 Sonar Soni Panigoro1*
Sonar Soni Panigoro1* Rafika Indah Paramita2,3,4*
Rafika Indah Paramita2,3,4* Fadilah Fadilah2,3,4
Fadilah Fadilah2,3,4 Septelia Inawati Wanandi4,5,6
Septelia Inawati Wanandi4,5,6 Aisyah Fitriannisa Prawiningrum3Linda Erlina2,3,4Wahyu Dian Utari2,3Ajeng Megawati Fajrin2
Aisyah Fitriannisa Prawiningrum3Linda Erlina2,3,4Wahyu Dian Utari2,3Ajeng Megawati Fajrin2- 1Surgical Oncology Division, Department of Surgery, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia
- 2Department of Medical Chemistry, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia
- 3Bioinformatics Core Facilities - IMERI, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia
- 4Master’s Programme in Biomedical Sciences, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia
- 5Department of Biochemistry and Molecular Biology, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia
- 6Molecular Biology and Proteomics Core Facilities-IMERI, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia
1 Introduction
Breast cancer remains the most prevalent form of cancer worldwide. Based on data from the Global Cancer Observatory (GLOBOCAN) in 2020, breast cancer ranks first in the category of new cases of cancer worldwide (11.7%) and ranks fifth as a cause of death (6.9%) (Sung et al., 2021). Mutations in the BRCA1 and BRCA2 genes have been extensively studied and are known to be associated with an increased risk of developing the disease (Momozawa et al., 2022). While BRCA1 and BRCA2 mutations are well-known germline mutations associated with an increased risk of breast cancer, several other non-BRCA genes can also harbor germline mutations linked to breast cancer susceptibility—for example, TP53, PTEN, STK11, PALB2, CHEK2, ATM, RAD51C, and RAD51D genes (Wang et al., 2021). The aforementioned genes play a crucial role in DNA repair, cell cycle regulation, and the inhibition of tumor formation (Yang et al., 2022). Identifying germline mutations, not only in BRCA genes but also in other genes, can have important implications for both affected individuals and their families, allowing for prevention and treatment strategies (Wang et al., 2018; Momozawa et al., 2018). In this cross-sectional study, we aimed to identify non-BRCA germline mutations found in breast cancer patients using a less-invasive method that could serve as a biomarker for breast cancer subtyping.
2 Methods
2.1 Sample collection and DNA purification
A total of 28 female individuals diagnosed with breast cancer participated in this study, and blood samples were obtained from each participant. DNA extraction was conducted using the Genomic DNA Mini Kit® (Geneaid, New Taipei City, Taiwan), following the manufacturer’s instructions. The purity of DNA isolates was assessed by measuring the 260/280 absorbance ratio using a NanoDrop instrument (Thermo Fisher Scientific, Waltham, MA, United States). The quantification of DNA isolates was performed using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, United States) on a Qubit® 4.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, United States).
2.2 Library preparation and sequencing
Library preparation was performed utilizing the Illumina AmpliSeq™ Cancer Hotspot Panel v2 (Illumina®, United States). The first step involved the amplification of specific areas within the DNA sample. The amplicons were subsequently subjected to partial digestion using the FuPa reagent. The indexes were ligated using the Ligate program on a thermal cycler. In order to purify the libraries, 30 μL of Agencourt AMPure XP beads (Beckman Coulter™, United States) was added to the reaction mixtures.
Amplification techniques were implemented in order to ensure a sufficient quantity of the libraries. The second round of purification was subsequently performed twice to remove high molecular weight DNA and excess primers, using Agencourt AMPure XP beads (Beckman Coulter™, United States). The libraries were diluted to a final loading concentration and subsequently subjected to sequencing utilizing the Illumina MiSeq technology. Sequencing yielded paired-end libraries in FASTQ format, with a read length of 150 base pairs (bp) for both ends. The data sequences have been deposited in the Sequence Read Archive (SRA) database under BioProject accession number PRJNA998562.
