 Famke Bäuerle1,2,3*
Famke Bäuerle1,2,3* Gwendolyn O. Döbel1,3,4
Gwendolyn O. Döbel1,3,4 Laura Camus2,4,5
Laura Camus2,4,5 Simon Heilbronner2,4,5,6*
Simon Heilbronner2,4,5,6* Andreas Dräger1,3,4,5*
Andreas Dräger1,3,4,5*- 1Computational Systems Biology of Infections and Antimicrobial-Resistant Pathogens, Institute for Bioinformatics and Medical Informatics (IBMI), Eberhard Karl University of Tübingen, Tübingen, Germany
- 2Interfaculty Institute of Microbiology and Infection Medicine Tübingen (IMIT), Eberhard Karl University of Tübingen, Tübingen, Germany
- 3Department of Computer Science, Eberhard Karl University of Tübingen, Tübingen, Germany
- 4German Center for Infection Research (DZIF), Partner Site Tübingen, Tübingen, Germany
- 5Cluster of Excellence “Controlling Microbes to Fight Infections (CMFI)”, Eberhard Karl University of Tübingen, Tübingen, Germany
- 6Faculty of Biology, Microbiology, Ludwig Maximilian University of Munich, Munich, Germany
Introduction: Genome-scale metabolic models (GEMs) are organism-specific knowledge bases which can be used to unravel pathogenicity or improve production of specific metabolites in biotechnology applications. However, the validity of predictions for bacterial proliferation in in vitro settings is hardly investigated.
Methods: The present work combines in silico and in vitro approaches to create and curate strain-specific genome-scale metabolic models of Corynebacterium striatum.
Results: We introduce five newly created strain-specific genome-scale metabolic models (GEMs) of high quality, satisfying all contemporary standards and requirements. All these models have been benchmarked using the community standard test suite Metabolic Model Testing (MEMOTE) and were validated by laboratory experiments. For the curation of those models, the software infrastructure refineGEMs was developed to work on these models in parallel and to comply with the quality standards for GEMs. The model predictions were confirmed by experimental data and a new comparison metric based on the doubling time was developed to quantify bacterial growth.
Discussion: Future modeling projects can rely on the proposed software, which is independent of specific environmental conditions. The validation approach based on the growth rate calculation is now accessible and closely aligned with biological questions. The curated models are freely available via BioModels and a GitHub repository and can be used. The open-source software refineGEMs is available from https://github.com/draeger-lab/refinegems.
1 Introduction
Human life and substantial cultural achievements depend on prokaryotes. While a minority of bacterial species cause invasive diseases, humans could not exist without beneficial microbes colonizing the gut and the body surfaces. Additionally, the metabolic capacities of microbes flavor and preserve our food, detoxify the environment, or allow the production of antibiotics to treat infections. Without the ability to identify and grow microbes under defined conditions within laboratory surroundings, many modern comforts would be unthinkable. Remarkably, we are only beginning to appreciate the enormous diversity of bacterial life, and even with state-of-the-art techniques to grow bacteria, the vast majority remains uncultivated (Lloyd et al., 2018).
As a huge metabolic capacity is slumbering within these “unculturable” bacteria, efforts for their cultivation should be intensified (Thrash, 2019; Bodor et al., 2020). Computer modeling of the nutritional needs of microorganisms holds great promise in this regard. The importance of computational modeling in biology has steadily increased over the last decades (Bordbar et al., 2014). In particular, genome-scale metabolic models, so-called GEMs, have proven beneficial in numerous application areas due to their facile mathematical manageability and predictive power (Gu et al., 2019).
Among other benefits, GEMs permit predicting an appropriate media composition that should allow proliferation for any organism. However, up to date, this potential seems hardly exploited as it needs combined efforts of bioinformaticians to make predictions and of microbiologists to test the same systematically. Most published models lack sufficient experimental validation, or the designators of the components contained in the models only allow preliminary conclusions to be drawn about tangible cell components. These shortcomings reduce the validity or interpretability of the predicted results. Only if modelers and experimenters work closely together can they check predictions made by the model and, if necessary, change the structure of the model or the experimental setup to gradually arrive at a meaningful representation of reality in the model that yields reliable results (Fitzpatrick and Stefan, 2022).
Herein we tested the ability of de novo created GEMs to predict in vitro growth characteristics of Corynebacterium striatum. At the time of writing, no genome-scale systems biology models are available that characterize this species. C. striatum is a Gram-positive, non-sporulating rod discovered in the early 20th century. It was considered a commensal within the healthy human skin microbiota for a long time (McMullen et al., 2017), and its pathogenic properties remained unknown. With increasing awareness of nosocomial infections, C. striatum was identified as a cause of diseases, particularly for immunocompromised patients. It may cause several diseases, including Chronic Obstructive Pulmonary Disease, COPD, or pneumonia (Shariff et al., 2018). In addition to the human respiratory tract, long-standing open wounds belong to its habitat (Chandran et al., 2016), leading to prolonged hospitalizations (Nudel et al., 2018). Compared to other Gram-positive members of the skin flora, C. striatum is particularly resistant to several antibiotics (McMullen et al., 2017), including ampicillin, penicillin, and tetracycline (Chandran et al., 2016). For these reasons, C. striatum constitutes an ideal example case for benchmarking and improving a systematic and semi-automatic modeling environment.
The individual work steps required to reconstruct high-quality models have been excellently characterized in the literature in numerous examples (van’t Hof et al., 2022; Feierabend et al., 2021; Dillard et al., 2023; Dahal et al., 2023). However, a substantial obstacle in this endeavor is the enormous complexity and the sheer number of those steps, which Thiele and Palsson put at 96 repetitive steps (Thiele and Palsson, 2010). In addition, this requires knowledge from many orthogonal scientific disciplines ranging from (bio)chemistry, biophysics, and bioinformatics to mathematics and their application in bioengineering and, depending on the use case, various fields such as microbiology, oncology, or biotechnology. Working with the underlying file formats and data standards also requires specialized knowledge in particular areas. It is easy to see why the modeling work is neither intuitive nor generally accessible. Fundamental programming skills are required to examine the resulting models just rudimentarily.
The file format SBML (Keating et al., 2020; Renz et al., 2020) is most widely used to encode systems biology models (Dräger and Waltemath, 2021). However, a particular extension, the Flux Balance Constraints (FBC) package (Olivier and Bergmann, 2018), is required to store constraint-based genome-scale models in it. Other packages open up additional features of the SBML format to users, such as linking metabolic maps directly to the computational model (Gauges et al., 2015; Bergmann et al., 2018), offering a wide range of visualization capabilities (Buchweitz et al., 2020; Holzapfel et al., 2022).
Such metabolic maps are often drawn using web-based programs such as Escher (King et al., 2015) or Newt (Balci et al., 2021), which are compatible with the Systems Biology Graphical Notation (SBGN) standard (Bergmann et al., 2020; Touré et al., 2021) and generate unambiguously represented biological network maps. The computational model can be created and edited using various programs, depending on the type of modeling. Constraint-Based Reconstruction and Analysis (COBRA) programs such as COBRApy (Ebrahim et al., 2013) (for Python users) or COBRA Toolbox (Heirendt et al., 2019) (for Matlab users) have proven particularly useful for genome-scale models. An initial model is, in turn, first created with programs such as CarveMe (Machado et al., 2018). For the publication of models, the BioModels database is usually available (Malik-Sheriff et al., 2020).
This short list is not exhaustive but should illustrate that modeling work is often discontinuous because numerous different programs are required in combination. In addition, the modeler needs to think about a suitable structure for versioning their models and save it in a way that is understandable to others. Many steps also require manual revision, sometimes done directly in the SBML files or using the command line and highly specific scripts. Much of the work required for modeling may be similar in its outcome but repetitively and independently developed. Not only does the resulting redundancy lead to endless hours of avoidable programming work. It also results in projects that are incompatible with each other but similar in their effect (Yurkovich et al., 2017).
The fewer users test a particular software project, the higher the probability of finding previously undiscovered programming errors. Ultimately, these also affect the model quality. Since each developer typically places the aspects relevant to its respective project in the foreground and cannot cover the curation of the models in sufficient depth, it can be easily explained why frequently certain aspects fall by the wayside: either due to a lack of time or the sheer overwhelming nature of the overall complexity.
The present work addresses such problems by providing a unified directory structure and a collection of directly executable programs within a Git-based version control system. To this end, the authors use their experience gained in numerous previous projects on systems biology modeling (Feierabend et al., 2021; Dahal et al., 2023), software development (Panchiwala et al., 2022; Glöckler et al., 2023) and laboratory work (Adolf et al., 2023; Krauss et al., 2023) to precisely reconstruct and experimentally validate multiple strains in parallel using currently common standard operating protocols for a bacterium that remains to be studied in systems biology. In this work, we constructed GEMs of five C. striatum model strains and used them to predict growth characteristics under defined nutritional conditions. These predictions were then tested in the laboratory. Interestingly, we found that the in vitro and in silico data for C. striatum largely overlap. In addition to the comparison between laboratory and in silico strain behavior, a program to enhance the creation of high-quality models is presented.
2 Materials and methods
2.1 Genome sequences
Both the National Center for Biotechnology Information (NCBI) and PATRIC were used to search for complete genome sequences of C. striatum strains. On NCBI the genome assembly ASM215680v1 of C. striatum KC-Na-01 with the accession reference GCF_002156805.1 was the most prominent. This sequence was used to create a first GEM. This strain was not available to order from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, translates to German Collection of Microorganisms and Cell Cultures). Thus, four additional strains with BacDrive sequence information were obtained from DSMZ (Table 1). All sequences were downloaded from NCBI (Table 1).
