 Shuang Zhao
Shuang Zhao He Zhang
He Zhang- General Hospital of Northern Theater Command of the Chinese People's Liberation Army, Shenyang, China
Background: This study aims to investigate potential biomarkers associated with NSCLC-BM and elucidate their regulatory roles in critical pathways involved in cerebral metastatic dissemination.
Methods: The identified DEGs were subjected to functional enrichment analysis. PPI networks were predicted using the STRING database and visualized with Cytoscape. Hub genes were subsequently screened from the PPI network to construct a transcription TF-miRNA regulatory network. Subsequent analyses included: survival analysis, immune infiltration assessment and comprehensive mutational profiling.
Results: Among the 56 identified DEGs, 19 were upregulated while 37 were downregulated. GOntology enrichment analysis revealed significant enrichment in immune response, signaling receptor binding, and extracellular region. KEGG pathway analysis demonstrated predominant involvement in cytokine-cytokine receptor interaction and chemokine signaling pathway. Through Cytoscape-based screening, we identified 10 hub genes: CD19, CD27, IL7R, SELL, CCL5, CCR5, PRF1, GZMK, GZMA, and TIGIT. The TF-miRNA regulatory network analysis uncovered 6 transcription factors (STAT5A/B, NFKB1, EGR1, RELA, and CTCF) and 4 miRNAs(hsa-miR-204, hsa-miR-148b, hsa-miR-618, and hsa-miR-103) as critical transcriptional and post-transcriptional regulators of DEGs.Integrated analyses including Kaplan-Meier survival curves, immune infiltration profiling, and comprehensive mutational analysis demonstrated significant associations with brain metastatic progression in the studied cohort.
Conclusion: This study provides novel biomarkers from a unique perspective for the diagnosis, prognosis, and development of molecular-targeted therapies or immunotherapies for brain metastasis in NSCLC.
Introduction
Lung cancer ranks as the second most commonly diagnosed malignancy worldwide, exceeded only by breast cancer in incidence. It represents the most frequent primary tumor type that metastasizes to the brain, followed by breast cancer and melanoma (Cagney et al., 2017). Non-small cell lung cancer (NSCLC) comprises approximately 85% of all lung cancer cases (Sung et al., 2021; Jonna and Subramaniam, 2019), with brain metastasis (BM) being particularly common in this subgroup. Between 10% and 20% of NSCLC patients present with BM at initial diagnosis (Waqar et al., 2018), and an additional 25%–40% will develop BM throughout the disease course (Page et al., 2020). The prognosis for NSCLC patients with BM remains poor, and symptomatic cases are often associated with rapid deterioration in quality of life (Matsui et al., 2022). Historical reports indicate a median survival of only 4–6 months (Cheng and Perez-Soler, 2018). More recent epidemiological studies show that 15%–20% of NSCLC patients are diagnosed with BM at initial presentation, a figure that increases to 25%–40% over time (Waqar et al., 2018; Nayak et al., 2012; Hubbs et al., 2010). This incidence is even higher among patients with stage IV adenocarcinoma, among whom 40%–50% have BM at diagnosis (Yang et al., 2019). NSCLC patients harboring EGFR or ALK mutations are especially prone to developing BM and exhibit a higher incidence of such events (Gillespie et al., 2023). While historical median survival was reported between several months to one year—and below 6 months without treatment (Ali et al., 2013)-contemporary series report improved outcomes, with a median survival of approximately 15 months in lung adenocarcinoma patients with BM (Sperduto et al., 2020). The Lung-molGPA index further stratifies prognosis, identifying a small subgroup (4%) of patients with scores of 3.5–4.0 who may achieve a median survival of nearly 4 years (Sperduto et al., 2017). This prognostic tool incorporates clinical variables such as age, performance status, number of metastases, and extracranial disease burden, alongside molecular markers including EGFR and ALK mutations (Sperduto et al., 2017). The pathogenesis of BM in NSCLC entails complex crosstalk among tumor cells, immune components, and the specialized brain tumor microenvironment (TME). Metastasis is not solely an intrinsic trait of certain tumors, but a multistep, multidimensional process shaped by mutational landscapes, epigenetic alterations, and growth factor signaling (Srinivasan et al., 2021). As illustrated in Figure 1, the metastatic cascade initiates with local invasion through the basement membrane of the primary lung tumor—a step involving epithelial-mesenchymal transition (EMT) and intravasation into blood or lymphatic vessels. This allows circulating tumor cells (CTCs) to circumvent host immune surveillance and survive in circulation. Nevertheless, the precise mechanisms driving BM remain inadequately characterized, impeding the development of more effective treatment approaches.
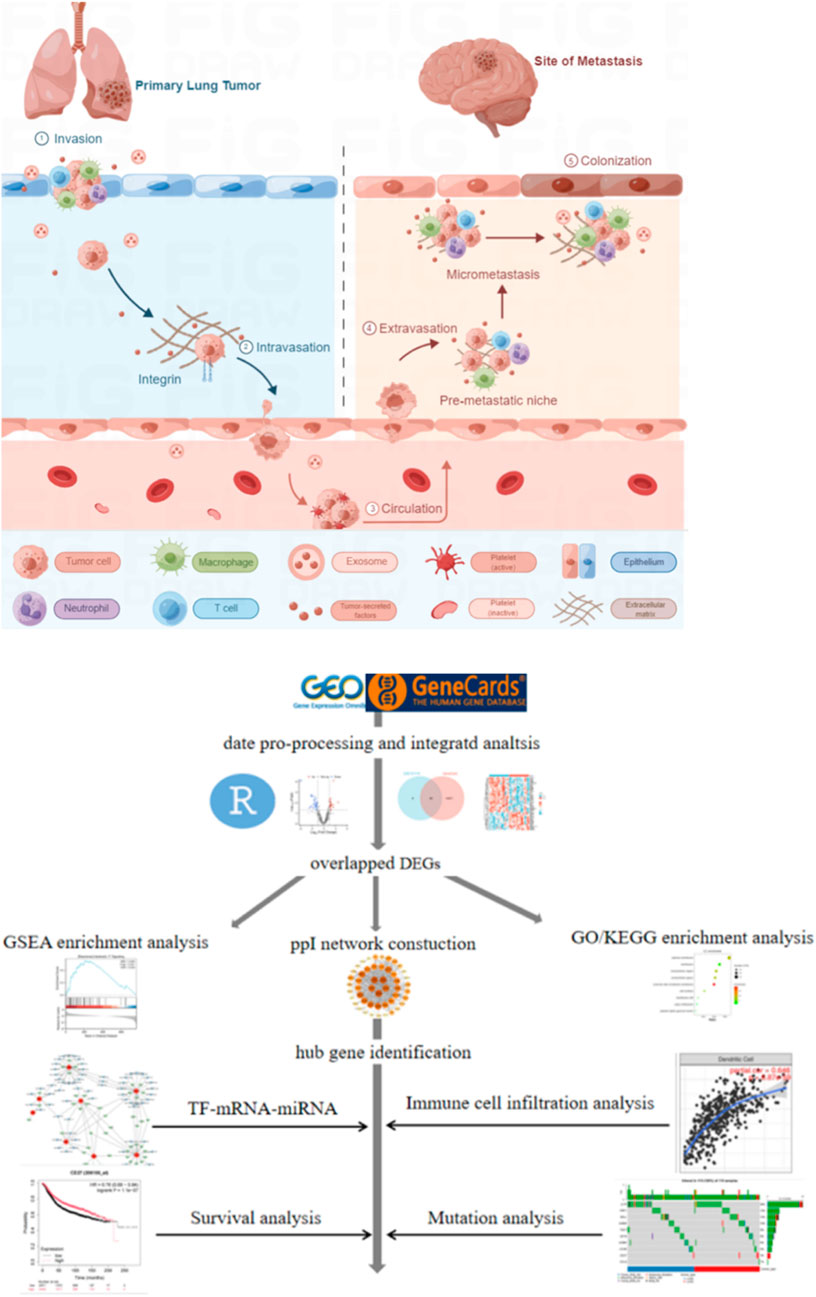
Figure 1. Schematic illustration of brain metastasis in NSCLC and the workflow of bioinformatic analysis.
