 Nicola Maggio1,2*
Nicola Maggio1,2* Menahem Segal1*
Menahem Segal1*
- 1 Department of Neurobiology, The Weizmann Institute of Science, Rehovot, Israel
- 2 Talpiot Medical Leadership Program, Department of Neurology, The Joseph Sagol Neuroscience Center, The Chaim Sheba Medical Center, Tel HaShomer, Israel
Several new observations have shifted the view of the hippocampus from a structure in charge of cognitive processes to a brain area that participates in the formation of emotional memories, in addition to its role in cognition. Specifically, while the dorsal hippocampus is involved in the processing of cognitive memories; the ventral sector is mainly associated with the control of behavioral inhibition, stress, and emotional memory. Stress is likely to cause this switch in control of hippocampal functions by modulating synaptic plasticity in the dorsal and ventral sectors of the hippocampus through the differential activation of mineralocorticosteroid or glucocorticosteroid receptors. Herein, we will review the effects of stress hormones on synaptic plasticity in the hippocampus and outline the outcomes on stress-related global functions of this structure. We propose that steroid hormones act as molecular switches: by changing the strength of synaptic connectivity in the hippocampus following stress, they regulate the routes by which the hippocampus is functionally linked to the rest of the brain. This hypothesis has profound implications for the pathophysiology of psychiatric disorders.
Introduction
Steroid hormones have been traditionally associated with regulation of peripheral organs, associated with stress (corticosterone) or with gonadal function (estrogen and androgens). Over the years, it became evident that these hormones also act within the hypothalamus, in a feedback regulatory loop, to affect the release of the neural factors that modulate production of the steroid hormones. More recently, several observations have elucidated new roles of steroid hormones in modulating higher CNS functions. Specifically, both stress and steroid hormones have been shown to affect synaptic receptors and ion channels and therefore regulate in several different ways synaptic transmission and neuronal plasticity. Consequently, stress hormones have been implicated in processes ranging from homeostatic to cognitive functions. Furthermore, in some disorders of the nervous system, hormones have been shown to play critical roles: favoring or halting the disease process. Thus, the interaction between peripheral hormones and central networks seem to be more intense than ever imagined before.
In the present study we review current knowledge on the effects of steroid hormones on synaptic plasticity and define their influence on hippocampal cognitive and emotional functions.
The Different Families of Corticosteroid Receptors in the Brain
Following the exposure to stressful stimuli, the steroid hormone corticosterone (cortisol in humans) is released from the adrenal glands in order to set up the best response to the challenge by acting on steroid receptors (de Kloet et al., 2005). These are distributed throughout the body and have a particularly dense distribution in the CNS (de Kloet et al., 2005). In the brain, the cellular and molecular targets for the action of corticosterone include, in addition to basic metabolic processes, an effect on excitatory (Karst and Joels, 2005) and inhibitory (Maggio and Segal, 2009a) synaptic transmission, as well as an effect on voltage-gated calcium channels (VGCC; Karst et al., 2000; Chameau et al., 2007). These effects are mediated by the activation of mineralocorticoid receptors (MRs) and glucocorticoid receptors (GRs; Joels, 1999, 2008; de Kloet et al., 2005). Initially, it was suggested that both receptors act as nuclear transcription factors that modify protein synthesis and produce a slow, persistent change in the function of the cell (de Kloet et al., 1993; Joels, 2001, 2008). More recently, the existence of a new family of membrane-bound MR and GR (mMR and mGR, respectively), which act through novel non-genomic pathways, has been reported (Karst et al., 2005; de Kloet et al., 2008). In this route, mMR and mGR can rapidly affect ionic conductances and thereby modify cell excitability and function (Karst et al., 2005; de Kloet et al., 2008). These membrane-bound receptors appear to differ from their intracellular cognates, not only in their location on the cell membrane, but also in their molecular structures (Joels et al., 2008), in their affinities for corticosterone, and in their downstream mechanisms of action which involve activation of G proteins (Joels et al., 2008). Specifically, intracellular MR (iMR) have a very high affinity for corticosterone and are highly expressed in all hippocampal subfields, as well as in cells of the central amygdala, lateral septum, and some motor nuclei in the brainstem (Joels, 2006). Intracellular GR (iGR) have a relatively low affinity, are widely distributed throughout the brain, and are expressed both in neurons and in glia (Joels, 2006). Consequently, it has been proposed that iMR hardly participate, if at all, in the fast response to stressful stimuli, due to their characteristic of being already saturated by the low ambient levels of corticosterone at rest (Joels, 2006, 2008). Conversely, iGR have been reported to become gradually activated by rising levels of corticosterone following a stressful event (Joels, 2006, 2008; Figure 1A). Therefore, under physiological conditions, cells that coexpress both receptor types, such as principal cells in the CA1 region, the dentate gyrus (DG), and the central amygdala, will shift between predominant iMR activation and concurrent mMR and iGR activation (Joels and Krugers, 2007).