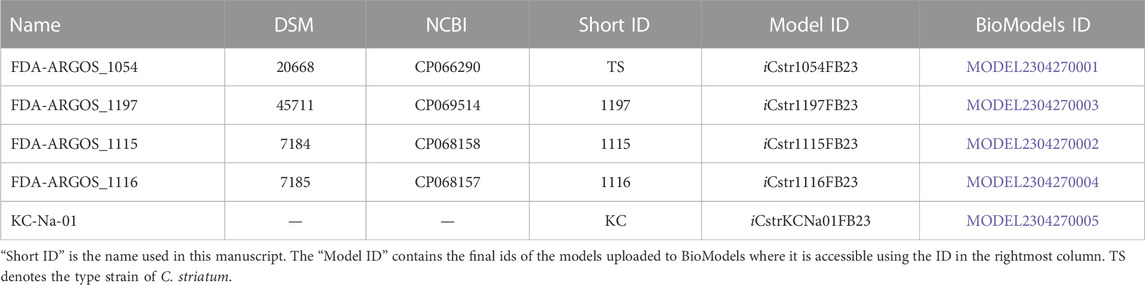
TABLE 1. Corynebacterium striatum strains used in this study, with the corresponding DSM-number, NCBI accession number, and identifier.
2.2 Existing model assessment
One GEM for C. striatum exists in the Virtual Metabolic Human (VMH) database. The VMH offers GEMs of organisms interesting for human microbiomes. Most models within the VMH database are used as a basis to build simulatable microbiomes. Additionally, all VMH GEMs use specific entity IDs of which some overlap with biochemically, genomically, and genetically structured (BiGG, Norsigian et al., 2019) IDss. The one existing GEM was assessed with MEMOTE and manually evaluated by looking at the Extensible Markup Language (XML) file.
2.3 Draft models
Draft models were created from the genome sequences annotated by the NCBI Prokaryotic Genome Annotation Pipeline using CarveMe (Machado et al., 2018). We used version 1.5.1 of the package, which we installed via pip on a MacBookPro running macOS Monterey version 12.3.1. CarveMe was run on the protein FASTA files of the sequences with the fbc2 flag. Fbc2 refers to the SBML package “Flux Balance Constraints” (Olivier and Bergmann, 2018) which extends models written in SBML by structures that enable flux bounds and optimization functions. These structures are necessary for growth simulations with flux balance analysis (FBA) which were used to compare the models to laboratory results.
2.4 Automated polishing
The drafts were polished with ModelPolisher (Römer et al., 2016) and with the script polish.py available with the refineGEMs toolbox (see Section 3.3). Automated polishing included moving entries from the notes section to the annotation section of an entity, annotating all entities with their respective BiGG (Norsigian et al., 2019) ID as identifiers.org link, and setting the models parameters to mmol_per_gDW_per_h. In addition, the GeneProducts were polished using refineGEMs: They were annotated with the NCBI Protein ID and renamed with the respective name indicated on NCBI.
2.5 Semi-automated curation
RefineGEMs was used to add charges to metabolites that had no denoted charge, some of the missing charges were extracted from the model of Pseudomonas putida KT2440 with the ID iJN1463. RefineGEMs was used to apply the SBOannotator tool (Leonidou et al., 2023) which allows for automated Systems Biology Ontology (SBO, Courtot et al., 2011) term annotation and specialization. The Python module MassChargeCuration (Finnem and Mostolizadeh, 2023) was used to further correct the charges and masses of the metabolites. RefineGEMs was used to synchronize annotations of metabolites in different compartments.
2.6 Manual curation
Manual curation was first focused on cleaning residues left from the automated draft reconstruction: Duplicate reactions and metabolites with different IDs were identified using MEMOTE (Lieven et al., 2020) and subsequently removed. Metabolites with only a few or no annotations were researched manually in multiple databases and annotated based on the results. Network gaps were identified by drawing pathways as Escher maps (King et al., 2015) and comparing those to Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway maps (Kanehisa et al., 2021).
2.7 Model quality assessment
The MEMOTE score (Lieven et al., 2020) serves as a tool for researchers to quickly evaluate the completeness and accuracy of GEMs and to compare models across different organisms and strains. It also helps to identify potential gaps in the models, making it easier to improve them in the future.
2.8 In silico growth rates
Growth rates and, thus, doubling times were determined by FBA. We used the routine implemented in refineGEMs which is based on the COBRApy optimize function. This maximized the flux through the biomass objective function (BOF). It assumes that the goal of any organism is to increase its biomass toward cell division. The returned objective value was then interpreted as growth rate in mmol
with r being the growth rate or objective value extracted from the FBA. All used media formulations can be found in the sbo_media_db.sql in refineGEMs.
2.9 Growth rate comparison
Growth rates from simulations with COBRApy are given in mmol
2.10 In vitro growth phenotypes
All media compositions are available in the Supplementary Material. Cultures were grown in Tryptic Soy Broth (TSB) overnight at 37°C and 150 rpm. After 10 min of centrifugation at 4,000 rpm, the remaining pellets were resuspended in the medium of interest. The samples were inoculated at an OD of 0.1. The OD 600 nm (OD600) was measured at t = 0 h and at t = 24 h. Fold changes were calculated by dividing OD600 (t = 24 h) by OD600 (t = 0 h).
2.11 In vitro growth rates
For each of the four strains, one colony grown on Tryptic Soy Agar (TSA) Blood plates was inoculated in 10 mL of TSB. Precultures were incubated overnight at 37°C and 150 rpm and diluted to an OD of 0.05 in TSB, Brain-Heart-Infusion Broth (BHI), Roswell Park Memorial Institute cell culture medium 1640 (RPMI), Lysogeny Broth (LB), M9 minimal medium (M9), and Corynebacterium glutamicum minimal medium version 12 (CGXII), Supplementary Table S1. 150 L of the suspensions was distributed in a Greiner Bio-One 96 flat bottom well plate and incubated at 37°C for 24 h in the BioTek Epoch 2 Microplate Reader. OD600 was monitored every 15 min after agitation at 600 rpm. All experiments were performed using three technical replicates per plate and at least three biological replicates.
2.12 Growth data analysis
Growth data were extracted from the plate reader in a Microsoft Excel® file and were then read using pandas (Reback et al., 2022). The logistic equation for growth was fitted using curve_fit from scipy.optimize (Virtanen et al., 2020). This uses a non-linear least squares approach to fit Eq. 2 to the growth data. Then the growth rate r can be extracted, and the doubling time or generation time Td is then calculated by Td = ln 2/r. A logistic function that results from a logistic model of bacterial growth is shown on Eq. 2:
Here, ODt is the OD at time t, K is the asymptote (usually the maximum OD), OD0 is the OD at time 0, d refers to the displacement along the x-axis. Multiple columns with OD values are possible. Coming from the plate reader, technical replicates were averaged before fitting. The wells representing the blank were also averaged and subtracted from the averaged technical replicates.
For the logistic fit, curve_fit was always initialized with p0 = np.asarray([0.2, 0.05, 0.05]), the function is defined in Python as
def logistic (t, K, y0, r):
return K/(1+((K-y0)/y0)*np.exp(-r*t))
2.13 Model monitoring
During the tests on different media, the models were continuously monitored, and simulations were run to obtain missing metabolites that might help to recover growth on the minimal media of interest. We noticed that the models had no exchange reactions for sodium which was changed to reflect the ability of sodium uptake of the respective organisms.
3 Results
3.1 Construction of five strain-specific GEMs
3.1.1 Available model and strains of C. striatum
To the authors’ best knowledge, no other manually curated high-quality GEM of C. striatum is currently available. However, within the gut microbiota resource of the VMH database (Magnúsdóttir et al., 2017), a model for the type strain ATCC 6940 exists (Heinken et al., 2023).
The VMH model for C. striatum has a MEMOTE score of 88 %, even though it contains erroneous annotations with NaN IDs and incorrect InChIKeys. All the GeneProduct objects lack annotation, and some of the reactions were only annotated with their respective SBO terms. Thus, within this study, we created manually curated high-quality GEMs for C. striatum.
We decided to work with multiple strains to get a comprehensive picture of the strain-specific properties of C. striatum. Apart from the type strain FDAARGOS_1054/ATCC 6940 [type strain (TS)], we used three more strains, namely, FDAARGOS_1197 (1197), FDAARGOS_1115 (1115) and FDAARGOS_1116 (1116), whose genome sequences have been completely assembled. In addition to these four strains, we also investigated the strain KC-Na-01 (KC), which is well characterized by the KEGG database, but unlike the other four strains, it is not available from the DSMZ. For this reason, for strain KC, we exclusively performed an in silico analysis.
The strain-specific models were named following the guidelines given by Carey et al. (2020). However, as there were no indications for naming strain-specific models we decided to elongate the species indicator by a strain indicator (in our case the FDA-ARGOS ID). As the iteration identifier the author’s initials (FB) and the year (23) were added.
3.1.2 Characteristics of the models created in this study
The models created in this study are composed of 1,053–1,382 metabolites, 1,541–2,002 reactions, and 719–772 genes. This is within the usual range of bacterial GEM [compare, e.g., with 1,183 genes and 2,276 reactions (Hawkey et al., 2022)]. Figure 1A and Supplementary Table S2 show that the model of strain TS has the biggest scope with respect to the number of entities that it holds. All GEMs have a metabolic coverage above 2 %. Usually, models with a high level of modeling detail will have a metabolic coverage above 1 %, which highlights the degree of detail of our models. All MEMOTE scores are above 84 %, which is higher than all scores of models evaluated for the MEMOTE meta-study in 2020 (Lieven et al., 2020) (compare SI Figure 30 of that study). All entities that are present in the models are annotated both with Uniform Resource Identifiers (URIs) and SBO terms. We used ten different databases for metabolite annotations, eight for reaction annotations, and two databases to annotate protein-encoding sequences which are added as Gene Protein Reactions to the models. KEGG metabolic pathways were added to all models. The models were checked for energy-generating cycles, which were eliminated, and orphaned metabolites were connected to the network. All models are stoichiometrically consistent.