Results
Identification of differentially expressed genes (DEGs)
Based on the GSE161116 microarray dataset (GPL19965 platform), this study employed a systematic bioinformatics pipeline for differentially expressed gene (DEG) identification. Data normalization: Raw expression profiles underwent background correction and quantile normalization via the RMA algorithm to eliminate batch effects. Differential analysis: The limma package was applied to identify DEGs between primary NSCLC (n = 14) and NSCLC-BM (n = 14), with thresholds set at |log2FC| >1 and FDR < 0.05, yielding 779 significant DEGs.Visualization:A volcano plot highlighted 56 robust DEGs (19 upregulated, 37 downregulated) (Figure 2A). Venn diagram analysis revealed 50 core overlapping genes between GSE161116 DEGs and the GeneCards BM-related gene set (Figure 2C; Supplementary Table S1). Hierarchical clustering heatmap analysis (pheatmap package) of these 50 intersecting genes demonstrated heterogeneous expression patterns across groups (Figure 2B).
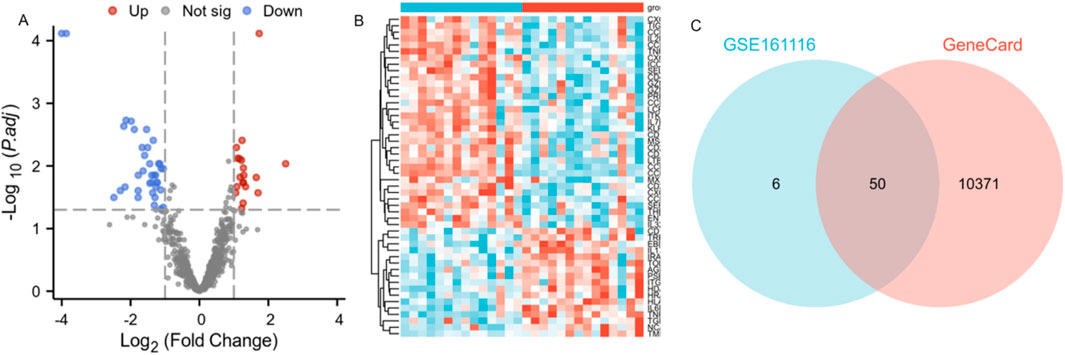
Figure 2. Identification of differentially expressed genes (DEGs) associated with brain metastasis in lung cancer patients. Note: (A) Volcano plot of DEGs in GSE161116. X-axis: log2FC; Y-axis: log10 (p-value). Blue: downregulated genes; red: upregulated genes; gray: non-significant genes. (B) Heatmap of DEGs in GSE161116. X-axis: samples; Y-axis: genes. Red: high expression; blue: low expression. NSCLC-BM and primary NSCLC samples were clearly separated into two distinct clusters. (C) Venn diagram showing overlapping DEGs between GSE161116 and GeneCards databases.
Enrichment analysis of DEGs
Our systematic analysis integrating GSEA and multidimensional functional annotation revealed distinct molecular regulatory characteristics of NSCLC brain metastasis (NSCLC-BM). Using the MSigDB database (C2: curated gene sets), we observed marked upregulation of the “interleukin-17 signaling pathway” (NES = 2.024, FDR = 0.024), suggesting that an IL-17-mediated proinflammatory microenvironment may facilitate central nervous system colonization through the TLR/NF-κB axis (Figure 3). To further interpret the functional implications of the differentially expressed genes (DEGs), we performed comprehensive functional enrichment analyses. Detailed results of the GO enrichment analysis are presented in Table1. The most significantly enriched biological process (BP) terms included immune response, signal transduction, inflammatory response, and cell surface receptor signaling pathway (Figure 4A). For molecular function (MF), the top enriched terms were signaling receptor binding, chemokine activity, cytokine activity, and transmembrane signaling receptor activity (Figure 4B). Notably, the key cellular component (CC) terms included plasma membrane, membrane, extracellular region, and extracellular space (Figure C). The KEGG pathway enrichment analysis further highlighted eighteen significantly enriched pathways (Table2), which are visualized in Figure 4D. Major pathways included Cytokine-cytokine receptor interaction, Chemokine signaling pathway, Viral protein interaction with cytokine and cytokine receptor, PI3K-Akt signaling pathway, and Human cytomegalovirus infection.
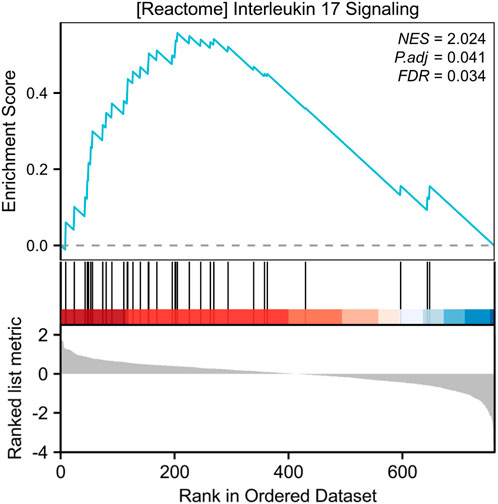
Figure 3. GSEA enrichment analysis results of DEGs between NSCLC and NSCLC-BM groups.
