 Bart Borghans†
Bart Borghans† Natalia Dmitrieva
Natalia Dmitrieva Christoph Fahlke
Christoph Fahlke- Institute of Biological Information Processing, Molekular- und Zellphysiologie (IBI-1), Jülich, Germany
Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system. After exocytotic release from presynaptic nerve terminals, glutamate diffuses across the synaptic cleft and opens postsynaptic ionotropic glutamate receptors, thus depolarizing the postsynaptic neuron. Synaptic activity is terminated by rapid and efficient uptake into surrounding neurons and glial cells. The function of a glutamatergic synapse thus critically depends on two distinct transport systems: vesicular and plasma membrane glutamate transporters. Vesicular glutamate transporters (VGLUTs) accumulate glutamate in synaptic vesicles and determine the amount of released glutamate. Plasma membrane glutamate transporters (excitatory amino acid transporters, EAATs) clear the synaptic cleft from glutamate, setting the time resolution and energy demand of glutamatergic synaptic signaling. Both classes of glutamate transporters are not only secondary-active transporters, but also function as chloride channels, with different roles in chloride and glutamate homeostasis. Despite similar transport functions, VGLUTs and EAATs are structurally diverse and employ different molecular mechanisms to overcome the same chemical challenges. We here review recent progress in understanding the molecular and cellular biophysics of vesicular glutamate transporters and compare their properties with plasma membrane glutamate transporters.
Introduction
At chemical synapses, neurotransmitters are released via the exocytosis of synaptic vesicles (Jahn and Fasshauer, 2012) that specifically accumulate neurotransmitters with the help of vesicular neurotransmitter transporter (Takamori et al., 2000; Blakely and Edwards, 2012; Omote et al., 2016). The driving force and the transport rates of vesicular neurotransmitter transporters are major determinants of the synaptic strength. In the mammalian brain, three vesicular glutamate transporters (VGLUT1, VGLUT2 and VGLUT3) are responsible for filling the synaptic vesicles with glutamate (Blakely and Edwards, 2012; Omote et al., 2016; Farsi et al., 2017; Eriksen et al., 2020). For effective and isosmotic glutamate accumulation, VGLUTs harness the electrochemical H+ gradient across the vesicular membrane and additionally assume an anion channel mode to permit Cl¯ efflux during synaptic vesicle filling (Schenck et al., 2009; Eriksen et al., 2016; Kolen et al., 2023).
After synaptic release, plasma membrane glutamate transporters terminate glutamatergic synaptic transmission by quickly taking up neurotransmitters into surrounding neuronal and glial cells (Danbolt, 2001). By ensuring low resting extracellular glutamate concentrations, they improve the time resolution, reduce the energy demand of synaptic transmission and prevent glutamate excitotoxicity. There exist five mammalian plasma membrane glutamate transporters, the excitatory amino acid transporters (EAATs) 1–5 (Danbolt, 2001; Vandenberg and Ryan, 2013; Rose et al., 2018). EAATs couple glutamate transport to the symport of three Na+ ions and 1 H+, in exchange with one K+. This complex transport stoichiometry allows accumulating glutamate against up to 106-fold concentration gradients and maintaining extracellular glutamate levels below the micromolar range (Zerangue and Kavanaugh, 1996). They are also anion-selective channels that open and close during transitions along the glutamate transport cycle (Fairman et al., 1995; Wadiche et al., 1995; Bergles et al., 2002; Fahlke et al., 2016), with various functions beyond glutamate uptake (Untiet et al., 2017; Kovermann et al., 2020; Engels et al., 2021; Gehlen et al., 2021).
In recent years, molecular cloning, heterologous expression, cellular electrophysiology, fluorescence spectroscopy, structural approaches, and molecular simulations have uncovered the molecular mechanisms of coupled transport, substrate selectivity, and anion conduction in VGLUTs as well as in EAAT glutamate transporters. We here review recent progress in understanding of these two classes of glutamate transporters.
VGLUTs are highly selective low affinity glutamate transporters
VGLUTs were initially characterized in secretory vesicle preparations using radiotracer flux methods (Naito and Ueda, 1985; Maycox et al., 1988; Tabb et al., 1992; Hartinger and Jahn, 1993; Moriyama and Yamamoto, 1995). These experiments already identified the key features of vesicular glutamate transport: electrochemical proton gradients as driving force, low affinity, with half-maximum transport at 1.6 mM [L-glutamate] (Naito and Ueda, 1985; Tabb et al., 1992), high glutamate-over-aspartate selectivity (Naito and Ueda, 1985; Maycox et al., 1988), independence from monovalent cation concentration (Naito and Ueda, 1985) and stimulation by luminal as well as by cytoplasmic [Cl¯] (Naito and Ueda, 1985; Tabb et al., 1992; Hartinger and Jahn, 1993). Molecular cloning (Bellocchio et al., 2000; Takamori et al., 2001; Takamori et al., 2002) made heterologous expression, purification and reconstitution possible (Kasahara and Hinkle, 1977; Omote and Moriyama, 2013). These novel techniques resulted in detailed analysis of the Cl¯ effects on VGLUT transport function, demonstrating that VGLUTs are not only allosterically activated by Cl¯ (Juge et al., 2010), but also function as Cl¯ channel/transporter (Schenck et al., 2009).
VGLUTs are dual function glutamate transporter/anion channels
The exclusive intracellular localization in neurons and in heterologous expression systems impedes the characterization of WT VGLUT with conventional cellular electrophysiology techniques. Eriksen et al. (2016) removed intracellular retention motifs by site-directed mutagenesis, generating a mutant VGLUT1 construct that predominantly inserts into the surface membrane. Expression of this mutant VGLUT in Xenopus oocytes and voltage clamp analysis unambiguously demonstrated that vesicular glutamate transporters can conduct Cl¯. The VGLUT Cl¯ conductance is not coupled to glutamate transport, but is activated by luminal H+ and Cl¯ in an allosteric manner, closely similar to the regulation of VGLUT glutamate transport. In a follow-up paper, Chang et al. (2018) applied electrophysiology on WT VGLUT1 by patching enlarged endosomes, demonstrating that the modification necessary for surface membrane insertion did not change functional properties of the VGLUT. Baraglia et al. recently used a computational model of synaptic vesicle acidification to describe how an integrated Cl¯ conductance of vesicular glutamate transporters modifies the dynamics of synaptic vesicle pH and [glutamate] (Baraglia et al., 2025).
Kolen et al. used whole-cell patch clamp and noise analysis on transfected mammalian cells with a slightly different surface–membrane optimized construct to analyze single channel properties of VGLUT1 anion channels (Kolen et al., 2023). They determined unitary transport rates of 2 × 105 s−1 at −160 mV; this value is above maximum rates expected for transporters and demonstrates that VGLUTs form an aqueous anion diffusion pathway across the membrane. They also provided an estimate of channel open times based on changes of apparent unitary current amplitudes with filtering (Silberberg and Magleby, 1993; Alekov and Fahlke, 2009). VGLUTs exhibit very low open times in the range of 25 µs (Kolen et al., 2023) indicating rapid flickering between open and closed states. Figure 1A depicts representative responses of a HEK293T cell transiently expressing EE6, 7AA/L11A/EE505,506AA/F510A/V511A VGLUT1-GFP (VGLUT1PM; with PM as abbreviation for plasma membrane) to voltage steps between −160 mV and +60 mV from a holding potential of 0 mV.
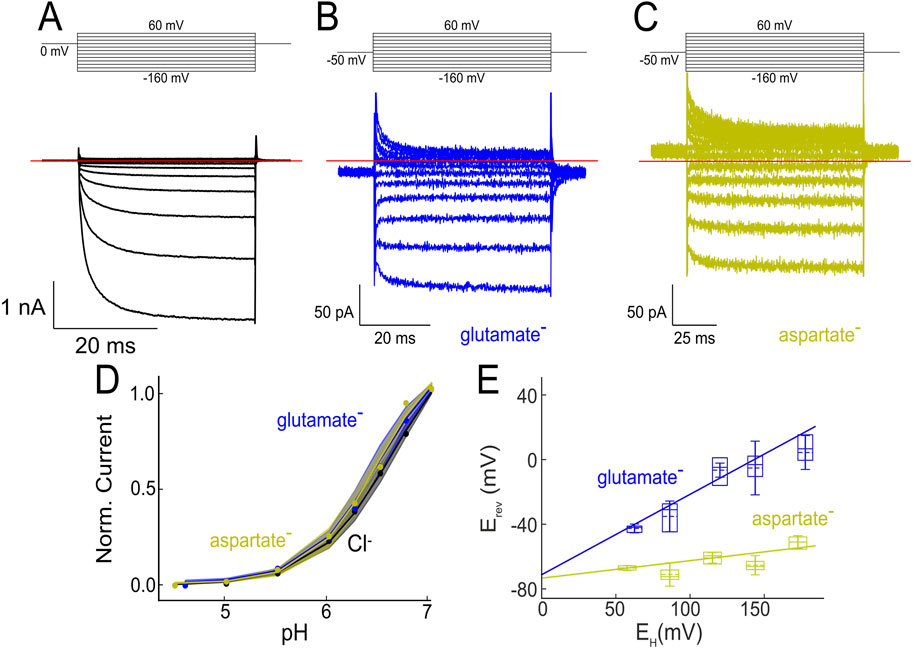
Figure 1. VGLUT can function as glutamate/aspartate transporter as well as Cl− channel. (A–C) Representative current responses from HEK293T cells expressing VGLUT1PM at external pH 5.5. Cells were held at 0 mV, and voltage steps were applied to voltages between −160 mV and +60 mV. The cell in (A) was dialyzed with a Cl−-containing solution; in (B) or (C) Cl− was fully substituted with glutamate (B) or aspartate (C). (D) VGLUT currents are activated by external pH; VGLUT channel and transporter currents exhibit identical pH dependences. Modified and reprinted from (Borghans et al., 2025), with permission. (E) Glutamate (blue boxes) and aspartate (yellow boxes) current reversal potentials versus H+ gradients given as Nernst potential (EH =
Since channel-mediated Cl− currents are much bigger than VGLUT transport currents, complete substitution of intracellular Cl¯ by glutamate- is necessary to record glutamate currents with such an approach (Figure 1B). VGLUT1PM glutamate currents depend on external/luminal pH and [Cl−], with similar concentration dependence as VGLUT1PM Cl¯ currents (Figure 1D). Such currents are highly specific; they could not be observed in non-transfected cells or cells expressing a non-functional mutant; they were inhibited by the VGLUT blocker Rose Bengal (Ogita et al., 2001), and current amplitudes were shown to be proportional to protein expression levels (Kolen et al., 2023). Comparison of Cl¯ and glutamate currents at identical expression levels provided unitary glutamate transport rates of around 600 s−1 at −160 mV (Kolen et al., 2023).
