 Mebrie Zemene Kinde
Mebrie Zemene Kinde Bizuayehu Kerisew2
Bizuayehu Kerisew2 Tegegne Eshetu
Tegegne Eshetu Abebe Tesfaye Gessese
Abebe Tesfaye Gessese- 1Department of Veterinary Biomedical Sciences, College of Veterinary Medicine and Animal Sciences, University of Gondar, Gondar, Ethiopia
- 2Department of Biology, Bahir Dar University, Bahir Dar, Ethiopia
- 3Department of Biomedical and Laboratory Science, College of Medicine and Health Science, University of Gondar, Gondar, Ethiopia
This study explored virulence genes, antibiotic resistance genes, and mobile genetic elements in 14 Listeria monocytogenes strains from milk and dairy products collected from different regions of Ethiopia. The strains were classified into two Multilocus Sequence Typing sequence types (ST2 and ST45) and further grouped into clonal complexes (CC2) and different cgMLST types. Twenty-nine virulence genes were identified across all 14 strains, with lplA1 detected at higher levels in all strains except SAMN28661660. All L. monocytogenes strains also carried four antibiotic resistance genes (fosX, lin, norB, mprF), contributing to their ability to withstand multiple antimicrobial agents. Notably, no plasmids or mobile genetic elements were detected. Stress resistance genes, including stress survival islet 1 (SSI1_lmo0447), lmo 1800, and lmo1799, were identified in all strains. However, genes encoding for disinfectant resistance were not identified from all strains. LGI-2 was found in all the strains and none of the studied strains harbored LGI-1 and LGI-3. Conserved CRISPR-Cas systems were found in some strains. KEGG pathway analysis revealed that inlA and inlB genes facilitate bacterial internalization through host actin polymerization. Overall, the study provided crucial insights into the genomic features of L. monocytogenes in the Ethiopian dairy chain. It is crucial to establish continuous monitoring of L. monocytogenes in dairy products, improve sanitation, enforce stricter antibiotic usage and food safety regulations, and raise public awareness of associated risks.
1 Introduction
Listeria monocytogenes (L. monocytogenes) is a Gram-positive, facultative anaerobic bacterium responsible for listeriosis, which carries significant public health and economic implications (Rogalla & Bomar, 2023; Matle et al., 2020). This bacterium affects both humans and animals, spreading primarily through contaminated food, particularly unpasteurized dairy (Maury et al., 2019; Ravindhiran et al., 2023). In animals, it can cause severe conditions like encephalitis, septicemia, and abortion, especially in cattle and sheep (Matle et al., 2020). Meanwhile, in humans, symptoms range from mild gastroenteritis to severe conditions such as septicemia and meningitis, with high fatality rates among vulnerable populations like pregnant women, neonates, the elderly, and immunocompromised individuals (Hansen et al., 2006; Rohilla et al., 2024; Ravindhiran et al., 2023).
The pathogenicity of L. monocytogenes is influenced by key genes, such as those encoding the internalins (InlA, InlB), listeriolysin O (LLO), and actin assembly-inducing protein (ActA). These genes facilitate host cell invasion, intracellular survival, and intercellular spread (Orsi et al., 2011). Notably, these virulence factors can be dispersed throughout the genome islands (Quereda et al., 2021). With advances in genotyping and whole-genome sequencing, researchers have revealed considerable variation in pathogenic potential among L. monocytogenes isolates (Kathariou et al., 2017). Additionally, adaptation to stress in food processing environments can further enhance L. monocytogenes pathogenicity, as stress exposure may induce lasting phenotypic changes (Quereda et al., 2021).
Although antimicrobial chemotherapy is the primary treatment for listeriosis (Luque-Sastre et al., 2018), the increasing emergence of multidrug-resistant L. monocytogenes presents a growing public health challenge, particularly in developing countries like Ethiopia (Dufailu et al., 2021). The frequent use of antibiotics in food animal production has contributed to this resistance, leading to transmission to humans through contaminated dairy and meat products (Odu and Okonko, 2017). Alarmingly, resistance to antibiotics commonly used to treat listeriosis, such as tetracycline, ampicillin, and gentamicin, has been reported (Luque-Sastre et al., 2018). This rise in resistance underscores a severe threat to routine medical procedures, with potential deaths escalating if effective antibiotics become unavailable (Naghavi et al., 2024).
In Ethiopia, phenotypic antimicrobial resistance studies have highlighted the emergence of resistant L. monocytogenes. For instance, isolates from raw cow milk exhibited 100% resistance to nalidixic acid and high resistance to erythromycin (88%) (Tola, 2024). Similarly, resistance rates in sheep meat were high chloramphenicol (88.9%). Furthermore, clinical isolates from pregnant women demonstrated significant resistance to penicillin G and clindamycin (66.7%) (Tola, 2024). These findings signal a major public health challenge in a resource-limited country like Ethiopia.
Compounding the issue, L. monocytogenes is particularly concerning due to its ability to form biofilms on surfaces such as stainless steel and plastic in food processing facilities. This ability protects the bacteria from environmental stress and cleaning agents (Quereda et al., 2021; Ravindhiran et al., 2023). Inadequate sanitation in Ethiopian dairy facilities and poor handling practices, like insufficient storage temperatures, further exacerbate food safety risks (Abdi Keba et al., 2020; Mebrate et al., 2020). The genetic adaptability of L. monocytogenes, including the acquisition and loss of genetic elements, influences traits such as antibiotic resistance and environmental fitness (Dobrindt et al., 2004; Ghaly and Gillings, 2022; Wein et al., 2021).
On a global scale, L. monocytogenes remains a significant foodborne pathogen. The WHO reports that 20%–30% of clinical listeria infections are fatal, with approximately 23,150 infections and 5,463 deaths worldwide (World Health Organization, 2015). Developed countries like the U.S. report around 1,600 cases annually, with a mortality rate of 15%–20% (Datta and Burall, 2018). In Africa, the prevalence of listeriosis is likely underestimated due to limited diagnostic capabilities and inadequate surveillance data from farm animals (Sibanda et al., 2023). For example, the 2017 South African outbreak linked to contaminated processed meats resulted in over 1,000 cases and more than 200 deaths, highlighting the urgent need for improved food safety measures across the continent (Thomas et al., 2020). In Ethiopia, contamination rates of L. monocytogenes in milk and dairy products have reached up to 60% in some studies (Tola, 2024; Seyoum et al., 2015). This indicates that widespread dairy consumption poses a significant public health risk.
Despite the clear public health risks associated with L. monocytogenes, genomic research on this pathogen in Ethiopia remains limited. Most studies have focused on isolation and phenotypic characterization and Multilocus Sequence Typing (MLST) (Wei et al., 2024), leaving a gap in understanding the genetic diversity, virulence factors, and resistance genes specific to the local context. Consequently, there is an urgent need for genomic analysis of L. monocytogenes isolates to better understand their virulence and antimicrobial resistance profiles. Insights gained from studying the pathogen’s virulence, resistance mechanisms, and adaptation to environmental stress could guide the development of effective, cost-efficient antimicrobial agents and biocontrol strategies. Hence, this study aimed to conduct a genomic analysis of L. monocytogenes isolates from milk and dairy products in Ethiopia, focusing on identification of virulence and antimicrobial resistance genes, and mobile genetic elements (MGEs, genetic materials that can move within or between genomes, including across different organisms) linked to virulence and resistance from whole genome sequence.
The study identified multiple virulence genes in the L. monocytogenes strains, including inlA and inlB, which are crucial for host cell invasion. Additionally, LGI-2 was found in all the strains and none of the studied strains harbored LGI-1 and LGI-3. The isolates were categorized into two sequence types (ST2 and ST45) and further grouped into clonal complexes (CC2). All strains harbored antibiotic resistance genes, such as fosX, lin, norB, and mprF. However, MGEs and genes related to metal resistance, Benzalkonium Chloride tolerance, and disinfectant resistance were not found in all strains, suggesting variability in environmental adaptability.
2 Materials and methods
2.1 Whole-genome sequencing and assembly
The raw sequence data of the strains in this study were obtained in a published study (Wei et al., 2024), from raw milk and pasteurized milk samples collected in three Ethiopian regions (i.e., Amhara, Oromia, and the Southern Nations, Nationalities, and Peoples’ Region (SNNPR)) between 2020 and 2021. Samples were collected from three stages of a dairy supply chain, including collectors, processors, and producers by Addis Ababa University. Samples were tested for Listeria spp., including L. monocytogenes by following the ISO 11290–1:2018 protocol and isolates were confirmed using a multiplex PCR (Chen and Knabel, 2007). The strains were sequenced by Michigan Department of Agriculture and Rural Development, submitted by Food and Drug Administration (FDA) GenomeTrakr Project and registered in the NCBI Sequence Read Archive (SRA) repository under the accession number PRJNA357724. For this study, the raw sequence data (which were saved as FASTQ file) for strains of 14 L. monocytogenes were retrieved from NCBI (BioProject: PRJNA357724), each strains was sequenced using the Illumina MiSeq instrument with v2 500 cycles kit (Illumina, San Diego, United States), producing paired-end reads. The quality of the raw sequence reads was assessed using fastQC v0.12.1. Sequencing data were processed to remove low-quality bases and adapter sequences with the optimized settings (LEADING:6 TRAILING:6 SLIDINGWINDOW:4:20 MINLEN:50) using Trimmomatic v0.39 (Bolger et al., 2014). After trimming, de novo assembly was performed using SPAdes v3.13.1 for downstream analysis such as identification of antimicrobial resistance and virulence genes. The quality of the assembly was determined using QUAST (Quality Assessment Tool for Genome Assemblies) and high-quality assemblies were included in the subsequent analyses.