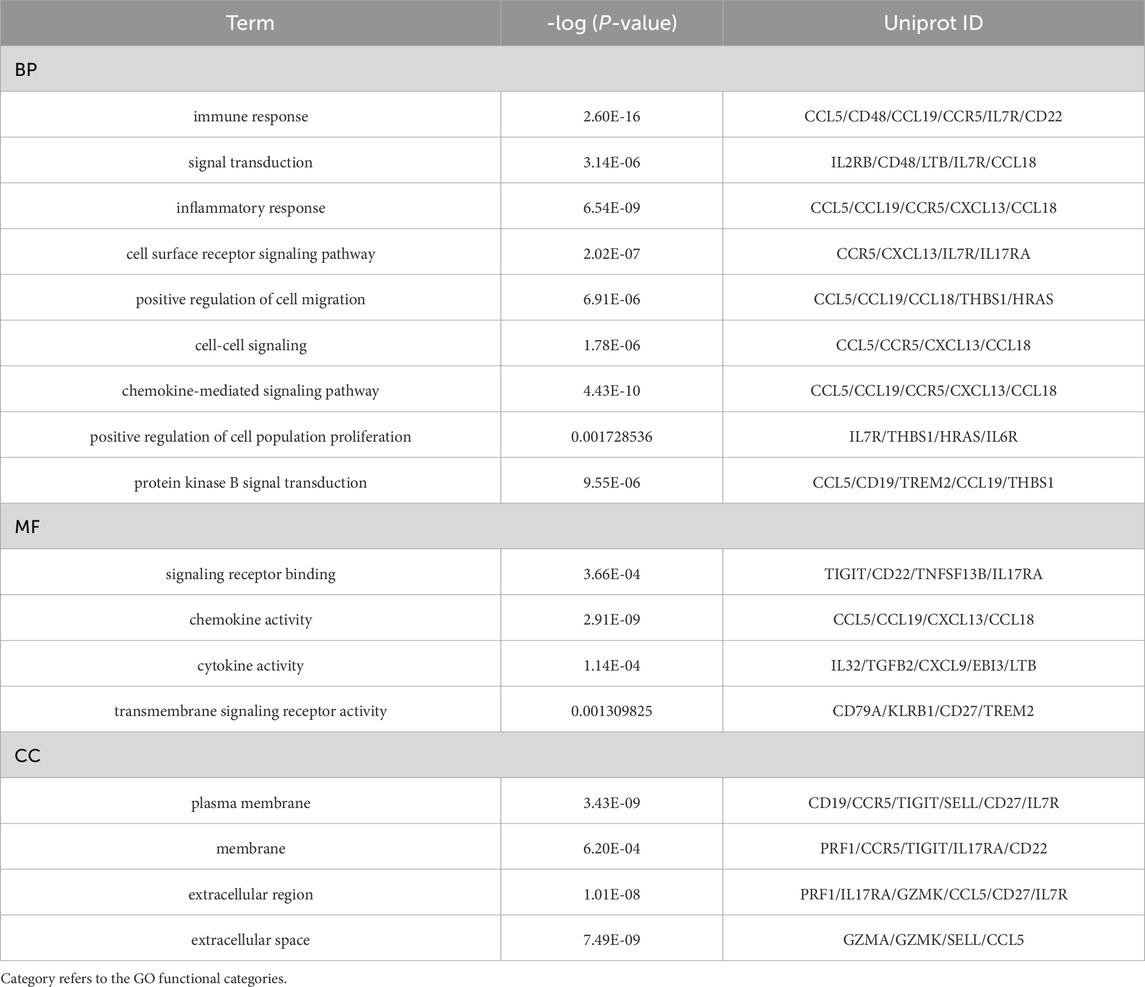
Table 1. GO functional enrichment analysis for the DEGs.
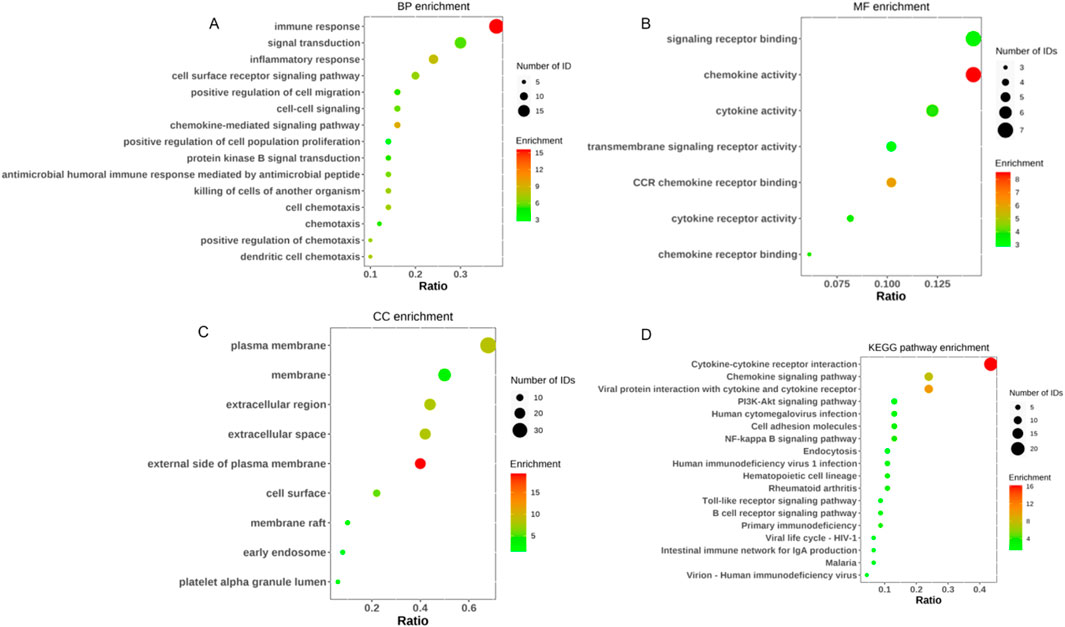
Figure 4. GO enrichment and KEGG pathway analysis of DEGs in NSCLC 和 NSCLC-BM group. (A) GO categories of BP. (B) GO categories of MF. (C) GO categories of CC. (D) KEGG pathway analysis of the DEGs.
Table 2. Pathway enrichment analysis for the DEGs.
PPI network construction and hub gene selection
We constructed a protein-protein interaction (PPI) network using the STRING database based on the 50 overlapping DEGs and visualized it with Cytoscape to identify highly interconnected hub proteins (hub-DEGs). The PPI network contained 64 nodes and 174 edges (Figure 5A). Among these genes, the top 10 proteins with the highest degree of interaction were identified as key hub genes: CD19, CD27, IL7R, SELL, CCL5, CCR5, PRF1, GZMK, GZMA, and TIGIT (Figure 5B). Literature mining revealed that these candidate genes are predominantly involved in: (1) immune synapse formation (CD27-IL7R axis), (2) T-cell exhaustion (TIGIT-PRF1 pathway), and (3) chemokine-mediated blood-brain barrier penetration (CCL5-CCR5 signaling). These findings suggest their potential role in driving brain metastasis progression through modulation of tumor-immune microenvironment interactions.
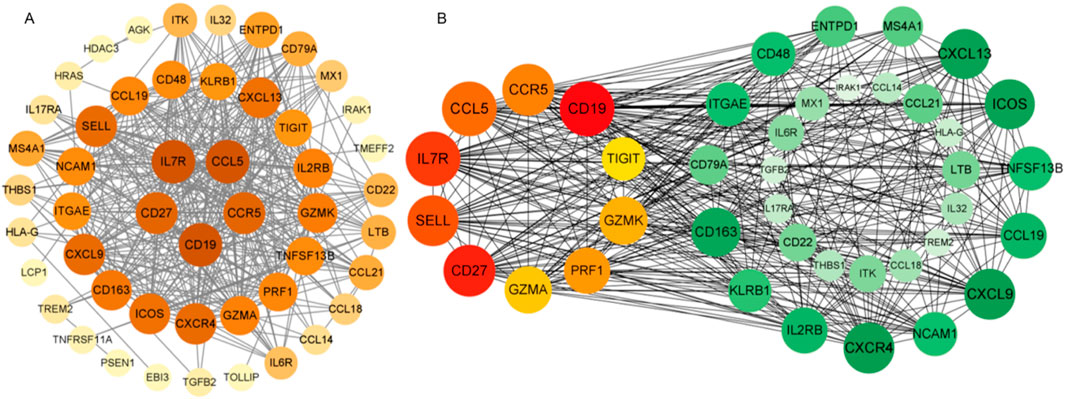
Figure 5. Protein-protein interaction (PPI) network visualized using Cytoscape software. Figure Note: (A) Node color intensity corresponds to degree value. (B) The network contains 40 nodes and 368 edges, with progressively redder hues indicating higher degree scores as measured by CytoHubba. Hub-DEGs in the PPI network are distinguished by unique coloring, while green nodes represent associated proteins.
Validation data analysis
The ten identified hub genes were validated using additional GEO datasets. The GSE248830 dataset, which includes 11 NSCLC and 11 NSCLC-BM samples, was employed to examine differential expression between primary and metastatic tumors. Preliminary analysis revealed that the expression levels of these ten hub genes were significantly downregulated following metastasis, a trend consistent with previous findings from the GSE161116 dataset. These results further support the reliability of our conclusions. The validation results are presented in Figure 6.