2.3 Quality control and data trimming
Quality control of the FASTQ data was conducted to evaluate the quality of each sample’s raw reads. FastQC software (Andrews, 2010) was used to perform FASTQ quality assessment. The total number of raw bases and Q30 percentage were determined using the q30 Python programs (https://github.com/dayedepps/q30/tree/master). If the quality of sequence reads was poor, quality-improvement steps were taken. Trimmomatic (Bolger et al., 2014) was used to trim the low-quality reads and remove adapters (ILLUMINACLIP: NexteraPE-PE.FA: 2:30:10, LEADING: 3, TRAILING: 3, SLIDINGWINDOW: 4:15, and MINLEN: 35). Read alignment was performed using BWA-MEM (Li and Durbin, 2009), with GRCh38. p13 as the human reference genome. After alignment, the amplicon mean depth, coverage uniformity, and the percentage of on-target rate were calculated using an in-house script containing Mosdepth (Pedersen and Quinlan, 2018), SAMtools (Danecek et al., 2021), and BEDTools (Danecek et al., 2021) software. The command-line scripts used to calculate the Q30 percentage, amplicon mean depth, coverage uniformity, and the percentage of on-target rate are provided in Supplementary Material.
2.4 Variant calling analysis
Variant calling analysis was performed to find likely pathogenic and pathogenic variants in all samples. The workflow followed the methods described by Panigoro et al. (2022) and included read alignment using BWA (Li and Durbin, 2009), SAM-to-BAM conversion using SAMTools (Li et al., 2009), variant calling using GATK (McKenna et al., 2010), and variant annotation using SnpEff and SnpSift (Cingolani et al., 2012). Germline variant classification was conducted using VarSome (Kopanos et al., 2019) (https://varsome.com/), which applies the ACMG classification guidelines. A total score is computed by summing the points from pathogenic rules and subtracting the points from benign rules. The total score is then compared with predefined thresholds to determine the final verdict: pathogenic if greater than or equal to 10, likely pathogenic if between 6 and 9 inclusive, uncertain significance if between 0 and 5, likely benign if between −6 and −1, and benign if less than or equal to −7. The command-line scripts used for variant calling analysis are available on GitHub: https://github.com/fikaparamita04/variant-calling. The most frequently observed pathogenic variants were visualized using MutationMapper (Vohra and Biggin, 2013) (https://www.cbioportal.org/mutation_mapper).
3 Data analysis
3.1 Patients
We successfully collected blood samples from 28 patients diagnosed with breast cancer at Cipto Mangunkusumo National Hospital, Jakarta. The patients ranged in age from 40 to 71 years (Table 1). The patients were categorized into four subtypes, namely, Luminal A, Luminal B, HER2-positive and triple-negative breast cancer (TNBC), with the total number of patients being 8, 9, 7, and 4, respectively. Among the subjects, four patients were diagnosed at stage IV.
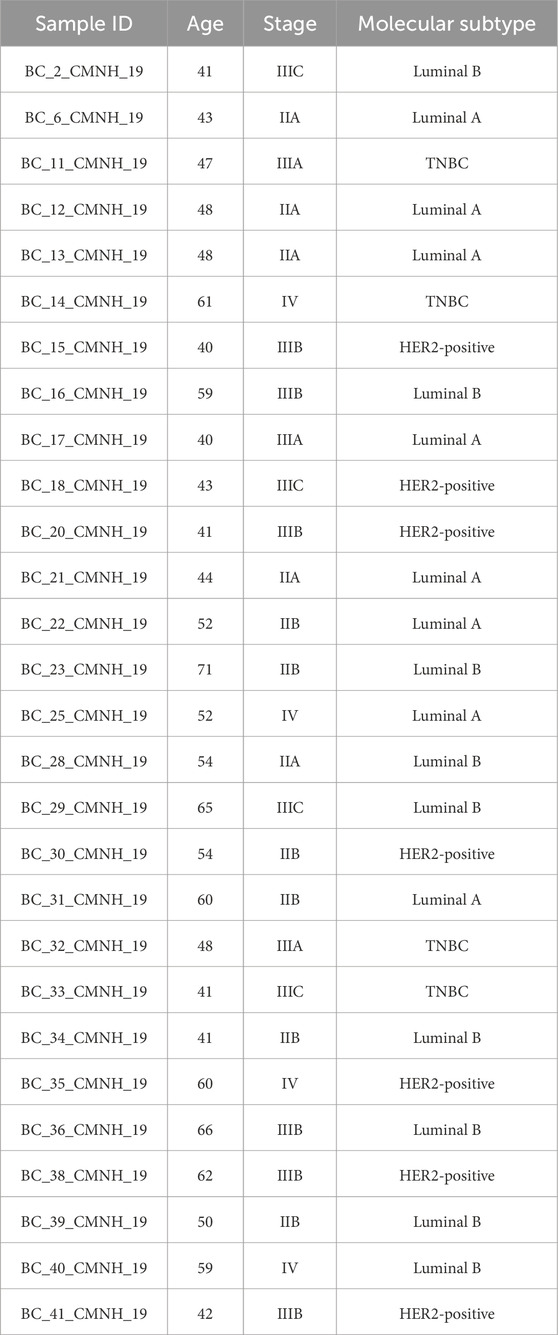
Table 1. Descriptive information of the patients.
