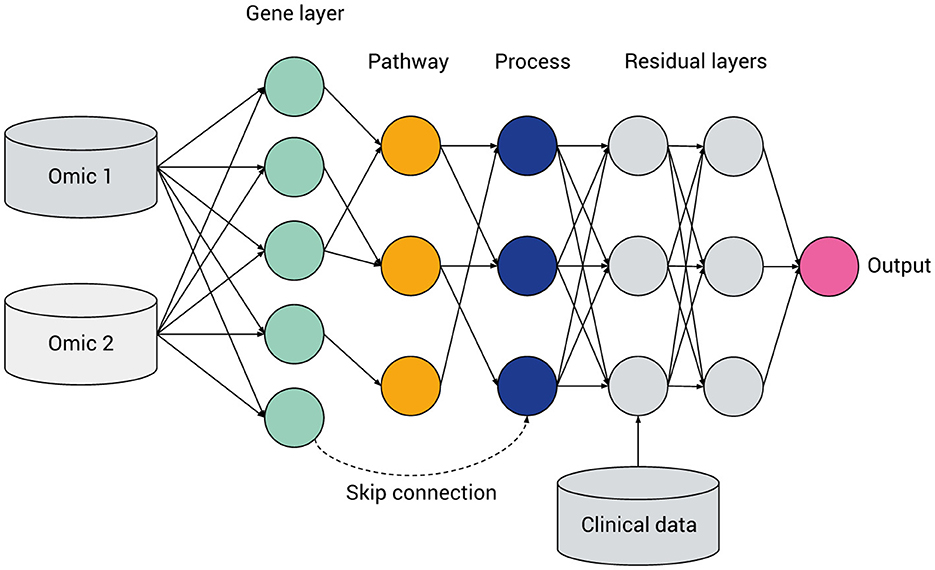
Figure 4. General hierarchical structure of a visible neural network, featuring multi-omics integration, a hierarchy of genes, multi-level pathways or biological processes (green, yellow, and blue, respectively), a ragged structure with skip connections and layers containing fully connected “residual” nodes. Data not mappable to pathways can be included via intermediate or late fusion.
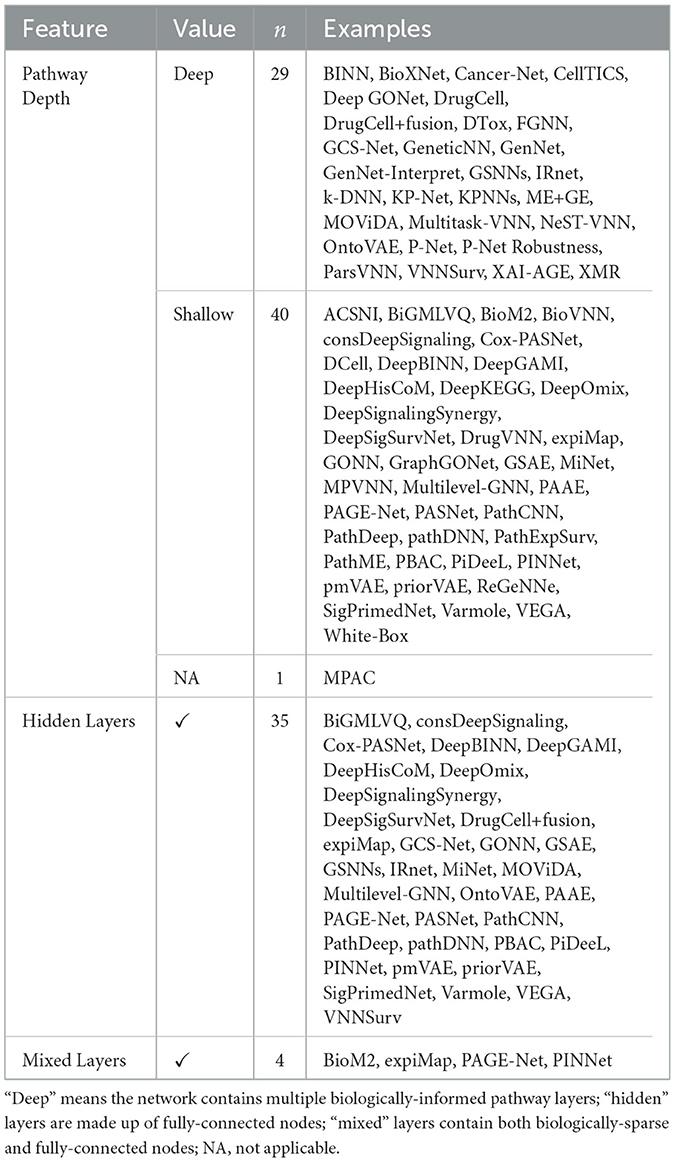
Table 3. Architectural features in included papers.
Some authors adopt hybrid approaches, for example: ReGeNNe (Sharma and Xu, 2023) combines GNN and CNN layers; GraphGONet (Bourgeais et al., 2022) claims to offer advantages of an FFNN and a GNN; DeepKEGG (Lan et al., 2024) introduces a “pathway self-attention module.” Biologically informed generalized matrix learning vector quantization (BiGMLVQ; Voigt et al., 2024) bills itself as a non-deep-learning approach but is nevertheless NN based. BioM2 (Zhang et al., 2024) performs pathway-level feature selection, concatenating features with filtered inputs unmapped to pathways. While this approach can be performed in a deep learning pipeline, the authors note it could also be performed using multi-stage logistic regression or other machine learning methods. In a more network focused approach, Kim et al. (2018, 2019) employ a random walk algorithm across an integrated pathway network based on the Kyoto Encyclopædia of Genes and Genomes (KEGG) pathways to derive pathway-level scores for downstream tasks such as survival prediction. However, this approach does not directly incorporate a biologically informed neural network structure. Uzunangelov et al. (2021) present AKLIMATE, an example of a stacked kernel learner incorporating pathway knowledge.
5.3 Data and applications
Figure 5 shows gene expression (transcriptomics/mRNA) is the dominant omics modality, featuring in more than half of papers, followed by copy number variations (CNV); DNA mutations, including single nucleotide polymorphisms (SNPs), were also a common data type. As shown in Table 2, 31 models integrated multi-omics data, with some models such as P-Net (Elmarakeby et al., 2021) ostensibly applicable to any number and combination of omics levels (in the paper itself they used just two).
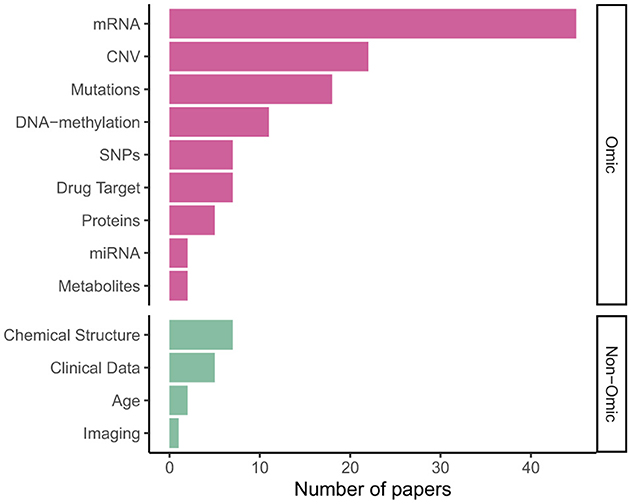
Figure 5. Input data types in published VNN models.