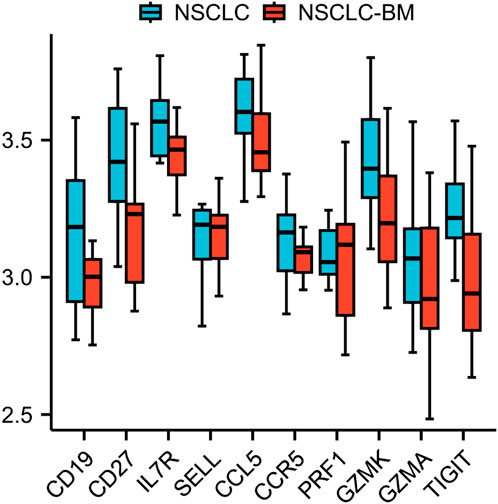
Figure 6. Grouped box plot analysis confirming differential expression.
TF regulatory network analysis of ten genes
We established an integrated TF-mRNA-miRNA regulatory network comprising 10 hub genes, 43 transcription factors (TFs), and 63 miRNAs (Figure 7). Comprehensive analysis of the TF-DEG and miRNA-DEG interaction networks revealed significant regulatory molecules. Notably, 8 of the 10 hub genes were embedded within this regulatory architecture. Subsequent subnetwork analysis identified key transcriptional regulators (STAT5A, STAT5B, NFKB1, EGR1, RELA, and CTCF) and post-transcriptional modulators (hsa-miR-204, hsa-miR-148b, hsa-miR-618, and hsa-miR-103) as pivotal biomolecules governing DEG expression. Mechanistically, 6 TFs (STAT5A/B, NFKB1, EGR1, RELA, and CTCF) emerged as central transcriptional regulators, while the miRNAs exhibited specific target interactions: hsa-miR-204: IL7R and PRF1/SELL; hsa-miR-148b: GZMK and SELL; hsa-miR-618: IL7R and GZMK; hsa-miR-103: CD19 and GZMK; These computational predictions require experimental validation to confirm their biological relevance in NSCLC-BM pathogenesis.
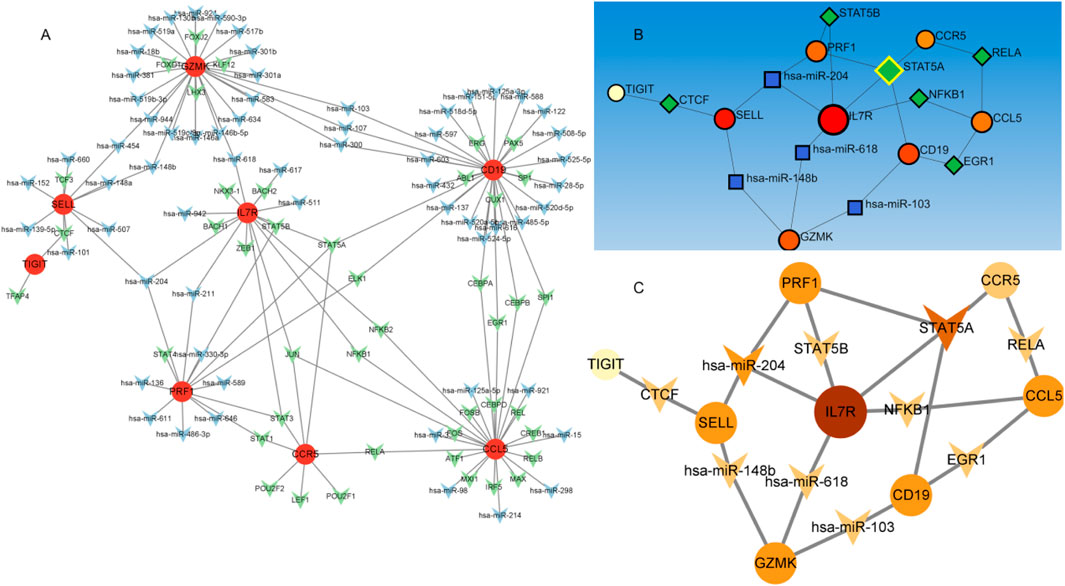
Figure 7. The TF-mRNA -miRNA regulatory network. Figure Note: (A) Regulatory network of hub genes. Red circles represent hub genes, green circles denote transcription factors (TFs), and blue circles indicate miRNAs. (B,C) Subnetworks of key TF-regulated genes, with node color intensity scaled according to degree values.
Survival impact of hub genes in brain metastasis
To investigate the prognostic significance of the ten hub genes in patients with brain metastasis (BM), we performed Kaplan-Meier survival analysis stratified by median gene expression levels (high vs. low expression groups). The results (Figure 8) demonstrated that decreased expression of these genes was significantly associated with shortened overall survival (OS) in patients with brain metastasis. Notably, prior studies have demonstrated that STAT5A promotes tumor invasion and metastasis by upregulatingCD44 (Szczepanik et al., 2019)—a cancer stem cell (CSC) marker linked to unfavorable prognosis in gastric cancer (GC). Our findings align with this mechanism, suggesting that the identified transcription factors (TFs) and hub genes may collectively accelerate brain metastasis through: Enhanced tumor cell invasiveness (via STAT5A-CD44 axis), Metastatic niche modulation, Post-metastatic transcriptional reprogramming (evidenced by expression downregulation post-metastasis) These results implicate the ten hub genes as critical mediators of lung cancer brain metastasis, potentially governing tumor cell dissemination and survival outcomes. Detailed results are shown in the Supplementary Figure S1.
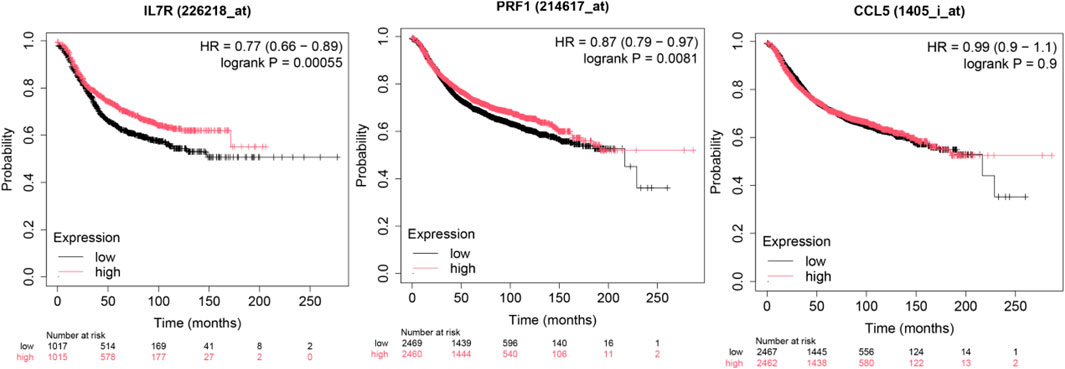
Figure 8. Overall survival analysis of 10 hub DEGs in kmplot website.
Immune cell infiltration analysis
To elucidate the relationship between the 10 hub genes and immune cell activity, we performed tumor-infiltrating immune cell (TIIC) profiling. Compositional analysis revealed significant positive correlations between the expression of hub genes (IL7R,PRF1, etc.) and activated immune subsets, including: Memory B cells, Activated CD4+T cells (correlation with IL7R:r = 0.409), CD8+T cells, Notably,IL7R exhibited the strongest associations: B cell activity (r = 0.374, Figure 9), CD4+T cell recruitment (r = 0.409). Additional hub gene–immune interactions are detailed in Supplementary Figure S2. These findings underscore the pivotal role of these genes in modulating B and T cell crosstalk within the tumor microenvironment (TME) of LUAD patients, suggesting their potential as immunomodulatory targets.