VGLUT1PM transport currents obtained by whole-cell patch clamp are not as selective as expected from radiotracer flux measurements (Naito and Ueda, 1985). In such experiments, VGLUT1PM currents of comparable amplitudes were observed in cells dialyzed with glutamate, aspartate, gluconate, MES or isethionate (Kolen et al., 2023). VGLUT1PM aspartate currents (Figure 1C) differ from VGLUT1PM glutamate (Figure 1B) currents in a more negative reversal potential (Kolen et al., 2023). An earlier study, which measured glutamate and aspartate currents in excised endosomal patches in a reduced voltage range, reported much smaller aspartate than glutamate currents (Chang et al., 2018). However, both experimental approaches, whole-cell and excised patch clamp recordings, revealed differences in the current reversal potential of glutamate and aspartate currents, with glutamate values shifted towards the H+ reversal potential.
Comparing glutamate currents at various proton gradients demonstrated a linear relationship between current reversal potential and the Nernst potential for protons (Figure 1E). Such a behavior is expected for H+-glutamate exchangers, with the electrochemical gradient for coupled exchange of n H+ ions for m glutamate ions being:
where F is Faraday’s constant, U the transmembrane voltage, R is the universal gas constant, and T the absolute temperature. Currents generated by stoichiometric H+-glutamate exchange reverse at:
A plot of experimentally obtained reversal potentials versus the Nernst potential of the applied H+ gradient can thus be described with a linear function, providing
There are arguments in favor of VGLUTs functioning as glutamate uniporter, however, also exclusively driven by the electrical gradient (Li et al., 2025). For example, electric gradients without pH gradients, but not pH gradients alone can drive glutamate uptake in reconstituted systems (Juge et al., 2006; Juge et al., 2010). The absent glutamate uptake upon mere pH gradients, however, can be explained by the pronounced voltage dependence of VGLUT glutamate currents (Figure 1B), that results in negligible glutamate uptake at 0 mV and prevents any conclusions from results without electrical gradients. In native synaptic vesicles, dissipation of the pH gradient can even increase glutamate accumulation in the synaptic vesicle (Goh et al., 2011), most likely via stimulating the vesicular H+ pumps that increase the electrical gradient and thus stimulates VGLUT transport. In contrast, VGLUT H+-glutamate exchange is not only supported by the electrophysiological experiments shown in Figure 1E (Kolen et al., 2023), but also by early experiments with bovine synaptic vesicles (Tabb et al., 1992) and pH measurements in synaptic vesicles of cultured neurons (Martineau et al., 2017). These experiments demonstrated that glutamate accumulation reduces the vesicular proton content in synaptic vesicles. Moreover, mathematical modeling (see below) demonstrates that H+-glutamate transport makes vesicular acidification even more effective.
Variable transport stoichiometry explains the observed transport selectivity of VGLUT
Aspartate does not compete in VGLUT 3H-L-glutamate uptake assays (Naito and Ueda, 1985; Bellocchio et al., 2000), and only glutamate, but not aspartate is present in synaptic vesicles (Burger et al., 1989). Both results support the idea that VGLUTs can only transport glutamate, but not aspartate. In contrast to this conclusion, cells expressing VGLUT1PM exhibit glutamate and aspartate whole-cell currents of comparable amplitude (Figure 1) (Kolen et al., 2023).
Mathematical modelling of synaptic vesicle accumulation helped understanding how–although VGLUT can transport aspartate–different glutamate and aspartate transport stoichiometries result in exclusive glutamate accumulation in synaptic vesicle (Kolen et al., 2023). The synaptic vesicle model contains VGLUTs, V-type ATPases, and H+ leak channels; the VGLUT anion channel function was described as Cl¯ conductance with the same pH and [Cl¯] dependence as VGLUT transport. Transmembrane ion fluxes are calculated with a set of differential equations, with neutral pH, high [Cl¯], and 0 mM glutamate/aspartate within the vesicle as starting point for vesicle filling after endocytosis. The larger driving force for H+-coupled exchange makes glutamate accumulation much more effective than aspartate accumulation. Modelling demonstrates that the different driving forces result in exclusive glutamate accumulation under conditions resembling VGLUT glutamate/aspartate transport in presynaptic nerve terminals, i.e., equal cytoplasmic amounts of glutamate and aspartate. Moreover, the model also correctly predicts glutamate-aspartate interactions in radiotracer flux measurements; an excess of external aspartate fails to reduce radioactive glutamate uptake (Kolen et al., 2023).
Mathematical modeling explains how VGLUT anion channel function affect glutamate uptake
Endosomal Cl¯ channels support vesicular acidification by maintaining electroneutrality of V-ATPase transport (Bae and Verkman, 1990; George, 1998), and the VGLUT anion conductance (Schenck et al., 2009) was originally proposed to fulfill a similar role. Neither VGLUT variants without Cl¯ channel activity nor blockers that separate VGLUT transport and channel functions have yet been identified; it is therefore currently not possible to experimentally test the physiological impact of the VGLUT anion channel function. Kolen et al. (2023) used the quantitative synaptic vesicle model described above to predict the consequences of increasing or decreasing synaptic vesicle Cl− conductances. The complete removal of the VGLUT Cl¯ conductance reduces glutamate accumulation by less than 50%; a 100-fold lower Cl¯ conductance results in slower glutamate accumulation to the same levels as normal values. Predicted membrane potentials of synaptic vesicles during glutamate accumulations revealed that VGLUT-mediated Cl¯ efflux supports glutamate accumulation by depolarizing synaptic vesicles. A reduction of the VGLUT anion conductance in the model makes higher primary active H+ accumulation necessary, indicating that the VGLUT anion channel function improves the energy efficiency of vesicular glutamate accumulation.
Allosteric regulation of VGLUTs by H+ and Cl−
Both VGLUT transport functions, glutamate transport and anion channel opening, are stimulated by luminal H+ and Cl¯, and this regulatory pathways have important physiological consequences. After endocytosis and synaptic vesicle formation, primary active H+ transport by V-ATPases acidifies the vesicles. Allosteric H+ regulation ensures full VGLUT function only at a significant H+ gradient across the vesicle, i.e., under conditions that provide much higher driving forces for H+-glutamate exchange than for aspartate uniport. The requirement for an acidic milieu also prevents VGLUT glutamate uptake through the surface membrane after synaptic vesicle exocytosis (Eriksen et al., 2016). The high driving force of H+-glutamate exchange will result in very effective glutamate accumulation in the synaptic vesicle that saturates at vesicular [glutamate] close to molar concentrations (Eriksen et al., 2020; Kolen et al., 2023). Such overfilling that would result in water influx into synaptic vesicles is prevented by allosteric Cl− regulation. Glutamate accumulation is associated with vesicular Cl¯ efflux, and block of VGLUT activity by low vesicular [Cl¯] prevents glutamate accumulation to such high values. Whole endosome recording in transfected cells and site-directed mutagenesis allowed identifying R176 as molecular determinant of VGLUT1 allosteric Cl¯ activation (Chang et al., 2018).
In ion channels, ions cross the membrane by diffusing along an aqueous conduction pathway that is opened and closed by conformational changes, the so-called channel gating. Ion conduction can be separated from ion channel gating by voltage clamp analysis (Hille, 2001). In transporters, ion transport is mediated by conformational changes that are driven by subsequent substrate association/dissociation steps. The separation of individual steps is complicated and requires complex kinetic modeling of the transporter cycle (Bergles et al., 2002). Borghans et al. (2025) combined measurements of VGLUT current transients upon pH and [Cl−] steps and mathematical modeling with extensive statistical testing to generate kinetic schemes that describe VGLUT glutamate/aspartate transport and ion channel gating. These analyses allowed the identification of changes in kinetic scheme parameters upon binding of Cl¯, glutamate, or aspartate or upon applying voltage. They described VGLUT Cl¯ channel gating with a kinetic scheme that includes two protonation sites and distinct opening and closing rates and Cl¯ binding rates for each protonation state (Figures 2A–E). This analysis suggest that Cl¯ binding promotes channel opening by modifying pKas of the protonation sites, as well as the rates of pore opening and closure (Borghans et al., 2025).
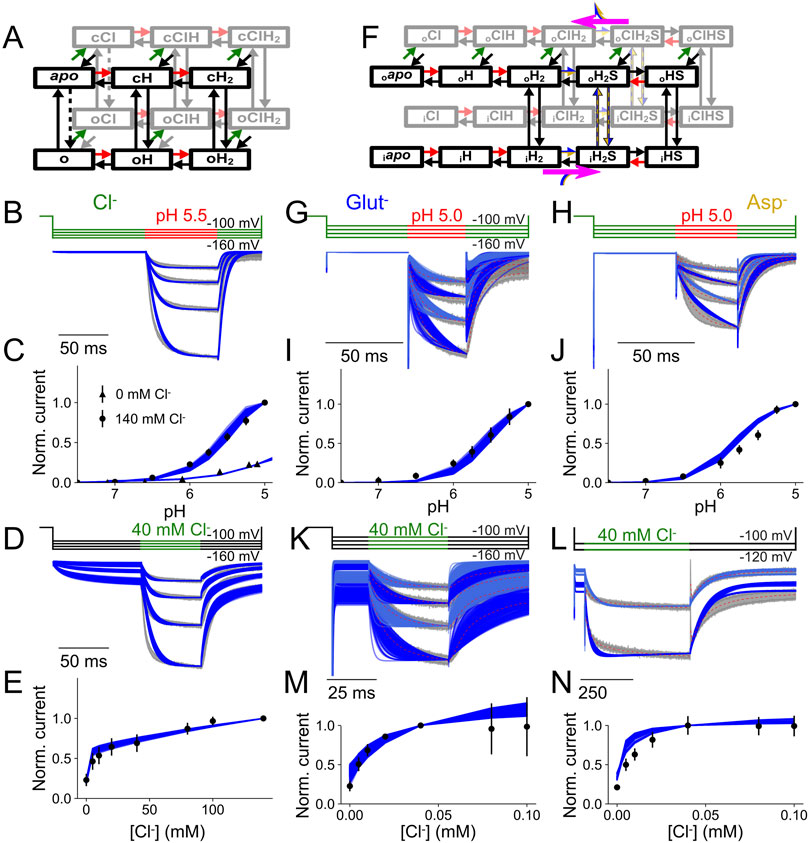
Figure 2. Kinetic description of VGLUT1 channel and transporter currents. (A,F) Kinetic schemes used to describe VGLUT1PM Cl− currents (A) or glutamate/aspartate transport functions (F). (B,G,H) Current responses and predictions from the kinetic schemes for rapid pH steps to pH 5.5 (B) or 5.0 (G,H) at [Cl−]o = 140 mM. (C,I,J) Experimental and predicted late current plots versus pH at [Cl−]o = 140 mM. (D,K,L) Current responses and predictions from the kinetic schemes for rapid Cl− steps to 140 mM at pH 5.0. (E,M,N) late current plots versus [Cl−]o. Modified and reprinted from (Borghans et al., 2025), with permission.
