 Abigail J. Hiller
Abigail J. Hiller Makoto Ishii
Makoto Ishii- 1Feil Family Brain and Mind Research Institute, Weill Cornell Medicine, Cornell University, New York, NY, United States
- 2Department of Neurology, Weill Cornell Medicine, Cornell University, New York, NY, United States
While cognitive decline and memory loss are the major clinical manifestations of Alzheimer’s disease (AD), they are now recognized as late features of the disease. Recent failures in clinical drug trials highlight the importance of evaluating and treating patients with AD as early as possible and the difficulties in developing effective therapies once the disease progresses. Since the pathological hallmarks of AD including the abnormal aggregation of amyloid-beta (Aβ) and tau can occur decades before any significant cognitive decline in the preclinical stage of AD, it is important to identify the earliest clinical manifestations of AD and elucidate their underlying cellular and molecular mechanisms. Importantly, metabolic and non-cognitive manifestations of AD such as weight loss and alterations of peripheral metabolic signals can occur before the onset of cognitive symptoms and worsen with disease progression. Accumulating evidence suggests that the major culprit behind these early metabolic and non-cognitive manifestations of AD is AD pathology causing dysfunction of the hypothalamus, a brain region critical for integrating peripheral signals with essential homeostatic physiological functions. Here, we aim to highlight recent developments that address the role of AD pathology in the development of hypothalamic dysfunction associated with metabolic and non-cognitive manifestations seen in AD. Understanding the mechanisms underlying hypothalamic dysfunction in AD could give key new insights into the development of novel biomarkers and therapeutic targets.
Introduction
Alzheimer’s disease (AD) is the leading cause of dementia in the elderly and remains an incurable and devastating neurodegenerative disease (Alzheimer’s Association, 2018). Though the exact pathogenesis of AD remains unclear, abnormal accumulation of amyloid-beta (Aβ) peptides and the microtubule-associated protein tau over time leads to neuronal and synaptic dysfunction and the neuropathological hallmarks of extracellular Aβ plaques and neurofibrillary tau tangles (Sala Frigerio and De Strooper, 2016). While cognitive symptoms are the most prominent feature of AD, they are now recognized as a late manifestation. Aβ and tau pathology can be detected by cerebrospinal fluid (CSF) analysis or positron emission tomography (PET) imaging in the preclinical stage of AD, decades prior to the cognitive impairment seen in mild cognitive impairment (MCI) or dementia (Dubois et al., 2016). Notably, various metabolic and non-cognitive manifestations of AD including weight loss and sleep and circadian rhythm disorders can precede the cognitive decline (Ishii and Iadecola, 2015). The hypothalamus, which serves as the brain’s integrator of peripheral metabolic signals and houses the central circadian pacemaker, the suprachiasmatic nucleus (SCN), is uniquely positioned to mediate many of these non-cognitive changes. Importantly, both Aβ and tau pathology have been found in the hypothalamus of AD brains (Table 1). Moreover, non-cognitive symptoms can worsen with disease progression and are associated with increased morbidity and mortality in AD, highlighting the importance of hypothalamic dysfunction in AD. In this review article, we discuss recent developments in understanding the relationship between AD pathobiology and select metabolic and non-cognitive manifestations that are putatively linked to hypothalamic dysfunction.

Table 1. Hypothalamus and Alzheimer’s disease (AD): select pathological and neuroimaging studies.
Body Weight and Systemic Metabolism
Late-Life Weight Loss: An Early Manifestation of AD
Weight loss has long been recognized as a clinical manifestation of AD and was considered a criteria consistent with the diagnosis of probable AD in the 1984 NINCDS-ADRDA work group report (McKhann et al., 1984). Importantly, weight loss in AD patients correlated with increased morbidity and mortality (White et al., 1998; Jang et al., 2015) and cortical Aβ load (Blautzik et al., 2018). Furthermore, MCI subjects who are underweight or lose weight have an increased risk for progressing to AD (Sobów et al., 2014; Joo et al., 2018). These studies collectively suggest that weight loss is an intrinsic feature of AD pathobiology.
While weight loss once dementia manifests could be attributed to impairments in appetite and eating behavior, epidemiological studies have found consistently that late-life weight loss can precede the cognitive decline in AD (Barrett-Connor et al., 1996; Buchman et al., 2005; Stewart et al., 2005; Johnson et al., 2006; Gao et al., 2011; Emmerzaal et al., 2015; Jimenez et al., 2017). Additionally, in a large community cohort study, late-life weight loss increased the risk for developing MCI regardless of mid-life body weight, suggesting that late-life weight loss is a clinical manifestation of early stages of AD regardless of mid-life metabolic risk factors (Alhurani et al., 2016). Furthermore, a recent study from the Dominantly Inherited Alzheimer Network (DIAN) found that asymptomatic carriers of gene mutations for autosomal dominant AD had significantly lower body mass index (BMI) compared to nonmutation carriers with weight loss starting more than a decade before onset of cognitive symptoms (Müller et al., 2017). Importantly, lower BMI was found to be associated with higher brain Aβ burden and lower scores on a delayed memory recall test in the asymptomatic AD mutation carriers.
Mounting evidence suggests that the adipocyte-derived hormone (adipokine) leptin is affected in AD. Leptin is produced in proportion to adiposity and serves as a critical negative afferent signal to the brain and in particular the hypothalamus to regulate body weight and systemic metabolism (Friedman, 2014). Low circulating leptin levels have been consistently found in AD subjects (Lieb et al., 2009; Bigalke et al., 2011; Khemka et al., 2014; Ma et al., 2016; Yu et al., 2018). While the underlying mechanisms for the early weight loss and low circulating leptin levels remain to be fully elucidated, dysfunction of the hypothalamus is likely to be a major driver. Compared to wild-type littermates, young transgenic mice with Aβ pathology (Tg2576 mice) prior to plaque formation or significant cognitive impairment exhibited low body weight/adiposity and low plasma leptin levels, which was associated with Aβ-mediated dysfunction of select hypothalamic neurons important for the regulation of body weight (Ishii et al., 2014). Taken together, these findings from animal and human studies raise the intriguing possibility that Aβ could interfere with hypothalamic sensors of peripheral metabolic signals such as leptin, preventing the brain from responding to signals of low body weight/adiposity resulting in pathologically low circulating leptin levels and unintentional weight loss.
Additionally, leptin is important for maintaining hippocampal structure and function (McGregor and Harvey, 2017) and exerting neuroprotective effects under a variety of neurotoxic conditions including Aβ (McGuire and Ishii, 2016). In humans, low circulating leptin levels have been associated with cognitive decline in the elderly (Holden et al., 2009) and decreased hippocampal gray matter volume (Narita et al., 2009). Similarly, studies using various transgenic mouse models of Aβ pathology have consistently found that leptin levels correlate positively with cognitive function and negatively with Aβ burden (Greco et al., 2010; Takeda et al., 2010; Pérez-González et al., 2014). Therefore, alterations in leptin signaling associated with unintentional weight loss may serve not only as a marker of early AD but may contribute to AD pathogenesis. While leptin has been the most extensively investigated adipokine in AD, a significant role for other adipokines and peripheral metabolic signals cannot be excluded (Kiliaan et al., 2014).