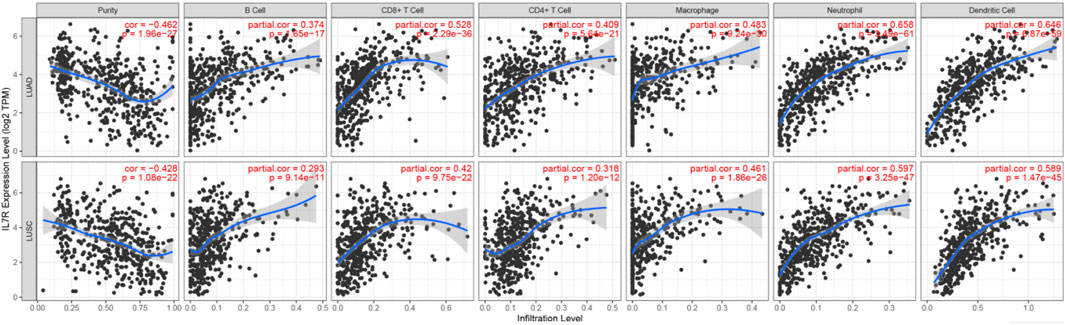
Figure 9. The correlation of IL7R with tumor purity and immune cells in the immune system shows the purity-corrected partial Spearman’s rho value and statistical significance. Log2 (TPM) is the log2 of the Transcript Count Per Million.
Mutation analysis of 10 crucial genes
We examined the mutation frequency and mutation types of these 10 hub genes in the GSCA database. The results revealed that IL7R exhibited the highest mutation frequency, followed by PRF1, with missense mutations accounting for 39% and 13% of the alterations in these genes, respectively. Additionally, copy number variation (CNV) analysis demonstrated that IL7R had the highest Figure 10. Notably, prior studies have reported that missense mutations in the perforin (PRF1) gene contribute to hereditary cancer predisposition (Chaudhry et al., 2016). Our findings suggest that mutations in these genes may play a role in cancer brain metastasis, potentially influencing tumor progression and metastatic potential.
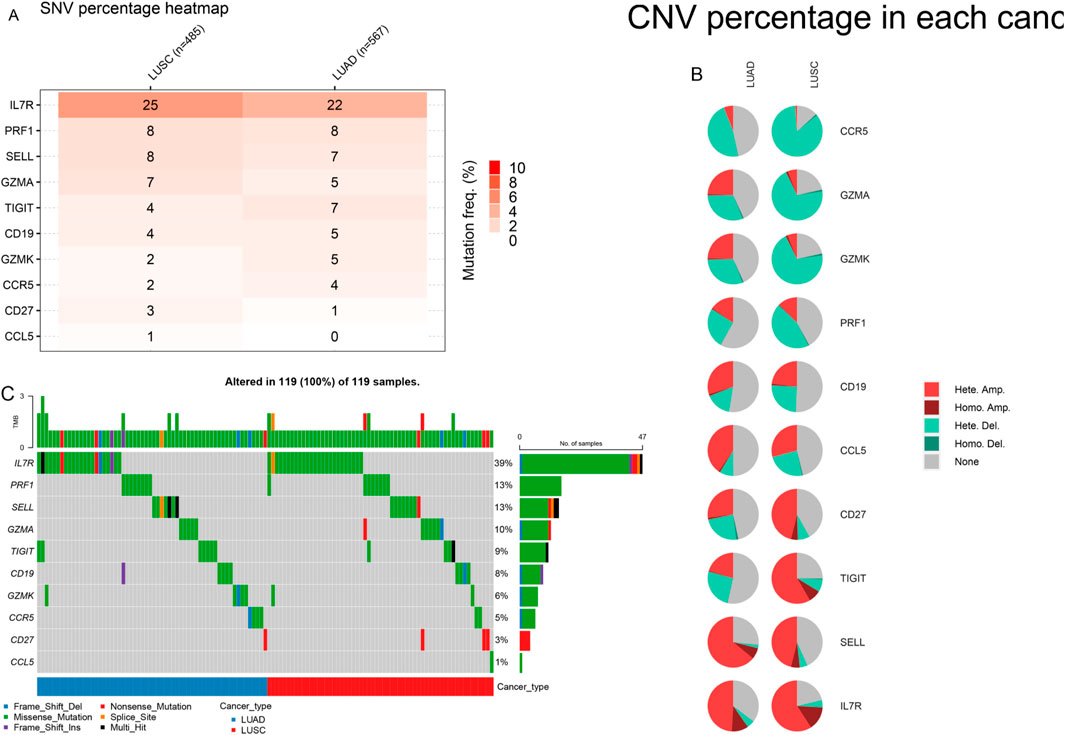
Figure 10. Variant frequency and mutation type of 10 genes in LUAD and LUSC. (A) The mutation rate of genes. (B) The CNV types of genes. (C) The mutation type of genes.
Discussion
The identification of biomarkers associated with lung cancer brain metastasis may provide deeper insights into the molecular mechanisms underlying metastatic progression. This study aimed to analyze NSCLC gene expression data to identify differentially expressed genes (DEGs), elucidate key molecular pathways, determine critical hub proteins, and characterize relevant regulatory biomolecules through a multi-omics data integration framework, with the ultimate goal of discovering potential therapeutic targets for NSCLC. Our gene expression profiling identified 56 DEGs, including 19 upregulated and 37 downregulated genes. Functional enrichment analysis revealed that these DEGs were significantly associated with several oncogenic molecular functions and pathways. GSEA results demonstrated marked upregulation of the “interleukin-17 signaling pathway.” Notably, the interleukin-17 (IL-17) signaling pathway has been previously established to contribute to the progression of lung cancer bone metastasis (Zhou et al., 2023). GO and KEGG enrichment analyses identified several critical biological processes and pathways, including immune response, signal transduction, inflammatory response, cell surface receptor signaling pathway, positive regulation of cell migration, cell-cell signaling, signaling receptor binding, chemokine activity, cytokine activity, transmembrane signaling receptor activity, plasma membrane, membrane, extracellular region, extracellular space, cytokine-cytokine receptor interaction, chemokine signaling pathway, viral protein interaction with cytokine and cytokine receptor, and PI3K-Akt signaling pathway. Existing evidence suggests that inflammatory chemokines and their receptors regulate tumor cell migration and participate in tumor growth, metastasis, angiogenesis, and invasion through interactions between mesenchymal and tumor cells (Cheng et al., 2016; Zhao et al., 2019). All these functions and pathways are significantly associated with cancer development and play crucial roles in the NSCLC microenvironment. Protein-protein interaction (PPI) network analysis has emerged as a promising approach for investigating the fundamental mechanisms of brain metastasis in lung cancer (Sevimoglu and Arga, 2014). Our PPI network analysis revealed hub proteins encoded by hub DEGs. The CCR5/CCL5 signaling axis has been shown to increase infiltration of regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) into the tumor microenvironment (TME), creating an immune effector cell desert that promotes cancer survival and progression (Sevimoglu and Arga, 2014), while potentially contributing to immunotherapy resistance. This pathway has also demonstrated prognostic and predictive value in metastatic colorectal cancer (CRC) (Suarez-Carmona et al., 2019; Schlechter and Stebbing, 2024). CD27, a member of the TNF receptor superfamily, is essential for T cell immunity generation and long-term maintenance; Pagès et al. (Pages et al., 2005) found that CD27 expression correlates with early metastasis in colorectal cancer. IL-7R has emerged as a potential prognostic marker in breast cancer patients, particularly in maintaining immunologically active states in the TME and promoting immune reconstitution (Yu et al., 2024). PRF1, a crucial cytotoxic molecule, plays a vital role in the killing functions of natural killer (NK) cells and cytotoxic T lymphocytes (CTLs) (Guan et al., 2024). GZMA, GZMK, and PRF1 (Tibbs and Cao, 2022; Paczek et al., 2022; Park et al., 2021; Lavergne et al., 2021) not only induce apoptosis and modulate immune responses within the TME but also exhibit other distinct functions. Inhibition of tumor growth has been associated with reduced expression of the immune checkpoint molecule TIGIT (Shaw et al., 2022). The subnetwork modules containing these hub genes provide strong evidence supporting their reliability as therapeutic targets. The TF–mRNA–miRNA regulatory network analysis identified six transcription factors (STAT5A, STAT5B, NFKB1, EGR1, RELA, and CTCF) and four miRNAs (hsa-miR-204, hsa-miR-148b, hsa-miR-618, and hsa-miR-103) as key transcriptional and post-transcriptional regulators of hub DEGs. Previous studies have reported that various tumor-associated genes are regulated by STAT5A/STAT5B, which maintain multiple cancer-related pathways (Erdogan et al., 2022). NFKB1 plays critical roles in tumor cell invasion and metastasis, with its expression linked to invasion and metastasis across various cancer types (Zhang et al., 2023). EGR1 acts as a pro-metastatic factor in pancreatic cancer cells, promoting cell migration and invasion through the SNAI2–EMT pathway (Wang et al., 2023). Currently, there are limited reports on the tumor-related effects of RELA and CTCF. hsa-miR-204 has been reported to be downregulated and function as a tumor suppressor in various cancers including colorectal cancer, papillary thyroid carcinoma, malignant melanoma, and hepatocellular carcinoma (Chu et al., 