 Guzel Valeeva1,2 Azat Abdullin1,2,3 Roman Tyzio1 Andrei Skorinkin2,4 Evgeny Nikolski2 Yehezkiel Ben-Ari1 Rustem Khazipov1*
Guzel Valeeva1,2 Azat Abdullin1,2,3 Roman Tyzio1 Andrei Skorinkin2,4 Evgeny Nikolski2 Yehezkiel Ben-Ari1 Rustem Khazipov1*
- 1 Institut de Neurobiologie de la Méditerranée-Institut National de la Santé et de la Recherche Médicale U90, Universite Mediterranean Aix-Marseille II, Marseille, France
- 2 Kazan Institute of Biochemistry and Biophysics, Russian Academy of Sciences, Kazan, Russia
- 3 Tatar State University of Humanities and Education, Kazan, Russia
- 4 Kazan Federal University, Kazan, Russia
In the developing hippocampus, GABA exerts depolarizing and excitatory actions and contributes to the generation of neuronal network driven giant depolarizing potentials (GDPs). Here, we studied spike time coding at immature GABAergic synapses and its impact on synchronization of the neuronal network during GDPs in the neonatal (postnatal days P2–6) rat hippocampal slices. Using extracellular recordings, we found that the delays of action potentials (APs) evoked by synaptic activation of GABA(A) receptors are long (mean, 65 ms) and variable (within a time window of 10–200 ms). During patch-clamp recordings, depolarizing GABAergic responses were mainly subthreshold and their amplification by persistent sodium conductance was required to trigger APs. AP delays at GABAergic synapses shortened and their variability reduced with an increase in intracellular chloride concentration during whole-cell recordings. Negative shift of the GABA reversal potential (EGABA) with low concentrations of bumetanide, or potentiation of GABA(A) receptors with diazepam reduced GDPs amplitude, desynchronized neuronal firing during GDPs and slowed down GDPs propagation. Partial blockade of GABA(A) receptors with bicuculline increased neuronal synchronization and accelerated GDPs propagation. We propose that spike timing at depolarizing GABA synapses is determined by intracellular chloride concentration. At physiological levels of intracellular chloride GABAergic depolarization does not reach the action potential threshold and amplification of GABAergic responses by non-inactivating sodium conductance is required for postsynaptic AP initiation. Slow and variable excitation at GABAergic synapse determines the level of neuronal synchrony and the rate of GDPs propagation in the developing hippocampus.
Introduction
Gamma aminobutyric acid is the main inhibitory neurotransmitter in the adult brain. Synchronous inhibition by hyperpolarization and shunt provided by GABAergic interneurons is instrumental for the generation of various network activity patterns in the adult brain (Freund and Buzsaki, 1996). However, early in development, GABA acting via chloride-permeable GABA(A) channels, exerts depolarizing and excitatory action due to elevated intracellular chloride concentration in immature neurons (Ben Ari et al., 2007). Depolarizing GABA is involved in the generation of primitive pattern of neuronal network activity in the immature hippocampus – so called GDPs (Ben-Ari et al., 1989; Khazipov et al., 2004; Dzhala et al., 2005; Sipila et al., 2005). During GDPs, both pyramidal cells and interneurons fire within a time window of a few hundreds of milliseconds. GDPs are initiated in CA3 hippocampal region and propagate, in a wave-like manner, to CA1, dentate gyrus, and along hippocampus in a septo-temporal direction. Slow wave-like propagation of GDPs along the intact hippocampus in vitro may take almost a second (Leinekugel et al., 1998). Excitation to pyramidal cells and interneurons during GDPs is brought by synergistic excitatory actions of GABA and glutamate (Menendez de la et al., 1996; Khazipov et al., 1997; Leinekugel et al., 1997; De la Prida et al., 1998; Bolea et al., 1999; Lamsa et al., 2000). By providing a coincidence between pre- and postsynaptic neurons, GDPs are thought to be involved in activity-dependent spike timing dependent plasticity (Kasyanov et al., 2004; Mohajerani and Cherubini, 2006), which is particularly robust during the early developmental period (Isaac et al., 1995; Durand et al., 1996). However, factors determining spatio-temporal characteristics of GPDs and spike timing during GDPs are not completely understood.
While excitatory actions of GABA on immature neurons have been well documented in virtually all brain structures using variety of electrophysiological and imaging techniques, an important question of postsynaptic action potential (AP) time coding at the GABAergic synapse has not been addressed. AP temporal coding is a major characteristic of excitatory synapse, which implies a delay of APs and its variability in the postsynaptic cell receiving an excitatory input. AP temporal coding is determined by kinetics of transmitter release, kinetics of postsynaptic receptor mediated excitatory currents, and postsynaptic membrane properties including voltage-gated conductances which shape and amplify or inhibit postsynaptic potentials (PSPs) (Fricker and Miles, 2000). In glutamatergic synapses, synchronous activation of multiple glutamatergic inputs generates suprathreshold EPSPs that evoke APs with short and reliable delays that synchronizes neuronal population giving rise to population spike. Upon removal of GABAergic inhibition, synchronization of neuronal population via recurrent collateral glutamatergic synapses also enables generation of high-frequency oscillations (Miles and Wong, 1986, 1987). However, small near-threshold excitatory postsynaptic potentials (EPSPs) evoke postsynaptic APs with long and variable delays due to activation of the voltage-gated potassium conductance and amplification of EPSPs by non-inactivating sodium conductance in pyramidal cells (Fricker and Miles, 2000). Thus, the AP timing at glutamatergic synapses strongly depends on the amount of glutamatergic conductance activated in the postsynaptic cell. In contrast, at immature GABAergic synapses EGABA is likely to be the critical factor determining the AP timing as EGABA is set by chloride transporters not far from the AP threshold (APthr) (Ben-Ari et al., 1989; Hollrigel et al., 1998; Sipila et al., 2005; Rheims et al., 2008). In the present study, we provide evidence that (i) GABAergic interneurons excite CA3 pyramidal cells with long and variable delays; (ii) the AP timing at GABAergic synapses depends on intracellular chloride concentration in the postsynaptic neuron; and (iii) in physiological conditions, GABAergic depolarization is mainly subthreshold and requires activation of persistent sodium conductance to trigger APs. We also show that slow and variable spike timing at depolarizing GABAergic synapses shapes GDPs by reducing neuronal synchronization and slowing down their propagation.
Materials and Methods
Hippocampal slices were prepared from Wistar rats of both sexes (Postnatal days [P]2–6). All animal use protocols conformed to the INSERM guidelines on the use of laboratory animals. Transverse (perpendicular to the longitudinal hippocampal axis) slices (350–500 μm) were cut from the middle third of hippocampus using Vibratome (VT 1000E; Leica, Nussloch, Germany). Slices were kept in oxygenated (95% O2/5% CO2) artificial cerebrospinal fluid (ACSF) of the following composition (in mM): NaCl 126, KCl 3.5, CaCl2 2.0, MgCl2 1.3, NaHCO3 25, NaH2PO4 1.2 and glucose 11 (pH 7.4) at room temperature (20–22°C) at least 1 h before use. For recordings, slices were placed into a conventional fully submerged chamber superfused with ACSF at a rate of 2–3 ml/min.
Extracellular field potentials and multiple unit activity (MUA) were recorded using 64-electrodes planar arrays (MCA, Germany) or 2–3 electrodes made from tungsten wire of diameter 50 μm (California Fine Wire, Grover Beach, CA, USA) placed in CA3 and CA1 pyramidal cell layer. Signals were amplified (×1000, bandpass 0.1 Hz–4 kHz) and digitized at 10 kHz. Patch-clamp recordings were performed using Axopatch 200A (Axon Instruments, Union City, CA, USA) and EPC-9 (HEKA Elektronik Dr. Schulze GmbH, Lambrecht/Pfalz, Germany) amplifiers. Patch electrodes were made from borosilicate glass capillaries (GC150F-15, Clark Electromedical Instruments). For recordings of single GABA(A) channels, patch pipette solution contained (in mM): NaCl 120, TEA–Cl 20, KCl 5, 4-aminopyridine 5, CaCl2 0.1, MgCl2 10, glucose 10, Hepes–NaOH 10 buffered to pH 7.2–7.3 and GABA (1–5 μM) was added at the day of experiment from 1 mM frozen stock solution. Driving force for GABA(A) receptor mediated currents was determined from the current–voltage relationships of the currents through single GABA(A) channels as described earlier (Tyzio et al., 2006) and corrected for an error of 2 mV (Tyzio et al., 2008). Membrane potential was estimated using cell-attached recordings of single NMDA channels as described before (Leinekugel et al., 1997; Tyzio et al., 2003). For recordings of single NMDA channels pipette solution contained nominally Mg2+ free ACSF with NMDA (10 μM), glycine (1 μM), and strychnine (1 μM). Patch pipette solution for whole-cell recordings contained (in mM): potassium gluconate (0–135) or KCl (0–135); MgCl2, 2; Hepes, 10; Mg–ATP, 4; Na–GTP, 0.3; equilibrated at pH 7.3 with KOH. In current-clamp recordings, membrane potential was maintained at values between −75 and −80 mV by injection of constant negative current. Synaptic GABA(A) receptor mediated responses were evoked by electrical stimulation in the presence of the antagonists of glutamate ionotropic receptors CNQX (10 μM) and d-APV (40 μM), and GABA(B) receptors CGP55845 (1 μM). Simulated GABA–PSPs were evoked by injection of GABA-PSCs waveform obtained by averaging one hundred spontaneous GABA-PSCs recorded in voltage-clamp mode in the presence of glutamate ionotropic receptor antagonists. The amplitude of GABA-PSPs was adjusted to evoke near- threshold responses. Action potential threshold (APthr) was determined offline using the custom-made software (IGOR Pro, WaveMetrics) as a value of membrane potential at which dU/dt exceeded 10 V/s (Stuart et al., 1997; Fricker et al., 1999). AP delays were measured as a time between the stimulus and AP peak. Recordings were digitized (10 kHz) online with Digidata 1200/1322 interface cards (Axon Instruments, Union City, CA, USA) and analyzed offline with Axon package, miniAnalysis (Synaptosoft), IGOR Pro (WaveMetrics) and Origin 5.0 (Microcal Software, Northampton, MA, USA). Group measures are expressed as means ± SE unless indicated; error bars also indicate SE. The statistical significance of differences was assessed with the Students t-test and Kolmogorov–Smirnov test. The level of significance was set at P < 0.05.
Results
Action Potential Delays at the Excitatory GABAergic Synapse
Experiments were performed in hippocampal slices obtained from P2–6 neonatal rats. To asses temporal characteristics of GABAergic excitation we determined the AP delays in CA3 pyramidal cells in response to synaptic activation of GABA(A) receptors. Synaptic GABA(A) receptor mediated responses were elicited by electrical slice stimulation using bipolar electrodes or glass pipette positioned in a vicinity of the recording site (separation distance <0.5 mm) in the presence of ionotropic glutamate (10 μM CNQX, 40 μM d-APV) and GABA(B) (1 μM CGP55845) receptor antagonists (Davies et al., 1990; Khazipov et al., 1993).
