Abstract
Ferroptosis is a unique regulated cell death defined by the intracellular iron overload and distinct biological features compared with other well-known programmed cell death. Ferroptosis can be triggered by many causes including decreased expression of glutathione (GSH), inhibition of the function of glutathione-dependent peroxidase 4 (GPX4), and system xc–, all of which finally lead to the over-accumulation of lipid peroxides in the cell. Ferroptosis has been reported to play an important role in the pathophysiological process of various cancers. In recent years, much evidence also proved that ferroptosis is involved in the progress of cerebral stroke. In this review, we summarized the characteristics of ferroptosis and the potential relationship between ferroptosis and ischemic and hemorrhagic stroke, to provide new targets and ideas for the therapy of stroke.
Introduction
Ferroptosis is a newly discovered iron-dependent regulated cell death, which has unique morphological and biochemical distinctions from other programmed cell death such as apoptosis, and necroptosis (Dixon et al., 2012). The characteristic morphological changes of ferroptosis are the shrunken mitochondria with a ruptured external membrane, reduced crista, a compressed internal membrane, and the intact cell nucleus (Dixon et al., 2012; Friedmann Angeli et al., 2014), while the apoptosis and necroptosis generally have swollen mitochondria and the broken nucleus (Dixon et al., 2012). Being different from the cell death caused by apoptosis due to activation of caspases and that caused by necroptosis due to activation of RIP1, RIP3, and MLKL (Kroemer et al., 2009; Galluzzi et al., 2012; Xie et al., 2016), the most essential biochemical attributes of ferroptosis are iron overload and the lethal accumulation of lipid peroxides and reactive oxygen species (ROS) within the cell (Stockwell et al., 2017), which can produce a large number of alkyl oxygen radicals, leading to fatal cell membrane damage and disorganization (Agmon et al., 2018). Ferroptosis has been proved to be closely related to a variety of diseases throughout the human body. For the central nervous system, it has been shown that ferroptosis plays an important role in many neurodegenerative diseases such as Alzheimer’s Disease (Cong et al., 2019; Masaldan et al., 2019), Parkinson’s Disease (Ayton et al., 2013, 2015), and Huntington’s Disease (Lee et al., 2011). Furthermore, in recent years, the potential roles of ferroptosis in the pathologic process of cerebral stroke are given with increasing focus (Zille et al., 2017; Karuppagounder et al., 2018). Thus, in this review, we intend to summarize the current knowledge and recent findings in the field of metabolic mechanisms of ferroptosis, its inducers and inhibitors, and the critical roles of ferroptosis in cerebral stroke pathophysiology.
The Cellular Metabolic Mechanisms of Ferroptosis
The occurrence of ferroptosis is associated with the metabolic processes of iron, amino acids, and lipid peroxide (Stockwell et al., 2017; Figure 1), which will be elaborated one by one as follows.
FIGURE 1
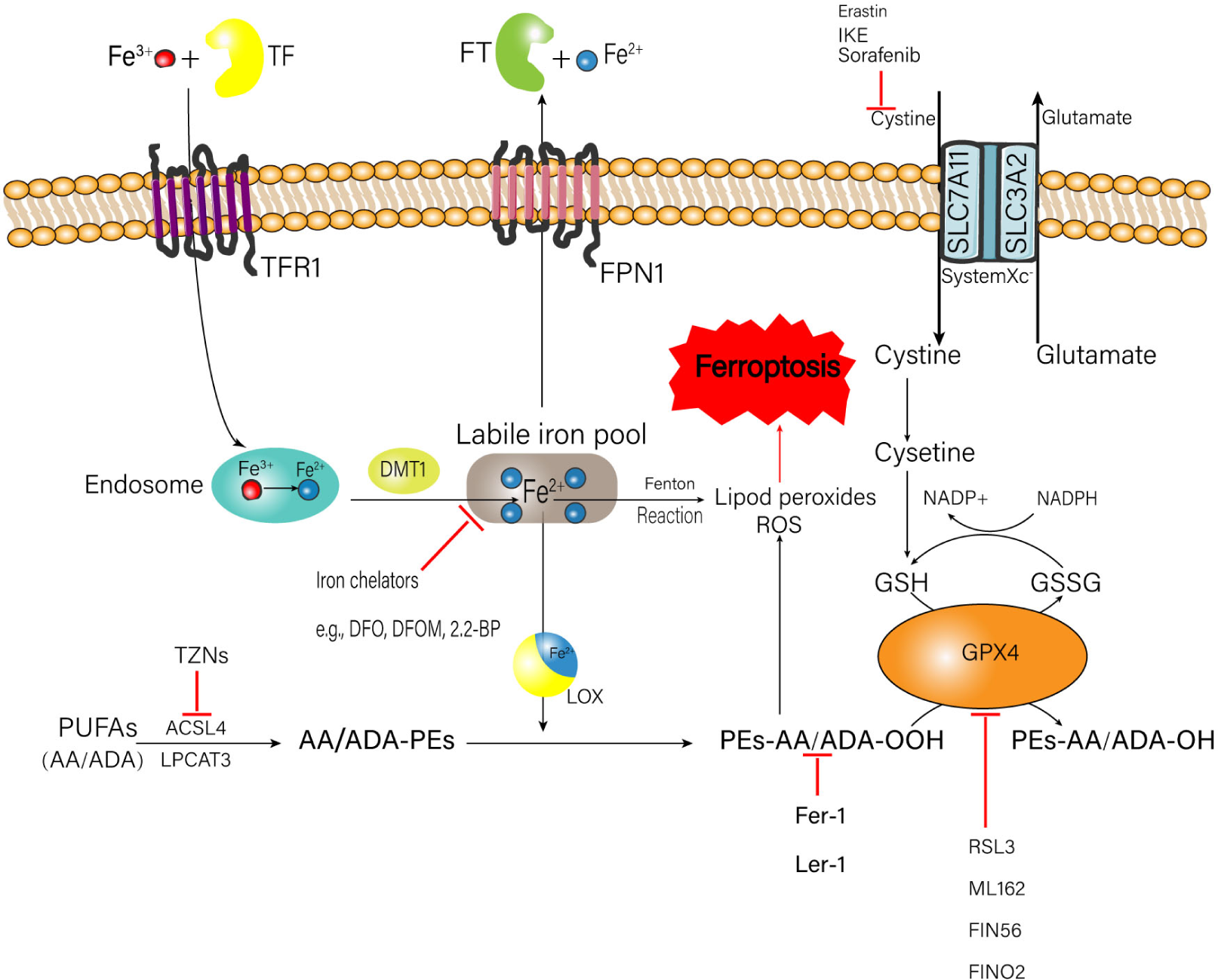
Metabolic processes of iron, amino acids, and lipid peroxide related to the happen of ferroptosis. A number of medicinal inducer have been shown to induce ferroptosis (e.g., erastin and RSL3). A variety of ferroptosis inhibitors also have been shown in the figure (e.g., Fer-1, lip-1, TZNs, and DFO). TF, transferrin; FT, ferritin; IKE, imidazole ketone erastin; TFR1, transferrin receptor 1; FPN1, ferroportin-1; SLC7A11, solute carrier family 7 member 11; SLC3A2, solute carrier family 3 member 2; DMT1, divalent metal transporter 1; ROS, reactive oxygen species; NADPH, nicotinamide adenine dinucleotide phosphate; GSH, glutathione; GSSG, oxidized glutathione; GPX4, glutathione peroxidase 4; DFO, deferoxamine; DFOM, deferoxamine mesylate; 2, 2-BP, 2, 2′-bipyridyl; PUFAs, polyunsaturated fatty acids; AA, arachidonic acid; ADA, adrenic acid; TZNs, thiazolidinediones; ACSL4, acyl-CoA synthetase long-chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3; LOX, lipoxygenase; Fer-1, ferrostatin-1; Lip-1, liproxstatin-1; RSL3, ras-selective lethal small molecules 3.