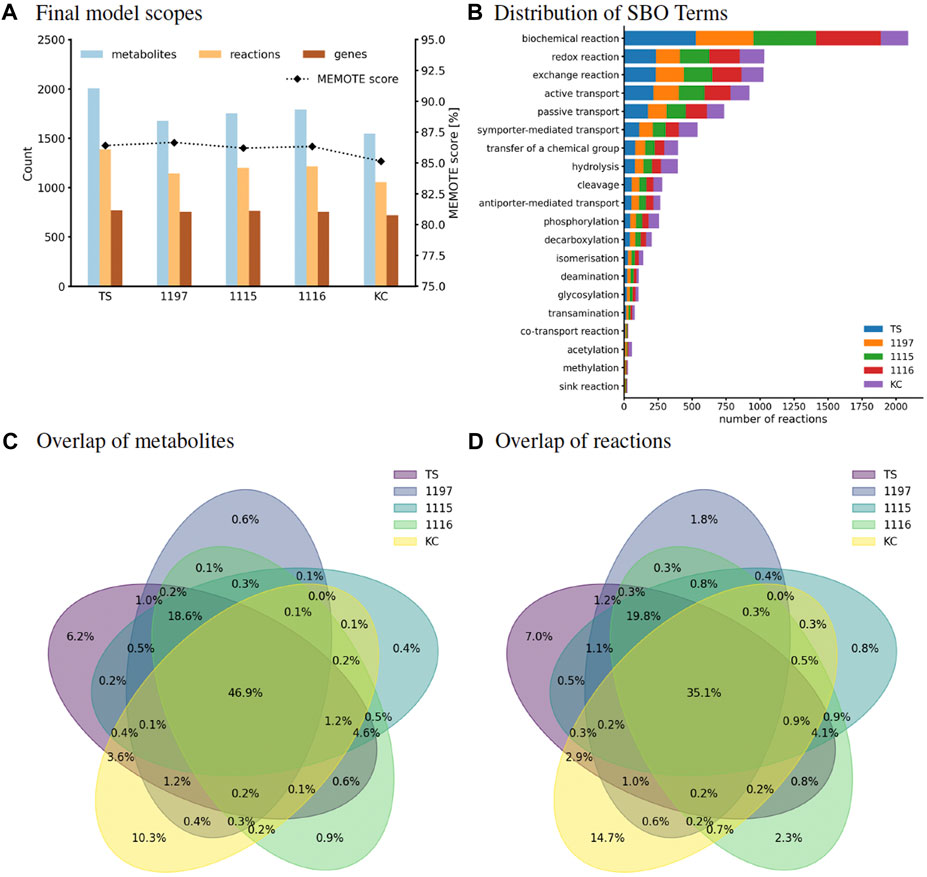
FIGURE 1. (A) Scopes of all curated C. striatum models. Number of reactions, metabolites, and genes are read on the left y-axis, MEMOTE scores are read on the right y-axis. This score is based on standardized and community-maintained metabolic tests for quality control and quality assurance of GEMs. (B) Classification of reaction types by assigned SBO terms. SBO terms were annotated using the SBOAnnotator (Leonidou et al., 2023) tool and thus are specialized. The distribution of different reactions is shown with stacked bars which represent the number of reactions that are classified with that SBO term. (C,D) Strain comparison based on the metabolic reconstructions. Venn diagrams showing the overlap of all metabolites (C) and all reactions (D) of the curated models in this study. These diagrams were created based on the metabolite/reaction identifiers using the pyvenn Python module.
Reactions can be classified into different types using the SBO terms. SBO analysis showed that all four models follow a similar reaction type distribution (Figure 1B), suggesting vastly overlapping metabolic capacities of the strains.
The metabolic reconstructions were used for a detailed comparison of the five strains. This showed an overlap of 47 % for all metabolites and an overlap of 35 % for all reactions across all models (Figures 1C, D). With 168 and 391 unique metabolites and reactions, respectively, the strain KC showed higher divergence compared to the other models. In comparison, strain 1115 showed 6 and 21 unique metabolites and reactions, respectively. This suggests a reduced metabolic fitness compared to strain KC.
3.2 Experimental validation of the strain-specific GEMs
GEMs can be used to predict growth characteristics of bacterial species under defined nutritional conditions (Dahal et al., 2023). This can be useful for laboratory experimentation, especially when little to no in vitro data for the microorganism of interest is available. However, the accuracy of GEMs in predicting in vitro characteristics is frequently unclear. Therefore, we decided to compare model predictions to in vitro growth characteristics under various nutritional conditions.
3.2.1 Growth characteristics under varying nutritional conditions
We used the complex, nutritionally rich medium LB (Bertani, 1951) for which a nutritional composition to be used in metabolic modeling is available (Machado et al., 2018). However, this medium contains the complex component yeast extract, making the precise composition of the medium in vitro unclear. As defined media, we used RPMI (Thermo Fisher Scientific, Inc, 2023), M9 (Bécard and Fortin, 1988; Merck & Co., Inc, 2023), and CGXII (Keilhauer et al., 1993; Unthan et al., 2014; Yang et al., 2021) since their precise chemical composition is known.
In vitro experiments allow for different experimental approaches. Besides changing incubation temperature, bacterial cultures can be grown in flasks with or without a baffle whose volumes can be adapted from microliters to liters. Alternatively, cultures can be grown in microtiter formats (100 µL⋅well−1 to 500 µL⋅well−1) or even in continuous culture systems where a constant refreshment of medium with an inflow of metabolites can be used. It is well known that varying growth conditions impact the achieved cell densities and growth rates significantly. However, the influence such conditions have on, e.g., gas exchange rates, are difficult to reflect within GEMs.
We, therefore, decided to test the accuracy of the model to predict the growth characteristics of the strains under two distinct growth conditions (Figure 2).
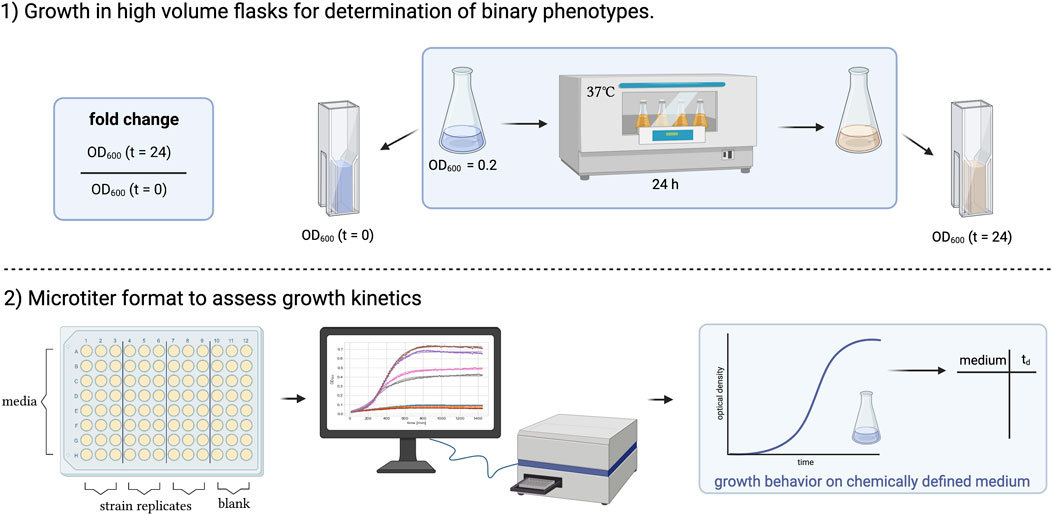
FIGURE 2. Graphical summary of two experimental approaches to study the growth of Corynebacterium striatum under defined nutritional conditions. 1) Growth in high volume flasks for binary assessment of growth. 2) Growth in microtiter plate format to assess growth kinetics. The figure was created using BioRender.com.
1. Growth in high volume flasks for determination of binary phenotypes. Higher volumes of media (10 mL in 50 mL flasks) were inoculated to an optical density of 0.2, and the optical density was assessed after 0 and 24 h to determine binary growth phenotypes (growth vs. no growth). Binary phenotypes are used to assess the accuracy of a GEM where a simulation can be run, e.g., on different carbon sources that can then be tested in the laboratory.
2. Microtiter format to assess growth kinetics. Besides binary phenotypes, the prediction of metabolites or additional nutrients that increase/decrease growth rates is of high relevance for the planning of in vitro experiments. We used a volume of 150 µL per medium in a 96-well plate and automated the assessment of OD600 over time to determine growth kinetics. From the growth curves captured via a plate reader the doubling time at the inflection point of the sigmoidal curve can be extracted and compared to those predicted by the GEM.