In the first experiment, we used extracellular recordings of MUA that is action currents from tens of neurons nearby the recording site (Cohen and Miles, 2000). Synaptic activation of GABA(A) receptors invariably increased MUA in CA3 pyramidal cell layer (Figure 1), and the response was suppressed by the GABA(A) receptor antagonist bicuculline (10 μM; see also Figure 4). In certain cases, short-latency spikes persisted in the presence of bicuculline, probably due to the direct stimulation of axons of the recorded neurons (not shown); these experiments were discarded from the analysis. In the experiments, in which bicuculline completely suppressed synaptic MUA response, MUA was cross-correlated with stimulus and the time course of MUA response has been analyzed. MUA peaked at 28 ± 6 ms after the stimulus and decayed with a decay time constant of 76 ± 18 ms; half-width of MUA elevation was of 71 ± 13 ms (mean ± S.D.; n = 8 slices). Mean value of APs delay was of 65 ± 12 ms (n = 8). In none of recorded slices, activation of GABA(A) receptors evoked population spikes, that are a characteristic response to the collective activation of glutamatergic synapses. These results indicate that the AP delays at excitatory GABAergic synapses in the immature CA3 pyramidal cells are long and variable.
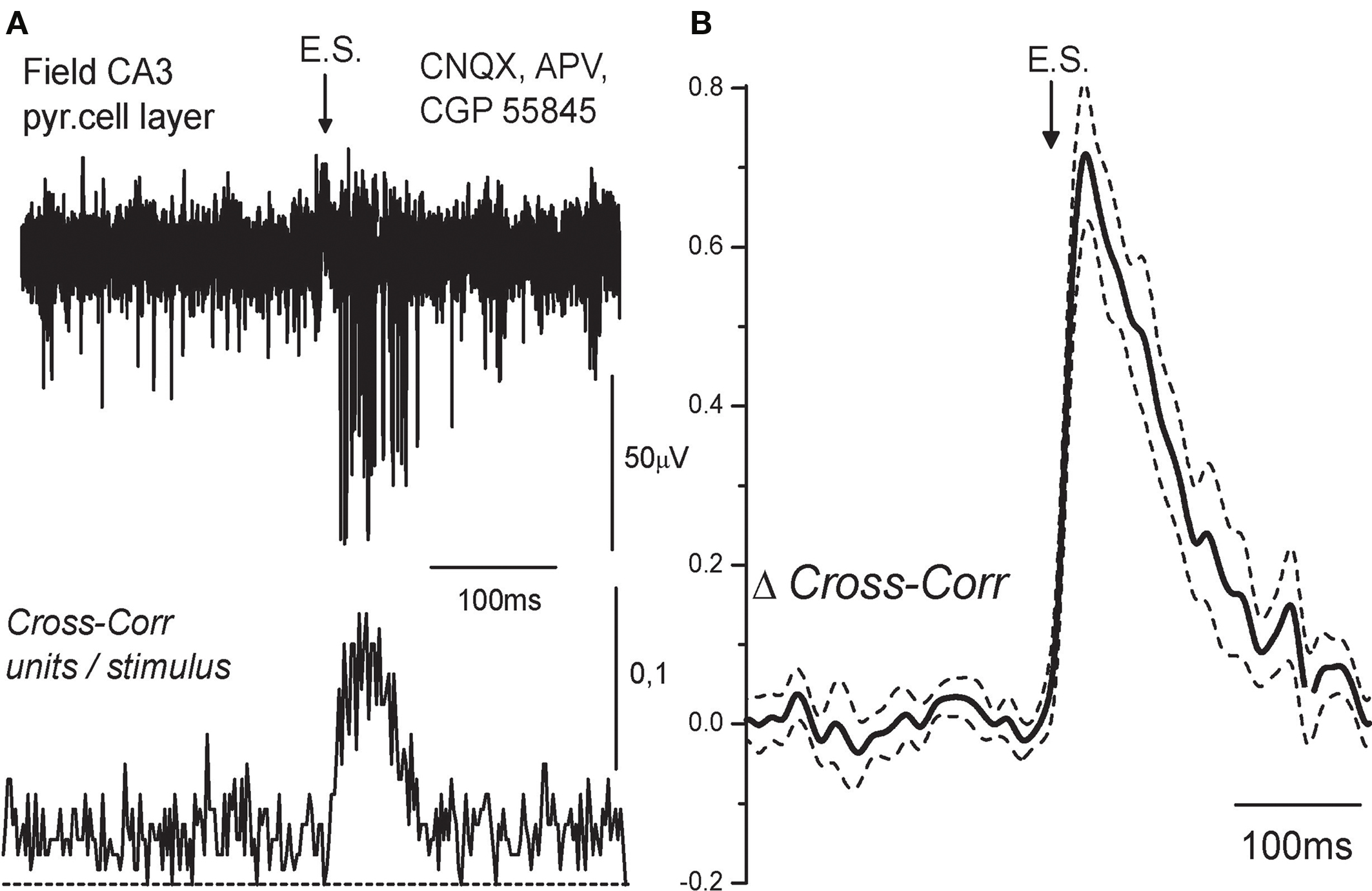
Figure 1. Temporal characteristics of excitation at GABAergic synapses using extracellular recordings. (A) Extracellular recordings of the action potentials (APs) evoked by electrical stimulation (ES) in the presence of the AMPA, NMDA, and GABA(B) receptor antagonists (10 μM CNQX, 40 μM D-APV, 1 μM CGP55845) in the CA3 layer of P5 rat hippocampus. Five traces are superimposed. Below: cross-correlogram of the APs times versus stimulus. (B) Average cross-correlogram of the APs versus stimulus obtained from eight slices (P2–5 rats). Dashed lines show SEM.
Dependence of the Temporal Characteristics of GABA-Mediated Excitation on [Cl−]I
We hypothesized that slow kinetics of GABAergic excitation results from relatively small driving force (DFGABA) of the GABA(A) receptor mediated responses. Therefore, we next studied the temporal characteristics of GABAergic excitation as a function of intracellular chloride concentration. In this aim, we investigated AP delays of evoked by synaptic GABA(A) receptor mediated responses using current-clamp whole-cell recordings from CA3 pyramidal cells with chloride concentration of [Cl−]p = 18, 30, 50, and 140 mM in the recording pipette (Figure 2). Membrane potential was maintained at −70/−80 mV by injection of the constant negative current to compensate for the leakage through the gigaseal contact between the membrane and pipette (Tyzio et al., 2003). Under these conditions, synaptic activation of GABA(A) receptors evoked depolarizing postsynaptic potentials (GABA-PSPs) which were completely blocked by bicuculline (20 μM). In keeping with the results of previous studies using intracellular recordings with sharp electrodes (Ben-Ari et al., 1989), GABA-PSPs often triggered APs in pyramidal cells. In each neuron, AP probability and delays were analyzed from a hundred of evoked GABA-PSPs. With [Cl−]p of 18 mM, GABA-PSPs evoked APs in two of 12 neurons. With [Cl−]p of 30, 50, and 140 mM probability of GABA-PSPs to evoke APs increased to 39 ± 8% (n = 11), 30 ± 2% (n = 2) and 70 ± 7% (n = 8), respectively. AP delays and their variability also strongly depended on [Cl−]p (Figure 3). At [Cl−]p of 18 mM, mean AP delay and dispersion attained 56 ± 8 and 14 ± 1 ms, respectively (n = 2; Figure 2A, left), that was close to the values obtained using extracellular recordings. At [Cl−]p = 30 mM, APs delays varied within a range from 8 to 145 ms, with a mean of 38 ± 21 ms (n = 14); AP delays were highly variable with standard deviation varying from 8 to 44 ms in individual cells, and with a mean SD of 17 ± 2 ms (n = 14). With symmetrical chloride ([Cl−]p = 140 mM), AP delays and their dispersion reduced to 12 ± 2 and 4 ± 0.5 ms, respectively (n = 8; Figure 3, right). Interestingly, at lower [Cl−]p values APs barely emerged at the rising phase or peak of GABA-PSPs but rather from an additional depolarization, which prolonged and amplified synaptic potentials. With elevated intracellular chloride concentration, APs typically emerged at the rising phase of GABA-PSPs.
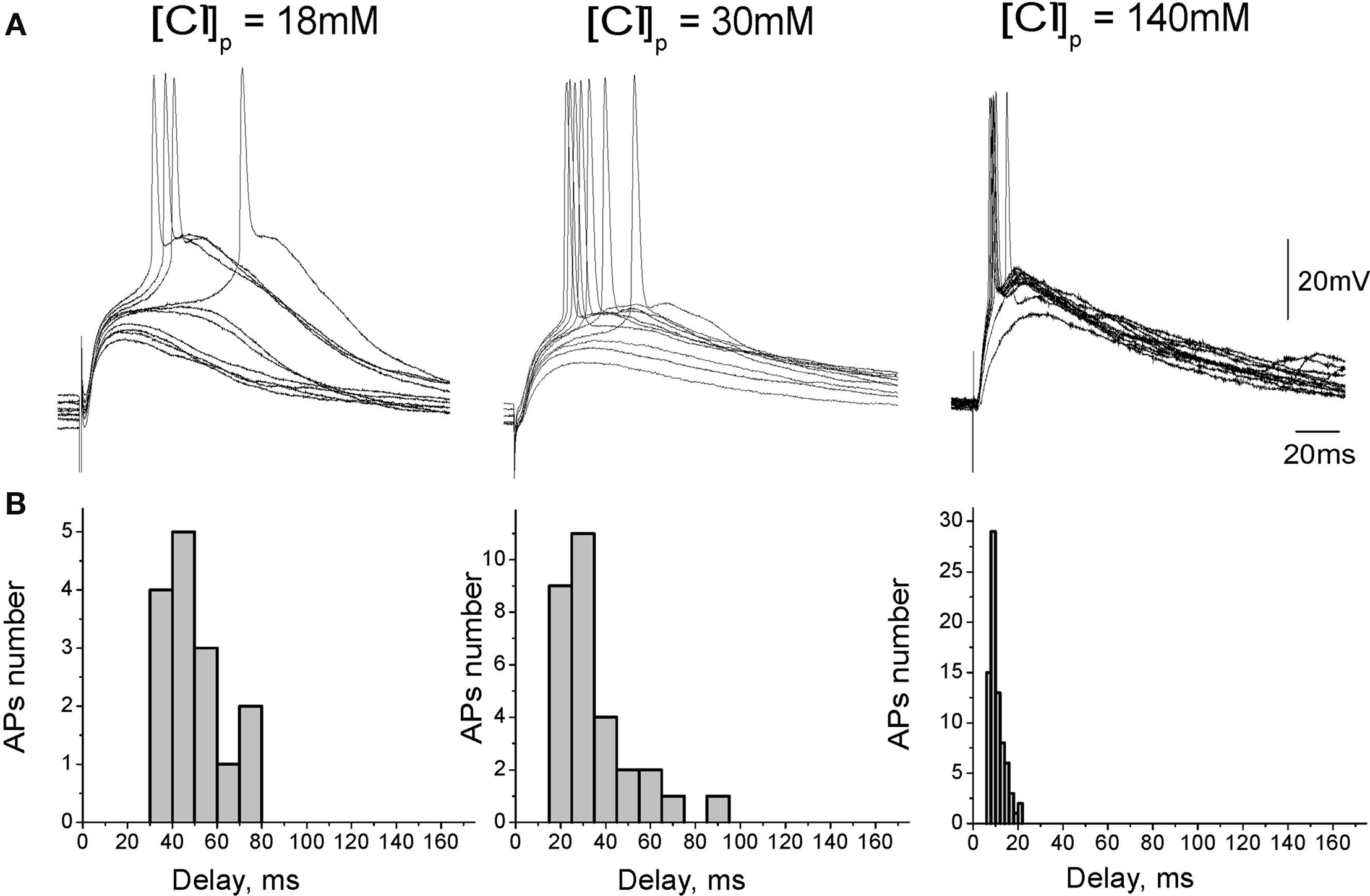
Figure 2. Dependence of depolarizing GABAergic postsynaptic responses on chloride concentration in the recording pipette. (A) GABA(A) receptor mediated postsynaptic potentials recorded from CA3 pyramidal cells in whole-cell current-clamp configuration at different [Cl−]p. GABA-PSPs were evoked by electrical stimulation in the presence of the glutamate and GABA(B) receptors blockers. (B) Corresponding histograms of the GABA-PSP-triggered AP delays.