Mid-Life Obesity and Diabetes: Risk Factors for Developing AD
In contrast to late-life weight loss, mid-life obesity and related comorbid conditions including insulin resistance and type 2 diabetes mellitus (T2DM) have been found in several epidemiological studies to be risk factors for cognitive decline and AD (Arvanitakis et al., 2004; Kivipelto et al., 2005; Biessels et al., 2014; McGuire and Ishii, 2016). In contrast, a large population study in the UK found that mid-life obesity decreased risk for dementia (Qizilbash et al., 2015); however, this study may have potential confounding factors including reverse causation bias and ill-defined range of mid-life (Gustafson, 2015; Kivimäki et al., 2015). While additional studies are clearly needed, the current evidence suggests that age is an important factor when considering body weight and adiposity changes in AD with mid-life obesity being a risk factor and late-life weight loss being an early manifestation of AD.
In light of the association between mid-life obesity and T2DM and AD, it has been noted that these metabolic disorders cause damage to the hypothalamus by similar mechanisms to those seen in AD (Clarke et al., 2018). Physiological consequences of obesity and T2DM, including chronic hyperinsulinemia and high circulating levels of free fatty acids have been shown to lead to hypothalamic insulin and leptin resistance (Thon et al., 2016), ER stress (Zhang et al., 2008; Mayer and Belsham, 2010) and pro-inflammatory intracellular cascades (Milanski et al., 2009) in hypothalamic neurons. Similarly, Aβ oligomers induced TNF-alpha mediated inflammation and ER stress in cultured hypothalamic neurons and the hypothalamus of mice and macaques (Clarke et al., 2015). Furthermore, an NMR-based metabolomics study of the transgenic amyloid precursor protein/presenilin 1 (APP/PS1) mouse model of Aβ pathology found that these mice had significant hypothalamic metabolic abnormalities prior to memory impairment (Zheng et al., 2018). These studies provide further support that hypothalamus dysfunction can occur early in the development of AD and is likely mediated by mid-life metabolic risk factors of obesity and T2DM.
There is also substantial evidence to suggest that obesity and T2DM related pathologies could directly promote early AD pathology. Studies in mouse models and humans have shown that hyperinsulinemia and insulin resistance can increase Aβ load by interfering with clearance mechanisms and increasing production of Aβ (Stanley et al., 2016; Ramos-Rodríguez et al., 2017; Benedict and Grillo, 2018). Additionally, obesity and T2DM lead to increased deposition of human islet APP (hIAPP or amylin) in not only pancreatic islets but in the brain parenchyma and cerebrovascular system, which may exacerbate AD pathology by causing neurotoxicity and decreased Aβ clearance (Jackson et al., 2013; Wijesekara et al., 2017). The cross-seeding of misfolded hIAPP and Aβ peptides has been hypothesized as a mechanism for shared disease pathogenesis between AD and T2DM (Moreno-Gonzalez et al., 2017). However, not all studies show that obesity and T2DM worsens AD pathology. For example, a mouse model of human tau pathology given a high-fat, high-sugar and high-cholesterol diet had no significant changes in hippocampal and cortical tau pathology (Gratuze et al., 2016).
Sleep and Circadian Rhythm Disorders
Sleep Disorders
Sleep disorders affect 25%–66% of AD patients and are a leading cause for institutionalization (Bianchetti et al., 1995; Moran et al., 2005; Guarnieri et al., 2012). Importantly, sleep quality in AD declines early in the disease and worsens with disease progression (Vitiello et al., 1990; Liguori et al., 2014). Furthermore, cognitively normal subjects with Aβ deposition by CSF measurements had worse sleep quality compared to those without Aβ deposition (Ju et al., 2013). This association between AD pathology and poor sleep quality has been recapitulated in multiple mouse models with increased Aβ deposition (Wisor et al., 2005; Roh et al., 2012; Sethi et al., 2015). Additionally, a single intracerebroventricular (ICV) infusion of Aβ oligomers disrupted sleep patterns in mice (Kincheski et al., 2017). Taken together, these findings provide evidence that AD pathology impacts sleep early in AD and may occur prior to the onset of cognitive symptoms.
Accumulating evidence suggests that hypothalamic dysfunction is responsible for the sleep dysfunction in AD. A recent study found reduced hypothalamic glucose uptake, as measured by 18F-flurodeoxyglucose PET, in AD subjects compared to non-demented control subjects, which was associated with sleep impairment and CSF AD biomarkers (Liguori et al., 2017). Evidence also exists for the involvement of specific hypothalamic nuclei in the sleep dysfunction in AD. The intermediate nucleus of the hypothalamus, the putative analog to the ventrolateral preoptic nucleus (VLPN) in rodents, contains neurons that are active in both rapid eye movement (REM) and non-REM (NREM) sleep (Chung et al., 2017; Saper and Fuller, 2017). A decrease of galanin-positive neurons in the intermediate nucleus was reported in postmortem AD brains (Lim et al., 2014). Because these neurons are active during sleep and inhibit wake-promoting neurons, loss of VLPN galanin neurons presents a potential mechanism for decreased NREM sleep and increased awakenings in AD (Saper and Fuller, 2017).
Another important hypothalamic nucleus in the regulation of sleep is the lateral hypothalamic area (LH), which contains neurons that synthesize the neuropeptide orexin (hypocretin). Orexin is critical for the maintenance of sleep-wake architecture by promoting arousal with orexin deficiency resulting in narcolepsy (Tsujino and Sakurai, 2013). In human studies, there are conflicting reports regarding orexin levels in AD with multiple studies reporting unchanged or decreased CSF and hypothalamic levels (Fronczek et al., 2012; Schmidt et al., 2013; Liguori et al., 2014). In contrast, more recent studies suggest that accumulating AD pathology is associated with increased CSF orexin levels and sleep disruption. In AD biomarker-defined MCI subjects, increased CSF orexin levels were associated with REM sleep disruption and sleep fragmentation (Liguori et al., 2016). In another study, higher CSF orexin levels were found in biomarker-defined AD subjects compared to MCI and control groups (Gabelle et al., 2017).
Substantial support also exists for sleep dysfunction worsening AD pathology and increasing the risk for developing dementia (Mander et al., 2016). A recent meta-analysis found that sleep disorders such as insomnia and sleep-disordered breathing increased the risk for developing AD (Shi et al., 2018). Prolonged sleep duration in older adults was also associated with increased development of dementia (Westwood et al., 2017). Therefore, abnormal sleep, regardless of the duration, is associated with increased dementia risk. Additionally, human and animal studies have found that poor sleep quality including deprivation can worsen AD pathology. In healthy human adults, a single night of lost sleep was associated with an increased Aβ load as measured by CSF and brain PET studies (Ooms et al., 2014; Shokri-Kojori et al., 2018). Furthermore, several studies have found various measures of poor sleep quality were associated with increased brain Aβ load in cognitively normal individuals (Spira et al., 2013; Branger et al., 2016) Consistent with these human studies, sleep deprivation or increased wakefulness in a Drosophila or transgenic mouse model of Aβ pathology increased Aβ burden (Kang et al., 2009; Roh et al., 2014; Tabuchi et al., 2015). The underlying mechanism behind the association between sleep and Aβ pathology has been hypothesized to be due to increased clearance of Aβ during sleep (Xie et al., 2013) or neuronal activity-dependent increases in Aβ secretion during wakefulness (Cirrito et al., 2005; Tabuchi et al., 2015). Despite some conflicting studies, the current evidence supports a bidirectional relationship where AD pathology can cause increased orexin levels and disruption of sleep, while disruption of sleep can lead to increased AD pathology.