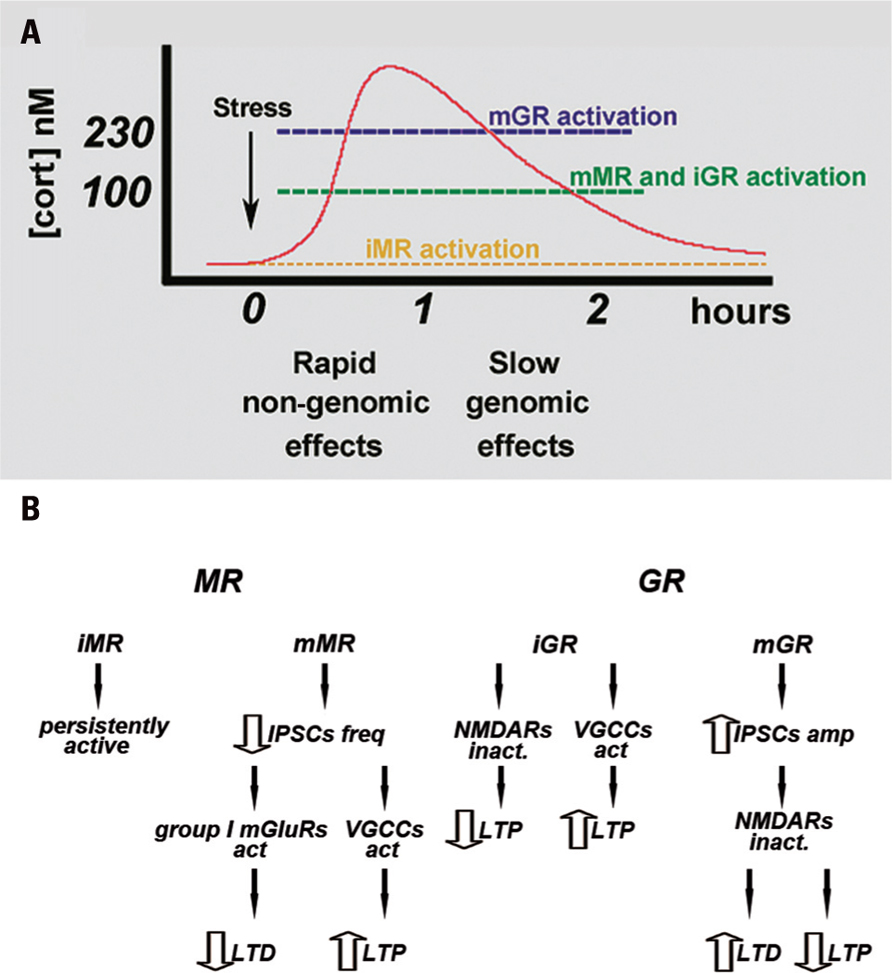
FIGURE 1. (A) Time course of MR and GR activation following stressful stimuli. At a resting level, iMR are already saturated by the baseline levels of corticosterone. Rising concentration of corticosterone activates both mMR and iGR, whereas an additional increase in corticosterone levels also activates mGR. mMR- and mGR-mediated effects appear in a faster time course than those mediated by the intracellular receptors. Modified from Maggio and Segal (2010). (B) Proposed mechanism by which corticosteroid receptors differently regulate LTP and LTD in the hippocampus. iMR are believed to be fully occupied at baseline level of corticosterone, therefore they might play a marginal role in synaptic plasticity. mMR might play a fundamental role in synaptic plasticity especially in VH: mMR activation reduces IPSC frequency. This determines an increase in the excitability of the pyramidal cells and raises the possibility of VGCC activation, thus enhancing LTP. In addition, a decrease in GABAergic inhibition can impair LTD through a group I mGluR-mediated mechanism. iGR are thought to both decrease NMDA-mediated LTP and increase VGCCs mediated LTP both in the hippocampus and amygdale. Their effects may occur at longer time scale due to their lower affinity to corticosterone. mGR might be involved in the regulation of synaptic plasticity mainly in DH: mGR activation increases IPSC amplitude and following hyperpolarization of the pyramidal cell membrane and inactivation of NMDA receptors, might impair LTP and enhance LTD. Modified from Maggio and Segal (2010).
