 Manuel Glöckler
Manuel Glöckler Andreas Dräger
Andreas Dräger Reihaneh Mostolizadeh
Reihaneh Mostolizadeh- 1Department of Computer Science, University of Tübingen, Tübingen, Germany
- 2Computational Systems Biology of Infections and Antimicrobial-Resistant Pathogens, Institute for Bioinformatics and Medical Informatics (IBMI), University of Tübingen, Tübingen, Germany
- 3German Center for Infection Research (DZIF), Partner Site Tübingen, Tübingen, Germany
- 4Cluster of Excellence “Controlling Microbes to Fight Infections”, University of Tübingen, Tübingen, Germany
The human upper respiratory tract is the reservoir of a diverse community of commensals and potential pathogens (pathobionts), including Streptococcus pneumoniae (pneumococcus), Haemophilus influenzae, Moraxella catarrhalis, and Staphylococcus aureus, which occasionally turn into pathogens causing infectious diseases, while the contribution of many nasal microorganisms to human health remains undiscovered. To better understand the composition of the nasal microbiome community, we create a workflow of the community model, which mimics the human nasal environment. To address this challenge, constraint-based reconstruction of biochemically accurate genome-scale metabolic models (GEMs) networks of microorganisms is mandatory. Our workflow applies constraint-based modeling (CBM), simulates the metabolism between species in a given microbiome, and facilitates generating novel hypotheses on microbial interactions. Utilizing this workflow, we hope to gain a better understanding of interactions from the metabolic modeling perspective. This article presents nasal community modeling workflow (NCMW)—a python package based on GEMs of species as a starting point for understanding the composition of the nasal microbiome community. The package is constructed as a step-by-step mathematical framework for metabolic modeling and analysis of the nasal microbial community. Using constraint-based models reduces the need for culturing species in vitro, a process that is not convenient in the environment of human noses.
Availability: NCMW is freely available on the Python Package Index (PIP) via pip install NCMW. The source code, documentation, and usage examples (Jupyter Notebook and example files) are available at https://github.com/manuelgloeckler/ncmw.
1 Introduction
The human nose community is far more than a convenient model system, while it plays vital roles in human health and global nutrient cycles (Widder et al., 2016). The previous work focusing on ecological interactions can identify whether the interacting partners promote or hinder each other’s growth (Carrara et al., 2015). Studying these interactions on their own does not reveal the mechanistic details of the nasal microbiome community or its composition and structure. The interaction mechanisms, either potential competition or cooperation of species in the community, can determine the species assembly processes in the community. Understanding the pair-wise or multispecies interactions in the human nose community is a starting point for manipulating microbiomes for therapeutic and prophylactic purposes. However, this is often made difficult due to the inability to culture and co-culture the various species of human nasal microbiota in vitro.
The study on human gut microorganisms and their community has received much attention compared to that on other communities. Different approaches such as high-throughput studies, modeling methodologies such as GEMs (Klitgord and Segrè, 2010; Freilich et al., 2011; Zomorrodi and Maranas, 2012; Seif et al., 2019; Diener et al., 2020), AGORA (Levy and Borenstein, 2013), community and systems-level interactive optimization (CASINO) (Levy and Borenstein, 2014), BacArena (Bauer et al., 2017), and GutLogo (Lin et al., 2018), and databases such as virtual metabolic human database (VMH) (Noronha et al., 2019), have been developed on microbial communities mainly focusing on the four human sites, such as gut, oral, skin, and vaginal. Although resources on human nose research have been limited, a direct application of existing modeling pipelines available for the gut is consequently difficult due to both aerobic and anaerobic living conditions for human nose bacteria (Man et al., 2017; Proctor and Relman, 2017). Some researches represent the role of nasal microbial inhabitants in inhibiting the pathogens or turning commensal into a pathogen (Yan et al., 2013; Liu et al., 2015; Bomar et al., 2018). However, factors that influence pathogens have not been defined yet. A synthetic nasal medium (SNM3) has been developed by Krismer et al. (2014) that permits consistent growth of S. aureus isolates. Yet, there are still members of the nasal microbiome that remain unculturable even in this medium. Using this medium simulates the bacterial growth within the human nose. In addition, GEMs and constraint-based modeling are used to biochemically and physicochemically predict the phenotype from genotype without the need for a bacterial culture of species in the lab (Shoaie et al., 2013; Cook and Nielsen, 2017; van der Ark et al., 2017).
Here, we developed a workflow to construct a predictive computational model of the human nose microbial community and its interactions. The workflow is highly flexible and can adapt to changes or updates whenever yet unexplored or modified models are added. This can be used for pair-wise interactions and a more extensive system with more species. However, the computational complexity increases with the number of species. In particular, this is the first time a comprehensive flux balance analysis workflow has been created for the nasal human microbial community. The workflow relies on a multilevel and multi-objective optimization problem to grasp commensal, competitive, or further interactions. Like OptCom (Zomorrodi and Maranas, 2012), a multilevel optimization framework for the metabolic modeling, we also considered species-level and community-level criteria in defining the objective function. Therefore, the optimal growth of each species in the community and the optimal growth of the entire community have been taken into account. Additionally, we explicitly included different weights in the definition of the objective function of the community based on the growth ratio of species in the community to estimate more realistic and quantitative metabolic predictions.
In NCMW, we integrate diverse types of available definitions for media those are subsets of the leading preferred media, e.g., SNM3. They may simulate environmental conditions that favor commensal or competitive interactions (Carrara et al., 2015). These different media definitions facilitate knowledge interchange within the theoretical, experimental, and computational definition of the human nose environment. This example demonstrates that systematic model-based analysis has the ability to detect such potential nutrients in a much more efficient way in adoption between wet and dry labs. In addition, adding any newly defined media to our workflow is feasible. Hence, the aforementioned one represents the compatibility of our package for different communities such as the gastrointestinal tract, skin, and vagina, in the case of the definition of respected media. This strategy is entirely implemented as a Python package and available as an open-source software package named NCMW. It facilitates integrating genome-scale metabolic models in the human nose environment for understanding the complexity of this community.
To validate our work and demonstrate the ability of NCMW, it was applied on the nasal microbial community involving Staphylococcus aureus and Dolosigranulum pigrum (Mostolizadeh et al., 2022). Next, NCMW is also employed to model the more complex system of the five abundant nasal microbial to show different applications of the package on a large community and documented as an example to the package.
Researchers can easily incorporate, access, modify, and extend our tool to develop their research protocols for scientific analysis on the community. The workflow discussed here lays the foundation for future testing of human nasal species as potential probiotics to prevent or antagonize colonization of the nares by pathogens, especially Staphylococcus aureus. We believe that NCMW can unravel some biological questions on how the nasal community shapes and the members can have widely different effects on each other.
2 Materials and Methods
The workflow’s essential methods and approaches depend on computational models for inferring fluxes in biochemical networks. In order to estimate the metabolic fluxes of the model organism, we use GEMs. The approach used here is therefore based on high-quality genome-scale models generated for species within the community, which we will discuss in more detail later.
2.1 Flux Balance Analysis and Flux Variability Analysis
Flux balance analysis (FBA) is a linear optimization method that maximizes a biologically motivated objective function. Biomass production, for instance, is a widely used objective that facilitates calculations of an organism’s growth rate. Other examples include the production rate of a biotechnologically relevant metabolite. A meaningful objective function enables flux balance analysis (FBA) to predict the flow of matter through the network (Orth et al., 2010).
Flux variability analysis (FVA) calculates the minimal and maximal possible flux through a reaction channel in the model. Simultaneously, flux variability analysis (FVA) maintains a specific network state. For instance, it might sustain 90% of the maximal possible biomass production rate. Such analysis is particularly useful for analyzing the robustness of metabolic models (Gudmundsson and Thiele, 2010).