Glutamate/aspartate transport was described with an alternating access model that assumes translocation of apo transporters in the doubly protonated state to the inward-facing conformation and return with bound amino acid substrate in either the singly protonated or doubly protonated state (Figures 2B–N). Glutamate, but not aspartate, promotes the release of one proton from inward-facing VGLUT1, resulting in preferential coupled H+–glutamate exchange (Borghans et al., 2025). Cl¯ stimulates glutamate transport by accelerating H+–glutamate exchange, mainly by making the glutamate-binding site accessible to the cytoplasm and by facilitating transitions to the inward-facing conformation after outward substrate release (Borghans et al., 2025).
Structural basis of VGLUT glutamate transport
Thus far, high resolution structures have been reported for two members of the SLC17 family, rat VGLUT2 (SLC17A3) (Li et al., 2020; Li et al., 2025) and human sialin (SLC17A5) (Hu et al., 2023; Schmiege et al., 2024), and for the bacterial model protein DgoT (Leano et al., 2019). All SLC17 proteins assume a typical MFS fold (Abramson et al., 2003) with two six transmembrane helix bundles in a pseudo 2-fold symmetry and two intracellular helices at the amino- and the carboxy-terminal end (Figure 3). Whereas cryo-EM data on VGLUT2 were exclusively obtained in the outward-facing conformation (Li et al., 2020; Li et al., 2025), Dgot was crystallized in inward- and outward-facing conformations (Leano et al., 2019). For sialin, cryogenic electron-microscopy structures have been reported in apo cytosol-open, apo lumen-open, as well as in complex with N-acetylaspartylglutamate (NAAG) (Hu et al., 2023; Schmiege et al., 2024). The comparison of inward and outward-facing conformations illustrates how SLC17 transporters translocate via the “rocker-switch” mechanism of MFS transporters, in which a rocking movement of the two halves of the transporter around a central binding site provides alternating access of the substrate from either of the two membrane sides (Forrest et al., 2011).
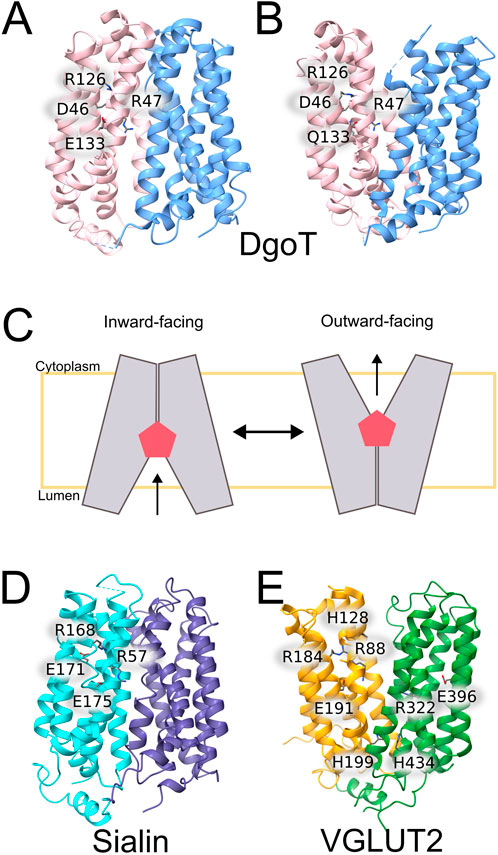
Figure 3. Three-dimensional structures of SLC17 proteins and bacterial model proteins. (A,B) X-ray crystal structures of WT (left) and E133Q (right) DgoT (Leano et al., 2019). (C) Schematic presentation of rocker-switch conformational changes in DgoT (D,E) Cryo-EM structures of rVGLUT2 (D) (Li et al., 2020) and human sialin (E) (Hu et al., 2023).
DgoT
DgoT mediates the symport of H+ and galactonate in a 2:1 transport stoichiometry (Dmitrieva et al., 2024). There are two H+ acceptors, D46 and E133, that interact in their deprotonated state with R47 and R126. Protonation of D46 partly opens the extracellular gate by releasing R126, and additional protonation of E133 results in full opening of the extracellular gate and most effective substrate binding. The disruption of the E133–R47 salt bridge permits interaction of R47 with negatively charged galactonate. Galactonate is coordinated by R47 and by various other polar side chains (Batarni et al., 2023), so that substrate binding brings TM1 and TM7 closer together and triggers the transition of DgoT into the inward-facing conformation. Whereas D46 and E133 can be simultaneously protonated in the outward-facing conformation, the D46 carboxyl group is sequestered in inward-facing conformations, making D46 deprotonation only possible via proton release to E133 after initial deprotonation of this residue. D46 deprotonation allows galactonate unbinding, either in its protonated or its non-protonated form, and subsequent proton release from E133 results in intracellular gate closure. Reorientation of the empty transporter to outward-facing conformation completes the cycle.
VGLUT2
From the vesicular glutamate transporters in the mammalian brain (VGLUT1-3), only rVGLUT2 has been structurally characterized, revealing an outward-facing conformation. There are two cryo-EM structures available, one of WT rVGLUT2 (Li et al., 2020) and the other one of R181Q/E191Q rVGLUT2 binding a cyclic glutamate analog, l-trans-1-amino-1,3-dicarboxy cyclopentane (ACPD), that binds with higher affinity than glutamate (Li et al., 2025). The VGLUTs contain two conserved arginine residues (R88 and R322 in VGLUT2) (Juge et al., 2006; Chang et al., 2018; Li et al., 2025) that form salt-bridges to unprotonated glutamate residues, E191-R88 and E396-R322 (Figure 3). It is tempting to speculate that protonation of these glutamates releases the arginines and permit glutamate binding and transport at acidic pH. At the luminal side, H128 is thought to function as the primary proton acceptor (Rostamipour et al., 2022). It is in close spatial proximity to a conserved arginine (R184 in VGLUT2), that was shown to bind Cl− (Chang et al., 2018). Experiments on endosomes showed Cl− independent anion currents by the R184A mutant (Chang et al., 2018), suggesting an allosteric chloride binding site formed by these residues and R88 (Rostamipour et al., 2022). On the other side of the transporter, the so-called histidine gate, formed by H199-H434 pair limits the central cavity at the cytosol site, thus regulating access to the glutamate binding site form the cell cytoplasm.
Sialin
Lysosomes decompose cellular and extracellular macromolecules via multiple hydrolases. The proteolytic endproducts need to be removed from the lysosome by various, mostly secondary-active transporters. SLC17A5/sialin mediates the H+-coupled symport of N-acetylneuraminic acid and glucuronic acid out of lysosomes (Verheijen et al., 1999; Hu et al., 2023; Schmiege et al., 2024). It has a broad substrate specificity and recognizes various sialic acids via electrostatic interaction with two conserved arginines, R168 and R57. Two glutamates, E171 and E175, serve as protonation sites and couple H+ and substrate binding using a similar mechanism as employed in DgoT and VGLUTs. Protonation of E171 releases R168 and permits its interaction with the substrate. Subsequent H+ transfer to E175 is followed by substrate transfer to a more cytoplasmic binding site close to R57 and translocation to the inward-facing conformation (Hu et al., 2023). Such coupling of substrate translocation to proton transfer results in the electroneutral co-export of 1 H+ and one sialic acid molecule from the lysosome.
Transport functions of EAAT glutamate transporters
Another class of glutamate transporters, the so-called excitatory amino acid transporters (EAATs), takes up glutamate from the synaptic cleft (Danbolt, 2001; Vandenberg and Ryan, 2013; Rose et al., 2018). There exist five mammalian EAAT isoforms, EAAT1 and EAAT2 are expressed in glia, and EAAT3, EAAT4 and EAAT5 in neurons, with distinct tissue distributions and cellular functions. EAAT2 is responsible for 95% of the brain glutamate uptake (Danbolt et al., 2016) and is therefore - among the EAAT isoforms - most important for brain glutamate homeostasis (Tanaka et al., 1997; Allen et al., 2013). It is not only expressed in glia, but also represents the major presynaptic glutamate transporter (Petr et al., 2015; Rimmele and Rosenberg, 2016). Its importance for normal brain function is illustrated by SLC1A2 mutations associated with epileptic encephalopathy (Allen et al., 2013; Guella et al., 2017; Kovermann et al., 2022; Kovermann et al., 2025). EAAT1 ablation in mice has only moderate consequences (Watase et al., 1998), but mutations in SLC1A3, the gene encoding EAAT1, causes Episodic Ataxia 6, a genetic condition combining ataxia, migraine and epilepsy (Jen et al., 2005; Jen et al., 2007; de Vries et al., 2009; Winter et al., 2012; Chivukula et al., 2020; Kovermann et al., 2020; Wu et al., 2022). EAAT3 is not only expressed in the majority of neurons, with selective localization in somata and dendrites (Holmseth et al., 2012), but also in the apical membrane of proximal tubule cells and required for clearing primary urine from glutamate (Peghini et al., 1997; Bailey et al., 2011). EAAT4 contributes to synaptic transmission at the climbing fiber-Purkinje cell synapse (Huang et al., 2004; Perkins et al., 2018), and EAAT5 is retina-specific and improves the temporal resolution in the retina (Gehlen et al., 2021).