Metabolism of Iron
Intracellular iron overload is a critical factor to trigger ferroptosis. Normally, the source of iron comes from two ways: (1) Food-derived iron including ferric iron (Fe3+) and ferrous iron (Fe2+); (2) Iron generated by the hemoglobin of senescent erythrocytes (Śliwińska et al., 2018). The absorbed Fe2+ is oxidized to Fe3+ in the small intestinal mucosa epithelial cells (Dong et al., 2008). Then the plasma Fe3+ is tightly bound to transferrin (TF) to form the complete TF, which can combine with the membrane protein transferrin receptor 1 (TFR1) to transport the Fe3+ into the endosome. After that, the Fe3+ is reduced to Fe2+ again by iron reductase (Hamaï and Mehrpour, 2017). Finally, the Fe2+ is transported into the labile iron pool in the cytoplasm through the divalent metal transporter 1 (DMT1), which is a commonly expressed protein with the ability to transfer a series of metal ions such as iron, crop, zinc, and cadmium (Garton et al., 2016) to perform their physio-pathology functions (Ji and Kosman, 2015; Knutson, 2019). Fe2+ can generate the ROS via the Fenton reaction, which is a type of reaction between ferrous iron and hydrogen peroxide (H2O2) referring to the creation of responsive varieties with the ability to oxidize a wide range of organic substrate (Winterbourn, 1995). In addition, Fe2+ is also an important component of the catalytic subunit of lipoxygenase (LOX), which can catalyze lipid peroxidation (Gaschler and Stockwell, 2017). Both mechanisms can trigger ferroptosis. Excessive Fe2+ is transported out of the cell through the ferroportin-1 (FPN1) on the cell membrane, to keep the intracellular Fe2+ concentration in a normal range. Then the Fe2+ outside the cell is stored in the ferritin (FT), to prevent the formation of hydroxyl radicals and ROS produced by H2O2 catalyzing (MacKenzie et al., 2008).
Metabolism of Lipid Peroxide
Excess lipid peroxidation is another key factor to induce ferroptosis. Polyunsaturated fatty acids (PUFAs) are fatty acids with more than one double bond in their structure (Liu et al., 2015). PUFAs, especially arachidonic acid (AA) and adrenic acid (ADA) are very vulnerable to be oxidized, subsequently leading to the accumulation of lipid hydroperoxide (LOOH) and ROS (Das, 2019); the enrichment and location of PUFAs intracellularly will determine the degree of ferroptosis. Before the oxidation happens, PUFAs are esterified into membrane phospholipids, mainly of which is phosphatidylethanolamines (PEs) (Latunde-Dada, 2017). Then the PEs are further oxidized to phospholipid hydroperoxides (PEs-AA/ADA-OOH) by LOX, inducing ferroptosis. During the formation of AA/ADA-PEs, acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3), which serve as the promotors for the esterification of AA and ADA into PEs, are thought to play an important role (Yuan et al., 2016; Kagan et al., 2017). Doll et al. reported that targeted inhibition of ACSL4 can relieve cell and tissue damage introduced by ferroptosis (Doll et al., 2017). Similarly, inhibition of LPCAT3 can decrease the insertion of AA/ADA into membrane phospholipids. This evidence suggests that the restriction of ACSL4 and LPCAT3 may be a feasible healing strategy to stop ferroptosis-related illness.