2.2 Community Approaches
There are different approaches for creating a community model. They are categorized as compartmentalized, pooled reactions, and nested analysis methods.
2.2.1 Pooled Community
This approach combines metabolic reactions and metabolites from all members of a species in one shared compartment. In cases where more than one species catalyzes the reaction, only one reaction is assigned. Although this community minimizes the computational burden, it has the weakness of not specifying which species use or produce a particular enzyme or metabolite (Taffs et al., 2009). As a result, the model can be used to investigate the community’s growth but not to analyze the interaction between community members.
2.2.2 Shuttle Community
In this approach, each exists in a separate compartment. Individual species then interact in an additional “external” compartment, representing the extracellular space. This shared external space allows for interspecies interaction through “shuttle reactions.” Thus, we can explicitly evaluate how bacterial species will interact with other community members as well as the surrounding environment. Furthermore, the shuttle reactions that identify shared reactions between species can also be defined based on our interests (Klitgord and Segrè, 2010).
2.2.3 Community Optimization
As standard, we interpret community growth as a linear combination of individual’s growth, i.e.,
where Si, Ssh and Sex denote the stoichiometry matrices to ensure mass-balance within any reaction. We denote a vector of reaction fluxes by ν.
The internal reactions νi are essentially unbounded. We use a lower bound of −1,000 mmol/(gDW⋅h) and an upper bound of 1,000 mmol/(gDW⋅h) as default. The lower bounds for the community exchange reactions νex are defined by the medium, e.g., typically −10 mmol/(gDW⋅h). For exchange reactions, we add an associated shuttle reaction that mediates the uptake from the external medium under mass balance constraints, i.e., the sum of individual fluxes must equal the community exchange flux. These reactions allow interaction between community members, i.e., model i can produce metabolites for model j. We assign each of them a lower bound of −50 mmol/(gDW⋅h) and an upper bound of 1,000 mmol/(gDW⋅h). As default for any metabolite that a model can uptake or secrete, we add associated shuttles. Nevertheless, we also specify the set of reactions for which shuttle reactions should be added (all others are modeled as regular exchanges). This kind of community is also employed in SteadyCom or MICOM (Chan et al., 2017; Diener et al., 2020).
Notice that if wigi = 0, we consider the community member i as “dead” and thus would be expected that all associated fluxes are zero, especially all shuttle reactions should have zero flux, as a “dead” community member should not be able to produce or consume metabolites into the external environment. Unfortunately, this behavior is not guaranteed by the constraints imposed. If wigi = 0, any pathway that does not provide biomass gi can be used to produce metabolites to increase the biomass of other models. This is unrealistic and should be prevented. To do so, we add an additional linear constraint to each shuttle reaction. We impose that
Consistently with Diener et al. (Diener et al., 2020), we observed that optimizing this objective can lead to solutions where only a few taxa grew with high growth rates, and all the other community members had growth near zero. Naively, we could enforce all growth gi ≥ gmin, yet if a single model is unable to obtain gmin, then there will be no feasible solution. We alternatively ensure that each community member must reach a certain percentage of the total community growth, i.e., wigi ≥ αG with α ∈ [0, 1]. Diener et al. (Diener et al., 2020) proposed the cooperative trade-off method, which first maximizes the community objective to obtain Gmax. For a given trade-off α ∈ [0, 1], the solution is then obtained by minimizing
In addition, by defining different weights in the optimization function for each species, an abundant profile for each species is scaled to predict requirements for inhibiting the pathogens (Chan et al., 2017; Mostolizadeh et al., 2019). This indeed enables the determination of the range of allowable fluxes to impose conditions for increasing or decreasing the commensal or pathogenic species’ growth rate and infers a reasonable presupposition for the experimental test (Chan et al., 2017; Mostolizadeh et al., 2019).
2.3 Media
A growth medium is a list of nutrients designed to support the growth of microorganisms. There are different types of media suitable for growing different types of cells. Here, we use a different type of media for growing microbial communities.
2.3.1 The Synthetic Nasal Medium
Krismer et al. (Krismer et al., 2014) developed a unique SNM3, which has been shown to provide all requirements for a variety of different nasal bacteria. SNM3 is an essential step toward an in vitro testing system for the human nose and is defined as the default medium in our Python package.
2.3.2 Competition-Inducing Medium and Cooperation-Inducing Medium
First, all exchange metabolites are provided at the minimal required amount computed by FVA. Then, the competition-inducing medium (COMPM) is defined as the ranges of fluxes of the exchange reactions that support its maximal biomass rate (MBR) while they are in the minimal amount (Freilich et al., 2011). Therefore, in this medium, each species is allowed for reaching its maximum biomass rate. However, within a community, any resource overlap should reduce the individual biomass rate.
Although, a cooperation-inducing Medium (COOPM) is defined as a set of metabolites that support only a small positive growth rate such that the removal of any metabolite from the set would force the system to have no such solution (Freilich et al., 2011). These two media are determined as rich and poor media, which are a subset of any main media of interest (based on our default setting, they are a subset of SNM3).
2.4 Similarity and Consistency
The nasal microbial community comprises various species living together and plays an essential role in human health. To identify the interaction of members, we would first focus on how the GEMs of species are qualitatively consistent and how similar they are. To detect the consistent part of a network or blocked reactions, an algorithm called FastCC is applied (Vlassis et al., 2014). FastCC is based on standard MatLAB (Vlassis et al., 2014), although this was also adjusted based on a pure LP implementation for cobra users1. In addition, the Jaccard coefficient can be used to measure the similarity between the two sets, in comparing the overlap of reactions and metabolites between species (Jaccard, 1912). The Jaccard coefficient can be defined as the size of the intersection of the two sets divided by the size of the union of the two sets (Tan et al., 2016). Therefore, if the Jaccard coefficient equals one, two sets are identical, while zero indicates two disjoint sets.
3 Results
3.1 Implementation
The core capabilities of NCMW are set up by the installation of the package via pip. All dependencies are installed as the Python package NCMW. The only requirement that is not installed (despite obvious ones such as Python) is some solver that can minimize quadratic objectives and is supported by pycobra (Ebrahim et al., 2013) (but also just for some operations).
The entire folder NCMW is shown in Figure 1 and is as follows:
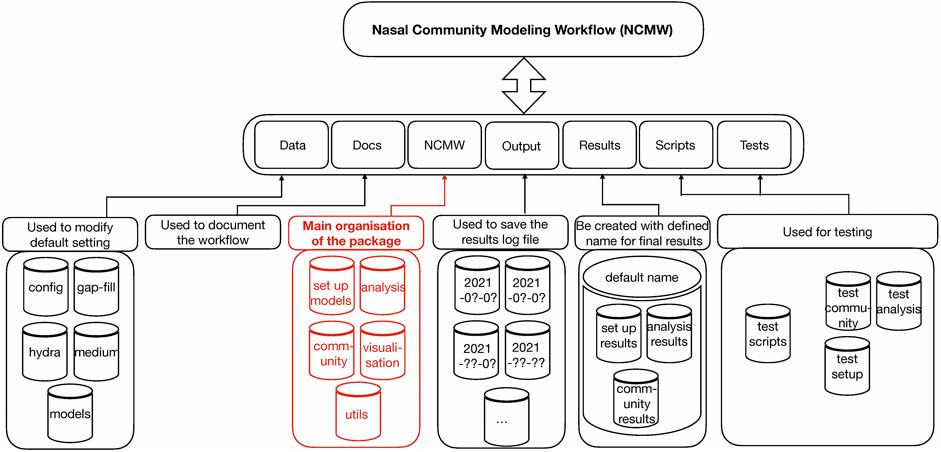
FIGURE 1. Entire nasal community modeling workflow (NCMW). In the first layer, the package includes seven folders. In the second layer, the tasks which each folder can do are represented. In the last layer, the sub-folders included in each folder are shown. The highlighted part, as the red part, is the main folder of the package. To run the package and get the results, we use command ncmw setup, ncmw analysis, and ncmw community in the command line.