All EAATs transport glutamate with a complex transport stoichiometry, i.e., one glutamate is co-transported with three Na+ and 1 H+, in countertransport with one K+ (Kanner and Bendahan, 1982; Zerangue and Kavanaugh, 1996). In clear contrast to the VGLUTs, the EAATs are high affinity transporter, with EAAT4 exhibiting the lowest transport KM of 2.5 µM (Mim et al., 2005). They do not distinguish between glutamate and aspartate, but between L- and D-glutamate. All EAATs can also function as anion channels (Fairman et al., 1995; Wadiche et al., 1995; Larsson et al., 1996), with the surprising ability to modulate their anion-to-cation selectivity (Melzer et al., 2005). In the two neurological diseases, Episodic Ataxia 6 and epileptic encephalopathy, which are associated with mutations in SLC1A2 or SLC1A3, some of the disease-causing mutations result in gain-of-function of EAAT1 or EAAT2 anion channels (Winter et al., 2012; Chivukula et al., 2020; Kovermann et al., 2022; Wu et al., 2022; Kovermann et al., 2025). Although the mechanisms, by which increased EAAT anion currents cause neurodegeneration and overexcitatbility are not fully understood (Kovermann et al., 2020), these human genetic diseases illustrate that the EAAT related anion conductance needs to be tightly regulated.
Structural basis of EAAT glutamate transport
Several mammalian EAATs and two prokaryotic model proteins, GltPh and GltTk, have been structurally studied providing high resolution structures in multiple conformations (Yernool et al., 2004; Boudker et al., 2007; Reyes et al., 2009; Verdon and Boudker, 2012; Jensen et al., 2013; Guskov et al., 2016; Arkhipova et al., 2019; Alleva et al., 2020; Qiu et al., 2021; Qiu and Boudker, 2023; Qiu and Boudker, 2025). EAAT/Glt proteins are assembled as trimers (Gendreau et al., 2004; Nothmann et al., 2011), with each protomer consisting of eight transmembrane helices (TMs) and two helical hairpins (HP1 and HP2) (Yernool et al., 2004). Each protomer contains two distinct domains: the trimerization domain, which mediates inter-subunit interactions, and the transport domain, which harbors the ion- and substrate binding sites (Figure 4A). Substrate transport is mediated by an elevator transport mechanism, in which the transport domain undergoes a large-scale (∼18 Å) rotational/translational movement along the trimerization domain (Crisman et al., 2009; Reyes et al., 2009). The translocation of the transport domain is gated by hairpin loop 2 (HP2), which regulates accessibility to the ligand-binding pocket (Boudker et al., 2007; Ewers et al., 2013; Garaeva et al., 2019). Open HP2 prevents translocation of the transport domain (Boudker et al., 2007; Jensen et al., 2013; Verdon et al., 2014; Guskov et al., 2016; Alleva et al., 2021), and the EAATs take advantage of this property to define and to adjust transport coupling (Alleva et al., 2021; Zhou et al., 2022). Moreover, the competitive blocker TBOA locks EAAT transporters in the OFC with an open HP2 (Boudker et al., 2007; Canul-Tec et al., 2017).
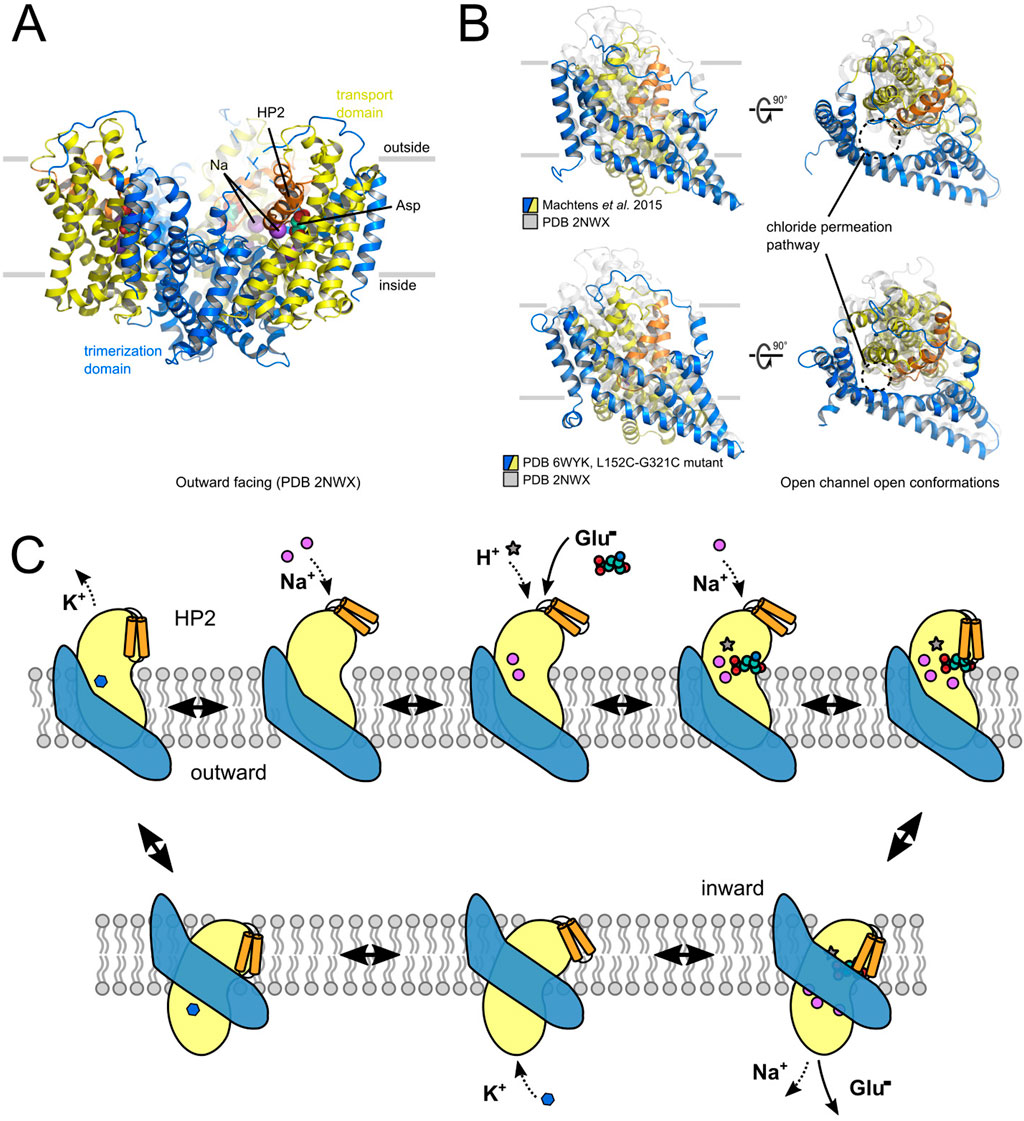
Figure 4. Structural determinants of EAAT channel and transporter functions. (A) X-ray crystal structure of GltPh in the outward-facing conformation. (B) Comparison of two potential anion-conducting states of GltPh, of the one proposed by Machtens et al. (2015) (upper part) or by Chen et al. (2021) (lower part). (C) Schematic presentation of the EAAT transport cycle.
Three different Na+-binding sites have been identified in the transport domain: whereas Na1 and Na3 need to be occupied before substrate binding, Na2 is formed by the closed HP2 and TM7 and thus requires bound glutamate (Boudker et al., 2007). Na+ association starts with the spontaneous opening of HP2, followed by a series of discrete conformational changes that result in maturation of the Na1 and Na3 binding sites: subsequent binding of Na+ to Na1 and Na3 stabilizes HP2 in an open state to permit association of the transport substrate (Alleva et al., 2020). As demonstrated in experiments with the bacterial homologue GltPh, the EAATs utilize an induced fit mechanism for substrate binding (Ewers et al., 2013). The stabilization of substrate binding by the closed HP2 permits selection of substrates that do not bind with highest affinity, but that enable the fastest closure of HP2. It optimizes GltPh for L-aspartate transport, which binds with a lower affinity than other substrates and is released easily on the other membrane side, thus combining high selectivity with high transport effectivity (Ewers et al., 2013).
The EAATs differ from bacterial homologues as well as from the structurally related ASCTs in their obligatory K+ dependence (Kanner and Bendahan, 1982; Kanner and Marva, 1982; Gaillard et al., 1996; Ryan et al., 2009), which is caused by an inability to return from the inward facing conformation in an apo state. Kortzak et al. (2019) studied K+ binding using atomistic molecular dynamics (MD) simulations and found identical K+-binding site in GltPh and in mammalian EAATs. EAATs and GltPh thus do not differ in their ability to bind K+, but in their requirement for K+ binding to permit HP2 closing. In GltPh, HP2 can close also in the apo state (Jensen et al., 2013), whereas HP2 is almost always open in apo EAAT1 (Kortzak et al., 2019), so that transmembrane translocation is only possible after K+ binding. The different K+ dependences of GltPh and EAAT illustrate how transport stoichiometries can be evolutionarily adjusted in elevator transporters by modifying the flexibility of HP2, without generating novel binding sites as observed in SLC6 transporters (Forrest et al., 2007; Zomot et al., 2007).
H+ coupling of mammalian EAATs was comprehensively studied by Watzke and colleagues using heterologous expression, whole-cell patch clamp recording, flash photolysis of caged glutamate and mathematical modeling (Watzke et al., 2000). They proposed an ordered binding model, in which one proton binds to a binding site with an apparent pK of eight to the outward-facing transporter prior to glutamate binding. After translocation, release to the internal medium is facilitated by a pK reduction to 6.5 in the inward-facing conformation. Subsequent mutagenesis experiments identified E373 (in EAAT3) as proton acceptor. This residue is conserved in mammalian EAATs, but not in the related Glts and ASCTs.
