 Anas Al-okaily
Anas Al-okaily Abdelghani Tbakhi
Abdelghani Tbakhi- 1Department of Cell Therapy and Applied Genomics, King Hussein Cancer Center, Amman, Jordan
- 2Department of Pathology and Molecular Medicine, McMaster University, Hamilton, ON, Canada
Suffix trees are fundamental data structures in stringology and have wide applications across various domains. In this work, we propose two linear-time algorithms for indexing strings under each internal node in a suffix tree while preserving the ability to track similarities and redundancies across different internal nodes. This is achieved through a novel tree structure derived from the suffix tree, along with new indexing concepts. The resulting indexes offer practical solutions in several areas, including DNA sequence analysis and approximate pattern matching.
1 Introduction
Numerous string-processing problems arise in several scientific fields, including biology and medicine. These problems include exact and approximate pattern matching, motif search, lowest common ancestor queries, and the detection of tandem repeats. The inputs for such problems can range from small documents and databases to DNA sequences and large-scale corporate data. To address string problems more efficiently, several data structures have been designed and are commonly used, including suffix trees (Weiner, 1973; McCreight, 1976; Ukkonen, 1995), suffix arrays (Abouelhoda et al., 2004), and the FM-index (Ferragina and Manzini, 2000).
Constructing suffix trees, suffix arrays, and FM-indexes can all be achieved in linear time and space. Although building suffix trees incurs a higher constant-factor overhead than building suffix arrays and FM-indexes, their structure is more flexible and dynamic. This flexibility arises from the ability of suffix trees to identify systematic redundancies among the suffixes in the input data—capabilities not offered by suffix arrays or FM-indexes. For instance, suffix trees make it easy to observe that a subtree rooted at an internal node is isomorphic or partially isomorphic to subtrees rooted at other internal nodes. Such structural observations are not possible with suffix arrays or FM-indexes. Once these redundancies are identified and abstracted, complex string problems can be solved more efficiently than using suffix arrays, FM-indexes, or even the standard suffix tree representation.
In this work, we introduce two algorithms that index strings under all internal nodes in suffix trees in linear time and space.
2 Methods
Let
Definition 1: Let
As an example, the

Figure 1. This diagram visualizes a suffix tree constructed from a string AGCCTAATTTAACTAAG$ using https://hwv.dk/st/?AGCATAATTTAACTAAG$. Each node is annotated with a unique identifier enclosed in a circle for ease of reference. The edges between nodes are labeled with substrings that represent segments of the original string along distinct suffix paths. Leaf nodes—those without children—are marked with red integers, indicating the starting positions (suffix indexes) of the corresponding suffixes in the original string. Green dotted arrows denote suffix links, which connect internal nodes according to standard suffix tree construction rules.
Observe the following properties:
Therefore, to compute this indexing scheme and traverse the suffix links recursively, the following tree structure must be designed and constructed.
2.1 Okaily-Sheehy-Huang-Rajasekaran (OSHR) tree structure
Given ST, the structure of the OSHR tree is defined as follows (the acronym “OSHR” is explained in the Acknowledgments section):
The directed edges in the OSHR tree, which are the reverse of suffix links, correspond to a simplified form of Weiner links in ST (as defined by Wellnitz (2021), Apostolico and Cunial (2014), Belazzougui et al. (2020). Due to the construction properties of ST and its suffix links, the OSHR tree forms a directed acyclic graph. The construction of the OSHR tree is carried out by traversing ST, and at each visited internal node
The OSHR tree differs from the suffix-tour graph (Starikovskaya and Vildhøj., 2015) and the suffix link tree (Starikovskaya and Vildhøj., 2015); (Apostolico and Cunial., 2014); (Belazzougui et al., 2020). Unlike the suffix-tour graph, the OSHR structure is acyclic. Compared to the suffix link tree, the edges in the OSHR tree are unlabeled, they do not include the leaf nodes of ST, and its leaf nodes correspond to internal nodes in ST that have no incoming suffix links.
2.2 Okaily-Tbakhi (OT) indexing
To identify all similarities and redundancies of strings under different internal nodes in an ST, a post-order traversal of the OSHR tree is required, during which both the ST and the OSHR tree structures are utilized.
Definition 2: We denote those strings, such as suffixes defined in the
The types of strings considered under each internal node
Definition 3: OT indexing (or OT processing) refers to the process of indexing or processing strings under each internal node (based on the ST structure, denoted as node
As a simple example, consider the task of performing OT indexing on the suffixes under each internal node (the set of suffixes as defined by function
Since AATTTAACTAAG$, and suffix 8 to TTAACTAAG$) and the base suffixes at this node are also
At this node, TAACTAAG$ and suffix 10 to AACTAAG$), and the base suffixes are again
For this node, AAG$). So, the suffix that now requires indexing is suffix 14 as the others were already indexed during the OT indexing process of nodes 15 and 21. Therefore, append 14 to
The second part of this work introduces the concepts of base suffixes and base paths and proposes both linear and nonlinear algorithms to identify them under each internal node in the ST.
2.3 Base suffixes
We begin by defining base suffixes and then describe linear and nonlinear algorithms for finding base suffixes under each internal node in the ST.