2018). Studies have demonstrated significant enrichment of hsa-miR-148b in cancer-related pathways including Wnt, MAPK, and Jak-STAT signaling pathways (Luo et al., 2015). Dysregulation of hsa-miR-618 has been associated with numerous cancers (Radanova et al., 2021). hsa-miR-103a can function as either a tumor promoter or suppressor, modulating tumor progression in various cancers (Li et al., 2021). Survival curve analysis clearly demonstrated significant differences in hub gene expression between high-risk and low-risk groups, indicating their important roles in patient survival. Furthermore, immune infiltration analysis revealed interactions between these key genes and B/T cells in LUAD patients, suggesting their influence on the TME. On the other hand, in cancer cells, gene mutations, amplifications, and deletions can lead to altered target proteins that fail to bind drugs, resulting in drug resistance. In particular, missense mutations may significantly affect protein function (Chen et al., 2004). Our mutation analysis of 10 genes in NSCLC revealed that IL7R and PRF1 had the highest missense mutation rates at 39% and 13%, respectively. It has been demonstrated that IL7R mutations activate downstream IL7R signaling independent of IL7 and promote cell transformation and tumor formation, indicating that IL7R exon 6 mutations are gain-of-function mutations (Kim et al., 2013). PRF1 plays important roles in various aspects of tumor cell development, immune escape mechanisms, cancer immunotherapy, and prognosis (Guan et al., 2024). However, the functional characteristics of most missense mutations in IL7R and PRF1 in tumors remain poorly characterized. Importantly, we found strong associations between IL7R/PRF1 mutation co-occurrence and immune-related pathways, particularly B cell and T cell signaling. The presence of infiltrating immune cells in these tumors, some of which inhibit or promote disease progression, further supports the involvement of immune pathways. In conclusion, although this study has certain limitations including a small sample size and lack of clinical sample validation, our current analysis has identified several key genes and pathways closely associated with NSCLC-BM that may enhance current understanding of its complex mechanisms. Notably, these findings warrant further investigation and experimental validation.
Conclusion
This study employed bioinformatics approaches to compare non-small cell lung cancer (NSCLC) and brain metastasis (NSCLC-BM) samples, leading to the preliminary identification of 56 differentially expressed genes (DEGs) potentially associated with metastatic progression. These genes were significantly enriched in key pathways including cytokine-cytokine receptor interaction, chemokine signaling pathway, viral protein-cytokine receptor interaction, and the PI3K-Akt signaling pathway. Among them, ten genes—CD19, CD27, IL7R, SELL, CCL5, CCR5, PRF1, GZMK, GZMA, and TIGIT—were selected as potential hub genes. Furthermore, predictions suggested that miRNAs such as hsa-miR-204 and hsa-miR-148b, along with certain transcription factors, may contribute to metastasis by modulating the tumor immune microenvironment. It is important to emphasize that the findings of this study are computational predictions, and all identified genes and regulatory molecules remain candidate biomarkers that have not been experimentally validated. None of the conclusions presented should be interpreted as established biomarkers or clinically applicable outcomes. These predictions require further experimental validation—including qRT-PCR, immunohistochemistry, independent patient cohort analyses, and in vitro/in vivo functional assays—to confirm their biological significance and translational potential. Future research will focus on experimental verification to evaluate the diagnostic or therapeutic value of these candidate molecules.
Methods
Microarray data
The GSE161116 dataset was obtained from the NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo/; GPL19965 platform). This study included 28NSCLC patients with brain metastasis (BM) who underwent surgery at Seoul National University Hospital (SNUH) between January 2013 and March 2018. Clinicopathological data—including age, sex, smoking history, tumor genetic status, treatment, and follow-up records—were retrieved from electronic medical records. Pathological staging was based on the 8th edition of the AJCC staging system (Song et al., 2021). Additionally, GeneCards (Stelzer et al., 2016) was searched using the keyword “lung cancer brain metastasis” to extract relevant target genes.
Data processing
Differentially expressed genes (DEGs) between primary NSCLC and NSCLC-BM were identified using the limma package (Suzuki et al., 2019) in R (version 4.2.1),with thresholds set at |logFC| > 1 and P < 0.05 (NSCLC without BM vs. NSCLC with BM). The GSE161116 dataset contained 14 NSCLC and 14 NSCLC-BM samples. Overlapping DEGs between GSE161116 and GeneCards (March 2025) were visualized using ggplot2 (Zeng et al., 2022) and VennDiagram packages in R.
DEG enrichment analysis
Functional enrichment analysis was performed using:Gene Set Enrichment Analysis (GSEA) with the MSigDB Collections (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp) as the reference gene set (500 permutations, significance threshold: p. adj<0.25). Results were visualized via ggplot2 (Zeng et al., 2022). DAVID (v6.8; http://david.ncifcrf.gov) for Gene Ontology (GO) and KEGG pathway analysis. GO terms included biological processes (BP), cellular components (CC), and molecular functions (MF). P < 0.05 was considered statistically significant.
PPI network construction and hub gene identification
Protein-protein interaction (PPI) networks were built using STRING (v10.0; http://string-db.org; interaction score cutoff: 0.4) and visualized in Cytoscape (v3.9.1). Hub genes were identified via CytoHubba and MCODE plugins, applying 12 ranking algorithms to select the top 10 nodes per method.
Hub gene-TF-miRNA regulatory network
A mRNA-TF-miRNA co-regulatory network was constructed using NetworkAnalyst (Xia et al., 2015), integrating data from TF-miRNA interaction databases, and visualized in Cytoscape.