A limited number of models—especially for survival analysis—integrated omics or multi-omics data with “non-omics” inputs not mappable to pathways: Cox-PASNet (Hao et al., 2018b) and MiNet (Hao et al., 2019), combine gene expression, DNA methylation and copy number variations with clinical data, such as patient age, via late fusion. DrugCell (Kuenzi et al., 2020; Greene and Costello, 2020) extended DCell (Ma et al., 2018) by integrating mutation data with embeddings of the chemical structure of different drugs, also via late fusion; however later analysis by Nguyen et al. (2024) found mid- or early fusion models performed better. Other examples of such drug–genotype modular networks include BioXNet (Yang et al., 2024), MOViDA (Ferraro et al., 2023) and XMR (Wang et al., 2023).
As shown in Appendix Table A2, oncology is a major application area for VNNs, with 46 papers; 13 were concerned with drug response (e.g., consDeepSignaling; Zhang et al., 2021); 4 with cellular processes; 3 with schizophrenia and 3 with COVID-19. While this shows cancer research is significant, VNNs are not limited to this disease.
Many authors used data from common sources, with omics data from the Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) particularly popular; see Figure 6. Some authors used their own collected data or else did not clearly specify an existing database as their source. Use of common data sources may make replication of results more straightforward (nearly every paper provided analysis code: Appendix Table A4) but may also raise questions about their real-world generalizability, given sampling biases present in these cohorts. (KEGG) and Reactome were equally commonly chosen as sources of biological ontology information, followed by Gene Ontology and MSigDB. All are publicly accessible databases. Kim and Lee (2023) highlighted differences in performance for the downstream task depending on the ontology used.
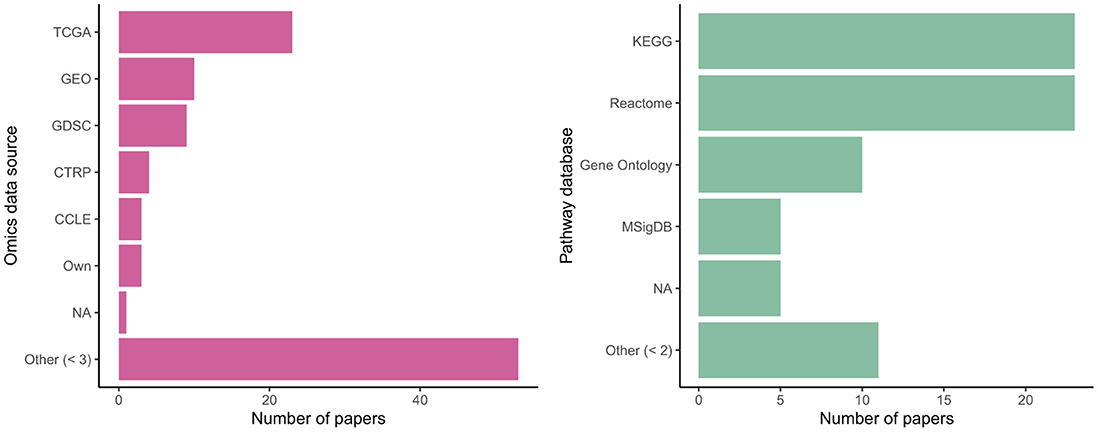
Figure 6. Sources of omics data and pathway knowledge in biologically-informed models.
5.4 Evaluating biologically informed architectures
Assessing the usability of VNNs requires comprehensive evaluation of predictive performance, interpretations, and robustness against appropriate baselines. Of included papers, 56 compared with traditional (non-NN) models and 54 compared with non-biologically-informed NNs, with just 22 comparing their proposed models with other BINN variants, meaning most VNNs are evaluated in isolation to other biologically-informed models.
5.4.1 VNNs perform similarly to denser models
Fortelny and Bock (2020) posited that BINNs operate as “information processing units” using network-based computations to regulate their states. Thus, by mirroring cellular regulatory systems, VNNs provide a deeper understanding of these mechanisms, and can offer similar prediction performance to “black box” fully-connected networks while being significantly more sparse. However, redundancy in the structure of VNNs leads to variability in edge weights, making explanations less robust—a problem that may be mitigated via dropout on hidden nodes during training. Similarly, Huang et al. (2021) argued that redundancy among nodes and edges can lead to overfitting and less robust explanations; they proposed a pruning mechanism to mitigate the issue, resulting in model simpler than DrugCell while offering superior accuracy: their ParsVNN model reduced computation time and memory footprint and number of included genes by up to 90%, with statistically significant increases in predictive performance across five datasets.
Kuenzi et al. (2020) compared their VNN to a “black-box” neural network with equivalent depth and sparsity, finding broadly similar performance and superior accuracy to a non-deep-learning model. In contrast, Pedersen et al. (2023) compared P-Net with a randomly-connected network. They observed a clear decrease in performance for the random connections in comparison to the BINN. Lin et al. (2017) also found biologically-sparse networks outperformed fully-connected models. However a fully-connected network with random masked nodes is not necessarily optimal for multi-omics if it assumes early fusion (Hauptmann and Kramer, 2023); VNNs have been shown sensitive to fusion stage (Nguyen et al., 2024), so but early-integrated baselines could be “strawmen.”
5.4.2 Performance is sensitive to architecture
Several studies have compared different biologically informed architectures on the same data to assess their relative benefits. For instance, PINNet (Kim and Lee, 2023) was compared against traditional ML models and dense NNs. They also compared two types of VNNs, informed by GO and KEGG respectively, which outperformed the non-biologically-informed models. Meanwhile, PBAC (Deng et al., 2024) conducted an ablation study on its architecture, revealing that removing the biological information mask and the attention layer, respectively, reduced the performance of drug response prediction. However, they did not benchmark their architecture against other models.
5.4.3 Robustness of interpretations
Since VNNs rely on the same biological knowledge as commonly used gene- or pathway-enrichment analysis frameworks, they inherit similar problems, such as balancing between specific pathways with few genes and broad pathways with many genes. Additionally, VNNs can be prone to overfitting and instability resulting from small sample sizes, leading to significant changes with different train-test splits or initializations (Esser-Skala and Fortelny, 2023). Common strategies for the training of VNNs are the introduction of dropout layers to stabilize training, in particular when overlapping ontology terms are used. Further, weights can be restricted e.g. by weight decay configuration or by restricting the direction (positive weight) to fix direction. Additionally, Esser-Skala and Fortelny (2023) assessed the robustness of interpretations in several models, including DTox and P-Net (Hao et al., 2022; Elmarakeby et al., 2021).
Meirer et al. (2024) raised the question of robustness of biomarker signatures found by BINNs. Their solution, DeepBINN, fits a sub-network per pathway—each a NN with a fixed number of hidden layers—whose output weight measures that pathway's importance. By comparing the ranks of pathways over successive initializations, the authors claim to yield a “robust” signature.
5.5 Architectural design
Even within the taxonomies listed in Table 2, VNN architectures differ considerably, based on different design considerations highlighted in Figure 4.