NCMW: ▸ Data.
This folder includes four sub folders, namely configs, hydra, medium, and models.
NCMW: ▸ Data ▸ Configs.
These are used for the default setting of maximum lower and upper bound of reactions −1,000 mmol/(gDW⋅h) to 1,000 mmol/(gDW⋅h).
NCMW: ▸ Data ▸ Hydra.
We use hydra (Yadan, 2019) to support, e.g., parameter overriding. This folder contains all default parameters for the scripts. All can be overwritten in the console or by modification of the file.
NCMW: ▸ Data ▸ Medium.
This folder contains the default SNM3. Any medium of user interest can be placed here, to the server as a new default medium. All derived media will then be based on the medium specified within the parameters.
NCMW: ▸ Data ▸ Models.
All models of participating species in the community must be accessible in this folder. One can either add additional models to the workflow or create a new folder and properly overwrite the corresponding parameters.
NCMW: ▸ Docs.
This folder contains all documents related to how to work with the workflow. The documents are readable via https://manuelgloeckler.github.io/ncmw/as well.
NCMW: ▸ NCMW.
The main folder is organized into five parts: setup, analysis, community, utils, and visualization.
NCMW: ▸ NCMW ▸setup.
This is used to set the default bounds as specified in data/configs and the medium as specified in data/medium. It is also used to gap-fill the model with reactions or the medium with metabolites, such that all models obtain growth on the specified medium.
NCMW: ▸ NCMW ▸ analysis.
Analysis performs flux variability analysis and visualizes results on all exchange reactions, as shown in Figure 2. This also shows the scaled growth behavior as shown in Figure 3. In addition, the uptake/secretion overlap between models is reanalyzed as shown in Figure 4. The similarity of models based on the number of shared metabolites/reactions as shown in Figure 5 is computed as well. COMPM, the medium in which all models are able to obtain their MBR, is computed.
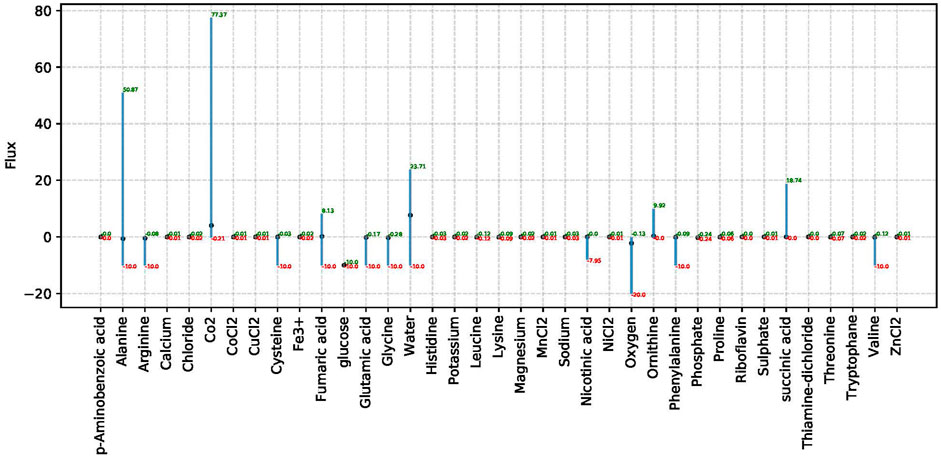
FIGURE 2. FVA results for non-zero exchange reactions in species Dolosigranulum pigrum. This analysis only takes non-zero exchange reactions into account based on the definition of the SNM3. The x-axis in this plot shows the associated metabolites to consider exchange reactions, and the y-axis shows the respective flux for the related exchange reaction.
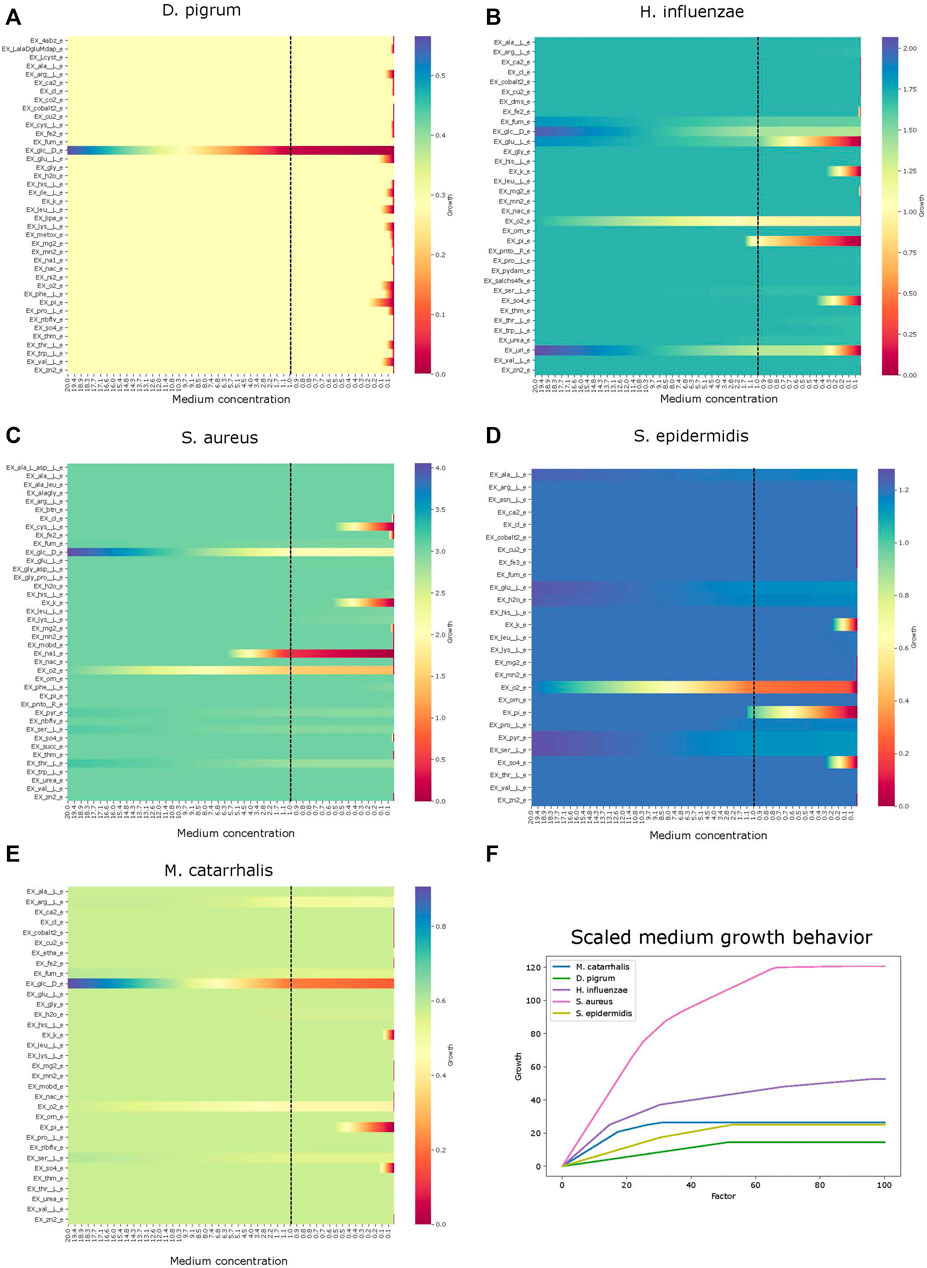
FIGURE 3. Effect of growth behavior on an increase of metabolites. Different models show how the growth scales are affected with abundance of metabolites. Panel (A) is species Dolosigranulum pigrum. Panel (B), Haemophilus influenzae. Panel (C), Staphylococcus aureus. Panel (D), Staphylococcus epidermidis. Panel (E), Moraxella catarrhalis. Panel (F), growth comparison.
