 Innocent Sibanda
Innocent Sibanda Geoff Nitschke
Geoff Nitschke- Department of Computer Science, University of Cape Town, Cape Town, South Africa
The goal of bioengineering in synthetic biology is to redesign, reprogram, and rewire biological systems for specific applications using standardized parts such as promoters and ribosomes. For example, bioengineered micro-organisms capable of cleaning up environmental pollution or producing antibodies de novo to defend against viral pandemics have been predicted. Artificial Life (ALife) facilitates the design and understanding of living systems, not just those found in nature, but life as it could be, while synthetic biology provides the means to realize life as it can be engineered. Despite significant advances, the synthesis of evolving, adaptable, and bioengineered problem-solving ALife has yet to achieve practical feasibility. This is primarily due to limitations in directed evolution, fitness landscape mapping, and fitness approximation. Thus, currently synthetic (biological) ALife does not continue to evolve and adapt to changing tasks and environments. This is in stark contrast to current digital based ALife that continues to adapt and evolve in simulated environments demonstrating the dictum of life as it could be. We posit that if the bioengineering (on-demand design) of problem solving ALife is to ever become a reality then open issues pervading the directed evolution of synthetic ALife must first be addressed. This review examines open challenges in directed evolution, genetic diversity generation, fitness mapping, and fitness estimation, and outlines future directions toward a hybrid synthetic ALife design methodology. This review provides a novel perspective for a singular (hybridized) evolutionary design methodology, combining digital (in silico) and synthetic (in vitro) evolutionary design methods drawn from various bioengineering, digital and robotic ALife applications, while addressing highlighted directed evolution deficiencies.
1 Introduction
Artificial Life (ALife) is primarily defined as the study and creation of systems that exhibit life-like properties, with the objective of understanding the fundamental principles of living systems, such as self-organization, adaptation, and reproduction, through synthetic means (Bedau et al., 2007). However, there is no universally accepted definition of ALife, largely due to its interdisciplinary nature, which spans biology, computer science, and robotics (Heudin, 2006). Langton (1986) introduced the term ALife, describing it as the investigation of possible forms of life beyond those observed in nature (Langton, 1986), asserting that ALife seeks to understand life by constructing it, whether in software (soft ALife), hardware (hard ALife), or biochemical systems (wet alife) (Bedau, 2003). In this review, we adopt the definition of ALife by Langton (1986).
Synthetic biology is a bioengineering approach that uses engineering principles in biology, focusing on designing, building, and programming components such as DNA circuits, metabolic pathways, and RNA-based devices like ribozymes (Peng et al., 2021). It extends beyond living cells to cell-free platforms and nucleic acid–based molecular circuits (Pardee et al., 2014). Standardized biological parts, computational design, and rapid DNA synthesis enables the creation of new or enhanced biological functions for use in medicine, biotechnology, and environmental applications (Khalil and Collins, 2010; Marris and Calvert, 2020), just to mention a few.
Integrating ALife and Synthetic Biology presents a profound synthesis of interdisciplinary approaches that combine computational, biological, and engineering perspectives (Madhavan and Mustafa, 2023). Coupled together, these fields can bridge the gap between simulation and synthesis, creating a unified framework for investigating how life might arise, evolve, and be engineered across digital and physical domains and providing the foundation for hybrid, evolvable systems capable of purposeful function (National Research Council, 2014).
1.1 Foundations: ALife and synthetic biology
ALife has provided a platform for the evolution of self-replicating computer programs capable of mutating, evolving, and adapting in simulated environments (digital organisms) (Aguilar et al., 2014), supporting the mantra of “life as it could be” in in silico (simulation) systems as envisioned by Langton (1986) (Bedau et al., 2007). The mantra of “life as it could be” defines the full design and understanding of living systems, not just exclusive to nature, but abstracted from any specific medium and in contrast to biology’s study of “life as we know it to be” on earth (Dorin and Stepney, 2024). An in vitro (physical) system counterpart to ALife is synthetic biology (Brooks and Alper, 2021), an engineering-driven approach to biology with a special focus on designing and constructing new biological parts, devices, and systems, or redesigning existing natural biological systems for useful purpose (Heinemann and Panke, 2006). Synthetic biology aims to assemble modular biological building blocks with defined functions and interfaces (standardized biological parts) such as promoters, ribosome and binding sites that are well-defined, reusable, characterized, and programmable (Katz et al., 2018) into problem-solving synthetic (ALife) organisms (Endy, 2005), similarly addressing the ALife mantra of “life as it could be” (Forbes, 2000). Both ALife and synthetic biology provide experimental platforms for studying the fundamental biological principles of life, such as heritable information, variation and selection leading to evolution, energy-driven self-maintenance (metabolism) (Gánti, 2003), and the emergence of complex systems and evolution (de Vladar et al., 2017). This demonstrates the potential of ALife in the digital and physical worlds and are thus inexorably related. For example, various types of ALife existing in silico are artificially evolved computational analogues of living systems, exhibiting behaviors adapted to compete for resources (such as computer processor cycles) within a simulation (Adami, 2006). Current biotechnological applications enabled by synthetic biology are primarily limited to in vitro (controlled laboratory environments) and, to a lesser extent, in vivo (living organisms) contexts (Slomovic et al., 2015). However, future applications are expected to address global humanitarian challenges (Brooks and Alper, 2021), including improvements in health, food and water security, sustainable energy production, and environmental restoration (Pachauri et al., 2009). On-demand bioengineering, which involves rapid and precise design, construction, and deployment of biological systems and materials as needed, offers significant potential for applications such as vaccines (Hart and Ferguson, 2019), regenerative medicines (Peng et al., 2023), novel biomaterials (Szymanski et al., 2023), and sustainable products such as biofuels and plant-based foods (Voigt, 2020). Natural processes often employ regulatory architectures similar to those found in control, electrical, and robotic engineering; this similarity has motivated the application of control-theoretic methods for designing and implementing synthetic biological systems (Steel et al., 2017). Foundational technologies, including engineered gene regulatory networks, provide essential design frameworks for these advancements (Baier et al., 2023). Furthermore, synthetic biology-driven platforms have been proposed as foundational elements for Mars mission architectures (Nangle et al., 2020), where biomanufacturing of food, materials, therapeutics, and waste reclamation could support a self-sustaining Martian infrastructure (Santomartino et al., 2023). Although on-demand bioengineering is partially achievable today, with the ability to design and produce simple biological components rapidly, a significant gap remains in achieving robust, adaptable, and safe deployment (Brooks and Alper, 2021). Key capabilities, such as automated DNA synthesis, cell-free expression, and biofoundries, contribute to the feasibility of on-demand bioengineering; however, challenges persist in terms of predictive reliability, scalability, and containment (Brookwell et al., 2021). ALife addresses these challenges by enabling in silico evolution and digital-twin stress testing, thereby improving the transition from design to deployment (Hale and Stanney, 2014).
1.2 Challenges in deploying artificial life
There are two key problems that confound the synthesis and deployment of ALife for real-world applications. First, current bioengineering platforms (Grozinger et al., 2019) are mainly intentionally limited to function within controlled laboratory (in vitro) environments, serving as a critical bio-safety measure to prevent unintended environmental release or ecological impact (Gao et al., 2025). However, in some instances, this limitation is unintentional rather than caused by technical constraints, specifically the inability to accurately translate in vitro functioning systems into robust, self-sustaining entities capable of reliable performance in complex, real-world settings (Hitchins, 2008). This persistent gap between laboratory success and operational viability outside the laboratory remains a significant barrier to the development of deployable artificial life systems (Brooks and Alper, 2021). Second, there is no gold-standard method to direct the evolutionary design of organisms for optimal adaptation to a given task environment (application) (Wang et al., 2021) because designing a globally optimal organism requires balancing trade-offs between multiple, often competing, objectives, such as efficiency, robustness, and adaptability, rather than optimizing a single property (Vincent, 2017). Consequently, the synthesis of problem-solving ALife depends on predicting evolutionary trajectories, which in turn requires sufficient knowledge of the organism’s underlying fitness landscape (Wortel et al., 2023). A fitness landscape is a conceptual framework that can be visualized as a multidimensional surface, where each genotype occupies a position in sequence space and its corresponding fitness value determines the height at that point (Poelwijk et al., 2007). At the molecular level, the sequence space is the combinatorial space of all possible nucleotide or amino acid sequences (Povolotskaya and Kondrashov, 2010). In this review, we specifically refer to proteins and nucleic acids, where the notion of sequence space is well defined. Fitness is a flexible and context-dependent measure of performance according to a specific function or goal, such as catalytic activity in ribozymes (Schoenle et al., 2018) and the fitness function is the specific rule or calculation used to assign a quantitative score that measures the goodness of any possible solution for a given problem (Wang et al., 2021). Fitness landscapes have been demonstrated as critical to optimization in various applications, beyond synthetic biology (Yi and Dean, 2019), including ecology (Sanchez-Gorostiaga et al., 2019) and biomedicine (Nichol et al., 2015). Mapping fitness landscapes has been demonstrated as notoriously difficult (Poelwijk et al., 2007), such that predicting and controlling the evolutionary trajectories of bioengineered agents remains one of the significant obstacle to synthetic (biological) ALife reaching its full potential (Lässig et al., 2017). Although understanding fitness landscapes is not the sole limitation in constructing synthetic life, it illustrates significant methodological and conceptual challenges (Brun-Usan et al., 2022). These challenges restrict the integration of molecular, cellular, and system-level processes into unified, evolving artificial systems (Rothschild et al., 2024). This article thus focuses on the methodological deficiencies in mapping fitness landscapes and directed evolution as core mechanisms driving the evolutionary design of synthetic ALife. Artificial evolutionary design, inspired by natural evolution as the core proponent of biological design, diversity and adaptivity, has been a constant source of novel and innovative solutions to a vast array of design and optimization problems (Eiben and Smith, 2015a).
1.3 A hybrid synthetic perspective on artificial life
To further the state-of-the-art of ALife evolutionary design methodologies, we propose future research guidelines for novel Hybrid ALife methodologies that combine in silico (computational) and in vitro (biological) design of synthetic problem-solving ALife. Various definitions of Hybrid ALife have been proposed (Baltieri et al., 2023), but such work does not account for experimental (in vitro) evaluation of biologically synthesized ALife agents. Historically, ALife design methodologies have been grounded in evolutionary design on digital (in silico) (Sims, 1994; Yaeger, 2009; Auerbach and Bongard, 2014; Joachimczak et al., 2016; Kriegman et al., 2018) or physical (robotic) (Lipson and Pollack, 2000; Brodbeck et al., 2015; Jelisavcic et al., 2017; Hale et al., 2019) experimental platforms with relatively little work in experimental validation of synthetic ALife (Blackiston et al., 2021; Bongard and Levin, 2021). If synthetic ALife is to be designed on-demand for various real-world applications (Brooks and Alper, 2021), then we will require something akin to a Hybrid-ALife tool-kit, integrating in silico and in vitro tools to enable directed evolutionary design of synthetic ALife. Specifically, just as synthetic biologists construct (bioengineer) biological devices by assembling standardized biological parts such as (BioBrick parts (iGEM Foundation, 2025)) into problem-solving biological systems (Stock and Gorochowski, 2024), we advocate extending current ALife research into a hybrid (computational and biological) evolutionary design methodologies that enable ALife researchers to automate the design of synthetic problem-solving ALife. BioBrick parts are standardized DNA sequences with specific functions that, when assembled, implement more complex behaviors (Shetty et al., 2008). Throughout this article, we refer to individual BioBrick parts as functional sequences, and assemblies of BioBrick parts as complex synthetic biological devices, which we treat as agents.
Adaptive walks play a critical role in the design of ALife, they are a stepwise evolutionary process that begins with an initial sequence, often referred to as the wild type or reference sequence and progresses toward sequences with higher fitness (Sarkisyan et al., 2016). From the starting sequence, adaptive walks explore the fitness landscape, a conceptual representation that maps genotypes to their corresponding fitness values (Greenbury et al., 2022). They sequentially accept beneficial mutations and move toward regions of higher fitness within the landscape, demonstrating the importance of the topography of the fitness landscape (De Visser and Krug, 2014). The fitness landscape topography is characterized by a pattern of regions representing high-fitness genotypes (peaks), low-fitness regions (valleys), and neutral zones (plateaus) and this pattern determines how easily evolution can proceed towards optimal solutions (Weinreich et al., 2018). Consequently, this strongly affects how readily organisms can adapt and how robust they are to mutations (Smerlak, 2021). To explain robustness and adaptation, theories of neutral (molecular changes that are neutral and have no effect on fitness) (Kimura, 1989) and nearly neutral (molecular changes have tiny-effect changes influenced by population size) (Ohta, 2002) evolution suggest that many genetic changes have little or no immediate effect on fitness. The presence of neutrality (vast majority of evolutionary changes at the molecular level are caused by random genetic drift acting on mutations that have no effect on the fitness of an organism) promotes broad search of the fitness landscape (exploration), allowing populations to move freely across the landscape and potentially discover new adaptive peaks, thereby fostering the emergence of new traits (evolutionary innovation) and the capacity to respond to changing environments (adaptability) (Gray et al., 2010; Draghi and Plotkin, 2011). Evidence supporting these ideas comes from experiments in computer-simulated evolutionary systems (digital evolution) (Franklin et al., 2019), as well as studies of RNA structures (Hayden et al., 2011), proteins (Podgornaia and Laub, 2015), and gene-regulatory networks (Baier et al., 2023). Despite these insights, there is no standardized method for directly reconstructing or inferring fitness landscapes from biological data, nor is there a formal mathematical expression of fitness applicable across systems (Wang D. et al., 2024). The challenge is heightened by the extremely high dimensionality of these landscapes, the complex epistatic interactions between genes (when the effect of one mutation depends on the presence of others), and the constantly changing nature of these interactions due to ongoing mutation and natural selection (De Visser and Krug, 2014). Moreover, limited empirical data on how mutations and epistasis reshape fitness landscapes continue to hinder accurate mapping and predictive models for directed evolution (Wang et al., 2021).
Given this, we first review directed evolution as it applies to bioengineering and open issues with directed evolution to be addressed if on-demand evolution of synthetic ALife is to become a reality. Second, we review fitness landscape mapping as it relates to directed evolution in bioengineering, specifically, the disconnect between empirical fitness landscapes mapped in silico and evolving fitness landscapes in nature (Fragata et al., 2019). Third, we propose future work guidelines for a Hybrid ALife methodology as a potential way forward for on-demand design (directed evolution) of synthetic ALife problem-solving agents. Throughout this review, we focus on in vitro, as opposed to in vivo methods as the biological substrate for validating directed evolution methods. This is because directed evolution methods operating in controlled in vitro (laboratory) environments enable fine-grained control of genetic sequence mutation and validation. Also there is no generalized method to assess generated sequence fitness in vivo, meaning the exact impact of sequences evolved in vivo across various biological substrates is difficult to ascertain (Vidal et al., 2023). Hence, we limit discussion in this review to the evolutionary design of synthetic ALife agents with potential applications limited to controlled and structured environments such as laboratories and factories. An important final caveat is that we purposely do not refer to, and thus review and critique, chemically synthesized biological ALife or wetware as it is popularly known in the ALife community (Aguilar et al., 2014). We instead defer to current bioengineering (synthetic biology) research, which addresses many of the same fundamental questions as wetware research. This is especially the case since this article’s purview is limited to evolutionary design for the purpose of synthesizing problem solving (ALife) agents for specific applications, which extends the scope of many bioengineering efforts (Voigt, 2020).
2 Directed evolution
Directed evolution (DE) is a closed loop evolutionary optimization that mimics the process of natural evolution, but on a shorter timescale, where a the specific rule or calculation used to assign a quantitative score that measures the goodness of any possible solution for a given problem) objectifies (fitness function) a specific target, and thus individuals are selected for improvement based on measured performance according to a specific function or goal, such as catalytic activity, binding affinity, growth rate and substrate specificity (fitness) (Knowles, 2010). Recent advances in machine learning have been applied to optimise directed evolution, leading to greater efficiency and predictive accuracy (James et al., 2024). However, most existing methodologies rely on DNA sequencing and synthesis, rendering them resource-intensive and less compatible with emerging in vivo mutagenesis techniques (James et al., 2024). In response to these limitations, James et al. (2024) explored the GB1 and TrpB empirical landscapes without sequencing information and demonstrated up to 19-fold and 7-fold increases, respectively, in the probability of reaching the global fitness peak. In the evolutionary design of ALife, directed evolution has produced a diverse range of simulated and robotic organisms, demonstrating the artificial evolution of various forms of “life as it could be” (Aguilar et al., 2014). In bioengineering, directed evolution enables the rapid selection of tiny building blocks of life (biomolecules) with properties suitable for applications such as protein engineering (Packer and Liu, 2015), isolating new enzyme variants with improved function (Currin et al., 2021), generating novel bio-catalysts (Coelho et al., 2013), and engineering genetic parts and synthetic gene circuits (Wang et al., 2021) as the biological components of various bioengineering applications (Cubillos-Ruiz et al., 2021). Directed evolution is becoming increasingly important as an experimental tool to bioengineer holistic genetic constructs containing multiple biological components (Vidal et al., 2023) (synthetic ALife). Since the first in vitro evolution experiments (Mills and Spiegelman, 1967), many techniques have been developed to address the two key steps of directed evolution: genetic diversification (library generation), and isolating genetic variants of interest (Vidal et al., 2023).
Directed evolution modalities can be classified into three categories: traditional directed evolution (Cheng et al., 2015), growth-coupled directed evolution (Chen et al., 2022), and AI-assisted directed evolution (Yang et al., 2025). Traditional and growth-coupled approaches rely primarily on experimental diversification and selection, whereas AI-assisted directed evolution augments or partially replaces empirical screening with model-guided design. Complementary strengths between these frameworks exist as traditional and growth-coupled methods offer robust, biology-native selection regimes but can be experimentally intensive, while AI-assisted DE can accelerate convergence and explore sequence space more efficiently, at the cost of increased dependence on data quality and modeling assumptions. Table 1 presents a comparison of the three modalities.

Table 1. Comparison of traditional DE, growth-coupled DE, and AI-assisted DE.
2.1 Computational and experimental directed evolution
In bioengineering, directed evolution is formulated as an algorithmic process that mimics natural selection to improve biomolecules with desired properties (Yang et al., 2019). It involves generating genetic (DNA) diversity for a specific individual, expressing the genetically diversified DNA through transcription and translation systems, screening the resulting sequences, and then selecting specific mutant sequences for the next round (Wang et al., 2021). During transcription the information encoded in DNA is converted into a transient carrier of information (mRNA), while translation is the process by which the mRNA code is converted into an amino acid polymer by ribosomes (RNA catalysts called ribozymes) (Taylor, 2006; Cech, 2000; Engstrom and Pfleger, 2017). Similarly, in in silico (Sims, 1994; Auerbach and Bongard, 2014; Joachimczak et al., 2016; Kriegman et al., 2018) and robotic (Lipson and Pollack, 2000; Brodbeck et al., 2015; Jelisavcic et al., 2017; Hale et al., 2019) ALife systems, evolutionary design is governed by evolutionary algorithms, which simulate the same principles of variation and selection (Bartz-Beielstein et al., 2014). Initialized with a population of randomly generated solutions, the algorithm enters a loop where it evaluates each generation of solutions, assigns a fitness (or solution quality), and applies variation operators (mutation and crossover) to the fittest subset to create the next-generation (Eiben and Smith, 2015a). The solution quality of directed evolution is driven by the selection (fitness function) and exploration of the search space (fitness landscape), which are determined by these variation operators (Wang et al., 2021). Genetic diversity methods in computational and bioengineering models are further discussed in Sections 3.1, 3.2. The importance and challenges of fitness landscape mapping are explained in detail in Section 5. Parallels between experimental and computational approaches to generating genetic diversity are summarized in Table 2.