Circadian Rhythm Disorders and Sundowning
Closely related to sleep disorders, circadian rhythm abnormalities including disrupted day-night activity patterns are common in AD patients (Musiek et al., 2015). In particular, aggressive behaviors in AD are often temporally dependent, worsening in the afternoon and evening, in a pattern that is clinically termed Sundown Syndrome or “sundowning” (Khachiyants et al., 2011). Importantly, agitation such as seen with sundowning in AD patients can precede significant adverse outcomes including institutionalization, accelerated cognitive decline and increased caregiver burden (Canevelli et al., 2016). Yet, current strategies for managing aggressive symptoms rely on pharmacological interventions including anti-psychotics that may not target the underlying pathways affected and can have significant adverse effects (Ballard and Corbett, 2013). Therefore, understanding the underlying mechanisms of sundowning would be critical for improving the clinical care of AD patients.
The hypothalamus has long been recognized as a major regulator of both circadian rhythm and aggressive behaviors, suggesting a potential role in sundowning. Dysfunction in the hypothalamic SCN, the central pacemaker, is a likely mediator of circadian rhythm disorders in AD (Van Erum et al., 2018). In AD patients, the SCN shows increased aging-related atrophy and neurodegeneration with evidence for neurofibrillary tangle accumulation (Swaab et al., 1985; Stopa et al., 1999). Additionally, in postmortem AD brains, blunted fluctuations in circadian motor activity and increased SCN amyloid plaque burden are reported to be correlated with reduction of two central circadian neurotransmitters, vasopressin and neurotensin (Stopa et al., 1999; Harper et al., 2008; Hu et al., 2013), although one study reported no change in SCN vasopressin levels in AD (Wang et al., 2015). Similarly, the hypothalamus has been long implicated in the role of aggressive behaviors. In the early 20th century, electrical stimulation of specific regions of the hypothalamus including the LH and the VMH promoted aggression in cats (Hess and Akert, 1955). These areas of the hypothalamus have been classically identified as “attack areas, ” and their stimulation in a variety of animal species has been linked with distinct aggressive behaviors (Haller, 2013). A recent study identified a hypothalamic circuit involving projections from the SCN to the VMH that regulated the daily rhythm in aggression propensity of male mice (Todd et al., 2018), suggesting that disruption of this hypothalamic circuit could lead to sundowing in AD.
Molecular and genetic studies in animal models further support the hypothalamus and in particular the SCN playing a central role in circadian rhythm disorders associated with AD pathology. A mouse model of Aβ pathology was found to have dampened SCN excitability rhythms, concurrent with circadian-associated behavioral disturbances and reduced daytime A-type potassium currents (Paul et al., 2018). In contrast, several Aβ mouse and Drosophila models exhibit circadian behavioral abnormalities despite normal central clock function, suggesting that Aβ-related circadian abnormalities may also stem from a “central clock output failure” in which the SCN fails to entrain brain-resident and peripheral clocks (Chauhan et al., 2017).
Conclusions
We have briefly reviewed select recent findings on the metabolic and non-cognitive manifestations of AD that can occur before the cognitive decline and focused specifically on disorders of body weight, sleep and circadian rhythm. We provide evidence that these metabolic and non-cognitive manifestations of AD are due to hypothalamic dysfunction caused by AD pathology and can be bidirectional and feed-forward in nature (Figure 1). Furthermore, while body weight and sleep/circadian rhythm may appear to act independently from each other, they often share common neurotransmitters (e.g., orexin, galanin) and brain regions (e.g., VMH, LH) in the hypothalamus, which can be modulated by peripheral circulating factors such as leptin and glucose (Fang et al., 2012; Tsujino and Sakurai, 2013; McGuire and Ishii, 2016). Therefore, seemingly disparate clinical manifestations of AD may be due to alterations of common hypothalamic pathways affected early in AD.
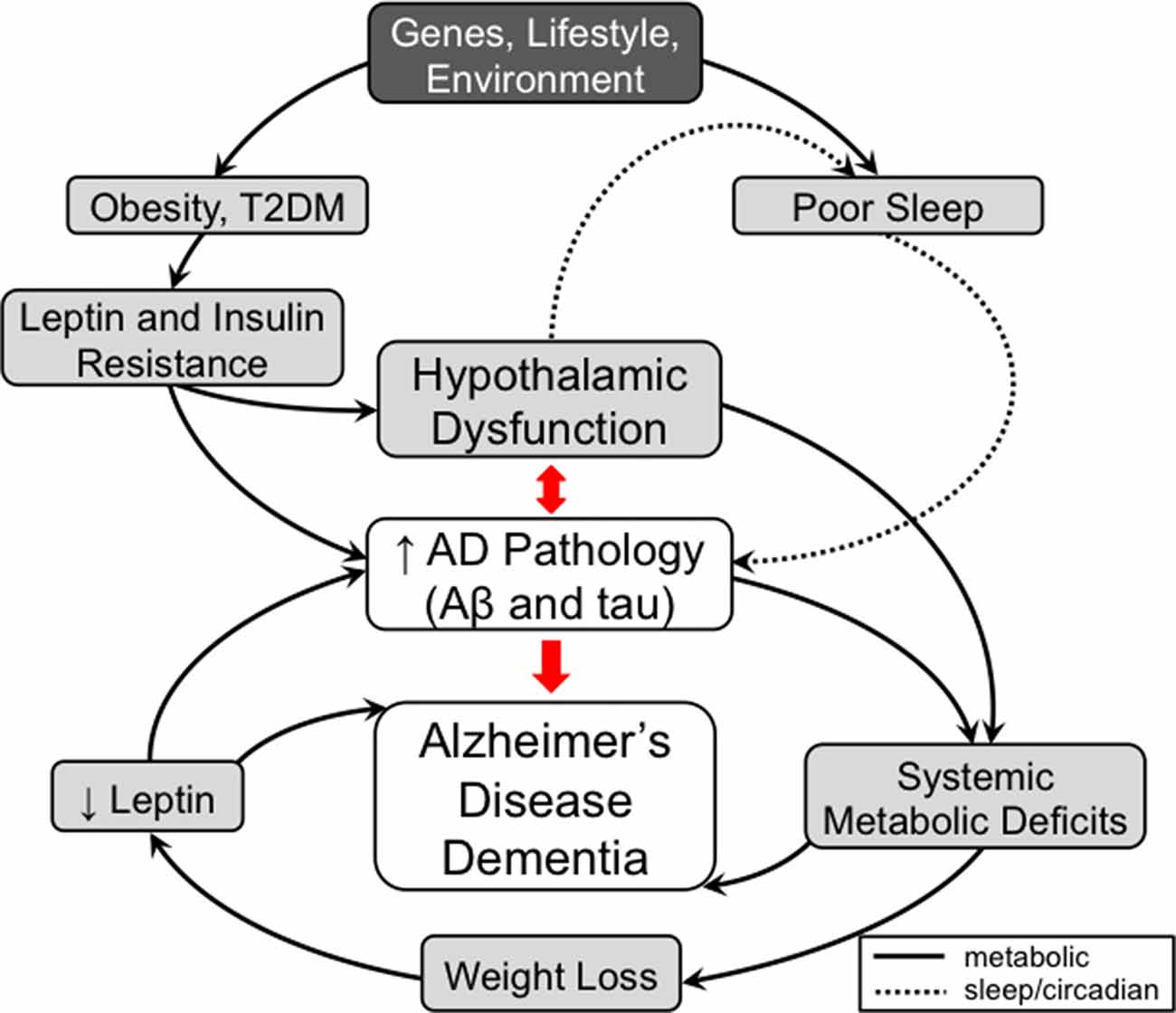
Figure 1. A model for the contribution of metabolic and non-cognitive factors (e.g., sleep/circadian rhythm) in the pathogenesis of Alzheimer’s disease (AD). In mid-life, obesity and type 2 diabetes mellitus (T2DM) are risk factors for AD. These conditions are associated with high circulating insulin and leptin levels leading to the development of hypothalamic dysfunction, including insulin and leptin resistance, as well as to worsening AD pathology directly. Development of hypothalamic insensitivity to peripheral metabolic signals in mid-life sets the stage for exacerbation of metabolic dysregulation in late-life AD, when accumulation of AD pathology can lead to further neuronal injury. A possible model to explain the correlation between late-life weight loss and AD posits that AD pathology-mediated neuronal injury in the hypothalamus leads to a hypermetabolic (catabolic) state, which results in weight loss and a pathologically low leptin state. As leptin has possible roles as a neuroprotective factor and a regulator of hippocampal structure and function, deficiency in leptin signaling could also contribute to cognitive impairment independent of hypothalamic signaling. Similar to metabolic dysfunction, sleep disorders play a role in AD pathogenesis. Poor sleep quality in mid-life has been associated with increased AD pathology. In late-life, hypothalamic dysfunction caused by AD pathology contributes to the sleep dysfunctions seen in AD. The worsening sleep disorders would then feed forward into the development of further AD pathology and eventually dementia. Therefore, disorders of metabolism and non-cognitive (e.g., sleep/circadian rhythm) factors mediated by hypothalamic dysfunction are both early risk factors and manifestations of AD that can contribute in a feed-forward manner that ultimately results in AD dementia. Solid lines represent metabolic pathways and dashed lines represent pathways related to sleep and circadian rhythms.