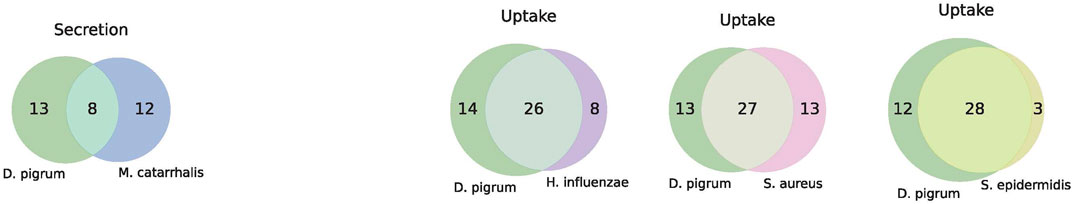
FIGURE 4. Major player within a community is what a model must uptake and what it can produce. Produced metabolites can benefit other community members, namely Dolosigranulum pigrum, Haemophilus influenzae, Staphylococcus aureus, Staphylococcus epidermidis, and Moraxella catarrhalis. However, too many shared uptakes can lead to competition within the community (resource overlap).
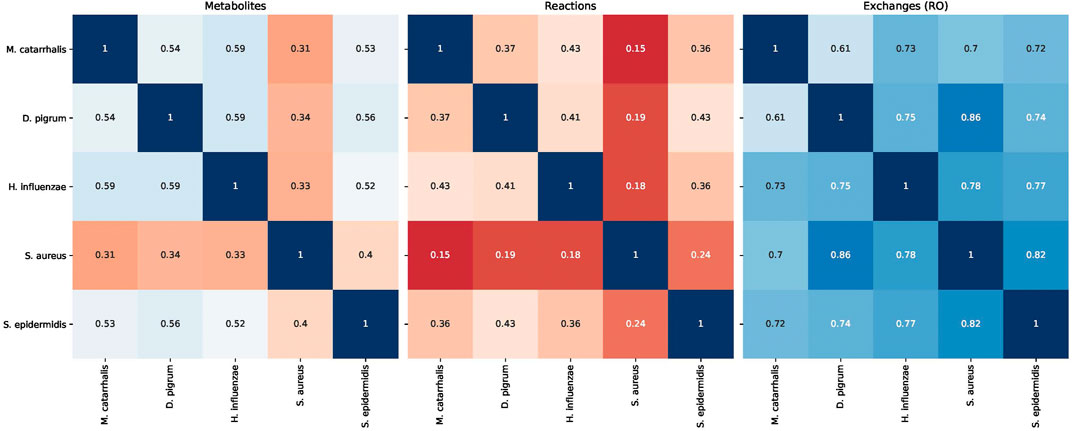
FIGURE 5. Similarity of different models of species Dolosigranulum pigrum, Haemophilus influenzae, Staphylococcus aureus, Staphylococcus epidermidis, and Moraxella catarrhalis based on the Jaccard distance.
NCMW: ▸ NCMW ▸ community.
Community creates several kinds of community models, which all are based on Ebrahim et al., (2013). This also computes the COOPM, which is the smallest medium such that the community achieves 10% of the MBR, which induces cooperation. In addition, it visualizes the observed interactions between models, for instance, in Figure 6 and investigates the dependence of community weight and observed growth. This visualizes how the community looks like and visualizes the similarity between species in the community in consumption or production of metabolites as shown in Figure 7. In addition, it shows the growth of each species in the community.
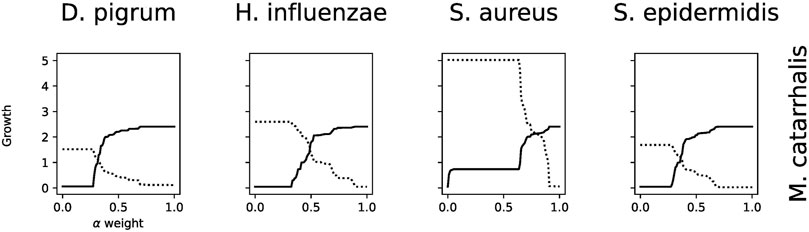
FIGURE 6. Growth of each model, given the weight α. This shows, e.g., at which weight the community is balanced between two species. For instance, the pair-wise interaction between species Moraxella catarrhalis and Dolosigranulum pigrum, Haemophilus influenzae, Staphylococcus aureus, and Staphylococcus epidermidis is plotted.
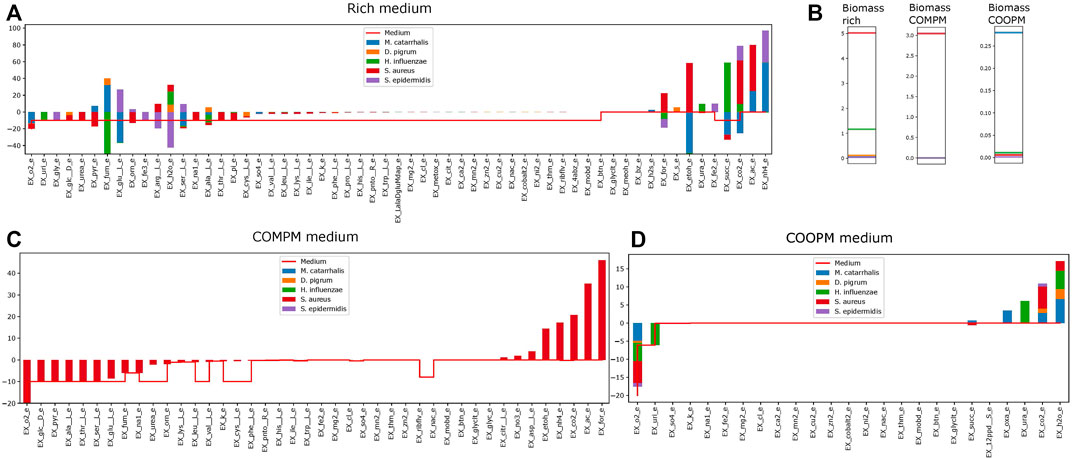
FIGURE 7. How much concentration of each metabolites are consumed or produced by each model when they are in the community for rich SNM3 [panel (A)], the COMPM [panel (C)], and the COOPM [panel (D)]. In panel (B), we show the individual biomass rates obtained.
NCMW: ▸ Output.
This folder contains all log files of each time run. A new folder that entitled the run date is automatically created and shows which command was applied while representing the log file.
NCMW: ▸ Results.
The results folder is automatically created with the default name defined. This is included in three main folders as setup, analysis, and community. Each contains the final results associated with the commands ncmw_setup, ncmw_analysis, and ncmw_community. Since the scripts automatically produce some results, NCMW, Data, Hydra helps the users to set up any block for each part of running scripts out of their interest.
NCMW: ▸ Tests.
Test folder includes test scripts related to commands ncmw_setup, ncmw_analysis, and ncmw_community.
3.2 Workflow
The NCMW scripts implement a series of tasks to carry out typical analysis procedures without any programming. We will discuss each component slightly based on the example of a five-member community consisting of D. pigrum, S. aureus, M. catarrhalis, S. epidermidis, and H. influenzae on the SNM3. To create the community of five members, the already existing GEMs of D. pigrum (Renz et al., 2021a) and S. aureus (Seif et al., 2019) were used. In addition, auto-generated GEMs of S. epidermidis, H. influenzae, and M. catarrhalis by CarveMe (Machado et al., 2018) version 1.2.2 in combination with DIAMOND (Buchfink et al., 2021) aligner version 0.9.26.127 were made. The information related to genome sequences of S. epidermidis, H. influenzae, and M. catarrhalis found in NCBI (Pruitt et al., 2005) is as follows:
1) S. epidermidis: The annotated genome sequence of S. epidermidis ATCC 12 228 (Zhang et al., 2003) included a NCBI Reference Sequence NC_004461.1 with the reference GCF_000007645.12 and a total sequence length of 2,564,615.