Algorithm 1. Non-Trivial algorithm for identifying base suffixes.
Definition 4: A base suffix is a suffix that occurs under an internal node in the ST structure, denoted as node
The examples from Figure 1 help illustrate the concept of base suffix. The base suffixes for node 26 are the set 14 (base suffix 14 corresponds to AAG$). The base suffixes for node 23 are {11, 15, 6, 1, 4} (base suffix 11 corresponds to ACTAAG$, 15 to AG$, 6 to ATTTAACTAAG$, 1 to GCATAATTTAACTAAG$, and 4 to TAATTTAACTAAG$). Because node 20 has no incoming suffix links CTAAG$, 16 to G$, and 7 to TTTAACTAAG$). Node 12 has no base suffixes as all suffixes under it are already covered under nodes of
Definition 5: If
For example, suffix 8 is a base suffix for node 15 (corresponds to TTAACTAAG$, starting from node 15 and ending at leaf node 6). The extended suffixes corresponding to this base suffix are the occurrences of TTAACTAAG$ under node 26 (ending at leaf node 11) and under the root node (ending at leaf node 10).
Observation 1: Based on definitions 4 and 5, the upper bound on the number of extended suffixes for any base suffix is
Observation 2: Based on definitions 4 and 5 and Observation 1, the base suffixes under all internal nodes in ST must be
In the example provided in Section 2.2, once the traversal reaches the root node, the
Therefore, once a base suffix is processed or indexed, this processing or indexing can be applied implicitly to all

Algorithm 2. Non-Trivial algorithm for identifying base suffixes.

Algorithm 3. Linear algorithm for finding base suffixes.
2.3.1 Finding base suffixes
To find base suffixes under each internal node in ST, we present four approaches: a trivial algorithm with
Trivially, all base suffixes under each internal node can be identified using the following algorithm. Build the OSHR tree (to mainly generate the
Given Observation 2, the following non-trivial algorithm, which requires auxiliary
The second non-trivial algorithm achieves
The linear algorithm was motivated by Observation 2. As the total number of base suffixes across all internal nodes in ST is equal to
Definition 6: Let
As shown in Figure 1, node 6 is a reference leaf node for node 21 and node 21 is an inbetween node for node 6.
Note that a reference leaf node can be associated with
Definition 7: Let
As illustrated in Figure 1, node 20 is a reference internal node for node 23 and node 23 is an inbetween node for node 20.
A reference internal node may have
The linear algorithm derives and identifies each base suffix in constant time using the inbetween nodes, reference leaf nodes, and reference internal nodes as stated in Algorithm 3. Since the upper bound on the number of reference leaf nodes and reference internal nodes is
Theorem 1. Finding all base suffixes under all internal nodes in a ST can be achieved in linear time and space
Once the base suffixes have been identified for each internal node in an ST in linear time, let us OT index the
After finding the base suffixes under all internal nodes in an ST in linear time, several applications become feasible, particularly when combined with OT indexing. One such application is illustrated by the following example.
Let the OT indexing of the base suffixes in an ST be applied to solve the problem of exact pattern matching (which is a fundamental problem in biological applications such as read alignment, motif search, and genome annotation). Suppose there is a pattern that exactly matches one of the base suffixes under some node
2.4 Base paths
The motivation for this indexing approach arises from the following observations. First, the primary source of complexity in a tree structure lies in the branching caused by internal nodes. Second, the tails of suffixes (i.e., the labels between a leaf node and its parent) are often very long, making their processing computationally expensive. Third, if a process reaches an internal node whose children are all leaf nodes, the computational cost for handling these leaves is bounded by the alphabet size
Next, we define the concept of base paths and present algorithms for identifying base paths under each internal node in an ST, with both linear and nonlinear costs.