Despite recent advances, there are significant gaps in our knowledge. The hypothalamus is a complex brain region comprised of numerous distinct molecular cell types with each potentially a part of multiple different pathways. While several studies using a candidate-based approach have identified select individual cell types affected by AD pathology (Ishii et al., 2014; Clarke et al., 2015), the exact cell types affected in the hypothalamus are not known. Therefore, large unbiased molecular screens such as with Drop-Seq and similar approaches will likely be needed (Campbell et al., 2017). Additionally, once the cell types affected by AD pathology are identified, the exact cellular mechanisms leading to the dysfunction of those neurons and whether they are similar to those seen in more extensively studied brain regions such as the hippocampus need to be elucidated. Finally, any mechanistic studies in cellular or animal models needs to be validated and verified in carefully conducted AD biomarker-defined human studies.
While cognitive manifestations have deservedly received the bulk of the attention in AD research, non-cognitive manifestations are often correlated with disease progression, increased morbidity including institutionalization, and increased mortality. These non-cognitive signs and symptoms could be developed as inexpensive and readily accessible markers of AD progression in a clinical setting. Moreover, elucidating the underlying molecular mechanisms for these early clinical manifestations of AD may yield important insights into novel pathways affected in AD, which could lead to the development of important new therapeutic targets.
Author Contributions
All authors participated in the study design, drafted/revised the manuscript, approved the final version and agreed to be accountable for all the aspects of the work.
Funding
This study was supported by the BrightFocus Foundation (A2015485S) and National Institute on Aging, NIH (K08AG051179).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alhurani, R. E., Vassilaki, M., Aakre, J. A., Mielke, M. M., Kremers, W. K., Machulda, M. M., et al. (2016). Decline in weight and incident mild cognitive impairment: mayo clinic study of aging. JAMA Neurol. 73, 439–446. doi: 10.1001/jamaneurol.2015.4756
Alzheimer’s Association. (2018). 2018 Alzheimer’s disease facts and figures. Alzheimers Dement. 14, 367–429. doi: 10.1016/j.jalz.2018.02.001
Arvanitakis, Z., Wilson, R. S., Bienias, J. L., Evans, D. A., and Bennett, D. A. (2004). Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol. 61, 661–666. doi: 10.1001/archneur.61.5.661
Ballard, C., and Corbett, A. (2013). Agitation and aggression in people with Alzheimer’s disease. Curr. Opin. Psychiatry 26, 252–259. doi: 10.1097/YCO.0b013e32835f414b
Barrett-Connor, E., Edelstein, S. L., Corey-Bloom, J., and Wiederholt, W. C. (1996). Weight loss precedes dementia in community-dwelling older adults. J. Am. Geriatr. Soc. 44, 1147–1152. doi: 10.1111/j.1532-5415.1996.tb01362.x
Benedict, C., and Grillo, C. A. (2018). Insulin resistance as a therapeutic target in the treatment of Alzheimer’s disease: a state-of-the-art review. Front. Neurosci. 12:215. doi: 10.3389/fnins.2018.00215
Bianchetti, A., Scuratti, A., Zanetti, O., Binetti, G., Frisoni, G. B., Magni, E., et al. (1995). Predictors of mortality and institutionalization in Alzheimer disease patients 1 year after discharge from an Alzheimer dementia unit. Dementia 6, 108–112. doi: 10.1159/000106930
Biessels, G. J., Strachan, M. W. J., Visseren, F. L. J., Kappelle, L. J., and Whitmer, R. A. (2014). Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions. Lancet Diabetes Endocrinol. 2, 246–255. doi: 10.1016/s2213-8587(13)70088-3
Bigalke, B., Schreitmüller, B., Sopova, K., Paul, A., Stransky, E., Gawaz, M., et al. (2011). Adipocytokines and CD34 progenitor cells in Alzheimer’s disease. PLoS One 6:e20286. doi: 10.1371/journal.pone.0020286
Blautzik, J., Kotz, S., Brendel, M., Sauerbeck, J., Vettermann, F., Winter, Y., et al. (2018). Relationship between body mass index, ApoE4 status, and PET-based amyloid and neurodegeneration markers in amyloid-positive subjects with normal cognition or mild cognitive impairment. J. Alzheimers Dis. 65, 781–791. doi: 10.3233/JAD-170064
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/bf00308809
Branger, P., Arenaza-Urquijo, E. M., Tomadesso, C., Mézenge, F., André, C., de Flores, R., et al. (2016). Relationships between sleep quality and brain volume, metabolism, and amyloid deposition in late adulthood. Neurobiol. Aging 41, 107–114. doi: 10.1016/j.neurobiolaging.2016.02.009
Buchman, A. S., Wilson, R. S., Bienias, J. L., Shah, R. C., Evans, D. A., and Bennett, D. A. (2005). Change in body mass index and risk of incident Alzheimer disease. Neurology 65, 892–897. doi: 10.1212/01.wnl.0000176061.33817.90
Callen, D. J. A., Black, S. E., Caldwell, C. B., and Grady, C. L. (2004). The influence of sex on limbic volume and perfusion in AD. Neurobiol. Aging 25, 761–770. doi: 10.1016/j.neurobiolaging.2003.08.011
Callen, D. J., Black, S. E., Gao, F., Caldwell, C. B., and Szalai, J. P. (2001). Beyond the hippocampus: MRI volumetry confirms widespread limbic atrophy in AD. Neurology 57, 1669–1674. doi: 10.1212/wnl.57.9.1669
Campbell, J. N., Macosko, E. Z., Fenselau, H., Pers, T. H., Lyubetskaya, A., Tenen, D., et al. (2017). A molecular census of arcuate hypothalamus and median eminence cell types. Nat. Neurosci. 20, 484–496. doi: 10.1038/nn.4495
Canevelli, M., Valletta, M., Trebbastoni, A., Sarli, G., D’Antonio, F., Tariciotti, L., et al. (2016). Sundowning in dementia: clinical relevance, pathophysiological determinants, and therapeutic approaches. Front. Med. 3:73. doi: 10.3389/fmed.2016.00073