2) H. influenzae: The annotated genome sequence of H. influenzae Rd KW 20 (Fleischmann et al., 1995; Tatusov et al., 1996) included a NCBI Reference Sequence NC_000907.1 with the reference GCF_000027305.13 and a total sequence length of 1,830,138.
3) M. catarrhalis: The annotated genome sequence of M. catarrhalis BBH 18 (de Vries et al., 2013) included a NCBI Reference Sequence NC_014147.1 with the reference GCA_000092265.14 and a total sequence length of 1,863,286.
3.2.1 Setup of the Models
This part of the workflow considers two main parts: to check the quality of genome-scale metabolic models and the growth on the given SNM3. Most genome scale models are developed for the gut environment; thus, they often do not grow on a nasal medium.
3.2.1.1 Quality Report of Genome-Scale Metabolic Models
The workflow starts with genome-scale metabolic models of the organism of interest, which have been already reconstructed, manually curated, and refined. The first step in the workflow is to check the quality of models. The models have been curated using the FastCC algorithm implemented in COBRApy (Vlassis et al., 2014). We further use Memote to create a quality report for each model (Lieven et al., 2020). A single improper model can strongly influence the community, i.e., a model that can produce much glucose out of nothing can enormously increase the biomass rates of all members to infinity [i.e., 1,000 mmol/(gDW⋅h)] within the community. Hence, this problematic behavior will become the dominant source of interaction and render the community useless for analysis. As a result, bad-quality models should be removed. The genome-scale metabolic models used here were all well curated and thus were kept for further analysis.
3.2.1.2 The Growth Report of Models on SNM3
It has been reported whether the models are able to grow on SNM3, a medium mimicking the environment of the human nose. The exchange bounds of those metabolites defined for SNM3 can be set to a lower and upper bound. The default setting would be −10 mmol/(gDW⋅h) to 1,000 mmol/(gDW⋅h) for all metabolites defined in SNM3 except for oxygen and iron, which were set at −20 mmol/(gDW⋅h) to 1,000 mmol/(gDW⋅h) and −0.1 mmol/(gDW⋅h) to 1,000 mmol/(gDW⋅h), respectively. As explained in implementation of configs, the setting for bounds can be changed to the user’s interest. In addition, the default medium is SNM3, as the main focus of the workflow is the human nose community. However, implementation of the medium makes the possibility for defining any new medium based on the user’s interest.
The growth/no growth on SNM3 is reported in this step. In case of no growth, the workflow search for the missing metabolites that can be supplemented to the media allowed the organism’s growth on SNM3. This step is called the gap-filled step. The models with growth on SNM3 will be saved as an XML file. This makes their usage for further steps in the workflow smoother.
Alternatively, instead of supplementing the medium, we can add new reactions to the models to achieve growth. This alternative gap-filling strategy leaves the medium unchanged, yet one should typically check if the added reactions have supporting biological evidence.
Indeed, the workflow detected that H. influenzae, D. pigrum, and M. catarrhalis did not grow on the default SNM3. However, the gap-filling strategy succeeded. M. catarrhalis achieved a reasonable biomass rate of 1.2 mmol/(gDW⋅h) if we substitute EX_fe3_e in the medium. H. influenzae requires EX_uri_e to obtain a reasonable biomass rate of 1.7 mmol/(gDW⋅h). Our workflow shows that D. pigrum requires three new metabolites within the medium such as EX_LalaDgluMdap_e, EX_ile__L_e, and EX_metox_e. Renz et al. (2021a) identified l-isoleucine and l-methionine as well as 2,6-diaminoheptanedioate, which is required for peptidoglycan metabolism of D. pigrum. Our result consists of some similar chemical derivatives. The solution obtained by the workflow is not unique; both solutions are optimal (minimal number of metabolites), yet there may exist other combinations of metabolites to obtain growth.
3.2.2 Analysis
This part majorly analyzes the models independently. This can give unique insights and understandings about the behavior within the community. The analysis focuses on exchange reactions, as internal reactions are less relevant for the community.
3.2.2.1 Flux Analysis
For each model, the FBA and FVA are implemented to mimic the environment of the human nose. The FVA results in the range of values for which a specific model has a MBR. We show an example in Figure 2. In addition, the flux boundary for each species is computed by FVA and saved as tables and plots to show all non-zero exchange fluxes. FVA yields the upper and lower bounds for the fluxes through every reaction. With this, FVA can estimate the optimal solution when paired with the right combination of other fluxes as almost 100% of the maximum growth rate is achieved. The reactions sustaining low flux variability are presumably of higher priority to an organism. Consequently, FVA is a suitable method for identifying crucial reactions.
3.2.2.2 Secretion Uptake
Using the FVA results, we can identify non-zero secreted and taken-up metabolites for each species, i.e., exchange reactions with strictly positive or negative flux. This step, as a starting point, helps the user determine how interactions can be. If we know what each model uptakes or secretes independently, we can draw certain conclusions about interactions within the community. If certain species have a similar set of uptake reactions, it may be reasonable to assume that they compete for common uptakes within the community. Alternatively, if one species secretes metabolites taken up by another, this may indicate a commensal interaction. To draw this conclusion, we must know if a particular metabolite, either added or removed, actually harms or benefits the growth rate of a species. This is shown in Figure 3, for each identified uptake reaction, we show the effect on the growth by changing the corresponding metabolite concentration within the medium. Figure 3 reveals that D. pigrum is highly glucose-dependent but requires many metabolites in small concentrations to obtain any growth. All other models admit more complex profiles, e.g., S. aureus also majorly depends on glucose, but growth can also be improved by, e.g., EX_pyr_e. Similarly, the growth of H. influenzae can be strongly improved by EX_glu__L_e, EX_fum_e, and EX_uri_e next to glucose.
3.2.2.3 Growth
The default setting for exchange bounds is between −10 mmol/(gDW⋅h) and 1,000 mmol/(gDW⋅h) except for oxygen and iron. We scale this value by a factor k ∈ {1, … , 110} to see how the growth changes, and the results are reported as a plot. This compares the growth rate of all species to find their potential growth on an increasingly rich medium. The result is shown in Figure 3 F. Clearly, S. aureus has the largest growth potential and is thus expected to outcompete other species within the community.
3.2.2.4 Similarity
These steps are ended by calculating the Jaccard index between species to find the similarity between reactions and metabolites of all species in one glance. Interestingly, all models are somewhat similar to each other, except S. aureus. This may explain the difference within the potential growths. Additionally, it shows the resource overlap, i.e., the Jaccard similarity of exchange reactions, e.g., D. pigrum and S. aureus have high similarity. Thus, we may expect that they compete for similar resources.
3.2.2.5 Medium
To design a community, we need to define which metabolites should be shared in the community since the interaction analysis would be feasible for experimental validation if not taking all the significant number of union metabolites into account. To this end, the COMPM defined in the Materials and Methods part is computed as a minimum of min FVA fluxes for those exchange reactions that are common and only take the corresponding min FVA flux for uncommon ones and are saved as a JavaScript Object Notation (JSON) file. This medium is a subset of the default medium, based on our setting, which is SNM3.
3.2.3 Community
Last but not least, we create a community model to analyze the interaction between species as a pooled or compartmentalized community with the shuttle reaction. Here, we focus on the compartmentalized community as it allows detailed investigation into metabolite exchange.