Trimmed sequences were also aligned against the reference genome, L. monocytogenes EGD-e genome (Li and Durbin, 2009) with bwa v0.7.18 with the default options and filtered out by samtools v1.13. Variant calling was carried out using freebayes v0.9.12. Single Nucleotide Polymorphisms (SNPs) were extracted from the VCF file after applying stringent quality filters to ensure the reliability of variant calls. The filtering criteria included a minimum mapping quality (MAPQ) of ≥30, base quality (BQ) of ≥20, and read depth (DP) of ≥10. Variants with an allele frequency (AF) of ≥0.01 and a minimum alternative allele count (AC) of ≥2 were retained to minimize false positives. The filtered SNPs were then used to construct a phylogenetic tree to infer evolutionary relationships among the strains.
Multiple sequences alignment was performed using ClustalW. The phylogenetic tree was reconstructed using the Maximum Likelihood method based on the General Time Reversible (GTR) model (Nei and Kumar, 2000). For heuristic searches, initial tree topologies were obtained using the Neighbor-Join and BioNJ algorithms, applied to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, with the topology showing the highest log likelihood selected. Bootstrapping of 1,000 was used for confidence estimation. The analysis included 14 nucleotide sequences, and all positions with less than 95% site coverage were eliminated (partial deletion option), allowing for fewer than 5% alignment gaps, missing data, and ambiguous bases at any position. All evolutionary analyses were conducted using MEGA11 (Tamura et al., 2021).
2.2 Subtyping and core genome multilocus sequence typing of Listeria monocytogenes
MLST was conducted as recommended by the Institut Pasteur with seven housekeeping genes (abcZ, bglA, cat, dapE, dat, ldh, and lhkA). Serogroup PCR, Sequence Type (ST), clonal complex (CC) and Core Genome Multilocus Sequence Typing (cgMLST) were assigned from the Listeria MLST database (accessed on 26 August 2024 at https://bigsdb.pasteur.fr/listeria/) managed by the Institut Pasteur and based on Ragon’s scheme (Ragon et al., 2008; Moura et al., 2016). In addition, to ensure the robustness of the clustering results, we used MLST 2.0 (https://cge.food.dtu.dk/services/MLST/). A minimum spanning tree was constructed by using the software GrapeTree 1.5.0 (Zhou et al., 2018), and we have used the clustering parameters based on a seven allelic difference, as described by Moura et al. (2016), to define the genetic relationships among the isolates.
2.3 Detection of virulence, antimicrobial resistance, and stress, metal, and disinfectant resistance genes
Virulence genes in the strains of Listeria monocytogesnes were identified using the virulence factor database (VFDB) and National Center for Biotechnology Information (NCBI) via a local installation of Abricate v.1.0.1. Antimicrobial-resistant genes were also screened through the Comprehensive Antibiotic Resistance Database (CARD) (Alcock et al., 2023) and NCBI using a local installation of Abricate v.1.0.1. Abricate was configured with a minimum coverage of 80% and a minimum identity of 80%. Heatmaps were constructed using ggplot2 version 3.5.1 (R version 4.4.1) for both virulence and antimicrobial resistance genes. The change between resistant genes and reference genome was visualized using IGV (Integrative Genomics Viewer) v2.18.2.
Additionally, genes encoding for stress resistance, and metal and disinfectant resistance were detected using BLASTN algorithm according to BIGSdb-Lm database (Moura et al., 2016; Jolley and Maiden, 2010), available at https://bigsdb.pasteur.fr/listeria/ [accessed on 27 August 2024].
2.4 Detection of plasmids and mobile genetic elements
The presence of plasmids in L. monocytogenes strains was assessed using PlasmidFinder 2.1 (Camacho et al., 2009; Carattoli et al., 2014), accessed on 26 August 2024 at https://cge.food.dtu.dk/services/PlasmidFinder/, with criteria of 95% identity and 60% coverage. Additionally, MGEs were detected using the MobileElementFinder v1.0.3 tool (Johansson et al., 2020), accessed on 26 August 2024 at https://cge.food.dtu.dk/services/MobileElementFinder/, with a minimum identity and coverage of 95% and 60%, respectively.
2.5 CRISPR-Cas systems in Listeria monocytogenes genome
The search for and characterization of CRISPR arrays and their association with Cas proteins was determined with CRISPRCasFinder v4.3.2 (Couvin et al., 2018; Zhang et al., 2018), which is available at https://crisprcas.i2bc.paris-saclay.fr and accessed on 1 October 2024. The following default parameters were used: 23–55 bp repeated sequence length, 25–60 bp spacer length, 0.6–2.5 spacer sequence size as a function of repeated sequence size, a gap size between repeats of 25–60 bp, 20% nucleotide mismatch between repeats, 33% nucleotide mismatch for truncated Repeat, 100 bp size of flanking regions for each analyzed CRISPR array, and 60% maximum percentage similarity between spacers.
2.6 Metabolic pathway analysis of virulence genes
A KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis was performed to investigate the metabolic pathways and mechanisms associated with the virulence genes identified in L. monocytogenes strains. This analysis was conducted using DAVID (Database for Annotation, Visualization, and Integrated Discovery), accessible at https://david.ncifcrf.gov/summary.jsp [accessed on 13/10/2024]. Only virulence genes present in the KEGG database (InlA and InlB) were included in the metabolic pathway analysis.
3 Results
3.1 MLST analysis
All strains of L. monocytogenes were belonged to phylogenetic lineage I, sublineage SL2, and PCR-serogroup IVb. In addition, two different MLST sequence types were identified: ST2 (92.86%, n = 13 strains), ST45 (7.14%, n = 1). ST 2 was found across multiple regions, including Amhara (seven strains), Oromia (four strains), and SNNPR (four strains). It was predominantly associated with both processed food (seven strains) and raw food (6 strains). The majority of strains with ST 2 were collected at processors and collectors, with six strains found at each collection site. In contrast, ST 145 was found in only one strain, which came from Oromia, associated with raw food, and collected by a collector. Furthermore, In addition, all strains of L. monocytogenes were grouped into CC2 (Table 1) (Wei et al., 2024).
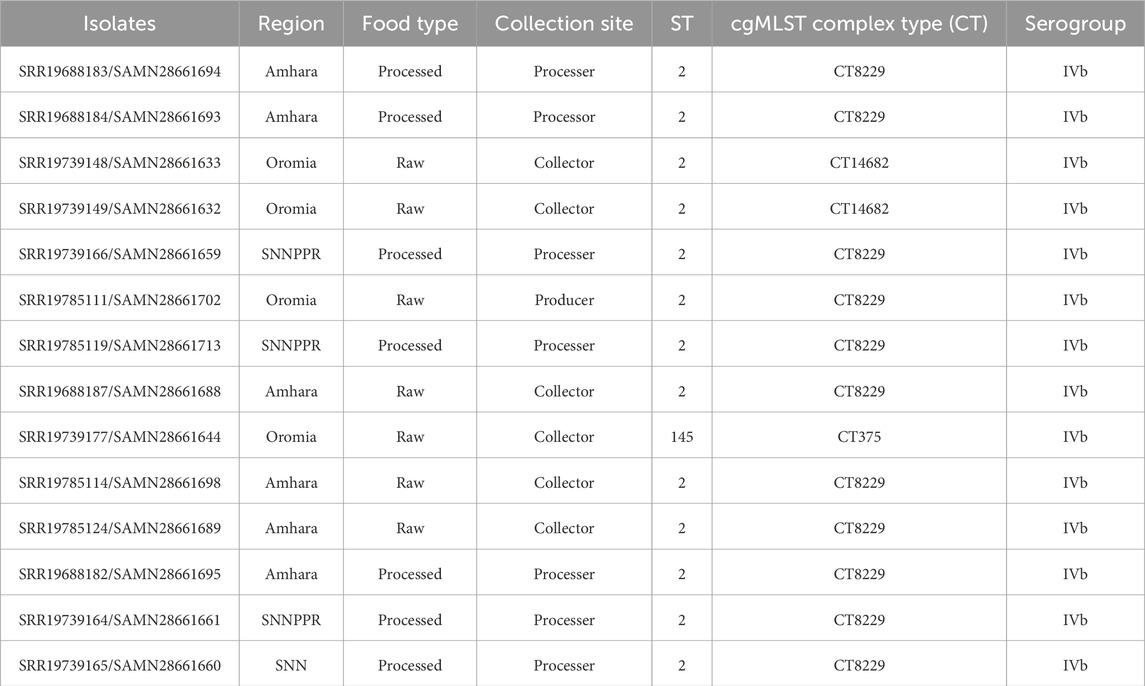
Table 1. Distribution of Listeria monocytogenes isolates in Region, food type and collection site of the food.