Metabolism of Glutathione and Amino Acid
Glutathione (GSH) is the major intercellular antioxidant against oxidative stress (Lu, 2013), which exerts an important duty on protecting cells from the ROS attack (Stockwell et al., 2017; Sun et al., 2018). It is suggested that cystine is the key to maintain the normal level of GSH (Imai et al., 2017). The system xc– on the cell membrane consisting of a single-pass transmembrane regulatory protein solute carrier family 3 member 2 (SLC3A2) and a 12-pass transmembrane protein transporter solute carrier family 7 member 11 (SLC7A11) can exchange the extracellular cystine and the intracellular glutamate by a ratio of 1:1 simultaneously (Yang and Stockwell, 2016; Imai et al., 2017). When the cystine enters the cell, it is decomposed into cysteine, which combines with glutamate to form γ-glutamylcysteine under the catalysis of γ-glutamylcysteine synthetase (γ-GCS) (Fujii et al., 2019). Finally, the γ-glutamylcysteine combines with glycine to form GSH. The natural synthesis of GSH is the critical factor for glutathione peroxidase 4 (GPX4) to conduct its biological activity. GPX4 converts two molecules of GSH to oxidized glutathione (GSSG) (Maiorino et al., 2018) and reduces L-OOH to Phospholipids-H (L-OH) simultaneously, to prevent the accumulation of toxic lipid oxidation (Maiorino et al., 2018; Ursini and Maiorino, 2020). Selective ablation of GPX4 in neurons of mice can provoke lethal ferroptosis and neurological dysfunction (Chen et al., 2015). GSH also acts as the binding legend of Fe2+ in the labile iron pool to decrease the concentration of Fe2+, which can abate the production of Fe2+-related lipid peroxides and ROS.
Inducers of Ferroptosis
As multiple mechanisms are involved in ferroptosis, according to different activated targets, ferroptosis inducers can be classified into three categories: (1) Inhibitors of GPX4, (2) Inhibitors of system xc–, (3) Small molecular inducers of iron loading.
Inhibitors of GPX4
Ras-selective lethal small molecules 3 (RSL3) is found using phenotypic small molecule screens based on its ability to selectively facilitate oncogenic RAS mutant cell death (Dolma et al., 2003). RSL3 can bind to GPX4 by targeting an enzyme having a nucleophilic active site such as serine and cysteine, and inhibit the biological activity of GPX4 (Yang et al., 2014). ML162 is another small molecule compound that also can inhibit the activity of GPX4 (Dixon et al., 2015). In addition, both RSL3 and ML162 can enhance the expression of ACSL4 and LPCAT3 at the genetic level, which increases the accumulation of lipid peroxides (Dixon et al., 2015). FIN56 is a ferroptosis inhibitor acquired from CIL56, which can accelerate the degradation of GPX4 enzymatic function based on the chemical activity of acetyl-CoA carboxylase (Liang et al., 2019). FINO2, a course of organic peroxides that has many similar functions with artemisinins (Liang et al., 2019), is capable of indirectly inhibiting GPX4 and directly oxidate the Fe2+, resulting in an excessive build-up of lipid peroxides (Gaschler et al., 2018).
Inhibitors of System xc–
Erastin is the classical ferroptosis inducers recognized by high-throughput screening of small-molecule libraries. Erastin could inactivate the function of SLC7A11 (xCT) in system xc– to prevent cystine intake from GSH synthesizing and interfere with protein folding. Because of this, incompletely folded proteins gather in cells and cause cellular stress, thus giving rise to ferroptosis (Liang et al., 2019). The exact principle through which erastin hinders xCT remains unidentified. One possibility is that it inhibits xCT directly (Hirschhorn and Stockwell, 2019). Furthermore, erastin can combine with the voltage-dependent anion channel (VADC), a channel on the external membrane of mitochondria for controlling calcium ion exchange, bringing about mitochondrial impairment and further production of ROS by nicotinamide adenine dinucleotide phosphate (NADPH) dependent pathway (Yagoda et al., 2007). Imidazole ketone erastin (IKE) has a more powerful inhibiting effect and metabolic stability than erastin, which makes IKE an excellent choice for research of ferroptosis triggering in vivo and in vitro (Hirschhorn and Stockwell, 2019). L-α-aminoadipate, an amino acid intermediate in chain length between glutamate and cystine, is also an effective substrate inhibitor of system xc– (Patel et al., 2004). The last common system xc– inhibitor is sorafenib, which has been reported to trigger the ferroptosis of cancer cells by depleting the GSH (Lachaier et al., 2014). The RAF/MEK/ERK signaling pathway may exert an essential role in sorafenib inducing ferroptosis in vitro (Liu et al., 2006).
Small Molecules Inducing Iron Loading
It has been shown that many small molecules such as ferrous chloride, ferrous ammonium sulfate, hemoglobin, and hemin trigger ferroptosis through iron overload, aggravating the Fenton reaction to generate more ROS, then promoting oxidation-related cell membrane damage.
Inhibitors of Ferroptosis
With more and more studies focusing on ferroptosis, the possible duty of ferroptosis in diseases has been gradually revealed. To better study how to treat ferroptosis, lots of specific and effective ferroptosis inhibitors have been developed. According to the different inhibitory targets, inhibitors can be divided into (1) iron chelators, (2) lipid peroxidation inhibitors, and (3) ACSL4 inhibitors.
Iron Chelators
The common iron chelators include deferoxamine (DFO), deferoxamine mesylate (DFOM), and 2, 2′-bipyridyl (2, 2-BP), among which DFO is the most widely used iron chelators approved by the Food and Drug Administration (FDA). It inhibits the acclamation of lipid peroxides by suppressing the Fenton reaction. DFO can effectively reduce stroke volume by increasing the expression of hypoxia-inducible factor 1 (HIF-1) (Baranova et al., 2007). It also has been reported that the DFO treatment can increase the resistance of neurons to impairment through regulating the microglial/macrophage heme oxygenase-1 (HO-1) expression in the SAH mouse model (LeBlanc et al., 2016). However, a randomized controlled trial regarding using DFO to treat intracerebral hemorrhage showed that there was no statistical difference in the clinical outcome after 90 days between the treatment group and the placebo group (Selim et al., 2019). Therefore, more studies are needed in the future to ascertain the role of DFO in the treatment of ferroptosis after stroke.