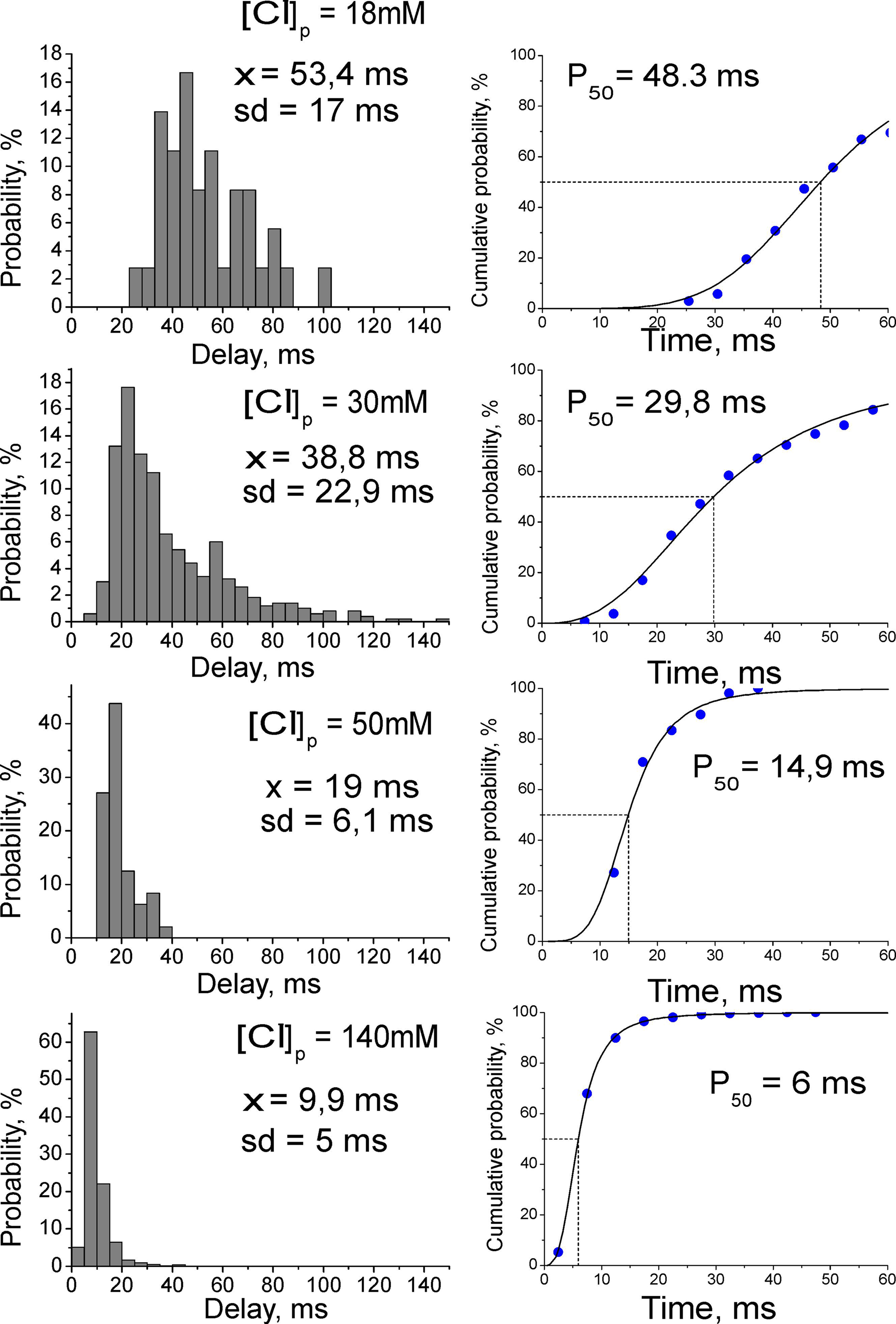
Figure 3. Dependence of spike timing at GABAergic synapse on intracellular chloride concentration. Left: Average histograms of the AP delays at different [Cl-]p in whole-cell recordings. Right: Cumulative curves for the AP delays fitted by Hill equation.
In the immature cortical neurons, elevated [Cl−]i is set by NKCC1 chloride co-transporter (Payne et al., 2003; Yamada et al., 2004; Dzhala et al., 2005; Tyzio et al., 2006). We therefore hypothesized that reduction of NKCC1 activity, via reduction in [Cl−]i and negative shift in EGABA, will increase GABA-evoked AP delays and their variability. Using extracellular recordings of MUA responses evoked by pharmacologically isolated GABA-PSPs we found that indeed, low concentrations of bumetanide (0.3–0.6 μM) cause significant increase in the AP delays and their variability, that was manifested by an increase in the mean AP delay, half-width and decay time constant of the MUA response. Bumetanide also reduced APs number in MUA response evoked by GABA-PSPs (Figure 4). Thus, the results of whole-cell recordings with different [Cl−]p and the effects of low-bumetanide on MUA responses indicate that AP timing at GABAergic synapses is strongly dependent on [Cl−]i in the postsynaptic neuron.
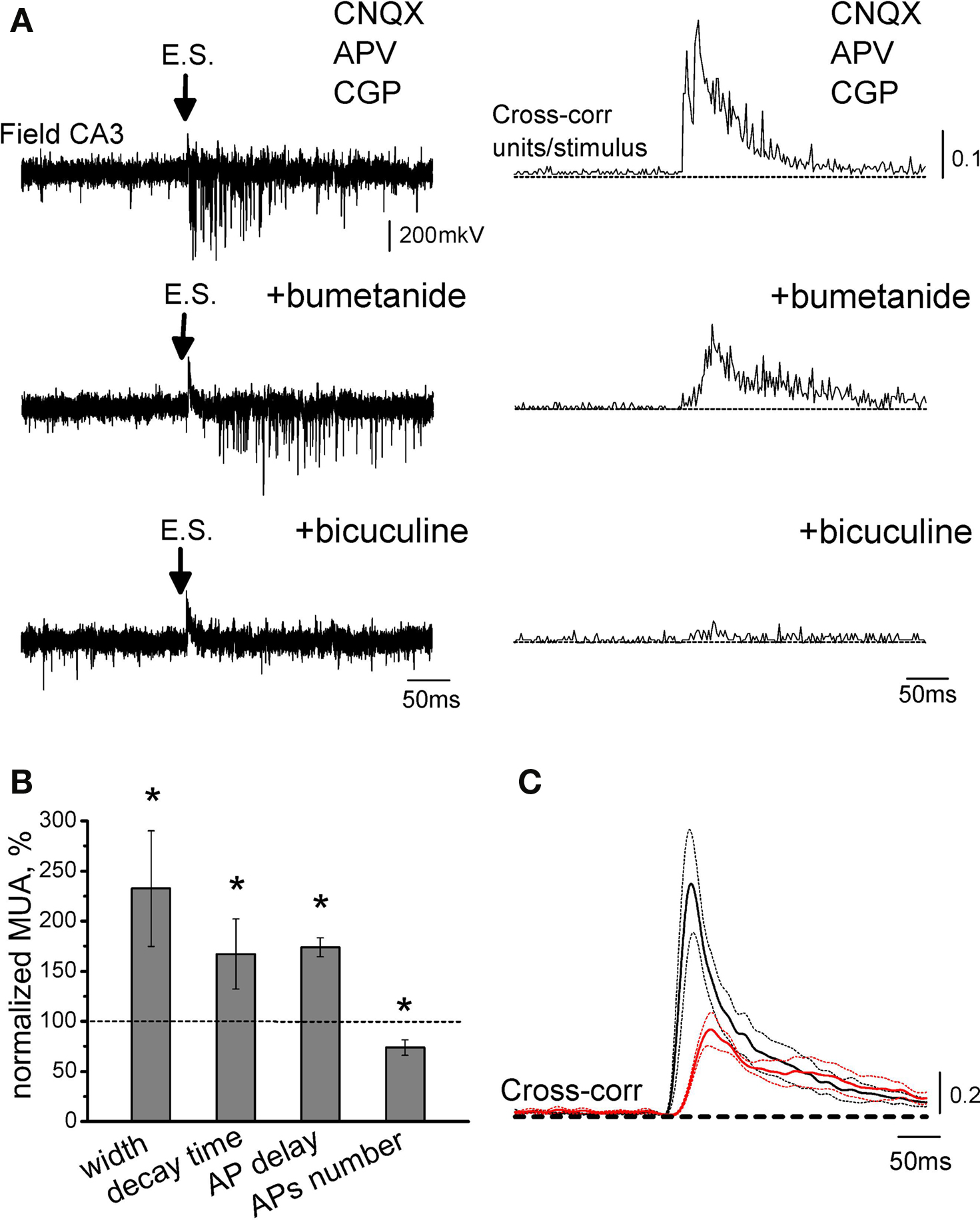
Figure 4. Effect of partial blockade of NKCC1 co-transporter on action potential delays at GABAergic synapses. (A) Extracellular recordings of MUA evoked by electrical stimulation (E.S.) in the presence of the glutamate and GABA(B) receptor antagonists from the CA3 layer of P5 rat hippocampus (top: control, middle: 0.3 μM bumetanide and bottom: 20 μM bicuculline). On the right are shown corresponding cross-correlograms of the AP times versus stimulus. (B) Summary plot of the effects of low-bumetanide on synaptic GABA-evoked MUA. (C) Average cross-correlograms in control conditions (black) and in the presence of 0.3–0.6 μM bumetanide (red) (n = 4 slices; P2–5 rats). Dashed lines show SEM. Note that partial blockade of NKCC1 co-transporter increases AP delays and their jitter, and decreases APs number.