Chauhan, R., Chen, K.-F., Kent, B. A., and Crowther, D. C. (2017). Central and peripheral circadian clocks and their role in Alzheimer’s disease. Dis. Model Mech. 10, 1187–1199. doi: 10.1242/dmm.030627
Chung, S., Weber, F., Zhong, P., Tan, C. L., Nguyen, T. N., Beier, K. T., et al. (2017). Identification of preoptic sleep neurons using retrograde labelling and gene profiling. Nature 545, 477–481. doi: 10.1038/nature22350
Cirrito, J. R., Yamada, K. A., Finn, M. B., Sloviter, R. S., Bales, K. R., May, P. C., et al. (2005). Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron 48, 913–922. doi: 10.1016/j.neuron.2005.10.028
Clarke, J. R., Lyra e Silva, N. M., Figueiredo, C. P., Frozza, R. L., Ledo, J. H., Beckman, D., et al. (2015). Alzheimer-associated A oligomers impact the central nervous system to induce peripheral metabolic deregulation. EMBO Mol. Med. 7, 190–210. doi: 10.15252/emmm.201404183
Clarke, J. R., Ribeiro, F. C., Frozza, R. L., De Felice, F. G., and Lourenco, M. V. (2018). Metabolic dysfunction in Alzheimer’s disease: from basic neurobiology to clinical approaches. J. Alzheimers Dis. 64, S405–S426. doi: 10.3233/jad-179911
Cross, D. J., Anzai, Y., Petrie, E. C., Martin, N., Richards, T. L., Maravilla, K. R., et al. (2013). Loss of olfactory tract integrity affects cortical metabolism in the brain and olfactory regions in aging and mild cognitive impairment. J. Nucl. Med. 54, 1278–1284. doi: 10.2967/jnumed.112.116558
Dubois, B., Hampel, H., Feldman, H. H., Scheltens, P., Aisen, P., Andrieu, S., et al. (2016). Preclinical Alzheimer’s disease: definition, natural history, and diagnostic criteria. Alzheimers Dement. 12, 292–323. doi: 10.1016/j.jalz.2016.02.002
Emmerzaal, T. L., Kiliaan, A. J., and Gustafson, D. R. (2015). 2003–2013: a decade of body mass index, Alzheimer’s disease, and dementia. J. Alzheimers Dis. 43, 739–755. doi: 10.3233/JAD-141086
Fang, P., Yu, M., Guo, L., Bo, P., Zhang, Z., and Shi, M. (2012). Galanin and its receptors: a novel strategy for appetite control and obesity therapy. Peptides 36, 331–339. doi: 10.1016/j.peptides.2012.05.016
Friedman, J. (2014). 20 years of leptin: leptin at 20: an overview. J. Endocrinol. 223, T1–T8. doi: 10.1530/joe-14-0405
Fronczek, R., van Geest, S., Frölich, M., Overeem, S., Roelandse, F. W. C., Lammers, G. J., et al. (2012). Hypocretin (orexin) loss in Alzheimer’s disease. Neurobiol. Aging 33, 1642–1650. doi: 10.1016/j.neurobiolaging.2011.03.014
Gabelle, A., Jaussent, I., Hirtz, C., Vialaret, J., Navucet, S., Grasselli, C., et al. (2017). Cerebrospinal fluid levels of orexin-A and histamine, and sleep profile within the Alzheimer process. Neurobiol. Aging 53, 59–66. doi: 10.1016/j.neurobiolaging.2017.01.011
Gao, S., Nguyen, J. T., Hendrie, H. C., Unverzagt, F. W., Hake, A., Smith-Gamble, V., et al. (2011). Accelerated weight loss and incident dementia in an elderly African-American cohort. J. Am. Geriatr. Soc. 59, 18–25. doi: 10.1111/j.1532-5415.2010.03169.x
Gratuze, M., Julien, J., Morin, F., Calon, F., Hébert, S. S., Marette, A., et al. (2016). High-fat, high-sugar, and high-cholesterol consumption does not impact tau pathogenesis in a mouse model of Alzheimer’s disease-like tau pathology. Neurobiol. Aging 47, 71–73. doi: 10.1016/j.neurobiolaging.2016.07.016
Greco, S. J., Bryan, K. J., Sarkar, S., Zhu, X., Smith, M. A., Ashford, J. W., et al. (2010). Leptin reduces pathology and improves memory in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 19, 1155–1167. doi: 10.3233/jad-2010-1308
Guarnieri, B., Adorni, F., Musicco, M., Appollonio, I., Bonanni, E., Caffarra, P., et al. (2012). Prevalence of sleep disturbances in mild cognitive impairment and dementing disorders: a multicenter Italian clinical cross-sectional study on 431 patients. Dement. Geriatr. Cogn. Disord. 33, 50–58. doi: 10.1159/000335363
Gustafson, D. (2015). BMI and dementia: feast or famine for the brain? PLoS Genet. 3, 397–398. doi: 10.1016/s2213-8587(15)00085-6
Haller, J. (2013). The neurobiology of abnormal manifestations of aggression—a review of hypothalamic mechanisms in cats, rodents, and humans. Brain Res. Bull. 93, 97–109. doi: 10.1016/j.brainresbull.2012.10.003
Harper, D. G., Stopa, E. G., Kuo-Leblanc, V., McKee, A. C., Asayama, K., Volicer, L., et al. (2008). Dorsomedial SCN neuronal subpopulations subserve different functions in human dementia. Brain 131, 1609–1617. doi: 10.1093/brain/awn049
Hess, W. R., and Akert, K. (1955). Experimental data on role of hypothalamus in mechanism of emotional behavior. AMA. Arch. Neurol. Psychiatry 73, 127–129. doi: 10.1001/archneurpsyc.1955.02330080005003
Holden, K. F., Lindquist, K., Tylavsky, F. A., Rosano, C., Harris, T. B., Yaffe, K., et al. (2009). Serum leptin level and cognition in the elderly: findings from the health ABC study. Neurobiol. Aging 30, 1483–1489. doi: 10.1016/j.neurobiolaging.2007.11.024
Hu, K., Harper, D. G., Shea, S. A., Stopa, E. G., and Scheer, F. A. J. L. (2013). Noninvasive fractal biomarker of clock neurotransmitter disturbance in humans with dementia. Sci. Rep. 3:2229. doi: 10.1038/srep02229
Ishii, M., and Iadecola, C. (2015). Metabolic and non-cognitive manifestations of Alzheimer’s disease: the hypothalamus as both culprit and target of pathology. Cell Metab. 22, 761–776. doi: 10.1016/j.cmet.2015.08.016
Ishii, M., Wang, G., Racchumi, G., Dyke, J. P., and Iadecola, C. (2014). Transgenic mice overexpressing amyloid precursor protein exhibit early metabolic deficits and a pathologically low leptin state associated with hypothalamic dysfunction in arcuate neuropeptide Y neurons. J. Neurosci. 34, 9096–9106. doi: 10.1523/jneurosci.0872-14.2014