3.2.3.1 Computation of the Maximal Biomass Rate of the Communities
The computation of the MBR for the community is possible by setting which approach for construction of the community is used and which defined medium is applied. Additionally, due to different growth between species, the weights used to compute objective functions in the community can be set manually. The weights can represent the (relative) abundances of each species within the community, yet this assumes that the biomass rate of each model is well calibrated concerning each other. Alternatively, we can see them as free parameters which can be set to achieve a realistic community behavior. Analyzing the community with uniform weights can be challenging if a single species can achieve a comparably high biomass rate. Thus, the community solution will only allow this community member to have growth. Hence, the workflow implements several methods to counteract this problem. First, we can set “fair” weights, which are inversely proportional to the individual biomass rates. Therefore, the growth within the community is not only majorly determined by individual biomass rates but also how good or bad the interaction with other community members is. Alternatively, we can change the community objective to either ensure that community members must reach a certain percentage of community growth or use cooperative trade-off (Diener et al., 2020).
3.2.3.2 Community Summary
The community summary report contains all exchanges between community members or the medium. To show how the community summary works, for instance, a community summary of five species is visualized in Figure 7, while uniform weights are applied. On the rich SNM3, the model predicts that S. aureus and H. influenzae admit significant growth rates, whereas all other community members are close to zero. As glucose majorly affects the growth of S. aureus, it indeed uptakes the majority of glucose within the medium as visualized in Figure 7. Glucose is only shared with D. pigrum, the second major glucose-dependent species. However, this glucose deficit seems to be compensated by S. aureus through the uptake of EX_pyr_e (which we identified previously as beneficial for growth (Mostolizadeh et al., 2022) and experimentally observed that pyruvate was consumed instead of glucose to produce lactate for balancing the reducing equivalents (NAD+) (Carvalho et al., 2017)). This excess amount is not provided by the medium but by M. catarrhalis. On the other hand, M. catarrhalis and D. pigrum provide EX_fum_e, which is the major contributor to the growth of H. influenzae, which then provides outstanding amounts of EX_succ_e for M. catarrhalis and S. aureus. This serves as one example of a complex commensal interaction cycle that can be revealed by metabolic community modeling.
The COMPM induces more competition for resources as indeed the standard community objective only supports the growth of S. aureus, which outcompetes all other species due to its high individual growth rate (see Figure 7).
The COOPM represents the smallest medium in which all community members can (slightly) grow. Notice that the medium does not contain any glucose. M. catarrhalis is now the dominant community member as it can achieve comparably high biomass rates even without any glucose.
3.2.3.3 Prediction of Pair-Wise Interaction
Freilich et al. (Freilich et al., 2011) designed two different formulas as a potential competition score (PCMS) and a potential cooperation score (PCPS), to quantify the level of competition and cooperation predicted among the species.
where VBM,x,y represents the MBR of species y in the community x. If the PCMS value equals 0, then this denotes no competition, while one indicates maximal competition. In addition, the negative PCMS values and positive PCPS values stand for cooperation, while negative PCPS values indicate competition. Therefore, the workflow determines the level of competition and cooperation between species by comparing their individual and combined biomass rates across simulated communities in the COMPM and COOPM.
These two equations Eq. 2 and Eq. 3 are applied to computationally predict the pair-wise interaction between species and compare our results with experimental ones. In Figure 8, we show the computationally obtained values. Notice that on the COMPM, only S. aureus achieved growth. Thus, no interactions can be determined. Nevertheless, by using cooperative trade-off, we obtain a less sparse solution as shown in Figure 8. Clearly, the competition among species increased. Here, the negative interaction of D. pigrum to S. aureus becomes more apparent. However, the interaction of S. aureus to D. pigrum is slightly commensal (Mostolizadeh et al., 2022). As shown in Mostolizadeh et al., (2022), S. aureus can provide relevant metabolites to D. pigrum, yet D. pigrum is a major competitor for glucose.
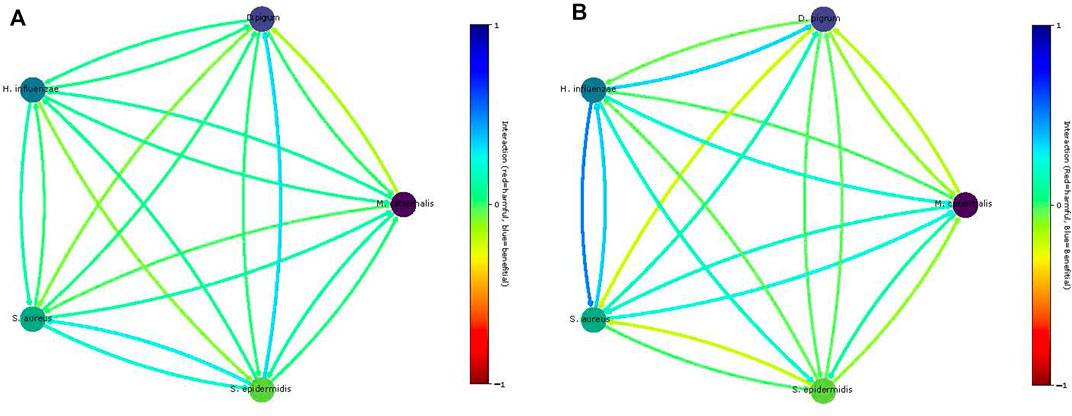
FIGURE 8. Predicted community interactions within different media. Panel (A) shows interaction on the rich SNM3. Panel (B) shows the interaction on the COMPM under cooperative trade-off.
3.2.3.4 Ability of the Model to Provide Metabolites for its Members’ Growth
Genome-scale modeling for each species creates a situation that rarely occurs in nature because it considers each species isolated. In real-life environments, species usually thrive in complex communities in which the growth of a single species depends on the interactions with other species in the population. Our workflow makes this analysis possible to find out how species can influence each other’s growth. To this end, first, essential metabolites necessary for the growth of each single species in SNM3 are found. Second, additional constraints are added to our community to allow the exchange of those metabolites between species if they are among the exchange reactions of other species. Those required metabolites that are not found among the exchange reactions of other species are manually added to the community since there are no species to provide it.
3.3 Availability and Requirements
Project name: NCMW.
Project documentary page: https://manuelgloeckler.github.io/ncmw/
Operating systems: MacOS, Windows, Linux.
Programming language: Python (version >3.7).
License: MIT License.
4 Discussion
The open-source package NCMW simulates the microbe–microbe interactions to show how important their roles in shaping the composition of human nasal microbiota are. However, the cultural capability of strains in microbial communities in the laboratory remains limited. Since this community research in the human nose provides explicit, testable hypotheses and potential targets for experimental verification, it provides a basis to examine the therapeutic potential of individual species as a typical nasal probiotic and possible novel discoveries.
We applied a combination of different optimization approaches to analyze the nasal microbial community to determine what a well-matched theoretical and experimental approach would be. Therefore, two stages are taken into account. One can be the optimization of the objective functions, and the other is the process of deciding what kind of trade-offs are appropriate from the decision-maker’s perspective. These two stages classify the techniques into three techniques: The decision-maker, the designer, is the set of optimal compromise solutions that an effective and complete search procedure must identify to carry out the best choice (Chiandussi et al., 2012). Therefore, these techniques are discussed by analyzing some of their advantages and disadvantages.
1) Priori technique: It takes decisions before searching and includes those approaches that assume that either a specific desired achievable goal or a certain preordering of the objectives can be performed by the decision-maker prior to the search.
2) Posteriori technique: It searches before making decisions and does not require prior preference information from the decision-maker.
3) Progressive technique: It integrates search and decision-making.