3.2 Quality control of FASTQ data
Raw FASTQ data were quality-checked to ensure high sequencing quality. Given that this was a targeted sequencing study, we evaluated the Q30 percentage, average amplicon depth, coverage uniformity, and target level percentage (Table 2). The Q30 result of 97.71% ± 0.44 indicates high-quality sequencing. The average amplicon depth was also strong, with a score of 1,076 ± 256.36, although variability between samples was relatively high. Coverage uniformity, which measures how evenly sequencing reads are distributed across the genome or a specific region of interest, was excellent across all samples. All samples showed a coverage uniformity score of 1, or close to 1 (0.9901 ± 0.01), indicating that all target bases were covered to the same extent, without regions of significantly higher or lower read depth. The target-level percentage, which reflects the proportion of bases within the targeted regions that were successfully sequenced, was also high at 95.57% ± 0.57. The raw data files in FASTQ format have been archived in the BioProject database under accession number PRJNA998562. These data may serve as a potentially valuable resource for screening gene mutation markers in breast cancer and could aid in predicting treatment efficacy related to specific mutations.
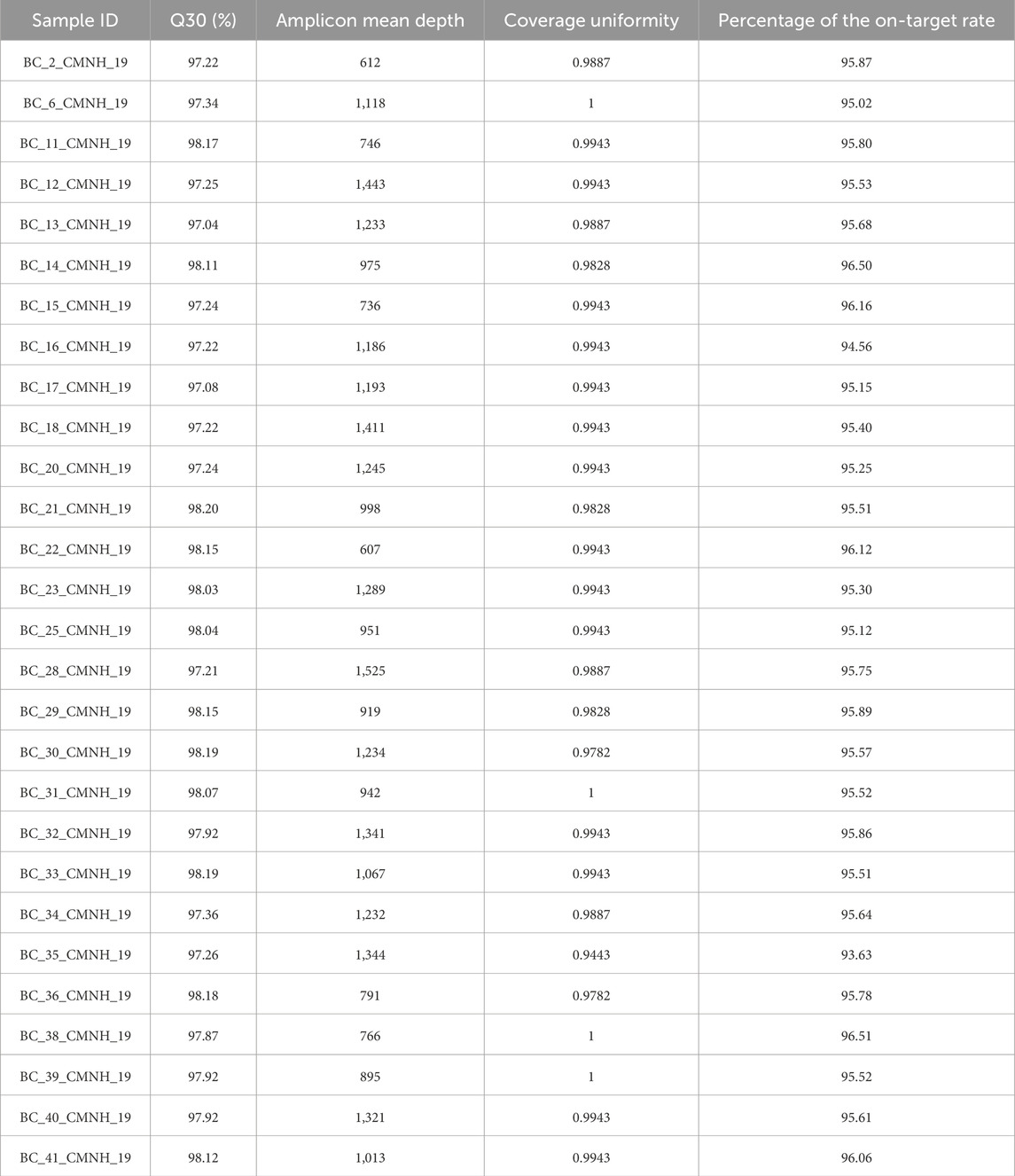
Table 2. Descriptive information of targeted sequencing evaluations.
3.3 Variant calling analysis
We found the highest germline frameshift mutation in the FBXW7 gene (4:g.152324246del), with a frequency of approximately 35.7%. This variant was predicted as likely pathogenic by VarSome (prediction score = 9), due to the loss of protein functions (Figure 1). Interestingly, this mutation was found in all Luminal B and one HER2-positive patient. FBXW7 is a tumor suppressor that modulates the degradation of oncogenic substrates, including c-Jun, c-Myc, the Notch1 intracellular domain (ICD), and cyclin E, by acting as the substrate recognition protein within the Skp1–Cullin–F-box (SCF) ubiquitin ligase complex. Deletion of chromosome 4q3, which encompasses FBXW7, occurs in approximately 30% of primary breast tumors (Meyer et al., 2020).
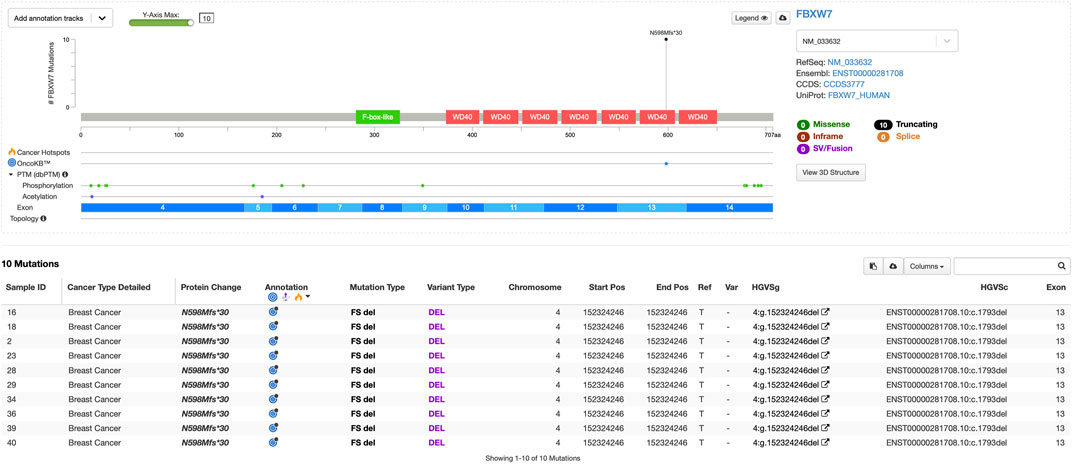
Figure 1. Lollipop plot of germline pathogenic variants in the FBXW7 gene (visualization using MutationMapper).