Jackson, K., Barisone, G. A., Diaz, E., Jin, L.-W., DeCarli, C., and Despa, F. (2013). Amylin deposition in the brain: a second amyloid in Alzheimer disease? Ann. Neurol. 74, 517–526. doi: 10.1002/ana.23956
Jang, H., Kim, J. H., Choi, S. H., Lee, Y., Hong, C. H., Jeong, J. H., et al. (2015). Body mass index and mortality rate in Korean patients with Alzheimer’s disease. J. Alzheimers Dis. 46, 399–406. doi: 10.3233/jad-142790
Jimenez, A., Pegueroles, J., Carmona-Iragui, M., Vilaplana, E., Montal, V., Alcolea, D., et al. (2017). Weight loss in the healthy elderly might be a non-cognitive sign of preclinical Alzheimer’s disease. Oncotarget 8, 104706–104716. doi: 10.18632/oncotarget.22218
Johnson, D. K., Wilkins, C. H., and Morris, J. C. (2006). Accelerated weight loss may precede diagnosis in Alzheimer disease. Arch. Neurol. 63, 1312–1317. doi: 10.1001/archneur.63.9.1312
Joo, S. H., Yun, S. H., Kang, D. W., Hahn, C. T., Lim, H. K., and Lee, C. U. (2018). Body mass index in mild cognitive impairment according to age, sex, cognitive intervention, and hypertension and risk of progression to Alzheimer’s disease. Front. Psychiatry 9:142. doi: 10.3389/fpsyt.2018.00142
Ju, Y.-E. S., McLeland, J. S., Toedebusch, C. D., Xiong, C., Fagan, A. M., Duntley, S. P., et al. (2013). Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 70, 587–593. doi: 10.1001/jamaneurol.2013.2334
Kang, J.-E., Lim, M. M., Bateman, R. J., Lee, J. J., Smyth, L. P., Cirrito, J. R., et al. (2009). Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007. doi: 10.1126/science.1180962
Khachiyants, N., Trinkle, D., Son, S. J., and Kim, K. Y. (2011). Sundown syndrome in persons with dementia: an update. Psychiatry Investig. 8, 275–287. doi: 10.4306/pi.2011.8.4.275
Khemka, V. K., Bagchi, D., Bandyopadhyay, K., Bir, A., Chattopadhyay, M., Biswas, A., et al. (2014). Altered serum levels of adipokines and insulin in probable Alzheimer’s disease. J. Alzheimers Dis. 41, 525–533. doi: 10.3233/jad-140006
Kiliaan, A. J., Arnoldussen, I. A. C., and Gustafson, D. R. (2014). Adipokines: a link between obesity and dementia? Lancet Neurol. 13, 913–923. doi: 10.1016/S1474-4422(14)70085-7
Kincheski, G. C., Valentim, I. S., Clarke, J. R., Cozachenco, D., Castelo-Branco, M. T. L., Ramos-Lobo, A. M., et al. (2017). Chronic sleep restriction promotes brain inflammation and synapse loss, and potentiates memory impairment induced by amyloid-β oligomers in mice. Brain Behav. Immun. 64, 140–151. doi: 10.1016/j.bbi.2017.04.007
Kivimäki, M., Singh-Manoux, A., Shipley, M. J., and Elbaz, A. (2015). Does midlife obesity really lower dementia risk? Lancet Diabetes Endocrinol. 3:498. doi: 10.1016/S2213-8587(15)00216-8
Kivipelto, M., Ngandu, T., Fratiglioni, L., Viitanen, M., Kåreholt, I., Winblad, B., et al. (2005). Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 62, 1556–1560. doi: 10.1001/archneur.62.10.1556
Lieb, W., Beiser, A. S., Vasan, R. S., Tan, Z. S., Au, R., Harris, T. B., et al. (2009). Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA 302, 2565–2572. doi: 10.1001/jama.2009.1836
Liguori, C., Chiaravalloti, A., Nuccetelli, M., Izzi, F., Sancesario, G., Cimini, A., et al. (2017). Hypothalamic dysfunction is related to sleep impairment and CSF biomarkers in Alzheimer’s disease. J. Neurol. 264, 2215–2223. doi: 10.1007/s00415-017-8613-x
Liguori, C., Nuccetelli, M., Izzi, F., Sancesario, G., Romigi, A., Martorana, A., et al. (2016). Rapid eye movement sleep disruption and sleep fragmentation are associated with increased orexin-A cerebrospinal-fluid levels in mild cognitive impairment due to Alzheimer’s disease. Neurobiol. Aging 40, 120–126. doi: 10.1016/j.neurobiolaging.2016.01.007
Liguori, C., Romigi, A., Nuccetelli, M., Zannino, S., Sancesario, G., Martorana, A., et al. (2014). Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol. 71, 1498–1505. doi: 10.1001/jamaneurol.2014.2510
Lim, A. S. P., Ellison, B. A., Wang, J. L., Yu, L., Schneider, J. A., Buchman, A. S., et al. (2014). Sleep is related to neuron numbers in the ventrolateral preoptic/intermediate nucleus in older adults with and without Alzheimer’s disease. Brain 137, 2847–2861. doi: 10.1093/brain/awu222
Loskutova, N., Honea, R. A., Brooks, W. M., and Burns, J. M. (2010). Reduced limbic and hypothalamic volumes correlate with bone density in early Alzheimer’s disease. J. Alzheimers Dis. 20, 313–322. doi: 10.3233/jad-2010-1364
Ma, J., Zhang, W., Wang, H.-F., Wang, Z.-X., Jiang, T., Tan, M.-S., et al. (2016). Peripheral blood adipokines and insulin levels in patients with Alzheimer’s disease: a replication study and meta-analysis. Curr. Alzheimer Res. 13, 223–233. doi: 10.2174/156720501303160217111434
Mander, B. A., Winer, J. R., Jagust, W. J., and Walker, M. P. (2016). Sleep: a novel mechanistic pathway, biomarker, and treatment target in the pathology of Alzheimer’s disease? Trends Neurosci. 39, 552–566. doi: 10.1016/j.tins.2016.05.002
Mayer, C. M., and Belsham, D. D. (2010). Palmitate attenuates insulin signaling and induces endoplasmic reticulum stress and apoptosis in hypothalamic neurons: rescue of resistance and apoptosis through adenosine 5’ monophosphate-activated protein kinase activation. Endocrinology 151, 576–585. doi: 10.1210/en.2009-1122
McDuff, T., and Sumi, S. M. (1985). Subcortical degeneration in Alzheimer’s disease. Neurology 35, 123–126. doi: 10.1212/wnl.35.1.123
McGregor, G., and Harvey, J. (2017). Leptin regulation of synaptic function at hippocampal TA-CA1 and SC-CA1 synapses: implications for health and disease. Neurochem. Res. doi: 10.1007/s11064-017-2362-1 [Epub ahead of print].