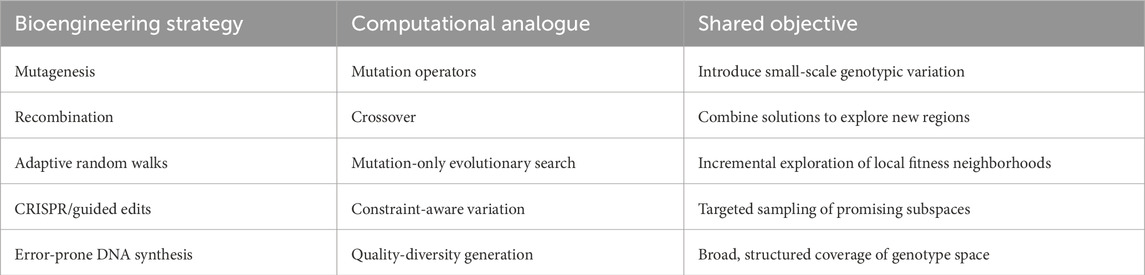
Table 2. Parallels in experimental and computational strategies for genetic diversity generation.
2.2 Fitness landscapes and adaptive dynamics
Selection refers to the process by which organisms possessing advantageous traits are more likely to survive and reproduce, thereby transmitting these traits to their offspring (Bell, 2008). This process serves as the primary mechanism driving the evolution of diverse forms, functions, complexity, and behaviors observed in ALife (Gershenson, 2023), where bioengineering relies upon a natural selection approximation (fitness) (Barton, 2022). Specifically, in the in vitro evolutionary environments of bioengineering laboratories, sequence fitness is equated with function performance such as relative catalytic rates of enzymes (Rotrattanadumrong and Yokobayashi, 2024) or observed fluorescence of protein genotypes (Sarkisyan et al., 2016), while ALife evolution in silico equates agent fitness with solution quality, represented as a numerical value. In both cases, a conceptual representation that maps genotypes to their corresponding fitness values (fitness landscape) (Hartl, 2014) is assumed to underlie evolutionary adaptation of individual agents, where the heritable differences in the genetic material and its arrangement among the population (genetic variation) driven by mechanisms such as mutation and recombination are coupled with selection, enabling the propagation of higher fitness individuals over evolutionary time (Mitchell-Olds et al., 2007).
An ongoing challenge in the evolutionary design of problem-solving ALife, for example, evolved robotic agents (Lipson and Pollack, 2000; Brodbeck et al., 2015; Jelisavcic et al., 2017; Hale et al., 2019), is premature convergence to sub-optimal solutions (for example, robotic forms and functions). Similarly, directed evolution of sequence design for various bioengineering applications (Currin et al., 2021) also suffer from convergence to sub-optimal solutions. This is typically a result of poor or highly localized search of the fitness landscape underlying the evolution of a genetic individual (sequence) (Firnberg et al., 2014). Recent studies of empirically mapped fitness landscapes demonstrate that epistatic interactions, particularly reciprocal sign epistasis, can create barriers that render certain fitness peaks inaccessible from specific ancestral genotypes, thereby constraining global optimization even in computationally or experimentally evolved systems (De Visser and Krug, 2014; Nishikawa et al., 2021; Poelwijk et al., 2011; Weinreich et al., 2005).
2.3 Exploration and exploitation in evolutionary search
As in the evolutionary design of ALife agents with desired forms and functions, bioengineering applications must similarly balance locally optimizing variants around known high-fitness sequences (exploitation) versus searching widely in sequence space for novel functional solutions (exploration) in the evolutionary search (Crepinsek et al., 2013) of the fitness landscape. For example, via enabling sufficient exploration of new solutions (via genetic variation operators) while concurrently exploiting (via selection operators) already discovered solutions (Wang et al., 2021). Practically, this means maintaining sufficient genetic diversity in solutions so as enough effective (high fitness) solutions can be screened, synthesized, tested and validated (Currin et al., 2021). Achieving an exploration (broad search) versus exploitation (local optimization) balance is especially prevalent in bioengineering given the expense of experimentally verifying solutions (in vitro), and added risks of wasting time and funds on synthesizing sub-optimal solutions. Realistically, as many sequences as practically possible should be screened and evaluated, meaning that any evolutionary search process enacted by bioengineering applications are orders of magnitude more expensive than their in silico evolutionary design counterparts. This expense is calculated in terms of computational run-time, the fiscal cost of producing synthetic sequences and the time taking for testing synthesized sequences (Reetz et al., 2008). This accentuates the critical importance of balancing exploration (broad search) versus exploitation (local optimization) in the intractable search space of genetic sequences (Currin et al., 2015). Thus, if hybrid ALife systems are to be efficient (minimizing experiment resources) in the evolutionary design of effective (problem-solving) ALife agents, then such an evolutionary design processes must suitably balance exploitation (optimization) versus exploration (broad search) of the fitness landscape underlying ALife agent design. An open issue that continues to pervade evolutionary design in ALife and bioengineering applications alike is the limited discovery of new genotypes or phenotypes that gives the population achieve higher fitness or new adaptive fitness function (novel solutions), where such limitations are determined by the capabilities of genetic variation operators, generating feasible novel solutions that can be selected and evaluated by the evolutionary design process.
3 The directed evolution problem: generating genetic diversity
There has been considerable progress in applying directed evolution to synthesis of fundamental biological components, for example, enabling de novo designs of functional proteins with new topologies, assemblies, binders, catalysts and materials (Cao et al., 2022). However, current macromolecule designs produced by directed evolution methods are still far from the complexity and variety of those found in nature (Ingraham et al., 2023). This is because such directed evolution methods rely on iterative search and incremental sampling of highly rugged and complex fitness landscapes (Wang et al., 2021). Given the limited sampling possibility in laboratory timescales, directed evolution typically explores only a small fraction of sequence space (Currin et al., 2015). While natural evolution itself is not necessarily globally optimal and is shaped by historical contingencies and co-evolutionary constraints, it has nonetheless explored vast regions of sequence space over billions of years, resulting in a high diversity of effective natural solutions (Koskella et al., 2017). Consequently, directed evolution may often rediscover or modestly improve upon natural functionalities rather than consistently surpass them (Turner, 2009). An overview of algorithmic applications in directed evolution is provided in Table 3.

Table 3. Algorithms and their applications in directed evolution.
3.1 Genetic diversity in evolutionary algorithms
In evolutionary design (computation) algorithms, as the fitness landscape dimensionality increases, the effectiveness of evolutionary search degrades, leading to sub-optimal solution convergence. Addressing such sub-optimal algorithmic convergence requires solution diversity maintenance. Diversity maintenance methods either maintain genotype (solution encodings on the fitness landscape), or phenotype diversity (solution diversity in the application space) (Eiben and Smith, 2015a). Genotypic and phenotypic diversity maintenance are well explored topics in evolutionary ALife design (Cully and Demiris, 2018; Miras and Ferrante, 2020; Nordmoen et al., 2021; Mkhatshwa and Nitschke, 2024). Some key strategies for maintaining and increasing genetic diversity include niching, fitness sharing, crowding, and multi-population models (Sareni and Krahenbuhl, 1998). Niching encourages subsets of solutions to specialise in distinct regions of the problem space, promoting the formation of diverse subpopulations (Li et al., 2016). Fitness sharing reduces the effective fitness of an individual by dividing its raw fitness by a factor representing the density of similar individuals nearby (Della et al., 2004). Crowding ensures that a new offspring replaces only the most similar individual in the current population (Sng et al., 2017). Finally, multi-population models prevent premature convergence by maintaining separate evolutionary lines that are less likely to become trapped in the same local optimum (Li et al., 2015). Together, these methods encourage broader exploration of the solution space. Phenotype diversity is essential for exploring a wider range of solutions and is closely linked with quality diversity (Mouret and Doncieux, 2012). Quality Diversity (QD) algorithms aim to generate not just high-performing but also varied solutions (Pugh et al., 2016). Popular examples include Novelty Search (Pugh et al., 2016), which rewards the discovery of new behaviors rather than optimizing performance directly, and MAP-Elites, which organizes and preserves diverse high-performing solutions across different behavioral dimensions (Mouret and Clune, 2015). In phenotype diversity maintenance the search for phenotypically diverse solutions replaces the fitness function, meaning an encoded solution (genotype) is more likely to be selected for evolutionary variation and propagation if its phenotype (behavior) is sufficiently different and fit compared to solutions discovered thus far (Pugh et al., 2016). Phenotypic diversity maintenance has been successfully applied to boost the quality of evolved ALife organism designs (Cully and Demiris, 2018; Miras and Ferrante, 2020; Nordmoen et al., 2021; Mkhatshwa and Nitschke, 2024). In either case, solution diversity is derived from mutation of an individual’s genotype.
3.2 Genetic diversity in bioengineering
Mutation serves as a fundamental source of genetic diversity, and facilitates the enhancement of a population’s evolvability and fitness (Wagner, 2023). Genetic stability in engineered cell populations remains a significant challenge in synthetic biology, as synthetic constructs impose burdens on host cells and promote mutations that can eliminate engineered functions (Sechkar and Steel, 2025). Previous mutation-control strategies have typically relied on gene-specific designs, which limit their general applicability (Sechkar and Steel, 2025). Sechkar and Steel (Sechkar and Steel, 2025) proposed a universal biomolecular controller that suppresses mutant growth regardless of circuit identity, demonstrating enhanced performance and adaptability through resource-aware modeling and simulation. A prevalent example is of mutations in the SARS-CoV-2 genotype leading to changes in the protein structure (genetic diversity) resulting in an increase in the infectiousness, fitness, and virulence of the SARSCoV-2 virus (DeGrace et al., 2022). In bioengineering, approaches for generating genetic diversity include adaptive random walks (Rotrattanadumrong and Yokobayashi, 2024) of the conceptual representation that maps genotypes to their corresponding fitness values (fitness landscape), reshuffling of hereditary information by exchanging segments between related sequences, creating new combinations (recombination) either through DNA shuffling (Stemmer, 1994), and unpredictable changes throughout a genetic sequence to create a wide variety of variants (random mutagenesis). The latter is done in vitro using specially designed alterations in synthetically derived DNA (Morrison et al., 2020), or in vivo via genotype engineering tools such as CRISPR (Naseri and Koffas, 2020). Such genetic diversity generation methods enable the sampling and testing of a broader range of genetically diverse sequences, which is tantamount to evolutionary computation diversity maintenance methods such as quality-diversity (Pugh et al., 2016), where both approaches are designed to improve fitness landscape exploration and thus solution quality in directed evolution (Wang et al., 2021).
Adaptive random walks which are a stepwise evolutionary process that normally start with the wild type or reference sequence, and progresses toward sequences with higher fitness are the simplest approach to fitness landscape exploration (broad search) and thus discovery of genetically diverse solutions (Emmeche, 1996). Such random walks explore the landscape via enabling a given sequence to move to a neighboring sequence, for example, via a single mutation, with a probability proportional to the fitness of that sequence (Papkou et al., 2023). Such an adaptive walk process models a low mutation rate where mutants of a given sequence have only a single mutation, though higher mutation rates enable greater exploration of the fitness landscape (Saha et al., 2024). Adaptive random walks are analogous to evolutionary algorithms using mutation only (Eiben and Smith, 2015a), and have been successfully applied to explore the fitness landscapes of synthetic Escherichia coli (Santo et al., 2023) and RNA sequences (Wagner, 2023). Another popular exploration (diversity generation) method is recombination (DNA shuffling), where homologous sequences are fragmented and reassembled (Stemmer, 1994). DNA shuffling has been applied as a means of to assist directed evolution of proteins, enzymes, and metabolites (Zhang et al., 2019), for example, using the Synthetic Chromosome Rearrangement and Modification by LoxP-mediated Evolution (SCRaMbLE) method (Ma et al., 2019), for in vitro genotype rearrangement. Similarly, random mutagenesis methods explore the fitness landscape of a genetic sequence with precise, controlled mutations of the given sequence (thus generating genetic diversity), using, for example, targeted mutagenesis and error-prone polymerase chain reaction (epPCR) (Wong et al., 2006).
Alternate methods for generating genetic diversity in vitro have been proposed in the form of DNA synthesis methods (Wang and Vasquez, 2023). For example, Wang B. et al. (2024) proposed a directed evolution approach for in vitro genetic diversification in artificial DNA synthesis, a foundational technology in synthetic biology based on the assembly of synthetic oligonucleotides into double-stranded DNA. One method is error-prone Artificial DNA Synthesis (epADS), where base errors generated during the chemical synthesis of oligonucleotides are treated as random mutations, enabling epADS to introduce diverse mutation types (including base substitutions and indels) for in vitro genetic diversification of synthetic DNA. Applications studied included genetic diversification of synthetic circuits, and microbial cells demonstrating various levels of phenotypic modification (Wang B. et al., 2024). Furthermore, these computational design methods support modularity in synthetic biology, allowing the generation of standardized biological parts and their assembly using widely adopted synthetic biology assembly workflows, ensuring high interoperability, scalability, and facilitate practical adoption.
3.3 From evolutionary limits to computational design
Although such fitness landscape exploration (genetic diversity generation) methods have been successfully demonstrated for various in vitro applications (Müller et al., 2005), the complex, rugged, high-dimensional topography of fitness landscapes presents significant challenges for their broader use (De Visser and Krug, 2014). Such exploration methods are still deficient for eliciting broad genetic diversity maintenance and thus an extensive fitness landscapes exploration (Müller et al., 2005). This failure largely results from the inherently high dimensionality and ruggedness of fitness landscapes, where local optimization and limited sampling prevent exhaustive exploration, while statistical or smart library designs further narrow the search to preselected sequence subsets (Van Cleve et al., 2015). Consequently, poor exploration of fitness landscapes significantly limits the quality of solutions discovered by directed evolution. Various experimental improvements to synthetic biology methods have been proposed to improve the efficacy of exploration, and they are highly compatible with synthetic biology because their modular structure aligns effectively with established assembly methodologies. These experiments include creating smart libraries (Tang et al., 2016) of reduced diversity sequences to restrict the search space to statistically designed variants, thus improving process efficiency by lowering the portion of mutated sequences (Currin et al., 2021). Prevalent examples include: CodonGenie (Swainston et al., 2017), ANT (Engqvist and Nielsen, 2015), SwiftLib (Jacobs et al., 2015), DYNAMCC (Halweg-Edwards et al., 2016), DC-Analyzer and MDC-Analyzer (Wang et al., 2015). However, while such digital libraries reduce the experimental workload via reducing the screening duplicate or unwanted variants, such approaches assume that the selected variant sequences (stored in smart libraries) are already near optimal for the tested conditions; however, their effectiveness may still vary depending on the specific genetic and environmental contexts in which the genetic components are implemented. Such in silico tools have demonstrably enhanced the entire experimental process, for example, reducing manual intervention in experiment setup via integrating assembly designs with laboratory automation scripts to increases throughput and accuracy (Carbonell et al., 2016).
However, optimal use of artificial evolution as the primary design mechanism in bioengineering applications remains elusive, given that directed evolution is often confounded by erroneous assumptions that evolutionary trajectories always follow adaptive walks (Williams, 1996), and unanswered questions about how evolutionary trajectories can span fitness landscape valleys (Steinberg and Ostermeier, 2016). To mitigate such open issues recent bioengineering applications have gone beyond the evolutionary operators of mutation and recombination, with the in vitro synthesis of new sequences, optimized in silico with predictive and generative machine learning (Wittmann BJ. et al., 2021). In such approaches, high fitness sequences are selected by the experimenter as a starting point for further optimization, synthesis, and testing in the design-build-test cycle of bioengineering (Carbonell et al., 2016).
4 Directed evolution: what now and what next?
Evolutionary design, employing various recombination and mutation variation operators (Eiben and Smith, 2015a), has been successfully applied to explore fitness landscapes and direct the evolution of in silico ALife for decades (Wilke et al., 2001). Similarly, various analogues of artificial evolution operators (Eiben and Smith, 2015a), such as random mutagenesis (Copp et al., 2014) and DNA shuffling (Stemmer, 1994), have been applied for fitness landscape exploration and directed evolution in various bioengineering applications (Currin et al., 2021). The simplest artificial evolution analogue in directed evolution for bioengineering is the adaptive random walk (Papkou et al., 2023), where the fitness landscape is explored via generating and evaluating the fitness of mutant sequences from a given high fitness wild-type sequence (Rotrattanadumrong and Yokobayashi, 2024). Though, such adaptive random walks do not currently employ equivalents of the myriad of selection operators (Eiben and Smith, 2015a) available to the directed evolution of simulated ALife (Aguilar et al., 2014). Rather, selection operators in the directed evolution of bioengineering applications are replaced with experimental selection using specially designed mutant screening (biochemical assay) processes (Duong-Trung et al., 2023).
4.1 The combinatorial constraint
Regardless of random walk functionality, few fitness landscapes can be completely (exhaustively) mapped (Rowe et al., 2010) as the underlying sequence space is astronomically large (Rowe et al., 2010), though partial mappings have provided valuable insights to landscape topography (ruggedness) for specific sequence types such as proteins (Currin et al., 2015). For instance, a protein of 100 amino acids could, in principle, adopt a vast number of possible sequences, far exceeding the number of atoms in the observable universe (Koonin et al., 2002). Similarly, a 100-nucleotide RNA has potential variants, and even considering only single and double mutants of a 300-residue protein already involves over 16 million sequences (Keller et al., 2018). This combinatorial explosion renders exhaustive mapping infeasible; nevertheless, partial mappings have provided valuable insights into the topography of fitness landscapes for specific sequence types such as proteins (Kondrashov and Kondrashov, 2015). Hence, just as the fitness landscape metaphor has guided the directed evolution of simulated and robotic forms of ALife (Lipson and Pollack, 2000; Joachimczak et al., 2016; Jelisavcic et al., 2017; Hale et al., 2019), fitness landscape mappings provide valuable observations that assist in guiding directed evolution methods and the efficacy of sequence design in bioengineering more generally (Castle et al., 2024). The problem of balancing exploration versus exploitation in any directed evolution method continues to confound simulated, robotic and synthetic ALife alike, though various genotype and phenotype diversity maintenance methods (Pugh et al., 2016), including non-objective based (novelty) search (Lehman and Stanley, 2011), and hybrids of non-objective and objective (fitness function) based evolutionary search (Nitschke and Didi, 2017), have demonstrated improved designs of various forms of ALife (Cully and Demiris, 2018; Mkhatshwa and Nitschke, 2024), including unexpected, non-intuitive designs (Lehman et al., 2020). Such diversity maintenance methods have also been proposed as means to boost fitness landscape exploration and thereby continually discover novel synthetic ALife designs for bioengineering applications (Stock and Gorochowski, 2024).