The Roles of Corticosteroid Receptors in the Regulation of Hippocampal LTP
The identification of the molecular cascades linked to the effects of corticosteroids in the brain resulted in a series of studies examining the role of corticosterone in neuronal plasticity as well as in the cellular mechanisms underlying learning and memory such as long-term potentiation (LTP) and long-term depression (LTD; Bliss and Collingridge, 1993). Initial studies indicated that induction of LTP in the hippocampal area CA1 is impaired in a rat exposed to behavioral stress, such as inescapable shock (Foy et al., 1987; Shors et al., 1989). Administration of high doses of corticosterone either in vivo (Diamond et al., 1992) or in vitro (Pavlides et al., 1996; Alfarez et al., 2002) produced the same effects, indicating that corticosterone is likely to mediate this action of stress. Specifically, corticosterone-induced impairment of LTP seems to be due to the activation of iGR, which depresses NMDA receptor-dependent LTP (Krugers et al., 2005; Figure 1B). Conversely, it was also shown that LTP could be enhanced in the presence of low to moderate concentrations of corticosterone, while in absence of corticosterone LTP induction was impaired (Diamond et al., 1992). These studies show that the effects of corticosteroids on LTP induction are dose-dependent and follow an inverted U-shaped relationship (Diamond et al., 1992; Joels, 2006).
Further studies, however, have presented a more complex picture of the effects of steroids on synaptic plasticity. Specifically, it seems that the same dosage of corticosterone that impairs NMDA-dependent LTP can indeed enhance VGCC-dependent LTP (Krugers et al., 2005). This species of LTP is found in the amygdale where it is believed to underlie the formation of fear memories (Blair et al., 2001; Bauer et al., 2002) and can be evoked in the hippocampus as well (Borroni et al., 2000; Figure 1B). Interestingly, in the hippocampus, corticosterone appears to enhance VGCC LTP through an iGR-dependent mechanism (Krugers et al., 2005). It has been proposed that this effect requires a genomic pathway, as it occurs after a long delay between the exposure to stress and/or corticosterone and the recordings (Krugers et al., 2005), thus probably depending on the binding of GR homodimers to DNA that causes an increase in calcium currents (Karst and Joels, 2005; Chameau et al., 2007). Recent data from our group have shown that MRs are also able to enhance VGCC LTP (Maggio and Segal, 2007b): either stress or physiological concentrations of corticosterone can enhance LTP in the ventral hippocampus (VH), while inhibiting it in the dorsal hippocampus (DH; Maggio and Segal, 2007b). In particular, corticosterone enhances LTP through MRs since a selective MR agonist, aldosterone, shares the same effect in the VH (Maggio and Segal, 2007b). The proposed mechanism excludes an interaction between MR and NMDA receptors, as aldosterone by itself does not increase NMDA-dependent synaptic potentials (Maggio and Segal, 2007b). Conversely, MR-induced LTP can be blocked by nifedipine, suggesting that VGCCs are likely responsible for this effect (Maggio and Segal, 2007b; Figure 1B). It is likely that MR activates VGCC by modulating ionic conductances or changing VGCC activation kinetics. In vivo experiments have shown that MR activation is able to increase LTP in the DH as well (Avital et al., 2006). Specifically, animals which were injected with a GR antagonist prior to the stressful exposure, such that only MR could be activated by stress, show a much larger LTP than controls. In contrast, those animals previously injected with an MR antagonist and then exposed to stress, allowing only GR activation, show a much lower LTP than controls (Avital et al., 2006). These recordings were performed in the DG and even though there could be differences in the effects of stress and steroids between the DG and CA1 (Joels and Krugers, 2007), MRs were still shown to mediate an enhancement of LTP.