3.2 cgMLST analysis
Based on the cgMLST analysis, L. monocytogenes strains were classified into three different CTs (Figure 1). The most prevalent one, cgMLST type CT8229, covered 11 strains obtained from three different regions (Amhara, Oromia and SNNPR), both raw and pasteurized milk, and three different milk sources (collector, processor and producer). The remaining cgMLST types were CT14682 (found in two strains), and CT375 (found in one strains), both of which were obtained only from Oromia region, raw milk, and milk collectors (Table 1).
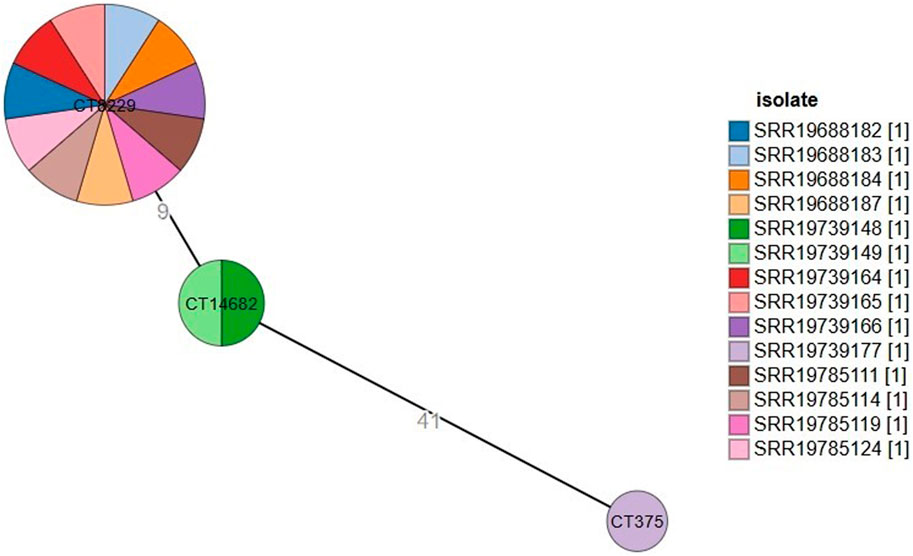
Figure 1. Minimum spanning tree constructed based on MLST allelic profiles of 14 Listeria monocytogenes isolates. Each circle clusters a cgMLST complex type, and colors distinguish isolates. Branch length indicates the difference in the number of alleles.
The minimum spanning tree (Figure 1) showed how various strains relate to one another in terms of genetic distance, using clustering parameters based on a seven allelic difference, as described by Moura et al. (2016). CT9229 forms a large and diverse cluster, suggesting that the strains within this cluster are genetically similar but show some variability. It contains many strains that are closely related, possibly indicating a recent common ancestor or a rapidly evolving population. CT14682 is smaller and contains fewer strains. The genetic distance of nine between CT9229 and CT14682 suggests that these two clusters are relatively close but distinct, possibly indicating separate evolutionary events or selective pressures. CT375 is isolated and has a large genetic distance of 41 from CT14682, indicating that this strain has evolved more independently.
3.3 Virulence genes in the studied Listeria monocytogenes strains
To investigate the virulence potentials of the 14 strains, an in silico detection of 29 virulence genes was carried out, main virulent genes in each strain were presented in Figure 2. All virulence genes were detected in all the 14 L. monocytogenes strains. However, lplA1 is detected in higher amount in all strains, except SAMN28661660, suggesting it may play a significant role in the virulence of these particular strains. This suggests that lplA1 is the most distinctively expressed virulence gene among the strains. Thus, the lplA1 could be the most effective genetic sequence for continuous environmental monitoring. This gene could serve as a valuable marker for rapid testing and surveillance, potentially aiding in early outbreak detection.
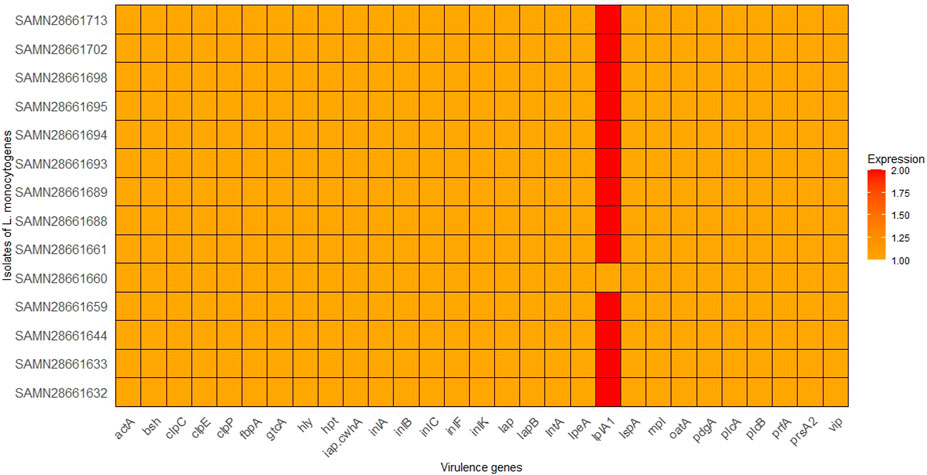
Figure 2. Distribution of virulence genes in the 14 isolates of L. monocytognes. Isolates are represented on the y-axis, the x-axis represents the virulence genes being analyzed, and the color intensity corresponds to the number of copies of each virulence gene found in each isolate.
3.4 Antibiotic resistance genes in the studied Listeria monocytogenes strains
Four antibiotic resistance genes were identified in this study (Figure 3). fosX (resistance to fosfomycin), lin (resistance to lincosamides), norB (resistance to quinolones), and Listeria_monocytogenes_mprF Cationic peptide (resistance to cationic antimicrobial peptides) were present in all studied strains of L. monocytogenes. fosX, Listeria_monocytogenes_mprF, and lin were detected in high level in all strains, except low level detection of lin in SAMN28661644. This suggests that these resistance genes are highly conserved and potentially critical for the resistance profile of these L. monocytogenes strains. norB was identified in a lower amount in all strains.
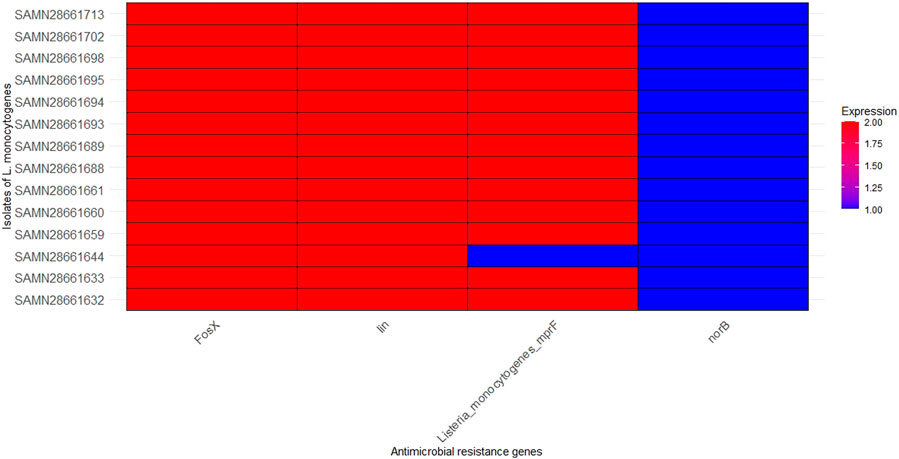
Figure 3. Distribution of antimicrobial resistance genes in the 14 isolates of L. monocytognes. Isolates are represented on the y-axis. The x-axis represents specific antimicrobial resistance genes involved in resistance mechanisms. The color intensity corresponds to the number of copies of each antimicrobial resistance genes found in each isolate.
3.5 Genes for stress, metal, and disinfectant resistance and genomic island
In this study, all 14 L. monocytogenes strains were detected for the presence of stress, metal, and disinfectant resistance adaptation associated genes or gene clusters. Stress resistance genes include stress survival islet 1 (SSI1_lmo0447), lmo 1800, and lmo1799 were identified in all strains of L. monocytogenes, whereas, none of the strains harboured Stress Survival Islet 2 (STI2). However, genes encoding for metal/Benzalkonium Chloride tolerance and disinfectant resistance were not identified from all 14 strains of L. monocytogenes. LGI-2 was found in all the strains and none of the studied strains harbored LGI-1 and LGI-3.
3.6 Plasmids and mobile genetic elements
The presence of plasmids in L. monocytogenes strains was tested using PlasmidFinder 2.1. However, plasmids were not detected in any of the 14 strains. Additionally, MGEs were assessed using MGEs, but none were found in any of the 14 strains.
3.7 Phylogenetic relationship of strains
SNP based phylogenetic tree (Figure 4) was constructed using MEGA 11 that represented the evolutionary relationships among the 14 strains of L. monocytogenes based on their genetic similarity. Closely related samples like SAMN28661688 and SAMN28661689 (with a genetic distance of 0.0022) likely share a recent common ancestor, whereas SAMN28661660 showed greater divergence, indicating it may have evolved separately from the other samples over a longer period.