5.5.1 Pathway nodes
Some VNN architectures have multiple hidden nodes per gene or pathway, representing their ability to perform multiple tasks concurrently. For example, DCell (Ma et al., 2018) and derivatives (Kuenzi et al., 2020; Ferraro et al., 2023, DrugCell, MOViDA;) have 6 nodes per pathway; OntoVAE (Doncevic and Herrmann, 2023) has 3. However, most other architectures map each entity to at most one node in the network, simplifying the representation—and its interpretation—at the possible expense of predictive power.
Perhaps the most crucial design choice is the selection of the source and type of biological knowledge to be incorporated in a VNN. However, surprisingly little efforts have been made in the field to analyse and study the impact on choosing Reactome pathways, KEGG, GO-terms, or other sources. Since these sources all have their strength and weaknesses in relation to coverage (number of genes represented), overlap between terms or curation quality, a big impact on VNNs and their architectural choices is expected. For example for VAEs authors state that overlap of genes across pathways is countered by dropout layers (Seninge et al., 2021), similarly reported in feed/forward VNNs by Fortelny and Bock (2020). ParsVNN (Huang et al., 2021) approaches the problem of redundant subsystems in VNN models by a pruning mechanism.
Lastly, the chosen database or ontology, as well as the level of hierarchy, ideally matches the purpose of the model e.g., using KEGG metabolic networks for insights into cancer cell metabolism or Reactome immune pathways for studying infections. Currently, most studies using VNNs are explorative and typically do not restrict incorporated biological knowledge in a purpose-driven way. However, PathExpSurv (Hou et al., 2023) uses a “pathway expansion” phase to adaptively adjust pathway-gene connections, potentially enhancing interpretability. Also expanding on the idea of including pathway information, MPVNN (Ghosh Roy et al., 2022) is uses mutation assays to build multiple (mutated) versions of a given pathway.
5.5.2 Auxiliary layers (layer-wise loss)
DCell also introduced “auxiliary layers,” a method for intermediate pathway layers (for models who have them) to contribute to the loss function and to allow attribution to particular hierarchical levels. P-Net adopts a similar approach of adding “predictive layers” with sigmoid activation after each hidden layer, with later layers weighted more highly in the loss function.
5.5.3 Hidden and mixed layers
Some architectures include fully-connected hidden layers that are not a priori biologically interpretable. These layers introduce additional complexity to the network, potentially improving predictive power but at the expense of interpretability. For example, models like PASNet (Hao et al., 2018a) and MiNet (Hao et al., 2019) include this structure. However, the rationale behind including such layers is not always clearly explained.
The inclusion of these hidden layers may allow the model to capture latent interactions not accounted for by existing biological knowledge, but this assumption requires further validation.
Other architectures include fully-connected nodes in the same layer as the interpretable nodes, a design we will call “mixed layers;” with the the additional nodes denoted “residual” nodes for their ostensible aid to model performance or simply “hidden” nodes due to their lack of biological interpretability.
In most works the authors do not mention why they chose a given design. However, some works have performed ablation studies with and without hidden layers or nodes in the architecture: Lin et al. (2017) compare multiple dense NNs and VNNs with one or two fully connected hidden layers, seeing the best overall performance in the networks with two additional hidden layers in a clustering task. They also found pre-training not beneficial in comparison to training from scratch for clustering cells, but it increased performance for cell type retrieval (classification task). In contrast in Voigt et al. (2024), BiGMLVQ the model with more prototype layers (classification layers) performed worse, which the authors attributed to overfitting. Fortelny and Bock (2020) showed that drop-out on pathway level, as well as input layers, increased robustness to redundancies and imbalances inherent to biological networks.
In particular, autoencoders using biological knowledge to guide representation learning rely heavily on the combination of fully-connected encoders and sparse decoders representing pathway or ontology relationships. However, only authors of VEGA (Seninge et al., 2021) studied other possible architectures and combinations while others like expiMap (Lotfollahi et al., 2023) or OntoVAE (Doncevic and Herrmann, 2023) adopted the same general structure and a theoretical analysis or broad benchmark of different encoder-decoder combinations has not been done to our knowledge. Neither are we aware of any work that performed a systematic architecture search with the pathway layers or additional fully connected layers on feed-forward VNNs.
5.5.4 Shallow and deep hierarchies
Biologically-informed sparse neural network architectures involve a kind of inductive bias, wherein the hidden layers represent biological entities or processes and the connections between the layers represent biological relations. In principle, such a network can be constructed by starting with a fully-connected network and then applying a masking matrix corresponding to the existence or strength of relations between the biological entities. However, given heterogeneous pathway databases and data processing pipelines, the method of converting a hierarchy of biological pathways into a neural network is by no means standardized. For example, a pathway may contain genes and other pathways: should the network include skip connections to account for this (see Figure 4)? How many hidden layers or nodes in each layer should be chosen before certain paths are “pruned” from the model?
Many architectures incorporate a single layer for pathways following the gene layer, but others extend this to multiple levels to represent a hierarchy of pathways. The methods for constructing these hierarchical relationships vary across models, and often it is not explicitly clear how a “top-level” pathway or biological process is defined. For instance, decisions on whether intermediate pathways should be merged or truncated are typically not well-documented. Ma and Zhang (2019) proposed factor graph neural networks (FGNNs), which include a single pathway layer, but may be “unrolled” to a deeper structure to add greater expressive power.
The early pathway-guided neural network architectures are rather complex, incorporating deep pathway hierarchies. ParsVNN (Huang et al., 2021) applied proximal alternative linearized minimization to the NP-hard problem of l0 norm and group lasso regularization in order to prune redundant edges between pathways, resulting in simpler structures.
Other authors argue for the simplicity and interpretability of a shallow network (Chen H.-I. H. et al., 2018; Voigt et al., 2024) with just one gene layer aggregating inputs from one or more omics levels, followed by a single pathway layer.
One technical consideration when building a deeper network is how to handle pathways that are not all the same depth of hierarchy. The bottom of a pathway ontology is not well defined, so networks such as P-Net (Elmarakeby et al., 2021) and later Cancer-Net (Pedersen et al., 2023) start from the most abstract level of “biological processes,” defined as Reactome pathways with no parents, successively adding layers for each generation of child pathways, stopping at a user-selected depth, for example, 6 layers. If a branch of the Reactome tree does not have so many levels, then “dummy nodes” are added to allow implementation in a layered machine learning framework like PyTorch. However, since each of these dummy nodes has a nonlinear activation, some information is inevitably lost or altered. In contrast, DrugCell (Kuenzi et al., 2020) employs direct skip connections, which may be more challenging to implement in common ML frameworks but offer greater flexibility in modeling complex biological relationships.
The notion of skip connections can be extended further. Whereas a VNN is typically formulated as a directed acyclic graph with discrete sequential layers, Evans et al. (2024) recently proposed “graph-structured neural networks” (GSNNs)—not to be confused with GNNs—an extension of VNNs that allows for cycles and self-loops. Like GNNs, GSNNs use an input graph as an inductive bias to constrain the information flow in the neural network. Unlike GNNs, GSNNs do not share weights across nodes; instead, each node is associated with its own distinct neural network.
5.6 Scientific discovery
VNNs being more performant than their densely-connected counterparts raises the possibility of discovering pathway structures through regularization. That is, if a VNN really is better than an equivalently sparse model, then neural architecture search, for example via pruning, could in principle learn such a structure without prior knowledge integration (Sprang et al., 2024). Such an approach to scientific knowledge discovery—which would enable prediction of new pathway relations, akin to knowledge graph edge prediction—has not yet been widely explored in the VNN literature.