GABA Driving Force, Resting Membrane Potential and Action Potential Threshold
We further measured three parameters on which depends excitatory action of GABA: GABA driving force (DFGABA), resting membrane potential (Em) and APthr in CA3 pyramidal cells (Figure 5). DFGABA was measured using cell-attached recordings of single GABA(A) channels, that do not affect intracellular chloride (Tyzio et al., 2006). Deduced from the current–voltage relationships of the currents through GABA(A) channels, DFGABA in P2–5 CA3 pyramidal cells was of 16.5 ± 1.6 mV (n = 32 cells; Figure 5A), which is close to the previously reported values (Tyzio et al., 2008). Resting potential was measured using NMDA channels in cell-attached recordings as voltage sensors (Tyzio et al., 2003). We found that Em in P2–5 CA3 pyramidal cells is of −78.8 ± 3.1 mV (n = 14; Figure 5B). Knowing these values we further estimated EGABA (EGABA = Em + DFGABA) in P2–5 CA3 pyramidal cells of −61.4 ± 5.4 mV. Corresponding Nernst values of [Cl−]i ranged from 7 to 30 mM (see also Tyzio et al., 2007, 2008).
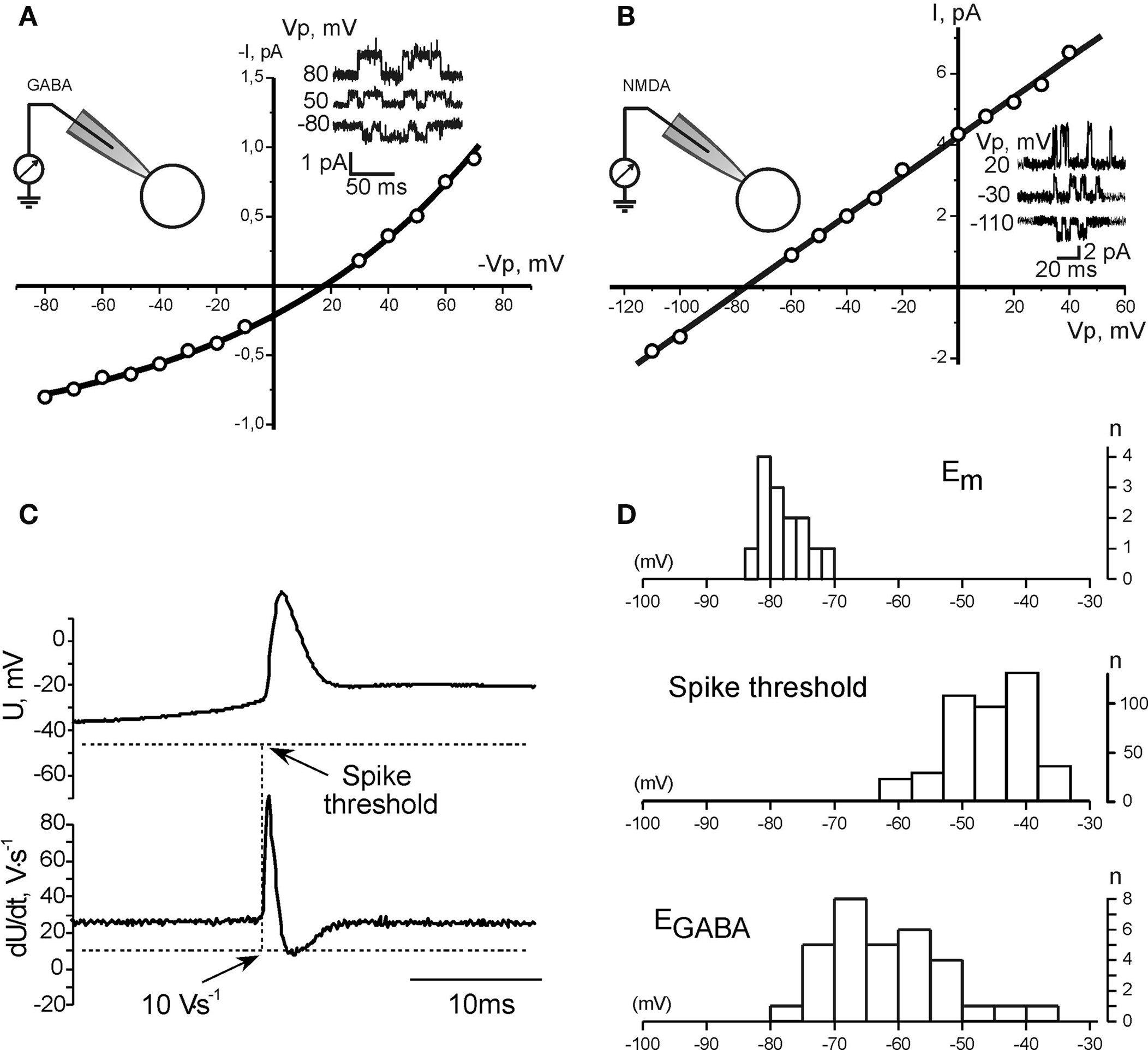
Figure 5. GABA driving force, resting membrane potential, and the action potential threshold in neonatal CA3 pyramidal cells. (A, B) Measurements of the GABA driving force (A) and membrane potential (B) using cell-attached recordings of GABA and NMDA channels, respectively. Example traces of single channel openings at different pipette potentials (Vp) are shown in insets. (C) Measurement of the action potential threshold in whole-cell current-clamp recordings. (D) Summary plot of the resting membrane potential, GABA reversal potential and the action potential threshold in P2–5 CA3 pyramidal neurons.
Threshold of APs triggered by GABA-PSPs was measured as a value of membrane potential at which dU/dt exceeded 10 V/s in whole-cell current-clamp recordings. APthr varied from −63 to −34 mV, with a mean of −45.9 ± 6.5 mV (n = 11 cells; Figure 5C). As summarized on Figure 5D, there is only small overlap between EGABA and Em, and in the vast majority of neurons EGABA is essentially subthreshold and barely attains APthr. These findings are in agreement with the results obtained using whole-cell recordings, which suggested that excitatory action of GABA requires activation of additional subthreshold conductance, which amplifies GABA-PSPs and brings potential to the APthr.
Role of Persistent Na+ Currents in the GABAergic Excitation
In the previous studies, persistent Na+ current (INap), which is activated at ≈−60 mV was shown to account for the slow regenerative depolarization triggering spontaneous bursting in the immature CA3 pyramidal neurons (Sipila et al., 2005, 2006). Because EGABA is close to the activation threshold of INap, we hypothesized that INap amplifies GABA-PSPs and contributes to the excitatory action of GABA. To test this hypothesis, we studied the effects of sodium channel blockers TTX and phenytoin on GABA-PSPs. Because blockade of sodium channels causes presynaptic inhibition, we examined simulated GABA-PSPs (simGABA-PSPs) evoked by injection of GABA-PSCs waveform, which was obtained in voltage-clamp recordings similar to the procedure previously described for glutamatergic synapses (Fricker and Miles, 2000). The amplitude of GABA-PSCs was adjusted in each neuron to evoke near-threshold responses. The experiments were performed in the presence of the GABA and glutamate receptor antagonists to suppress ongoing synaptic activity. As illustrated by Figure 6, simulated GABA responses were amplified and prolonged by voltage-gated conductance, and triggered APs with long and variable delays similarly to synaptic GABA-PSPs with a mean AP delay of 69 ± 4 ms and standard deviation of 10 ± 1 ms (n = 20). Application of voltage-gated sodium channels blocker TTX (1 μM) not only inhibited APs evoked by simGABA-PSPs but also suppressed the amplification of simGABA-PSPs by subthreshold voltage-gated conductance (Figure 7A). Integral response calculated as a surface of simGABA-PSPs was reduced by TTX from 4851 ± 591 μVs (calculated for selected simGABA-PSPs which did not evoke APs) to 3536 ± 574 μVs, that is to 70 ± 5% of the control values (n = 6; P = 0.004). TTX also reduced the half-width of simGABA-PSPs from 132 ± 15 to 110 ± 11 ms, that is to 85 ± 5% of the control values (n = 6; P = 0.0003). Antiepileptic drug phenytoin (200 μM) that preferentially blocks repeated Na+ cannel openings and INap (Kuo and Bean, 1994; Segal and Douglas, 1997) also blocked amplification of simGABA-PSPs, reducing the integral response to 70 ± 5% (from 7585 ± 549 to 5239 ± 555 μVs; n = 3; P = 0.005) and the half-width from 182 ± 10 to 146 ± 2 ms; n = 3; P = 0.003; Figure 7B). Phenytoin completely suppressed APs evoked by simGABA-PSPs. Phenytoin also inhibited APs triggered by synaptically evoked GABA-PSPs (n = 4, Figure 7C). However, phenytoin also increased the number of failures of the synaptically evoked responses suggesting an additional presynaptic effect. Taken together, these results indicate that INap participates in the amplification of depolarizing subthreshold GABA-PSPs and that this amplification is involved in excitatory action of GABA in immature CA3 pyramidal neurons. It should be noted that simGABA-PSPs under standard current-clamp conditions may differ from natural responses as DFGABA decreases during depolarizing GABA-PSPs; this should result in an acquisition of a “plateau” shape of the response and favor the role of near-threshold activated conductances. Use of dynamic clamp would be useful to address these questions in future studies.