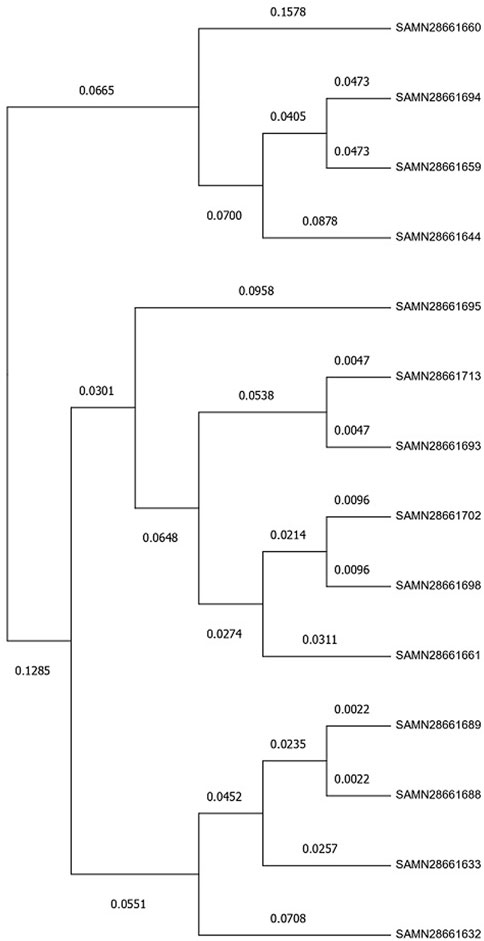
Figure 4. Evolutionary analysis by Maximum Likelihood method. The evolutionary history was inferred by using the Maximum Likelihood method and General Time Reversible model (Nei and Kumar, 2000). The tree with the highest log likelihood (−8,483.46) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (five categories (+G, parameter = 9.4062)). The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 0.00% sites). This analysis involved 14 nucleotide sequences. All positions with less than 95% site coverage were eliminated, i.e., fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position (partial deletion option). There were a total of 1,513 positions in the final dataset. Evolutionary analyses were conducted in MEGA11 (Tamura et al., 2021).
3.8 CRISPR-Cas systems
Analysis of the CRISPR-Cas system of strains revealed that most strains have two CRISPR arrays located at similar genome positions, with consistent repeat consensus sequences: “TTTCCACCACCACCTACGGATGAAGAGTTAAGACTTGCTTTGCCAGAGAC” and “GAACTCGTATCAAAACTGCTTAA.” 14.28% (2/14) of the strains also carry the Cas gene csa5, indicating a functional Cas system (CAS-TypeIA) (Table 2). The presence of csa5 in a subset of strains indicates potential variability in CRISPR-Cas activity, with some strains possibly retaining an active defense mechanism against foreign genetic elements, such as phages and plasmids, while others may rely on alternative defense strategies. The overall conservation of CRISPR arrays and their structural features suggests that this system may play a significant role in genome stability and strain adaptation, potentially influencing their evolutionary trajectory and resistance to horizontal gene transfer.
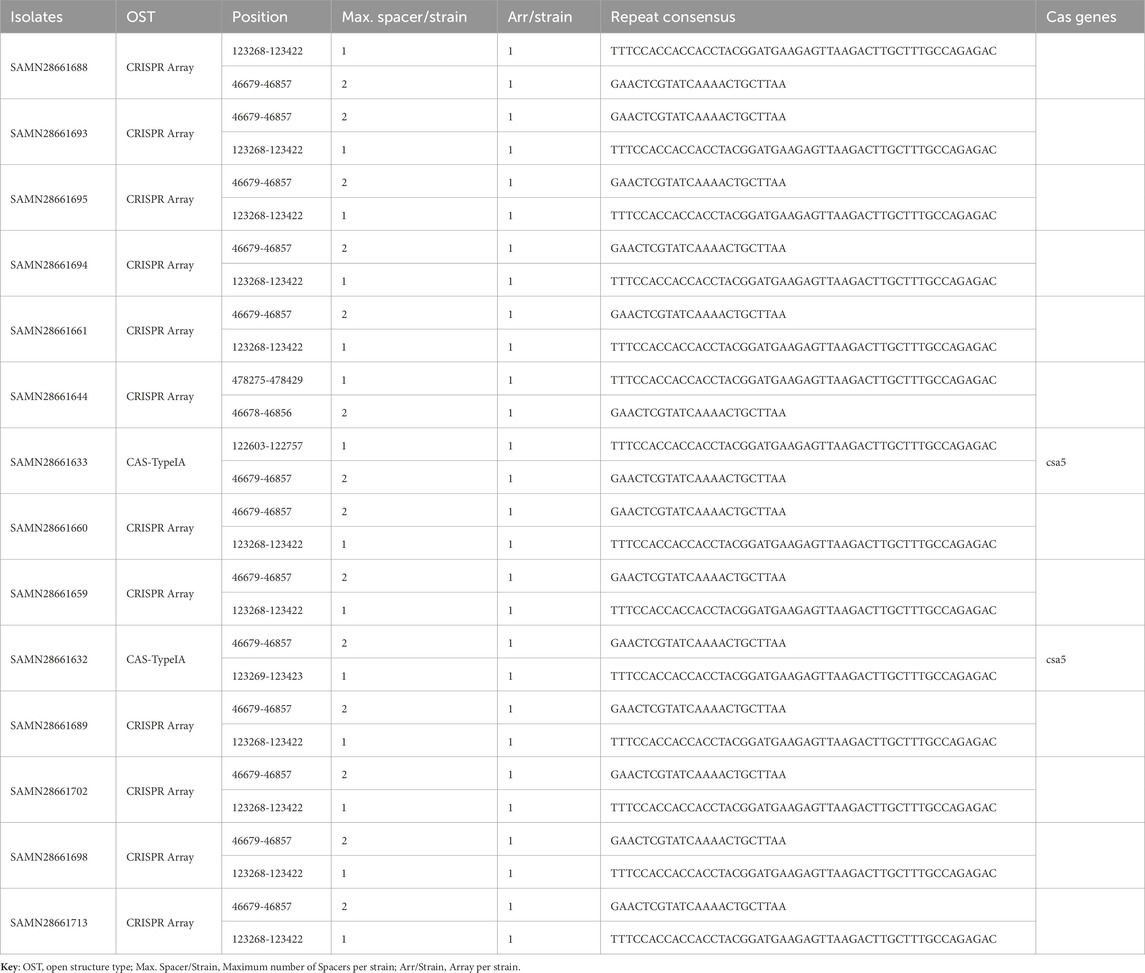
Table 2. CRISPR-Cas systems identified in Listeria monocytogenes genomes.
3.9 Metabolic pathway analysis
A KEGG pathway analysis was performed to investigate the metabolic pathways and mechanisms associated with the virulence genes identified in L. monocytogenes strains using DAVID. InlA and InlB are used by L. monocytogenes as invasins to bind receptors like E-cadherin and Met on the surface of intestinal epithelial cells. This interaction triggers actin polymerization through a cascade of signaling molecules, including PI3K, Rho GTPases (Cdc42, Rac), N-WASP, and the Arp2/3 complex, leading to internalization of the bacteria. The actin cytoskeleton is regulated to facilitate bacterial entry and vacuole formation (Figure 5). The enrichment of L. monocytogenes virulence genes in the actin polymerization pathway indicates that actin dynamics are essential for the bacteria’s ability to enter host cells and cause infection. Understanding how the bacteria manipulate this pathway can lead to the development of more accurate diagnostic tools, such as biomarkers specific to this pathway, enabling sensitive detection of L. monocytogenes infections. Additionally, targeting key components of the actin polymerization pathway could provide new therapeutic strategies to prevent bacterial entry and control infection.
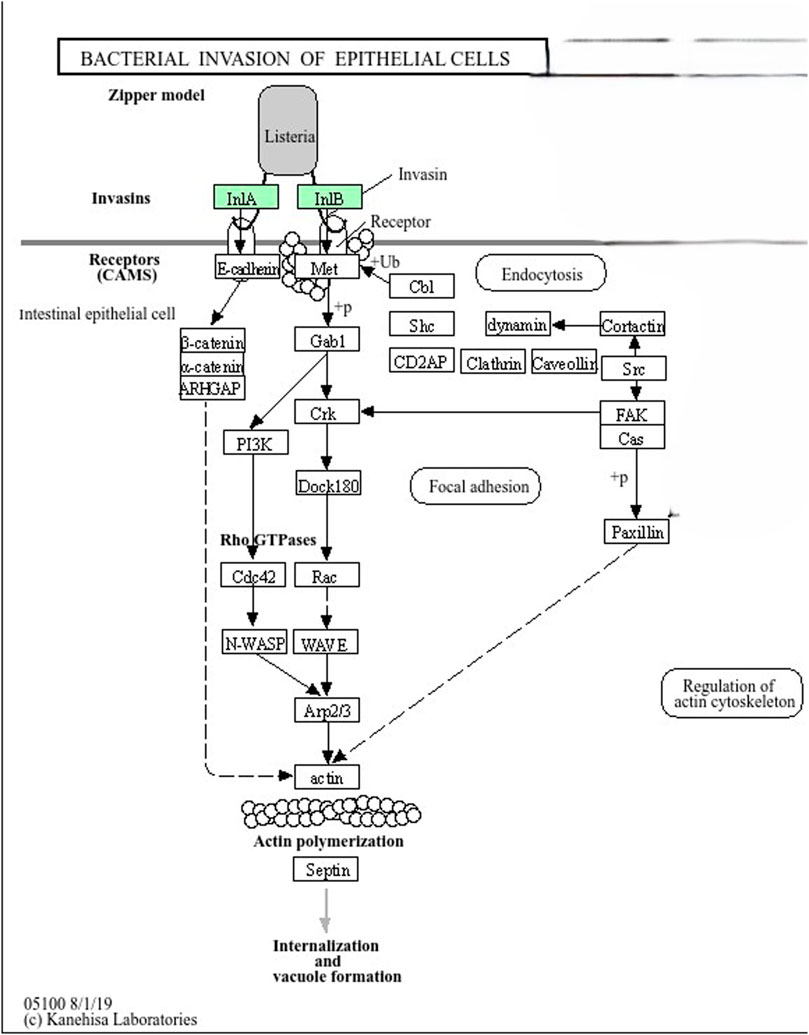
Figure 5. InlA and InlB in Zipper Model (Listeria Invasion Pathway). Listeria monocytogenes uses InlA and InlB (colored in green) proteins as invasins to bind receptors like E-cadherin and Met on the surface of intestinal epithelial cells. This interaction triggers actin polymerization through a cascade of signaling molecules, including PI3K, Rho GTPases (Cdc42, Rac), N-WASP, and the Arp2/3 complex, leading to internalization of the bacterium. The actin cytoskeleton is regulated to facilitate bacterial entry and vacuole formation.