Though ParsVNN (Huang et al., 2021) prunes irrelevant pathways, it starts with a biologically-informed structure. Similarly DeepHisCoM (Park et al., 2022) and DeepBINN (Meirer et al., 2024) explore non-linear relationships only among existing pathways. PathExpSurv (Hou et al., 2023) extended pathway knowledge integration by performing “pathway expansion” to include pathways that may not exist in the original database. First a biologically-sparse network is trained, then fine-tuned with dense connections; weights that are not regularized in the second stage may be indicative of undiscovered pathways. Mixed layers (see Table 3), containing both interpretable and non-interpretable nodes, might allow models to harness predictive performance beyond the constraints of the pathway database, but so far this has not been applied to discovery of new pathways.
5.7 Resources and tools
Most papers introducing methods provide open-source code for reproducibility (Appendix Table A4), however, the re-usability and maintainability of some of these codebases is not necessarily guaranteed, especially if raw data or data preprocessing code are not provided or the repository hard-coded for a specific dataset or file system. Nonetheless, Pedersen et al. (2023) was able to reproduce the results of P-Net (Elmarakeby et al., 2021), updating the code to newer ML frameworks. van Hilten et al. (2021) released their GenNet framework, which has since been extended with modules for interpretability (van Hilten et al., 2024b).
A recently developed package called binn (Hartman et al., 2023), focused on proteomics, offers the capability to build VNNs with a given input pathway set. Autoencodix (Joas et al., 2024) is a framework for building different autoencoder architectures, which includes one ontology-based architecture as an option.
There remains a gap, however, for a user-friendly, general-purpose package that supports multi-omics data inputs and different pathway databases and allows exploration of different design decisions to ensure the robustness of the yielded predictions and explanations. Especially interesting would be a highly general package capable of modeling non-biological ontologies (such as in chemistry or social sciences) using a sparse VNN framework.
6 Discussion
VNNs have seen increasing adoption since their emergence around 2017–18. Their versatility is evidence in applications spanning protein classification, survival analysis, diagnosis, and drug-interaction prediction (Kulmanov et al., 2018; Hao et al., 2018a,b; Ma et al., 2018). Compared to full-connected neural networks and other ML algorithms, VNNs often demonstrate comparable or superior performance. Studies such as Pedersen et al. (2023) highlight that randomized sparse networks of comparable size underperform in comparison, underscoring the value of integrating biological knowledge. This integration enhances neural networks' ability to extract signals from relatively small and tabular datasets. However, many studies fail to benchmark against traditional ML methods, which can outperform neural networks [e.g., SVMs in Kim and Lee (2023), see also Borisov et al. (2022)], do not compare with non-biologically-informed NNs to quantify the value of knowledge integration, or do not compare their implementation with existing VNN frameworks.
Interpretability is a key advantage of VNNs over “black box” dense neural networks. By constraining networks with biological pathways, VNNs reduce the function space, trading universality for an inductive bias that promotes faster convergence and enables pathway-level insights via node activations (Kerg et al., 2022). Despite this potential, further theoretical exploration of these inductive biases is required. Emerging evidence suggests that node activations in VNNs can be mapped to their biological counterparts to yield novel insights verifiable in the laboratory (e.g., Elmarakeby et al., 2021, validated biomarkers in vitro) but no studies have conclusively shown that these activations are both data-driven and shaped by the inductive bias. Sparse network randomizations studies remain insufficient to fully elucidate the function spaces of these models. Additionally, the reliance on simplified gene-to-pathway mappings often neglects directional and regulatory relationships, limiting real-world applicability. Flexible architectures with less restrictive activation functions may better capture the multi-layered complexity of cellular biology, bridging trends in explainability and the training of molecular foundation models (Ma et al., 2024; Hao et al., 2024; Cui et al., 2024).
Another limitation lies in the completeness and quality of pathway databases. While these resources are invaluable, they provide an incomplete picture of cellular processes, particularly for non-human or non-model organisms where database curation is often automated and less robust. Only a few studies, such as Park et al. (2022); Hou et al. (2023), have explored the use of VNNs for pathway expansion or the discovery of new biological relationships, leaving this area largely untapped.
Similarly, the impact of different biological databases or knowledge graphs on performance remains underexplored. Variations in database focus and quality could significantly influence VNN results, but most studies do not benchmark their models across multiple databases.
Finally, nearly all VNN architectures rely on early fusion for multi-omics data, which can result in the loss of structural information and diminished performance. For example, Pedersen et al. (2023) found that fusion strategies had a greater impact on performance than randomized sparse connections. Intermediate fusion techniques, as proposed by Hauptmann and Kramer (2023), may offer a more effective approach. However, there remains a gap in developing VNN architectures that incorporate these advanced strategies.
7 Conclusion
Visible neural networks (VNNs) represent a promising advancement in multi-omics data integration, providing a unique combination of predictive performance and biological interpretability. This systematic review has highlighted critical trends, including the importance of sparse architectures informed by biological priors, the impact of design choices on model robustness, and the variability in performance across datasets and ontologies.
While the field has grown substantially since 2020, challenges remain in standardizing terminology, benchmarking methodologies and improving reproducibility. While many works use the nomenclature “BINNs” or “VNNs,” other works use different conventions or do not offer any specific name. Some authors test models extensively against other neural networks, both sparse and dense, and classical ML methods, others perform fewer or no such comparisons, motivating a comprehensive benchmarking framework.
Future research should prioritize systematic evaluations of VNN architectures, particularly regarding the robustness of pathway-level interpretations and the interplay between sparsity and prediction accuracy. Expanding the use of pathway databases, integrating multi-omics modalities with more flexible fusion strategies and exploring novel applications beyond oncology could further enhance their utility. Additionally, developing general-purpose, user-friendly software frameworks for constructing and evaluating VNNs would foster broader adoption and reproducibility. Discovering novel pathway relations through neural architecture search remains an untapped opportunity.
By addressing these challenges, VNNs have the potential to unlock novel biological insights, bridge gaps in multi-omics research and contribute to advances in personalized medicine.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
DS: Data curation, Project administration, Validation, Visualization, Methodology, Formal analysis, Conceptualization, Software, Investigation, Funding acquisition, Writing – review & editing, Resources, Supervision, Writing – original draft. RJ: Writing – original draft, Writing – review & editing, Investigation, Formal analysis, Data curation. MS: Writing – original draft, Writing – review & editing, Conceptualization. GG: Writing – review & editing, Writing – original draft. HR: Writing – original draft, Writing – review & editing. NM: Writing – original draft, Writing – review & editing, Data curation. DP: Writing – review & editing, Writing – original draft, Data curation. JE: Writing – original draft, Writing – review & editing. SV: Funding acquisition, Supervision, Software, Writing – review & editing, Project administration, Writing – original draft, Formal analysis, Resources, Visualization, Data curation, Methodology, Investigation, Validation, Conceptualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the German Federal Ministry of Education and Research (BMBF), grant number 03ZU1202JA in the frame of Clusters4Future project curATime. JE acknowledges support by the Sächsische Staatsministerium für Wissenschaft, Kultur und Tourismus (SMWK) under the frame of ERA PerMed (MIRACLE, 2021-055).