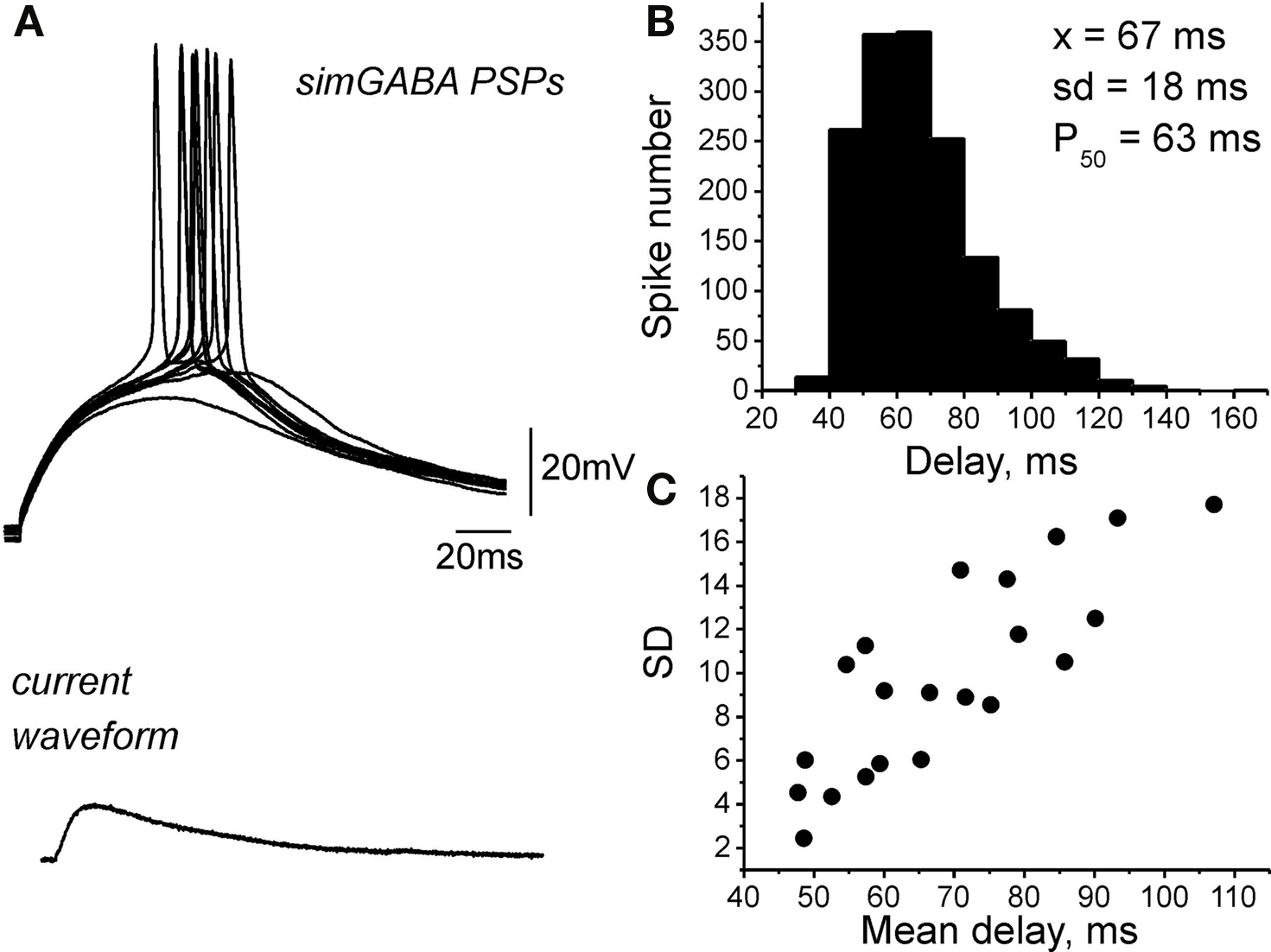
Figure 6. Responses elicited by injection of synaptic GABAergic current waveform. (A) Example traces of the responses (top traces) elicited by injection of the current waveform of GABA-PSCs (bottom trace) recorded in current-clamp mode in the presence of the GABA and glutamate receptor antagonists. (B) Histogram of AP delays distribution over 20 cells. (C) Plot of standard deviation (SD) versus mean AP delay value; each point corresponds to an individual neuron.
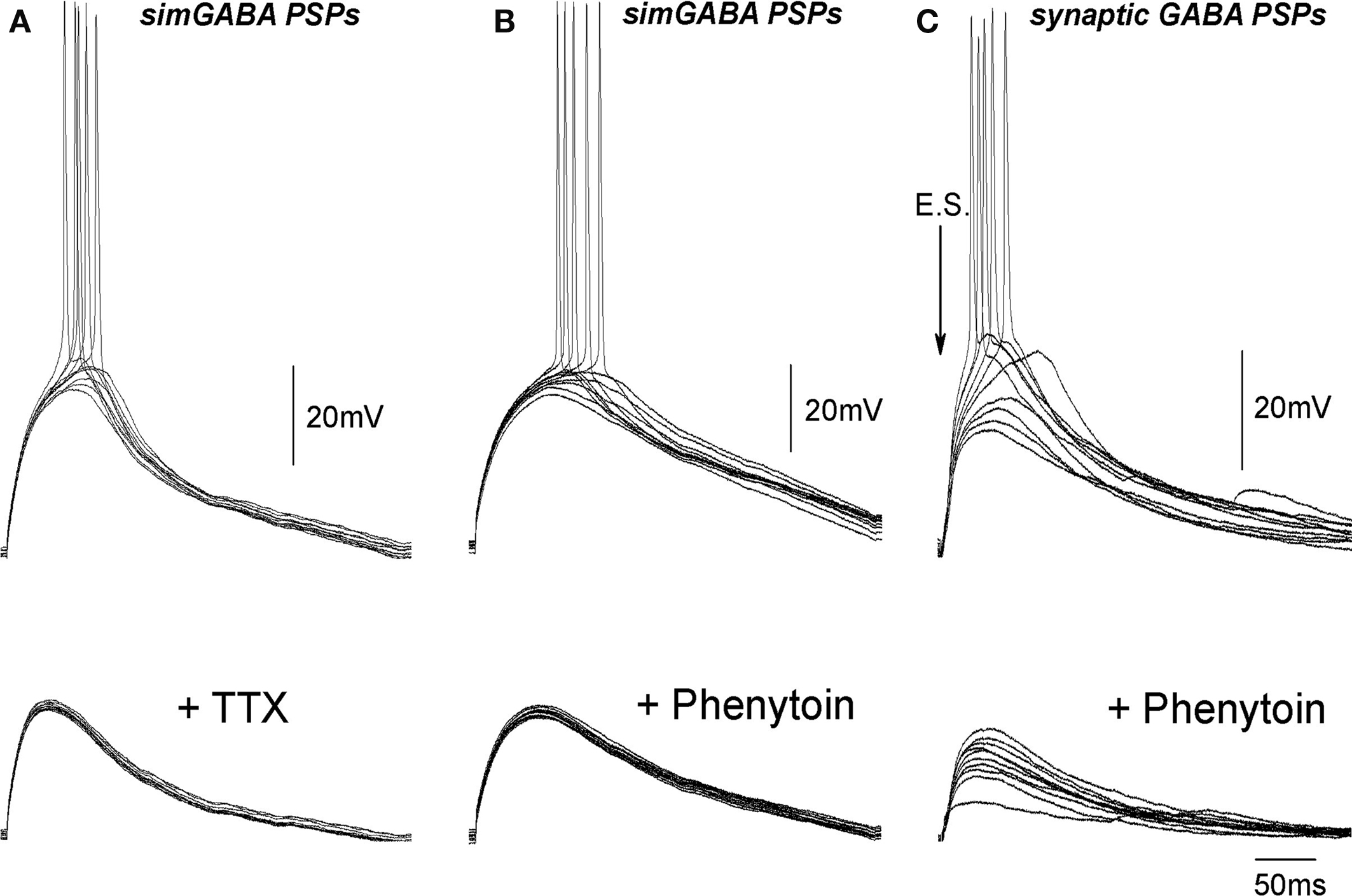
Figure 7. Na+ channel blockers inhibit subthreshold voltage-gated conductance and the action potentials evoked by simulated and synaptic GABA-PSPs. (A, B) Example traces of simGABA-PSPs evoked by injection of GABA-PSC waveform in control conditions (upper traces) and in the presence of 1 μM TTX (A) and 200 μM phenytoin (B) (bottom traces). (C) Synaptic GABA-PSPs in control conditions (upper traces) and in the presence of 100 μM phenytoin (bottom traces).
Implication of “Slow” GABA(A)-Mediated Excitation for GDPs
Essential features of GDPs are relatively low level of neuronal firing synchrony within the temporal window of GDPs (Ben-Ari et al., 1989; Khazipov et al., 1997; Strata et al., 1997; Hollrigel et al., 1998; Menendez de la and Sanchez-Andres, 2000; Sipila et al., 2006) and slow wave-like propagation of GDPs between the hippocampal regions and along the hippocampus (Leinekugel et al., 1998; Menendez de la et al., 1998). We suggested that low level of synchrony and slow propagation of GDPs involves slow and asynchronous spiking at excitatory GABAergic synapses. To verify our assumption, we tested effects of drugs that modulate EGABA (low-bumetanide) and efficacy of GABAergic transmission (bicuculline, diazepam) on synchronization of neuronal network during generation of GDPs. Extracellular recordings were performed using two to three wire electrodes placed in CA3 and CA1 pyramidal cell layer with 300–600 μm separation distance or 64-sites recordings using planar electrode arrays with a 200-μm separation distance. Neuronal synchrony was quantified by measuring: (i) GDPs parameters at a single recording site in CA3 pyramidal cell layer including amplitude and duration of the local field potential, duration of MUA bursts associated with GDPs and GDPs frequency, and (ii) velocity of GDPs propagation (Figure 8).