4 Discussion
In Ethiopia, L. monocytogenes contamination in milk and dairy products is alarmingly high, with some studies reporting rates up to 60% (Tola, 2024; Seyoum et al., 2015). This poses a significant public health risk, given the widespread consumption of dairy, particularly raw milk, in both rural and urban areas as well as inadequate sanitation in dairy processing and poor handling practices (Abdi Keba et al., 2020; Mebrate et al., 2020). Its ubiquity, persistence under adverse environmental conditions, pathogenicity (Hurley et al., 2019; Chen et al., 2020), and ability to develop multidrug resistance (Dufailu et al., 2021) contribute to its impact. Understanding the pathogen’s virulence, antimicrobial resistance mechanisms, and environmental stress adaptation in the region/Ethiopia is crucial for developing novel, efficient, and cost-effective antimicrobial agents and biocontrol methods. In this study, we applied WGS to strains isolated from milk and dairy products in Ethiopia to examine their virulence gene profiles, antimicrobial resistance genes, MGEs linked to virulence and resistance, and genetic relatedness.
A total of 14 L. monocytogenes strains originating from milk and dairy products were typed and characterized for the presence of virulence, antimicrobial resistance, and stress response genetic determinants. MLST typing in this study revealed a dominance of sequence type ST2 (92.86%, n = 13) over ST45 (7.14%, n = 1), aligning with previous research suggesting that certain clones, such as ST2, are more prevalent in food-related environments, including dairy products (Amarasekara et al., 2024). The widespread occurrence of ST2 in food sources is further supported by Tricoli et al. (2024), who reported the circulation of ST2, a clinically relevant strain linked to suspected food contamination.
All strains belonged to PCR-serogroup IVb and were classified into clonal complex (CC) 2, further supporting the notion that ST2 strains are well adapted to persist in food production settings (Koopmans et al., 2023). CC2, identified in the studied strains, is one of the dominant clonal complexes responsible for listeriosis in different regions of the world (Chenal-Francisque et al., 2011). This complex is known to confer hypervirulence by enhancing bacterial invasion into the central nervous system (CNS) and the placenta, which significantly increases the risk of severe infections (Maury et al., 2016).
While ST45 was less common in this study, it has been reported in China as spreading across different regions, demonstrating remarkable virulence potential. Specifically, ST45 strains exhibited high hemolysin activity and significant virulence capacity, particularly in bloodstream infections (Wang et al., 2019). These findings emphasize the importance of continuous monitoring and characterization of L. monocytogenes strains in dairy products to better understand their potential risks to public health.
The cgMLST analysis revealed three distinct core genome sequence types (CTs) with the most prevalent type, L1-SL2-ST2-CT8229, comprising 11 strains from diverse geographical regions (Amhara, Oromia, South Nation Nationalities), different types of milk (raw and pasteurized), and various sources within the dairy supply chain (collectors, processors, and producers). This widespread distribution of CT8229 suggests a highly adaptive clone that may be circulating widely in Ethiopia’s milk production chain. The broad presence of this cgMLST type across multiple regions and milk sources highlights the potential for cross-contamination at different points in the supply chain, indicating gaps in food safety practices that require attention (Palaiodimou et al., 2021). The less prevalent cgMLST types, L1-SL2-ST2-CT14682 and L1-SL2-ST45-CT375, were found only in raw milk from the Oromia region, specifically from milk collectors. This suggests a more localized and specific distribution pattern, possibly linked to regional practices in milk collection and handling (Maury et al., 2016).
The minimum spanning tree (MST) analysis (Figure 1) shows that CT8229 forms a large and diverse cluster, indicating genetic homogeneity with slight variability. This genetic closeness may suggest a recent common ancestor or rapid clonal expansion in response to selective pressures within the dairy production environment. Such expansion may be due to environmental resilience, increased fitness, or adaptation to selective pressures like pasteurization or other food processing measures (Henri et al., 2016). The genetic distance of nine between CT8229 and CT14682 indicates that these clusters are genetically close, possibly reflecting micro-evolutionary changes or selective pressures that have shaped their genomic characteristics. CT14682 may represent a slightly divergent subgroup of ST2, indicating the evolution of sub-clonal populations that could respond differently to environmental pressures or interventions. In contrast, the genetic distance of 41 between CT14682 and CT375, which belongs to ST45, suggests that this strain has evolved more independently. The significant divergence of CT375 may reflect distinct evolutionary events, potentially driven by different ecological niches or selective pressures in the dairy environment (Osek et al., 2022).
The detection of all 29 virulent and virulence-related genes across all 14 L. monocytogenes strains underscores the virulent potential of these strains. This result is consistent with previous studies where the core virulence genes of L. monocytogenes, such as prfA, hly, actA, and inlA, were present in all strains regardless of their source (Maury et al., 2016). A particularly interesting finding is the elevated detection of lplA1 in all strains except SAMN28661660. lplA1 is associated with lipid metabolism and has been implicated in the virulence of L. monocytogenes during host infection, contributing to intracellular survival and growth (Harter et al., 2019). Its high prevalence in the strains may indicate a critical role in the virulence of these strains, potentially aiding in the persistence and pathogenicity of L. monocytogenes in different environments. This also suggests that lplA1 is the most distinctively expressed virulence gene among the strains. Thus, the lplA1 could be the most effective genetic sequence for continuous environmental monitoring. This gene could serve as a valuable marker for rapid testing and surveillance, potentially aiding in early outbreak detection. The absence of this gene in SAMN28661660 may indicate differences in its virulence potential, warranting further investigation into how lplA1 influences the overall virulence and fitness of L. monocytogenes in various ecological niches.
Four antibiotic resistance genes—fosX, lin, norB, and mprF—were identified across all strains. The consistent presence of fosX (fosfomycin resistance), mprF (cationic antimicrobial peptide resistance), and lin (lincosamide resistance) suggests these genes are highly conserved in L. monocytogenes populations, potentially contributing to the persistence of antibiotic resistance in the environment (Morvan et al., 2010). The lower copy number detection of lin in SAMN28661644 could reflect a varying degree of resistance expression or a genetic variation specific to this strain. Meanwhile, norB (quinolone resistance) was detected at a lower copy number across all strains, indicating it may not be as critical for resistance as the other genes, but its presence still poses a concern for treatment efficacy, especially in clinical settings where quinolones are used (Parra-Flores et al., 2022). These findings reflect the importance of continuous monitoring of antimicrobial resistance genes in L. monocytogenes to mitigate the risk of resistance spread. In agreement to this study, Parra-Flores et al. (2022) reported the presence of resistance genes with mechanisms of antibiotic efflux (norB), antibiotic target alteration (mprF), and antibiotic inactivation (lin, fosX). However, in addition, their study reported genes that confer resistance to tetracycline (tetA and tetC).
All strains harbored stress survival islet 1 (SSI1_lmo0447) and the lmo1799 and lmo1800 genes, which are known to confer stress resistance, particularly in low pH or osmotic stress conditions that are common in food processing environments (Bergholz et al., 2012). This widespread presence indicates that these strains are well-equipped to survive harsh conditions, which is a concern for food safety as L. monocytogenes can persist even in processed food products. However, the absence of stress survival islet 2 (SSI2) in all strains is noteworthy since SSI2 is linked to survival under alkaline and oxidative stress conditions. Its absence may suggest that the strains in this study are less adapted to these specific stresses, possibly due to the particular conditions of the milk production environment from which they were isolated (Harter et al., 2017).
The absence of genes encoding resistance to metals and disinfectants, including benzalkonium chloride, in all strains is a positive finding. Benzalkonium chloride is commonly used in food production as a disinfectant, and its effectiveness against L. monocytogenes in strains of this study suggests that these strains may not have developed resistance mechanisms that could undermine food safety efforts (Romanova et al., 2006). This finding emphasizes the continued relevance of current disinfection practices in dairy production facilities for controlling L. monocytogenes.