Molecular basis of the EAAT anion channel mode
EAATs/Gltx can also function as anion channels (Fairman et al., 1995; Wadiche et al., 1995; Larsson et al., 1996; Otis and Jahr, 1998; Bergles et al., 2002). EAAT anion channel opening is stimulated by Na+ and glutamate (Wadiche et al., 1995; Wadiche and Kavanaugh, 1998; Grewer et al., 2000; Watzke and Grewer, 2001), and certain isoforms also exhibit anion channel activity in the mere presence of K+ or even in the apo state (Leinenweber et al., 2011; Divito et al., 2017). Thus far, two different conformations have been proposed as potential structural basis of EAATs/Glt anion conduction (Machtens et al., 2015; Chen et al., 2021).
Machtens et al. (2015) observed the formation of an anion conduction pathway in unbiased atomistic MD simulations on GltPh. In simulations initiating from translocation intermediates, the lateral movement of the transport domain opens a water-filled pore with relatively wide diameter (minimum diameter of 5.6 Å) that permits Cl¯ permeation in a partially hydrated state, but is impermeable to glutamate or aspartate. It is perfectly selective for anions over cations and accounts for the experimentally observed lyotropic anion selectivity (Wadiche and Kavanaugh, 1998; Melzer et al., 2003) and unitary current amplitudes in EAATs (Kovermann et al., 2010; Machtens et al., 2011). The comparison of in silico and experimental mutagenesis results on predicted pore-forming residues provided perfect agreement (Machtens et al., 2015). Tryptophan insertion mutagenesis and experimental testing of collisional quenching with I− demonstrated that predicted pore-forming GltPh residues indeed come in close contact with permeating anions. The molecular basis of anion selectivity of this channel is remarkably simple: a single positively charged pore-forming residue, arginine R276 at the tip of HP1 in GltPh, or in the EAATs, an arginine residue at the position corresponding to M395 in TM8 of GltPh (Machtens et al., 2015). This residue projects its side chain into the anion conduction pathway, and its neutralization abolishes anion selectivity (Machtens et al., 2015; Cater et al., 2016).
In a subsequent study, Chen et al. (2021) resolved the cryo-EM structure of L152C/G351C GltPh after disulfide crosslinking. In MD simulations with this structure, the authors observed the formation of a continuous water pathway in the presence of an external electric field. Free-energy profiles for potential Cl¯ permeation events along this water pore were determined using umbrella sampling, and residues that interact with Cl¯ were identified as energy minima. To test whether this pore can account for experimentally observed anion currents, mutant EAAT1, which carry substitutions in candidate pore-forming residues, were expressed and functionally characterized in Xenopus oocytes. This approach resulted in the identification of similar pore residues as identified in (Machtens et al., 2015), although the experimental structure was not identical to the computational structure.
The two proposed anion channel conformation do not differ substantially, and - at present - neither computational nor experimental approaches permit ranking the impact of the two conformations for EAAT anion currents. Most likely, there exist multiple anion-conducting conformations.
Conclusion
VGLUTs and EAATs are two classes of glutamate transporters with clear differences in structure and cellular function. The EAATs are expressed in the surface membrane, and EAAT transport is driven by Na+ gradients established by Na+-K+ ATPases. In contrast, the main driving force for the VGLUTs are H+ gradients generated by vesicular H+ pumps. The VGLUTs distinguish between glutamate and aspartate, and synaptic vesicles almost exclusively contain glutamate (Burger et al., 1989; Herring et al., 2015); the EAATs can transport both.
VGLUTs and EAATs can both assume anion-selective channel modes. Within the SLC17 family, the association with an anion channel mode appears to be specific for the VGLUT glutamate transporters, with the structurally closely related SLC17A5/sialin or the bacterial galactonate transporter DgoT not functioning as anion channels. In contrast, within the SLC1 family, not only the EAAT glutamate transporters, but also ASCT neutral amino acid transporters assume anion-conducting channel modes (Zerangue and Kavanaugh, 1995). Since SLC1 anion conduction is due to a single positively charged residue - with distinct sequence localization in prokaryotic and mammalian EAATs (Machtens et al., 2015), it is surprising that there exist no unselective leak conductance in any related structure.
VGLUT anion channels fulfil obvious and clear roles supporting glutamate accumulation in the synaptic vesicles. VGLUT-mediated Cl− efflux depolarizes the synaptic vesicle and thus increases the driving force for glutamate filling. Moreover, the VGLUT anion currents decrease the amount of primary active H+ transport necessary for vesicle acidification and glutamate accumulation, thus decreasing the energy demand for synaptic vesicle filling (Kolen et al., 2023). The cellular functions of EAAT anion channel function are less obvious. Originally, EAAT anion channels were proposed to reduce membrane depolarization by electrogenic glutamate transport; however, for the two glial EAATs, which are most important for glutamate homeostasis, this function is clearly not necessary because of the high K+ conductance of glial cells. The retinal isoform EAAT5 appears to play a direct role as anion channel (Gehlen et al., 2021). It is expressed at output synapses in rod bipolar cells, and its activation upon glutamate release will activate presynaptic EAAT5 and cause hyperpolarization due to Cl− influx and reduced glutamate release. In glial cells, EAAT1 and EAAT2 anion channels have been shown to contribute to the glial chloride homeostasis (Untiet et al., 2017; Engels et al., 2021). The purpose of this regulation remains unclear under healthy conditions.
Open questions
Glutamate is the most important excitatory neurotransmitter in the mammalian central nervous system, and the mechanisms underlying glutamate release and reuptake have been extensively studied for more than half a century. However, there are a number of open questions, concerning both - transport and channel - functions of VGLUTs and EAATs.
The main function of vesicular glutamate transporters is glutamate accumulation in synaptic vesicles. However, thus far, no agreement about the transport stoichiometry of VGLUT glutamate transport has been reached, with experimental support for glutamate uniport as well as for H+-glutamate exchange (Li et al., 2025; Kolen et al., 2023). Apart from its apparent physiological importance, this controversy illustrates the complexity of determining transport stoichiometries in coupled transporters, especially in multi-functional transporters with complex voltage dependence.
For both classes of glutamate transporters, cellular roles of the associated anion conductances remain insufficiently understood. For the vesicular glutamate transporters, a direct role of VGLUT Cl¯ channels in glutamate accumulation has been suggested (Martineau et al., 2017; Kolen et al., 2023). At present, we do not know whether this feature contributes to differences in glutamate loading between species or whether changes in the VGLUT anion conductances–via alterations in presynaptic [Cl¯] or via activity dependent changes in VGLUT anion channel activity - might allow adjusting vesicular glutamate accumulation in synaptic plasticity. The idea that Cl¯ efflux is necessary for effective glutamate accumulation appears convincing, but why there is no cation conductance associated with vesicular monoamine transporters (VMATs) that fill synaptic vesicles with positively charged neurotransmitter such as dopamine or serotonin? After endocytosis, synaptic vesicles contain even higher Na+ than Cl¯ concentrations.
For the EAAT Cl− channels, a role in glial [Cl¯] regulation is firmly established (Untiet et al., 2017; Kovermann et al., 2020; Engels et al., 2021), but a possible involvement in neuronal anion homeostasis has not yet been addressed. The impact of genetic ablation of EAATs appears to be mainly determined by the contribution of transporters to glutamate uptake, and it is still impossible to test the exclusive ablation of EAAT anion channels, while preserving transport functions. It therefore remains unclear how important this particular EAAT transport function is for neuronal and glial physiology. However, gain-of-EAAT anion channel function has severe neurological consequence in patients carrying SLC1A2 and SLC1A3 mutations, resulting in neurodegeneration and therapy-resistant epilepsy (Allen et al., 2013; Allen et al., 2016; Guella et al., 2017; Kovermann et al., 2022; Kovermann et al., 2025). How can a transport function have such severe consequences under pathological, but not play an important role under normal conditions?
For both classes of transporters, there are open questions concerning the structural basis of anion conduction. For the VGLUTs, no anion-conducting conformation has been reported so far. For the EAATs, two different anion channel conformations have been proposed (Machtens et al., 2015; Chen et al., 2021). In both cases, the transporters assume such conformations not from outward- or inward-facing, but from intermediate structures. They can therefore not account for the Na+ activation of the EAAT anion conductance in absence of glutamate (Wadiche and Kavanaugh, 1998; Watzke et al., 2001; Kovermann et al., 2010), since this condition confines EAAT transporters in its outward-facing conformation. Moreover, how can such an intermediate conformation account for the drastic increase in EAAT anion channel activity in EAAT transporteropathies (Winter et al., 2012; Kovermann et al., 2022; Wu et al., 2022; Kovermann et al., 2025)?