4.2 Continuous evolution platforms
Rapid advances in continuous directed evolution have led to many new experimental platforms that closely combine in vivo diversification, automated selection, and real-time measurement. Among these, PACE, OrthoRep, EvolvR, and automated continuous-evolution systems are the most advanced for sustained, hands-free evolution of biomolecules. These platforms utilize various methods to connect genetic diversification to organism growth or phage propagation, thereby reducing manual work. They offer strong tools for closed-loop optimization through the build–test–select cycle with automation.
Phage-assisted continuous evolution (PACE) is considered the first broadly applicable system for fully continuous in vivo (in living organisms) directed evolution (Esvelt et al., 2011). Evolving genes are carried on filamentous phage (viruses that infect bacteria) that propagate through E. coli (a common laboratory bacterium) in a chemostat-like (continuous culture) setup, where phage infectivity, and thus replication rate, is directly tied to the activity of the biomolecule under selection (Arnold, 1998). This architecture enables numerous evolutionary rounds with minimal intervention, significantly accelerating and deepening evolutionary searches compared to batch (stepwise) methods (Arnold, 1998). PACE has been utilized to evolve diverse protein functions, including altered protease (enzyme) and DNA-binding specificities, and deep sequencing (analyzing DNA with high coverage) has revealed the underlying adaptation pathways (Miller et al., 2020). Together, these advances establish PACE as a mature, partially automated platform well-suited for integration with computational design and analysis.
OrthoRep is a yeast-based continuous evolution system that utilizes an orthogonal DNA polymerase plasmid pair to drive mutation rates ∼ 105-fold above those of the yeast genome, while preserving genomic stability (Ravikumar et al., 2018). Genes placed on the orthogonal plasmid undergo continuous error-prone replication during routine culturing, enabling scalable evolution through simple serial (Ravikumar et al., 2014). OrthoRep has been used to evolve drug-resistant malarial DHFRs, probe large ortholog sequence spaces, and optimize enzymes for plant metabolic engineering, demonstrating deep, multi-lineage evolutionary searches with minimal handling (Rix et al., 2020). Overall, OrthoRep is a robust platform for continuous gene diversification and selection, supporting computational frameworks that propose targets, lineages, or selection strategies.
EvolvR is a CRISPR-guided (Cas protein and guide RNA-based), locus-specific (targeting exact DNA sites) continuous diversification system that can target either genomic (organismal DNA) or plasmid (circular DNA) sequences (Halperin et al., 2018). It utilizes a Cas9 nickase (a DNA-cutting protein that creates single-strand breaks) fused to an error-prone polymerase (a mutation-adding enzyme), with guide RNAs directing mutations to user-defined loci within a tunable window around the nick site (the location of the DNA cut) (Halperin et al., 2018). In this way, EvolvR generates focused mutational clouds (clusters of variants) during growth (Halperin et al., 2018). This localized diversification enables parallel evolution of multiple genetic elements and, importantly, can be re-targeted as design goals or inferred fitness landscapes (relationships between genetic changes and organismal function) change (Halperin et al., 2018). In subsequent work, EvolvR has been positioned within a broader class of CRISPR-based evolution tools (Abbott and Qi, 2018). This highlights its potential for closed-loop (iterative, feedback-driven) optimization of genetic parts, regulatory sequences, and enzymes when combined with automated assays and computational design.
Automated continuous-evolution (ACE) systems combine in vivo mutagenesis (mutation induction within living cells) with programmable hardware to enable hands-free, feedback-controlled evolution over long timescales (Zhong et al., 2020). The ACE platform links OrthoRep’s targeted hypermutation in yeast to the eVOLVER system, an array of independently controlled culture vessels equipped with automated dilution (automatic culture thinning), temperature regulation, and media switching (changing growth liquid), enabling software-defined adjustments of selection pressures (factors determining survival) and culture conditions (Heins et al., 2019). These setups have evolved enzymes with altered temperature optima (best activity temperatures) and catalytic properties over hundreds of generations with minimal intervention, demonstrating how continuous evolution, real-time monitoring, and programmable feedback can operate as a unified platform.
Most continuous evolution platforms optimize only phenotypes that are readily measurable, such as simplen growth - infectivity linked traits. As a result, these systems tend to fine-tune narrow functions rather than reveal complex or context-dependent behaviors. They also depend heavily on specific chassis organisms and fixed culture conditions, which can further constrain evolution to a particular cellular environment. This dependence reduces the portability of evolved variants and limits the exploration of diverse biological contexts. Additionally, the prevailing mutation modes and population dynamics promote dense local search rather than large evolutionary leaps, often causing systems to become trapped on local fitness peaks and overlook rare but innovative solutions. Collectively, these constraints enable rapid optimization within a defined niche but restrict these platform’s capacity to generate truly novel and broadly adaptable functions.
4.3 Machine learning as a catalyst for evolutionary discovery
Predictive and generative machine learning methods provide a complementary approach to boost genetic diversity and address the exploration versus exploitation problem in directed evolution via discovering new functional sequences (Brya et al., 2021), where combinatorial mutations generate greater genetic diversity, accelerating the discovery of novel traits and optimizing adaptive potential in evolving organisms (Schlötterer et al., 2015). However, genetic sequence information is sometimes poorly represented, since non-functional patterns and high levels of expression are often under-represented in training data (Fernandez-de Cossi et al., 2021), which limits exploration of the fitness landscape and reduces overall benefit of machine learning methods in directed evolution. However, machine learning can increase the accuracy and efficiency of directed evolution methods via generating new genetically diverse sequences and predicting the efficacy of such newly generated sequences, such as proteins (Notin et al., 2024) and enzymes (Landwehr et al., 2020). For example, Large Language Models have been applied to protein discovery (Ingraham et al., 2023), generated to user specifications (Lin Z. et al., 2023). A study by Biswas et al. (2018) used predictive machine learning to address issue of directed evolution in protein engineering, where evolved proteins often became trapped in local solution space maxima, and yielding only marginal improvements (Romero and Arnold, 2009). They encoded protein sequences as learned amino acid embeddings and used them to train Linear Regression, Feed-Forward Networks, and a novel Composite Residues model with the goal to predict log fluorescence via mean-squared error loss, and the models jointly learned embeddings and parameters through empirical tuning. Multiple train-dev-test splits were implemented to evaluate the model’s generalization. Performance was benchmarked using the mean squared error and false discovery rate, with an emphasis on robustness in identifying functional variants. Biswas et al. (2018) demonstrated via combining supervised machine learning, DNA synthesis, and high-throughput screening that data-driven machine learning can predict novel, functional protein variants, thus sufficiently exploring the fitness landscape, and aiding directed evolution in optimizing protein design. The proposed approach by Biswas et al. (2018) provides an efficient directed evolution strategy for exploring unseen regions of the fitness landscape, escaping local maxima, and increasing the likelihood of finding useful protein variants, which traditional methods can struggle to find. However, despite its potential to improve the speed and efficiency of protein engineering, this model depends on the sufficiency of high-quality training data and the model’s capability to generalize beyond known variants is limited. In the absence of such resources, model generalization beyond known variants is limited, and predictions may fail to accurately represent biological reality (Ching et al., 2018). Furthermore, as demonstrated in large language models applied to biological data, generative and predictive models may produce plausible yet incorrect outputs, underscoring the necessity for rigorous benchmarking and experimental validation prior to deployment in practical protein design applications (Zhang et al., 2025).
One prevalent example is the work of Ding et al. (2019) used generative machine learning to address open issues with a protein inference method, heuristic phylogeny reconstruction, including the method’s failure to capture high-order epistasis effects. Using both simulated and experimental data, Ding et al. (2019) demonstrated that latent space models, trained using Variational Auto-Encoders (VAEs) and information within multiple sequence alignments of protein families, can help capture phylogenetic relationships, boost exploration of protein fitness landscapes, and thus aid in predicting protein stability change upon single-site mutations. Variational autoencoders were trained on one-hot encoded multiple sequence alignment data. Each sequence was mapped to a Gaussian posterior in a low-dimensional latent space, and the decoder produced per-position amino acid probabilities. For training, they employed the reparameterization trick with stochastic variational inference and mini-batch optimization. Sequence weights derived from the multiple sequence alignment were incorporated during model training. Modeling the protein sequence space using Variational Auto-Encoders (VAEs) created a condensed search space, increasing fitness landscape exploration and the likelihood of finding beneficial mutations. However, the VAEs summarize complex protein data meaning resulting representations often lack biological interpretability where this lack of interpretability hinders an explainable design process in directed evolution.
Another emblematic is where Angenent-Mari et al. (Wang et al., 2020) applied predictive machine learning to address open problems with effective design and validation of RNA molecules in the bioengineering of RNA molecules with targeted biological functions (Pardee et al., 2016). Specifically, Angenent-Mari et al. (Wang et al., 2020) developed a high-throughput DNA synthesis, sequencing, and deep-learning pipeline to design and analyze a programmable RNA switch, achieving a ten-fold improvement in functional prediction compared to traditional thermodynamic and kinetic models. The Deep Neural Networks (DNNs) pipeline proposed by Angenent-Mari et al. (Wang et al., 2020), generated attention visualizations (VIS4Map), providing a way to visualize sequence parts that contribute to the success or failure of the RNA switches, enhancing understanding of the underlying functionality and identification of critical features or motifs that drive RNA functionality. However, the DNN pipeline is based on sequence-function relationships, meaning it only accounts for some of the complex interactions present in RNA molecules, leading to potential over-simplifications in predicted (designed) RNA sequences.
Hence, even though there has been some cross-fertilization between evolutionary design, machine learning, and the design-build-test processes of bioengineering, synthetic biology still lacks an equivalent to the autonomous, iterative evolutionary processes used in simulated and robotic ALife, where systems evolve new functions without explicit human-guided design (Lipson and Pollack, 2000; Aguilar et al., 2014; Joachimczak et al., 2016; Jelisavcic et al., 2017; Hale et al., 2019). Though, various bioengineering case studies (Biswas et al., 2018; Ding et al., 2019; Wang et al., 2020) highlight a deficiency in methods to generate genetic diversity (discovering new genetic sequences) and overcome the confounding exploration versus exploitation problem of directed evolution. We envisage future synthetic ALife design-build-test processes as encapsulating complements of predictive and generative machine learning (Wittmann BJ. et al., 2021), hybrid (objective and non-objective based) diversity maintenance operators and adaptive random walks using recombination and mutagenesis. Design, would thus be novel sequence discovery (generated genetic diversity) achieved via generative machine learning or evolutionary operators (recombination or mutagenesis). The probable efficacy (fitness) of discovered sequences could be gauged by data-driven predictive machine learning and experimentally built (in vitro), to test the efficacy of built sequences. Fitness information could then be fed back into a library of sequences, representing an in silico empirically mapped fitness landscape. The fitness landscape comprehensiveness and thus quality of ALife solutions produced by directed evolution, could then be incrementally improved with each iteration of the design-build-test process (Carbonell et al., 2016).
However, the success of directed evolution presupposes a suitably mapped fitness landscape, where such a mapping successfully bridges the fitness landscape disconnect − between a biological agent’s natural fitness landscape and its empirically mapped digital counterpart. Given this, we next review methods for mapping fitness landscapes as a means to effectively guide directed evolution.
5 Mapping fitness landscapes
Fitness landscape mapping aims to determine the effects of variations in an organism’s genetic blueprint (genotype), such as sequence changes or design parameters, on the phenotype or performance (fitness) (Greenbury et al., 2022). This is achieved by quantifying how different types of mutations or structural modifications influence overall function in an organism, molecule, or digital organism (Wilke and Adami, 2002). As the sequence length of the genotype increases, the possible variants also increase exponentially, creating a potentially large combinatorial space (Alkan et al., 2011). Mapping such landscapes, therefore, poses a significant technical challenge, not only because of the vast amount of quantifiable data required to adequately sample this space (Vaishnav et al., 2022), but also due to the difficulty of obtaining high-quality, reproducible measurements (Hietpas et al., 2011). Experimental and computational methods must cope with noise, measurement bias, and limited throughput, all of which can obscure the true topography of the landscape (Eling et al., 2019). Despite significant advances that have made in the complete empirical mappings of conceptual representation that maps genotypes to their corresponding fitness values (molecular fitness landscapes), such as that of tRNA (Domingo et al., 2018) and ribozymes (Pressman et al., 2019), enabled by in vitro selection and advances in high-throughput sequencing, the intractable size of sequence space limits purely experimental investigations, especially for complete genotype sequences (North et al., 2021). Machine learning is increasingly utilized as a data-driven approach to characterize fitness landscapes and improve directed evolution strategies (Towers et al., 2024). Towers et al. (2024) demonstrated that machine learning models can surpass traditional evolutionary control methods, particularly in highly complex and rugged environments. These models are also practical for laboratory use because they require only population-average data, rather than individual-level measurements, thereby providing a scalable and accessible tool for guiding directed evolution experiments (Towers et al., 2024). This section examines the significance of fitness landscape mapping and the methodologies employed to study fitness landscapes.
5.1 Conceptual foundations: why fitness landscapes matter
In evolutionary design, a fitness landscape represents the solution space of a given problem, where the solution representation (genotype encoding) used by the evolutionary algorithm defines the level of detail at which data and features are sampled, represented, and distinguished by the algorithm (granularity) and how the different solutions are connected to each other (topology) of the landscape (Jones, 1995). The specific rule or calculation for assigning a quantitative score that measures the goodness of any possible solution for a given problem (fitness function) serves to select and propagate encoded solutions (genotypes) such that the population of solutions coalesces at optimal regions (fitness peaks) and the algorithm thus generates fit (high-quality) solutions (Eiben and Smith, 2015b). Evolutionary design and optimization has a diverse set of applications, ranging from satellite-antenna design (Hornby et al., 2011), robotic controller design (Doncieux et al., 2015), robotic swarm control (Vásárhelyi et al., 2018), architectural (Kicinger et al., 2005) and building material design (Collins et al., 2016), optimizing ligands against polypharmacological profiles (Besnard et al., 2012) to evolving ALife digital organisms (Wilke et al., 2001). All depend on an evolutionary design algorithm that adequately represents the space of possible solutions as the fitness landscape and then effectively explores the fitness landscape for high quality solutions (Vikhar, 2016). Beyond the digital realm, suitably representing the fitness landscapes of robotic ALife that evolves in physical environments has remained challenging, since the fitness landscape underlying online robot evolution corresponds to body-brain couplings that adapt in response to dynamic environments (Doncieux et al., 2015). Thus, with few exceptions (Brodbeck et al., 2015; Hale et al., 2019; Nygaard et al., 2021), most examples of robotic ALife are evolved offline in simulation and then physical versions are built for counterpart real-world environments as validation of simulated evolution (Lipson and Pollack, 2000; Jelisavcic et al., 2017).
Similarly, across bioengineering applications, genetic sequences are usually evolved in controlled laboratory environments with the goal of being deployed in uncontrolled (non-laboratory) environments (Cubillos-Ruiz et al., 2021). In such applications, fitness landscapes represent the space of all possible sequences, where specific sequences (genotypes) correspond to desired solutions (phenotypes) (Currin et al., 2015). The relationships between genotype and phenotype in the fitness landscape are the foundation of evolutionary design in bioengineering and, more generally, in natural evolution. For example, RNA enzymes (ribozymes), play critical roles in the RNA world hypothesis, positing that RNA comprises the metabolic architecture of the earliest cells (Pressman et al., 2015). As such, understanding the topography of the underlying fitness landscapes provides valuable insights into molecular evolution, such as how emergent catalytic properties evolve into complex living systems (Kun and Szathmary, 2015).
There have been similar insights for ALife evolved in silico and underlying fitness landscapes in simulated evolutionary systems (Franklin et al., 2019), where the concept of a fitness landscape is of critical importance to all directed evolution applications (Castle et al., 2024). For example, comprehensively mapping the protein fitness landscapes of SARS-CoV-2 variants elucidates potential paths of viral evolution thus informing the development of effective anti-SARS-CoV-2 drugs (Flynn et al., 2022). Various independent studies have determined that mostly biological fitness landscapes are rugged, meaning that they can be dynamic and complex with multiple peaks and valleys created by interactions between genes with even single mutations having the potential to cause strong epistatic effects are rugged and dynamic, for example, as demonstrated for viruses (Quadeer et al., 2020), where a high degree of complexity (ruggedness and dynamism) persists across the evolutionary process (Domingo et al., 2023). Despite the seemingly insurmountable complexity of fitness landscapes, recent work indicates that in some biological systems, such as bacterial (Rodrigues et al., 2016), viral (Chéron et al., 2016) and norovirus (Rotem et al., 2018), evolving in response to antibiotic and antiviral treatments, the underlying fitness landscapes can be systematically and quantitatively mapped. For example, Wang D. et al. (2024) found that the fitness landscape of the SARS-CoV-2 virus receptor bindings, evolving versus neutralizing antibodies, can be systematically described through biophysical properties such as antibody binding affinity and protein folding stability.
5.2 Simulation-based mapping of fitness landscapes
Simulation-based approaches play a crucial role in evolutionary dynamics by reconstructing or predicting fitness and the examination of adaptive trajectories and evolutionary outcomes landscapes in silico (Hoban et al., 2012). Lou et al. (2024) developed a computational method, Beth-1, to forecast influenza virus evolution and thus inform efficacious influenza vaccine design. Beth-1 predicts influenza evolution via modeling site-wise (virus genotype segments) mutation fitness, where site-wise fitness dynamics are used to predict the fitness of future influenza variants. Through adaptive estimation of fitness by genotypic site and time, and tracking all advantageous mutations, Beth-1 projects a probable (future) fitness landscape of the virus population. The method demonstrated promising prediction performances in both retrospective and prospective applications using pH1N1 and H3N2 virus data, demonstrating a clear advantage of using biological data to map fitness landscapes of influenza viruses. Given such fitness landscape mapping one can feasibly understand the underlying evolutionary dynamics of viruses and use predicted viral evolution to assist in vaccine design. A key limitation to this study was the sequence sample size and sparse evolution time intervals in the data-sets. This impacted the comprehensiveness of the mapped fitness landscape, reflecting that constructed fitness landscapes are only as good as the sampled evolutionary data, meaning predicted evolutionary trajectories may be inaccurate.