Acknowledgments
We are grateful for the input of Derian Boer, whose helpful suggestions improved the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frai.2025.1595291/full#supplementary-material
References
Abbasi, A. F., Asim, M. N., Ahmed, S., Vollmer, S., and Dengel, A. (2024). Survival prediction landscape: an in-depth systematic literature review on activities, methods, tools, diseases, and databases. Front. Artif. Intell. 7:1428501. doi: 10.3389/frai.2024.1428501
Avelar, P. H. d. C, Wu, M., and Tsoka, S. (2023). Incorporating prior knowledge in deep learning models via pathway activity autoencoders. arXiv:2306.05813.
Borisov, V., Leemann, T., Seßler, K., Haug, J., Pawelczyk, M., and Kasneci, G. (2022). Deep neural networks and tabular data: a survey. IEEE Trans. Neural Netw. Learn. Syst. 35, 7499–7519. doi: 10.1109/TNNLS.2022.3229161
Bourgeais, V., Zehraoui, F., and Hanczar, B. (2022). GraphGONet: a self-explaining neural network encapsulating the Gene Ontology graph for phenotype prediction on gene expression. Bioinformatics 38, 2504–2511. doi: 10.1093/bioinformatics/btac147
Breiman, L. (2001). Statistical modeling: the two cultures (with comments and a rejoinder by the author). Statist. Sci. 16, 199–231. doi: 10.1214/ss/1009213726
Cao, Z.-J., and Gao, G. (2022). Multi-omics single-cell data integration and regulatory inference with graph-linked embedding. Nat. Biotechnol. 40, 1458–1466. doi: 10.1038/s41587-022-01284-4
Chamberlain, S., Zhu, H., Jahn, N., Boettiger, C., and Ram, K. (2022). rcrossref: client for Various CrossRef APIs. R package version 1.2.0.
Chandak, P., Huang, K., and Zitnik, M. (2023). Building a knowledge graph to enable precision medicine. Sci. Data 10:67. doi: 10.1038/s41597-023-01960-3
Chen, H.-I. H., Chiu, Y.-C., Zhang, T., Zhang, S., Huang, Y., and Chen, Y. (2018). GSAE: an autoencoder with embedded gene-set nodes for genomics functional characterization. BMC Syst. Biol. 12:142. doi: 10.1186/s12918-018-0642-2
Chen, R. T., Rubanova, Y., Bettencourt, J., and Duvenaud, D. K. (2018). “Neural ordinary differential equations,” in Advances in Neural Information Processing Systems, 31.
Chen, V., Yang, M., Cui, W., Kim, J. S., Talwalkar, A., and Ma, J. (2024). Applying interpretable machine learning in computational biology—pitfalls, recommendations and opportunities for new developments. Nat. Methods 21, 1454–1461. doi: 10.1038/s41592-024-02359-7
Ciallella, H. L., Russo, D. P., Aleksunes, L. M., Grimm, F. A., and Zhu, H. (2021). Revealing adverse outcome pathways from public high-throughput screening data to evaluate new toxicants by a knowledge-based deep neural network approach. Environ. Sci. Technol. 55, 10875–10887. doi: 10.1021/acs.est.1c02656
Crawford, J., and Greene, C. S. (2020). Incorporating biological structure into machine learning models in biomedicine. Curr. Opin. Biotechnol. 63, 126–134. doi: 10.1016/j.copbio.2019.12.021
Croft, D., O'Kelly, G., Wu, G., Haw, R., Gillespie, M., Matthews, L., et al. (2011). Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 39, D691–D697. doi: 10.1093/nar/gkq1018
Cui, H., Wang, C., Maan, H., Pang, K., Luo, F., Duan, N., et al. (2024). SCGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nat. Methods 21, 1470–1480. doi: 10.1038/s41592-024-02201-0
Dash, T., Chitlangia, S., Ahuja, A., and Srinivasan, A. (2022). A review of some techniques for inclusion of domain-knowledge into deep neural networks. Sci. Rep. 12:1040. doi: 10.1038/s41598-021-04590-0
Deng, D., Xu, X., Cui, T., Xu, M., Luo, K., Zhang, H., et al. (2024). PBAC: a pathway-based attention convolution neural network for predicting clinical drug treatment responses. J. Cell. Mol. Med. 28:e18298. doi: 10.1111/jcmm.18298
Dhillon, A., Singh, A., and Bhalla, V. K. (2023). A systematic review on biomarker identification for cancer diagnosis and prognosis in multi-omics: from computational needs to machine learning and deep learning. Arch. Comput. Methods Eng. 30, 917–949. doi: 10.1007/s11831-022-09821-9
Doncevic, D., and Herrmann, C. (2023). Biologically informed variational autoencoders allow predictive modeling of genetic and drug-induced perturbations. Bioinformatics 39:btad387. doi: 10.1093/bioinformatics/btad387
Elmarakeby, H. A., Hwang, J., Arafeh, R., Crowdis, J., Gang, S., Liu, D., et al. (2021). Biologically informed deep neural network for prostate cancer discovery. Nature 598, 348–352. doi: 10.1038/s41586-021-03922-4
Esser-Skala, W., and Fortelny, N. (2023). Reliable interpretability of biology-inspired deep neural networks. NPJ Syst. Biol. Appl. 9, 1–8. doi: 10.1038/s41540-023-00310-8
Evans, N. J., Mills, G. B., Wu, G., Song, X., and McWeeney, S. (2024). Graph Structured Neural Networks for Perturbation Biology. bioRxiv: 2024.02.28.582164. doi: 10.1101/2024.02.28.582164
Ferraro, L., Scala, G., Cerulo, L., Carosati, E., and Ceccarelli, M. (2023). MOViDA: multiomics visible drug activity prediction with a biologically informed neural network model. Bioinformatics 39:btad432. doi: 10.1093/bioinformatics/btad432
Fortelny, N., and Bock, C. (2020). Knowledge-primed neural networks enable biologically interpretable deep learning on single-cell sequencing data. Genome Biol. 21:190. doi: 10.1186/s13059-020-02100-5
Galluzzo, Y. (2022). “A review: biological insights on knowledge graphs,” in European Conference on Advances in Databases and Information Systems (Springer), 388–399. doi: 10.1007/978-3-031-15743-1_36
Gazestani, V. H., and Lewis, N. E. (2019). From genotype to phenotype: augmenting deep learning with networks and systems biology. Curr. Opin. Syst. Biol. 15, 68–73. doi: 10.1016/j.coisb.2019.04.001
Ghosh Roy, G., Geard, N., Verspoor, K., and He, S. (2022). MPVNN: Mutated Pathway Visible Neural Network architecture for interpretable prediction of cancer-specific survival risk. Bioinformatics 38, 5026–5032. doi: 10.1093/bioinformatics/btac636
Greene, C. S., and Costello, J. C. (2020). Biologically informed neural networks predict drug responses. Cancer Cell 38, 613–615. doi: 10.1016/j.ccell.2020.10.014
Grinsztajn, L., Oyallon, E., and Varoquaux, G. (2022). “Why do tree-based models still outperform deep learning on typical tabular data?” Advances in Neural Information Processing Systems, 507–520.