Algorithm 4. Non-trivial algorithm for finding base paths.
Definition 8: Let
For example, in Figure 1 (noting that the
Definition 9: If
For instance, the path between the root and node 25 is an extended path of the base path between nodes 23 and 14.
Observation 3: Based on definitions 8 and 9, the upper bound on the number of extended paths for any base path is
Observation 4: Based on definitions 8 and 9 and Observation 3, any path from the root node to an internal node can be the final extended path of a base path; hence, the total number of base paths is bounded by

Algorithm 5. Linear algorithm for finding base paths.
2.6 Finding base paths
All base paths in an ST can be identified using a straightforward (trivial) algorithm with time complexity
Algorithm 4, which is analogous to Algorithm 2, can find base paths under all internal nodes with a time complexity of
Since the total number of base paths is no more than
Theorem 2: All base paths under all internal nodes in an ST can be found in linear time and space
Once base paths are computed for each internal node in an ST, any index or process
The following is an example of OT indexing base paths. Let the OT index be constructed to resolve the pattern matching problem, as discussed in the example at the end of Section 2.3.1, where the pattern here is an exact match of one of the base paths under node
3 Results
To assess the correctness and effectiveness of the proposed algorithms, we evaluated them on the genomes of the following organisms, with genome sizes ranging from
In the preprocessing step, header lines and newline characters were removed from each FASTA file, and all lowercase nucleotides were converted to uppercase. As a result, each genome was converted to a single-line sequence with all nucleotides in uppercase. The Python script used for this preprocessing step is available at the repository: https://github.com/aalokaily/Finding_base_suffixes_and_base_paths_in_suffix_trees.
All five algorithms presented in this study were implemented in Python and are publicly available in the aforementioned repository. Notably, the non-trivial algorithm (Algorithm 2) was excluded from the comparative analysis because it is both theoretically and empirically slower than the other non-trivial algorithm (Algorithm 1), as demonstrated by preliminary tests (data not shown). Regarding base suffix identification, the results obtained using the linear algorithm (Algorithm 3) perfectly matched those of its non-trivial counterpart (Algorithm 1) for each internal node in the ST. Across all tested genomes, the total number of base suffixes under all internal nodes is equal to