Lipid Peroxidation Inhibitors
Lipid peroxidation inhibitors can be categorized into lipophilic radical-trapping antioxidants (RTAs) and LOX inhibitors according to their mechanism of action. RTAs are a class of molecules that is able to break the autoxidation of chain-propagating peroxyl radicals. The widely used RTAs are ferrostatin-1 (Fer-1) and liproxstatin-1 (Lip-1), which can inhibit lipid peroxidation linked to ferroptosis. Fer-1 can prevent erastin triggered-ferroptosis in HT1080 cells and is presently thought of as a probe for researching ferroptosis in various situations (Han et al., 2020). The activity of ferrostain-1 relies on the primary aromatic amine, which particularly hinders the accumulation of ROS from lipid oxidation (Xie et al., 2016). Lip-1 is a spiroquinoxalinamine by-product (Feng et al., 2019) and has a similar effect with Fer-1, but it only requires a lower dose to function due to the excellent absorption and distribution (Han et al., 2020). A-tocopherol, an analog of vitamin E (Ranard and Erdman, 2018), is also a potent type of RTAs to suppress ferroptosis in vitro (Angeli et al., 2017) and in vivo (Dalleau et al., 2013). It has been reported that A-tocopherol can inhibit breast cancer cell death induced by autophagy (Ma et al., 2017), which implies that ferroptosis may have a similar mechanism on cell death compared with autophagy. In addition, A-tocopherol can inhibit the function of 15-LOX, a subtype of LOX in the human body, by converting the non-heme iron of 15-LOX from the active Fe3+ state to the non-active Fe2+ state (Hinman et al., 2018). Another powerful kind of RTAs is tetrahydronaphthyridinols (TINs), of which the structure can significantly maintain the electron-rich phenols against autoxidation (Angeli et al., 2017). LOX inhibitors prevent ferroptosis through inhibiting the lipid peroxidation. LOXBlock-1 (LB1) is an inhibitor of 12/15-LOX, which is the dominant isoform of LOX in the brain (Singh and Rao, 2019). Karatas et al. (2018) reported that LB1 treatment decreased the infarct volume and the hemorrhage area in MCAO mice model. Similarly, Yigitkanli et al. (2013) found that LB1 could reduced infarct sizes and tissue plasminogen activator-associated hemorrhage in ischemia rat model. These studies imply that ferroptosis is closely related to the brain damage after stroke.
ACSL4 Inhibitors
Acyl-CoA synthetase long-chain family member 4 is a critical enzyme to esterify AA and ADA, making it highly possible to be an inhibitory target for ferroptosis. Thiazolidinediones (TZNs), a class of drugs suggested for treating type 2 diabetes mellitus, have been shown to specifically suppress the ACSL4 activity and prevent ferroptosis (Kim et al., 2001; Doll et al., 2017). This effect may be a result of the chromanol ring, which endows tocopherols with antioxidant activity. Thus, TZNs provide a new perspective for pharmacological inhibition of lipid peroxidation and ferroptosis by suppressing the activity of ACSL4. Apart from that, rosiglitazone, one of the synthetic agonists for proliferator-activated receptor γ (PPARγ), is found to have the ability to selectively inhibit the activity of ACSL4 in cerebral ischemia rat models, thereby reducing neuronal lipid peroxidation and oxidative stress damage, and protecting neurological function (Sayan-Ozacmak et al., 2012).
The Role of Ferroptosis in Cerebral Stroke
Cerebral stroke, including ischemic stroke and hemorrhagic stroke, is one of the most common causes of mortality and disability in modern society, which often results in devastating and irreversible brain impairment (Cordonnier et al., 2017; Steliga et al., 2020). Lots of research has proved that ferroptosis is closely allied to the pathophysiology of ischemic and hemorrhagic stroke and inhibition of ferroptosis can alleviate the secondary brain injury after stroke (Weiland et al., 2019; Chen et al., 2020), indicating that ferroptosis is a potential and critical therapeutic target for stroke.
Ferroptosis in Ischemic Stroke
Ischemic stroke is the dominant subtype of stroke, defined by the unexpected cessation of cerebral blood flow (CBF) to an area of the brain, consequently producing a homologous loss of neurologic function. Ischemic stroke contains two important events: ischemia and reperfusion injury (IRI), the outcomes of which are able to prompt serious cellular impairment responsible for the poor prognosis of ischemic stroke (Yan et al., 2020). The specific signaling pathways and molecular mechanisms related to IRI are still not well recognized and are greatly disputed. A series of studies have reported that ferroptosis is involved in IRI in recent years (Xie et al., 2016; Doll and Conrad, 2017). First, iron accumulation has emerged after ischemia through inhibiting the expression of tau, which is a protein being able to form neuronal tangles in Parkinson’s disease and promote neuronal iron outflow (Tuo et al., 2017). Then the excess iron flows into the brain parenchyma through the interrupted BBB, boosting the generation of ROS via Fenton reaction, which promotes nucleic, proteomic, and membrane damage, ultimately triggering ferroptosis-related cell death (Liu et al., 2020). Moreover, ferroptotic cell death is found to be alleviated with the treatment of ferroptosis inhibitors Lip-1 and Fer-1 (Tuo et al., 2017). Apart from that, Guan et al. (2019) reported that carvacrol, a plentiful monoterpenic phenol in the essential oil of oregano and thyme, can rescue the IRI-induced hippocampal neuronal damage via suppressing ferroptosis by enhancing the expression of GPX4. Similarly, Lanet et al., found that the compound Chinese herbal medicine: Naotaifang, can also mitigate neuron ferroptosis after ischemic stroke via the TFR1/DMT1 and SCL7A11/GPX4 pathways (Lan et al., 2020). This research has highly proven that ferroptosis is closely linked to the pathology of ischemic stroke and may regulate the impairment after IRI.