These experiments raise several issues. It could be argued that the experiments in the VH were conducted using an in vitro preparation where ambient corticosterone maintained normally through the circulation is washed out. Consequently, MRs are not occupied, and are ready to be activated by the superfused drug and produce LTP enhancement in the VH. This might not reflect the situation in the intact animal, where the brain is constantly exposed to fluctuating concentrations of corticosterone. In fact, MR should be already saturated by the resting concentration of corticosterone and should not respond to the stress-induced rise of corticosterone in the presence of a GR blockade. This, however, does not seem to be the case (Avital et al., 2006). Furthermore, even though both MR and GR are expressed in the VH, corticosterone action is mediated by activation of MR rather than GR. This reflects the observation that in the VH, MR concentration is double that of GR (Robertson et al., 2005). If so, according to the U-shaped curve model of corticosterone effects, MR should be saturated rapidly by the rising concentration of corticosterone and their effect should fade away faster in favor of the slower GR activation. This is in contrast with the experimental evidence. Altogether, it seems that the simple, dose-dependent, inverted, U-shaped curve does not fully explain the modulatory functions of MR and GR on LTP in the different sectors of the hippocampus, therefore calling for the involvement of other factors.
A possible mechanism that may clarify the MR-dependent enhancement of LTP should take into consideration the activation of mMR. These receptors act through a faster mechanism (de Kloet et al., 2008) and have lower affinities for corticosterone compared to their intracellular cognates (Joels, 2008) and similar to that of the iGR (Joels, 2008). In addition, MR activation enhances LTP in the VH within 1 h, too short time window to be accounted for by activation of genomic mechanisms (Joels and Krugers, 2007; Joels, 2008), but compatible with the faster time course of the non-genomic routes. Thus, mMR could be the preferential target for rising concentrations of corticosterone in the VH if one takes into account the similar affinities for corticosterone between mMR and iGR, and the denser distribution of the former over the latter (Robertson et al., 2005; Figure 1).
Mineralocorticoid receptors are likely to enhance LTP through activation of VGCC. In our experiments, we could not detect any effect of iGR on VGCC LTP. This could most likely be due to the shorter time window of observation in our experiments compared to those done by others (Krugers et al., 2005). In any case, both MR and GR were reported to increase VGCC LTP (Krugers et al., 2005; Maggio and Segal, 2007b). This apparent contrast could probably be explained by considering the different time courses of MR and GR enhancement of VGCC LTP. Specifically, MR has an earlier effect than GR and it could be that in the VH stress mediates a fast enhancement of LTP by MR followed by a second, slow increase in LTP due to GR activation. This proposal is compatible with the proposed role of the VH as a key player in the pathway that conveys stressful information to the hypothalamus and the amygdale so as to organize the stress response (Moser and Moser, 1998; Maggio and Segal, 2010; Segal et al., 2010).
Corticosteroid Regulation of Synaptic Plasticity Regulates Hippocampal Functions
The regulation of LTP by corticosterone in the hippocampus has profound system implications. Following stress, the quick MR-mediated increase in LTP facilitates the flow of the information related to stress from the VH to the ventral hypothalamus and other lower brain centers, so that the autonomic response to stress can be organized. Later on, the MR-mediated response fades away and the effect of GR dominates. As previously mentioned, GR enhancement of VGCC LTP has been shown to have a role in the formation of fear memories in the amygdale (Blair et al., 2001; Bauer et al., 2002). In this respect, GR could play the same function in the VH: the formation of the memory for the stressful event at the VH–amygdala pathway. Indeed, the evidence that MR and GR act on the same mechanism can have different purposes due to the time window of the respective outcomes that take place. Considering this, it could be interesting to study the relationship between the MR and GR responses in the VH.