The big advantage of the package is its flexibility in creating the different types of communities to convey the way for analysis. In addition, it builds a community in the de facto standard format SBML Level 3 Version 1 (Hucka et al., 2018; Keating et al., 2020; Renz et al., 2021b) with flux balance constraints (FBC) extension version 2 (Olivier and Bergmann, 2018), making any further analysis on the community smoother. Although the workflow was reconstructed for the nasal microbiome community, it is also possible to apply it for any model in other communities. The package automatically creates media defined in the input model and saves it as a JSON file to proceed with the research. As a disadvantage, when more models are added to the community, more running time is expected. However, to shorten the running time, one can use high-quality models to ignore using quality checks of models. In addition, defining a small medium that included only a few metabolites makes understanding a complex community smoother but far from a realistic way.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Author Contributions
RM and AD developed the conceptual idea. RM derived the mathematical method and supervised the work. RM provided the auto-generated GEM of H. influenzae. MG implemented the method under the guidance of RM. RM and MG performed the analysis and wrote the manuscript. AD critically revised the manuscript and the figures. All authors have revised, read, and accepted the manuscript in its final form.
Funding
This research was funded by the German Center for Infection Research (DZIF, doi: 10.13039/100009139) within the Deutsche Zentren der Gesundheitsforschung (BMBF-DZG, German Centers for Health Research of the Federal Ministry of Education and Research), under grant No. 8020708703 and supported by infrastructural funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), Cluster of Excellence EXC 2124 Controlling Microbes to Fight Infections. The parts of this work were funded by the Federal Ministry of Education and Research (BMBF, Germany) and the Baden-Württemberg Ministry of Science as part of the Excellence Strategy of the German Federal and State Governments.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Alina Renz for providing the auto-generated GEMs of M. catarrhalis and S. epidermidis for the example test of the community in this article. The authors acknowledge the support by the Open Access Publishing Fund of the University of Tübingen (https://uni-tuebingen.de/en/216529).
Footnotes
1https://cobrapy.readthedocs.io/en/devel/autoapi/cobra/flux_analysis/fastcc/index.html.
2https://www.ncbi.nlm.nih.gov/assembly/GCF_000007645.1.
3https://www.ncbi.nlm.nih.gov/assembly/GCF_000027305.1.
4https://www.ncbi.nlm.nih.gov/assembly/GCA_000092265.1.
References
Bauer, E., Zimmermann, J., Baldini, F., Thiele, I., and Kaleta, C. (2017). Bacarena: Individual-Based Metabolic Modeling of Heterogeneous Microbes in Complex Communities. Plos Comput. Biol. 13, e1005544. doi:10.1371/journal.pcbi.1005544
Bomar, L., Brugger, S. D., and Lemon, K. P. (2018). Bacterial Microbiota of the Nasal Passages across the Span of Human Life. Curr. Opin. Microbiol. 41, 8–14. doi:10.1016/j.mib.2017.10.023
Buchfink, B., Reuter, K., and Drost, H. G. (2021). Sensitive Protein Alignments at Tree-Of-Life Scale Using diamond. Nat. Methods 18, 366–368. doi:10.1038/s41592-021-01101-x
Carrara, F., Giometto, A., Seymour, M., Rinaldo, A., and Altermatt, F. (2015). Inferring Species Interactions in Ecological Communities: a Comparison of Methods at Different Levels of Complexity. Methods Ecol. Evol. 6, 895–906. doi:10.1111/2041-210x.12363
Carvalho, S. M., de Jong, A., Kloosterman, T. G., Kuipers, O. P., and Saraiva, L. M. (2017). The Staphylococcus aureus α-Acetolactate Synthase ALS Confers Resistance to Nitrosative Stress. Front. Microbiol. 8, 1273. doi:10.3389/fmicb.2017.01273
Chan, S. H. J., Simons, M. N., and Maranas, C. D. (2017). Steadycom: Predicting Microbial Abundances while Ensuring Community Stability. Plos Comput. Biol. 13, e1005539. doi:10.1371/journal.pcbi.1005539
Chiandussi, G., Codegone, M., Ferrero, S., and Varesio, F. E. (2012). Comparison of Multi-Objective Optimization Methodologies for Engineering Applications. Comput. Maths. Appl. 63, 912–942. doi:10.1016/j.camwa.2011.11.057
Cook, D. J., and Nielsen, J. (2017). Genome-scale Metabolic Models Applied to Human Health and Disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 9, e1393. doi:10.1002/wsbm.1393
de Vries, S. P., Eleveld, M. J., Hermans, P. W., and Bootsma, H. J. (2013). Characterization of the Molecular Interplay between moraxella Catarrhalis and Human Respiratory Tract Epithelial Cells. PloS one 8, e72193. doi:10.1371/journal.pone.0072193
Diener, C., Gibbons, S. M., and Resendis-Antonio, O. (2020). MICOM: Metagenome-Scale Modeling to Infer Metabolic Interactions in the Gut Microbiota. MSystems 5, e00606. doi:10.1128/mSystems.00606-19
Ebrahim, A., Lerman, J. A., Palsson, B. O., and Hyduke, D. R. (2013). Cobrapy: Constraints-Based Reconstruction and Analysis for python. BMC Syst. Biol. 7, 74. doi:10.1186/1752-0509-7-74
Fleischmann, R. D., Adams, M. D., White, O., Clayton, R. A., Kirkness, E. F., Kerlavage, A. R., et al. (1995). Whole-genome Random Sequencing and Assembly of haemophilus Influenzae Rd. Science 269, 496–512. doi:10.1126/science.7542800
Freilich, S., Zarecki, R., Eilam, O., Segal, E. S., Henry, C. S., Kupiec, M., et al. (2011). Competitive and Cooperative Metabolic Interactions in Bacterial Communities. Nat. Commun. 2, 589. doi:10.1038/ncomms1597
Gudmundsson, S., and Thiele, I. (2010). Computationally Efficient Flux Variability Analysis. BMC Bioinformatics 11, 489. doi:10.1186/1471-2105-11-489
Hucka, M., Bergmann, F. T., Dräger, A., Hoops, S., Keating, S. M., Le Novère, N., et al. (2018). The Systems Biology Markup Language (SBML): Language Specification for Level 3 Version 1 Core. J. Integr. Bioinformatics 15, 1. doi:10.1515/jib-2017-0080
Jaccard, P. (1912). The Distribution of the Flora in the Alpine Zone.1. New Phytol. 11, 37–50. doi:10.1111/j.1469-8137.1912.tb05611.x
Keating, S. M., Waltemath, D., König, M., Zhang, F., Dräger, A., Chaouiya, C., et al. (2020). SBML Level 3: an Extensible Format for the Exchange and Reuse of Biological Models. Mol. Syst. Biol. 16, e9110. doi:10.15252/msb.20199110
Klitgord, N., and Segrè, D. (2010). Environments that Induce Synthetic Microbial Ecosystems. Plos Comput. Biol. 6, e1001002. doi:10.1371/journal.pcbi.1001002
Krismer, B., Liebeke, M., Janek, D., Nega, M., Rautenberg, M., Hornig, G., et al. (2014). Nutrient Limitation Governs Staphylococcus aureus Metabolism and Niche Adaptation in the Human Nose. Plos Pathog. 10, e1003862. doi:10.1371/journal.ppat.1003862
Levy, R., and Borenstein, E. (2013). Metabolic Modeling of Species Interaction in the Human Microbiome Elucidates Community-Level Assembly Rules. Proc. Natl. Acad. Sci. U S A. 110, 12804–12809. doi:10.1073/pnas.1300926110
Levy, R., and Borenstein, E. (2014). Metagenomic Systems Biology and Metabolic Modeling of the Human Microbiome: from Species Composition to Community Assembly Rules. Gut Microbes 5, 265–270. doi:10.4161/gmic.28261
Lieven, C., Beber, M. E., Olivier, B. G., Bergmann, F. T., Ataman, M., Babaei, P., et al. (2020). MEMOTE for Standardized Genome-Scale Metabolic Model Testing. Nat. Biotechnol. 38, 272–276. doi:10.1038/s41587-020-0446-y
Lin, C., Culver, J., Weston, B., Underhill, E., Gorky, J., and Dhurjati, P. (2018). Gutlogo: Agent-Based Modeling Framework to Investigate Spatial and Temporal Dynamics in the Gut Microbiome. PloS one 13, e0207072. doi:10.1371/journal.pone.0207072
Liu, C. M., Price, L. B., Hungate, B. A., Abraham, A. G., Larsen, L. A., Christensen, K., et al. (2015). Staphylococcus aureus and the Ecology of the Nasal Microbiome. Sci. Adv. 1, e1400216. doi:10.1126/sciadv.1400216
Machado, D., Andrejev, S., Tramontano, M., and Patil, K. R. (2018). Fast Automated Reconstruction of Genome-Scale Metabolic Models for Microbial Species and Communities. Nucleic Acids Res. 46, 7542–7553. doi:10.1093/nar/gky537
Man, W. H., de Steenhuijsen Piters, W. A., and Bogaert, D. (2017). The Microbiota of the Respiratory Tract: Gatekeeper to Respiratory Health. Nat. Rev. Microbiol. 15, 259–270. doi:10.1038/nrmicro.2017.14
Mostolizadeh, R., Glöckler, M., and Dräger, A. (2022). Workflow for Interaction between Metabolic Community Models in the Human Nose Applied to Dolosigranulum Pigrum and Staphylococcus aureus within the Human Nose.