McGuire, M. J., and Ishii, M. (2016). Leptin dysfunction and Alzheimer’s disease: evidence from cellular, animal, and human studies. Cell. Mol. Neurobiol. 36, 203–217. doi: 10.1007/s10571-015-0282-7
McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D., and Stadlan, E. M. (1984). Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group* under the auspices of department of health and human services task force on Alzheimer’s disease. Neurology 34, 939–944. doi: 10.1212/wnl.34.7.939
Milanski, M., Degasperi, G., Coope, A., Morari, J., Denis, R., Cintra, D. E., et al. (2009). Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J. Neurosci. 29, 359–370. doi: 10.1523/JNEUROSCI.2760-08.2009
Moran, M., Lynch, C. A., Walsh, C., Coen, R., Coakley, D., and Lawlor, B. A. (2005). Sleep disturbance in mild to moderate Alzheimer’s disease. Sleep Med. 6, 347–352. doi: 10.1016/j.sleep.2004.12.005
Moreno-Gonzalez, I., Edwards Iii, G., Salvadores, N., Shahnawaz, M., Diaz-Espinoza, R., and Soto, C. (2017). Molecular interaction between type 2 diabetes and Alzheimer’s disease through cross-seeding of protein misfolding. Mol. Psychiatry 22, 1327–1334. doi: 10.1038/mp.2016.230
Müller, S., Preische, O., Sohrabi, H. R., Gräber, S., Jucker, M., Dietzsch, J., et al. (2017). Decreased body mass index in the preclinical stage of autosomal dominant Alzheimer’s disease. Sci. Rep. 7:1225. doi: 10.1038/s41598-017-01327-w
Musiek, E. S., Xiong, D. D., and Holtzman, D. M. (2015). Sleep, circadian rhythms and the pathogenesis of Alzheimer Disease. Exp. Mol. Med. 47:e148. doi: 10.1038/emm.2014.121
Narita, K., Kosaka, H., Okazawa, H., Murata, T., and Wada, Y. (2009). Relationship between plasma leptin level and brain structure in elderly: a voxel-based morphometric study. Biol. Psychiatry 65, 992–994. doi: 10.1016/j.biopsych.2008.10.006
Nestor, P. J., Fryer, T. D., Smielewski, P., and Hodges, J. R. (2003). Limbic hypometabolism in Alzheimer’s disease and mild cognitive impairment. Ann. Neurol. 54, 343–351. doi: 10.1002/ana.10669
Ooms, S., Overeem, S., Besse, K., Rikkert, M. O., Verbeek, M., and Claassen, J. A. H. R. (2014). Effect of 1 night of total sleep deprivation on cerebrospinal fluid β-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol. 71, 971–977. doi: 10.1001/jamaneurol.2014.1173
Paul, J. R., Munir, H. A., van Groen, T., and Gamble, K. L. (2018). Behavioral and SCN neurophysiological disruption in the Tg-SwDI mouse model of Alzheimer’s disease. Neurobiol. Dis. 114, 194–200. doi: 10.1016/j.nbd.2018.03.007
Pérez-González, R., Alvira-Botero, M. X., Robayo, O., Antequera, D., Garzón, M., Martín-Moreno, A. M., et al. (2014). Leptin gene therapy attenuates neuronal damages evoked by amyloid-β and rescues memory deficits in APP/PS1 mice. Gene. Ther. 21, 298–308. doi: 10.1038/gt.2013.85
Qizilbash, N., Gregson, J., Johnson, M. E., Pearce, N., Douglas, I., Wing, K., et al. (2015). BMI and risk of dementia in two million people over two decades: a retrospective cohort study. Lancet Diabetes Endocrinol. 3, 431–436. doi: 10.1016/s2213-8587(15)00033-9
Ramos-Rodríguez, J. J., Spires-Jones, T., Pooler, A. M., Lechuga-Sancho, A. M., Bacskai, B. J., and García-Alloza, M. (2017). Progressive neuronal pathology and synaptic loss induced by prediabetes and type 2 diabetes in a mouse model of Alzheimer’s disease. Mol. Neurobiol. 54, 3428–3438. doi: 10.1007/s12035-016-9921-3
Roh, J. H., Jiang, H., Finn, M. B., Stewart, F. R., Mahan, T. E., Cirrito, J. R., et al. (2014). Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer’s disease. J. Exp. Med. 211, 2487–2496. doi: 10.1084/jem.20141788
Roh, J. H., Huang, Y., Bero, A. W., Kasten, T., Stewart, F. R., Bateman, R. J., et al. (2012). Disruption of the sleep-wake cycle and diurnal fluctuation of β-amyloid in mice with Alzheimer’s disease pathology. Sci. Transl. Med. 4:150ra122. doi: 10.1126/scitranslmed.3004291
Rudelli, R. D., Ambler, M. W., and Wisniewski, H. M. (1984). Morphology and distribution of Alzheimer neuritic (senile) and amyloid plaques in striatum and diencephalon. Acta Neuropathol. 64, 273–281. doi: 10.1007/bf00690393
Sala Frigerio, C., and De Strooper, B. (2016). Alzheimer’s disease mechanisms and emerging roads to novel therapeutics. Annu. Rev. Neurosci. 39, 57–79. doi: 10.1146/annurev-neuro-070815-014015
Saper, C. B., and Fuller, P. M. (2017). Wake-sleep circuitry: an overview. Curr. Opin. Neurobiol. 44, 186–192. doi: 10.1016/j.conb.2017.03.021
Saper, C. B., and German, D. C. (1987). Hypothalamic pathology in Alzheimer’s disease. Neurosci. Lett. 74, 364–370. doi: 10.1016/0304-3940(87)90325-9
Schmidt, F. M., Kratzsch, J., Gertz, H.-J., Tittmann, M., Jahn, I., Pietsch, U.-C., et al. (2013). Cerebrospinal fluid melanin-concentrating hormone (MCH) and hypocretin-1 (HCRT-1, orexin-A) in Alzheimer’s disease. PLoS One 8:e63136. doi: 10.1371/journal.pone.0063136
Schultz, C., Ghebremedhin, E., Braak, E., and Braak, H. (1999). Sex-dependent cytoskeletal changes of the human hypothalamus develop independently of Alzheimer’s disease. Exp. Neurol. 160, 186–193. doi: 10.1006/exnr.1999.7185
Sethi, M., Joshi, S. S., Webb, R. L., Beckett, T. L., Donohue, K. D., Murphy, M. P., et al. (2015). Increased fragmentation of sleep-wake cycles in the 5XFAD mouse model of Alzheimer’s disease. Neuroscience 290, 80–89. doi: 10.1016/j.neuroscience.2015.01.035
Shi, L., Chen, S.-J., Ma, M.-Y., Bao, Y.-P., Han, Y., Wang, Y.-M., et al. (2018). Sleep disturbances increase the risk of dementia: a systematic review and meta-analysis. Sleep Med. Rev. 40, 4–16. doi: 10.1016/j.smrv.2017.06.010
Shokri-Kojori, E., Wang, G.-J., Wiers, C. E., Demiral, S. B., Guo, M., Kim, S. W., et al. (2018). β-amyloid accumulation in the human brain after one night of sleep deprivation. Proc. Natl. Acad. Sci. U S A 115, 4483–4488. doi: 10.1073/pnas.1721694115