In the DH, the reduction of LTP is mediated by GR (Maggio and Segal, 2007b). This effect seems to occur in less than 1 h, a relatively quick response that is unlikely to be mediated by a genomic mechanism. GR could reduce NMDA-mediated LTP either by a direct or an indirect mechanism. As far as it concerns the indirect mechanism hypothesis, we have demonstrated that a GR agonist, dexamethasone, increases IPSCs and mIPSCs amplitude in the DH within 10 min (Maggio and Segal, 2009a, 2012), consistent with the possible activation of mGR. Therefore, the increase in GABAA conductance could hyperpolarize the membrane, thus preventing the cell from reaching the threshold of depolarization that unlocks NMDA receptors from the Mg2+ block (Figure 1B). All in all, our experiments indicate that GR affect LTP through a fast, probably non-genomic mechanism. Even though this hypothesis needs to be explored further, the fast suppression of LTP in the DH can underlie the switch in the weight between the DH and VH; by reducing DH LTP and simultaneously enhancing LTP in the VH, the stressful stimuli could temporarily suppress the cognitive route of the hippocampus to cortical structures and enable the transmission of the emotional information through the VH to the amygdala.
Conversely, LTD induction is facilitated by behavioral stress, through a mechanism that requires GR (Pavlides et al., 1995; Xu et al., 1997, 1998) and their effect on NMDA receptors (Kim et al., 1996; Yang et al., 2005). We replicated previous experiments where both stress and corticosterone facilitate LTD through a GR-dependent mechanism in the DH, but we have also shown that LTD is impaired in the VH through a MR-dependent mechanism (Maggio and Segal, 2009b). Specifically in the latter case, LTD is transformed into a slow-onset LTP following the exposure to stressful stimulation (Maggio and Segal, 2009b). As is the case for LTP, changes in LTD either in the DH or VH were observed at approximately 1 h after the exposure to the stress, a time window that could be compatible with non-genomic mechanisms. The MR-induced conversion of LTD to LTP in the VH could be due to the activation of VGCC, which will further facilitate the ventral route to the amygdale (Figure 1B). Group I mGluR have been shown to enhance LTD in CA1 (Fitzjohn et al.,2001; Rammes et al., 2003), but, interestingly, they have been reported to induce a slow-onset potentiation in the DG (Manahan-Vaughan and Reymann, 1996). In a previous study, we showed that, in the VH, application of DHPG, a group I mGluR agonist, increases the population spike amplitude in response to a baseline stimulation (Maggio and Segal, 2007a). Taken together, these observations suggest that in the VH, a decrease in GABAergic inhibition can shift LTD to a slow-onset LTP through a group I mGluR-mediated mechanism (Figure 1B).
Conclusion
In conclusion, corticosteroid regulation of synaptic plasticity in the hippocampus is affected by several factors. An inverted U-shape effect of corticosterone partially explains the observed modulation of LTP. Indeed, this hypothesis mainly refers to the activation of intracellular corticosteroid receptors and does not take into account the contribution of membrane-bound steroid receptors. In fact, mMR, which bears a similar corticosterone affinity to that of iGR, will be activated at similar steroid concentrations. This implies that the effect of mMR appears earlier than that of iGR, thus inducing an enhancement of LTP instead of LTD. This might be the case in the VH. An additional factor to be considered is the distribution of MR and GR in specific brain areas, and the ratio of membrane-bound to intracellular receptors expressed therein. This is because at the same affinity value for corticosterone concentration, the receptor that is highly expressed will lead the effects on synaptic plasticity. The molecular structure of corticosterone receptors seems to be important. MRs, for example, exist in different molecular configurations (Joels, 2008), thus these receptors can be very diverse. This diversity in molecular structure could be linked to diverse intracellular pathways that differently influence neuronal functions. Another issue that has to be considered is the clusters of brain areas that are involved in a particular stress situation. Various brain regions have specific properties and are incorporated into unique networks, so that even if