Hancox-Li, L. (2020). “Robustness in machine learning explanations: does it matter?” in Proceedings of the 2020 Conference on Fairness, Accountability, and Transparency, 640–647. doi: 10.1145/3351095.3372836
Hanczar, B., Zehraoui, F., Issa, T., and Arles, M. (2020). Biological interpretation of deep neural network for phenotype prediction based on gene expression. BMC Bioinform. 21:501. doi: 10.1186/s12859-020-03836-4
Hao, J., Kim, Y., Kim, T.-K., and Kang, M. (2018a). PASNet: pathway-associated sparse deep neural network for prognosis prediction from high-throughput data. BMC Bioinform. 19:510. doi: 10.1186/s12859-018-2500-z
Hao, J., Kim, Y., Mallavarapu, T., Oh, J. H., and Kang, M. (2018b). “Cox-PASNet: pathway-based sparse deep neural network for survival analysis,” in 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) (IEEE), 381–386. doi: 10.1109/BIBM.2018.8621345
Hao, J., Masum, M., Oh, J. H., and Kang, M. (2019). “Gene- and pathway-based deep neural network for multi-omics data integration to predict cancer survival outcomes,” in Bioinformatics Research and Applications, eds. Z. Cai, P. Skums, and M. Li (Cham: Springer International Publishing), 113–124. doi: 10.1007/978-3-030-20242-2_10
Hao, M., Gong, J., Zeng, X., Liu, C., Guo, Y., Cheng, X., et al. (2024). Large-scale foundation model on single-cell transcriptomics. Nat. Methods 21, 1481–1491. doi: 10.1038/s41592-024-02305-7
Hao, Y., Romano, J. D., and Moore, J. H. (2022). Knowledge-guided deep learning models of drug toxicity improve interpretation. Patterns 3:100565. doi: 10.1016/j.patter.2022.100565
Hartman, E., Scott, A. M., Karlsson, C., Mohanty, T., Vaara, S. T., Linder, A., et al. (2023). Interpreting biologically informed neural networks for enhanced proteomic biomarker discovery and pathway analysis. Nat. Commun. 14:5359. doi: 10.1038/s41467-023-41146-4
Hasin, Y., Seldin, M., and Lusis, A. (2017). Multi-omics approaches to disease. Genome Biol. 18:83. doi: 10.1186/s13059-017-1215-1
Hauptmann, T., and Kramer, S. (2023). A fair experimental comparison of neural network architectures for latent representations of multi-omics for drug response prediction. BMC Bioinformatics 24:45. doi: 10.1186/s12859-023-05166-7
Hogan, A., Blomqvist, E., Cochez, M., D'amato, C., Melo, G. D., Gutierrez, C., et al. (2021). Knowledge graphs. ACM Comput. Surv. 54, 1–37. doi: 10.1145/3447772
Hou, Z., Leng, J., Yu, J., Xia, Z., and Wu, L.-Y. (2023). Pathexpsurv: pathway expansion for explainable survival analysis and disease gene discovery. BMC Bioinform. 24:434. doi: 10.1186/s12859-023-05535-2
Hu, J., Yu, W., Dai, Y., Liu, C., Wang, Y., and Wu, Q. (2022). A deep neural network for gastric cancer prognosis prediction based on biological information pathways. J. Oncol. 2022, 1–9. doi: 10.1155/2022/2965166
Huang, X., Huang, K., Johnson, T., Radovich, M., Zhang, J., Ma, J., et al. (2021). ParsVNN: parsimony visible neural networks for uncovering cancer-specific and drug-sensitive genes and pathways. NAR Gen. Bioinform. 3:lqab097. doi: 10.1093/nargab/lqab097
Jia, X., Willard, J., Karpatne, A., Read, J., Zwart, J., Steinbach, M., et al. (2019). “Physics guided rnns for modeling dynamical systems: A case study in simulating lake temperature profiles,” in Proceedings of the 2019 SIAM International Conference on Data Mining (SIAM), 558–566. doi: 10.1137/1.9781611975673.63
Joas, M., Jurenaite, N., Prascevic, D., Scherf, N., and Ewald, J. (2024). A generalized and versatile framework to train and evaluate autoencoders for biological representation learning and beyond: Autoencodix. bioRxiv. doi: 10.1101/2024.12.17.628906
Kadonaga, J. T. (1998). Eukaryotic transcription: an interlaced network of transcription factors and chromatin-modifying machines. Cell 92, 307–313. doi: 10.1016/S0092-8674(00)80924-1
Kanehisa, M. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Karniadakis, G. E., Kevrekidis, I. G., Lu, L., Perdikaris, P., Wang, S., and Yang, L. (2021). Physics-informed machine learning. Nat. Rev. Phys. 3, 422–440. doi: 10.1038/s42254-021-00314-5
Kerg, G., Mittal, S., Rolnick, D., Bengio, Y., Richards, B., and Lajoie, G. (2022). On neural architecture inductive biases for relational tasks. arXiv preprint arXiv:2206.05056.
Kim, S. Y., Kim, T. R., Jeong, H.-H., and Sohn, K.-A. (2018). Integrative pathway-based survival prediction utilizing the interaction between gene expression and DNA methylation in breast cancer. BMC Med. Gen. 11:68. doi: 10.1186/s12920-018-0389-z
Kim, T. R., Jeong, H.-H., and Sohn, K.-A. (2019). Topological integration of RPPA proteomic data with multi-omics data for survival prediction in breast cancer via pathway activity inference. BMC Med. Gen. 12:94. doi: 10.1186/s12920-019-0511-x
Kim, Y., and Lee, H. (2023). PINNet: a deep neural network with pathway prior knowledge for Alzheimer's disease. Front. Aging Neurosci. 15:1126156. doi: 10.3389/fnagi.2023.1126156
Klinger, E., Motta, A., Marr, C., Theis, F. J., and Helmstaedter, M. (2021). Cellular connectomes as arbiters of local circuit models in the cerebral cortex. Nat. Commun. 12:2785. doi: 10.1038/s41467-021-22856-z
Kuenzi, B. M., Park, J., Fong, S. H., Sanchez, K. S., Lee, J., Kreisberg, J. F., et al. (2020). Predicting drug response and synergy using a deep learning model of human cancer cells. Cancer Cell 38, 672–684.e6. doi: 10.1016/j.ccell.2020.09.014
Kulmanov, M., Khan, M. A., and Hoehndorf, R. (2018). DeepGO: predicting protein functions from sequence and interactions using a deep ontology-aware classifier. Bioinformatics 34, 660–668. doi: 10.1093/bioinformatics/btx624
Lagergren, J. H., Nardini, J. T., Baker, R. E., Simpson, M. J., and Flores, K. B. (2020). Biologically-informed neural networks guide mechanistic modeling from sparse experimental data. PLoS Comput. Biol. 16:e1008462. doi: 10.1371/journal.pcbi.1008462
Lan, W., Liao, H., Chen, Q., Zhu, L., Pan, Y., and Chen, Y.-P. P. (2024). DeepKEGG: a multi-omics data integration framework with biological insights for cancer recurrence prediction and biomarker discovery. Brief. Bioinform. 25:bbae185. doi: 10.1093/bib/bbae185
Lee, D., and Kim, S. (2022). Knowledge-guided artificial intelligence technologies for decoding complex multiomics interactions in cells. Clin. Exper. Pediatr. 65, 239–249. doi: 10.3345/cep.2021.01438
Leng, D., Zheng, L., Wen, Y., Zhang, Y., Wu, L., Wang, J., et al. (2022). A benchmark study of deep learning-based multi-omics data fusion methods for cancer. Genome Biol. 23:171. doi: 10.1186/s13059-022-02739-2
Lin, C., Jain, S., Kim, H., and Bar-Joseph, Z. (2017). Using neural networks for reducing the dimensions of single-cell RNA-Seq data. Nucleic Acids Res. 45:e156. doi: 10.1093/nar/gkx681
Liu, B., Rosenhahn, B., Illig, T., and DeLuca, D. S. (2024). A variational autoencoder trained with priors from canonical pathways increases the interpretability of transcriptome data. PLoS Comput. Biol. 20:e1011198. doi: 10.1371/journal.pcbi.1011198
Liu, P., Page, D., Ahlquist, P., Ong, I. M., and Gitter, A. (2024). MPAC: a computational framework for inferring cancer pathway activities from multi-omic data. bioRxiv: 2024.06.15.599113. doi: 10.1101/2024.06.15.599113
Lotfollahi, M., Rybakov, S., Hrovatin, K., Hediyeh-zadeh, S., Talavera-López, C., Misharin, A. V., et al. (2023). Biologically informed deep learning to query gene programs in single-cell atlases. Nat. Cell Biol. 25, 337–350. doi: 10.1038/s41556-022-01072-x
Ma, J., Yu, M. K., Fong, S., Ono, K., Sage, E., Demchak, B., et al. (2018). Using deep learning to model the hierarchical structure and function of a cell. Nat. Methods 15, 290–298. doi: 10.1038/nmeth.4627
Ma, Q., Jiang, Y., Cheng, H., and Xu, D. (2024). Harnessing the deep learning power of foundation models in single-cell omics. Nat. Rev. Molec. Cell Biol. 25, 593–594. doi: 10.1038/s41580-024-00756-6
Ma, T., and Zhang, A. (2019). Incorporating biological knowledge with factor graph neural network for interpretable deep learning. arXiv:1906.00537.