Sobów, T., Fendler, W., and Magierski, R. (2014). Body mass index and mild cognitive impairment-to-dementia progression in 24 months: a prospective study. Eur. J. Clin. Nutr. 68, 1216–1219. doi: 10.1038/ejcn.2014.167
Spira, A. P., Gamaldo, A. A., An, Y., Wu, M. N., Simonsick, E. M., Bilgel, M., et al. (2013). Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA Neurol. 70, 1537–1543. doi: 10.1001/jamaneurol.2013.4258
Stanley, M., Macauley, S. L., Caesar, E. E., Koscal, L. J., Moritz, W., Robinson, G. O., et al. (2016). The effects of peripheral and central high insulin on brain insulin signaling and amyloid-β in young and old APP/PS1 mice. J. Neurosci. 36, 11704–11715. doi: 10.1523/JNEUROSCI.2119-16.2016
Stewart, R., Masaki, K., Xue, Q.-L., Peila, R., Petrovitch, H., White, L. R., et al. (2005). A 32-year prospective study of change in body weight and incident dementia: the Honolulu-Asia aging study. Arch. Neurol. 62, 55–60. doi: 10.1001/archneur.62.1.55
Stief, A. (1927). über die anatomischen grundlagen der vegetativen störungen bei geisteskrankheiten. Dtsch. Z. Nervenheilkd. 97, 112–132. doi: 10.1007/bf01667908
Stopa, E. G., Volicer, L., Kuo-Leblanc, V., Harper, D., Lathi, D., Tate, B., et al. (1999). Pathologic evaluation of the human suprachiasmatic nucleus in severe dementia. J. Neuropathol. Exp. Neurol. 58, 29–39. doi: 10.1097/00005072-199901000-00004
Swaab, D. F., Fliers, E., and Partiman, T. S. (1985). The suprachiasmatic nucleus of the human brain in relation to sex, age and senile dementia. Brain Res. 342, 37–44. doi: 10.1016/0006-8993(85)91350-2
Tabuchi, M., Lone, S. R., Liu, S., Liu, Q., Zhang, J., Spira, A. P., et al. (2015). Sleep interacts with aβ to modulate intrinsic neuronal excitability. Curr. Biol. 25, 702–712. doi: 10.1016/j.cub.2015.01.016
Takeda, S., Sato, N., Uchio-Yamada, K., Sawada, K., Kunieda, T., Takeuchi, D., et al. (2010). Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Aβ deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. U S A 107, 7036–7041. doi: 10.1073/pnas.1000645107
Thon, M., Hosoi, T., and Ozawa, K. (2016). Possible integrative actions of leptin and insulin signaling in the hypothalamus targeting energy homeostasis. Front. Endocrinol. 7:138. doi: 10.3389/fendo.2016.00138
Todd, W. D., Fenselau, H., Wang, J. L., Zhang, R., Machado, N. L., Venner, A., et al. (2018). A hypothalamic circuit for the circadian control of aggression. Nat. Neurosci. 21, 717–724. doi: 10.1038/s41593-018-0126-0
Tsujino, N., and Sakurai, T. (2013). Role of orexin in modulating arousal, feeding, and motivation. Front. Behav. Neurosci. 7:28. doi: 10.3389/fnbeh.2013.00028
van de Nes, J. A., Kamphorst, W., Ravid, R., and Swaab, D. F. (1998). Comparison of β-protein/A4 deposits and Alz-50-stained cytoskeletal changes in the hypothalamus and adjoining areas of Alzheimer’s disease patients: amorphic plaques and cytoskeletal changes occur independently. Acta Neuropathol. 96, 129–138. doi: 10.1007/s004010050872
Van Erum, J., Van Dam, D., and De Deyn, P. P. (2018). Sleep and Alzheimer’s disease: a pivotal role for the suprachiasmatic nucleus. Sleep Med. Rev. 40, 17–27. doi: 10.1016/j.smrv.2017.07.005
Vitiello, M. V., Prinz, P. N., Williams, D. E., Frommlet, M. S., and Ries, R. K. (1990). Sleep disturbances in patients with mild-stage Alzheimer’s disease. J. Gerontol. 45, M131–M138. doi: 10.1093/geronj/45.4.m131
Wang, J. L., Lim, A. S., Chiang, W.-Y., Hsieh, W.-H., Lo, M.-T., Schneider, J. A., et al. (2015). Suprachiasmatic neuron numbers and rest-activity circadian rhythms in older humans. Ann. Neurol. 78, 317–322. doi: 10.1002/ana.24432
Westwood, A. J., Beiser, A., Jain, N., Himali, J. J., DeCarli, C., Auerbach, S. H., et al. (2017). Prolonged sleep duration as a marker of early neurodegeneration predicting incident dementia. Neurology 88, 1172–1179. doi: 10.1212/WNL.0000000000003732
White, H., Pieper, C., and Schmader, K. (1998). The association of weight change in Alzheimer’s disease with severity of disease and mortality: a longitudinal analysis. J. Am. Geriatr. Soc. 46, 1223–1227. doi: 10.1111/j.1532-5415.1998.tb04537.x
Wijesekara, N., Ahrens, R., Sabale, M., Wu, L., Ha, K., Verdile, G., et al. (2017). Amyloid-β and islet amyloid pathologies link Alzheimer’s disease and type 2 diabetes in a transgenic model. FASEB J. 31, 5409–5418. doi: 10.1096/fj.201700431R
Wisor, J. P., Edgar, D. M., Yesavage, J., Ryan, H. S., McCormick, C. M., Lapustea, N., et al. (2005). Sleep and circadian abnormalities in a transgenic mouse model of Alzheimer’s disease: a role for cholinergic transmission. Neuroscience 131, 375–385. doi: 10.1016/j.neuroscience.2004.11.018
Xie, L., Kang, H., Xu, Q., Chen, M. J., Liao, Y., Thiyagarajan, M., et al. (2013). Sleep drives metabolite clearance from the adult brain. Science 342, 373–377. doi: 10.1126/science.1241224
Yu, S., Liu, Y.-P., Liu, H.-L., Li, J., Xiang, Y., Liu, Y.-H., et al. (2018). Serum protein-based profiles as novel biomarkers for the diagnosis of Alzheimer’s disease. Mol. Neurobiol. 55, 3999–4008. doi: 10.1007/s12035-017-0609-0
Zhang, X., Zhang, G., Zhang, H., Karin, M., Bai, H., and Cai, D. (2008). Hypothalamic IKKβ/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135, 61–73. doi: 10.1016/j.cell.2008.07.043
Keywords: hypothalamus, obesity, diabetes, sleep, circadian rhythm, dementia, amyloid-beta, tau
Citation: Hiller AJ and Ishii M (2018) Disorders of Body Weight, Sleep and Circadian Rhythm as Manifestations of Hypothalamic Dysfunction in Alzheimer’s Disease. Front. Cell. Neurosci. 12:471. doi: 10.3389/fncel.2018.00471
Received: 31 August 2018; Accepted: 19 November 2018;
Published: 05 December 2018.
Edited by:
Marco Mainardi, Scuola Normale Superiore di Pisa, ItalyReviewed by:
Cláudia Pereira, Universidade de Coimbra, PortugalInes Moreno-Gonzalez, University of Texas Health Science Center at Houston, United States
Copyright © 2018 Hiller and Ishii. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Makoto Ishii, bWlzaGlpQG1lZC5jb3JuZWxsLmVkdQ==