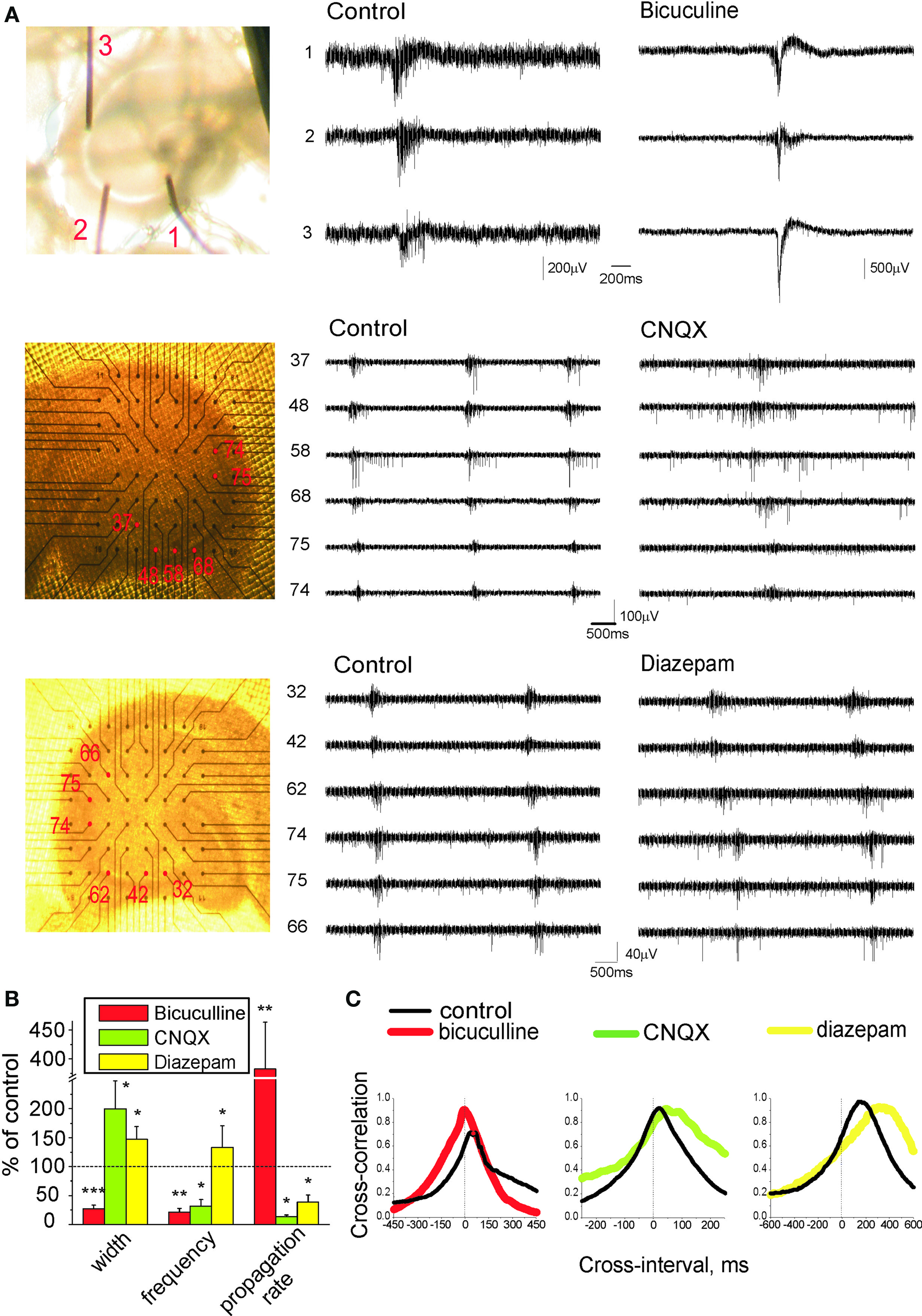
Figure 8. Effects of drugs modulating efficacy of GABAergic and glutamatergic transmission on network synchronization during GDPs. (A) Left: recording field electrodes arrangement in pyramidal layer of hippocampus for concomitant recordings from different sites. Middle and right: traces representing extracellular GDP recordings in control conditions and after drug application (top traces, 3 μM bicuculline; middle traces, 1 μM CNQX; bottom traces, 2 μM diazepam). (B) GDPs half-width, frequency, and propagation rate in the presence of drugs normalized to the control values. (C) Cross-correlograms of unitary APs between two distinct CA regions (CA3 and CA1, or within CA3) in control conditions (black) and in the presence of the drug. (B, C) represent pooled data obtained in the presence of bicuculline (n = 7); CNQX (n = 5); diazepam (n = 4).
In control conditions, field GDPs had 149 ± 35 μV amplitude, 101 ± 6 ms half-width and occurred at 0.2 ± 0.03 s−1. GDPs typically originated from CA3a or CA3b and propagated towards CA1 and CA3c, at a rate of 25 ± 5 mm/s (n = 20). Because of variability in GDPs parameters between slices and rats, the effects of drugs were studied in paired experiments, and the results were normalized to control values in each given slice. Partial blockade of GABA(A) receptors with low doses of bicuculline (2–5 μM), that causes nearly half-reduction of the GABA-PSCs in the hippocampal slices (Whittington et al., 1995), caused transformation of GDPs into super synchronous neuronal network discharges with tremendously high amplitude (916 ± 315% of control values; P < 0.05) and short duration (57 ± 12 ms versus 226 ± 22 ms in control conditions; P < 0.001). This was accompanied by five-fold shortening of delays of GDP in neighboring recording sites (21 ± 6% of control values; P < 0.01), therefore increasing propagation rate by 282 ± 81% (P < 0.01; n = 7). Yet the frequency of GDPs decreased when GABA receptors were partially blocked, probably because of more powerful activation of the inhibitory conductances (GABA(B) and Ca2+-activated potassium channels) which control inter-GDPs intervals (Gaiarsa et al., 1995; Sipila et al., 2006). In contrast to the effects caused by suppression of GABA(A) receptors, enhancement of GABA action by positive allosteric modulator of GABA(A) receptors diazepam (2 μM) had desynchronizing effect on GDPs. Local field potentials associated with GDPs reduced so that they could be hardly measured from the baseline noise, and duration of MUA bursts associated with GDPs increased to 147 ± 22% of control values. Propagation of GDPs was delayed by 282 ± 73% (P < 0.05; n = 4). These results suggest that GABAergic synapses desynchronize hippocampal network during generation of GDPs, and slow down GDPs propagation.
GABAergic and glutamatergic synapses are co-activated in CA3 pyramidal cells during GDPs (Leinekugel et al., 1997; Bolea et al., 1999; Lamsa et al., 2000). To determine the role of fast AMPA receptor mediated synaptic transmission in the network synchronization during GDPs we tested low doses of the AMPA receptor antagonist CNQX (1 μM, that causes nearly 50% reduction in the amplitude of field EPSPs (Andreasen et al., 1989). After CNQX application, field GDPs reduced to baseline noise levels and duration of associated with GDPs MUA bursts prolonged by 100 ± 49% (P < 0.05), while GDP frequency and propagation rate decreased respectively by 78 ± 11 and 86 ± 3% (P < 0.05; n = 5).
We further investigated how modulation of EGABA affects GDPs using NKCC1 inhibitor bumetanide (Figure 9). At 5–10 μM, bumetanide completely blocked GDPs in agreement with previous reports (Dzhala et al., 2005; Nardou et al., 2009; Sipila et al., 2009). At lower doses (0.3–1 μM), at which bumetanide caused an increase in AP delays and their variability (Figure 4), we observed a decrease in GDPs frequency and the level of neuronal firing during GDPs. Field GDPs reduced to baseline noise levels, duration of GDPs-associated MUA bursts increased from 228 ± 47 to 278 ± 52 ms (n = 6; P < 0.01) and propagation rate of GDPs decreased by 63 ± 6% (n = 6; P < 0.05). In the presence of low-bumetanide, we often observed compartmentalized GDPs that were restricted to one electrode and failed to propagate to the neighboring sites (not shown).
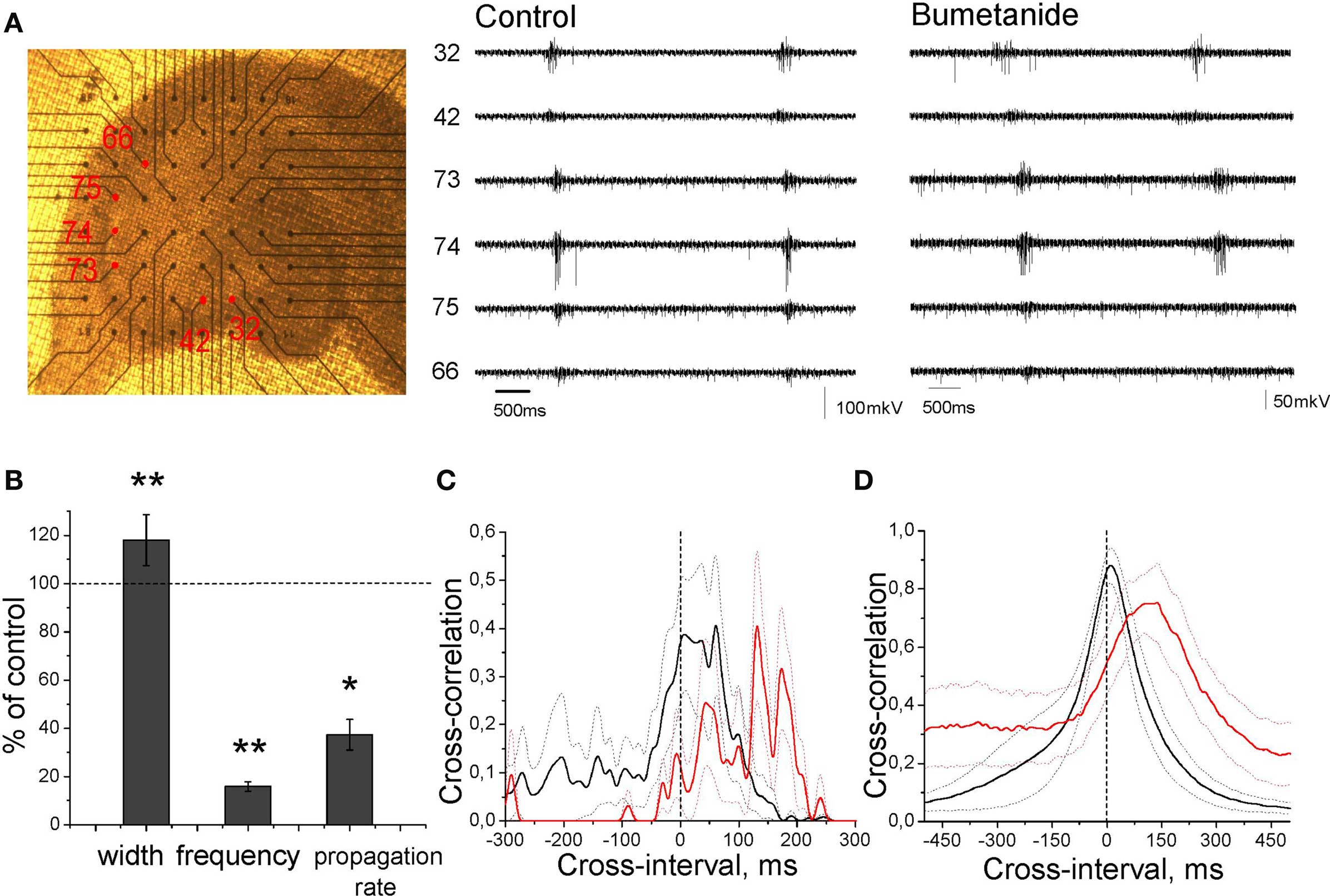
Figure 9. Effect of partial blockade of NKCC1 on GDPs. (A) Photograph of the recording setup and extracellular GDP recordings in control conditions (left traces) and in the presence of 0.3 μM bumetanide (right traces). (B) Normalized GDPs width, frequency, and propagation rate in the presence of 0.3 μM bumetanide. (C, D) Cross-correlograms of unitary APs (C) and GDPs (D) between two distinct CA regions (CA3 and CA1, or within CA3) in control conditions (black) and in the presence of bumetanide (red). Dashed lines show SEM. (B–D) Pooled data from four slices.