Radford et al. (2023) developed a computational method to map the fitness landscapes of synthetic bacterial ribosome (orthogonal tethered ribosome), and permit mutagenesis of nucleotides located in the ribosomal peptidyl transfer center (PTC) which is the RNA-based active site in the large ribosomal subunit (50S/60S) that forms peptide bonds (Radford et al., 2023). The ribosome is a macromolecular machine essential for protein synthesis in all organisms (Nissen et al., 2000). Thus understanding ribosome function via mapping underlying its fitness landscape is essential for evolutionary design of synthetic ribosomes for new bio-materials and therapeutics. Radford et al. (2023) reconstructed the fitness landscape of oRiboTs using next-generation sequencing data, where the fitness predictions from analyses of PTC libraries were validated experimentally. Such analyses measured the viability of mutations at individual PTC nucleotides and identified epistatic interactions between positions (nucleobases) within the PTC. This enabled the design of ribosome libraries based on an empirically constructed fitness landscape. Though experimentally validated, mapped fitness landscapes were not used to aid directed evolution for ribosome design. This implores the possibility of feeding back experimentally validated (synthesized) ribosomes to update the fitness landscape and thus guide ribosome evolutionary design.
5.3 Empirical mapping of biological fitness landscapes
Empirical mapping aims to experimentally quantify fitness values across defined genotypic variants, providing tangible insight into how mutations shape adaptive landscapes (De Visser and Krug, 2014). Biological fitness landscapes (containing a fixed number of mutational variants of a given molecule or organism) have been partially mapped to study the incidence and distribution of fitness effects of mutations and resultant evolutionary dynamics. For example, Wagner (2023) modeled adaptive evolution on a protein fitness landscape of an E. coli toxin-antitoxin system, comprising fitness values for 7,882 antitoxin protein genotypes, and an RNA fitness landscape of 4,176 yeast (Saccharomyces cerevisiae) transfer RNA (tRNA) genotypes.
Advances in high-throughput approaches are extending classical deep mutational scanning (DMS) into the single-cell domain, and facilitating direct and high-resolution mapping of genotype–phenotype–fitness relationships (Gantz et al., 2023). By coupling pooled variant libraries with single-cell readouts such as scRNA-seq, these methods capture variant effects across diverse cellular states rather than only bulk averages (Cuomo et al., 2023). For example, Zhao et al. (Qiu et al., 2020) developed scMPRA, which links thousands of regulatory variants to cell–type–specific transcriptional outputs within mixed populations, revealing how context shapes fitness effects (Science, 2022). Similarly, Lindenhofer et al. (2025) introduced SDR-seq, a single-cell DNA–RNA co-profiling approach that connects endogenous genetic variants to their transcriptional phenotypes at cellular resolution. Together, these technologies move DMS from scalar enrichment scores toward multidimensional, state-aware fitness landscapes that better capture pleiotropy and context dependence in molecular evolution.
Adaptive evolution is modeled as adaptive random walks, starting with a randomly chosen genotype, where the random walk followed genotype variants, changed via point mutations. Wagner (2023) analysis demonstrated that evolvability and fitness enhancing mutations are present in these (protein and RNA) fitness landscapes. However, the analysis only considered a small number of variable sites in mapping the protein and RNA fitness landscapes and relatively few mutations (thus modeling short-term adaptive evolution). This was due to the impracticality of constructing combinatorially complete adaptive fitness landscapes for entire proteins or genotypes for many mutations (long-term adaptive evolution). Though, this limitation holds for adaptive walks of many fitness landscapes mapped from biological datasets (O’Brien et al., 2024). Importantly, conclusions drawn from the modeled short-term adaptive evolution process are not directly comparable across fitness landscapes, because of varying fitness metrics. This is another limitation that continues to frustrate the analysis of evolutionary dynamics across fitness landscapes for various types of genotypes.
5.4 Machine-learning-based mapping of fitness landscapes
The enormous size of fitness landscapes corresponding to most genetic sequences has motivated recent work on predictive, data-driven (supervised machine learning) methods to assist in mapping fitness landscapes (Dechant and He, 2021). Such methods essentially interpolate (predict) new data points (functional sequences) given a current library of sequences (training dataset) (Khakzad et al., 2023). Such methods have already demonstrated the efficacy of predicting current and probable future fitness landscapes for entire genotype sequences (influenza A pH1N1 and H3N2 virus populations) (Lou et al., 2024), and for directed evolution in protein engineering (Wittmann B. J. et al., 2021). Supervised machine learning has also been applied to map the fitness landscapes of catalytic RNA, which is of special interest since RNA molecules can store genetic information (for example, RNA viruses) and catalyze chemical reactions (for example, ribozymes). Also, the relatively small sequence spaces of RNA makes it suitable for fitness landscape mappings and indispensable for bioengineering applications (Saha et al., 2024). Various supervised machine learning methods using data-driven interpolation and extrapolation over sequence spaces have also been proposed (Saha et al., 2024). Such methods enable prediction of potential evolutionary paths (sets of mutant sequences) between fitness peaks. However, the rarity of functional genotypes means that random sampling of a ribozyme fitness landscape would yield a dataset that is highly biased toward deleterious (non-functional) variants. Thus, supervised machine learning methods are challenged by data bias, since the majority of data will comes from low or moderate fitness landscapes, and where a scarcity of labeled data, that is, sequences with an associated measurement of the target property, continues to significantly limit the predictive capabilities of supervised machine learning applied to fitness landscape mapping (Wittmann B. J. et al., 2021).
Rotrattanadumrong and Yokobayashi (2024) addressed these problems by using in silico selection, recombination, and mutation to guide an adaptive walk along evolutionary paths in an RNA sequence space (F1*U ligase ribozyme). This generated a dataset with a more balanced distribution of neutral and deleterious mutants. These data were used to train a deep neural network to predict and identify functional mutational variants that have comparable fitness (relative ligation activity) to a wild-type (a synthesized ribozyme with relatively high fitness). Information about functional variants in distant regions of the fitness landscape was learned by the deep neural network using data acquired from a few mutational steps (starting from the wild-type). This enabled the mapping of the fitness landscape topography around a selected wild-type. The authors discovered an extensive neutral network (van Nimwegen, 2006), a set of genotypes connected by single mutations that sharing the same phenotype (for example, structure, catalytic activity), between the structurally and functionally similar ribozymes (F1*U, F1*U m). This indicated that neutral networks might be common among similar ribozymes, meaning such fitness landscapes could readily be traversed without being blocked by deleterious mutants, that is, evolving populations could travel large mutational distances without detrimental effects on fitness (Knezic et al., 2022). Given this, one could potentially use artificial evolution to engineer ribozymes that adopt a new structure while retaining its function, or acquiring a new function (Portillo et al., 2021). A key contribution of Rotrattanadumrong and Yokobayashi (2024), was integrating information from multiple rounds of in vitro selection, so underlying data were not biased solely toward deleterious mutants, thus enabling effective sequence prediction using a deep-learning method. However, supervised approaches require substantial quantities of labeled experimental data, the acquisition of which is both resource-intensive and time-consuming.
The time consuming nature of generating labeled data via experimental means (Rotrattanadumrong and Yokobayashi, 2024), has prompted the application of unsupervised machine learning methods trained on unlabeled sequence data to predict if given sequences will be functional or not. This alleviates the need for labeled datasets while still enabling elimination of non-functional sequences (Riesselman et al., 2018). Such methods include Generative Adversarial Networks (GANs) trained on a latent space underlying the sequence space to generate novel functional variant sequences (Ziegler et al., 2023). In such cases, the latent space is an abstract vector space that positions chemically similar sequences near each other (Ding et al., 2019). For example, GANs have successfully generated functional enzyme mutants, with differences of up to 106 point mutations from an original sequence of the malate dehydrogenase enzyme (Repecka et al., 2021), where the efficacy of newly discovered sequences can be verified experimentally with in vitro methods. For instance the ProteinGAN generated 55 malate dehydrogenase variants, of which 13 (approximately 24%) were both soluble and catalytically active demonstrating experimentally validated functional sequence generation (Repecka et al., 2021). Similarly the PepGAN produced antimicrobial peptides, and 5 of 6 synthesized candidates demonstrated confirmed antimicrobial activity and one peptide achieved a minimum inhibitory concentration of 3.1 µg/mL,which is comparable to ampicillin (Lin TT. et al., 2023). Generative models have also shown promise for novel RNA sequence discovery given their capability to learn underlying patterns and generate novel sequences for testing. For example, Variational Auto-Encoders (VAEs), can encode sequences into a low-dimensional latent space and subsequently decode the latent space back into a sequence (Klys et al., 2018). Assuming the latent space accurately represents the fitness landscape, then decoding the latent space assists in mapping high fitness peaks (Iwano et al., 2022). Validation of VAEs in biological sequence design has also been reported, as seen in Hawkins-Hooker et al. (2021), where a VAE trained on luciferase-like oxidoreductase sequences generated novel luxA variants that retained measurable luminescence activity in vitro (Hawkins-Hooker et al., 2021). Also, RaptGen employed a variational autoencoder (VAE) architecture trained on Systematic Evolution of Ligands by Exponential Enrichment (SELEX) data to generate RNA aptamers. Several aptamers generated in this study were experimentally confirmed to bind their intended targets with high affinity (Iwano et al., 2022).
Such predictive and generative machine learning methods are invaluable for identifying potentially functional sequences, unlikely to be found via experimental means due to the intractable size of the sequence search space (Biswas et al., 2021). However, as the number of possible combinations grows exponentially with the number of variables, data sparsity and exploration become inefficient (curse of dimensionality), which then pervades evolutionary design and optimization (Eiben and Smith, 2015a), similarly confounds bioengineering applications, since the total number of sequences and possible functional combinations is many orders of magnitude larger than what can be practically synthesized in vitro. For example, even representing a small 100 amino acid protein yields a sequence space larger than the number of atoms in the observable universe (Kondrashov and Kondrashov, 2015). Furthermore, biological fitness landscapes are in constant state of flux resulting from the complex interplay between evolving organisms, changing environments and evolutionary phenomena such as epistasis, where the impact of one mutation is dependent on other mutations (Storz, 2018).
Thus, mapping fitness landscapes for the purposes of directed evolution poses major technical challenges due to extremely large amounts of quantifiable data required (Saha et al., 2024). The intractable complexity of biological fitness landscapes highlights the need for universal adoption of canonical in silico fitness landscape mapping procedures to ensure consistency in mapping across disparate fitness landscapes (Alvarez et al., 2024). Furthermore, experimental in vitro procedures enable experimenters to determine if given mutations, for example, of machine learning generated or predicted sequences, are actually enhancing fitness (Wagner, 2023). For example, bioengineering applications can maintain a digital library of genetically diverse sequence mutants, where the fitness of all mutants has been experimentally evaluated for the purpose of empirically mapping to a fitness landscape (Yang et al., 2024). Hence, suitably mapped fitness landscapes can be used as predictive and prognostic tools enabling one to predict (given trackable mutations) evolutionary trajectories of sequences in given and related fitness landscapes. For example, the SARS-CoV-2 fitness landscape mapping of Wang D. et al. (2024) is hypothesized to be generally applicable to evolving influenza viruses (Gong et al., 2016; Fonville et al., 2014). Similarly, other fitness landscapes mapping underlying virus evolution, have provided insights into the evolutionary dynamics of viruses enabling more accurate prediction of viral evolution (Luksza and Lassig, 2014). Broad accessibility of fitness-landscape datasets is essential because these datasets support model training, benchmarking, and comparative evolutionary analyses (Pitzer and Affenzeller, 2012). Protein and enzyme fitness data are typically deposited in MaveDB, UniProt, and structural archives such as the Protein Data Bank (PDB) (Notin et al., 2023). Complementary kinetic information is available in BRENDA and Expasy ENZYME (Schomburg et al., 2012). Repositories including Rfam, RNAcentral, and miRBase provide standardized frameworks for sequence and structural annotation of RNA and regulatory elements (The RNAcentral Consortium, 2019). Genomic and viral data are distributed through GenBank, GEO, NCBI Virus, and GISAID (Bernasconi et al., 2021). Consistent use of public databases and metadata standards facilitates reproducible, interoperable, and integrative mapping of biological fitness landscapes across molecular systems (Brancato et al., 2024).
Though overall, such fitness landscape mapping studies continue to highlight the disconnect between fitness landscapes empirically mapped from sampled biological data and those that underlie evolving organisms in nature. Given this, we next discuss the fitness landscape disconnect that continues to frustrate the evolutionary design of synthetic counterparts to digital ALife.
6 The fitness landscape disconnect: what to do?
In the broad scope of evolutionary ALife there is a fundamental disconnect between the fitness landscapes that inform directed evolution of digital (in silico), physical (robotic), and synthetic (in vitro) organisms. For example, ALife evolved in simulation and then built as counterpart physical designs cannot continue to evolve their form (morphology) or function (behavior) in real-world environments due to a disconnect between the fitness landscape underlying digital evolution in simulation versus the fitness landscape underlying robotic evolution in a physical environment (Hauser, 2019). Similarly, in bioengineering, there is a disconnect between the artificial fitness landscapes underlying sequences evolved in vitro (in laboratory environments), and fitness landscapes underlying sequences evolved in vivo (in natural environments) (De Visser and Krug, 2014).
6.1 Consequences of the disconnect
Populations (sequences) evolved in vitro are rarely functionally transferable beyond specially controlled laboratories to real-world environments (Brooks and Alper, 2021). This limited transferability highlights a broader challenge of generalisation (Walker et al., 2010). Models or experimental systems designed with narrowly defined objectives or task-specific feature sets often capture context-dependent correlations instead of general principles (Kejriwal, 2021). As a result, their predictive or functional performance declines in variable or less-controlled environments (Cavuoto and Nussbaum, 2014). Recent research on general or foundation models aims to identify invariant features across diverse datasets, which may enhance robustness and external validity (Awais et al., 2025). Furthermore, the fitness functions constructed for such experiments are an approximation defined by a specific experiment setup (Luksza and Lassig, 2014). The sheer diversity of fitness definitions for in vitro experiments further supports the notion of a disconnect between fitness approximated under controlled experimental conditions and fitness in nature (Livesey and Marsh, 2020). That is, such fitness approximations rarely account for the many additional properties that contribute to fitness in nature. For example, in protein evolution, mutations that improve functional stability or evolvability are considered concomitant with increased fitness (Hie et al., 2024). This fitness function mismatch between controlled laboratory and uncontrolled real-world environments remains a key reason why organisms evolved in vitro are rarely transferable to real-world environments for continued evolution.
6.2 Empirical insights and partial remedies
Despite the disconnect between empirically mapped and natural fitness landscapes, which will inevitably persist for complex (multi-cellular) organisms due to the enormous complexity of fitness landscapes and confounding factors such as epistasis (Claudia, 2022), suitably (partially) mapped fitness landscapes are still a powerful tool for informing directed evolution for bioengineering applications (Currin et al., 2015). Empirically mapped (computational) fitness landscapes constructed from fitness data (gathered via specially formulated in vitro experiments) remain essential for three key reasons. First, a sufficiently large digital library of sequences, derived, for example, via generating with artificial evolution or generative machine learning, N mutational variants from a set of initial wild-types, enables researchers to infer (using predictive machine learning) the relative competitive advantage (fitness) per sequence and thus define a suitably mapped fitness landscape (Hie et al., 2024). Second, a sufficiently large sequence library enables researchers to observe the short-term evolution (relatively few generations) of disparate sequences under specific, consistent and controlled laboratory conditions (Yilmaz et al., 2021). Third, such controlled laboratory conditions enable researchers to readily manipulate (direct) and observe the evolutionary trajectories of diverse sequences (in terms of genetic coding and fitness) thereby allowing causal relationships between genetic variation and fitness to be investigated (Van den Bergh et al., 2018).
Considerable progress has already been made in mapping combinatorially complete fitness landscapes corresponding to the complete genotypes of simple single celled micro-organisms such as yeast (Saccharomyces Cerevisiae) and bacteria (Escherichia Coli (Faure et al., 2022). Such fitness landscapes have then been used as the basis of directed evolution efforts (Wang et al., 2021). In terms of bioengineering entire synthetic (micro) organisms, there are already established directed evolution frameworks operating on entire genotypes. For example, Adaptive Laboratory Evolution, is a directed evolution method where microorganisms are cultured in controlled laboratory environments, enabling in vitro genotype variation, generational propagation and selection of phenotypes that exhibit improved growth in their environment (Lee and Kim, 2020). For example, one application is the selection of Escherichia Coli strains with increased resistance to high temperatures across multiple generations (Tenaillon et al., 2012). Another prevalent example is the genotype-scale directed evolution using Multiplex Automated Genome Engineering, which has been applied to develop novel variant organisms such as a new antioxidant producing E. coli strain (Wang et al., 2009). More recent approaches use RNA interference and CRISPR/Cas9 systems for genotype manipulation within a single cell, demonstrating effective selection of yeast strains with multiple phenotypes (Si et al., 2017). Thus, suitably mapped fitness landscapes (sufficiently large sequence libraries) facilitate reproducible experiments necessary for continued, progressively comprehensive, fitness landscape mappings that are expected to facilitate the development of new methods for the directed evolutionary design of multicellular organisms (Yi and Dean, 2019).
6.3 Toward integrative evolutionary design
Given prevailing unknowns as to how fitness landscapes adapt in concert with changing environments (Yi and Dean, 2019), fitness landscape mappings, at least in the short term, are unlikely to be applicable to all levels of evolution. Even though fitness landscape mappings are currently inadequate for describing complex organism evolution (accounting for the interplay and cyclic feedback between an organism’s genotype, phenotype and environment), the fitness landscape remains an unparalleled concept that succinctly encapsulates molecular evolution, and is thus an indispensable model for evolutionary design in ALife research. Furthermore, significant promise can be observed in recent work that bridges the computational and biological fitness landscape disconnect via mapping large and combinatorially complete fitness landscapes in micro-organisms such as Escherichia Coli (Papkou et al., 2023). Such work provides valuable insights into generally applicable evolutionary traits such as demonstrating that fitness landscape ruggedness does not preclude evolving populations from accessing high fitness peaks.
Furthermore, significant promise can be observed in recent work that bridges the computational–biological disconnect by experimentally mapping combinatorially dense fitness landscapes in microbes. For example, a complete five-mutation landscape of the TEM-1 β-lactamase in E. coli revealed that sign epistasis makes most mutational routes to high-resistance genotypes inaccessible, yet evolution can still reach global fitness peaks through a few specific adaptive paths (Weinreich et al., 2006). Similarly, large-scale mapping of the green fluorescent protein (GFP) fitness landscape, measuring the function of tens of thousands of variants expressed in bacteria, revealed a narrow, epistasis-rich landscape in which most single substitutions are deleterious, enabling predictive modeling of genotype–phenotype relationships (Sarkisyan et al., 2016). Together, these studies illustrate that ruggedness does not preclude access to high peaks but instead constrains the set of viable trajectories, offering generalizable principles for evolutionary prediction. In this context, success is often defined by achieving near-complete mapping of genotype–fitness relationships, demonstrating reproducible epistatic interactions, and developing predictive models that generalize to unmeasured genotypes or related systems.
Thus, fitness landscape mappings remain invaluable models of a genetic sequence space, enabling insights as to how each sequence corresponds to its activity and can be used to guide the evolutionary design of synthetic agents. The final section of this review thus brings together the highlighted potential of directed evolution methods that suitably balance exploitation versus exploration of the sequence space, with bridging the fitness landscape disconnect via combining in silico fitness landscape mapping with in vitro sequence fitness evaluation in an iterative design-build-test cycle for on-demand evolutionary design of synthetic ALife problem-solvers (agents).