Ferroptosis in Hemorrhagic Stroke
Hemorrhagic stroke refers to an unexpected rupture of cerebral vessels that causes blood to flow into subarachnoid space, brain parenchyma, or ventricular system (Zhao et al., 2018). Therefore, a hemorrhagic stroke can be categorized into subarachnoid hemorrhage (SAH), intracerebral hemorrhage (ICH), and intraventricular hemorrhage (IVH). Compared with ischemic stroke, hemorrhagic stroke has a higher rate of mortality and morbidity due to severe neuronal death (Weiland et al., 2019). It has been shown that ferroptosis is involved in neuronal death after ICH in vitro and in vivo (Li et al., 2017; Karuppagounder et al., 2018; Zhang et al., 2018; Chen et al., 2019). Inhibition of the ferroptosis after ICH via ferrostatin-1 can alleviate neuronal death and enhance neurologic function (Li et al., 2017). Ferroptosis also plays an important role in early brain injury after SAH (Li et al., 2021). Cao et al. (2021) recently reported that the treatment with lip-1 protected HT22 cells against hemin-induced injury and reduced neurological deficits and neuroinflammation after SAH in mice. Additionally, earlier research has verified that the NF-E2-related factor 2 (Nrf2) pathway, a multifunctional signaling pathway that can control more than 250 genetics (Chang et al., 2014), is activated and provides neuroprotection after SAH (Chen et al., 2020). The expression level of Nrf2 is directly related to the occurrence of ferroptosis: an increase in Nrf2 expression inhibits ferroptosis; on the contrary, a decrease of it promotes ferroptosis (Fan et al., 2017). The mechanism by which Nrf2 inhibits ferroptosis involves two accepts: (1) Nrf2 upregulates the expression of xCT, thereby promoting the synthesis of GSH and GPX4 and enhancing the function of the antioxidant system (Fan et al., 2017); (2) Nrf2 promotes the expression of FT and FPN1 to store and export free iron, thereby reducing the accumulation of intracellular iron to prevent ferroptosis (Yang et al., 2017). Apart from that, Selenium is reported to be able to inhibit ferroptosis and protect neurons by promoting the expression of antioxidant GPX4 (Alim et al., 2019). These findings strongly suggest that ferroptosis aggravates the progress of hemorrhagic stroke and the inhibition of ferroptosis can reduce cell death and complications after a hemorrhagic stroke.
The Possible Regulatory Mechanism of Ferroptosis in Stroke
Since the metabolisms of lipid peroxide, amino acids, and iron are closely related to ferroptosis, the regulatory mechanism of stroke ferroptosis may also be involved in these three aspects.
Metabolism of Lipid Oxidation in Stroke
Inhibition of LOX Can Alleviate the Impairment Induced by Stroke
As we discussed above, LOX is the critical enzyme to catalyze PEs to form lipid peroxides. There are at least six subtypes of LOX in the human, among which the 12/15-LOX is a specific one due to its ability to directly oxidize lipid membranes containing PUFAs without the prior action of phospholipase and directly attack the mitochondria (van Leyen et al., 2014). It was reported that using the 12/15-LOX inhibitor can improve the neurological outcome and reduce the edema after ischemic stroke (Jin et al., 2008). This study provides evidence that 12/15-LOX is overexpressed and leads to both neuronal cell death and blood-brain barrier disruption after a stroke hit (Jin et al., 2008). Similarly, Gaberel et al. (2019) found that the expression of 12/15-LOX is upregulated in macrophages after SAH in mice, and inhibition of the 12/15-LOX pathway reduces EBI and protects the neurological function. In addition, 5-LOX, another subtype of LOX, is also found to have the ability to produce toxic lipids inducing ferroptosis. Treatment with N-acetylcysteine (NAC), a clinically approved thiol-containing redox modulatory compound that can inhibit the function of 5-LOX, is able to reverse brain injury and alleviate neuronal death (Karuppagounder et al., 2018). All the research indicates that LOX, especially 12/15-LOX is an innovative healing target to restrict brain injury after stroke.
ACSL4 Can Regulate Ferroptosis After Stroke
Acyl-CoA synthetase long-chain family member 4 has been found to be upregulated after ischemia and involved in ischemia-reperfusion injury (Gubern et al., 2013). This overexpression of ACSL4 is possibly controlled by miR-347, which is increased after ischemic stroke and upregulates ACSL4 at the transcriptional or post-transcriptional level (Gubern et al., 2013). Latunde-Dada et al., reported that inhibition of ACSL4 can alleviate ferroptosis and cell death in an ischemic rat model, and the special protein 1 (Sp1) is considered to be an essential transcription element for promoting the expression of ACSL4 via binding to the ACSL4 promoter region (Latunde-Dada, 2017; Li et al., 2019). However, studies related to this field remain limited. Thus in the future, more studies should be done to reveal the specific mechanism concerning the role of ACSL4 in stroke.