In line with previous studies, the deletion mutation in the FBXW7 gene closely resembles the human breast cancer luminal B subtype, characterized by ERα+, PR-, and elevated Ki67 staining (Meyer et al., 2020). Furthermore, Luminal B tumors exhibit the lowest FBXW7 mRNA expression among breast cancer subtypes. Lower FBXW7 expression is associated with a high Ki-67 labeling index and positive cyclin E protein expression, both indicators of proliferation. Breast cancer patients with the greatest FBXW7 gene expression have a longer disease-free survival rate (Yeh et al., 2018). The process by which FBXW7 regulates breast cancer growth, cell cycle, and metastasis involves many signaling pathways and gene interactions. For example, FBXW7-deficient breast tumors inhibit the NF-κB signaling pathway, which normally involves E3 ubiquitin ligase binding and degradation. This inhibition results in enhanced NF-κB DNA-binding activity, promoting tumor development and metastasis (Chen et al., 2023).
As shown in Figure 1, the mutation found in the FBXW7 gene is also reported in the OncoKB database (Chakravarty et al., 2017). According to the database, FBXW7 N598Mfs*30 is a truncating mutation in a tumor suppressor gene and is, therefore, considered likely oncogenic. There is promising scientific and anecdotal clinical evidence supporting the use of lunresertib and camonsertib in patients with FBXW7-mutated solid tumors. Lunresertib is an orally available, small-molecule PKMYT1 inhibitor, while camonsertib is an orally available, small-molecule ATR inhibitor. In the Phase I MYTHIC trial of lunresertib plus camonsertib in patients with advanced tumors harboring CCNE1 amplifications, FBXW7 deleterious mutations, or PPP2R1A deleterious mutations, the lunresertib + camonsertib cohort (n = 59 [n = 17 endometrial; n = 13 colorectal; n = 11 ovarian; n = 3, breast; n = 3, lung; n = 12, other]) showed an overall response rate of 23.6% among all evaluable patients across tumor types (n = 55) (Yap et al., 2023). However, future studies with larger patient populations and integration of multi-omics approaches are needed for precise subtyping and personalized therapy. We hope that our small contribution can help advance precision therapy for breast cancer, particularly in Indonesia.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at https://www.ncbi.nlm.nih.gov/, PRJNA998562.
Ethics statement
The studies involving humans were approved by the Faculty of Medicine Universitas Indonesia Ethical Committee (approval number: 0450/UN2.F1/ETIK/2018). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
SP: Writing – original draft, Writing – review and editing, Conceptualization, and Supervision. RP: Writing – review and editing, Formal Analysis, Methodology, Writing – original draft, Software, Conceptualization, and Data curation. FF: Writing – original draft and Supervision. SW: Writing – original draft and Supervision. AP: Methodology, Software, and Writing – original draft. LE: Writing – original draft, Data curation, and Methodology. WU: Writing – original draft, Software, and Methodology. AF: Writing – original draft and Methodology.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by PUTI Pascasarjana 2023 Grant from Universitas Indonesia [grant number: NKB-150/UN2. RST/HKP.05.00/2023].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbinf.2025.1620025/full#supplementary-material
References
Andrews, S. (2010). FastQC - a quality control tool for high throughput sequence data. Babraham Bioinforma. Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (Accessed January 15, 2025).
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi:10.1093/bioinformatics/btu170
Chakravarty, D., Gao, J., Phillips, S. M., Kundra, R., Zhang, H., Wang, J., et al. (2017). OncoKB: a precision oncology knowledge base HHS public access. Available online at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5586540/pdf/nihms897314.pdf.
Chen, S., Leng, P., Guo, J., and Zhou, H. (2023). FBXW7 in breast cancer: mechanism of action and therapeutic potential, 42. Journal of Experimental and Clinical Cancer Research. BioMed Central Ltd.