7 A bioengineered future: hybrid ALife
The multi-disciplinary fields of synthetic biology (Voigt, 2020) and ALife (Aguilar et al., 2014) are closely related and can be viewed as synthetic and digital counterparts addressing the same goal of deriving new principles and methodologies governing the bottom-up synthesis of living systems on either digital or robotic (ALife) or bioengineering (synthetic biology) experimental platforms. Both fields were founded on the premise of synthesizing either digital, robotic or biological agents in controlled environments to better understand the origins, evolution and development of living systems, learning from life as it could be, while addressing the core challenge of consistently and predictably synthesize biological systems with desired problem solving behaviors.
Given unresolved issues with fitness landscape mapping, directed evolution, and appropriate fitness evaluation that continue to confound the bioengineering design-build-test cycle (Carbonell et al., 2016), this section proposes guidelines for future synthetic ALife research that complements such current deficiencies with in silico machine learning and in vitro fitness evaluation. Specifically, we propose extensions the current diverse array of bioengineering methodologies (Grozinger et al., 2019; Hart and Ferguson, 2019; Szymanski et al., 2023; Baier et al., 2023; Voigt, 2020), into a unified and principled counterpart (hybrid ALife design methodology), similar to what has been proposed for the evolutionary design of physical ALife agents (Hale et al., 2019). As with previous evolutionary design methodologies for simulated and physical forms of ALife, for example, in evolutionary robotics (Doncieux et al., 2015), such a hybrid ALife methodology would follow a design-build-test cycle that includes directed evolution (exploitation: improving solution quality), and genetic diversity maintenance (exploration: evaluating novel solutions), where a suitable balance between exploration and exploitation enables improved fitness landscape mappings.
7.1 Towards a hybrid synthetic ALife methodology
Figure 1 presents our vision of the hybrid synthetic ALife design process. Innovation and novelty in synthetic biology are primarily driven by designers; for example, the R3C ligase ribozyme was first created through in vitro evolution (Stock and Gorochowski, 2024). Due to the designer-driven innovation approach in synthetic biology, the ability to predict how genetic information (genotype) leads to observable traits (phenotype) is weak and highly dependent on specific conditions, as engineered traits often face evolutionary instability (Wortel et al., 2023). Secondly, standard directed evolution (DE) methods often restrict exploration to genetic variants similar to those already tested, which can trap progress in locally optimal solutions even if other, superior solutions exist elsewhere in sequence space (Wang et al., 2021). Thirdly, the Design-Build-Test-Learn (DBTL) cycle, where ideas are designed, constructed, evaluated, and iteratively improved, remains unbalanced, as the stages after Design (Build, Test, and Learn) lag behind, particularly in terms of data quality and automation (Liao et al., 2022). To address these challenges, we propose an approach that combines both goal-driven (objective) and exploration-driven (novelty, quality diversity) search strategies with adaptive, coevolution-aware constraints and active learning loops (methods where the system iteratively improves based on acquired data). This integration broadens exploration while keeping compatibility with current DBTL workflows. To realize such a design methodology, we propose that future experiments first incorporate advanced forms of directed (objective and non-objective based) evolutionary search (Figure 1, right-center) of in silico fitness landscapes (sequence spaces, Figure 1, left-top). Evolutionary search enables the discovery of new sequences using diversity generation and maintenance methods such as recombination (Stemmer, 1994), already established in evolutionary design of physical and simulated ALife systems (Eiben and Smith, 2015a), and well known mutagenesis methods, often used in bioengineering applications (Currin et al., 2021). This extends previous related work on quantitative computational models that direct the evolutionary design of robotic agents (Brodland, 2015), with objective and non-objective (Nitschke and Didi, 2017) evolutionary search to balance exploration versus exploitation, that is solution quality-diversity (Pugh et al., 2016), in the evolving agent design space. In this context, our framework does not replace existing DBTL methodologies. Rather, it adapts these approaches to guide experiment selection in DE-driven synthetic artificial life systems. Critically, beyond a limited set of simulated ALife studies (Gomes et al., 2013; Gomes et al., 2015; Nitschke and Didi, 2017), the impact of hybridized (objective and non-objective) evolutionary search remains untested in the directed evolution of synthetic agents. Consequently, we presuppose that any future synthetic ALife design process will require a careful balance between open-endedness (Packard et al., 2019) and directed (exploitative) evolutionary search as the first key methodological component within an extended DBTL framework.
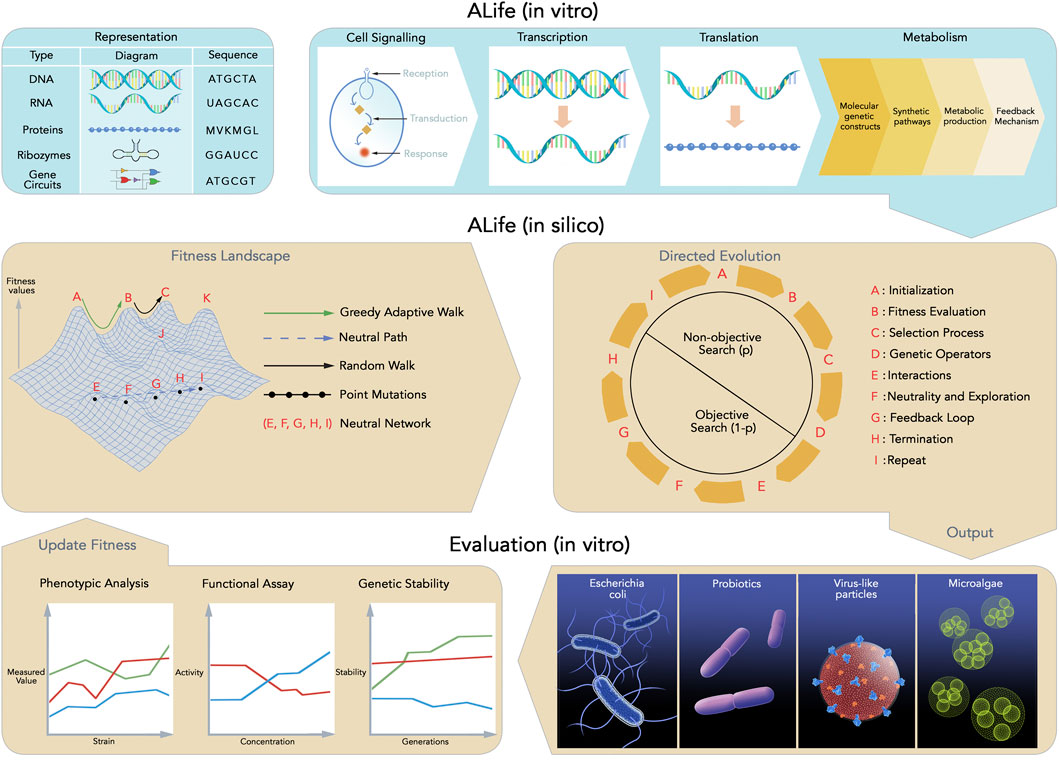
Figure 1. The iterative design process of a hybrid Artificial Life (ALife) methodology, combining synthetic (in vitro) and digital (in silico) evolution. Biological individuals have various potential representations including DNA, RNA, proteins, ribozymes and gene circuits. Directed evolution is applied to biological individuals (sequences), where the viability of each is represented on an in silico fitness landscape. Directed evolution combines non-objective (exploratory), objective (exploitative) evolutionary search and predictive machine learning to select individuals for variation. Individuals selected in silico are synthesized (varied) and evaluated in vitro. Techniques to evaluate the fitness of varied individuals include: phenotypic analysis, functional assays and genetic stability. The fitness landscape is updated, reflecting newly evaluated individuals and used for the next iteration of in silico selection, variation, in vitro genetic variation and fitness evaluation.
A second key methodological component of our envisaged synthetic ALife design process, surpassing most ALife design methodologies (Aguilar et al., 2014), but staying inline with current bioengineering design-build-test processes (Carbonell et al., 2016), is experimental (in vitro) fitness evaluation. Thus, we advocate controlled laboratory experimental evaluation to validate sequence efficacy (fitness) (Blackiston et al., 2021) (Figure 1, right-bottom), where such experimental validation enables continued, high-precision fitness landscape mapping (Figure 1, left-center). The appropriate validation approach is determined by the class of model predictions and the experimentally measurable aspects of system behavior. Rigorous testing necessitates standardized and quantitative assays, replication, and comprehensive uncertainty analysis to establish confidence in model predictions. Large-scale and automated validation is increasingly performed in robotic biofoundries, where liquid-handling systems and high-throughput measurement platforms enable reproducible and data-rich Build–Test–Learn cycles. This addresses the reality gap (Koos et al., 2013) problem that has continued to pervade the design of physical ALife agents with desired functionality (Lipson and Pollack, 2000; Brodbeck et al., 2015; Jelisavcic et al., 2017; Hale et al., 2019). Importantly, both quality-diversity driven evolutionary design and fitness landscape mapping can also be complemented by predictive and generative machine learning (Wittmann BJ. et al., 2021), already established as an effective means for interpolating highly probable novel sequences on evolving fitness landscapes (Brya et al., 2021; Notin et al., 2024; Landwehr et al., 2020). The first and second components of our envisioned synthetic ALife design process will potentially mitigate open issues with directed evolution (balancing exploratory and exploitative evolutionary search with quality-diversity maintenance) and fitness landscape disconnects (validation of evolved solutions with in vitro fitness evaluation). This process completes the experimental loop such that model predictions identify candidate experiments, biofoundry testing provides calibrated, high-throughput measurements, and uncertainty-aware learning refines both the landscape model and future design objectives.
The third key methodological component of our envisaged synthetic ALife agent design methodology is integrated predictive and generative machine learning to compute the probable efficacy of novel sequences (Wittmann BJ. et al., 2021). Critically, such machine learning based sequence discovery and validation mitigates the pervasive bioengineering impracticalities of an intractable computational and time expense of in vitro validation of all viable sequences. The choice of learning strategy depends on the specific application and model type, which may involve data-driven statistical learners for sequence to function inference or mechanistic and agent-based surrogates that generalize behavioral or ecological dynamics. In each scenario, the Learning phase involves iterative model refinement based on feedback from experimental testing, guided by uncertainty estimation, active learning, and error-driven retraining. This approach completes the DBTL cycle by converting experimental outcomes into updated priors and refined design objectives. All three components extend previous work on current design-build-test processes, where computational design trials translate to counterpart biological trials (Carbonell et al., 2016), for the purpose of providing a unified methodology for synthetic ALife design. An end goal is thus to provide bioengineering and ALife communities with a singular, coherent principled approach for on-demand evolutionary design of synthetic (problem solving) agents. Such a methodology would constitute an improved counterpart to current robotic (Doncieux et al., 2015) and simulated ALife agent (Aguilar et al., 2014) evolutionary design methods, where such methods remain ad hoc and disparate without a single unifying methodology for ALife design. A coherent evolutionary design process that suitably integrates these three methodological components will be necessary if synthetic ALife agents are to be evolved as solutions to challenging and practical applications.
7.2 Applications and controlled environments
Specifically, we anticipate such a hybrid ALife design methodology would most practically be for bioengineering synthetic ALife agents that operate in controlled environments such as indoor farms, factories and manufacturing plants. For example, producing sustainable bio-fuels, plant-based meats and enabling sustainable industry using renewable bio-based raw materials (Voigt, 2020). One can envisage the evolutionary design of synthetic agents (biological organisms) that operate within structured environments. For example, microorganism chemical factories that synthesize bio-fuels and plant-based food (Hillson et al., 2017), but adapt to unexpected errors and changing tasks, or genetically modified organisms bioengineered to mitigate the spread of disease. For example, genetically engineered malaria free mosquitoes that mate with wild populations and spread genetic traits that reduce malaria transmission (Robinson and Nadal, 2025). Such an operational capacity constraint is deemed necessary (at least for the near future), given the enormity of challenges and complexities in bioengineering synthetic ALife agents that would continue to evolve and adapt outside of strictly controlled laboratory environments (Brooks and Alper, 2021). Solving the critical predicament of synthesizing agents that perpetually evolve, adapt and function outside of the laboratory environment, in nature, is tantamount to solving the reality gap problem that continues to confounded robotic ALife systems (Koos et al., 2013). Furthermore, controlled environments would enable experimenters to focus their research efforts to evolving agent designs for specific tasks, increasing the likelihood of discovering beneficial sequences in the solution space, leading to the directed evolution of desired agents.
7.3 Emerging tools and future directions
Furthermore, in the coming decades it is predicted that increased affordability of genetic sequencing and synthesis will mitigate current experimental screening and validation fiscal and time cost bottlenecks (Robinson and Nadal, 2025). Reduced experiment costs are posited to be aided by emerging plant and animal cell bioengineering tools. For example, 3D Organoids, have been proposed as in vitro models that would eventually replace animals as the experimental platforms in bioengineering research (van Berlo et al., 2021), where such advances are predicted to enable the eventual development of living computers that integrate biological and digital components (Robinson and Nadal, 2025). However, if such living computer platforms are to be practical advanced problem solvers, they must be coupled with similarly advanced computational methodologies to direct solution design. Recent advancements in laboratory automation, including robotic liquid handling, high-throughput DNA assembly, microfluidic testing platforms, and integrated biofoundries, are projected to substantially enhance experimental throughput (Ma et al., 2024). These automated systems enable rapid construction and parallel testing of large construct libraries, effectively merging design and validation phases into continuous, data-driven design-build-test-learn cycles (Gurdo et al., 2023). Thus our proposed hybrid synthetic ALife design methodology, combining evolutionary design in silico and experimental validation in vitro, is a step towards the realization of such living computer systems, which would simply be experimental platforms for the design of synthetic ALife agents.
Our proposed hybrid synthetic ALife design methodology builds directly on earlier ALife work that combined computational design with physical evaluation, most notably in robotics, where evolutionary algorithms were paired with rapid-prototyping technologies to produce and test robots designed in silico (Lipson and Pollack, 2000). More recently, analogous principles have been applied in bioengineering: for example, Blackiston et al. (2021) evolved biological robots using an integrated in vitro and in silico design pipeline. By adapting and generalizing these ideas to the domain of synthetic biology, our framework aims to unify such disparate ALife methodologies and extend contemporary bioengineering design-build-test cycles (Carbonell et al., 2016) into an evotype design space (Castle et al., 2021). In doing so, we outline how proven iterative design processes can be leveraged to derive complete genotypes for novel synthetic products (Foo and Chang, 2018), positioning hybrid ALife as a coherent and extensible design paradigm for future synthetic biological systems.
Author contributions
IS: Writing – original draft, Writing – review and editing. GN: Writing – review and editing, Writing – original draft.