Metabolism of Iron in Stroke
Iron Overload Leads to Ferroptosis After Stroke
Before the concept of ferroptosis is defined, people have already recognized that the accumulation of iron in the brain will contribute to secondary brain injury (Wu et al., 2003; Lou et al., 2009; Liu et al., 2019). Currently, iron overload is thought to be a critical cause to trigger ferroptosis after ischemic stroke owning to its role of increasing mitochondrial oxidative damage and infarct volume (Lipscomb et al., 1998; Chi et al., 2000; Castellanos et al., 2002). Additionally, a similar phenomenon is also observed in hemorrhagic stroke. For example, iron overload can induce mitochondrial damage in hippocampal neurons (Park et al., 2015) and increase significant perihematomal edema after ICH (Xi et al., 2006; Garton et al., 2016). The occurrence of iron overload after stroke is involved in two aspects: iron influx and iron efflux. The iron influx and efflux greatly depend on the iron metabolism-related proteins including TF, TFR1, DMT1, FT, and FPN1, as we mentioned above. These proteins are mostly controlled by the concentration of iron (Gill et al., 2018). Iron regulatory proteins (IRPs) are a class of proteins that can bind to iron-responsive elements (IREs) in specific mRNAs of iron metabolism-related proteins and control their usage. When the iron supply is decreased in the cell, IRPs will effectively bind to the IRE in mRNA of FT and FPN1, inhibiting the translation of FT and FPN1 (Eisenstein and Blemings, 1998). Conversely, when iron overload happens, the RNA binding function of IRPs is inactive in FT and FPN1 mRNA but active in TF mRNA, so the translation of FT and FPN1 is increased whereas the translation of TF is decreased, thereby preventing the iron import and prompting iron export (Eisenstein and Blemings, 1998; Silva and Faustino, 2015). However, studies found that the expression of TF is upregulated after stroke (Wu et al., 2003; DeGregorio-Rocasolano et al., 2018). This contradictory phenomenon indicates that there must be another undiscovered mechanism regulating TF. Apart from that, Hepcidin, a cysteine-rich polypeptide encoded by the human HAMP gene and secreted by the liver (Reichert et al., 2017), also has been identified as an important regulator of iron homeostasis recently (Almutairi et al., 2019). Hepcidin can bind to the iron efflux protein FPN1, thereby inhibiting its activity and promoting its degradation, finally suppressing the iron excretion (Gulec et al., 2014) and causing iron overload and oxidative stress damage in both ischemic and hemorrhagic stroke (Słomka et al., 2015; Tan et al., 2016; Xiong et al., 2016). It has been illustrated that iron overload in hepatocytes can activate the BMP6 signaling pathway and increase Hepcidin expression by regulating samd transcription factors (Ramos et al., 2011); inflammatory factors such as IL-6 can increase Hepcidin expression by activating the STAT3 signaling pathway (Wrighting and Andrews, 2006; Ding et al., 2011); hypoxia can inhibit Hepcidin expression by inducing erythropoiesis or secretion of PDGF-BB (Sonnweber et al., 2014). Furthermore, histone deacetylase (HDAC), a critical factor in regulating gene transcription closely related to cell proliferation and cell death (Gallinari et al., 2007), may play an important role in controlling the transcription of the HAMP gene. Drug screening experiments confirmed that HDAC inhibitors Pan have the potential to increase Hepcidin expression (Gaun et al., 2014), and in vitro studies also proved that hepatocytes could increase the expression of HAMP and Hepcidin (Mleczko-Sanecka et al., 2017). Therefore, it is highly possible that HDAC regulates iron overload and ferroptosis at the gene level of Hepcidin after stroke.
Metabolism of Amino Acids in Stroke
GPX4 Inhibits Ferroptosis After Stroke
The expression of GPX4 is closely related to ferroptosis after stroke as it is able to inhibit lipid peroxidation. It has been shown that the upregulation of GPX4 can alleviate ferroptosis in both ischemic and hemorrhagic stroke. Alim et al. (2019) found that Selenium could promote the expression of antioxidant GPX4 to inhibit ferroptosis and protect neurons by activating the transcription factors TFAP2c and Sp1 in a hemorrhagic stroke rat model. Zhang et al. (2018) found that the genetic-upregulation of GPX4 efficiently relieved the secondary brain injury after ICH in the rat model including brain edema, blood-brain barrier leakage, and neuronal dysfunction. On the contrary, knocking out of GPX4 gene will aggravate brain injury (Zhang et al., 2018). Belayev et al. (2011) reported that Docosahexaenoic acid (DHA), an essential omega-3 fatty acid particularly abundant in nerve cell membrane phospholipids, may modulate the expression of the GPX4 gene via upregulating the Gpx4 Cytoplasmic Intron-sequence Retaining Transcripts (CIRT), which is an innovative Gpx4 splicing variant (Casañas-Sánchez et al., 2015). Overexpression of CIRT can harbor part of the initial intronic region, thereby increasing the capability of GPX4 to offer neuronal protection against oxidation based on the “sentinel RNA theory” (Casañas-Sánchez et al., 2015).
Overexpression of System xc– Will Aggravate the Ferroptosis After Stroke
As is aforementioned, system xc– serves as a glutamate/cysteine antiporter to produce GSH and GPX4, therefore it has a positive effect on inhibiting ferroptosis. The glutamate/cysteine exchange relies on the gradient of glutamate toward the cell rather than consuming adenosine triphosphate (ATP). When a stroke occurs, the concentration of extracellular glutamate will be escalated due to the lower glutamate uptake, increased exocytotic vesicular liberation, and non-vesicular glutamate release (Seki et al., 1999; Jabaudon et al., 2000), thereby leading to glutamate toxicity (Mou et al., 2019). As a result, the exchange will be blocked (Ignowski et al., 2018) and the production of GPX4 will be inhibited, finally initiating ferroptosis (Massie et al., 2015). The dysfunction of system xc– may be triggered by the inactivation of its specific subunit xCT. However, the upregulation of system xc– degree will result in greater glutamate release and excitotoxic impairment to oligodendrocytes (Pampliega et al., 2011). Similarly, astrocytic system xc– can aggravate cerebral ischemic injury via regulating the deleterious and excitotoxic effects of IL-1β. Also, the upregulation of xCT can induce long-lasting glutamate excitotoxicity in an ischemic rat model (Hsieh et al., 2017). Taken together, these studies indicate that when the concentration of system xc– reaches a certain threshold, it may aggravate the glutamate toxicity and ferroptosis rather than inhibit it. However, more studies are needed to confirm the specific role of system xc– in stroke ferroptosis.
Conclusion and Prospects
There is no doubt that ferroptosis plays a critical role in the progression and toxicity of ischemic stroke and hemorrhagic stroke, however, some confusing questions still need to be elucidated. First, although iron overload is thought to be the key factor to trigger ferroptosis, whether other metal ions are involved in ferroptosis remains unclear, as the latest research has revealed that copper and calcium may be also associated with the initiation of ferroptosis (Lewerenz et al., 2018; Maher et al., 2018; Weiland et al., 2019). Second, although people have known the important role of GPX and system xc– in ferroptosis, the specific mechanism of regulating the change of GPX4 and system xc– at the gene level is still not clear. Furthermore, while a series of studies have proved that ferroptosis can aggravate secondary brain injury after stroke, the underlying mechanisms and signaling pathways involved are still not understood. Finally, the relationship between ferroptosis, apoptosis, and autophagy remains unclear. It has been shown that ferroptosis, autophagy, and apoptosis will occur together in neuronal death after ICH via ultrastructural analysis (Li et al., 2018), however, whether ferroptosis can promote apoptosis or autophagy, or they can suppress each other after stroke requires to be studied in the years to come.