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly. (Austin). 6 (2), 80–92. doi:10.4161/fly.19695
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience 10 (2), giab008–4. doi:10.1093/gigascience/giab008
Kopanos, C., Tsiolkas, V., Kouris, A., Chapple, C. E., Albarca Aguilera, M., Meyer, R., et al. (2019). “VarSome: the human genomic variant search engine.” Bioinformatics. Editor J. Wren 35, 1978–1980. doi:10.1093/bioinformatics/bty897
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi:10.1101/gr.107524.110
Meyer, A. E., Furumo, Q., Stelloh, C., Minella, A. C., and Rao, S. (2020). Loss of Fbxw7 triggers mammary tumorigenesis associated with E2F/c-Myc activation and Trp53 mutation. Neoplasia (United States) 22 (11), 644–658. doi:10.1016/j.neo.2020.07.001
Momozawa, Y., Iwasaki, Y., Parsons, M. T., Kamatani, Y., Takahashi, A., Tamura, C., et al. (2018). Germline pathogenic variants of 11 breast cancer genes in 7,051 Japanese patients and 11,241 controls. Nat. Commun. 9 (1), 4083. doi:10.1038/s41467-018-06581-8
Momozawa, Y., Sasai, R., Usui, Y., Shiraishi, K., Iwasaki, Y., Taniyama, Y., et al. (2022). Expansion of cancer risk profile for BRCA1 and BRCA2 pathogenic variants. JAMA Oncol. 8 (6), 871–878. doi:10.1001/jamaoncol.2022.0476
Panigoro, S. S., Paramita, R. I., Siswiandari, K. M., and Fadilah, F. (2022). Targeted sequencing of germline breast cancer susceptibility genes for discovering pathogenic/likely pathogenic variants in the jakarta population. Diagnostics 12 (9), 2241. doi:10.3390/diagnostics12092241
Pedersen, B. S., and Quinlan, A. R. (2018). Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics 34 (5), 867–868. doi:10.1093/bioinformatics/btx699
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71 (3), 209–249. doi:10.3322/caac.21660
Vohra, S., and Biggin, P. C. (2013). Mutationmapper: a tool to aid the mapping of protein mutation data. PLoS One 8 (8), e71711. doi:10.1371/journal.pone.0071711
Wang, Y. A., Jian, J. W., Hung, C. F., Peng, H. P., Yang, C. F., Cheng, H. C. S., et al. (2018). Germline breast cancer susceptibility gene mutations and breast cancer outcomes. BMC Cancer 18 (1), 315–13. doi:10.1186/s12885-018-4229-5
Wang, J., Singh, P., Yin, K., Zhou, J., Bao, Y., Wu, M., et al. (2021). Disease spectrum of breast cancer susceptibility genes. Front. Oncol. 11 (April), 663419–10. doi:10.3389/fonc.2021.663419
Yang, L., Xie, F., Liu, C., Zhao, J., Hu, T., Wu, J., et al. (2022). Germline variants in 32 cancer-related genes among 700 Chinese breast cancer patients by next-generation sequencing: a clinic-based, observational study. Int. J. Mol. Sci. 23 (19), 11266. doi:10.3390/ijms231911266
Yap, T. A., Schram, A., Lee, E. K., Simpkins, F., Weiss, M. C., LoRusso, P., et al. (2023). MYTHIC: first-in-human (FIH) biomarker-driven phase I trial of PKMYT1 inhibitor lunresertib (lunre) alone and with ATR inhibitor camonsertib (cam) in solid tumors with CCNE1 amplification or deleterious alterations in FBXW7 or PPP2R1A. Mol. Cancer Ther. 22 (12_Suppl. ment), PR008. doi:10.1158/1535-7163.targ-23-pr008
Keywords: breast cancer, FASTQ data, next-generation sequencing, non-BRCA sequencing panels, pathogenic mutation
Citation: Panigoro SS, Paramita RI, Fadilah F, Wanandi SI, Prawiningrum AF, Erlina L, Utari WD and Fajrin AM (2025) Germline mutation profiling of breast cancer patients using a non-BRCA sequencing panel. Front. Bioinform. 5:1620025. doi: 10.3389/fbinf.2025.1620025
Received: 29 April 2025; Accepted: 28 July 2025;
Published: 02 September 2025.
Edited by:
David W. Ussery, Oklahoma State University, United StatesReviewed by:
Anderson Rodrigues dos Santos, Federal University of Uberlandia, BrazilYang Zhang, Carnegie Mellon University, United States
Copyright © 2025 Panigoro, Paramita, Fadilah, Wanandi, Prawiningrum, Erlina, Utari and Fajrin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sonar Soni Panigoro, c29uYXIuc29uaUB1aS5hYy5pZA==; Rafika Indah Paramita, cmFmaWthaW5kYWhAdWkuYWMuaWQ=