corticosterone evokes the same effect at the single cell level, this would not always result in the same effect on network functions such as LTP. For instance, both CA1 pyramidal neurons and granule cells in the DG highly express MR as well as GR (Joels, 2008). In the DH, corticosterone and stress consistently suppress the induction of CA1 LTP in vivo and in vitro, unlike the case for the DG. High concentration of corticosteroid (Pavlides et al., 1993) or tail shocks (Shors and Dryver, 1994) can indeed suppress LTP; however, in other situations, either no effect (Bramham et al., 1998; Gerges et al., 2001; Alfarez et al., 2003) or enhancement of LTP has been reported (Kavushansky et al., 2006). This is because LTP in the DG seems to be more dependent on indirect inputs from the amygdale (Akirav and Richter-Levin, 2002; Kavushansky and Richter-Levin, 2006). Finally, the response to a stressor is also determined by the history of the organism. For instance, the induction of LTP is impaired in animals that have been exposed to repetitive stress in the weeks prior to the experiment, even if corticosterone levels, at the time of LTP induction, are compatible with the expression of a normal LTP (Alfarez et al., 2003). Studies on the effect of maternal care on synaptic plasticity report that animals that received very little maternal care have poor LTP when they are adult, as opposed to animals that received high maternal care (Champagne et al., 2008). Interestingly, while LTP is suppressed by corticosterone in the latter group, it is enhanced in the former (Champagne et al., 2008). All in all, corticosteroid modulation of synaptic plasticity in the hippocampus seems to be more complex than previously thought and additional experiments are needed to address the role of membrane-bound as well as intracellular receptors on LTP/LTD regulation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akirav, I., and Richter-Levin, G. (2002). Mechanisms of amygdala modulation of hippocampal plasticity. J. Neurosci. 22, 9912–9921.
Alfarez, D. N., Joels, M., and Krugers, H. J. (2003). Chronic unpredictable stress impairs long-term potentiation in rat hippocampal CA1 area and dentate gyrus in vitro. Eur. J. Neurosci. 17, 1928–1934.
Alfarez, D. N., Wiegert, O., Joels, M., and Krugers, H. J. (2002). Corticosterone and stress reduce synaptic potentiation in mouse hippocampal slices with mild stimulation. Neuroscience 115, 1119–1126.
Avital, A., Segal, M., and Richter-Levin, G. (2006). Contrasting roles of corticosteroid receptors in hippocampal plasticity. J. Neurosci. 26, 9130–9134.
Bauer, E. P., Schafe, G. E., and Ledoux, J. E. (2002). NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J. Neurosci. 22, 5239–5249.
Blair, H. T., Schafe, G. E., Bauer, E. P., Rodrigues, S. M., and Ledoux, J. E. (2001). Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn. Mem. 8, 229–242.
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39.
Borroni, A. M., Fichtenholtz, H., Woodside, B. L., and Teyler, T. J. (2000). Role of voltage-dependent calcium channel long-term potentiation (LTP) and NMDA LTP in spatial memory. J. Neurosci. 20, 9272–9276.
Bramham, C. R., Southard, T., Ahlers, S. T., and Sarvey, J. M. (1998). Acute cold stress leading to elevated corticosterone neither enhances synaptic efficacy nor impairs LTP in the dentate gyrus of freely moving rats. Brain Res. 789, 245–255.
Chameau, P., Qin, Y., Spijker, S., Smit, A. B., and Joels, M. (2007). Glucocorticoids specifically enhance L-type calcium current amplitude and affect calcium channel subunit expression in the mouse hippocampus. J. Neurophysiol. 97, 5–14.
Champagne, D. L., Bagot, R. C., van Hasselt, F., Ramakers, G., Meaney, M. J., de Kloet, E. R., Joels, M., and Krugers, H. (2008). Maternal care and hippocampal plasticity: evidence for experience-dependent structural plasticity, altered synaptic functioning, and differential responsiveness to glucocorticoids and stress. J. Neurosci. 28, 6037–6045.
de Kloet, E. R., Joels, M., and Holsboer, F. (2005). Stress and the brain: from adaptation to disease. Nat. Rev. Neurosci. 6, 463–475.
de Kloet, E. R., Karst, H., and Joels, M. (2008). Corticosteroid hormones in the central stress response: quick-and-slow. Front. Neuroendocrinol. 29, 268–272.
de Kloet, E. R., Oitzl, M. S., and Joels, M. (1993). Functional implications of brain corticosteroid receptor diversity. Cell. Mol. Neurobiol. 13, 433–455.