Author contributions
BB: Writing – original draft, Writing – review and editing. ND: Writing – original draft, Writing – review and editing. AN: Writing – original draft, Writing – review and editing. CF: Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) to CF (FA 301/13-1) as part of the Research Unit FOR 2795 (Synapses under stress).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abramson, J., Smirnova, I., Kasho, V., Verner, G., Kaback, H. R., and Iwata, S. (2003). Structure and mechanism of the lactose permease of Escherichia coli. Science 301, 610–615. doi:10.1126/science.1088196
Alekov, A., and Fahlke, C. (2009). Channel-like slippage modes in the human anion/proton exchanger ClC-4. J. General Physiology 133, 485–496. doi:10.1085/jgp.200810155
Alleva, C., Kovalev, K., Astashkin, R., Berndt, M. I., Baeken, C., Balandin, T., et al. (2020). Na+-dependent gate dynamics and electrostatic attraction ensure substrate coupling in glutamate transporters. Sci. Adv. 6 (47), eaba9854. doi:10.1126/sciadv.aba9854
Alleva, C., Machtens, J. P., Kortzak, D., Weyand, I., and Fahlke, C. (2021). Molecular basis of coupled transport and anion conduction in excitatory amino acid transporters. Neurochem. Res. 47, 9–22. doi:10.1007/s11064-021-03252-x
Arkhipova, V., Trinco, G., Ettema, T. W., Jensen, S., Slotboom, D. J., and Guskov, A. (2019). Binding and transport of D-aspartate by the glutamate transporter homolog GltTk. Elife 8, e45286. doi:10.7554/eLife.45286
Bae, H. R., and Verkman, A. S. (1990). Protein kinase A regulates chloride conductance in endocytic vesicles from proximal tubule. Nature 348 (6302), 637–639. doi:10.1038/348637a0
Bailey, C. G., Ryan, R. M., Thoeng, A. D., Ng, C., King, K., Vanslambrouck, J. M., et al. (2011). Loss-of-function mutations in the glutamate transporter SLC1A1 cause human dicarboxylic aminoaciduria. J. Clin. Invest. 121 (1), 446–453. doi:10.1172/jci44474
Baraglia, E. C., Fattorini, G., Chattaraj, S., Pasqualini, F., and Conti, F. (2025). An open-science computational model of organelle acidification to integrate putative mechanisms of synaptic vesicle acidification and filling. Neurochem. Res. 50 (3), 181. doi:10.1007/s11064-025-04432-9
Batarni, S., Nayak, N., Chang, A., Li, F., Hareendranath, S., Zhou, L., et al. (2023). Substrate recognition and proton coupling by a bacterial member of solute carrier family 17. J. Biol. Chem. 299 (5), 104646. doi:10.1016/j.jbc.2023.104646
Bellocchio, E. E., Reimer, R. J., Fremeau, R. T., and Edwards, R. H. (2000). Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science 289 (5481), 957–960. doi:10.1126/science.289.5481.957
Bergles, D. E., Tzingounis, A. V., and Jahr, C. E. (2002). Comparison of coupled and uncoupled currents during glutamate uptake by GLT-1 transporters. J. Neurosci. 22 (23), 10153–10162. doi:10.1523/JNEUROSCI.22-23-10153.2002
Blakely, R. D., and Edwards, R. H. (2012). Vesicular and plasma membrane transporters for neurotransmitters. Cold Spring Harb. Perspect. Biol. 4 (2), a005595. doi:10.1101/cshperspect.a005595
Borghans, B., Kortzak, D., Longo, P., Kolen, B., Machtens, J. P., and Fahlke, C. (2025). Allosteric modulation of proton binding confers Cl- activation and glutamate selectivity to vesicular glutamate transporters. PLoS Comput. Biol. 21, e1013214. doi:10.1371/journal.pcbi.1013214
Boudker, O., Ryan, R. M., Yernool, D., Shimamoto, K., and Gouaux, E. (2007). Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 445, 387–393. doi:10.1038/nature05455
Burger, P. M., Mehl, E., Cameron, P. L., Maycox, P. R., Baumert, M., Lottspeich, F., et al. (1989). Synaptic vesicles immunoisolated from rat cerebral cortex contain high levels of glutamate. Neuron 3, 715–720. doi:10.1016/0896-6273(89)90240-7
Canul-Tec, J. C., Assal, R., Cirri, E., Legrand, P., Brier, S., Chamot-Rooke, J., et al. (2017). Structure and allosteric inhibition of excitatory amino acid transporter 1. Nature 544 (7651), 446–451. doi:10.1038/nature22064
Cater, R. J., Vandenberg, R. J., and Ryan, R. M. (2016). Tuning the ion selectivity of glutamate transporter-associated uncoupled conductances. J. General Physiology 148 (1), 13–24. doi:10.1085/jgp.201511556
Chang, R., Eriksen, J., and Edwards, R. H. (2018). The dual role of chloride in synaptic vesicle glutamate transport. Elife 7, e34896. doi:10.7554/eLife.34896
Chen, I., Pant, S., Wu, Q., Cater, R. J., Sobti, M., Vandenberg, R. J., et al. (2021). Glutamate transporters have a chloride channel with two hydrophobic gates. Nature 591, 327–331. doi:10.1038/s41586-021-03240-9
Chivukula, A. S., Suslova, M., Kortzak, D., Kovermann, P., and Fahlke, C. (2020). Functional consequences of SLC1A3 mutations associated with episodic ataxia 6. Hum. Mutat. 41, 1892–1905. doi:10.1002/humu.24089
Crisman, T. J., Qu, S., Kanner, B. I., and Forrest, L. R. (2009). Inward-facing conformation of glutamate transporters as revealed by their inverted-topology structural repeats. Proc. Natl. Acad. Sci. U. S. A. 106, 20752–20757. doi:10.1073/pnas.0908570106
Danbolt, N. C. (2001). Glutamate uptake. Prog. Neurobiol. 65, 1–105. doi:10.1016/s0301-0082(00)00067-8
Danbolt, N. C., Furness, D. N., and Zhou, Y. (2016). Neuronal vs glial glutamate uptake: resolving the conundrum. Neurochem. Int. 98, 29–45. doi:10.1016/j.neuint.2016.05.009
de Vries, B., Mamsa, H., Stam, A. H., Wan, J., Bakker, S. L., Vanmolkot, K. R., et al. (2009). Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Arch. Neurol. 66, 97–101. doi:10.1001/archneurol.2008.535
Divito, C. B., Borowski, J. E., Glasgow, N. G., Gonzalez-Suarez, A. D., Torres-Salazar, D., Johnson, J. W., et al. (2017). Glial and neuronal glutamate transporters differ in the Na+ requirements for activation of the substrate-independent anion conductance. Front. Mol. Neurosci. 10, 150. doi:10.3389/fnmol.2017.00150
Dmitrieva, N., Gholami, S., Alleva, C., Carloni, P., Alfonso-Prieto, M., and Fahlke, C. (2024). Transport mechanism of DgoT, a bacterial homolog of SLC17 organic anion transporters. Embo J. 43, 6740–6765. doi:10.1038/s44318-024-00279-y
Engels, M., Kalia, M., Rahmati, S., Petersilie, L., Kovermann, P., van Putten, M. J. A. M., et al. (2021). Glial chloride homeostasis under transient ischemic stress. Front. Cell. Neurosci. 15, 735300. doi:10.3389/fncel.2021.735300
Epi4K ConsortiumEpilepsy Phenome/Genome Project Cossette, P., Delanty, N., Dlugos, D., Eichler, E. E., et al. (2013). De novo mutations in epileptic encephalopathies. Nature 501 (7466), 217–221. doi:10.1038/nature12439
Eriksen, J., Chang, R., McGregor, M., Silm, K., Suzuki, T., and Edwards, R. H. (2016). Protons regulate vesicular glutamate transporters through an allosteric mechanism. Neuron 90, 768–780. doi:10.1016/j.neuron.2016.03.026
Eriksen, J., Li, F., and Edwards, R. H. (2020). The mechanism and regulation of vesicular glutamate transport: coordination with the synaptic vesicle cycle. Biochimica Biophysica Acta (BBA) - Biomembr. 1862 (12), 183259. doi:10.1016/j.bbamem.2020.183259
Ewers, D., Becher, T., Machtens, J. P., Weyand, I., and Fahlke, C. (2013). Induced fit substrate binding to an archeal glutamate transporter homologue. Proc. Natl. Acad. Sci. U. S. A. 110, 12486–12491. doi:10.1073/pnas.1300772110
Fahlke, C., Kortzak, D., and Machtens, J. P. (2016). Molecular physiology of EAAT anion channels. Pflügers Archiv - Eur. J. Physiology 468, 491–502. doi:10.1007/s00424-015-1768-3
Fairman, W. A., Vandenberg, R. J., Arriza, J. L., Kavanaught, M. P., and Amara, S. G. (1995). An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 375, 599–603. doi:10.1038/375599a0
Farsi, Z., Jahn, R., and Woehler, A. (2017). Proton electrochemical gradient: driving and regulating neurotransmitter uptake. Bioessays 39, 1600240. doi:10.1002/bies.201600240
Forrest, L. R., Tavoulari, S., Zhang, Y. W., Rudnick, G., and Honig, B. (2007). Identification of a chloride ion binding site in Na+/Cl--dependent transporters. Proc. Natl. Acad. Sci. U. S. A. 104, 12761–12766. doi:10.1073/pnas.0705600104
Forrest, L. R., Kramer, R., and Ziegler, C. (2011). The structural basis of secondary active transport mechanisms. Biochimica Biophysica Acta (BBA) - Bioenergetics 1807, 167–188. doi:10.1016/j.bbabio.2010.10.014
Gaillard, I., Slotboom, D. J., Knol, J., Lolkema, J. S., and Konings, W. N. (1996). Purification and reconstitution of the glutamate carrier GltT of the thermophilic bacterium Bacillus stearothermophilus. Biochemistry 35, 6150–6156. doi:10.1021/bi953005v
Garaeva, A. A., Guskov, A., Slotboom, D. J., and Paulino, C. (2019). A one-gate elevator mechanism for the human neutral amino acid transporter ASCT2. Nat. Commun. 10, 3427. doi:10.1038/s41467-019-11363-x
Gehlen, J., Aretzweiler, C., Mataruga, A., Fahlke, C., and Müller, F. (2021). Excitatory amino acid transporter EAAT5 improves temporal resolution in the retina. eNeuro 8, ENEURO.0406. doi:10.1523/eneuro.0406-21.2021