3.2.2 Growth in LB medium is predicted correctly, and growth rates for three strains in LB medium in silico are reflected in vitro
GEM analysis predicted binary growth of TS, 1197, and 1115 while growth of strain 1116 was not possible under the modeled nutritional composition of LB medium (Table 2). Investigation of this phenomenon revealed that strain 1116 lacks a part of the nicotinate metabolism (Supplementary Figure S2) making the strain auxotrophic for the essential enzymatic cofactor nicotinamide d-ribonucleotide (nmn). A putative nmn transporter is predicted by the GEM, supporting the existence of an auxotrophy to this compound. In contrast, transporters for the precursors of nmn (nicotinate or nicotinamide) were not predicted. This caused the GEM to predict a specific need for nmn which was not available in the in silico formulation of LB. Interestingly, all strains were able to grow in LB in in vitro experiments (Figure 3A). This shows that the auxotrophy of 1116 is not relevant in LB. This might have two reasons. Either nmn might be present in sufficient amounts in LB medium, or its precursors (nicotinate, nicotinamide) are present and taken up by unidentified transport systems in C. striatum. To further compare in silico modeling with in vitro results, we used growth curve analysis to extract in vitro generation times. Of note, automated extraction of generation times relies on fitting a logistic equation to the growth curve, which is error-prone when OD values do not show evident sigmoidal characteristics. Accordingly, we have to mention that the generation times of 1115 and TS are more reliable than those of 1115 and 1197 (Figure 3B). Additionally, it has to be noted that the generation time is calculated for the period of exponential growth (even when very short) and does not necessarily reflect the reached final ODs of the strains. Thus, the generation times calculated herein have to be interpreted with care. However, we used them as a proxy to assess the GEM predictions. In LB medium, strain 1197 grew significantly slower (81.13 min⋅generation−1) than strains 1115 and 1116 (45.92 min⋅generation−1 and 40.38 min⋅generation−1 respectively) (Figure 3B). These differences were not predicted by the respective GEMs, suggesting differences in the metabolisms of the strains that are currently not reflected within the models. Strain 1116 differed from strain 1115 in the reached ODs of around 0.2 and 0.6, respectively, but not in the doubling times of 40–45 min. The doubling times extracted from the growth curves only differed for strain 1197 notably from those predicted by the model, most likely due to the non-sigmoidal growth of the strain in our experiment.
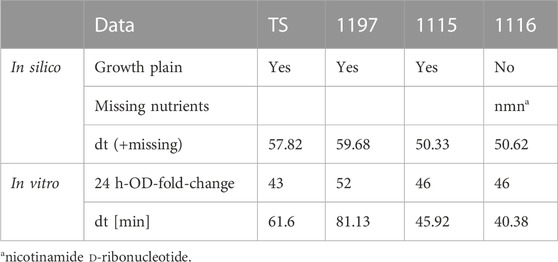
TABLE 2. Growth behavior in silico and in vitro of all strains in LB.
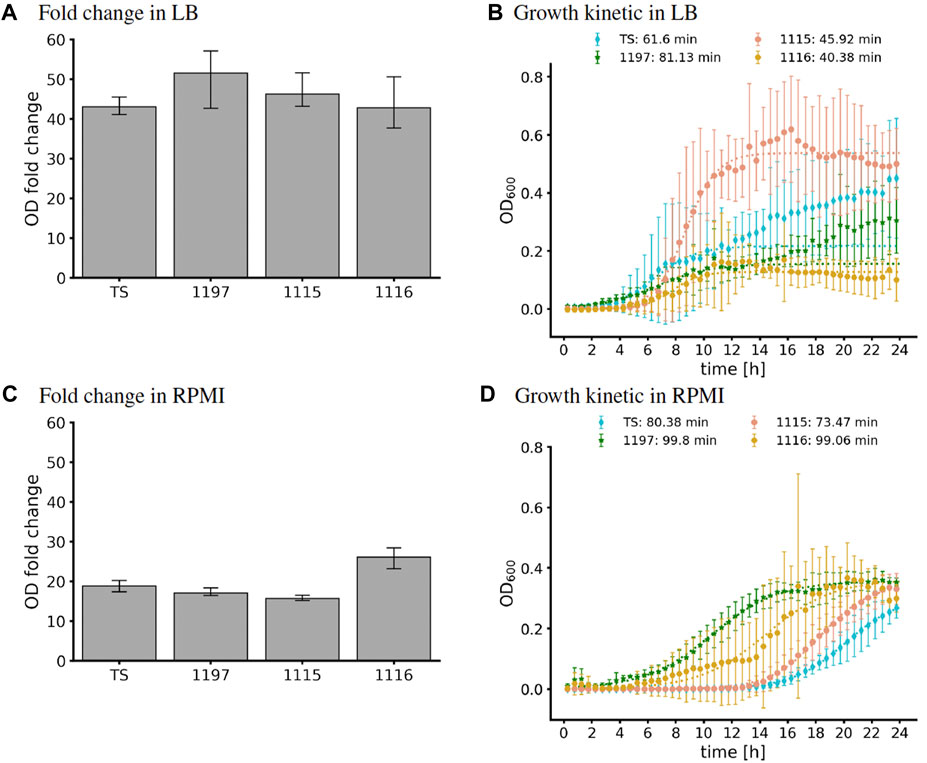
FIGURE 3. OD fold changes and in vitro growth curves in LB (A,B) and RPMI (C,D) of the strains TS, 1197, 1115, and 1116. (A,C) 10 mL medium in 50 mL flasks were inoculated to OD600 = 0.2. After 24 h of incubation, the OD600 was measured again, and the fold-change was calculated as a measure for bacterial growth. Shown are the mean and SD of three independent experiments. (B,D) Growth in Microtiter plates. 150 mL of medium in a 96 well plate were inoculated and OD600 was assessed automatically for 24 h using an Epoch2 plate reader. The logistic fit was calculated using curve_fit (dotted lines). The doubling times extracted from the logistic fit are indicated in the legend. For strain TS and 1197, we only fitted data up to 12.5 h to avoid fitting to diphasic growth.
3.2.3 Model optimization allows congruent in silico and in vitro growth in RPMI medium
None of the strains showed in silico proliferation on the nutritional composition of RPMI (as detailed by the supplier). Investigating this phenomenon showed a lack of the trace elements (Co2+, Cu2+, Fe2+, Mn2+ and Zn2+) for all strains while 1116 lacked additionally nmn as observed before. However, all strains grew, effectively in flasks in vitro (Figure 3C). This shows the presence of sufficient amounts of trace elements in the medium to allow bacterial growth. Consequently, modification of the in silico composition of RPMI by the addition of trace elements is needed to optimize the congruency of in silico and in vitro analysis. Furthermore, RPMI contains nicotinamide. This strongly suggests that nicotinamide can be taken up by strain 1116 and enables synthesis of nmn.
Growth curve analysis resulted in final optical densities of 0.4 for all strains. However, the curves were characterized by long lag phases and did not show sigmoidal appearances, complicating the extraction of the doubling times. Nevertheless, doubling times ranged from 73 min (strain 1115) to 100 min (strain 1197; Figure 3D). All strains grew slower than predicted (Table 3).
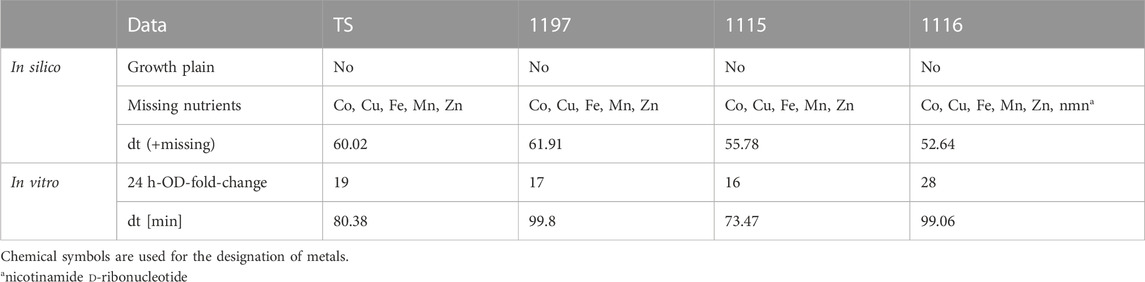
TABLE 3. Growth behavior in silico and in vitro of all strains in RPMI.
3.2.4 Growth in CGXII medium requires enrichment to allow proliferation in vitro
CGXII medium is used as an optimal, chemically defined medium to grow Corynebacterium glutamicum (Unthan et al., 2014). Therefore, we decided to test the ability of our strains to proliferate in this medium. In silico, none of the strains were predicted to grow in standard CGXII composition. Analysis of this phenomenon revealed that all strains need the addition of the trace metal cobalt. This allowed in silico the proliferation of the strain TS while all other strains showed auxotrophies for l-Cysteine and pantothenic acid (strain 1197 and 1115) or nmn and pantothenic acid (strain 1116). The addition of the respective nutrients in silico allowed simulated proliferation for all strains (see Table 4). Interestingly, none of the strains, not even the strain TS showed proliferation in CGXII (supplemented with cobalt) containing flasks in vitro. As the reasons for this discrepancy were unclear, we tested if the addition of other nutrients might stimulate growth. Firstly, we added 0.2 % Tween 80 (Tw) which is known to have growth stimulatory effects on Corynebacteria (Chevalier et al., 1987). This increased the reached OD after 24 h slightly (Figure 4A). However, we observed strongly improved growth of strain TS when Tween 80 as well as a complex mixture of amino acids (casamino acids—CasA) was added to CGXII.