Diamond, D. M., Bennett, M. C., Fleshner, M., and Rose, G. M. (1992). Inverted-U relationship between the level of peripheral corticosterone and the magnitude of hippocampal primed burst potentiation. Hippocampus 2, 421–430.
Fitzjohn, S. M., Palmer, M. J., May, J. E., Neeson, A., Morris, S. A., and Collingridge, G. L. (2001). A characterisation of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro. J. Physiol. 537, 421–430.
Foy, M. R., Stanton, M. E., Levine, S., and Thompson, R. F. (1987). Behavioral stress impairs long-term potentiation in rodent hippocampus. Behav. Neural Biol. 48, 138–149.
Gerges, N. Z., Stringer, J. L., and Alkadhi, K. A. (2001). Combination of hypothyroidism and stress abolishes early LTP in the CA1 but not dentate gyrus of hippocampus of adult rats. Brain Res. 922, 250–260.
Joels, M. (1999). Effects of corticosteroid hormones in the hippocampus. Acta Physiol. Scand. 167, A3.
Joels, M. (2006). Corticosteroid effects in the brain: U-shape it. Trends Pharmacol. Sci. 27, 244–250.
Joels, M. (2008). Functional actions of corticosteroids in the hippocampus. Eur. J. Pharmacol. 583, 312–321.
Joels, M., Karst, H., DeRijk, R., and de Kloet, E. R. (2008). The coming out of the brain mineralocorticoid receptor. Trends Neurosci. 31, 1–7.
Karst, H., Berger, S., Turiault, M., Tronche, F., Schutz, G., and Joels, M. (2005). Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc. Natl. Acad. Sci. U.S.A. 102, 19204–19207.
Karst, H., and Joels, M. (2005). Corticosterone slowly enhances miniature excitatory postsynaptic current amplitude in mice CA1 hippocampal cells. J. Neurophysiol. 94, 3479–3486.
Karst, H., Karten, Y. J., Reichardt, H. M., de Kloet, E. R., Schutz, G., and Joels, M. (2000). Corticosteroid actions in hippocampus require DNA binding of glucocorticoid receptor homodimers. Nat. Neurosci. 3, 977–978.
Kavushansky, A., and Richter-Levin, G. (2006). Effects of stress and corticosterone on activity and plasticity in the amygdala. J. Neurosci. Res. 84, 1580–1587.
Kavushansky, A., Vouimba, R. M., Cohen, H., and Richter-Levin, G. (2006). Activity and plasticity in the CA1, the dentate gyrus, and the amygdala following controllable vs. uncontrollable water stress. Hippocampus 16, 35–42.
Kim, J. J., Foy, M. R., and Thompson, R. F. (1996). Behavioral stress modifies hippocampal plasticity through N-methyl-D-aspartate receptor activation. Proc. Natl. Acad. Sci. U.S.A. 93, 4750–4753.
Krugers, H. J., Alfarez, D. N., Karst, H., Parashkouhi, K., Van Gemert, N., and Joels, M. (2005). Corticosterone shifts different forms of synaptic potentiation in opposite directions. Hippocampus 15, 697–703.
Maggio, N., and Segal, M. (2007a). Unique regulation of long term potentiation in the rat ventral hippocampus. Hippocampus 17, 10–25.
Maggio, N., and Segal, M. (2007b). Striking variations in corticosteroid modulation of long-term potentiation along the septotemporal axis of the hippocampus. J. Neurosci. 27, 5757–5765.
Maggio, N., and Segal, M. (2009a). Differential corticosteroid modulation of inhibitory synaptic currents in the dorsal and ventral hippocampus. J. Neurosci. 29, 2857–2866.
Maggio, N., and Segal, M. (2009b). Differential modulation of long-term depression by acute stress in the rat dorsal and ventral hippocampus. J. Neurosci. 29, 8633–8638.