Gendreau, S., Voswinkel, S., Torres-Salazar, D., Lang, N., Heidtmann, H., Detro-Dassen, S., et al. (2004). A trimeric quaternary structure is conserved in bacterial and human glutamate transporters. J. Biol. Chem. 279, 39505–39512. doi:10.1074/jbc.M408038200
George, A. L. (1998). Chloride channels and endocytosis: ClC-5 makes a dent. Proc. Natl. Acad. Sci. U. S. A. 95, 7843–7845. doi:10.1073/pnas.95.14.7843
Goh, G. Y., Huang, H., Ullman, J., Borre, L., Hnasko, T. S., Trussell, L. O., et al. (2011). Presynaptic regulation of quantal size: K+/H+ exchange stimulates vesicular glutamate transport. Nat. Neurosci. 14, 1285–1292. doi:10.1038/nn.2898
Grewer, C., Watzke, N., Wiessner, M., and Rauen, T. (2000). Glutamate translocation of the neuronal glutamate transporter EAAC1 occurs within milliseconds. Proc. Natl. Acad. Sci. U. S. A. 97, 9706–9711. doi:10.1073/pnas.160170397
Guella, I., McKenzie, M. B., Evans, D. M., Buerki, S. E., Toyota, E. B., Van Allen, M. I., et al. (2017). De novo mutations in YWHAG cause early-onset epilepsy. Am. J. Hum. Genet. 101, 300–310. doi:10.1016/j.ajhg.2017.07.004
Guskov, A., Jensen, S., Faustino, I., Marrink, S. J., and Slotboom, D. J. (2016). Coupled binding mechanism of three sodium ions and aspartate in the glutamate transporter homologue GltTk. Nat. Commun. 7, 13420. doi:10.1038/ncomms13420
Hartinger, J., and Jahn, R. (1993). An anion binding site that regulates the glutamate transporter of synaptic vesicles. J. Biol. Chem. 268, 23122–23127. doi:10.1016/s0021-9258(19)49435-0
Herring, B. E., Silm, K., Edwards, R. H., and Nicoll, R. A. (2015). Is aspartate an excitatory neurotransmitter? J. Neurosci. 35, 10168–10171. doi:10.1523/jneurosci.0524-15.2015
Holmseth, S., Dehnes, Y., Huang, Y. H., Follin-Arbelet, V. V., Grutle, N. J., Mylonakou, M. N., et al. (2012). The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J. Neurosci. 32, 6000–6013. doi:10.1523/jneurosci.5347-11.2012
Hu, W., Chi, C., Song, K., and Zheng, H. (2023). The molecular mechanism of sialic acid transport mediated by Sialin. Sci. Adv. 9, eade8346. doi:10.1126/sciadv.ade8346
Huang, Y. H., Dykes-Hoberg, M., Tanaka, K., Rothstein, J. D., and Bergles, D. E. (2004). Climbing fiber activation of EAAT4 transporters and kainate receptors in cerebellar Purkinje cells. J. Neurosci. 24, 103–111. doi:10.1523/JNEUROSCI.4473-03.2004
Jahn, R., and Fasshauer, D. (2012). Molecular machines governing exocytosis of synaptic vesicles. Nature 490, 201–207. doi:10.1038/nature11320
Jen, J. C., Wan, J., Palos, T. P., Howard, B. D., and Baloh, R. W. (2005). Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology 65, 529–534. doi:10.1212/01.wnl.0000172638.58172.5a
Jen, J. C., Graves, T. D., Hess, E. J., Hanna, M. G., Griggs, R. C., and Baloh, R. W. (2007). Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain 130, 2484–2493. doi:10.1093/brain/awm126
Jensen, S., Guskov, A., Rempel, S., Hänelt, I., and Slotboom, D. J. (2013). Crystal structure of a substrate-free aspartate transporter. Nat. Struct. Mol. Biol. 20, 1224–1226. doi:10.1038/nsmb.2663
Juge, N., Yoshida, Y., Yatsushiro, S., Omote, H., and Moriyama, Y. (2006). Vesicular glutamate transporter contains two independent transport machineries. J. Biol. Chem. 281, 39499–39506. doi:10.1074/jbc.M607670200
Juge, N., Gray, J. A., Omote, H., Miyaji, T., Inoue, T., Hara, C., et al. (2010). Metabolic control of vesicular glutamate transport and release. Neuron 68, 99–112. doi:10.1016/j.neuron.2010.09.002
Kanner, B. I., and Bendahan, A. (1982). Binding order of substrates to the (sodium + potassium ion)-coupled L-glutamic acid transporter from rat brain. Biochemistry 21, 6327–6330. doi:10.1021/bi00267a044
Kanner, B. I., and Marva, E. (1982). Efflux of L-glutamate by synaptic plasma membrane vesicles isolated from rat brain. Biochemistry 21, 3143–3147. doi:10.1021/bi00256a017
Kasahara, M., and Hinkle, P. C. (1977). Reconstitution and purification of the D-glucose transporter from human erythrocytes. J. Biol. Chem. 252 (20), 7384–7390. doi:10.1016/s0021-9258(19)66976-0
Kolen, B., Borghans, B., Kortzak, D., Lugo, V., Hannack, C., Guzman, R. E., et al. (2023). Vesicular glutamate transporters are H+-anion exchangers that operate at variable stoichiometry. Nat. Commun. 14, 2723. doi:10.1038/s41467-023-38340-9
Kortzak, D., Alleva, C., Weyand, I., Ewers, D., Zimmermann, M. I., Franzen, A., et al. (2019). Allosteric gate modulation confers K+ coupling in glutamate transporters. EMBO J. 38, e101468. doi:10.15252/embj.2019101468
Kovermann, P., Machtens, J. P., Ewers, D., and Fahlke, C. (2010). A conserved aspartate determines pore properties of anion channels associated with excitatory amino acid transporter 4 (EAAT4). J. Biol. Chem. 285, 23676–23686. doi:10.1074/jbc.M110.126557
Kovermann, P., Untiet, V., Kolobkova, Y., Engels, M., Baader, S., Schilling, K., et al. (2020). Increased glutamate transporter-associated anion currents cause glial apoptosis in episodic ataxia 6. Brain Commun. 2, fcaa022. doi:10.1093/braincomms/fcaa022
Kovermann, P., Kolobkova, Y., Franzen, A., and Fahlke, C. (2022). Mutations associated with epileptic encephalopathy modify EAAT2 anion channel function. Epilepsia 63, 388–401. doi:10.1111/epi.17154
Kovermann, P., Bayat, A., Fenger, C. D., Leeuwen, L., Borovikov, A., Sharkov, A., et al. (2025). The severity of SLC1A2-associated neurodevelopmental disorders correlates with transporter dysfunction. EBioMedicine 114, 105648. doi:10.1016/j.ebiom.2025.105648
Larsson, H. P., Picaud, S. A., Werblin, F. S., and Lecar, H. (1996). Noise analysis of the glutamate-activated current in photoreceptors. Biophysical J. 70, 733–742. doi:10.1016/S0006-3495(96)79613-3
Leano, J. B., Batarni, S., Eriksen, J., Juge, N., Pak, J. E., Kimura-Someya, T., et al. (2019). Structures suggest a mechanism for energy coupling by a family of organic anion transporters. PLoS Biol. 17, e3000260. doi:10.1371/journal.pbio.3000260
Leinenweber, A., Machtens, J. P., Begemann, B., and Fahlke, C. (2011). Regulation of glial glutamate transporters by C-terminal domains. J. Biol. Chem. 286, 1927–1937. doi:10.1074/jbc.M110.153486
Li, F., Eriksen, J., Finer-Moore, J., Chang, R., Nguyen, P., Bowen, A., et al. (2020). Ion transport and regulation in a synaptic vesicle glutamate transporter. Science 368, 893–897. doi:10.1126/science.aba9202
Li, F., Eriksen, J., Oses-Prieto, J. A., Gomez, Y. K., Xu, H., Hareendranath, S., et al. (2025). Substrate recognition and allosteric regulation of synaptic vesicle glutamate transporter VGLUT2. Nat. Struct. Mol. Biol. 32, 1479–1487. doi:10.1038/s41594-025-01568-8
Machtens, J. P., Fahlke, C., and Kovermann, P. (2011). Noise analysis to study unitary properties of transporter-associated ion channels. Channels (Austin.) 5, 468–474. doi:10.4161/chan.5.6.17453
Machtens, J. P., Kortzak, D., Lansche, C., Leinenweber, A., Kilian, P., Begemann, B., et al. (2015). Mechanisms of anion conduction by coupled glutamate transporters. Cell 160, 542–553. doi:10.1016/j.cell.2014.12.035
Martineau, M., Guzman, R. E., Fahlke, C., and Klingauf, J. (2017). VGLUT1 functions as a glutamate/proton exchanger with chloride channel activity in hippocampal glutamatergic synapses. Nat. Commun. 8, 2279. doi:10.1038/s41467-017-02367-6
Maycox, P. R., Deckwerth, T., Hell, J. W., and Jahn, R. (1988). Glutamate uptake by brain synaptic vesicles. Energy dependence of transport and functional reconstitution in proteoliposomes. J. Biol. Chem. 263, 15423–15428. doi:10.1016/s0021-9258(19)37605-7
Melzer, N., Biela, A., and Fahlke, C. (2003). Glutamate modifies ion conduction and voltage-dependent gating of excitatory amino acid transporter-associated anion channels. J. Biol. Chem. 278, 50112–50119. doi:10.1074/jbc.M307990200
Melzer, N., Torres-Salazar, D., and Fahlke, C. (2005). A dynamic switch between inhibitory and excitatory currents in a neuronal glutamate transporter. Proc. Natl. Acad. Sci. U. S. A. 102, 19214–19218. doi:10.1073/pnas.0508837103
Mim, C., Balani, P., Rauen, T., and Grewer, C. (2005). The glutamate transporter subtypes EAAT4 and EAATs 1-3 transport glutamate with dramatically different kinetics and voltage dependence but share a common uptake mechanism. J. General Physiology 126 (6), 571–589. doi:10.1085/jgp.200509365
Moriyama, Y., and Yamamoto, A. (1995). Vesicular L-Glutamate transporter in microvesicles from bovine pineal glands. J. Biol. Chem. 270, 22314–22320. doi:10.1074/jbc.270.38.22314
Myers, C., McMahon, J., Schneider, A., Petrovski, S., Allen, A., Carvill, G., et al. (2016). De novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am. J. Hum. Genet. 99 (2), 287–298. doi:10.1016/j.ajhg.2016.06.003
Naito, S., and Ueda, T. (1985). Characterization of glutamate uptake into synaptic vesicles. J. Neurochem. 44, 99–109. doi:10.1111/j.1471-4159.1985.tb07118.x
Nothmann, D., Leinenweber, A., Torres-Salazar, D., Kovermann, P., Hotzy, J., Gameiro, A., et al. (2011). Hetero-oligomerization of neuronal glutamate transporters. J. Biol. Chem. 286, 3935–3943. doi:10.1074/jbc.M110.187492