Discussion
The main findings of the present study are the following: (i) delays of APs evoked by synaptic GABA in neonatal CA3 pyramidal cells are long and variable, and depend on intracellular chloride concentration; (ii) at physiological conditions, depolarization produced by GABA is typically more negative than the APthr, and activation of subthreshold voltage-gated INap is required to trigger APs; (iii) while depolarizing action of GABA is required for GDPs generation, slow and variable AP delays in GABAergic synapses reduce neuronal synchrony during generation of GDPs and slow down GDPs propagation.
Although excitatory actions of GABA in the immature neurons have been well documented in various brain structures (Ben Ari et al., 2007), the temporal AP coding aspect of GABAergic excitation until present study has not been analyzed. Our central finding is that transmission of excitation in the immature depolarizing GABAergic synapses is slow and highly variable. This was observed using non-invasive extracellular MUA recordings, that affect neither membrane potential nor intracellular chloride, and whole-cell recordings with elevated chloride concentration in the pipette solution. These observations are in agreement with the results of previous studies, in which representative traces show similar long and variable latency APs evoked by stimulation of synaptic GABAergic inputs, using cell-attached recordings from CA3 pyramidal cells (Leinekugel et al., 1997), interneurons (Khazipov et al., 1997), and neocortical pyramidal and stellate cells (Rheims et al., 2008). These findings are important because GABAergic synapses are the first synapses established in the hippocampal neurons, both in rodents and primates (Tyzio et al., 1999; Khazipov et al., 2001) and GABAergic conductance is predominant in GDPs (Ben-Ari et al., 1989; Khazipov et al., 1997; Leinekugel et al., 1997; Hollrigel et al., 1998). We have shown that the temporal coding at GABA synapse is strongly dependent on intracellular chloride concentration in the postsynaptic neuron: AP delays and their variability strongly decreased with an artificial elevation of [Cl−]i during whole-cell recordings, and increased in the conditions of partial blockade of NKCC1 using low concentration of bumetanide. Comparison of the physiological values of Em, DFGABA and APthr lead us to conclusion that at physiological levels of [Cl−]i, depolarizing GABA-PSPs do not attain the action potential threshold, and that the activation of intermediate subthreshold voltage-gated conductance is required to trigger APs. This is in keeping with findings in immature granular cells, in which EGABA is slightly, by ∼5 mV, is more negative than the action potential threshold (−46 and −42 mV, respectively), yet granular cells discharge, typically one action potential, during pure GABAergic GDPs (Hollrigel et al., 1998; see also Brickley et al., 1996). This intermediate step in AP initiation is likely the key factor contributing to slow AP delays in GABAergic synapse. Several lines of evidence indicate that persistent Na+ current INap contributes to the amplification of GABA-PSPs and excitatory action of GABA. INap is present in the immature CA3 pyramidal cells (McBain and Dingledine, 1992) and promotes their spontaneous bursting (Sipila et al., 2005, 2006). Activation threshold of INap is near −60 mV (Sipila et al., 2006), that is close to the depolarization attained by GABA-PSPs. In adult CA1 pyramidal cells, which also express INap (Azouz et al., 1996), small glutamatergic EPSPs evoked from subthreshold potentials initiate firing with long and variable latencies due to INap-mediated plateau potentials that amplify and prolong EPSPs (Fricker and Miles, 2002). Thus, present findings significantly modify traditional view on GABAergic excitation which assumed that GABA reaches APthr and directly activates APs (Ben Ari, 2002; Ben Ari et al., 2007). It appears that in P2–5 CA3 pyramidal cells, GABAergic depolarization per se typically does not attain APthr, and that amplification of GABA-PSPs by INap is required for postsynaptic excitation. That GDPs are inhibited by INap blockers phenytoin and riluzole (Sipila et al., 2006) is in agreement with these findings. Interestingly, similar mechanisms may operate in the immature neocortex, in which delays of GABA-evoked APs are also long and variable, and EGABA barely attains APthr (Rheims et al., 2008). Variability of AP delays, which is also observed using simGABA-PSPs, likely reflects dynamic changes in the state of the voltage-gated conductances (INap, potassium channels, Ih, etc.). At the level of population, variability is multiplied by heterogeneity in [Cl−]i that also strongly affects AP delays. However, we do not claim that amplification of GABA-PSPs by intrinsic membrane conductances is a general mechanism required for GABAergic excitation. It is plausible that a situation with EGABA ≥ APthr may also take place, for instance in cells with very positive EGABA at very early developmental stages (LoTurco et al., 1995) or as a result of epileptogenic process (Cohen et al., 2002; Khalilov et al., 2003; Huberfeld et al., 2007), trauma (van den Pol et al., 1996; Pieraut et al., 2007), or in the neurons with relatively negative APthr, such as neocortical interneurons (Rheims et al., 2008).
Present results suggest that slow and variable AP delays at GABAergic synapses have major impact on synchronization of neuronal network during generation and propagation of GDPs. Decrease of depolarizing driving force for GABA by low-bumetanide desynchronized neuronal network during generation of GDPs. On the other hand, reduction of GABAergic conductance with low-bicuculline and its augmentation with diazepam increased and decreased neuronal synchronization during GDPs, respectively. In keeping with the major contribution of GABAergic conductance to GDPs (Ben-Ari et al., 1989; Khazipov et al., 1997; Leinekugel et al., 1997; Strata et al., 1997), slow and variable AP delays at GABAergic synapses seem to play an important role in determining the level of neuronal synchrony during GDPs and the rate of their propagation. Yet GABA is not the only factor contributing to neuronal excitation during GDPs. Less prominent, but possessing stronger driving force glutamatergic component provided by recurrent collateral synapses plays equally important role in GDPs generation (Khazipov et al., 1997; Leinekugel et al., 1997; Bolea et al., 1999), as glutamate receptor antagonists reduce neuronal synchrony, according to present study, and block GDPs when glutamate receptors are fully blocked (Ben-Ari et al., 1989). After partial blockade of GABA receptors, increase in neuronal synchronization is likely determined by glutamatergic synapses which provide faster and less variable conduction of excitation than GABAergic synapses. The difference in excitation provided by GABA and glutamate is likely due to the difference in the level of depolarization, which is secondary to the amount and driving force of these two conductances. Small glutamatergic EPSPs evoked from just subthreshold potentials are amplified by INap-mediated plateau potentials and evoke postsynaptic spikes with long and variable delays in adult pyramidal cells (Fricker and Miles, 2002), that is similar to the present observations with GABA-PSPs. Because glutamate-driving force is close to 0 mV, increase in glutamatergic conductance provides stronger depolarization which exceeds APthr and postsynaptic cells fire APs at short delays with little variability. By an order higher input resistance and smaller capacitance of the immature neurons (Tyzio et al., 2003) also should be taken into account, because both factors should provide larger depolarization in response to even weak input. In contrast, GABAergic depolarization is limited by small driving force, and its level can not exceed EGABA despite of the large GABAergic conductance activated during GDPs. GABAergic depolarization also potentiates NMDA receptors via attenuation of their voltage-dependent magnesium block, and in keeping with their slow kinetics, NMDA receptor mediated depolarization could be an additional factor contributing to delayed firing (Khazipov et al., 1995; Leinekugel et al., 1997).
Present results suggest that slow and variable excitation in GABAergic synapses is an important factor controlling spike timing during GDPs. By providing a coincidence between pre- and postsynaptic neurons, GDPs are thought to be involved in activity-dependent plasticity in the hippocampal network (Kasyanov et al., 2004; Mohajerani and Cherubini, 2006). Synaptic plasticity is particularly robust during the early developmental period (Isaac et al., 1995; Durand et al., 1996), and similarly to adult forms of plasticity, it depends on timing of firing in pre- and postsynaptic neurons both in glutamatergic and GABAergic synapses. Because all neurons fire action potentials during GDPs, entire network is activated and all synaptic inputs are imposed to plasticity, which direction (potentiation or depression) should depend on the spike timing in the synaptically coupled neurons. In the future studies it would also be important to determine the role of slow spike timing at GABAergic synapses in the generation of the early patterns of hippocampal activity in the intact animals in vivo.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Financial support from EU (EC contract number LSH-CT-2006-037315 (EPICURE) FP6 -Thematic priority LIFESCIHEALTH), INSERM, ANR, FRM, LFCE, Rothschild/Inserm “interface” program and “co-tutelle” grant by the French Government and RFBR are acknowledged.
References
Andreasen, M., Lambert, J. D. C., and Jensen, M. S. (1989). Effects of new non-N-methyl-D-aspartate antagonists on synaptic transmission in the in vitro rat hippocampus. J. Physiol. (Lond.) 414, 317–336.
Azouz, R., Jensen, M. S., and Yaari, Y. (1996). Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J. Physiol (Lond.) 492, 211–223.
Ben Ari, Y. (2002). Excitatory actions of GABA during development: the nature of the nurture. Nat. Rev. Neurosci. 3, 728–739.
Ben-Ari, Y., Cherubini, E., Corradetti, R., and Gaïarsa, J.-L. (1989). Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. (Lond.) 416, 303–325.
Ben Ari, Y., Gaiarsa, J. L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284.
Bolea, S., Avignone, E., Berretta, N., Sanchez-Andres, J. V., and Cherubini, E. (1999). Glutamate controls the induction of GABA-mediated giant depolarizing potentials through AMPA receptors in neonatal rat hippocampal slices. J. Neurophysiol. 81, 2095–2102.
Brickley, S. G., Cull-Candy, S. G., and Farrant, M. (1996). Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J. Physiol. (Lond.) 497, 753–759.
Cohen, I., and Miles, R. (2000). Contributions of intrinsic and synaptic activities to the generation of neuronal discharges in in vitro hippocampus. J. Physiol. (Lond.) 524, 485–502.
Cohen, I., Navarro, V., Clemenceau, S., Baulac, M., and Miles, R. (2002). On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science 298, 1418–1421.
Davies, C. H., Davies, S. N., and Collingridge, G. L. (1990). Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. J. Physiol. (Lond.) 424, 513–531.
De la Prida, L. M., Bolea, S., and Sanchez-Andres, J. V. (1998). Origin of the synchronized network activity in the rabbit developing hippocampus. Eur. J. Neurosci. 10, 899–906.
Durand, G. M., Kovalchuk, Y., and Konnerth, A. (1996). Long-term potentiation and functional synapse induction in developing hippocampus. Nature 381, 71–75.
Dzhala, V. I., Talos, D. M., Sdrulla, D. A., Brumback, A. C., Mathews, G. C., Benke, T. A., Delpire, E., Jensen, F. E., and Staley, K. J. (2005). NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 11, 1205–1213.
Fricker, D., and Miles, R. (2000). EPSP amplification and the precision of spike timing in hippocampal neurons. Neuron 28, 559–569.
Fricker, D., and Miles, R. (2002). EPSP amplification and the precision of spike timing in hippocampal neurons. Neuron 28, 559–569.
Fricker, D., Verheugen, J. A., and Miles, R. (1999). Cell-attached measurements of the firing threshold of rat hippocampal neurones. J. Physiol. (Lond.) 517, 791–804.