All strains carried the Listeria genomic island 2 (LGI-2), while none harbored LGI-1 or LGI-3. LGI-2 is known to contribute to heavy metal resistance, antibiotic resistance, and stress adaptation (Kuenne et al., 2010), which could explain the persistence of these strains in the dairy environment. Despite the presence of LGI-2 in these strains, the absence of predicted heavy metal resistance genes indicates that these particular strains have not yet developed mechanisms to overcome this specific disinfectant. This highlights a critical aspect of microbial evolution, where resistance to certain agents may develop independently and is influenced by selective pressures (Christaki et al., 2020). However, the absence of plasmids, as revealed by PlasmidFinder, suggests that plasmid-mediated transfer of resistance or virulence factors is not occurring in this population. Similarly, no MGEs were identified, which is an encouraging sign as MGEs, including transposons and integrative conjugative elements, play a key role in the horizontal transfer of resistance genes (Pricope et al., 2013). This might indicate that the genetic content of the strains is relatively stable, with no immediate threat of gene transfer to other bacterial populations.
The SNP-based phylogenetic tree constructed using MEGA 11 illustrates the evolutionary relationships between the 14 L. monocytogenes strains. The close genetic distance between samples SAMN28661688 and SAMN28661689 suggests a recent common ancestor, which may imply shared evolutionary pressures or sources of contamination. In contrast, SAMN28661660 shows greater genetic divergence, suggesting it evolved separately from the other strains over a longer period. Phylogenetic trees based on SNP data have been widely used to infer the evolutionary relationships and trace the transmission patterns of L. monocytogenes in both clinical and environmental settings (Chen et al., 2020). This tree further underscores the genetic heterogeneity present in the studied strains, which can have implications for their virulence, resistance, and ability to survive in diverse environments.
CRISPR-Cas systems are acquired immunity systems that allow bacteria and archaea to acquire exogenous material from bacteriophages and plasmids (Hupfeld et al., 2018). The CRISPRCas systems is a possible involved in the regulation of gene expression, including virulence genes, which have been described in a number of pathogens (Louwen et al., 2014). In this study, the analysis of the CRISPR-Cas system revealed that most strains harbor two CRISPR arrays with conserved repeat sequences, indicating a relatively stable CRISPR system in these strains. CRISPR systems, particularly type IA, play important roles in bacterial immunity by providing resistance to foreign genetic elements, such as bacteriophages or plasmids (Marraffini, 2013). The presence of the Cas gene csa5 in 14.28% of the strains suggests a functional CRISPR-Cas system, which could enhance the adaptability of these strains by defending against horizontal gene transfer. This functionality may contribute to the evolutionary success of L. monocytogenes, as it can protect against phage attacks or acquisition of harmful MGEs while maintaining virulence potential. CRISPR-Cas system plays multiple roles beyond adaptive immunity, i.e., gene regulation and virulence (Faure et al., 2019). Understanding the structure and function of CRISPRCas system contributes to develop useful technology and products to control pathogen, i.e., phages of Listeria offer novel tools for detection, differentiation, CRISPR-Cas-assisted phage engineering, diagnostics, and biocontrol (Hagens and Loessner, 2014; Hupfeld et al., 2018; Meile et al., 2020).
The KEGG pathway analysis highlights the molecular mechanisms through which L. monocytogenes invades host cells. The virulence genes inlA and inlB were shown to play central roles in ability of the pathogen to bind to host cell receptors like E-cadherin and Met. This interaction activates a signaling cascade involving PI3K, Rho GTPases, N-WASP, and the Arp2/3 complex, which promotes actin polymerization and leads to bacterial internalization into host cells (Pizarro-Cerdá et al., 2012). The manipulation of the host actin cytoskeleton is a well-established strategy used by L. monocytogenes to evade immune responses and spread within the host, making it a key factor in the pathogen’s ability to cause infection. The conserved nature of these virulence mechanisms across the strains suggests that targeting these pathways could be a potential avenue for therapeutic intervention in L. monocytogenes infections. On the other hand, research indicates that while intact inlA genes are prevalent in clinical strains, truncations often occur in isolates from food processing environments, primarily due to premature stop codons (PMSCs) (Gorski et al., 2016). In food isolates, five out of seven cultures exhibited truncated inlA profiles, contrasting with the intact profiles found in clinical isolates (Tamburro et al., 2015).
5 Limitation of the study
The key limitations of this study is the relatively small sample size of L. monocytogenes strains analyzed, which includes only 14 strains from milk and dairy products in Ethiopia. This limited number of strains may not fully represent the genetic diversity of L. monocytogenes in different regions, production methods, or types of dairy products across the country. As a result, the findings may not be entirely generalizable to the broader population of L. monocytogenes in Ethiopia or other regions with similar agricultural and dairy production environments. Future studies should incorporate more diverse geographic and environmental samples to strengthen conclusions. Such an approach would provide a more comprehensive understanding of L. monocytogenes diversity, antimicrobial resistance patterns, and potential public health risks.
Despite our genomic findings provide valuable insights regarding antibiotic resistance genes, there was no experimental validation (e.g., antimicrobial susceptibility testing) to confirm resistance phenotypes. Therefore, future studies incorporating phenotypic validation will be necessary to confirm the functional resistance conferred by these genes. In addition, although this study indicates the presence of CRIPR-Cas system, experimental verification of CRISPR activity there was not performed. As a result, future work using functional assays, such as phage challenge experiments or plasmid interference assays, would be necessary to confirm the protective role of CRISPR-Cas in L. monocytogenes.
6 Conclusion and recommendations
This study highlights the significant genomic diversity among L. monocytogenes strains from dairy products in Ethiopia. The strains were categorized into two sequence types (ST2 and ST45), with ST2 being the dominant type. All 14 strains examined contained a complete set of 29 virulence genes, suggesting they may have a potential for pathogenicity. Additionally, antibiotic resistance genes associated with fosfomycin, lincosamides, and quinolones were identified, suggests a potential for antibiotic resistance in L. monocytogenes within the dairy sector. All strains harbored LGI-2, while none contained LGI-1 or LGI-3, nor did they possess genes related to metal resistance, disinfectant tolerance, or MGEs. The presence of the CRISPR-Cas system in some strains may contribute to phage defense and the preservation of genetic integrity. Additionally, KEGG pathway analysis illuminated the molecular mechanisms underlying host cell invasion, emphasizing the pathogen’s virulence potential. While this study provides important insights, it was conducted using a relatively small sample size of only 14 strains. Future research should expand the scope by including a larger number of isolates to better understand the full diversity of L. monocytogenes in dairy products. Further studies should investigate the role of CRISPR-Cas systems and other genetic elements in bacterial virulence and antimicrobial resistance. Moreover, continuous monitoring of L. monocytogenes in the dairy sector, coupled with stricter sanitation practices, food safety regulations, and targeted public health interventions, is crucial to mitigate the risks of contamination and resistance spread.
Data availability statement
The datasets used and/or analyzed during the current study are available in the NCBI Sequence Read Archive (SRA) repository under the accession number PRJNA357724.
Author contributions
MK: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review and editing. BK: Supervision, Writing – review and editing. TE: Writing – review and editing. AG: Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We thank the Institut Pasteur teams for the curation and maintenance of BIGSdb-Pasteur databases at https://bigsdb.pasteur.fr/
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
BLASTN, Basic Local Alignment Search Tool for Nucleotide Sequences; CARD, Comprehensive Antibiotic Resistance Database; CC, clonal complex; CgMLST, Core Genome Multilocus Sequence Typing; CRISPR-Cas, Clustered Regularly Interspaced Short Palindromic Repeats-CRISPR-associated proteins; DAVID, Database for Annotation, Visualization, and Integrated Discovery; EGD, Epidemic Group D; FASTQ, Fast Quality Format; IGV, Integrative Genomics Viewer; KEGG, Kyoto Encyclopedia of Genes and Genomes; L. monocytogenes, Listeria monocytogenes; LGI-1, Listeria Genomic Island 1; LGI-2, Listeria Genomic Island 2; LGI-3, Listeria Genomic Island 3; MGEs, Mobile Genetic Elements; MLST, Multilocus Sequence Typing; NCBI, National Center for Biotechnology Information; QUAST, Quality Assessment Tool for Genome Assemblies; SNNPR, Southern Nations, Nationalities, and Peoples’ Region; SNP, Single-nucleotide polymorphism; SRA, Sequence Read Archive; ST, Sequence Type; VCF, Variant Call Format; VFDB, virulence factor database; WGS, whole-genome sequencing.