Ogita, K., Hirata, K., Bole, D. G., Yoshida, S., Tamura, Y., Leckenby, A. M., et al. (2001). Inhibition of vesicular glutamate storage and exocytotic release by Rose Bengal. J. Neurochem. 77, 34–42. doi:10.1046/j.1471-4159.2001.t01-1-00200.x
Omote, H., and Moriyama, Y. (2013). Vesicular neurotransmitter transporters: an approach for studying transporters with purified proteins. Physiology 28, 39–50. doi:10.1152/physiol.00033.2012
Omote, H., Miyaji, T., Hiasa, M., Juge, N., and Moriyama, Y. (2016). Structure, function, and drug interactions of neurotransmitter transporters in the postgenomic era. Annu. Rev. Pharmacol. Toxicol. 56, 385–402. doi:10.1146/annurev-pharmtox-010814-124816
Otis, T. S., and Jahr, C. E. (1998). Anion currents and predicted glutamate flux through a neuronal glutamate transporter. J. Neurosci. 18, 7099–7110. doi:10.1523/jneurosci.18-18-07099.1998
Peghini, P., Janzen, J., and Stoffel, W. (1997). Glutamate transporter EAAC-1-deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. Embo. J. 16, 3822–3832. doi:10.1093/emboj/16.13.3822
Perkins, E. M., Clarkson, Y. L., Suminaite, D., Lyndon, A. R., Tanaka, K., Rothstein, J. D., et al. (2018). Loss of cerebellar glutamate transporters EAAT4 and GLAST differentially affects the spontaneous firing pattern and survival of Purkinje cells. Hum. Mol. Genet. 27, 2614–2627. doi:10.1093/hmg/ddy169
Petr, G. T., Sun, Y., Frederick, N. M., Zhou, Y., Dhamne, S. C., Hameed, M. Q., et al. (2015). Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J. Neurosci. 35, 5187–5201. doi:10.1523/jneurosci.4255-14.2015
Qiu, B., and Boudker, O. (2023). Symport and antiport mechanisms of human glutamate transporters. Nat. Commun. 14, 2579. doi:10.1038/s41467-023-38120-5
Qiu, B., and Boudker, O. (2025). Structural basis of excitatory amino acid transporter 3 substrate recognition. Proc. Natl. Acad. Sci. U.S.A. 122, e2501627122. doi:10.1073/pnas.2501627122
Qiu, B., Matthies, D., Fortea, E., Yu, Z., and Boudker, O. (2021). Cryo-EM structures of excitatory amino acid transporter 3 visualize coupled substrate, sodium, and proton binding and transport. Sci. Adv. 7, eabf5814. doi:10.1126/sciadv.abf5814
Reyes, N., Ginter, C., and Boudker, O. (2009). Transport mechanism of a bacterial homologue of glutamate transporters. Nature 462, 880–885. doi:10.1038/nature08616
Rimmele, T. S., and Rosenberg, P. A. (2016). GLT-1: the elusive presynaptic glutamate transporter. Neurochem. Int. 98, 19–28. doi:10.1016/j.neuint.2016.04.010
Rose, C. R., Ziemens, D., Untiet, V., and Fahlke, C. (2018). Molecular and cellular physiology of sodium-dependent glutamate transporters. Brain Res. Bull. 136, 3–16. doi:10.1016/j.brainresbull.2016.12.013
Rostamipour, K., Talandashti, R., and Mehrnejad, F. (2022). Atomistic insight into the luminal allosteric regulation of vesicular glutamate transporter 2 by chloride and protons: an all-atom molecular dynamics simulation study. Proteins 90, 2045–2057. doi:10.1002/prot.26396
Ryan, R. M., Compton, E. L., and Mindell, J. A. (2009). Functional characterization of a Na+-dependent aspartate transporter from Pyrococcus horikoshii. J. Biol. Chem. 284, 17540–17548. doi:10.1074/jbc.M109.005926
Schenck, S., Wojcik, S. M., Brose, N., and Takamori, S. (2009). A chloride conductance in VGLUT1 underlies maximal glutamate loading into synaptic vesicles. Nat. Neurosci. 12, 156–162. doi:10.1038/nn.2248
Schmiege, P., Donnelly, L., Elghobashi-Meinhardt, N., Lee, C. H., and Li, X. (2024). Structure and inhibition of the human lysosomal transporter Sialin. Nat. Commun. 15, 4386. doi:10.1038/s41467-024-48535-3
Silberberg, S. D., and Magleby, K. L. (1993). Preventing errors when estimating single channel properties from the analysis of current fluctuations. Biophysical J. 65, 1570–1584. doi:10.1016/s0006-3495(93)81196-2
Tabb, J. S., Kish, P. E., Van Dyke, R., and Ueda, T. (1992). Glutamate transport into synaptic vesicles. Roles of membrane potential, pH gradient, and intravesicular pH. J. Biol. Chem. 267, 15412–15418. doi:10.1016/s0021-9258(19)49549-5
Takamori, S., Rhee, J. S., Rosenmund, C., and Jahn, R. (2000). Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 407, 189–194. doi:10.1038/35025070
Takamori, S., Rhee, J. S., Rosenmund, C., and Jahn, R. (2001). Identification of differentiation-associated brain-specific phosphate transporter as a second vesicular glutamate transporter (VGLUT2). J. Neurosci. 21, Rc182. doi:10.1523/JNEUROSCI.21-22-j0002.2001
Takamori, S., Malherbe, P., Broger, C., and Jahn, R. (2002). Molecular cloning and functional characterization of human vesicular glutamate transporter 3. EMBO Rep. 3, 798–803. doi:10.1093/embo-reports/kvf159
Tanaka, K., Watase, K., Manabe, T., Yamada, K., Watanabe, M., Takahashi, K., et al. (1997). Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276, 1699–1702. doi:10.1126/science.276.5319.1699
Untiet, V., Kovermann, P., Gerkau, N. J., Gensch, T., Rose, C. R., and Fahlke, C. (2017). Glutamate transporter-associated anion channels adjust intracellular chloride concentrations during glial maturation. Glia 65, 388–400. doi:10.1002/glia.23098
Vandenberg, R. J., and Ryan, R. M. (2013). Mechanisms of glutamate transport. Physiol. Rev. 93, 1621–1657. doi:10.1152/physrev.00007.2013
Verdon, G., and Boudker, O. (2012). Crystal structure of an asymmetric trimer of a bacterial glutamate transporter homolog. Nat. Struct. Mol. Biol. 19, 355–357. doi:10.1038/nsmb.2233
Verdon, G., Oh, S., Serio, R. N., and Boudker, O. (2014). Coupled ion binding and structural transitions along the transport cycle of glutamate transporters. Elife 3, e02283. doi:10.7554/eLife.02283
Verheijen, F. W., Verbeek, E., Aula, N., Beerens, C. E., Havelaar, A. C., Joosse, M., et al. (1999). A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nat. Genet. 23, 462–465. doi:10.1038/70585
Wadiche, J. I., and Kavanaugh, M. P. (1998). Macroscopic and microscopic properties of a cloned glutamate transporter/chloride channel. J. Neurosci. 18, 7650–7661. doi:10.1523/JNEUROSCI.18-19-07650.1998
Wadiche, J. I., Amara, S. G., and Kavanaugh, M. P. (1995). Ion fluxes associated with excitatory amino acid transport. Neuron 15, 721–728. doi:10.1016/0896-6273(95)90159-0
Watase, K., Hashimoto, K., Kano, M., Yamada, K., Watanabe, M., Inoue, Y., et al. (1998). Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur. J. Neurosci. 10, 976–988. doi:10.1046/j.1460-9568.1998.00108.x
Watzke, N., and Grewer, C. (2001). The anion conductance of the glutamate transporter EAAC1 depends on the direction of glutamate transport. FEBS Lett. 503, 121–125. doi:10.1016/s0014-5793(01)02715-6
Watzke, N., Rauen, T., Bamberg, E., and Grewer, C. (2000). On the mechanism of proton transport by the neuronal excitatory amino acid carrier 1. J. Gen. Physiol. 116, 609–622. doi:10.1085/jgp.116.5.609
Watzke, N., Bamberg, E., and Grewer, C. (2001). Early intermediates in the transport cycle of the neuronal excitatory amino acid carrier EAAC1. J. General Physiology 117 (6), 547–562. doi:10.1085/jgp.117.6.547
Winter, N., Kovermann, P., and Fahlke, C. (2012). A point mutation associated with episodic ataxia 6 increases glutamate transporter anion currents. Brain 135, 3416–3425. doi:10.1093/brain/aws255
Wu, Q., Akhter, A., Pant, S., Cho, E., Zhu, J. X., Garner, A., et al. (2022). Ataxia-linked SLC1A3 mutations alter EAAT1 chloride channel activity and glial regulation of CNS function. J. Clin. Investigation 132, e154891. doi:10.1172/jci154891
Yernool, D., Boudker, O., Jin, Y., and Gouaux, E. (2004). Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 431, 811–818. doi:10.1038/nature03018
Zerangue, N., and Kavanaugh, M. P. (1996). ASCT-1 is a neutral amino acid exchanger with chloride channel activity. J. Biol. Chem. 271 (45), 27991–27994. doi:10.1074/jbc.271.45.27991
Zerangue, N., and Kavanaugh, M. P. (1996). Flux coupling in a neuronal glutamate transporter. Nature 383, 634–637. doi:10.1038/383634a0
Zhou, W., Trinco, G., Slotboom, D. J., Forrest, L. R., and Faraldo-Gómez, J. D. (2022). On the role of a conserved methionine in the Na+-coupling mechanism of a neurotransmitter transporter homolog. Neurochem. Res. 47, 163–175. doi:10.1007/s11064-021-03253-w
Keywords: vesicular glutamate transporter, excitatory amino acid transporters, anionchannels, galactonate transporter, dual function proteins, anion channel function
Citation: Borghans B, Dmitrieva N, Nikiforov A and Fahlke C (2025) Vesicular and plasma membrane glutamate transporters. Front. Biophys. 3:1693508. doi: 10.3389/frbis.2025.1693508
Received: 27 August 2025; Accepted: 13 October 2025;
Published: 30 October 2025.
Edited by:
Maddalena Comini, University of Oxford, United KingdomReviewed by:
Ulf Strauss, Charité University Medicine Berlin, GermanyFiorenzo Conti, Marche Polytechnic University, Italy
Salah El Mestikawy, McGill University, Canada
Copyright © 2025 Borghans, Dmitrieva, Nikiforov and Fahlke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christoph Fahlke, Yy5mYWhsa2VAZnotanVlbGljaC5kZQ==
†These authors have contributed equally to this work