Gaiarsa, J. L., McLean, H., Congar, P., Leinekugel, X., Khazipov, R., Tseeb, V., and Ben-Ari, Y. (1995). Postnatal maturation of gamma-aminobutyric acidA and B-mediated inhibition in the CA3 hippocampal region of the rat. J. Neurobiol. 26, 339–349.
Hollrigel, G. S., Ross, S. T., and Soltesz, I. (1998). Temporal patterns and depoarizing actions of spontaneous GABAA receptor activation in granule cells of the early postnatal dentate gyrus. J. Neurophysiol. 80, 2340–2351.
Huberfeld, G., Wittner, L., Clemenceau, S., Baulac, M., Kaila, K., Miles, R., and Rivera, C. (2007). Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 27, 9866–9873.
Isaac, J. T., Nicoll, R. A., and Malenka, R. C. (1995). Evidence for silent synapses: implications for the expression of LTP. Neuron 15, 427–434.
Kasyanov, A. M., Safiulina, V. F., Voronin, L. L., and Cherubini, E. (2004). GABA-mediated giant depolarizing potentials as coincidence detectors for enhancing synaptic efficacy in the developing hippocampus. Proc. Natl. Acad. Sci. U.S.A. 101, 3967–3972.
Khalilov, I., Holmes, G. L., and Ben Ari, Y. (2003). In vitro formation of a secondary epileptogenic mirror focus by interhippocampal propagation of seizures. Nat. Neurosci. 6, 1079–1085.
Khazipov, R., Bregestovski, P., and Ben-Ari, Y. (1993). Hippocampal inhibitory interneurons are functionally disconnected from excitatory inputs by anoxia. J. Neurophysiol. 70, 2251–2259.
Khazipov, R., Esclapez, M., Caillard, O., Bernard, C., Khalilov, I., Tyzio, R., Hirsch, J., Dzhala, V., Berger, B., and Ben-Ari, Y. (2001). Early development of neuronal activity in the primate hippocampus in utero. J. Neurosci. 21, 9770–9781.
Khazipov, R., Khalilov, I., Tyzio, R., Morozova, E., Ben Ari, Y., and Holmes, G. L. (2004). Developmental changes in GABAergic actions and seizure susceptibility in the rat hippocampus. Eur. J. Neurosci. 19, 590–600.
Khazipov, R., Leinekugel, X., Khalilov, I., Gaïarsa, J.-L., and Ben-Ari, Y. (1997). Synchronization of GABAergic interneuronal network in CA3 subfield of neonatal rat hippocampal slices. J. Physiol. (Lond.) 498, 763–772.
Khazipov, R., Ragozzino, D., and Bregestovski, P. (1995). Kinetics and Mg2+ block of N-methyl-D-aspartate receptor channels during postnatal development of hippocampal CA3 pyramidal neurons. Neuroscience 69, 1057–1065.
Kuo, C. C., and Bean, B. P. (1994). Na+ channels must deactivate to recover from inactivation. Neuron 12, 819–829.
Lamsa, K., Palva, J. M., Ruusuvuori, E., Kaila, K., and Taira, T. (2000). Synaptic GABA(A) activation inhibits AMPA-kainate receptor-mediated bursting in the newborn (P0-P2) rat hippocampus. J. Neurophysiol. 83, 359–366.
Leinekugel, X., Khalilov, I., Ben-Ari, Y., and Khazipov, R. (1998). Giant depolarizing potentials: the septal pole of the hippocampus paces the activity of the developing intact septohippocampal complex in vitro. J. Neurosci. 18, 6349–6357.
Leinekugel, X., Medina, I., Khalilov, I., Ben-Ari, Y., and Khazipov, R. (1997). Ca2+ oscillations mediated by the synergistic excitatory actions of GABAA and NMDA receptors in the neonatal hippocampus. Neuron 18, 243–255.
LoTurco, J. J., Owens, D. F., Heath, M. J., Davis, M. B., and Kriegstein, A. R. (1995). GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 15, 1287–1298.
McBain, C., and Dingledine, R. (1992). Dual-component miniature excitatory synaptic currents in rat hippocampal CA3 pyramidal neurons. J. Neurophysiol. 68, 16–27.
Menendez de la, P. L., and Sanchez-Andres, J. V. (2000). Heterogenous populations of cells mediate spontaneous synchronous bursting in the developing hippocampus though a frequency-dependent mechanism. Neuroscience 97, 227–241.
Menendez, de la, P. L., Bolea, S., and Sanchez-Andres, J. V. (1996). Analytical characterization of spontaneous activity evolution during hippocampal development in the rabbit. Neurosci. Lett. 218, 185–187.
Menendez, de la, P. L., Bolea, S., and Sanchez-Andres, J. V. (1998). Origin of the synchronized network activity in the rabbit developing hippocampus. Eur. J. Neurosci. 10, 899–906.
Miles, R., and Wong, R. K. S. (1986). Excitatory synaptic interactions between CA3 neurones in the guinea-pig hippocampus. J. Physiol. (Lond.) 373, 397–418.
Miles, R., and Wong, R. K. S. (1987). Inhibitory control of local excitatory circuits in the guinea- pig hippocampus. J. Physiol. (Lond.) 388, 611–629.
Mohajerani, M. H., and Cherubini, E. (2006). Role of giant depolarizing potentials in shaping synaptic currents in the developing hippocampus. Crit. Rev. Neurobiol. 18, 13–23.
Nardou, R., Ben-Ari, Y., and Khalilov, I. (2009). Bumetanide, an NKCC1 antagonist, does not prevent formation of epileptogenic focus but blocks epileptic focus seizures in immature rat hippocampus. J. Neurophysiol. 101, 2878–2888.
Payne, J. A., Rivera, C., Voipio, J., and Kaila, K. (2003). Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 26, 199–206.
Pieraut, S., Laurent-Matha, V., Sar, C., Hubert, T., Mechaly, I., Hilaire, C., Mersel, M., Delpire, E., Valmier, J., and Scamps, F. (2007). NKCC1 phosphorylation stimulates neurite growth of injured adult sensory neurons. J. Neurosci. 27, 6751–6759.
Rheims, S., Minlebaev, M., Ivanov, A., Represa, A., Khazipov, R., Holmes, G. L., Ben Ari, Y., and Zilberter, Y. (2008). Excitatory GABA in rodent developing neocortex in vitro. J. Neurophysiol. 100, 609–619.
Segal, M. M., and Douglas, A. F. (1997). Late sodium channel openings underlying epileptiform activity are preferentially diminished by the anticonvulsant phenytoin. J. Neurophysiol. 77, 3021–3034.
Sipila, S. T., Huttu, K., Soltesz, I., Voipio, J., and Kaila, K. (2005). Depolarizing GABA acts on intrinsically bursting pyramidal neurons to drive giant depolarizing potentials in the immature hippocampus. J. Neurosci. 25, 5280–5289.
Sipila, S. T., Huttu, K., Voipio, J., and Kaila, K. (2006). Intrinsic bursting of immature CA3 pyramidal neurons and consequent giant depolarizing potentials are driven by a persistent Na current and terminated by a slow Ca-activated K current. Eur. J. Neurosci. 23, 2330–2338.
Sipila, S. T., Huttu, K., Yamada, J., Afzalov, R., Voipio, J., Blaesse, P., and Kaila, K. (2009). Compensatory enhancement of intrinsic spiking upon NKCC1 disruption in neonatal hippocampus. J. Neurosci. 29, 6982–6988.
Strata, F., Atzori, M., Molnar, M., Ugolini, G., Tempia, F., and Cherubini, E. (1997). A pacemaker current in dye-coupled hilar interneurons contributes to the generation of giant GABAergic potentials in developing hippocampus. J. Neurosci. 17, 1435–1446.
Stuart, G., Schiller, J., and Sakmann, B. (1997). Action potential initiation and propagation in rat neocortical pyramidal neurons. J. Physiol. (Lond.) 505, 617–632.
Tyzio, R., Cossart, R., Khalilov, I., Minlebaev, M., Hubner, C. A., Represa, A., Ben Ari, Y., and Khazipov, R. (2006). Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science 314, 1788–1792.
Tyzio, R., Holmes, G. L., Ben-Ari, Y., and Khazipov, R. (2007). Timing of the developmental switch in GABA(A) mediated signalling from excitation to inhibition in CA3 rat hippocampus using gramicidin perforated patch and extracellular recordings. Epilepsia 48, 96–105.
Tyzio, R., Ivanov, A., Bernard, C., Holmes, G. L., Ben Ari, Y., and Khazipov, R. (2003). Membrane potential of CA3 hippocampal pyramidal cells during postnatal development. J. Neurophysiol. 90, 2964–2972.
Tyzio, R., Minlebaev, M., Rheims, S., Ivanov, A., Jorquera, I., Holmes, G. L., Zilberter, Y., Ben Ari, Y., and Khazipov, R. (2008). Postnatal changes in somatic gamma-aminobutyric acid signalling in the rat hippocampus. Eur. J. Neurosci. 27, 2515–2528.
Tyzio, R., Represa, A., Jorquera, I., Ben-Ari, Y., Gozlan, H., and Aniksztejn, L. (1999). The establishment of GABAergic and glutamatergic synapses on CA1 pyramidal neurons is sequential and correlates with the development of the apical dendrite. J. Neurosci. 19, 10372–10382.
van den Pol, A. N., Obrietan, K., and Chen, G. (1996). Excitatory actions of GABA after neuronal trauma. J. Neurosci. 16, 4283–4292.
Whittington, M. A., Traub, R. D., and Jefferys, J. G. R. (1995). Erosion of inhibition contributes to the progression of low magnesium bursts in rat hippocampal slices. J. Physiol. (Lond.) 486, 723–734.
Keywords: gamma aminobutyric acid, development, neonatal, hippocampus
Citation: Valeeva G, Abdullin A, Tyzio R, Skorinkin A, Nikolski E, Ben-Ari Y and Khazipov R (2010) Temporal coding at the immature depolarizing GABAergic synapse. Front. Cell. Neurosci. 4:17. doi: 10.3389/fncel.2010.00017
Received: 22 February 2010;
Paper pending published: 26 March 2010;
Accepted: 27 April 2010;
Published online: 14 July 2010
Edited by:
Enrico Cherubini, International School for Advanced Studies, ItalyReviewed by:
Enrico Cherubini, International School for Advanced Studies, ItalyIvan Soltesz, University of California at Irvine, USA
Copyright: © 2010 Valeeva, Abdullin, Tyzio, Skorinkin, Nikolski, Ben-Ari and Khazipov. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Rustem Khazipov, Institut de Neurobiologie de la Méditerranée-Institut National de la Santé et de la Recherche Médicale U901, 163 route de Luminy, Marseille 13273, France. e-mail:a2hhemlwb3ZAaW5tZWQudW5pdi1tcnMuZnI=