References
Abdi Keba, M., Rolon, L., Tamene, A., Dessie, K., Vipham, J., Kovac, J., et al. (2020). Review of the prevalence of foodborne pathogens in milk and dairy products in Ethiopia. Int. Dairy J. 109, 104762. doi:10.1016/j.idairyj.2020.104762
Alcock, B., Huynh, W., Chalil, R., Smith, K. W., Raphenya, A. R., Wlodarski, M. A., et al. (2023). CARD 2023: expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 51 (D1), D1–D6. doi:10.1093/nar/gkac1009
Amarasekara, N. R., Swamy, A. S., Paudel, S. K., Jiang, W., Li, K., Shen, C., et al. (2024). Hypervirulent clonal complex (CC) of Listeria monocytogenes in fresh produce from urban communities. Front. Microbiol. 15, 1307610. doi:10.3389/fmicb.2024.1307610
Bergholz, T. M., Bowen, B., Wiedmann, M., and Boor, K. J. (2012). Listeria monocytogenes shows temperature-dependent and -independent responses to salt stress, including responses that induce cross-protection against other stresses. Appl. Environ. Microbiol. 78 (8), 2602–2612. doi:10.1128/AEM.07658-11
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi:10.1093/bioinformatics/btu170
Camacho, C., Coulouris, G., Avagyan, V., Papadopoulos, J., Ma, N., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinforma. 10 (1), 421. doi:10.1186/1471-2105-10-421
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58 (7), 3895–3903. doi:10.1128/AAC.02412-14
Chen, Y., Chen, Y., Pouillot, R., Dennis, S., Xian, Z., Luchansky, J. B., et al. (2020). Genetic diversity and profiles of genes associated with virulence and stress resistance among isolates from the 2010-2013 interagency Listeria monocytogenes market basket survey. PLoS ONE 15 (6), e0231393. doi:10.1371/journal.pone.0231393
Chen, Y., and Knabel, S. J. (2007). Multiplex PCR for simultaneous detection of bacteria of the genus Listeria, Listeria monocytogenes, and major serotypes and epidemic clones of L. monocytogenes. Appl. Environ. Microbiol. 73 (19), 6299–6304. doi:10.1128/AEM.00961-07
Chenal-Francisque, V., Lopez, J., Cantinelli, T., Caro, V., Tran, C., Leclercq, A., et al. (2011). Worldwide distribution of major clones of Listeria monocytogenes. Emerg. Infect. Dis. 17 (6), 1110–1112. doi:10.3201/eid1706.101778
Christaki, E., Marcou, M., and Tofarides, A. (2020). Antimicrobial resistance in bacteria: mechanisms, evolution, and persistence. J. Mol. Evol. 88 (1), 26–40. doi:10.1007/s00239-019-09914-3
Couvin, D., Bernheim, A., Toffano-Nioche, C., Touchon, M., Michalik, J., Néron, B., et al. (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance, and integrates search for CAS proteins. Nucleic Acids Res. 46 (1), 246–251. doi:10.1093/nar/gky425
Datta, A., and Burall, L. (2018). Current trends in foodborne human listeriosis. Food Saf. (Tokyo) 6 (1), 1–6. doi:10.14252/foodsafetyfscj.2017020
Dobrindt, U., Hochhut, B., Hentschel, U., and Hacker, J. (2004). Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2 (6), 414–424. doi:10.1038/nrmicro884
Dufailu, O. A., Yaqub, M. O., Owusu-Kwarteng, J., and Addy, F. (2021). Prevalence and characteristics of Listeria species from selected African countries. Trop. Dis. Travel Med. Vaccines 7, 1–9. doi:10.1186/s41182-021-00425-5
Faure, G., Makarova, K. S., and Koonin, E. V. (2019). CRISPR-Cas: complex functional networks and multiple roles beyond adaptive immunity. J. Mol. Biol. 431 (1), 3–20. doi:10.1016/j.jmb.2018.08.030
Ghaly, T. M., and Gillings, M. R. (2022). New perspectives on mobile genetic elements: a paradigm shift for managing the antibiotic resistance crisis. Philosophical Trans. R. Soc. B Biol. Sci. 377 (1842), 20200462. doi:10.1098/rstb.2020.0462
Gorski, L., Parker, C. T., Liang, A. S., Walker, S., and Romanolo, K. F. (2016). The majority of genotypes of the virulence gene inlA are intact among natural watershed isolates of Listeria monocytogenes from the central California coast. PLoS ONE 11 (12), e0167566. doi:10.1371/journal.pone.0167566
Hagens, S., and Loessner, M. J. (2014). Phages of Listeria offer novel tools for diagnostics and biocontrol. Front. Microbiol. 5, 159. doi:10.3389/fmicb.2014.00159
Hansen, C. H., Vogel, B. F., and Gram, L. (2006). Prevalence and survival of Listeria monocytogenes in Danish aquatic and fish-processing environments. J. Food Prot. 69 (9), 2113–2122. doi:10.4315/0362-028x-69.9.2113
Harter, E., Lassnig, C., Wagner, E. M., Zaiser, A., Wagner, M., and Rychli, K. (2019). The novel internalins InlP1 and InlP4 and the internalin-like protein InlP3 enhance the pathogenicity of Listeria monocytogenes. Front. Microbiol. 10, 1644. doi:10.3389/fmicb.2019.01644
Harter, E., Wagner, E. M., Zaiser, A., Halecker, S., Wagner, M., and Rychli, K. (2017). Stress Survival Islet 2, predominantly present in Listeria monocytogenes strains of sequence type 121, is involved in the alkaline and oxidative stress responses. Appl. Environ. Microbiol. 83 (16), e00827-17. doi:10.1128/AEM.00827-17
Henri, C., Félix, B., Guillier, L., Leekitcharoenphon, P., Michelon, D., Mariet, J. F., et al. (2016). Population genetic structure of Listeria monocytogenes strains as determined by pulsed-field gel electrophoresis and multilocus sequence typing. Appl. Environ. Microbiol. 82 (18), 5720–5728. doi:10.1128/AEM.00583-16
Hupfeld, M., Trasanidou, D., Ramazzini, L., Klumpp, J., Loessner, M. J., and Kilcher, S. (2018). A functional type II-A CRISPR-Cas system from Listeria enables efficient genome editing of large non-integrating bacteriophage. Nucleic Acids Res. 46 (12), 6920–6933. doi:10.1093/nar/gky544
Hurley, D., Luque-Sastre, L., Parker, C. T., Huynh, S., Eshwar, A. K., Nguyen, S. V., et al. (2019). Whole-genome sequencing-based characterization of 100 Listeria monocytogenes isolates collected from food processing environments over a four-year period. mSphere 4 (3), e00252-19. doi:10.1128/mSphere.00252-19
Johansson, M. H., Bortolaia, V., Tansirichaiya, S., Aarestrup, F. M., Roberts, A. P., and Petersen, T. N. (2020). Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J. Antimicrob. Chemother. 76 (1), 101–109. doi:10.1093/jac/dkaa390
Jolley, K. A., and Maiden, M. C. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinforma. 11, 595. doi:10.1186/1471-2105-11-595
Kathariou, S., Evans, P., and Dutta, V. (2017). “Strain-specific virulence differences in Listeria monocytogenes: current perspectives in addressing an old and vexing issue,” in Foodborne pathogens. Food microbiology and food safety. Editors J. Gurtler, M. Doyle, and J. Kornacki (Springer), 35–63. doi:10.1007/978-3-319-56836-2_3
Koopmans, M. M., Brouwer, M. C., Vázquez-Boland, J. A., and van de Beek, D. (2023). Human listeriosis. Clin. Microbiol. Rev. 36 (1), e0006019. doi:10.1128/cmr.00060-19
Kuenne, C., Voget, S., Pischimarov, J., Oehm, S., Goesmann, A., Daniel, R., et al. (2010). Comparative analysis of plasmids in the genus Listeria. PLOS ONE 5 (9), e12511. doi:10.1371/journal.pone.0012511
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Louwen, R., Staals, R. H., Endtz, H. P., van Baarlen, P., and van der Oost, J. (2014). The role of CRISPR-Cas systems in virulence of pathogenic bacteria. Microbiol. Mol. Biol. Rev. 78 (1), 74–88. doi:10.1128/MMBR.00039-13
Luque-Sastre, L., Arroyo, C., Fox, E. M., McMahon, B. J., Bai, L., Li, F., et al. (2018). Antimicrobial resistance in Listeria species. Microbiol. Spectr. 6 (5). ARBA-0031-2017. doi:10.1128/microbiolspec.arba-0031-2017
Marraffini, L. A. (2013). CRISPR-Cas immunity against phages: its effects on the evolution and survival of bacterial pathogens. PLOS Pathog. 9 (12), e1003765. doi:10.1371/journal.ppat.1003765
Matle, I., Mbatha, K. R., and Madoroba, E. (2020). A review of Listeria monocytogenes from meat and meat products: epidemiology, virulence factors, antimicrobial resistance, and diagnosis. Onderstepoort J. Veterinary Res. 87 (1), e1–e20. doi:10.4102/ojvr.v87i1.1869
Maury, M. M., Bracq-Dieye, H., Huang, L., Vales, G., Lavina, M., Thouvenot, P., et al. (2019). Hypervirulent Listeria monocytogenes clones’ adaption to mammalian gut accounts for their association with dairy products. Nat. Commun. 10, 2488. doi:10.1038/s41467-019-10380-0
Maury, M. M., Tsai, Y. H., Charlier, C., Touchon, M., Chenal-Francisque, V., Leclercq, A., et al. (2016). Uncovering Listeria monocytogenes hypervirulence by harnessing its biodiversity. Nat. Genet. 48 (3), 308–313. doi:10.1038/ng.3501
Mebrate, G., Tewodros, A., and Etagegnehu, B. (2020). Review on milk and milk product handling practices, utilization, and microbial quality in Ethiopia. Int. J. Dairy Sci. Technol. 4 (1), 218–224.