Funding
The authors declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declared that generative AI was not used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abbott, T. R., and Qi, L. S. (2018). Evolution at the cutting edge: crispr-Mediated directed evolution. Mol. Cell 72, 402–403. doi:10.1016/j.molcel.2018.10.027
Adami, C. (2006). Digital genetics: unravelling the genetic basis of evolution. Nat. Rev. Genet. 7, 109–118. doi:10.1038/nrg1771
Aguilar, W., Santamaría-Bonfil, G., Froese, T., and Gershenson, C. (2014). The past, present, and future of artificial life. Front. Robotics AI 1, 8. doi:10.3389/frobt.2014.00008
Alkan, C., Coe, B. P., and Eichler, E. E. (2011). Genome structural variation discovery and genotyping. Nat. Reviews Genetics 12, 363–376. doi:10.1038/nrg2958
Alvarez, S., Nartey, C. M., Mercado, N., de la Paz, J. A., Huseinbegovic, T., and Morcos, F. (2024). In vivo functional phenotypes from a computational epistatic model of evolution. Proc. Natl. Acad. Sci. 121, e2308895121. doi:10.1073/pnas.2308895121
Arnold, F. H. (1998). Design by directed evolution. Accounts Chemical Research 31, 125–131. doi:10.1021/ar960017f
Auerbach, J. E., and Bongard, J. C. (2014). Environmental influence on the evolution of morphological complexity in machines. PLOS Comput. Biol. 10, e1003399. doi:10.1371/journal.pcbi.1003399
Awais, M., Naseer, M., Khan, S., Anwer, R. M., Cholakkal, H., Shah, M., et al. (2025). Foundation models defining a new era in vision: a survey and outlook. IEEE Trans. Pattern Analysis Mach. Intell. 47, 2245–2264. doi:10.1109/TPAMI.2024.3506283
Baier, F., Gauye, F., Perez-Carrasco, R., Payne, J. L., and Schaerli, Y. (2023). Environment-dependent epistasis increases phenotypic diversity in gene regulatory networks. Sci. Adv. 9, eadf1773. doi:10.1126/sciadv.adf1773
Baltieri, M., Iizuka, H., Witkowski, O., Sinapayen, L., and Suzuki, K. (2023). Hybrid life: integrating biological, artificial, and cognitive systems. WIREs Cognitive Sci. 14, e1662. doi:10.1002/wcs.1662
Barton, N. (2022). The “New Synthesis”. Proc. Natl. Acad. Sci. 119, e2122147119. doi:10.1073/pnas.2122147119
Bartz-Beielstein, T., Branke, J., Mehnen, J., and Mersmann, O. (2014). Evolutionary algorithms. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 4, 178–195. doi:10.1002/widm.1124
Bedau, M. A. (2003). Artificial life: organization, adaptation and complexity from the bottom up. Trends Cognitive Sci. 7, 505–512. doi:10.1016/j.tics.2003.09.012
Bedau, M. A. (2007). “Artificial life,” in Philosophy of biology (amsterdam: north-holland), handbook of the philosophy of science. Editors M. Matthen, and C. Stephens, 585–603. doi:10.1016/B978-044451543-8/50027-7
Bernasconi, A., Canakoglu, A., Masseroli, M., Pinoli, P., and Ceri, S. (2021). A review on viral data sources and search systems for perspective mitigation of covid-19. Briefings Bioinformatics 22, 664–675. doi:10.1093/bib/bbaa359
Besnard, J., Ruda, G. F., Setola, V., Abecassis, K., Rodriguiz, R. M., Huang, X. P., et al. (2012). Automated design of ligands to polypharmacological profiles. Nature 492, 215–220. doi:10.1038/nature11691
Biswas, S., Kuznetsov, G., Ogden, P., Conway, N., Adams, R., and Church, G. (2018). Toward machine-guided design of proteins. BioRxiv, 337154. doi:10.1101/337154
Biswas, S., Khimulya, G., Alley, E. C., Esvelt, K. M., and Church, G. M. (2021). Low-n protein engineering with data-efficient deep learning. Nat. Methods 18, 389–396. doi:10.1038/s41592-021-01100-y
Blackiston, D., Lederer, E., Kriegman, S., Garnier, S., Bongard, J., and Levin, M. (2021). A cellular platform for the development of synthetic living machines. Sci. Robotics 1, eabf1571. doi:10.1126/scirobotics.abf1571
Bongard, J., and Levin, M. (2021). Living things are not (20th century) machines: updating mechanism metaphors in light of the modern science of machine behavior. Front. Ecol. Evol. 9, 650726. doi:10.3389/fevo.2021.650726
Brancato, V., Esposito, G., Coppola, L., Cavaliere, C., Mirabelli, P., Scapicchio, C., et al. (2024). Standardizing digital biobanks: integrating imaging, genomic, and clinical data for precision medicine. J. Translational Medicine 22, 136. doi:10.1186/s12967-024-04891-8
Brodbeck, L., Hauser, S., and Iida, F. (2015). Morphological evolution of physical robots through model-free phenotype development. PLOS One 10, e0128444. doi:10.1371/journal.pone.0128444
Brodland, W. (2015). How computational models can help unlock biological systems. Seminars Cell and Dev. Biol. 47, 62–73. doi:10.1016/j.semcdb.2015.07.001
Brooks, S., and Alper, H. S. (2021). Applications, challenges, and needs for employing synthetic biology beyond the lab. Nat. Commun. 12, 1390. doi:10.1038/s41467-021-21740-0
Brookwell, A., Oza, J. P., and Caschera, F. (2021). Biotechnology applications of cell-free expression systems. Life 11, 1367. doi:10.3390/life11121367
Brun-Usan, M., Zimm, R., and Uller, T. (2022). Beyond genotype-phenotype maps: toward a phenotype-centered perspective on evolution. BioEssays 44, 2100225. doi:10.1002/bies.202100225
Bryant, D. H., Bashir, A., Sinai, S., Jain, N. K., Ogden, P. J., Riley, P. F., et al. (2021). Deep diversification of an AAV capsid protein by machine learning. Nat. Biotechnol. 39, 691–696. doi:10.1038/s41587-020-00793-4
Cao, L., Coventry, B., Goreshnik, I., Huang, B., Sheffler, W., Park, J. S., et al. (2022). Design of protein-binding proteins from the target structure alone. Nature 605, 551–560. doi:10.1038/s41586-022-04654-9
Carbonell, P., Currin, A., Jervis, A. J., Rattray, N. J. W., Swainston, N., Yan, C., et al. (2016). Bioinformatics for the synthetic biology of natural products: integrating across the design–build–test cycle. Nat. Product. Rep. 33, 925–932. doi:10.1039/c6np00018e
Castle, S. D., Grierson, C. S., and Gorochowski, T. E. (2021). Towards an engineering theory of evolution. Nat. Commun. 12, 3326. doi:10.1038/s41467-021-23573-3
Castle, S., Stock, M., and Gorochowski, T. (2024). Engineering is evolution: a perspective on design processes to engineer biology. Nat. Commun. 15, 3640. doi:10.1038/s41467–024–48000–1
Cavuoto, L. A., and Nussbaum, M. A. (2014). The influences of obesity and age on functional performance during intermittent upper extremity tasks. J. Occup. Environ. Hyg. 11, 583–590. doi:10.1080/15459624.2014.887848
Chen, J., Wang, Y., Zheng, P., and Sun, J. (2022). Engineering synthetic auxotrophs for growth-coupled directed protein evolution. Trends Biotechnol. 40, 773–776. doi:10.1016/j.tibtech.2022.01.010
Cheng, F., Zhu, L., and Schwaneberg, U. (2015). Directed evolution 2.0: improving and deciphering enzyme properties. Chem. Commun. 51, 9760–9772. doi:10.1039/c5cc01594d
Chéron, N., Serohijos, A. W. R., Choi, J. M., and Shakhnovich, E. I. (2016). Evolutionary dynamics of viral escape under antibodies stress: a biophysical model. Protein Sci. 25, 1332–1340. doi:10.1002/pro.2915
Ching, T., Himmelstein, D. S., Beaulieu-Jones, B. K., Kalinin, A. A., Do, B. T., Way, G. P., et al. (2018). Opportunities and obstacles for deep learning in biology and medicine. J. Royal Society Interface 15, 20170387. doi:10.1098/rsif.2017.0387
Claudia, B. (2022). Epistasis and adaptation on fitness landscapes. Annu. Rev. Ecol. Evol. Syst. 53, 457–479. doi:10.1146/annurev-ecolsys-102320-112153
Coelho, P. S., Brustad, E. M., Kannan, A., and Arnold, F. H. (2013). Cyclopropanation via carbene transfer catalyzed by engineered cytochrome P450 enzymes. Science 339, 307–310. doi:10.1126/science.1231434
Collins, S. P., Daff, T. D., Piotrkowski, S. S., and Woo, T. K. (2016). Materials design by evolutionary optimization of functional groups in metal-organic frameworks. Sci. Adv. 2, eaax1950. doi:10.1126/sciadv.1600954
Copp, J. N., Hanson-Manful, P., Ackerley, D. F., and Patrick, W. M. (2014). Error-prone PCR and effective generation of gene variant libraries for directed evolution. Methods Mol. Biol. 1179, 3–22. doi:10.1007/978-1-4939-1053-3_1
Crepinsek, M., Liu, S., and Mernik, M. (2013). Exploration and exploitation in evolutionary algorithms. ACM Comput. Surv. 45, 1–33. doi:10.1145/2480741.2480752
Cubillos-Ruiz, A., Guo, T., Sokolovska, A., Miller, P. F., Collins, J. J., Lu, T. K., et al. (2021). Engineering living therapeutics with synthetic biology. Nat. Rev. Drug Discov. 20, 941–960. doi:10.1038/s41573-021-00285-3
Cully, A., and Demiris, Y. (2018). Improving evolvability of morphologies and controllers of developmental soft-bodied robots with novelty search. IEEE Trans. Evol. Comput. 22, 245–259. doi:10.1109/tevc.2017.2704781
Cuomo, A. S., Nathan, A., Raychaudhuri, S., MacArthur, D. G., and Powell, J. E. (2023). Single-cell genomics meets human genetics. Nat. Rev. Genet. 24, 535–549. doi:10.1038/s41576-023-00599-5
Currin, A., Swainston, N., Day, P. J., and Kell, D. B. (2015). Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently. Chem. Soc. Rev. 44, 1172–1239. doi:10.1039/c4cs00351a
Currin, A., Parker, S., Robinson, C. J., Takano, E., Scrutton, N. S., and Breitling, R. (2021). The evolving art of creating genetic diversity: from directed evolution to synthetic biology. Biotechnol. Adv. 50, 107762. doi:10.1016/j.biotechadv.2021.107762
De Visser, JAGM, and Krug, J. (2014). Empirical fitness landscapes and the predictability of evolution. Nat. Rev. Genet. 15, 480–490. doi:10.1038/nrg3744
de Vladar, H. P., Santos, M., and Szathmáry, E. (2017). Grand views of evolution. Trends Ecol. Evol. 32, 324–334. doi:10.1016/j.tree.2017.01.008
Dechant, P., and He, Y. (2021). Machine-learning a virus assembly fitness landscape. PLoS ONE 16, e0250227. doi:10.1371/journal.pone.0250227
DeGrace, M. M., Ghedin, E., Frieman, M. B., Krammer, F., Grifoni, A., Alisoltani, A., et al. (2022). Defining the risk of SARSCoV-2 variants on immune protection. Nature 605, 640–652. doi:10.1038/s41586-022-04690-5
Della, C. A., De Stefano, C., and Marcelli, A. (2004). On the role of population size and niche radius in fitness sharing. IEEE Trans. Evol. Comput. 8, 580–592. doi:10.1109/tevc.2004.837341
Ding, X., Zou, Z., and Brooks, C. (2019). Deciphering protein evolution and fitness landscapes with latent space models. Nat. Communications 10, 5644. doi:10.1038/s41467–019–13633–0
Domingo, J., Diss, G., and Lehner, B. (2018). Pairwise and higher-order genetic interactions during the evolution of a trna. Nature 558, 117–121. doi:10.1038/s41586-018-0170-7
Domingo, E., García-Crespo, C., Soria, M. E., and Perales, C. (2023). Viral fitness, population complexity, host interactions, and resistance to antiviral agents. Curr. Top. Microbiol. Immunol. 439, 197–235. doi:10.1007/978-3-031-15640-3_6
Doncieux, S., Bredeche, N., Mouret, J., and Eiben, A. (2015). Evolutionary robotics: what, why, and where to. Front. Robotics AI 2, 4. doi:10.3389/frobt.2015.00004
Dorin, A., and Stepney, S. (2024). What is artificial life today, and where should it go? Artif. Life 30, 1–15. doi:10.1162/artl_e_00435
Draghi, J., and Plotkin, J. (2011). Hidden diversity sparks adaptation. Nature 474, 45–46. doi:10.1038/474045a
Duong-Trung, N., Born, S., Kim, J. W., Schermeyer, M. T., Paulick, K., Borisyak, M., et al. (2023). When bioprocess engineering meets machine learning: a survey from the perspective of automated bioprocess development. Biochem. Eng. J. 190, 108764. doi:10.1016/j.bej.2022.108764
Eiben, A. E., and Smith, J. E. (2015a). From evolutionary computation to the evolution of things. Nature 521, 476–482. doi:10.1038/nature14544
Eiben, A. E., and Smith, J. E. (2015b). Introduction to evolutionary computing. 2nd edn. Berlin, Germany: Springer.
Eling, N., Morgan, M. D., and Marioni, J. C. (2019). Challenges in measuring and understanding biological noise. Nat. Rev. Genet. 20, 536–548. doi:10.1038/s41576-019-0130-6
Engqvist, M., and Nielsen, J. (2015). Ant: software for generating and evaluating degenerate codons for natural and expanded genetic codes. ACS Synth. Biol. 4, 935–938. doi:10.1021/acssynbio.5b00057
Engstrom, M., and Pfleger, B. (2017). Transcription control engineering and applications in synthetic biology. Synthetic Syst. Biotechnol. 2, 176–191. doi:10.1016/j.synbio.2017.09.003
Esvelt, K. M., Carlson, J. C., and Liu, D. R. (2011). A system for the continuous directed evolution of biomolecules. Nature 472, 499–503. doi:10.1038/nature09929
Faure, A. J., Domingo, J., Schmiedel, J. M., Hidalgo-Carcedo, C., Diss, G., and Lehner, B. (2022). Mapping the energetic and allosteric landscapes of protein binding domains. Nature 604, 175–183. doi:10.1038/s41586-022-04586-4
Fernandez-de Cossio-Diaz, J., Uguzzoni, G., and Pagnani, A. (2021). Unsupervised inference of protein fitness landscape from deep mutational scan. Mol. Biol. Evol. 38, 318–328. doi:10.1093/molbev/msaa204
Firnberg, E., Labonte, J. W., Gray, J. J., and Ostermeier, M. (2014). A comprehensive, high-resolution map of a gene’s fitness landscape. Mol. Biol. Evol. 31, 1581–1592. doi:10.1093/molbev/msu081
Flynn, J. M., Samant, N., Schneider-Nachum, G., Barkan, D. T., Yilmaz, N. K., Schiffer, C. A., et al. (2022). Comprehensive fitness landscape of SARS-CoV-2 mpro reveals insights into viral resistance mechanisms. eLife 11, e77433. doi:10.7554/eLife.77433
Fonville, J. M., Wilks, S., James, S. L., Fox, A., Ventresca, M., Aban, M., et al. (2014). Antibody landscapes after influenza virus infection or vaccination. Science 346, 996–1000. doi:10.1126/science.1256427
Foo, J., and Chang, M. (2018). Synthetic yeast genome reveals its versatility. Nature 557, S11–S648. doi:10.1038/d41586-018-05164-3
Forbes, N. (2000). Life as it could be: alife attempts to simulate evolution. IEEE Intelligent Syst. Their Appl. 15, 2–7. doi:10.1109/5254.895847
Fragata, I., Blanckaert, A., Louro, M. A. D., Liberles, D. A., and Bank, C. (2019). Evolution in the light of fitness landscape theory. Trends Ecol. and Evol. 34, 69–82. doi:10.1016/j.tree.2018.10.009
Franklin, J., LaBar, T., and Adami, C. (2019). Mapping the peaks: fitness landscapes of the fittest and the flattest. Artif. Life 25, 250–262. doi:10.1162/artl_a_00296
Gantz, M., Neun, S., Medcalf, E. J., van Vliet, L. D., and Hollfelder, F. (2023). Ultrahigh-throughput enzyme engineering and discovery in in vitro compartments. Chem. Rev. 123, 5571–5611. doi:10.1021/acs.chemrev.2c00910
Gao, W., Wu, Z., Zuo, K., Xiang, Q., Chen, X., Zhang, L., et al. (2025). Biosafety concept: origins, evolution, and prospects. Biosaf. Health 7, 209–217. doi:10.1016/j.bsheal.2025.07.003
Gershenson, C. (2023). Emergence in artificial life. Artif. Life 29, 153–167. doi:10.1162/artl_a_00397
Gomes, J., Urbano, P., and Christensen, A. (2013). Evolution of swarm robotics systems with novelty search. Swarm Intell. 7, 115–144. doi:10.1007/s11721-013-0081-z
Gomes, J., Mariano, P., and Christensen, A. (2015). “Devising effective novelty search algorithms: a comprehensive empirical study,” in Proceedings of the genetic evolutionary computation conference (Madrid, Spain: ACM), 943–950.
Gong, L., Suchard, M., and Bloom, J. (2016). Stability-mediated epistasis constrains the evolution of an influenza protein. eLife 2, e00631. doi:10.7554/eLife.00631
Gray, M. W., Lukeš, J., Archibald, J. M., Keeling, P. J., and Doolittle, W. F. (2010). Irremediable complexity? Science 330, 920–921. doi:10.1126/science.1198594
Greenbury, S., Louis, A., and Ahnert, S. (2022). The structure of genotype-phenotype maps makes fitness landscapes navigable. Nat. Ecol. and Evol. 6, 1742–1752. doi:10.1038/s41559-022-01867-z
Grozinger, L., Amos, M., Gorochowski, T. E., Carbonell, P., Oyarzún, D. A., Stoof, R., et al. (2019). Pathways to cellular supremacy in biocomputing. Nat. Commun. 10, 5250. doi:10.1038/s41467-019-13232-z
Gurdo, N., Volke, D. C., McCloskey, D., and Nikel, P. I. (2023). Automating the design-build-test-learn cycle towards next-generation bacterial cell factories. New Biotechnol. 74, 1–15. doi:10.1016/j.nbt.2023.01.002
Hale, K. S., and Stanney, K. M. (2014). Handbook of virtual environments: design, implementation, and applications. Boca Raton, FL: CRC Press.
Hale, M. F., Buchanan, E., Winfield, A. F., Timmis, J., Hart, E., Eiben, A. E., et al. (2019). “The are robot fabricator: how to (re)produce robots that can evolve in the real world,” in Proceedings of the conference on artificial life (Newcastle, UK: MIT Press), 95–102.
Halperin, S. O., Tou, C. J., Wong, E. B., Modavi, C., Schaffer, D. V., and Dueber, J. E. (2018). Crispr-guided dna polymerases enable diversification of all nucleotides in a tunable window. Nature 560, 248–252. doi:10.1038/s41586-018-0384-8
Halweg-Edwards, A. L., Pines, G., Winkler, J. D., Pines, A., and Gill, R. T. (2016). A web interface for codon compression. ACS Synth. Biol. 5, 1021–1023. doi:10.1021/acssynbio.6b00026
Hart, G. R., and Ferguson, A. L. (2019). Computational design of hepatitis c virus immunogens from host–pathogen dynamics over empirical viral fitness landscapes. Phys. Biol. 16, 016004. doi:10.1088/1478-3975/aaeec0
Hartl, D. (2014). What can we learn from fitness landscapes? Curr. Opin. Microbiol. 21, 51–57. doi:10.1016/j.mib.2014.08.001
Hauser, H. (2019). Resilient machines through adaptive morphology. Nat. Mach. Intell. 1, 338–339. doi:10.1038/s42256-019-0076-6
Hawkins-Hooker, A., Depardieu, F., Baur, S., Couairon, G., Chen, A., and Bikard, D. (2021). Generating functional protein variants with variational autoencoders. PLoS Computational Biology 17, e1008736. doi:10.1371/journal.pcbi.1008736
Hayden, E. J., Ferrada, E., and Wagner, A. (2011). Cryptic genetic variation promotes rapid evolutionary adaptation in an rna enzyme. Nature 474, 92–95. doi:10.1038/nature10083
Heinemann, M., and Panke, S. (2006). Synthetic biology—putting engineering into biology. Bioinformatics 22, 2790–2799. doi:10.1093/bioinformatics/btl469
Heins, Z. J., Mancuso, C. P., Kiriakov, S., Wong, B. G., Bashor, C. J., and Khalil, A. S. (2019). Designing automated, high-throughput continuous cell growth experiments using evolver. J. Visualized Experiments JoVE, 10–3791. doi:10.3791/59652
Heudin, J. C. (2006). “Artificial life and the sciences of complexity: history and future,” in Self-organization and emergence in life sciences. Berlin: Springer, 227–247.
Hie, B. L., Shanker, V. R., Xu, D., Bruun, T. U., Weidenbacher, P. A., Tang, S., et al. (2024). Efficient evolution of human antibodies from general protein language models. Nat. Biotechnol. 42, 275–283. doi:10.1038/s41587-023-01763-2
Hietpas, R. T., Jensen, J. D., and Bolon, D. N. (2011). Experimental illumination of a fitness landscape. Proc. Natl. Acad. Sci. 108, 7896–7901. doi:10.1073/pnas.1016024108
Hillson, N., Caddick, M., Cai, Y., Carrasco, J. A., Chang, M. W., Curach, N. C., et al. (2017). Building a global alliance of biofoundries. Nat. Commun. 10, 2040. doi:10.1038/s41467–019–10079–2
Hitchins, D. K. (2008). Systems engineering: a 21st century systems methodology. John Wiley and Sons.
Hoban, S., Bertorelle, G., and Gaggiotti, O. E. (2012). Computer simulations: tools for population and evolutionary genetics. Nat. Rev. Genet. 13, 110–122. doi:10.1038/nrg3130
Hornby, G., Lohn, J., and Linden, D. (2011). Computer-automated evolution of an X-Band antenna for NASA’s space technology 5 mission. Evol. Comput. 19, 1–23. doi:10.1162/EVCO_a_00005
Ingraham, J. B., Baranov, M., Costello, Z., Barber, K. W., Wang, W., Ismail, A., et al. (2023). Illuminating protein space with a programmable generative model. Nature 623, 1070–1078. doi:10.1038/s41586-023-06728-8
Iwano, N., Adachi, T., Aoki, K., Nakamura, Y., and Hamada, M. (2022). Generative aptamer discovery using RaptGen. Nat. Comput. Sci. 2, 378–386. doi:10.1038/s43588-022-00249-6
Jacobs, T. M., Yumerefendi, H., Kuhlman, B., and Leaver-Fay, A. (2015). SwiftLib: rapid degenerate-codon-library optimization through dynamic programming. Nucleic Acids Res. 43, e34. doi:10.1093/nar/gku1323
James, J., Towers, S., Foerster, J., and Steel, H. (2024). Optimisation strategies for directed evolution without sequencing. PLOS Comput. Biol. 20, e1012695. doi:10.1371/journal.pcbi.1012695
Jelisavcic, M., De Carlo, M., Hupkes, E., Eustratiadis, P., Orlowski, J., Haasdijk, E., et al. (2017). Real-world evolution of robot morphologies: a proof of concept. Artif. Life 23, 206–235. doi:10.1162/ARTL/TNQDotTNQ/a/TNQDotTNQ/00231
Joachimczak, M., Suzuki, R., and Arita, T. (2016). Artificial metamorphosis: evolutionary design of transforming, soft-bodied robots. Artif. Life 22, 271–298. doi:10.1162/ARTL/TNQDotTNQ/a/TNQDotTNQ/00207
Jones, T. (1995). Evolutionary algorithms, fitness landscapes and search (Albuquerque, NM: University of New Mexico). Ph.D. thesis.