Table 1. Results from the evaluation and comparison of algorithms 1 and 3 (for base suffix identification) and algorithms 4 and 5 (for base path identification).
Finally, a statistical analysis was conducted to evaluate the scalability and performance differences among the proposed algorithms. The execution time for each algorithm was plotted against the genome size, as shown in Figure 2. Linear regression confirmed a strong linear relationship between genome size and runtime for linear algorithms 3 and 5
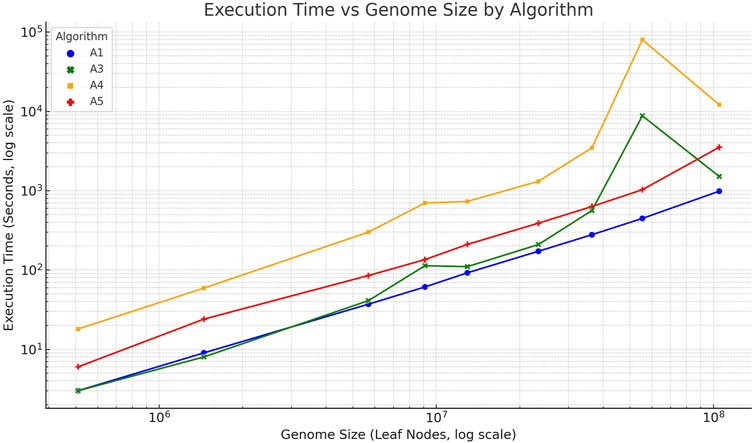
Figure 2. Execution time (log–log scale) of algorithms 1 (blue), 3 (green), 4 (orange), and 5 (red) plotted against genome size (number of nucleotide/leaf nodes). The linear trend observed for algorithms 3 and 5 confirms their linear-time behavior, while algorithms 1 and 4 exhibit superlinear growth.
4 Conclusion
The primary contribution of the OT indexing of base suffixes and base paths is their linear time and space cost for indexing all suffixes and paths under all internal nodes in an ST. This property is not achievable using existing suffix tree construction algorithms (such as Ukkonen’s algorithm (Ukkonen., 1995) or McCreight’s algorithm (McCreight, 1976)) or other approaches related to suffix trees. The resulting linear OT index enables indexing all suffixes or paths under all internal nodes with a complexity factor of
Data availability statement
Source code of the algorithms are available at https://github.com/aalokaily/Finding_base_suffixes_and_base_paths_in_suffix_trees. Further inquiries can be directed to the corresponding author.
Author contributions
AA: Conceptualization, Investigation, Methodology, Software, Validation, Writing – original draft, Writing – review and editing. AT: Investigation, Project administration, Supervision, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The term “OSHR” tree is derived from the last names of the first author and his PhD committee members at the University of Connecticut, Department of Computer Science, in 2016. The committee included Chun-Hsi Huang (Major Advisor), Sanguthevar Rajasekaran, and Don Sheehy. The name Okaily–Sheehy–Huang–Rajasekaran (OSHR) honors their kind, influential, and professional guidance throughout the first author’s doctoral studies. Additionally, the abbreviation “OT” corresponds to the last names of the authors of this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abouelhoda, M. I., Kurtz, S., and Ohlebusch, E. (2004). Replacing suffix trees with enhanced suffix arrays. J. discrete algorithms 2, 53–86. doi:10.1016/s1570-8667(03)00065-0
Apostolico, A., and Cunial, F. (2014). Suffix trees and arrays. J. Encycl. Algorithms, 1–10. doi:10.1007/978-3-642-27848-8_627-1
Belazzougui, D., Cunial, F., Kärkkäinen, J., and Mäkinen, V. (2020). Linear-time string indexing and analysis in small space. ACM Trans. Algorithms (TALG) 16, 1–54. doi:10.1145/3381417
Ferragina, P., and Manzini, G. (2000). “Opportunistic data structures with applications,” in Proceedings 41st annual symposium on foundations of computer science (IEEE), 390–398.
Guo, P., Li, Y., Wang, R., Chen, X., Kim, S., and Park, H. J. (2024). Deep neural network learning biological condition information refines gene-expression-based cell subtypes. Briefings Bioinforma. 25, bbad512. doi:10.1093/bib/bbad512
Hu, J., Wang, Z., Sun, Z., Hu, B., Ayoola, A. O., Liang, F., et al. (2024). NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biol. 25, 107. doi:10.1186/s13059-024-03252-4
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with bwa-mem. arXiv 1303.3997. doi:10.48550/arXiv.1303.3997
McCreight, E. M. (1976). A space-economical suffix tree construction algorithm. J. ACM (JACM) 23, 262–272. doi:10.1145/321941.321946
Starikovskaya, T., and Vildhøj, H. W. (2015). A suffix tree or not a suffix tree? J. Discrete Algorithms 32, 14–23. doi:10.1016/j.jda.2015.01.005
Ukkonen, E. (1995). On-line construction of suffix trees. Algorithmica 14, 249–260. doi:10.1007/bf01206331
Wang, S., Dong, K., Liang, D., Zhang, Y., Li, X., and Song, T. (2024a). Mippis: protein–protein interaction site prediction network with multi-information fusion. BMC Bioinforma. 25, 345. doi:10.1186/s12859-024-05964-7
Wang, T., Zhang, Y., Wang, H., Zheng, Q., Yang, J., Zhang, T., et al. (2024b). Fast and accurate dnaseq variant calling workflow composed of lush toolkit. Hum. Genomics 18: (1), 114. doi:10.1186/s40246-024-00666-w
Weiner, P. (1973). “Linear pattern matching algorithms,” in 14th annual symposium on Switching and Automata Theory (swat 1973) (IEEE), 1–11.
Wellnitz, P. (2021). Counting patterns in strings and graphs. Saarbrücken, Germany: Saarländische Universitäts-und Landesbibliothek. Ph.D. thesis.
Yue, J., Peng, B., Chen, Y., Jin, J., Zhao, X., Shen, C., et al. (2024a). 3dsmiles-gpt: 3d molecular pocket-based generation with token-only large language model. Chem. Sci. 15 (—), 13727–13740. doi:10.1039/d4sc03744h
Yue, J., Peng, B., Chen, Y., Jin, J., Zhao, X., Shen, C., et al. (2024b). Unlocking comprehensive molecular design across all scenarios with large language model and unordered chemical language. Chem. Sci. 15, 13727–13740. doi:10.1039/D4SC03744H
Zhao, B.-W., Su, X.-R., Hu, P.-W., Ma, Y. P., Zhou, X., and Hu, L. (2022). A geometric deep learning framework for drug repositioning over heterogeneous information networks. Briefings Bioinforma. 23, bbac384. doi:10.1093/bib/bbac384
Zhao, B.-W., Su, X.-R., Yang, Y., Li, D.-X., Li, G.-D., Hu, P.-W., et al. (2024). A heterogeneous information network learning model with neighborhood-level structural representation for predicting lncrna–mirna interactions. Comput. Struct. Biotechnol. J. 22, 2924–2933. doi:10.1016/j.csbj.2024.06.032
Keywords: suffix trees, strings indexing, approximate pattern matching, reads alignment, motif search
Citation: Al-okaily A and Tbakhi A (2025) A novel linear indexing method for strings under all internal nodes in a suffix tree. Front. Bioinform. 5:1577324. doi: 10.3389/fbinf.2025.1577324
Received: 15 February 2025; Accepted: 07 August 2025;
Published: 04 September 2025.
Edited by:
Kang Ning, Huazhong University of Science and Technology, ChinaReviewed by:
Bo-Wei Zhao, Zhejiang University, ChinaOsman Ali Sadek Ibrahim, Minia University, Egypt
Alok Misra, Lovely Professional University, Phagwara, Punjab, India
Copyright © 2025 Al-okaily and Tbakhi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anas Al-okaily , YWEuMTI2ODJAa2hjYy5qbyYjeDAyMDBhOw==