Meile, S., Sarbach, A., Du, J., Schuppler, M., Saez, C., Loessner, M. J., et al. (2020). Engineered reporter phages for rapid bioluminescence-based detection and differentiation of viable Listeria cells. Appl. Environ. Microbiol. 86 (7), e00442-20. doi:10.1128/AEM.00442-20
Morvan, A., Moubareck, C., Leclercq, A., Hervé-Bazin, M., Bremont, S., Lecuit, M., et al. (2010). Antimicrobial resistance of Listeria monocytogenes strains isolated from humans in France. Antimicrob. Agents Chemother. 54 (6), 2728–2731. doi:10.1128/AAC.01557-09
Moura, A., Criscuolo, A., Pouseele, H., Maury, M. M., Leclercq, A., Tarr, C., et al. (2016). Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat. Microbiol. 2, 16185. doi:10.1038/nmicrobiol.2016.185
Naghavi, M., Vollset, S. E., Ikuta, K. S., Swetschinski, L. R., Gray, A. P., Wool, E., et al. (2024). Global burden of bacterial antimicrobial resistance 1990–2021: a systematic analysis with forecasts to 2050. Lancet 404 (10459), 1199–1226. doi:10.1016/S0140-6736(24)01867-1
Odu, N., and Okonko, I. (2017). Prevalence and antibiotic susceptibility of Listeria monocytogenes in retailed meats in Port Harcourt metropolis, Nigeria. Public Health Res. 7, 91–99. doi:10.5923/j.phr.20170704.02
Orsi, R. H., Den Bakker, H. C., and Wiedmann, M. (2011). Listeria monocytogenes lineages: Genomics, evolution, ecology, and phenotypic characteristics. Int. J. Med. Microbiol. 301 (1), 79–96. doi:10.1016/j.ijmm.2010.05.002
Osek, J., Lachtara, B., and Wieczorek, K. (2022). Listeria monocytogenes - how this pathogen survives in food-production environments? Front. Microbiol. 13, 866462. doi:10.3389/fmicb.2022.866462
Palaiodimou, L., Fanning, S., and Fox, E. M. (2021). Genomic insights into persistence of Listeria species in the food processing environment. J. Appl. Microbiol. 131 (5), 2082–2094. doi:10.1111/jam.15089
Parra-Flores, J., Holý, O., Bustamante, F., Lepuschitz, S., Pietzka, A., Contreras-Fernández, A., et al. (2022). Virulence and antibiotic resistance genes in Listeria monocytogenes strains isolated from ready-to-eat foods in Chile. Front. Microbiol. 12, 796040. doi:10.3389/fmicb.2021.796040
Pizarro-Cerdá, J., Kühbacher, A., and Cossart, P. (2012). Entry of Listeria monocytogenes in mammalian epithelial cells: an updated view. Cold Spring Harb. Perspect. Med. 2 (11), a010009. doi:10.1101/cshperspect.a010009
Pricope, L., Nicolau, A., Wagner, M., and Rychli, K. (2013). The effect of sublethal concentrations of benzalkonium chloride on invasiveness and intracellular proliferation of Listeria monocytogenes. Food control. 31 (1), 230–235. doi:10.1016/j.foodcont.2012.09.031
Quereda, J. J., Morón-García, A., Palacios-Gorba, C., Dessaux, C., García-Del Portillo, F., Pucciarelli, M. G., et al. (2021). Pathogenicity and virulence of Listeria monocytogenes: a trip from environmental to medical microbiology. Virulence 12 (1), 2509–2545. doi:10.1080/21505594.2021.1975526
Ragon, M., Wirth, T., Hollandt, F., Lavenir, R., Lecuit, M., Le Monnier, A., et al. (2008). A new perspective on Listeria monocytogenes evolution. PLOS Pathog. 4 (9), e1000146. doi:10.1371/journal.ppat.1000146
Ravindhiran, R., Sivarajan, K., Sekar, J. N., Murugesan, R., and Dhandapani, K. (2023). Listeria monocytogenes: an emerging pathogen: a comprehensive overview on listeriosis, virulence determinants, detection, and anti-listerial interventions. Microb. Ecol. 86, 2231–2251. doi:10.1007/s00248-023-02269-9
Rogalla, D., and Bomar, P. A. (2023). “Listeria monocytogenes,” in StatPearls (Treasure Island, FL: StatPearls Publishing). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK534838/.
Rohilla, A., Kumar, V., and Ahire, J. J. (2024). Unveiling the persistent threat: recent insights into Listeria monocytogenes adaptation, biofilm formation, and pathogenicity in foodborne infections. J. Food Sci. Technol. 61, 1428–1438. doi:10.1007/s13197-023-05918-6
Romanova, N. A., Wolffs, P. F., Brovko, L. Y., and Griffiths, M. W. (2006). Role of efflux pumps in adaptation and resistance of Listeria monocytogenes to benzalkonium chloride. Appl. Environ. Microbiol. 72 (5), 3498–3503. doi:10.1128/AEM.72.5.3498-3503.2006
Seyoum, E. T., Woldetsadik, D. A., Mekonen, T. K., Gezahegn, H. A., and Gebreyes, W. A. (2015). Prevalence of Listeria monocytogenes in raw bovine milk and milk products from central highlands of Ethiopia. J. Infect. Dev. Ctries. 9 (11), 1204–1209. doi:10.3855/jidc.6211
Sibanda, T., Ntuli, V., Hudaa, S., Habib, I., Njage, P. M. K., Kunadu, A. P.-H., et al. (2023). Listeria monocytogenes at the food–human interface: a review of risk factors influencing transmission and consumer exposure in Africa. Int. J. Food Sci. Technol. 58, 4114–4126. doi:10.1111/ijfs.16540
Tamburro, M., Sammarco, L., Ammendolia, M. G., Fanelli, I., Minelli, F., and Ripabelli, G. (2015). Evaluation of transcription levels of inlA, inlB, hly, bsh, and prfA genes in Listeria monocytogenes strains using quantitative reverse-transcription PCR and ability of invasion into human Caco-2 cells. FEMS Microbiol. Lett. 362 (6), fnv018–7. doi:10.1093/femsle/fnv018
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA 11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38 (7), 3022–3027. doi:10.1093/molbev/msab120
Thomas, J., Govender, N., McCarthy, K. M., Erasmus, L. K., Doyle, T. J., Allam, M., et al. (2020). Outbreak of listeriosis in South Africa associated with processed meat. N. Engl. J. Med. 382 (7), 632–643. doi:10.1056/nejmoa1907462
Tola, E. Y. (2024). Prevalence, antimicrobial resistance, and characterization of Listeria spp. isolated from various sources in Ethiopia: a comprehensive review. Veterinary Med. Res. Rep. 15, 109–116. doi:10.2147/vmrr.s451837
Tricoli, M. R., Massaro, C., Arrigo, I., Diquattro, O., Di Bernardo, F., Galia, E., et al. (2024). Characterization of Listeria monocytogenes strains isolated in Palermo (sicily and Italy) during the years 2018–2020 from severe cases of listeriosis. Antibiotics 13, 57. doi:10.3390/antibiotics13010057
Wang, X., Wu, X., and Hu, H. (2019). A review of Listeria monocytogenes contamination in food and environmental samples, and its antimicrobial resistance. Food control. 98, 391–399. doi:10.1016/j.foodcont.2018.11.045
Wei, X., Hassen, A., McWilliams, K., Pietrzen, K., Chung, T., Acevedo, M. M., et al. (2024). Genomic characterization of Listeria monocytogenes and Listeria innocua isolated from milk and dairy samples in Ethiopia. BMC Genomic Data 25 (1), 12. doi:10.1186/s12863-024-01195-0
Wein, T., Wang, Y., Barz, M., Stücker, F. T., Hammerschmidt, K., Dagan, T., et al. (2021). Essential gene acquisition destabilizes plasmid inheritance. PLoS Genet 17 (7), e1009656. doi:10.1371/journal.pgen.1009656
World Health Organization (WHO) (2015). Estimates of the global burden of foodborne diseases: foodborne disease burden epidemiology reference group 2007-2015. Geneva, Switzerland: WHO.
Zhang, F., Zhao, S., Ren, C., Zhu, Y., Zhou, H., Lai, Y., et al. (2018). CRISPRminer is a knowledge base for exploring CRISPR-Cas systems in microbe and phage interactions. Commun. Biol. 1, 180. doi:10.1038/s42003-018-0184-6
Keywords: antibiotic resistance genes, dairy products, Listeria monocytogenes, mobile genetic elements, virulence genes
Citation: Kinde MZ, Kerisew B, Eshetu T and Gessese AT (2025) Genomic analysis of Listeria monocytogenes strains from dairy products in Ethiopia. Front. Bioinform. 5:1572241. doi: 10.3389/fbinf.2025.1572241
Received: 06 February 2025; Accepted: 31 March 2025;
Published: 16 April 2025.
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
Anna Giammanco, University of Palermo, ItalyYoutao Lu, University of Pennsylvania, United States
Bo-Wei Zhao, Chinese Academy of Sciences (CAS), China
Copyright © 2025 Kinde, Kerisew, Eshetu and Gessese. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mebrie Zemene Kinde, emVtZW5lbWVicmllQGdtYWlsLmNvbQ==