Meirer, J., Wittwer, L. D., Revol, V., and Heinemann, T. (2024). “DeepBINN: a tailored biologically-informed neural network for robust biomarker identification,” in 2024 11th IEEE Swiss Conference on Data Science (SDS), 246–249. doi: 10.1109/SDS60720.2024.00044
Miao, N. (2024). Incorporating inductive biases into machine learning algorithms. PhD thesis, University of Oxford.
Ng, S., Masarone, S., Watson, D., and Barnes, M. R. (2023). The benefits and pitfalls of machine learning for biomarker discovery. Cell Tissue Res. 394, 17–31. doi: 10.1007/s00441-023-03816-z
Nguyen, T., Campbell, A., Kumar, A., Amponsah, E., Fiterau, M., and Shahriyari, L. (2024). Optimal fusion of genotype and drug embeddings in predicting cancer drug response. Brief. Bioinform. 25:bbae227. doi: 10.1093/bib/bbae227
Novakovsky, G., Dexter, N., Libbrecht, M. W., Wasserman, W. W., and Mostafavi, S. (2023). Obtaining genetics insights from deep learning via explainable artificial intelligence. Nat. Rev. Genet. 24, 125–137. doi: 10.1038/s41576-022-00532-2
Oh, J. H., Choi, W., Ko, E., Kang, M., Tannenbaum, A., and Deasy, J. O. (2021). PathCNN: interpretable convolutional neural networks for survival prediction and pathway analysis applied to glioblastoma. Bioinformatics 37, i443–i450. doi: 10.1093/bioinformatics/btab285
Page, M. J., McKenzie, J. E., Bossuyt, P. M., Boutron, I., Hoffmann, T. C., Mulrow, C. D., et al. (2021). The prisma 2020 statement: an updated guideline for reporting systematic reviews. BMJ 372:n71. doi: 10.1136/bmj.n71
Pai, S., and Bader, G. D. (2018). Patient similarity networks for precision medicine. J. Molec. Biol. 430, 2924–2938. doi: 10.1016/j.jmb.2018.05.037
Park, C., Kim, B., and Park, T. (2022). DeepHisCoM: deep learning pathway analysis using hierarchical structural component models. Brief. Bioinform. 23:bbac171. doi: 10.1093/bib/bbac171
Pedersen, C., Tesileanu, T., Wu, T., Golkar, S., Cranmer, M., Zhang, Z., et al. (2023). Reusability report: Prostate cancer stratification with diverse biologically-informed neural architectures. arXiv:2309.16645.
Peng, J., Wang, X., and Shang, X. (2019). Combining gene ontology with deep neural networks to enhance the clustering of single cell RNA-Seq data. BMC Bioinform. 20:284. doi: 10.1186/s12859-019-2769-6
Przedborski, M., Smalley, M., Thiyagarajan, S., Goldman, A., and Kohandel, M. (2021). Systems biology informed neural networks (sbinn) predict response and novel combinations for pd-1 checkpoint blockade. Commun. Biol. 4:877. doi: 10.1038/s42003-021-02393-7
Rahnenführer, J., De Bin, R., Benner, A., Ambrogi, F., Lusa, L., Boulesteix, A.-L., et al. (2023). Statistical analysis of high-dimensional biomedical data: a gentle introduction to analytical goals, common approaches and challenges. BMC Med. 21:182. doi: 10.1186/s12916-023-02858-y
Retzlaff, C. O., Angerschmid, A., Saranti, A., Schneeberger, D., Roettger, R., Mueller, H., et al. (2024). Post-hoc vs ante-hoc explanations: xAI design guidelines for data scientists. Cogn. Syst. Res. 86:101243. doi: 10.1016/j.cogsys.2024.101243
Rodríguez, A., Cui, J., Ramakrishnan, N., Adhikari, B., and Prakash, B. A. (2023). “Einns: epidemiologically-informed neural networks,” in Proceedings of the AAAI conference on artificial intelligence, 14453–14460. doi: 10.1609/aaai.v37i12.26690
Samal, B. R., Loers, J. U., Vermeirssen, V., and De Preter, K. (2022). Opportunities and challenges in interpretable deep learning for drug sensitivity prediction of cancer cells. Front. Bioinform. 2:1036963. doi: 10.3389/fbinf.2022.1036963
Selby, D. A., Sprang, M., Ewald, J., and Vollmer, S. J. (2025). Beyond the black box with biologically informed neural networks. Nat. Rev. Genet. 26, 371–372. doi: 10.1038/s41576-025-00826-1
Seninge, L., Anastopoulos, I., Ding, H., and Stuart, J. (2021). VEGA is an interpretable generative model for inferring biological network activity in single-cell transcriptomics. Nat. Commun. 12:5684. doi: 10.1038/s41467-021-26017-0
Sharma, D., and Xu, W. (2023). ReGeNNe: genetic pathway-based deep neural network using canonical correlation regularizer for disease prediction. Bioinformatics 39:btad679. doi: 10.1093/bioinformatics/btad679
Sidorova, J., and Lozano, J. J. (2024). Review: deep learning-based survival analysis of omics and clinicopathological data. Inventions 9:59. doi: 10.3390/inventions9030059
Slack, D., Hilgard, A., Singh, S., and Lakkaraju, H. (2021). “Reliable post hoc explanations: Modeling uncertainty in explainability,” in Advances in Neural Information Processing Systems, 9391–9404.