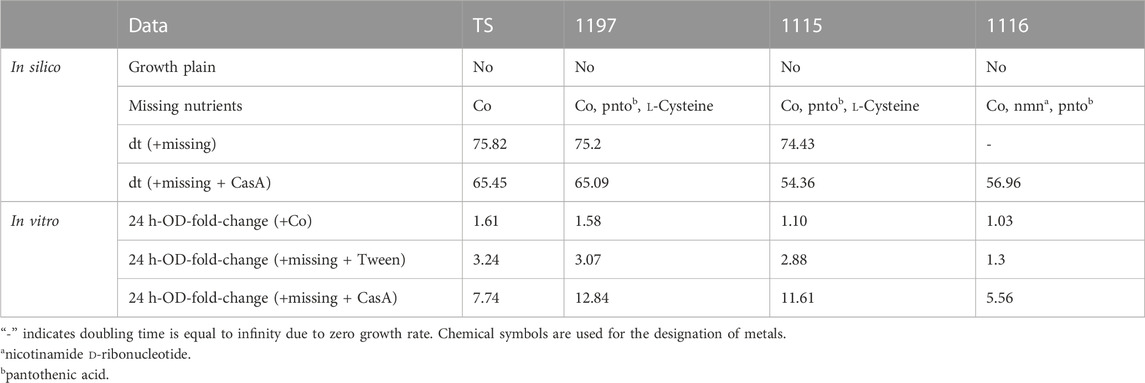
TABLE 4. Growth behavior in silico and in vitro of all strains in CGXII.
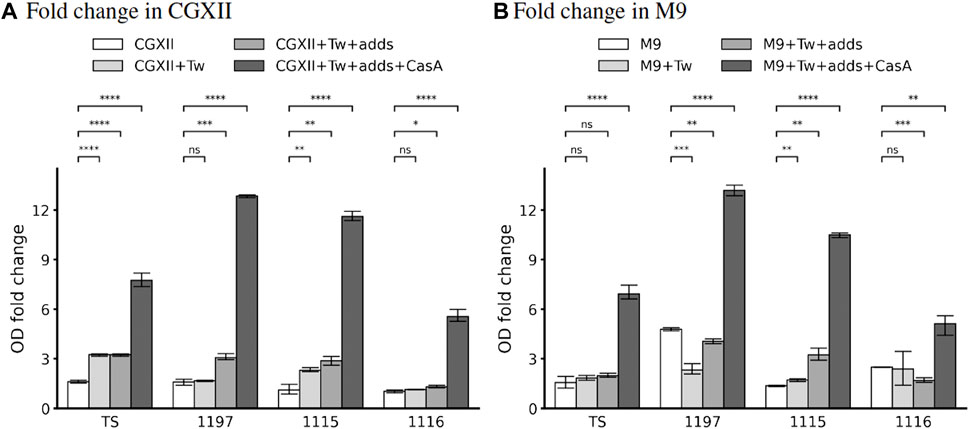
FIGURE 4. In vitro binary growth phenotypes in CGXII (A) and M9 (B) of the strains TS, 1197, 1115, and 1116. 10 mL medium in 50 mL flasks were inoculated to OD600 =0.2. After 24 h of incubation, the OD600 was measured again, and the fold-change was calculated as a measure for bacterial growth. Shown are the mean and SD of three independent experiments. “Tw” corresponds to the addition of 0.2 % Tween 80, “adds” indicates the addition of predicted missing metabolites (see Tables 4, 5), “CasA” indicates that 0.1 % casamino acids were added. The concentrations of the added predicted missing metabolites are denoted in Supplementary Table S1.
Similarly, all other strains failed to grow in cobalt supplemented CGXII and the addition of Tween 80 in combination with strain-specific metabolites (see Table 4) allowed only minor growth improvement. Interestingly, the addition of CasA improved the growth of all strains, but the growth of strain 1116 was improved to a lesser extent compared to the other strains.
None of the strains showed growth in microtiter plate format in CGXII, a phenotype that did not change upon the addition of further nutrients as described above. Accordingly, the growth kinetics of the different strains in CGXII could not be assessed.
3.2.5 Growth in M9 medium requires enrichment to allow proliferation in vitro
M9 is a widely used minimal medium. For growth in M9 our models predicted that all strains needed the addition of trace metals as well as strain-dependent metabolites such as l-Cysteine, nicotinamide d-ribonucleotide and pantothenic acid (Table 5).
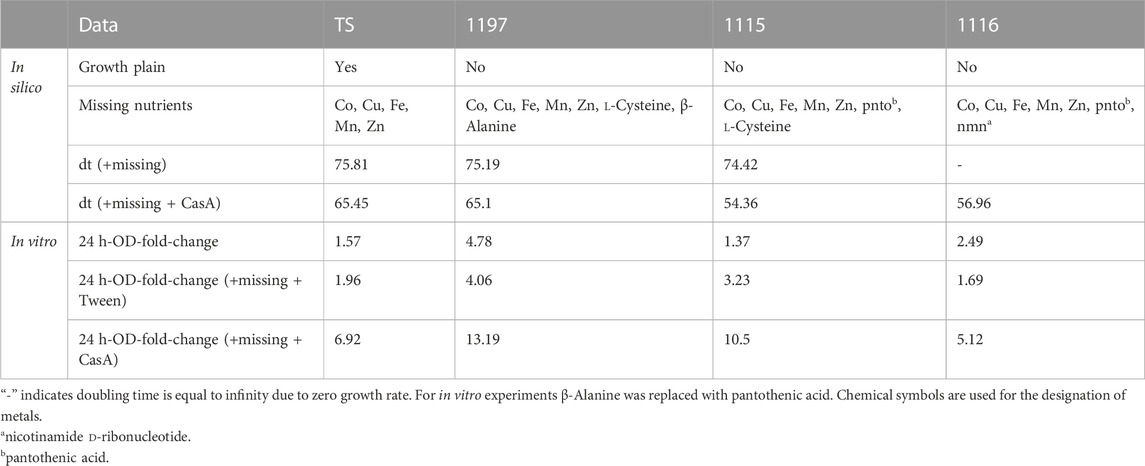
TABLE 5. Growth behavior in silico and in vitro of all strains in M9.
Again, the in vitro experiments did not reflect the in silico prediction. Growth in M9 medium (supplemented with all trace metals) was poor for all strains (Figure 4B). The addition of Tw as well as of the respective “missing” metabolites did only improve the growth for all strains but the addition of CasA increased the proliferation (Figure 4B). Similar to growth in CGXII, we failed to detect growth in M9 in microtiter plate format which prevented growth rate analysis.
3.3 RefineGEMs: a toolbox for faster curation and analysis
The work on reconstructing multiple strain-specific models of opportunistic bacterial pathogens resulted in a new, more general software toolbox called refineGEMs. It combines, integrates, and extends COBRApy and the libSBML (Bornstein et al., 2008) Python package for faster and more accessible GEM curation and analysis in a standardized repository structure.
The toolbox offers various features to help in the investigation of any GEM. It enables the user to load GEMs from SBML files, build a report containing key entities such as charge unbalanced reactions and numbers of reactions, and compare genes present in the model to those found in the KEGG Database given a GFF file and the KEGG ID of the organism. The charges and masses of the metabolites in the model can also be compared to those found in the ModelSEED database (Seaver et al., 2021). The toolbox facilitates performing reproducible growth simulations.
In addition to investigating GEMs, refineGEMs can also be used to curate a given model. For instance, if a model was created with CarveMe version 1.5.1, refineGEMs can transfer relevant information from the notes field within model components to their respective annotation section and can automatically annotate GeneProducts in the model from their IDs using the NCBI IDs. It also enables the addition of KEGG Pathways as groups (using the SBML groups extension) and SBO term annotation refinement via its integrated SBOannotator (Leonidou et al., 2023). Other important functionalities are updating the annotation of metabolites and extending the model with missing reactions based on a table filled in by the user during manual research.
RefineGEMs can be used in two different ways: a) as a standalone script using the main.py script and the corresponding config.yml file, which is available on GitHub b) it can be installed via pip and the functions can be accessed individually Comprehensive documentation is available online, which is accessible via GitHub. The scope of the different modules of refineGEMs is outlined in Table 6, giving an overview of the capabilities of the toolbox. All growth simulations and parts of the model refinement described in the results above were done using refineGEMs.
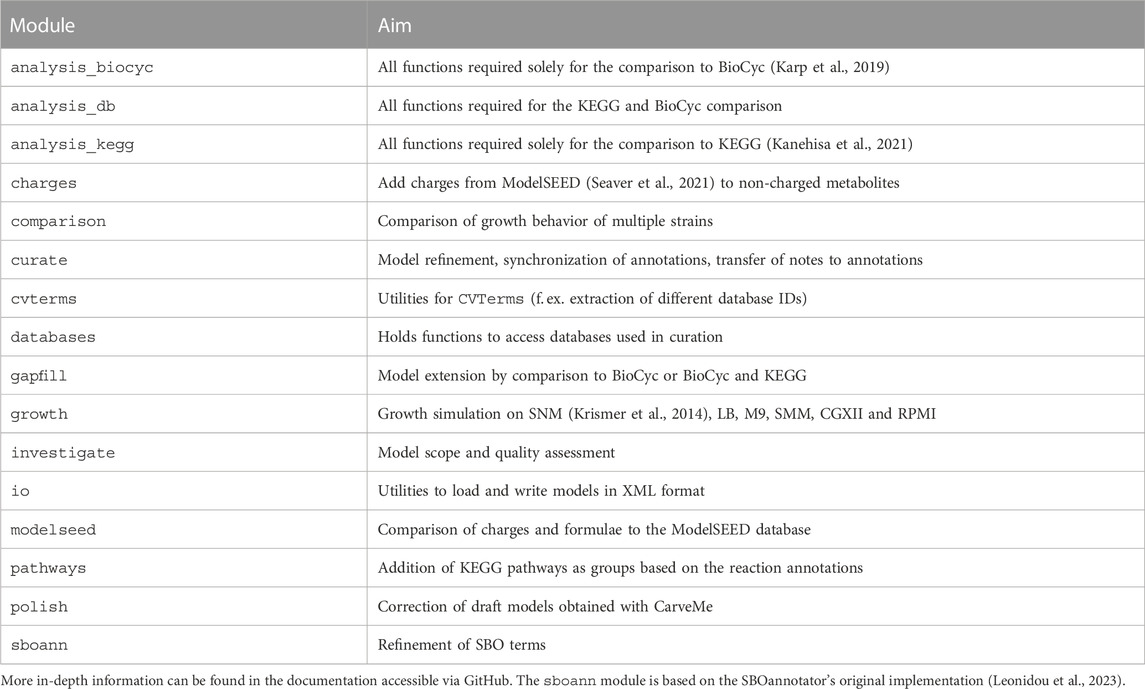
TABLE 6. Overview of refineGEMs modules and their scope.