Maggio, N., and Segal, M. (2010). Corticosteroid regulation of synaptic plasticity in the hippocampus. Sci. World J. 10, 462–469.
Maggio, N., and Segal, M. (2012). Stress and corticosteroid modulation of seizures and synaptic inhibition in the hippocampus. Exp. Neurol. 234, 200–207.
Manahan-Vaughan, D., and Reymann, K. G. (1996). Metabotropic glutamate receptor subtype agonists facilitate long-term potentiation within a distinct time window in the dentate gyrus in vivo. Neuroscience 74, 723–731.
Moser, M. B., and Moser, E. I. (1998). Functional differentiation in the hippocampus. Hippocampus 8, 608–619.
Pavlides, C., Kimura, A., Magarinos, A. M., and McEwen, B. S. (1995). Hippocampal homosynaptic long-term depression/depotentiation induced by adrenal steroids. Neuroscience 68, 379–385.
Pavlides, C., Ogawa, S., Kimura, A., and Mcewen, B. S. (1996). Role of adrenal steroid mineralocorticoid and glucocorticoid receptors in long-term potentiation in the CA1 field of hippocampal slices. Brain Res. 738, 229–235.
Pavlides, C., Watanabe, Y., and McEwen, B. S. (1993). Effects of glucocorticoids on hippocampal long-term potentiation. Hippocampus 3, 183–192.
Rammes, G., Palmer, M., Eder, M., Dodt, H. U., Zieglgansberger, W., and Collingridge, G. L. (2003). Activation of mGlu receptors induces LTD without affecting postsynaptic sensitivity of CA1 neurons in rat hippocampal slices. J. Physiol. 546(Pt. 2), 455–460.
Robertson, D. A., Beattie, J. E., Reid, I. C., and Balfour, D. J. (2005). Regulation of corticosteroid receptors in the rat brain: the role of serotonin and stress. Eur. J. Neurosci. 21, 1511–1520.
Segal, M., Richter-Levin, G., and Maggio, N. (2010). Stress-induced dynamic routing of hippocampal connectivity: a hypothesis. Hippocampus 20, 1332–1338.
Shors, T. J., and Dryver, E. (1994). Effect of stress and long-term potentiation (LTP) on subsequent LTP and the theta burst response in the dentate gyrus. Brain Res. 666, 232–238.
Shors, T. J., Seib, T. B., Levine, S., and Thompson, R. F. (1989). Inescapable versus escapable shock modulates long-term potentiation in the rat hippocampus. Science 244, 224–226.
Xu, L., Anwyl, R., and Rowan, M. J. (1997). Behavioural stress facilitates the induction of long-term depression in the hippocampus. Nature 387, 497–500.
Xu, L., Holscher, C., Anwyl, R., and Rowan, M. J. (1998). Glucocorticoid receptor and protein/RNA synthesis-dependent mechanisms underlie the control of synaptic plasticity by stress. Proc. Natl. Acad. Sci. U.S.A. 95, 3204–3208.
Keywords: hippocampus, synaptic plasticity, stress, LTP, corticosterone receptors
Citation: Maggio N and Segal M (2012) Steroid modulation of hippocampal plasticity: switching between cognitive and emotional memories. Front. Cell. Neurosci. 6:12. doi: 10.3389/fncel. 2012.00012
Received: 29 December 2011; Paper pending published: 07 February 2012;
Accepted: 09 March 2012; Published online: 20 March 2012.
Edited by:
Harmen J. Krugers, Universiteit van Amsterdam, NetherlandsReviewed by:
Oliver Von Bohlen Und Halbach, Universitätsklinikum Greifswald der Ernst-Moritz-Arndt-Universität, GermanyJean-Marc Goaillard, INSERM, France
Copyright: © 2012 Maggio and Segal. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Nicola Maggio and Menahem Segal, Department of Neurobiology, The Weizmann Institute of Science, 234 Herzl Street, Rehovot 76100, Israel. e-mail:bmljb2xhLm1hZ2dpb0BzaGViYS5oZWFsdGguZ292Lmls;bWVuYWhlbS5zZWdhbEB3ZWl6bWFubi5hYy5pbA==