Katz, L., Chen, Y. Y., Gonzalez, R., Peterson, T. C., Zhao, H., and Baltz, R. H. (2018). Synthetic biology advances and applications in the biotechnology industry: a perspective. J. Industrial Microbiol. Biotechnol. 45, 449–461. doi:10.1007/s10295-018-2056-y
Kejriwal, M. (2021). Essential features in a theory of context for enabling artificial general intelligence. Appl. Sci. 11, 11991. doi:10.3390/app112411991
Keller, M. W., Rambo-Martin, B. L., Wilson, M. M., Ridenour, C. A., Shepard, S. S., Stark, T. J., et al. (2018). Direct rna sequencing of the coding complete influenza a virus genome. Sci. Reports 8, 14408. doi:10.1038/s41598-018-32615-8
Khakzad, H., Igashov, I., Schneuing, A., Goverde, C., Bronstein, M., and Correia, B. (2023). A new age in protein design empowered by deep learning. Cell Syst. 14, 925–939. doi:10.1016/j.cels.2023.10.006
Khalil, A. S., and Collins, J. J. (2010). Synthetic biology: applications come of age. Nat. Rev. Genet. 11, 367–379. doi:10.1038/nrg2775
Kicinger, R., Arciszewski, T., and De Jong, K. (2005). Evolutionary computation and structural design: survey of the state of the art. Comput. Struct. 83, 1943–1978. doi:10.1016/j.compstruc.2005.03.002
Kimura, M. (1989). The neutral theory of molecular evolution and the world view of the neutralists. Genome 31, 24–31. doi:10.1139/g89-009
Klys, J., Snell, J., and Zemel, R. (2018). “Learning latent subspaces in variational autoencoders,” in Advances in neural information processing systems 31 (NeurIPS 2018). Editors S. Bengio, H. Wallach, H. Larochelle, K. Grauman, N. Cesa-Bianchi, and R. Garnett (Red Hook, NY: Curran Associates, Inc.), 6434–6444.
Knezic, B., Keyhani-Goldau, S., and Schwalbe, H. (2022). Mapping the conformational landscape of the neutral network of RNA sequences that connect two functional distinctly different ribozymes. ChemBioChem 23, e202200022. doi:10.1002/cbic.202200022
Knowles, J. (2010). Closed-loop evolutionary multiobjective optimization. IEEE Comput. Intell. Mag. 4, 77–91. doi:10.1109/mci.2009.933095
Kondrashov, D., and Kondrashov, F. (2015). Topological features of rugged fitness landscapes in sequence space. Trend Genet. 31, 24–33. doi:10.1016/j.tig.2014.09.009
Koonin, E. V., Wolf, Y. I., and Karev, G. P. (2002). The structure of the protein universe and genome evolution. Nature 420, 218–223. doi:10.1038/nature01256
Koos, S., Mouret, J., and Doncieux, S. (2013). The transferability approach: crossing the reality gap in evolutionary robotics. IEEE Trans. Evol. Comput. 17, 122–145. doi:10.1109/TEVC.2012.2185849
Koskella, B., Hall, L. J., and Metcalf, C. J. E. (2017). The microbiome beyond the horizon of ecological and evolutionary theory. Nat. Ecology and Evolution 1, 1606–1615. doi:10.1038/s41559-017-0340-2
Kriegman, S., Cheney, N., and Bongard, J. (2018). How morphological development can guide evolution. Sci. Rep. 8, 13934. doi:10.1038/s41598-018-31868-7
Kun, A., and Szathmary, E. (2015). Fitness landscapes of functional rnas. Life 5, 1497–1517. doi:10.3390/life5031497
Landwehr, G. M., Bogart, J. W., Magalhaes, C., Hammarlund, E. G., Karim, A. S., and Jewett, M. C. (2020). Accelerated enzyme engineering by machine-learning guided cell-free expression. Nat. Commun. 16, 865. doi:10.1038/s41467–024–55399–0
Langton, C. G. (1986). Studying artificial life with cellular automata. Phys. D. Nonlinear Phenom. 22, 120–149. doi:10.1016/0167-2789(86)90237-x
Lässig, M., Mustonen, V., and Walczak, A. M. (2017). Predicting evolution. Nat. Ecology and Evolution 1, 0077. doi:10.1038/s41559-017-0077
Lee, S., and Kim, P. (2020). Current status and applications of adaptive laboratory evolution in industrial microorganisms. J. Microbiol. Biotechnol. 30, 793–803. doi:10.4014/jmb.2003.03072
Lehman, J., and Stanley, K. (2011). Abandoning objectives: evolution through the search for novelty alone. Evol. Comput. 19, 189–223. doi:10.1162/EVCO_a_00025
Lehman, J., Clune, J., Misevic, D., Adami, C., Altenberg, L., Beaulieu, J., et al. (2020). The surprising creativity of digital evolution: a collection of anecdotes from the evolutionary computation and artificial life research communities. Artif. Life 26, 274–306. doi:10.1162/artl_a_00319
Li, C., Nguyen, T. T., Yang, M., Mavrovouniotis, M., and Yang, S. (2015). An adaptive multipopulation framework for locating and tracking multiple optima. IEEE Transactions Evolutionary Computation 20, 590–605. doi:10.1109/tevc.2015.2504383
Li, X., Epitropakis, M. G., Deb, K., and Engelbrecht, A. (2016). Seeking multiple solutions: an updated survey on niching methods and their applications. IEEE Trans. Evol. Comput. 21, 518–538. doi:10.1109/tevc.2016.2638437
Liao, X., Ma, H., and Tang, Y. J. (2022). Artificial intelligence: a solution to involution of design–build–test–learn cycle. Curr. Opinion Biotechnology 75, 102712. doi:10.1016/j.copbio.2022.102712
Lin, Z., Akin, H., Rao, R., Hie, B., Zhu, Z., Lu, W., et al. (2023a). Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130. doi:10.1126/science.ade2574
Lin, T. T., Yang, L. Y., Lin, C. Y., Wang, C. T., Lai, C. W., Ko, C. F., et al. (2023b). Intelligent de novo design of novel antimicrobial peptides against antibiotic-resistant bacteria strains. Int. Journal Molecular Sciences 24, 6788. doi:10.3390/ijms24076788
Lindenhofer, D., Bauman, J. R., Hawkins, J. A., Fitzgerald, D., Yildiz, U., Jung, H., et al. (2025). Functional phenotyping of genomic variants using joint multiomic single-cell dna–rna sequencing. Nat. Methods 22, 1–10. doi:10.1038/s41592-025-02805-0
Lipson, H., and Pollack, J. (2000). Automatic design and manufacture of robotic lifeforms. Nature 406, 974–978. doi:10.1038/35023115
Livesey, B., and Marsh, J. (2020). Using deep mutational scanning to benchmark variant effect predictors and identify disease mutations. Mol. Syst. Biol. 16, e9380. doi:10.15252/msb.20199380
Lou, J., Liang, W., Cao, L., Hu, I., Zhao, S., Chen, Z., et al. (2024). Predictive evolutionary modelling for influenza virus by site-based dynamics of mutations. Nat. Commun. 15, 2546. doi:10.1038/s41467–024–46918–0
Luksza, M., and Lassig, M. (2014). A predictive fitness model for influenza. Nature 507, 57–61. doi:10.1038/nature13087
Ma, L., Li, Y., Chen, X., Ding, M., Wu, Y., and Yuan, Y. J. (2019). Scramble generates evolved yeasts with increased alkali tolerance. Microb. Cell Factories 18, 52. doi:10.1186/s12934–019–1102–4
Ma, Y., Zhang, Z., Jia, B., and Yuan, Y. (2024). Automated high-throughput dna synthesis and assembly. Heliyon 10, e26967. doi:10.1016/j.heliyon.2024.e26967
Madhavan, M., and Mustafa, S. (2023). Systems Biology—the transformative approach to integrate sciences across disciplines: systems biology integrating biological sciences. Phys. Sci. Rev. 8, 2523–2545. doi:10.1515/psr-2021-0102
Marris, C., and Calvert, J. (2020). Science and technology studies in policy: the UK synthetic biology roadmap. Sci. Technol. Hum. Values 45, 34–61. doi:10.1177/0162243919828107
Miller, S. M., Wang, T., and Liu, D. R. (2020). Phage-assisted continuous and non-continuous evolution. Nat. Protocols 15, 4101–4127. doi:10.1038/s41596-020-00410-3
Mills, P. R. D., and Spiegelman, S. (1967). An extracellular Darwinian experiment with a self-duplicating nucleic acid molecule. Proc. Natl. Acad. Sci. 58, 217–224. doi:10.1073/pnas.58.1.217
Miras, K., and Ferrante, A. E. E. (2020). Environmental influences on evolvable robots. PLoS ONE 15, e0233848. doi:10.1371/journal.pone.0233848
Mitchell-Olds, T., Willis, J. H., and Goldstein, D. B. (2007). Which evolutionary processes influence natural genetic variation for phenotypic traits? Nat. Rev. Genet. 8, 845–856. doi:10.1038/nrg2207
Mkhatshwa, S., and Nitschke, G. (2024). Body and brain quality-diversity in robot swarms. ACM Trans. Evol. Learn. Optim. 6, 1–27. doi:10.1145/3664656
Morrison, M., Podracky, C., and Liu, D. (2020). The developing toolkit of continuous directed evolution. Nat. Chem. Biol. 16, 610–619. doi:10.1038/s41589-020-0532-y
Mouret, J. B., and Clune, J. (2015). Illuminating search spaces by mapping elites. Corr. abs/1504.04909. arXiv:1504.04909.
Mouret, J., and Doncieux, S. (2012). Encouraging behavioral diversity in evolutionary robotics: an empirical study. Evol. Comput. 20, 91–133. doi:10.1162/EVCO_a_00048
Müller, K. M., Stebel, S. C., Knall, S., Zipf, G., Bernauer, H. S., and Arndt, K. M. (2005). Nucleotide exchange and excision technology (NExT) DNA shuffling: a robust method for DNA fragmentation and directed evolution. Nucleic Acids Res. 33, e117. doi:10.1093/nar/gni116
Nangle, S. N., Wolfson, M. Y., Hartsough, L., Ma, N. J., Mason, C. E., Merighi, M., et al. (2020). The case for biotech on Mars. Nat. Biotechnol. 38, 401–407. doi:10.1038/s41587-020-0485-4
Naseri, G., and Koffas, M. (2020). Application of combinatorial optimization strategies in synthetic biology. Nat. Commun. 11, 2446. doi:10.1038/s41467–020–16175–y
National Research Council (2014). Division on Earth and life studies, board on life sciences, and committee on key challenge areas for convergence in Convergence: facilitating transdisciplinary integration of life sciences, physical sciences, engineering, and beyond (Washington, D.C.: National Academies Press).
Nichol, D., Jeavons, P., Fletcher, A. G., Bonomo, R. A., Maini, P. K., Paul, J. L., et al. (2015). Steering evolution with sequential therapy to prevent the emergence of bacterial antibiotic resistance. PLOS Comput. Biol. 11, e1004493. doi:10.1371/journal.pcbi.1004493
Nishikawa, K. K., Hoppe, N., Smith, R., Bingman, C., and Raman, S. (2021). Epistasis shapes the fitness landscape of an allosteric specificity switch. Nat. Commun. 12, 5562. doi:10.1038/s41467-021-25826-7
Nissen, P., Hansen, J., Ban, N., Moore, P. B., and Steitz, T. A. (2000). The structural basis of ribosome activity in peptide bond synthesis. Science 289, 920–930. doi:10.1126/science.289.5481.920
Nitschke, G., and Didi, S. (2017). Evolutionary policy transfer and search methods for boosting behavior quality: robocup keep-away case study. Front. Robotics AI 4, 62. doi:10.3389/frobt.2017.00062
Nordmoen, J., Veenstra, F., Ellefsen, K., and Glette, K. (2021). MAP-elites enables powerful stepping stones and diversity for modular robotics. Front. Robotics AI 8, 639173. doi:10.3389/frobt.2021.639173
North, H., McGaughran, A., and Jiggins, C. (2021). Insights into invasive species from whole-genome resequencing. Mol. Ecol. 30, 6289–6308. doi:10.1111/mec.15999
Notin, P., Kollasch, A., Ritter, D., Van Niekerk, L., Paul, S., Spinner, H., et al. (2023). Proteingym: large-scale benchmarks for protein fitness prediction and design. Adv. Neural Inf. Process. Syst. 36, 64331–64379. doi:10.5555/3666122.3668932
Notin, P., Rollins, N., Gal, Y., Sander, C., and Marks, D. (2024). Machine learning for functional protein design. Nat. Biotechnol. 42, 216–228. doi:10.1038/s41587-024-02127-0
Nygaard, T. F., Martin, C. P., Howard, D., Torresen, J., and Glette, K. (2021). Environmental adaptation of robot morphology and control through real-world evolution. Evol. Comput. 29, 441–461. doi:10.1162/evco_a_00291
Ohta, T. (2002). Near-neutrality in evolution of genes and gene regulation. Proc. Natl. Acad. Sci. 99, 16134–16137. doi:10.1073/pnas.252626899
O’Brien, N. L., Holland, B., Engelstädter, J., and Ortiz-Barrientos, D. (2024). The distribution of fitness effects during adaptive walks using a simple genetic network. PLOS Genet. 20, e1011289. doi:10.1371/journal.pgen.1011289
Pachauri, R. K., Gnacadja, L., Steiner, A., Cutajar, M. Z., Ogwu, J., Briceño, S., et al. (2009). Facing global environmental change: environmental, human, energy, food, health and water security concepts, 4. Springer Science and Business Media.