Sprang, M., Selby, D., and Vollmer, S. (2024). “Interpretable AutoML for biological knowledge discovery from multi-modal data,” in AI for Scientific Discovery Workshop (Ingelheim, Germany: Gutenberg Workshops. Poster).
Subramanian, I., Verma, S., Kumar, S., Jere, A., and Anamika, K. (2020). Multi-omics data integration, interpretation, and its application. Bioinform. Biol. Insights 14:1177932219899051. doi: 10.1177/1177932219899051
Sun, Y. V., and Hu, Y.-J. (2016). Integrative analysis of multi-omics data for discovery and functional studies of complex human diseases. Adv. Genet. 93, 147–190. doi: 10.1016/bs.adgen.2015.11.004
The Gene Ontology Consortium, Aleksander, S. A., Balhoff, J., Carbon, S., Cherry, J. M., and Drabkin, H. J.. (2023). The gene ontology knowledgebase in 2023. Genetics 224:iyad031. doi: 10.1093/genetics/iyad031
Uzunangelov, V., Wong, C. K., and Stuart, J. M. (2021). Accurate cancer phenotype prediction with AKLIMATE, a stacked kernel learner integrating multimodal genomic data and pathway knowledge. PLoS Comput. Biol. 17:e1008878. doi: 10.1371/journal.pcbi.1008878
Van Harmelen, F., and Ten Teije, A. (2019). A boxology of design patterns for hybrid learning and reasoning systems. J. Web Eng. 18, 97–123. doi: 10.13052/jwe1540-9589.18133
van Hilten, A., Katz, S., Saccenti, E., Niessen, W. J., and Roshchupkin, G. V. (2024a). Designing interpretable deep learning applications for functional genomics: a quantitative analysis. Brief. Bioinform. 25:bbae449. doi: 10.1093/bib/bbae449
van Hilten, A., Kushner, S. A., Kayser, M., Ikram, M. A., Adams, H. H., Klaver, C. C., et al. (2021). GenNet framework: interpretable deep learning for predicting phenotypes from genetic data. Commun. Biol. 4:1094. doi: 10.1038/s42003-021-02622-z
van Hilten, A., Melograna, F., Fan, B., Niessen, W., Steen, K., et al. (2024b). Detecting genetic interactions with visible neural networks. bioRxiv: 2024.02.27.582086. doi: 10.1101/2024.02.27.582086
Voigt, J., Saralajew, S., Kaden, M., Bohnsack, K., Reuss, L., and Villmann, T. (2024). “Biologically-informed shallow classification learning integrating pathway knowledge,” in Proceedings of the 17th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2024), 357–367. doi: 10.5220/0012420700003657
Wagle, M. M., Long, S., Chen, C., Liu, C., and Yang, P. (2024). Interpretable deep learning in single-cell omics. Bioinformatics 40:btae374. doi: 10.1093/bioinformatics/btae374
Wang, Z., Zhou, Y., Zhang, Y., Mo, Y. K., and Wang, Y. (2023). XMR: an explainable multimodal neural network for drug response prediction. Front. Bioinform. 3:1164482. doi: 10.3389/fbinf.2023.1164482
Wei, J., Turbé, H., and Mengaldo, G. (2024). Revisiting the robustness of post-hoc interpretability methods. arXiv preprint arXiv:2407.19683.
Winter, D. J. (2017). Rentrez: an R package for the NCBI eUtils API. R J. 9, 520–526. doi: 10.32614/RJ-2017-058
Wysocka, M., Wysocki, O., Zufferey, M., Landers, D., and Freitas, A. (2023). A systematic review of biologically-informed deep learning models for cancer: fundamental trends for encoding and interpreting oncology data. BMC Bioinform. 24:198. doi: 10.1186/s12859-023-05262-8
Xiao, S., Lin, H., Wang, C., Wang, S., and Rajapakse, J. C. (2023). Graph neural networks with multiple prior knowledge for multi-omics data analysis. IEEE J. Biomed. Health Inform. 27, 4591–4600. doi: 10.1109/JBHI.2023.3284794
Yamazaki, K., Vo-Ho, V.-K., Bulsara, D., and Le, N. (2022). Spiking neural networks and their applications: a review. Brain Sci. 12:863. doi: 10.3390/brainsci12070863
Yan, H., Weng, D., Li, D., Gu, Y., Ma, W., and Liu, Q. (2024). Prior knowledge-guided multilevel graph neural network for tumor risk prediction and interpretation via multi-omics data integration. Brief. Bioinform. 25:bbae184. doi: 10.1093/bib/bbae184
Yang, J., Wu, W. K. K., Hui, R. Y. M., Wong, I. C. K., and Zhang, Q. (2024). BioXNet: a biologically inspired neural network for deciphering anti-cancer drug response in precision medicine. bioRxiv: 2024.01.29.576766. doi: 10.1101/2024.01.29.576766
Yang, J. H., Wright, S. N., Hamblin, M., McCloskey, D., Alcantar, M. A., Schrübbers, L., et al. (2019). A white-box machine learning approach for revealing antibiotic mechanisms of action. Cell 177, 1649–1661. doi: 10.1016/j.cell.2019.04.016
Yeger-Lotem, E., Sattath, S., Kashtan, N., Itzkovitz, S., Milo, R., Pinter, R. Y., et al. (2004). Network motifs in integrated cellular networks of transcription-regulation and protein-protein interaction. Proc. Nat. Acad. Sci. 101, 5934–5939. doi: 10.1073/pnas.0306752101
Yi, H.-C., You, Z.-H., Huang, D.-S., and Kwoh, C. K. (2022). Graph representation learning in bioinformatics: trends, methods and applications. Brief. Bioinform. 23:bbab340. doi: 10.1093/bib/bbab340
Zahra, N., Vagiona, A.-C., Uddin, R., and Andrade-Navarro, M. A. (2023). Selection of multi-drug targets against drug-resistant mycobacterium tuberculosis xdr1219 using the hyperbolic mapping of the protein interaction network. Int. J. Mol. Sci. 24:14050. doi: 10.3390/ijms241814050
Zhang, H., Chen, Y., and Li, F. (2021). Predicting anticancer drug response with deep learning constrained by signaling pathways. Front. Bioinform. 1:639349. doi: 10.3389/fbinf.2021.639349
Keywords: multi-omics integration, deep learning, explainable AI, machine learning, interpretable models, gene regulatory networks, pathways, neural networks
Citation: Selby DA, Jakhmola R, Sprang M, Großmann G, Raki H, Maani N, Pavliuk D, Ewald J and Vollmer S (2025) Visible neural networks for multi-omics integration: a critical review. Front. Artif. Intell. 8:1595291. doi: 10.3389/frai.2025.1595291
Received: 17 March 2025; Accepted: 26 May 2025;
Published: 17 July 2025.
Edited by:
Qiao Xu, Shandong University, ChinaReviewed by:
Hasi Hays, University of Arkansas, United StatesJai Chand Patel, University of Nebraska Medical Center, United States
Copyright © 2025 Selby, Jakhmola, Sprang, Großmann, Raki, Maani, Pavliuk, Ewald and Vollmer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sebastian Vollmer, dm9sbG1lckBycHR1LmRl; David Antony Selby, ZGF2aWQuc2VsYnlAZGZraS5kZQ==
†These authors have contributed equally to this work