To recap, ferroptosis stands for a distinct type of programmed cell death, with a lot of its physiological functions yet to be specified. We believe that with the development of new biological technologies, more and more pathophysiological and physiological roles of ferroptosis will be gradually revealed.
Statements
Author contributions
YZ, XL, and BT manuscript writing. WL and TL manuscript revision and final approval of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 82001278) and the Fund for Young Doctors with The First People’s Hospital of Yunnan Province (Grant No. KHBS-2020-014).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Agmon E. Solon J. Bassereau P. Stockwell B. R. (2018). Modeling the effects of lipid peroxidation during ferroptosis on membrane properties.Sci. Rep.8:5155.
2
Alim I. Caulfield J. T. Chen Y. Swarup V. Geschwind D. H. Ivanova E. et al (2019). Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke.Cell1771262–1279.e25.
3
Almutairi M. M. A. Xu G. Shi H. (2019). Iron pathophysiology in stroke.Adv. Exp. Med. Biol.1173105–123. 10.1007/978-981-10-1430-7_9
4
Angeli J. P. F. Shah R. Pratt D. A. Conrad M. (2017). Ferroptosis inhibition: mechanisms and opportunities.Trends Pharmacol. Sci.38489–498. 10.1016/j.tips.2017.02.005
5
Ayton S. Lei P. Duce J. A. Wong B. X. Sedjahtera A. Adlard P. A. et al (2013). Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease.Ann. Neurol.73554–559. 10.1002/ana.23817
6
Ayton S. Lei P. Hare D. J. Duce J. A. George J. L. Adlard P. A. et al (2015). Parkinson’s disease iron deposition caused by nitric oxide-induced loss of β-amyloid precursor protein.J. Neurosci.353591–3597. 10.1523/jneurosci.3439-14.2015
7
Baranova O. Miranda L. F. Pichiule P. Dragatsis I. Johnson R. S. Chavez J. C. (2007). Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia.J. Neurosci.276320–6332. 10.1523/jneurosci.0449-07.2007
8
Belayev L. Khoutorova L. Atkins K. D. Eady T. N. Hong S. Lu Y. et al (2011). Docosahexaenoic Acid therapy of experimental ischemic stroke.Transl. Stroke Res.233–41. 10.1007/s12975-010-0046-0
9
Cao Y. Li Y. He C. Yan F. Li J. R. Xu H. Z. et al (2021). Selective ferroptosis inhibitor liproxstatin-1 attenuates neurological deficits and neuroinflammation after subarachnoid hemorrhage.Neurosci. Bull.10.1007/s12264-020-00620-5[Epub ahead of print].
10
Casañas-Sánchez V. Pérez J. A. Fabelo N. Quinto-Alemany D. Díaz M. L. (2015). Docosahexaenoic (DHA) modulates phospholipid-hydroperoxide glutathione peroxidase (Gpx4) gene expression to ensure self-protection from oxidative damage in hippocampal cells.Front. Physiol.6:203. 10.3389/fphys.2015.00203
11
Castellanos M. Puig N. Carbonell T. Castillo J. Martinez J. Rama R. et al (2002). Iron intake increases infarct volume after permanent middle cerebral artery occlusion in rats.Brain Res.9521–6. 10.1016/s0006-8993(02)03179-7
12
Chang C. F. Cho S. Wang J. (2014). (-)-Epicatechin protects hemorrhagic brain via synergistic Nrf2 pathways.Ann. Clin. Transl. Neurol.1258–271. 10.1002/acn3.54
13
Chen B. Chen Z. Liu M. Gao X. Cheng Y. Wei Y. et al (2019). Inhibition of neuronal ferroptosis in the acute phase of intracerebral hemorrhage shows long-term cerebroprotective effects.Brain Res. Bull.153122–132. 10.1016/j.brainresbull.2019.08.013
14
Chen J. Wang Y. Wu J. Yang J. Li M. Chen Q. (2020). The potential value of targeting ferroptosis in early brain injury after acute CNS disease.Front. Mol. Neurosci.13:110. 10.3389/fnmol.2020.00110
15
Chen L. Hambright W. S. Na R. Ran Q. (2015). Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis.J. Biol. Chem.29028097–28106. 10.1074/jbc.m115.680090
16
Chi S. I. Wang C. K. Chen J. J. Chau L. Y. Lin T. N. (2000). Differential regulation of H- and L-ferritin messenger RNA subunits, ferritin protein and iron following focal cerebral ischemia-reperfusion.Neuroscience100475–484. 10.1016/s0306-4522(00)00317-1
17
Cong L. Dong X. Wang Y. Deng Y. Li B. Dai R. (2019). On the role of synthesized hydroxylated chalcones as dual functional amyloid-β aggregation and ferroptosis inhibitors for potential treatment of Alzheimer’s disease.Eur. J. Med. Chem.16611–21. 10.1016/j.ejmech.2019.01.039
18
Cordonnier C. Sprigg N. Sandset E. C. Pavlovic A. Sunnerhagen K. S. Caso V. (2017). Stroke in women - from evidence to inequalities.Nat. Rev. Neurol.13521–532. 10.1038/nrneurol.2017.95
19
Dalleau S. Baradat M. Guéraud F. Huc L. (2013). Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance.Cell Death Differ.201615–1630. 10.1038/cdd.2013.138
20
Das U. N. (2019). Saturated fatty acids, MUFAs and PUFAs regulate ferroptosis.Cell Chem. Biol.26309–311. 10.1016/j.chembiol.2019.03.001
21