Packard, N., Bedau, M. A., Channon, A., Ikegami, T., Rasmussen, S., Stanley, K. O., et al. (2019). An overview of open-ended evolution. Artif. Life 25, 93–103. doi:10.1162/artl_a_00291
Packer, M., and Liu, D. (2015). Methods for the directed evolution of proteins. Nat. Rev. Genet. 16, 379–394. doi:10.1038/nrg3927
Papkou, A., Garcia-Pastor, L., Escudero, J. A., and Wagner, A. (2023). A rugged yet easily navigable fitness landscape. Science 382, eadh3860. doi:10.1126/science.adh3860
Pardee, K., Green, A. A., Ferrante, T., Cameron, D. E., DaleyKeyser, A., Yin, P., et al. (2014). Paper-based synthetic gene networks. Cell 159, 940–954. doi:10.1016/j.cell.2014.10.004
Pardee, K., Green, A. A., Takahashi, M. K., Braff, D., Lambert, G., Lee, J. W., et al. (2016). Rapid, low-cost detection of zika virus using programmable biomolecular components. Cell 165, 1255–1266. doi:10.1016/j.cell.2016.04.059
Peng, H., Latifi, B., Müller, S., Lupták, A., and Chen, I. A. (2021). Self-cleaving ribozymes: substrate specificity and synthetic biology applications. RSC Chem. Biol. 2, 1370–1383. doi:10.1039/d0cb00207k
Peng, M., Zhao, Q., Wang, M., and Du, X. (2023). Reconfigurable scaffolds for adaptive tissue regeneration. Nanoscale 15, 6105–6120. doi:10.1039/d3nr00281k
Pitzer, E., and Affenzeller, M. (2012). A comprehensive survey on fitness landscape analysis. Recent Advances Intelligent Engineering Systems, 161–191. doi:10.1007/978-3-642-23229-9_8
Podgornaia, A., and Laub, M. (2015). Pervasive degeneracy and epistasi in a protein-protein interface. Science 347, 673–677. doi:10.1126/science.1257360
Poelwijk, F. J., Kiviet, D. J., Weinreich, D. M., and Tans, S. J. (2007). Empirical fitness landscapes reveal accessible evolutionary paths. Nature 445, 383–386. doi:10.1038/nature05451
Poelwijk, F. J., Tănase-Nicola, S., Kiviet, D. J., and Tans, S. J. (2011). Reciprocal sign epistasis is a necessary condition for multi-peaked fitness landscapes. J. Theoretical Biology 272, 141–144. doi:10.1016/j.jtbi.2010.12.015
Portillo, X., Huang, Y. T., Breaker, R. R., Horning, D. P., and Joyce, G. F. (2021). Witnessing the structural evolution of an RNA enzyme. eLife 10, e71557. doi:10.7554/eLife.71557
Povolotskaya, I. S., and Kondrashov, F. A. (2010). Sequence space and the ongoing expansion of the protein universe. Nature 465, 922–926. doi:10.1038/nature09105
Pressman, A., Blanco, C., and Chen, I. (2015). The RNA world as a model system to study the origin of life. Curr. Biol. 25, 953–963. doi:10.1016/j.cub.2015.06.016
Pressman, A. D., Liu, Z., Janzen, E., Blanco, C., Muller, U. F., Joyce, G. F., et al. (2019). Mapping a systematic ribozyme fitness landscape reveals a frustrated evolutionary network for self-aminoacylating RNA. J. Am. Chem. Soc. 141, 6213–6223. doi:10.1021/jacs.8b13298
Pugh, K., Soros, L., and Stanley, K. (2016). Quality diversity: a new frontier for evolutionary computation. Front. Robotics AI 3. doi:10.3389/frobt.2016.00040
Qiu, Q., Hu, P., Qiu, X., Govek, K. W., Cámara, P. G., and Wu, H. (2020). Massively parallel and time-resolved rna sequencing in single cells with scnt-seq. Nat. Methods 17, 991–1001. doi:10.1038/s41592-020-0935-4
Quadeer, A. A., Barton, J. P., Chakraborty, A. K., and McKay, M. R. (2020). Deconvolving mutational patterns of poliovirus outbreaks reveals its intrinsic fitness landscape. Nat. Commun. 11, 377. doi:10.1038/s41467–019–14174–2
Radford, F., Rinehart, J., and Isaacs, F. (2023). Mapping the in vivo fitness landscape of a tethered ribosome. Sci. Adv. 9, eade8934. doi:10.1126/sciadv.ade8934
Ravikumar, A., Arrieta, A., and Liu, C. C. (2014). An orthogonal dna replication system in yeast. Nat. Chemical Biology 10, 175–177. doi:10.1038/nchembio.1439
Ravikumar, A., Arzumanyan, G. A., Obadi, M. K., Javanpour, A. A., and Liu, C. C. (2018). Scalable, continuous evolution of genes at mutation rates above genomic error thresholds. Cell 175, 1946–1957. doi:10.1016/j.cell.2018.10.021
Reetz, M., Kahakeaw, D., and Lohmer, R. (2008). Addressing the numbers problem in directed evolution. ChemBioChem 9, 1797–1804. doi:10.1002/cbic.200800298
Repecka, D., Jauniskis, V., Karpus, L., Rembeza, E., Rokaitis, I., Zrimec, J., et al. (2021). Expanding functional protein sequence spaces using generative adversarial networks. Nat. Mach. Intell. 3, 324–333. doi:10.1038/s42256-021-00310-5
Riesselman, A. J., Ingraham, J. B., and Marks, D. S. (2018). Deep generative models of genetic variation capture the effects of mutations. Nat. Methods 15, 816–822. doi:10.1038/s41592-018-0138-4
Rix, G., Watkins-Dulaney, E. J., Almhjell, P. J., Boville, C. E., Arnold, F. H., and Liu, C. C. (2020). Scalable continuous evolution for the generation of diverse enzyme variants encompassing promiscuous activities. Nat. Commun. 11, 5644. doi:10.1038/s41467-020-19539-6
Robinson, D., and Nadal, D. (2025). Synthetic biology in focus: policy issues and opportunities in engineering life. OECD Sci. Technol. Industry Work. Pap. 3. doi:10.1787/3e6510cf–en
Rodrigues, J. V., Bershtein, S., Li, A., Lozovsky, E. R., Hartl, D. L., and Shakhnovich, E. I. (2016). Biophysical principles predict fitness landscapes of drug resistance. Proc. Natl. Acad. Sci. 113 E1470, E1478. doi:10.1073/pnas.1603613113
Romero, P., and Arnold, F. (2009). Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 10, 866–876. doi:10.1038/nrm2805
Rotem, A., Serohijos, A. W., Chang, C. B., Wolfe, J. T., Fischer, A. E., Mehoke, T. S., et al. (2018). Evolution on the biophysical fitness landscape of an RNA virus. Mol. Biol. Evol. 35, 2390–2400. doi:10.1093/molbev/msy131
Rothschild, L. J., Averesch, N. J., Strychalski, E. A., Moser, F., Glass, J. I., Cruz Perez, R., et al. (2024). Building synthetic cells from the technology infrastructure to cellular entities. ACS Synth. Biol. 13, 974–997. doi:10.1021/acssynbio.3c00724
Rotrattanadumrong, R., and Yokobayashi, Y. (2024). Experimental exploration of a ribozyme neutral network using evolutionary algorithm and deep learning. Nat. Commun. 13, 4847. doi:10.1038/s41467-022-32538-z
Rowe, W., Platt, M., Wedge, D. C., Day, P. J., Kell, D. B., and Knowles, J. (2010). Analysis of a complete DNA-protein affinity landscape. J. R. Soc. Interface 7, 397–408. doi:10.1098/rsif.2009.0193
Saha, R., Vázquez-Salazar, A., Nandy, A., and Chen, I. A. (2024). Fitness landscapes and evolution of catalytic RNA. Annu. Rev. Biophysics 53, 109–125. doi:10.1146/annurev-biophys-030822-025038
Sanchez-Gorostiaga, A., Bajić, D., Osborne, M. L., Poyatos, J. F., and Sanchez, A. (2019). High-order interactions distort the functional landscape of microbial consortia. PLOS Biol. 17, e3000550. doi:10.1371/journal.pbio.3000550
Santos-Moreno, J., Tasiudi, E., Kusumawardhani, H., Stelling, J., and Schaerli, Y. (2023). Robustness and innovation in synthetic genotype networks. Nat. Commun. 14, 2454. doi:10.1038/s41467–023–38033–3
Santomartino, R., Averesch, N. J. H., Bhuiyan, M., Cockell, C. S., Colangelo, J., Gumulya, Y., et al. (2023). Toward sustainable space exploration: a roadmap for harnessing the power of microorganisms. Nat. Commun. 14, 1391. doi:10.1038/s41467-023-37070-2
Sareni, B., and Krahenbuhl, L. (1998). Fitness sharing and niching methods revisited. IEEE Trans. Evol. Comput. 2, 97–106. doi:10.1109/4235.735432
Sarkisyan, K. S., Bolotin, D. A., Meer, M. V., Usmanova, D. R., Mishin, A. S., Sharonovr, G. V., et al. (2016). Local fitness landscape of the green fluorescent protein. Nature 533, 397–401. doi:10.1038/nature17995
Schlötterer, C., Kofler, R., Versace, E., Tobler, R., and Franssen, S. (2015). Combining experimental evolution with next-generation sequencing: a powerful tool to study adaptation from standing genetic variation. Heredity 114, 431–440. doi:10.1038/hdy.2014.86
Schoenle, L. A., Zimmer, C., and Vitousek, M. N. (2018). Understanding context dependence in glucocorticoid–fitness relationships: the role of the nature of the challenge, the intensity and frequency of stressors, and life history. Integr. Comp. Biol. 58, 777–789. doi:10.1093/icb/icy046
Schomburg, I., Chang, A., Placzek, S., Söhngen, C., Rother, M., Lang, M., et al. (2012). Brenda in 2013: integrated reactions, kinetic data, enzyme function data, improved disease classification: new options and contents in brenda. Nucleic Acids Research 41, D764–D772. doi:10.1093/nar/gks1049
Sechkar, K., and Steel, H. (2025). Model-guided gene circuit design for engineering genetically stable cell populations in diverse applications. J. R. Soc. Interface 22, 20240602. doi:10.1098/rsif.2024.0602
Shetty, R., Endy, D., and Knight, T. (2008). Engineering BioBrick vectors from BioBrick parts. J. Biol. Eng. 2, 5. doi:10.1186/1754–1611–2–5
Si, T., Chao, R., Min, Y., Wu, Y., Ren, W., and Zhao, H. (2017). Automated multiplex genome-scale engineering in yeast. Nat. Commun. 8, 15187. doi:10.1038/ncomms15187
Sims, K. (1994). Evolving 3d morphology and behavior by competition. Artif. Life 1, 353–372. doi:10.1162/artl.1994.1.4.353
Slomovic, S., Pardee, K., and Collins, J. J. (2015). Synthetic biology devices for in vitro and in vivo diagnostics. Proc. Natl. Acad. Sci. 112, 14429–14435. doi:10.1073/pnas.1508521112
Smerlak, M. (2021). Neutral quasispecies evolution and the maximal entropy random walk. Sci. Adv. 7, eabb2376. doi:10.1126/sciadv.abb2376
Sng, O., Neuberg, S. L., Varnum, M. E., and Kenrick, D. T. (2017). The crowded life is a slow life: population density and life history strategy. J. Personality Soc. Psychol. 112, 736–754. doi:10.1037/pspi0000086
Steel, H., Lillacci, G., Khammash, M., and Papachristodoulou, A. (2017). “Challenges at the interface of control engineering and synthetic biology,” in 2017 IEEE 56th annual conference on decision and control (CDC) (IEEE), 1014–1023.
Steinberg, B., and Ostermeier, M. (2016). Environmental changes bridge evolutionary valleys. Sci. Adv. 2, e1500921. doi:10.1126/sciadv.1500921
Stemmer, W. (1994). Rapid evolution of a protein in vitro by DNA shuffling. Nature 370, 389–391. doi:10.1038/370389a0
Stock, M., and Gorochowski, T. (2024). Open-endedness in synthetic biology: a route to continual innovation for biological design. Sci. Adv. 10, eadi3621. doi:10.1126/sciadv.adi3621
Storz, J. (2018). Compensatory mutations and epistasis for protein function. Curr. Opin. Struct. Biol. 50, 18–25. doi:10.1016/j.sbi.2017.10.009
Swainston, N., Currin, A., Green, L., Breitling, R., Day, P. J., and Kell, D. B. (2017). CodonGenie: optimised ambiguous codon design tools. PeerJ - Comput. Sci. 3, e120. doi:10.7717/peerj-cs.120
Szymanski, N. J., Rendy, B., Fei, Y., Kumar, R. E., He, T., Milsted, D., et al. (2023). An autonomous laboratory for the accelerated synthesis of novel materials. Nature 624, 86–91. doi:10.1038/s41586-023-06734-w
Tang, L., Gao, H., Zhu, X., Wang, X., Zhou, M., and Jiang, R. (2016). Construction of “Small, Intelligent” focused mutagenesis libraries using well-designed combinatorial degenerate primers. BioTechniques 52, 149–158. doi:10.2144/000113820
Taylor, W. R. (2006). Transcription and translation in an rna world. Philosophical Trans. R. Soc. B Biol. Sci. 361, 1751–1760. doi:10.1098/rstb.2006.1910
Tenaillon, O., Rodríguez-Verdugo, A., Gaut, R. L., McDonald, P., Bennett, A. F., Long, A. D., et al. (2012). The molecular diversity of adaptive convergence. Science 335, 457–461. doi:10.1126/science.1212986
The RNAcentral Consortium (2019). Rnacentral: a hub of information for non-coding rna sequences. Nucleic Acids Res. 47, D221–D229. doi:10.1093/nar/gky1034
Towers, S., James, J., Steel, H., and Kempf, I. (2024). Learning-based estimation of fitness landscape ruggedness for directed evolution. BioRxiv. doi:10.1101/2024.02.28.582468
Turner, N. J. (2009). Directed evolution drives the next generation of biocatalysts. Nat. Chemical Biology 5, 567–573. doi:10.1038/nchembio.203
Vaishnav, E. D., de Boer, C. G., Molinet, J., Yassour, M., Fan, L., Adiconis, X., et al. (2022). The evolution, evolvability and engineering of gene regulatory DNA. Nature 603, 455–463. doi:10.1038/s41586-022-04506-6
van Berlo, D., Nguyen, V. V. T., Gkouzioti, V., Leineweber, K., Verhaar, M. C., and van Balkom, B. W. M. (2021). Stem cells, organoids, and Organ-on-a-chip models for personalized in vitro drug testing. Curr. Opin. Toxicol. 28, 7–14. doi:10.1016/j.cotox.2021.08.006
Van Cleve, J., and Weissman, D. B. (2015). Measuring ruggedness in fitness landscapes. Proc. Natl. Acad. Sci. 112, 7345–7346. doi:10.1073/pnas.1507916112
Van den Bergh, B., Swings, T., Fauvart, M., and Michiels, J. (2018). Experimental design, population dynamics, and diversity in microbial experimental evolution. Microbiol. Mol. Biol. Rev. 82, 10–1128. doi:10.1128/MMBR.00008-18
van Nimwegen, E. (2006). Influenza escapes immunity along neutral networks. Science 314, 1884–1886. doi:10.1126/science.1137300
Vásárhelyi, G., Virágh, C., Somorjai, G., Nepusz, T., Eiben, A. E., and Vicsek, T. (2018). Optimized flocking of autonomous drones in confined environments. Sci. Robotics 3, eaat3536. doi:10.1126/scirobotics.aat3536
Vidal, L. S., Isalan, M., Heap, J. T., and Ledesma-Amaro, R. (2023). A primer to directed evolution: current methodologies and future directions. RSC Chem. Biol. 4, 271–291. doi:10.1039/d2cb00231k
Vikhar, P. A. (2016). “Evolutionary algorithms: a critical review and its future prospects,” in 2016 international conference on global trends in signal processing, information computing and communication (ICGTSPICC) (IEEE), 261–265.
Vincent, J. F. V. (2017). The trade-off: a central concept for biomimetics. Bioinspired, Biomim. Nanobiomaterials 6, 67–76. doi:10.1680/jbibn.16.00005
Voigt, C. A. (2020). Synthetic biology 2020–2030: six commercially available products that are changing our world. Nat. Commun. 11, 6379. doi:10.1038/s41467-020-20122-2
Wagner, A. (2023). Evolvability-enhancing mutations in the fitness landscapes of an RNA and a protein. Nat. Commun. 14, 3624. doi:10.1038/s41467–023–39321–8
Walker, D. G., Teerawattananon, Y., Anderson, R., and Richardson, G. (2010). Generalisability, transferability, complexity and relevance. Evidence-Based Decis. Econ. Health Care, Social Welfare, Education Criminal Justice, 56–66. doi:10.1002/9781444320398.ch5
Wang, G., and Vasquez, K. (2023). Dynamic alternative DNA structures in biology and disease. Nat. Rev. Genet. 24, 211–234. doi:10.1038/s41576-022-00539-9
Wang, H. H., Isaacs, F. J., Carr, P. A., Sun, Z. Z., Xu, G., Forest, C. R., et al. (2009). Programming cells by multiplex genome engineering and accelerated evolution. Nature 460, 894–898. doi:10.1038/nature08187
Wang, X., Lin, H., Zheng, Y., Feng, J., Yang, Z., and Tang, L. (2015). MDC-analyzer-facilitated combinatorial strategy for improving the activity and stability of halohydrin dehalogenase from Agrobacterium radiobacter AD1. J. Biotechnol. 206, 1–7. doi:10.1016/j.jbiotec.2015.04.002
Wang, D., Huot, M., Mohanty, V., Shakhnovich, E., and Collins, J. J. (2020). A deep learning approach to programmable RNA switches. Nat. Commun. 11, 5057. doi:10.1038/s41467–020–18677–1
Wang, Y., Xue, P., Cao, M., Yu, T., Lane, S. T., and Zhao, H. (2021). Directed evolution: methodologies and applications. Chem. Rev. 121, 12384–12444. doi:10.1021/acs.chemrev.1c00260
Wang, D., Huot, M., Mohanty, V., and Shakhnovich, E. (2024a). Biophysical principles predict fitness of SARS-CoV-2 variants. Proc. Natl. Acad. Sci. 121, e2314518121. doi:10.1073/pnas.2314518121
Wang, B., Liu, Y., Bai, X., Tian, H., Wang, L., Feng, M., et al. (2024b). In vitro generation of genetic diversity for directed evolution by error-prone artificial DNA synthesis. Commun. Biol. 7, 628. doi:10.1038/s42003–024–06340–0
Weinreich, D. M., Watson, R. A., and Chao, L. (2005). Perspective: sign epistasis and genetic costraint on evolutionary trajectories. Evolution 59, 1165–1174. doi:10.1111/j.0014-3820.2005.tb01768.x
Weinreich, D. M., Delaney, N. F., DePristo, M. A., and Hartl, D. L. (2006). Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 312, 111–114. doi:10.1126/science.1123539
Weinreich, D. M., Lan, Y., Jaffe, J., and Heckendorn, R. B. (2018). The influence of higher-order epistasis on biological fitness landscape topography. J. Statistical Physics 172, 208–225. doi:10.1007/s10955-018-1975-3
Wilke, C. O., and Adami, C. (2002). The biology of digital organisms. TRENDS Ecology and Evolution 17, 528–532. doi:10.1016/s0169-5347(02)02612-5
Wilke, C. O., Wang, J. L., Ofria, C., Lenski, R. E., and Adami, C. (2001). Evolution of digital organisms at high mutation rates leads to survival of the flattest. Nature 412, 331–333. doi:10.1038/35085569
Williams, G. (1996). Adaptation and natural selection: a critique of some current evolutionary thought. Princeton, USA: Princeton University Press.
Wittmann, B. J., Johnston, K. E., Wu, Z., and Arnold, F. H. (2021a). Advances in machine learning for directed evolution. Curr. Opin. Struct. Biol. 69, 11–18. doi:10.1016/j.sbi.2021.01.008
Wittmann, B. J., Yue, Y., and Arnold, F. H. (2021b). Informed training set design enables efficient machine learning-assisted directed protein evolution. Cell Syst. 12, 1026–1045. doi:10.1016/j.cels.2021.07.008
Wong, T. S., Roccatano, D., Zacharias, M., and Schwaneberg, U. (2006). A statistical analysis of random mutagenesis methods used for directed protein evolution. J. Mol. Biol. 355, 858–871. doi:10.1016/j.jmb.2005.10.082
Wortel, M. T., Agashe, D., Bailey, S. F., Bank, C., Bisschop, K., Blankers, T., et al. (2023). Towards evolutionary predictions: current promises and challenges. Evol. Appl. 16, 3–21. doi:10.1111/eva.13513
Yang, K. K., Wu, Z., and Arnold, F. H. (2019). Machine-learning-guided directed evolution for protein engineering. Nat. Methods 16, 687–694. doi:10.1038/s41592-019-0496-6
Yang, J., Li, F., and Arnold, F. (2024). Opportunities and challenges for machine learning-assisted enzyme engineering. ACS Central Sci. 10, 226–241. doi:10.1021/acscentsci.3c01275
Yang, J., Lal, R. G., Bowden, J. C., Astudillo, R., Hameedi, M. A., Kaur, S., et al. (2025). Active learning-assisted directed evolution. Nat. Commun. 16, 714. doi:10.1038/s41467-025-55987-8
Yi, X., and Dean, A. (2019). Adaptive landscapes in the age of synthetic biology. Mol. Biol. Evol. 36, 890–907. doi:10.1093/molbev/msz004
Yilmaz, B., Mooser, C., Keller, I., Li, H., Zimmermann, J., Bosshard, L., et al. (2021). Long-term evolution and short-term adaptation of microbiota strains and sub-strains in mice. Cell Host and Microbe 29, 650–663. doi:10.1016/j.chom.2021.02.001
Zhang, H., Chu, W., Sun, J., Liu, Z., Huang, W. C., Xue, C., et al. (2019). Combining cell surface display and DNA-shuffling technology for directed evolution of streptomyces phospholipase D and synthesis of phosphatidylserine. J. Agric. Food Chem. 67, 13119–13126. doi:10.1021/acs.jafc.9b05394
Zhang, Q., Ding, K., Lv, T., Wang, X., Yin, Q., Zhang, Y., et al. (2025). Scientific large language models: a survey on biological and chemical domains. ACM Comput. Surv. 57, 1–38. doi:10.1145/3715318
Zhong, Z., Wong, B. G., Ravikumar, A., Arzumanyan, G. A., Khalil, A. S., and Liu, C. C. (2020). Automated continuous evolution of proteins in vivo. ACS Synthetic Biology 9, 1270–1276. doi:10.1021/acssynbio.0c00135
Keywords: Artificial life, sythetic biology, directed evolution, fitness landscape, evolutionary algorithms
Citation: Sibanda I and Nitschke G (2025) Bioengineering hybrid artificial life. Front. Bioinform. 5:1676359. doi: 10.3389/fbinf.2025.1676359
Received: 23 September 2025; Accepted: 27 November 2025;
Published: 16 December 2025.
Edited by:
Budheswar Dehury, Manipal Academy of Higher Education, IndiaReviewed by:
Matthieu Bultelle, Imperial College London, United KingdomIdris Kempf, University of Oxford, United Kingdom
Joseph Breeden, Deep Future Analytics LLC, United States
Copyright © 2025 Sibanda and Nitschke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geoff Nitschke, Z25pdHNjaGtlQGNzLnVjdC5hYy56YQ==