4 Discussion
This study aimed at building strain-specific genome-scale metabolic models (GEMs) for Corynebacterium striatum. Only strains whose genome has been entirely sequenced and available in the German Collection of Microorganisms and Cell Cultures (DSMZ) were selected as strains to be studied. In doing so, this work comprises three principal objectives: First, developing a unified infrastructure for creating systems biology models was and making it publicly available. Second, using this infrastructure to create GEMs for selected strains of the bacterium C. striatum and to make them freely accessible. Finally, experimentally verifying and using these strain-specific GEMs for their further refinement based on generated predictions of bacterial growth in the presence of defined nutritional environments.
The GEMs’ prediction of bacterial growth have enormous potential for laboratory-based microbiology as it might help to quickly adjust culture conditions to optimize growth rates and growth yields, to optimize fermentation processes or even to enable growth of so far not-culturable bacteria. However, while GEMs are nowadays created for many different species and even for multiple strains of the same species, their ability to correctly predict biological phenotypes is hardly investigated.
In a typical scenario, published computer models are initially developed as a theoretical foundation and only checked against previously published data, if available (Renz et al., 2021a). In other cases, previously published models get revised based on recent developments (Renz and Dräger, 2021; Dahal et al., 2023). Other research groups can perform a comprehensive analysis and validation once these models are publicly available. In the case of the coronavirus pandemic, host-virus models initially developed purely theoretically (Renz et al., 2021b) could be used as the basis for later experimental studies that enabled the identification of potential antiviral agents (Renz et al., 2022). However, it is not always possible for researchers working on systems biology modeling to have their models tested in the laboratory, but ideally, this should be standard practice. To close this gap we used a tightly integrated collaboration between bioinformatics and systems biology, on the one hand, and microbiology, on the other hand. We created novel GEMs for several C. striatum strains and used them to make predictions about proliferation of the strains under defined nutritional conditions. These predictions were then compared to biological data sets gained in the laboratory. All models are available for download from the BioModels database.
Our experiments identified several pitfalls reducing accuracy of model prediction. We found several examples of “false negative” predictions, referring to in vitro growth while GEMs predicted a growth failure. This phenomenon can have two different underlying reasons. Firstly, it is possible that certain nutrients are present in vitro that were not included in in silico formulation of the nutritional composition. This highlights that the in silico assembly of the nutritional composition of laboratory media needs to be approached deliberately. Trace metals are important to mention in this regard. These nutrients are hardly mentioned on ingredient lists of commercial media (e.g., RPMI). Similarly, not all trace metals are purposely added to defined media (CGXII, M9) produced in laboratories around the world. However, the amount needed is extremely low and physiologically relevant concentrations are available in most media as long as special action for their removal (e.g., addition of chelators) is avoided. Accordingly, media formulations used with GEMs should reflect that trace metals are most likely not limiting factors in experimental setups.
Furthermore, for complex media ingredients such as yeast, tryptic soy or meat extracts that are frequently used in microbiological practice, the precise composition of these extracts are unknown. However, they contain excess of amino acids, vitamins, ribonucleotides, etc. which should be included in the in silico composition of media to increase the accuracy of growth prediction.
Secondly, false negative predictions can also be caused by inappropriate transport activities within GEMs. One example in this regard is nicotinic acid transport. The GEM of strain 1116 predicts nmn to be a crucial metabolite for this strain. Supplementation of the medium with the precursors nicotinic acid or nicotinamide is regarded as insufficient as specific transporters for their import are not predicted within the genome. In contrast to this prediction, we found the strain to proliferate in the presence of nicotinamide (growth in RPMI) as well as in the presence of nicotinic acid (M9 medium), strongly suggesting that both intermediates can be acquired by the bacterium and enable the synthesis of nmn. However, additional experimental evidence is needed to validate this hypothesis and the responsible transport systems remain to be identified.
We also observed “false positive” predictions, referring to GEMs predicting growth, while proliferation is not observed in vitro. These inconsistencies are more problematic, as they reduce the usefulness of GEMs for laboratory-based experiments. Unfortunately, the reasons for this are less clear and the issues are more difficult to address. However, one reason might be that GEMs are based on genetic information while information about expression of the respective metabolic pathways is missing. Although genes for biosynthesis of aminoacids or vitamins might be present within a strain of interest, a failure to express the same might entail physiological auxotrophies. This phenomenon is known for the amino acids Leucin and Valin for Staphylococcus aureus (Onoue and Mori, 1997; Kaiser et al., 2018). Along this line we found that the addition of casamino acids improved proliferation of C. striatum in CGXII and M9 media.
Additionally, in silico assumptions are frequently not directly transferable to in vitro conditions. GEMs assume homogeneously mixed cells under isothermal and isobaric conditions whose compartments maintain constant volumes. Molecular concentrations are assumed abundant enough to be effectively continuous. In addition, for simplicity, some physicochemical factors, such as osmotic pressure or electroneutrality, are not considered. The modeled cells would also remain steady, resulting in constant molecular concentrations. These assumptions are by far not met in vitro which can also account for discrepancies between model experimental results. Taken together, GEMs have high potential to support experimental microbiologists. However, close interactions and repetitive cycles of prediction experimental validation and model refinements are needed to improve accuracy of the predictions.
Inclusion of experimental observations into GEMs represents another task for the future. Exemplarily, it is known that addition of Tween 80 improves in vitro growth of Corynebacteria. It is currently impossible to incorporate such findings into GEMs.
Since all individual steps for reconstructing these five strain-specific C. striatum models were performed by standardized Python scripts in a directory structure based on version control and made available, the modeling process can be traced in detail. Thus, a software infrastructure comprehensively tested on several genuine and relevant case studies is available, which can be applied to further reconstruction projects.
The tools and models created in this work through close collaboration between the dry and wet laboratories provide a valuable working basis for subsequent studies. The software infrastructure developed and the models created have been extensively tested and validated using standard methods.
However, since no computer model, no matter how carefully developed, can ever exhaustively represent all processes of a cell type under investigation, subsequent work will be necessary to elucidate further the metabolism of all five C. striatum strains described herein in order to provide even more accurate and thus more meaningful predictions.
Concerning the underlying model assumptions, extensions for gene expression would also be beneficial. Modeling approaches are already available as ME models [for Metabolism and Expression (O’Brien et al., 2013)]. On the other hand, it may be interesting to shed light on the effects of single gene mutations on enzyme efficiency and, thus, metabolism as a whole, for which so-called GEM-PRO models could be used (Brunk et al., 2016). This would be particularly important for clinical isolates. With the increasing availability of genome sequencing, individual patient germs could be characterized.
Further developments on the experimental side would also be beneficial: since the predictive power improves the more precisely the media composition is known, the substance concentrations of their components should be clarified more precisely for model development using human microbiota. Due to biovariability, only average values can be expected, but knowledge of the standard deviation across subjects also allows conclusions to be drawn and enables computer experiments under variation with higher accuracy.
Since no biological system can live in isolation, the interaction with other members of the microbiota remains an essential aspect for subsequent research to understand the diverse interactions with commensals, other pathogens, and the human host in a larger picture. Basic approaches in this direction are already available, with which the models developed here can also be combined (Glöckler et al., 2022; Mostolizadeh et al., 2022; Glöckler et al., 2023).
The models, Python programs, and Git-based version management working template presented with this work can be directly applied and used for subsequent projects immediately and can be independently further developed. Thus, an infrastructure extensively tested on several relevant reconstruction efforts is available and can be applied to any GEM.
Data availability statement
All models are available on BioModels and were submitted according to the standards set in the community to enable the curation of those models within the database. The models were submitted in a Computational Modeling in Biology Network (COMBINE) archive Open Modeling EXchange format (OMEX) format including the respective MEMOTE and FVA, Reaction deletion, Objective function values, Gene deletion fluxes (FROG) reports. Annotations of all models were formatted adhering to community standards. All models are also available via GitHub where future changes and additions will be continuously implemented. The GitHub repository was also used during GEM reconstruction and thus holds previous versions of the models. Its structure is based on the standard-GEM format. The MEMOTE diff report can be found on the GitHub repository as well.
Author contributions
FB created and curated the models, developed the binary growth phenotype experimental protocol, conducted the experiments and introduced the refineGEMs toolbox. GD extended the refineGEMs toolbox. LC was involved in in vitro study design, and supervised the laboratory work. SH supervised the in vitro study. AD supervised the in silico study. FB, SH, and AD wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the German Center for Infection Research (DZIF, doi: 10.13039/100009139) within the Deutsche Zentren der Gesundheitsforschung (BMBF-DZG, German Centers for Health Research of the Federal Ministery of Education and Research), grant [№ = 1] 8020708703 and supported by infrastructural funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), Cluster of Excellence EXC 2124—390838134 Controlling Microbes to Fight Infections. The authors acknowledge support from the Open Access Publishing Fund of the University of Tübingen (https://uni-tuebingen.de/de/216529).