Survival analysis
The Kaplan-Meier Plotter (http://kmplot.com/analysis/) assessed the impact of hub genes on overall survival (OS). Patients were stratified into high- and low-expression groups based on median expression levels (Tang et al., 2019). Differences were evaluated via log-rank test (P < 0.05).
Immune infiltration analysis
The TIMER 2.0 platform (https://cistrome.shinyapps.io/timer/) analyzed correlations between hub gene expression and immune cell abundance using purity-adjusted Spearman’s rank correlation.
Mutation analysis
Genomic alterations (mutations and copy number variations, CNVs) in hub genes were analyzed via GSCA (http://gsca.bio-data.cn) to determine mutation frequencies and functional impacts.
Statistical analysis
To account for multiple hypothesis testing in the identification of differentially expressed genes, the false discovery rate (FDR) was controlled using the Benjamini–Hochberg procedure. The statistical thresholds were set at |log2FC|>1 and adjusted p-value (FDR) < 0.05.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by This study did not involve human participants, animal experiments, or primary data collection requiring ethical approval. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
SZ: Writing – original draft. HZ: Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Establishment of a Pancreatic Cancer Xenograft Model Library and Research on Biomarker Screening.
Acknowledgments
We extend our gratitude to the contributors and curators of all publicly available datasets utilized in this study. We also acknowledge the patients and research teams whose efforts in generating and sharing these data have made this research possible.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbinf.2025.1625664/full#supplementary-material
References
Ali, A., Goffin, J. R., Arnold, A., and Ellis, P. (2013). Survival of patients with non-small-cell lung cancer after a diagnosis of brain metastases. Curr. Oncol. 20 (4), e300–e306. doi:10.3747/co.20.1481
Cagney, D. N., Martin, A. M., Catalano, P. J., Redig, A. J., Lin, N. U., Lee, E. Q., et al. (2017). Incidence and prognosis of patients with brain metastases at diagnosis of systemic malignancy: a population-based study. Neuro Oncol. 19 (11), 1511–1521. doi:10.1093/neuonc/nox077
Chaudhry, M. S., Gilmour, K. C., House, I. G., Layton, M., Panoskaltsis, N., Sohal, M., et al. (2016). Missense mutations in the perforin (PRF1) gene as a cause of hereditary cancer predisposition. Oncoimmunology 5 (7), e1179415. doi:10.1080/2162402x.2016.1179415
Chen, J., Lipska, B. K., Halim, N., Ma, Q. D., Matsumoto, M., Melhem, S., et al. (2004). Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am. J. Hum. Genet. 75 (5), 807–821. doi:10.1086/425589
Cheng, H., and Perez-Soler, R. (2018). Leptomeningeal metastases in non-small-cell lung cancer. Lancet Oncol. 19 (1), e43–e55. doi:10.1016/s1470-2045(17)30689-7
Cheng, Z., Shi, Y., Yuan, M., Xiong, D., Zheng, J. h., and Zhang, Z. y. (2016). Chemokines and their receptors in lung cancer progression and metastasis. J. Zhejiang Univ. Sci. B 17 (5), 342–351. doi:10.1631/jzus.b1500258
Chu, Y., Jiang, M., Du, F., Chen, D., Ye, T., Xu, B., et al. (2018). miR-204-5p suppresses hepatocellular cancer proliferation by regulating homeoprotein SIX1 expression. FEBS Open Bio 8 (2), 189–200. doi:10.1002/2211-5463.12363
Erdogan, F., Radu, T. B., Orlova, A., Qadree, A. K., de Araujo, E. D., Israelian, J., et al. (2022). JAK-STAT core cancer pathway: an integrative cancer interactome analysis. J. Cell Mol. Med. 26 (7), 2049–2062. doi:10.1111/jcmm.17228
Gillespie, C. S., Mustafa, M. A., Richardson, G. E., Alam, A. M., Lee, K. S., Hughes, D. M., et al. (2023). Genomic alterations and the incidence of brain metastases in advanced and metastatic NSCLC: a systematic review and meta-analysis. J. Thorac. Oncol. 18 (12), 1703–1713. doi:10.1016/j.jtho.2023.06.017
Guan, X., Guo, H., Guo, Y., Han, Q., Li, Z., and Zhang, C. (2024). Perforin 1 in cancer: mechanisms, therapy, and outlook. Biomolecules 14 (8), 910. doi:10.3390/biom14080910
Hubbs, J. L., Boyd, J. A., Hollis, D., Chino, J. P., Saynak, M., and Kelsey, C. R. (2010). Factors associated with the development of brain metastases: analysis of 975 patients with early stage nonsmall cell lung cancer. Cancer 116 (21), 5038–5046. doi:10.1002/cncr.25254
Jonna, S., and Subramaniam, D. S. (2019). Molecular diagnostics and targeted therapies in non-small cell lung cancer (NSCLC): an update. Discov. Med. 27 (148), 167–170.
Kim, M. S., Chung, N. G., Kim, M. S., Yoo, N. J., and Lee, S. H. (2013). Somatic mutation of IL7R exon 6 in acute leukemias and solid cancers. Hum. Pathol. 44 (4), 551–555. doi:10.1016/j.humpath.2012.06.017
Lavergne, M., Hernandez-Castaneda, M. A., Mantel, P., Martinvalet, D., and Walch, M. (2021). Oxidative and non-oxidative antimicrobial Activities of the granzymes. Front. Immunol. 12, 750512. doi:10.3389/fimmu.2021.750512
Li, H., Huhe, M., and Lou, J. (2021). MicroRNA-103a-3p promotes cell proliferation and invasion in non-small-cell lung cancer cells through akt pathway by targeting PTEN. Biomed. Res. Int. 2021, 7590976. doi:10.1155/2021/7590976
Luo, Y., Zhang, C., Tang, F., Zhao, J., Shen, C., Wang, C., et al. (2015). Bioinformatics identification of potentially involved microRNAs in Tibetan with gastric cancer based on microRNA profiling. Cancer Cell Int. 15, 115. doi:10.1186/s12935-015-0266-1
Matsui, J. K., Perlow, H. K., Baiyee, C., Ritter, A. R., Mishra, M. V., Bovi, J. A., et al. (2022). Quality of life and cognitive function Evaluations and Interventions for patients with brain metastases in the radiation oncology clinic. Cancers (Basel) 14 (17), 4301. doi:10.3390/cancers14174301
Nayak, L., Lee, E. Q., and Wen, P. Y. (2012). Epidemiology of brain metastases. Curr. Oncol. Rep. 14 (1), 48–54. doi:10.1007/s11912-011-0203-y
Paczek, S., Lukaszewicz-Zajac, M., and Mroczko, B. (2022). Granzymes-their role in colorectal cancer. Int. J. Mol. Sci. 23 (9), 5277. doi:10.3390/ijms23095277
Page, S., Milner-Watts, C., Perna, M., Janzic, U., Vidal, N., Kaudeer, N., et al. (2020). Systemic treatment of brain metastases in non-small cell lung cancer. Eur. J. Cancer 132, 187–198. doi:10.1016/j.ejca.2020.03.006
Pages, F., Berger, A., Camus, M., Sanchez-Cabo, F., Costes, A., Molidor, R., et al. (2005). Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 353 (25), 2654–2666. doi:10.1056/nejmoa051424