Mostolizadeh, R., Dräger, A., and Jamshidi, N. (2019). “Insights into Dynamic Network States Using Metabolomic Data,” in High-Throughput Metabolomics (Berlin, Germany: Springer), 243–258. doi:10.1007/978-1-4939-9236-2_15
Noronha, A., Modamio, J., Jarosz, Y., Guerard, E., Sompairac, N., Preciat, G., et al. (2019). The Virtual Metabolic Human Database: Integrating Human and Gut Microbiome Metabolism with Nutrition and Disease. Nucleic Acids Res. 47, D614–D624. doi:10.1093/nar/gky992
Olivier, B. G., and Bergmann, F. T. (2018). SBML Level 3 Package: Flux Balance Constraints Version 2. J. Integr. Bioinform 15, 20170082. doi:10.1515/jib-2017-0082
Orth, J. D., Thiele, I., and Palsson, B. Ø. (2010). What Is Flux Balance Analysis? Nat. Biotechnol. 28, 245–248. doi:10.1038/nbt.1614
Proctor, D. M., and Relman, D. A. (2017). The Landscape Ecology and Microbiota of the Human Nose, Mouth, and Throat. Cell Host Microbe 21, 421–432. doi:10.1016/j.chom.2017.03.011
Pruitt, K. D., Tatusova, T., and Maglott, D. R. (2005). Ncbi Reference Sequence (Refseq): a Curated Non-redundant Sequence Database of Genomes, Transcripts and Proteins. Nucleic Acids Res. 33, D501–D504. doi:10.1093/nar/gki025
Renz, A., Widerspick, L., and Dräger, A. (2021). First Genome-Scale Metabolic Model of Dolosigranulum pigrum Confirms Multiple Auxotrophies. Metabolites 11, 232. doi:10.3390/metabo11040232
Renz, A., Mostolizadeh, R., and Dräger, A. (2021). “Clinical Applications of Metabolic Models in SBML Format,” in Systems Medicine. Editor O. Wolkenhauer (Oxford: Academic Press), Vol. 3, 362–371. doi:10.1016/B978-0-12-801238-3.11524-7
Seif, Y., Monk, J. M., Mih, N., Tsunemoto, H., Poudel, S., Zuñiga, C., et al. (2019). A Computational Knowledge-Base Elucidates the Response of Staphylococcus aureus to Different media Types. Plos Comput. Biol. 15, e1006644. doi:10.1371/journal.pcbi.1006644
Shoaie, S., Karlsson, F., Mardinoglu, A., Nookaew, I., Bordel, S., and Nielsen, J. (2013). Understanding the Interactions between Bacteria in the Human Gut through Metabolic Modeling. Sci. Rep. 3, 2532. doi:10.1038/srep02532
Taffs, R., Aston, J. E., Brileya, K., Jay, Z., Klatt, C. G., McGlynn, S., et al. (2009). In Silico approaches to Study Mass and Energy Flows in Microbial Consortia: a Syntrophic Case Study. BMC Syst. Biol. 3, 114–116. doi:10.1186/1752-0509-3-114
Tan, P. N., Steinbach, M., and Kumar, V. (2016). Introduction to Data Mining. Chennai: Pearson Education India.
Tatusov, R. L., Mushegian, A. R., Bork, P., Brown, N. P., Hayes, W. S., Borodovsky, M., et al. (1996). Metabolism and Evolution of haemophilus Influenzae Deduced from a Whole-Genome Comparison with escherichia Coli. Curr. Biol. 6, 279–291. doi:10.1016/s0960-9822(02)00478-5
van der Ark, K. C. H., van Heck, R. G. A., Martins Dos Santos, V. A. P., Belzer, C., and de Vos, W. M. (2017). More Than Just a Gut Feeling: Constraint-Based Genome-Scale Metabolic Models for Predicting Functions of Human Intestinal Microbes. Microbiome 5, 78–13. doi:10.1186/s40168-017-0299-x
Vlassis, N., Pacheco, M. P., and Sauter, T. (2014). Fast Reconstruction of Compact Context-specific Metabolic Network Models. Plos Comput. Biol. 10, e1003424. doi:10.1371/journal.pcbi.1003424
Widder, S., Allen, R. J., Pfeiffer, T., Curtis, T. P., Wiuf, C., Sloan, W. T., et al. (2016). Challenges in Microbial Ecology: Building Predictive Understanding of Community Function and Dynamics. ISME J. 10, 2557–2568. doi:10.1038/ismej.2016.45
Yadan, O. (2019). Hydra - a Framework for Elegantly Configuring Complex Applications. San Francisco, CA, USA: Github.
Yan, M., Pamp, S. J., Fukuyama, J., Hwang, P. H., Cho, D. Y., Holmes, S., et al. (2013). Nasal Microenvironments and Interspecific Interactions Influence Nasal Microbiota Complexity and s. aureus Carriage. Cell Host Microbe 14, 631–640. doi:10.1016/j.chom.2013.11.005
Zhang, Y. Q., Ren, S. X., Li, H. L., Wang, Y. X., Fu, G., Yang, J., et al. (2003). Genome-based Analysis of Virulence Genes in a Non-biofilm-forming staphylococcus Epidermidis Strain (Atcc 12228). Mol. Microbiol. 49, 1577–1593. doi:10.1046/j.1365-2958.2003.03671.x
Keywords: microbial communities, nasal microbiome, computational biology, genome-scale modeling, constraint-based modeling
Citation: Glöckler M, Dräger A and Mostolizadeh R (2022) NCMW: A Python Package to Analyze Metabolic Interactions in the Nasal Microbiome. Front. Bioinform. 2:827024. doi: 10.3389/fbinf.2022.827024
Received: 01 December 2021; Accepted: 19 January 2022;
Published: 25 February 2022.
Edited by:
Miguel Rocha, University of Minho, PortugalReviewed by:
Vítor Pereira, University of Minho, PortugalShrikant Pawar, Yale University, United States
Copyright © 2022 Glöckler, Dräger and Mostolizadeh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Reihaneh Mostolizadeh, cmVpaGFuZWgubW9zdG9saXphZGVoQHVuaS10dWViaW5nZW4uZGU=