Acknowledgments
The authors thank Libera Lo Presti for manuscript reviewing and proof-reading.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor MO declared a past collaboration with the author AD.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbinf.2023.1214074/full#supplementary-material
References
Adolf, L. A., Müller-Jochim, A., Kricks, L., Puls, J.-S., Lopez, D., Grein, F., et al. (2023). Functional membrane microdomains and the hydroxamate siderophore transporter atpase fhuc govern isd-dependent heme acquisition in staphylococcus aureus. Elife 12, e85304. doi:10.7554/elife.85304
Balci, H., Siper, M. C., Saleh, N., Safarli, I., Roy, L., Kilicarslan, M., et al. (2021). Newt: a comprehensive web-based tool for viewing, constructing and analyzing biological maps. Bioinforma. Oxf. Engl. 37, 1475–1477. doi:10.1093/bioinformatics/btaa850
Bécard, G., and Fortin, J. A. (1988). Early events of vesicular–arbuscular mycorrhiza formation on Ri T-DNA transformed roots. New Phytol. 108, 211–218. doi:10.1111/j.1469-8137.1988.tb03698.x
Bergmann, F. T., Czauderna, T., Dogrusoz, U., Rougny, A., Dräger, A., Touré, V., et al. (2020). Systems biology graphical notation markup language (SBGNML) version 0.3. J. Integr. Bioinforma. 17, 20200016. doi:10.1515/jib-2020-0016
Bergmann, F. T., Keating, S. M., Gauges, R., Sahle, S., and Wengler, K. (2018). SBML level 3 package: render, version 1, release 1. J. Integr. Bioinforma. 15, 20170078. doi:10.1515/jib-2017-0078
Bertani, G. (1951). Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62, 293–300. doi:10.1128/jb.62.3.293-300.1951
Bodor, A., Bounedjoum, N., Vincze, G. E., Erdeiné Kis, Á., Laczi, K., Bende, G., et al. (2020). Challenges of unculturable bacteria: environmental perspectives. Rev. Environ. Sci. Bio/Technology 19, 1–22. doi:10.1007/s11157-020-09522-4
Bordbar, A., Monk, J. M., King, Z. A., and Palsson, B. O. (2014). Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 15, 107–120. doi:10.1038/nrg3643
Bornstein, B. J., Keating, S. M., Jouraku, A., and Hucka, M. (2008). LibSBML: an API library for SBML. Bioinformatics 24, 880–881. doi:10.1093/bioinformatics/btn051
Brunk, E., Mih, N., Monk, J., Zhang, Z., O’Brien, E. J., Bliven, S. E., et al. (2016). Systems biology of the structural proteome. BMC Syst. Biol. 10, 26. doi:10.1186/s12918-016-0271-6
Buchweitz, L. F., Yurkovich, J. T., Blessing, C., Kohler, V., Schwarzkopf, F., King, Z. A., et al. (2020). Visualizing metabolic network dynamics through time-series metabolomic data. BMC Bioinforma. 21, 130. doi:10.1186/s12859-020-3415-z
Carey, M. A., Dräger, A., Beber, M. E., Papin, J. A., and Yurkovich, J. T. (2020). Community standards to facilitate development and address challenges in metabolic modeling. Mol. Syst. Biol. 16, e9235. doi:10.15252/msb.20199235
Chandran, R., Puthukkichal, D. R., Suman, E., and Mangalore, S. K. (2016). Diphtheroids-important nosocomial pathogens. J. Clin. diagnostic Res. JCDR 10, DC28–DC31. doi:10.7860/JCDR/2016/19098.9043
Chevalier, J., Pommier, M. T., and Crémieux, A. (1987). Rôle du Tween-80 utilisé dans la culture des corynébactéries cutanées (groupe JK) sur la composition en acides gras cellulaires. Ann. De. l’Institut Pasteur. Microbiol. 138, 427–437. doi:10.1016/0769-2609(87)90060-3
Courtot, M., Juty, N., Knüpfer, C., Waltemath, D., Zhukova, A., Dräger, A., et al. (2011). Controlled vocabularies and semantics in systems biology. Mol. Syst. Biol. 7, 543. doi:10.1038/msb.2011.77
Dahal, S., Renz, A., Dräger, A., and Yang, L. (2023). Genome-scale model of Pseudomonas aeruginosa metabolism unveils virulence and drug potentiation. Commun. Biol. 6, 165. doi:10.1038/s42003-023-04540-8
Dillard, L. R., Glass, E. M., Lewis, A. L., Thomas-White, K., and Papin, J. A. (2023). Metabolic network models of the gardnerella pangenome identify key interactions with the vaginal environment. mSystems 8, e0068922. doi:10.1128/msystems.00689-22
Dräger, A., and Waltemath, D. (2021). “Overview: standards for modeling in systems medicine,” in Systems medicine. Editor O. Wolkenhauer (Oxford: Academic Press), 345–353. doi:10.1016/B978-0-12-816077-0.00001-7
Ebrahim, A., Lerman, J. A., Palsson, B. O., and Hyduke, D. R. (2013). COBRApy: COnstraints-based reconstruction and analysis for Python. BMC Syst. Biol. 7, 74. doi:10.1186/1752-0509-7-74
Feierabend, M., Renz, A., Zelle, E., Nöh, K., Wiechert, W., and Dräger, A. (2021). High-quality genome-scale reconstruction of Corynebacterium glutamicum ATCC 13032. Front. Microbiol. 12, 750206. doi:10.3389/fmicb.2021.750206
Finnem, , and Mostolizadeh, R. (2023). Biomathsys/MassChargeCuration: V0.1. Zenodo. doi:10.5281/zenodo.7525185
Fitzpatrick, R., and Stefan, M. I. (2022). Validation through collaboration: encouraging team efforts to ensure internal and external validity of computational models of biochemical pathways. Neuroinformatics 20, 277–284. doi:10.1007/s12021-022-09584-5
Gauges, R., Rost, U., Sahle, S., Wengler, K., and Bergmann, F. T. (2015). The systems biology Markup Language (SBML) level 3 package: layout, version 1 core. J. Integr. Bioinforma. 12, 550–602. doi:10.1515/jib-2015-267
Glöckler, M., Dräger, A., and Mostolizadeh, R. (2023). Hierarchical modelling of microbial communities. Bioinformatics 39, btad040. doi:10.1093/bioinformatics/btad040
Glöckler, M., Dräger, A., and Mostolizadeh, R. (2022). NCMW: a Python package to analyze metabolic interactions in the nasal microbiome. Front. Bioinforma. 2, 827024. doi:10.3389/fbinf.2022.827024
Gu, C., Kim, G. B., Kim, W. J., Kim, H. U., and Lee, S. Y. (2019). Current status and applications of genome-scale metabolic models. Genome Biol. 20, 121. doi:10.1186/s13059-019-1730-3
Hawkey, J., Vezina, B., Monk, J. M., Judd, L. M., Harshegyi, T., López-Fernández, S., et al. (2022). A curated collection of Klebsiella metabolic models reveals variable substrate usage and gene essentiality. Genome Res. 32, 1004–1014. doi:10.1101/gr.276289.121
Heinken, A., Hertel, J., Acharya, G., Ravcheev, D. A., Nyga, M., Okpala, O. E., et al. (2023). Genome-scale metabolic reconstruction of 7,302 human microorganisms for personalized medicine. Nat. Biotechnol. 1, 1320–1331. doi:10.1038/s41587-022-01628-0
Heirendt, L., Arreckx, S., Pfau, T., Mendoza, S. N., Richelle, A., Heinken, A., et al. (2019). Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 14, 639–702. doi:10.1038/s41596-018-0098-2
Holzapfel, C., Hoene, M., Zhao, X., Hu, C., Weigert, C., Niess, A., et al. (2022). FluxomicsExplorer: differential visual analysis of flux sampling based on metabolomics. Comput. Graph. 108, 11–21. doi:10.1016/j.cag.2022.08.008
Kaiser, J. C., King, A. N., Grigg, J. C., Sheldon, J. R., Edgell, D. R., Murphy, M. E. P., et al. (2018). Repression of branched-chain amino acid synthesis in Staphylococcus aureus is mediated by isoleucine via CodY, and by a leucine-rich attenuator peptide. PLoS Genet. 14, e1007159. doi:10.1371/journal.pgen.1007159
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M., and Tanabe, M. (2021). KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 49, D545–D551. doi:10.1093/nar/gkaa970
Karp, P. D., Billington, R., Caspi, R., Fulcher, C. A., Latendresse, M., Kothari, A., et al. (2019). The BioCyc collection of microbial genomes and metabolic pathways. Briefings Bioinforma. 20, 1085–1093. doi:10.1093/bib/bbx085
Keating, S. M., Waltemath, D., König, M., Zhang, F., Dräger, A., Chaouiya, C., et al. (2020). SBML Level 3: an extensible format for the exchange and reuse of biological models. Mol. Syst. Biol. 16, e9110. doi:10.15252/msb.20199110
Keilhauer, C., Eggeling, L., and Sahm, H. (1993). Isoleucine synthesis in Corynebacterium glutamicum: molecular analysis of the ilvB-ilvN-ilvC operon. J. Bacteriol. 175, 5595–5603. doi:10.1128/jb.175.17.5595-5603.1993
King, Z. A., Dräger, A., Ebrahim, A., Sonnenschein, N., Lewis, N. E., and Palsson, B. O. (2015). Escher: a web application for building, sharing, and embedding data-rich visualizations of biological pathways. PLOS Comput. Biol. 11, e1004321. doi:10.1371/journal.pcbi.1004321
Krauss, S., Harbig, T. A., Rapp, J., Schaefle, T., Franz-Wachtel, M., Reetz, L., et al. (2023). Horizontal transfer of bacteriocin biosynthesis genes requires metabolic adaptation to improve compound production and cellular fitness. Microbiol. Spectr. 11, 031766–e3222. doi:10.1128/spectrum.03176-22
Krismer, B., Liebeke, M., Janek, D., Nega, M., Rautenberg, M., Hornig, G., et al. (2014). Nutrient limitation governs Staphylococcus aureus metabolism and niche adaptation in the human nose. PLoS Pathog. 10, e1003862. doi:10.1371/journal.ppat.1003862
Leonidou, N., Fritze, E., Renz, A., and Dräger, A. (2023). SBOannotator: a Python tool for the automated assignment of systems biology Ontology terms. Bioinformatics 39, btad437. doi:10.1093/bioinformatics/btad437
Lieven, C., Beber, M. E., Olivier, B. G., Bergmann, F. T., Ataman, M., Babaei, P., et al. (2020). MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 38, 272–276. doi:10.1038/s41587-020-0446-y
Lloyd, K. G., Steen, A. D., Ladau, J., Yin, J., and Crosby, L. (2018). Phylogenetically novel uncultured microbial cells dominate earth microbiomes. mSystems 3, e00055-18. doi:10.1128/mSystems.00055-18
Machado, D., Andrejev, S., Tramontano, M., and Patil, K. R. (2018). Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 46, 7542–7553. doi:10.1093/nar/gky537
Magnúsdóttir, S., Heinken, A., Kutt, L., Ravcheev, D. A., Bauer, E., Noronha, A., et al. (2017). Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 35, 81–89. doi:10.1038/nbt.3703
Malik-Sheriff, R. S., Glont, M., Nguyen, T. V. N., Tiwari, K., Roberts, M. G., Xavier, A., et al. (2020). BioModels-15 years of sharing computational models in life science. Nucleic Acids Res. 48, D407–D415. doi:10.1093/nar/gkz1055
McMullen, A. R., Anderson, N., Wallace, M. A., Shupe, A., and Burnham, C.-A. D. (2017). When good bugs go bad: epidemiology and antimicrobial resistance profiles of Corynebacterium striatum, an emerging multidrug-resistant, opportunistic pathogen. Antimicrob. Agents Chemother. 61, e01111-17. doi:10.1128/AAC.01111-17
Merck & Co., Inc. (2023). M9 Minimal Salts, 5X, 5X powder, minimal microbial growth medium – sigma-Aldrich. Available at: https://www.sigmaaldrich.com/US/en/substance/m9minimalsalts5x1234598765 (Accessed March 24, 2023).