Park, S., Anderson, N. L., Canaria, D. A., and Olson, M. R. (2021). Granzyme-producing CD4 T cells in cancer and autoimmune disease. Immunohorizons 5 (12), 909–917. doi:10.4049/immunohorizons.2100017
Radanova, M., Mihaylova, G., Mihaylova, Z., Ivanova, D., Tasinov, O., Nazifova-Tasinova, N., et al. (2021). Circulating miR-618 has prognostic significance in patients with metastatic colon cancer. Curr. Oncol. 28 (2), 1204–1215. doi:10.3390/curroncol28020116
Schlechter, B. L., and Stebbing, J. (2024). CCR5 and CCL5 in metastatic colorectal cancer. J. Immunother. Cancer 12 (5), e008722. doi:10.1136/jitc-2023-008722
Sevimoglu, T., and Arga, K. Y. (2014). The role of protein interaction networks in systems biomedicine. Comput. Struct. Biotechnol. J. 11 (18), 22–27. doi:10.1016/j.csbj.2014.08.008
Shaw, G., Cavalcante, L., Giles, F. J., and Taylor, A. (2022). Elraglusib (9-ING-41), a selective small-molecule inhibitor of glycogen synthase kinase-3 beta, reduces expression of immune checkpoint molecules PD-1, TIGIT and LAG-3 and enhances CD8(+) T cell cytolytic killing of melanoma cells. J. Hematol. Oncol. 15 (1), 134. doi:10.1186/s13045-022-01352-x
Song, S. G., Kim, S., Koh, J., Yim, J., Han, B., Kim, Y. A., et al. (2021). Comparative analysis of the tumor immune-microenvironment of primary and brain metastases of non-small-cell lung cancer reveals organ-specific and EGFR mutation-dependent unique immune landscape. Cancer Immunol. Immunother. 70 (7), 2035–2048. doi:10.1007/s00262-020-02840-0
Sperduto, P. W., Yang, T. J., Beal, K., Pan, H., Brown, P. D., Bangdiwala, A., et al. (2017). Estimating survival in patients with lung cancer and brain metastases: an update of the graded prognostic assessment for lung cancer using molecular markers (Lung-molGPA). JAMA Oncol. 3 (6), 827–831. doi:10.1001/jamaoncol.2016.3834
Sperduto, P. W., Mesko, S., Li, J., Cagney, D., Aizer, A., Lin, N. U., et al. (2020). Survival in patients with brain metastases: summary report on the updated diagnosis-specific Graded prognostic assessment and Definition of the Eligibility Quotient. J. Clin. Oncol. 38 (32), 3773–3784. doi:10.1200/jco.20.01255
Srinivasan, E. S., Deshpande, K., Neman, J., Winkler, F., and Khasraw, M. (2021). The microenvironment of brain metastases from solid tumors. Neurooncol Adv. 3 (Suppl. 5), v121–v132. doi:10.1093/noajnl/vdab121
Stelzer, G., Rosen, N., Plaschkes, I., Zimmerman, S., Twik, M., Fishilevich, S., et al. (2016). The GeneCards suite: from gene data mining to disease Genome Sequence analyses. Curr. Protoc. Bioinforma. 54, 1–30. doi:10.1002/cpbi.5
Suarez-Carmona, M., Chaorentong, P., Kather, J. N., Rothenheber, R., Ahmed, A., Berthel, A., et al. (2019). CCR5 status and metastatic progression in colorectal cancer. Oncoimmunology 8 (9), e1626193. doi:10.1080/2162402x.2019.1626193
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global cancer Statistics 2020: GLOBOCAN Estimates of incidence and Mortality worldwide for 36 cancers in 185 Countries. CA Cancer J. Clin. 71 (3), 209–249. doi:10.3322/caac.21660
Suzuki, A., Horie, T., and Numabe, Y. (2019). RETRACTED ARTICLE: investigation of molecular biomarker candidates for diagnosis and prognosis of chronic periodontitis by bioinformatics analysis of pooled microarray gene expression datasets in Gene Expression Omnibus (GEO). BMC Oral Health 19 (1), 52. doi:10.1186/s12903-019-0738-0
Szczepanik, A., Sierzega, M., Drabik, G., Pituch-Noworolska, A., Kołodziejczyk, P., and Zembala, M. (2019). CD44(+) cytokeratin-positive tumor cells in blood and bone marrow are associated with poor prognosis of patients with gastric cancer. Gastric Cancer 22 (2), 264–272. doi:10.1007/s10120-018-0858-2
Tang, D., Zhao, X., Zhang, L., Wang, Z., and Wang, C. (2019). Identification of hub genes to regulate breast cancer metastasis to brain by bioinformatics analyses. J. Cell Biochem. 120 (6), 9522–9531. doi:10.1002/jcb.28228
Tibbs, E., and Cao, X. (2022). Emerging Canonical and non-Canonical roles of granzyme B in health and disease. Cancers (Basel) 14 (6), 1436. doi:10.3390/cancers14061436
Wang, Y., Qin, C., Zhao, B., Li, Z., Li, T., Yang, X., et al. (2023). EGR1 induces EMT in pancreatic cancer via a P300/SNAI2 pathway. J. Transl. Med. 21 (1), 201. doi:10.1186/s12967-023-04043-4
Waqar, S. N., Samson, P. P., Robinson, C. G., Bradley, J., Devarakonda, S., Du, L., et al. (2018). Non-small-cell lung cancer with brain metastasis at presentation. Clin. Lung Cancer 19 (4), e373–e379. doi:10.1016/j.cllc.2018.01.007
Xia, J., Gill, E. E., and Hancock, R. E. W. (2015). NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 10 (6), 823–844. doi:10.1038/nprot.2015.052
Yang, B., Lee, H., Um, S., Kim, K., Zo, J. I., Shim, Y. M., et al. (2019). Incidence of brain metastasis in lung adenocarcinoma at initial diagnosis on the basis of stage and genetic alterations. Lung Cancer 129, 28–34. doi:10.1016/j.lungcan.2018.12.027
Yu, X., Zhang, H., Liu, J., Hou, C., and Yang, Z. (2024). IL-7R expression correlates with prognosis in breast cancer. Comb. Chem. High. Throughput Screen 28, 973–987. doi:10.2174/0113862073293963240409040110
Zeng, C., Lin, M., Jin, Y., and Zhang, J. (2022). Identification of key genes associated with brain metastasis from breast cancer: a bioinformatics analysis. Med. Sci. Monit. 28, e935071. doi:10.12659/msm.935071
Zhang, L., Ludden, C. M., Cullen, A. J., Tew, K. D., Branco de Barros, A. L., and Townsend, D. M. (2023). Nuclear factor kappa B expression in non-small cell lung cancer. Biomed. Pharmacother. 167, 115459. doi:10.1016/j.biopha.2023.115459
Zhao, X., Wang, N., Chidanguro, T., Gu, H., Li, Y., Cao, H., et al. (2019). Candidate genes and pathways associated with brain metastasis from lung cancer compared with lymph node metastasis. Exp. Ther. Med. 18 (2), 1276–1284. doi:10.3892/etm.2019.7712
Keywords: brain metastasis, non-small-cell lung cancer, biomarker, signaling pathway, gene
Citation: Zhao S and Zhang H (2025) Key genes associated with brain metastasis in non-small cell lung cancer: novel insights from bioinformatics analysis. Front. Bioinform. 5:1625664. doi: 10.3389/fbinf.2025.1625664
Received: 09 May 2025; Accepted: 01 September 2025;
Published: 18 September 2025.
Edited by:
Guoqing Zhang, First Affiliated Hospital of Zhengzhou University, ChinaReviewed by:
Zeynep Tokcaer Keskin, Acıbadem University, TürkiyeMarwa Mohanad, Misr University for Science and Technology, Egypt
Copyright © 2025 Zhao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: He Zhang, YWx3YXlzemhoQDE2My5jb20=
†These authors have contributed equally to this work