Mostolizadeh, R., Glöckler, M., and Dräger, A. (2022). Towards the human nasal microbiome: simulating D. pigrum and S. aureus. Front. Cell. Infect. Microbiol. 12, 925215. doi:10.3389/fcimb.2022.925215
Norsigian, C. J., Pusarla, N., McConn, J. L., Yurkovich, J. T., Dräger, A., Palsson, B. O., et al. (2019). BiGG Models 2020: multi-strain genome-scale models and expansion across the phylogenetic tree. Nucleic Acids Res. 48, D402–D406. doi:10.1093/nar/gkz1054
Nudel, K., Zhao, X., Basu, S., Dong, X., Hoffmann, M., Feldgarden, M., et al. (2018). Genomics of Corynebacterium striatum, an emerging multi-drug resistant pathogen of immunocompromised patients. Clin. Microbiol. Infect. 24, 1016.e7–1016.e13. doi:10.1016/j.cmi.2017.12.024
O’Brien, E. J., Lerman, J. A., Chang, R. L., Hyduke, D. R., and Palsson, B. O. (2013). Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol. Syst. Biol. 9, 693. doi:10.1038/msb.2013.52
Olivier, B. G., and Bergmann, F. T. (2018). SBML level 3 package: flux balance constraints version 2. J. Integr. Bioinforma. 15, 20170082. doi:10.1515/jib-2017-0082
Onoue, Y., and Mori, M. (1997). Amino acid requirements for the growth and enterotoxin production by Staphylococcus aureus in chemically defined media. Int. J. Food Microbiol. 36, 77–82. doi:10.1016/s0168-1605(97)01250-6
Panchiwala, H., Shah, S., Planatscher, H., Zakharchuk, M., König, M., and Dräger, A. (2022). The systems biology simulation core library. Bioinformatics 38, 864–865. doi:10.1093/bioinformatics/btab669
Reback, J., McKinney, W., Jbrockmendel, , Van Den Bossche, J., Roeschke, M., Augspurger, T., et al. (2022). Pandas-dev/pandas: pandas 1.4.3. Zenodo. doi:10.5281/ZENODO.3509134
Renz, A., and Dräger, A. (2021). Curating and comparing 114 strain-specific genome-scale metabolic models of Staphylococcus aureus. npj Syst. Biol. Appl. 7, 30. doi:10.1038/s41540-021-00188-4
Renz, A., Hohner, M., Breitenbach, M., Josephs-Spaulding, J., Dürrwald, J., Best, L., et al. (2022). Metabolic modeling elucidates phenformin and atpenin A5 as broad-spectrum antiviral drugs. Tech. rep. Preprints. doi:10.20944/preprints202210.0223.v1
Renz, A., Mostolizadeh, R., and Dräger, A. (2020). “Clinical applications of metabolic models in SBML format,” in Systems medicine. Editor O. Wolkenhauer (Oxford: Academic Press), 3, 362–371. doi:10.1016/B978-0-12-801238-3.11524-7
Renz, A., Widerspick, L., and Dräger, A. (2021a). First genome-scale metabolic model of Dolosigranulum pigrum confirms multiple auxotrophies. Metabolites 11, 232. doi:10.3390/metabo11040232
Renz, A., Widerspick, L., and Dräger, A. (2021b). Genome-scale metabolic model of infection with SARS-CoV-2 mutants confirms guanylate kinase as robust potential antiviral target. Genes 12, 796. doi:10.3390/genes12060796
Römer, M., Eichner, J., Dräger, A., Wrzodek, C., Wrzodek, F., and Zell, A. (2016). ZBIT bioinformatics toolbox: a web-platform for systems biology and expression data analysis. PLOS ONE 11, e0149263. doi:10.1371/journal.pone.0149263
Seaver, S. M. D., Liu, F., Zhang, Q., Jeffryes, J., Faria, J. P., Edirisinghe, J. N., et al. (2021). The ModelSEED biochemistry database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 49, D575–D588. doi:10.1093/nar/gkaa746
Shariff, M., Aditi, A., and Beri, K. (2018). Corynebacterium striatum: an emerging respiratory pathogen. J. Infect. Dev. Ctries. 12, 581–586. doi:10.3855/jidc.10406
Thermo Fisher Scientific, Inc (2023). Technical resources - media formulations: 11875 - RPMI 1640 - de. Available at: https://www.thermofisher.com/de/de/home/technical-resources/media-formulation.114.html (Accessed March 22, 2023).
Thiele, I., and Palsson, B. Ø. (2010). A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 5, 93–121. doi:10.1038/nprot.2009.203
Thrash, J. C. (2019). Culturing the uncultured: risk versus reward. mSystems 4, 001300–e219. doi:10.1128/mSystems.00130-19
Touré, V., Dräger, A., Luna, A., Dogrusoz, U., and Rougny, A. (2021). “The systems biology graphical notation: current status and applications in systems medicine,” in Systems medicine. Editor O. Wolkenhauer (Oxford: Academic Press), 372–381. doi:10.1016/B978-0-12-801238-3.11515-6
Unthan, S., Grünberger, A., van Ooyen, J., Gätgens, J., Heinrich, J., Paczia, N., et al. (2014). Beyond growth rate 0.6: what drives Corynebacterium glutamicum to higher growth rates in defined medium. Biotechnol. Bioeng. 111, 359–371. doi:10.1002/bit.25103
van’t Hof, M., Mohite, O. S., Monk, J. M., Weber, T., Palsson, B. O., and Sommer, M. O. A. (2022). High-quality genome-scale metabolic network reconstruction of probiotic bacterium Escherichia coli Nissle 1917. BMC Bioinforma. 23, 566. doi:10.1186/s12859-022-05108-9
Virtanen, P., Gommers, R., Oliphant, T. E., Haberland, M., Reddy, T., Cournapeau, D., et al. (2020). SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272. doi:10.1038/s41592-019-0686-2
Yang, P., Chen, Y., and Gong, A.-d. (2021). Development of a defined medium for Corynebacterium glutamicum using urea as nitrogen source. 3 Biotech. 11, 405. doi:10.1007/s13205-021-02959-6
Yurkovich, J. T., Yurkovich, B. J., Dräger, A., Palsson, B. O., and King, Z. A. (2017). A padawan programmer’s guide to developing software libraries. Cell Syst. 5, 431–437. doi:10.1016/j.cels.2017.08.003
Glossary

Keywords: Corynebacterium striatum, genome-scale metabolic models, model-driven discovery, strain-specific model, opportunistic pathogen, software engineering
Citation: Bäuerle F, Döbel GO, Camus L, Heilbronner S and Dräger A (2023) Genome-scale metabolic models consistently predict in vitro characteristics of Corynebacterium striatum. Front. Bioinform. 3:1214074. doi: 10.3389/fbinf.2023.1214074
Received: 28 April 2023; Accepted: 02 October 2023;
Published: 23 October 2023.
Edited by:
Marek Ostaszewski, University of Luxembourg, LuxembourgCopyright © 2023 Bäuerle, Döbel, Camus, Heilbronner and Dräger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Famke Bäuerle, ZmFta2UuYmFldWVybGVAZ21haWwuY29t; Simon Heilbronner, c2ltb24uaGVpbGJyb25uZXJAYmlvLmxtdS5kZQ==; Andreas Dräger, YW5kcmVhcy5kcmFlZ2VyQHVuaS10dWViaW5nZW4uZGU=