DeGregorio-Rocasolano N. Martí-Sistac O. Ponce J. Castelló-Ruiz M. Millán M. Guirao V. et al (2018). Iron-loaded transferrin (Tf) is detrimental whereas iron-free Tf confers protection against brain ischemia by modifying blood Tf saturation and subsequent neuronal damage.Redox Biol.15143–158. 10.1016/j.redox.2017.11.026
22
Ding H. Yan C. Z. Shi H. Zhao Y. S. Chang S. Y. Yu P. et al (2011). Hepcidin is involved in iron regulation in the ischemic brain.PLoS One6:e25324. 10.1371/journal.pone.0025324
23
Dixon S. J. Lemberg K. M. Lamprecht M. R. Skouta R. Zaitsev E. M. Gleason C. E. et al (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death.Cell1491060–1072. 10.1016/j.cell.2012.03.042
24
Dixon S. J. Winter G. E. Musavi L. S. Lee E. D. Snijder B. Rebsamen M. et al (2015). Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death.ACS Chem. Biol.101604–1609. 10.1021/acschembio.5b00245
25
Doll S. Conrad M. (2017). Iron and ferroptosis: a still ill-defined liaison.IUBMB Life69423–434. 10.1002/iub.1616
26
Doll S. Proneth B. Tyurina Y. Y. Panzilius E. Kobayashi S. Ingold I. et al (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition.Nat. Chem. Biol.1391–98. 10.1038/nchembio.2239
27
Dolma S. Lessnick S. L. Hahn W. C. Stockwell B. R. (2003). Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells.Cancer Cell3285–296. 10.1016/s1535-6108(03)00050-3
28
Dong X. P. Cheng X. Mills E. Delling M. Wang F. Kurz T. et al (2008). The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel.Nature455992–996. 10.1038/nature07311
29
Eisenstein R. S. Blemings K. P. (1998). Iron regulatory proteins, iron responsive elements and iron homeostasis.J. Nutr.1282295–2298. 10.1093/jn/128.12.2295
30
Fan Z. Wirth A. K. Chen D. Wruck C. J. Rauh M. Buchfelder M. et al (2017). Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis.Oncogenesis6:e371. 10.1038/oncsis.2017.65
31
Feng Y. Madungwe N. B. Imam Aliagan A. D. Tombo N. Bopassa J. C. (2019). Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels.Biochem. Biophys. Res. Commun.520606–611. 10.1016/j.bbrc.2019.10.006
32
Friedmann Angeli J. P. Schneider M. Proneth B. Tyurina Y. Y. Tyurin V. A. Hammond V. J. et al (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice.Nat. Cell Biol.161180–1191. 10.1038/ncb3064
33
Fujii J. Homma T. Kobayashi S. (2019). Ferroptosis caused by cysteine insufficiency and oxidative insult.Free Radic. Res.[Epub ahead of print].
34
Gaberel T. Gakuba C. Zheng Y. Lépine M. Lo E. H. van Leyen K. (2019). Impact of 12/15-lipoxygenase on brain injury after subarachnoid hemorrhage.Stroke50520–523. 10.1161/strokeaha.118.022325
35
Gallinari P. Di Marco S. Jones P. Pallaoro M. Steinkuhler C. (2007). HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics.Cell Res.17195–211. 10.1038/sj.cr.7310149
36
Galluzzi L. Vitale I. Abrams J. M. Alnemri E. S. Baehrecke E. H. Blagosklonny M. V. et al (2012). Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012.Cell Death Differ.19107–120. 10.1038/cdd.2011.96
37
Garton T. Keep R. F. Hua Y. Xi G. (2016). Brain iron overload following intracranial haemorrhage.Stroke Vasc. Neurol.1172–184. 10.1136/svn-2016-000042
38
Gaschler M. M. Andia A. A. Liu H. Csuka J. M. Hurlocker B. Vaiana C. A. et al (2018). FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation.Nat. Chem. Biol.14507–515. 10.1038/s41589-018-0031-6
39
Gaschler M. M. Stockwell B. R. (2017). Lipid peroxidation in cell death.Biochem. Biophys. Res. Commun.482419–425.
40
Gaun V. Patchen B. Volovetz J. Zhen A. W. Andreev A. Pollastri M. P. et al (2014). A chemical screen identifies small molecules that regulate hepcidin expression.Blood Cells Mol. Dis.53231–240. 10.1016/j.bcmd.2014.06.002
41
Gill D. Monori G. Tzoulaki I. Dehghan A. (2018). Iron status and risk of stroke.Stroke492815–2821. 10.1161/strokeaha.118.022701
42
Guan X. Li X. Yang X. Yan J. Shi P. Ba L. et al (2019). The neuroprotective effects of carvacrol on ischemia/reperfusion-induced hippocampal neuronal impairment by ferroptosis mitigation.Life Sci235:116795. 10.1016/j.lfs.2019.116795
43
Gubern C. Camós S. Ballesteros I. Rodríguez R. Romera V. G. Cañadas R. et al (2013). miRNA expression is modulated over time after focal ischaemia: up-regulation of miR-347 promotes neuronal apoptosis.FEBS J.2806233–6246. 10.1111/febs.12546
44
Gulec S. Anderson G. J. Collins J. F. (2014). Mechanistic and regulatory aspects of intestinal iron absorption.Am. J. Physiol. Gastrointest. Liver Physiol.307G397–G409.
45
Hamaï A. Mehrpour M. (2017). [Autophagy and iron homeostasis].Med. Sci.33260–267.
46
Han C. Liu Y. Dai R. Ismail N. Su W. Li B. (2020). Ferroptosis and its potential role in human diseases.Front. Pharmacol.11:239. 10.3389/fphar.2020.00239
47
Hinman A. Holst C. R. Latham J. C. Bruegger J. J. Ulas G. McCusker K. P. et al (2018). Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase.PLoS One13:e0201369. 10.1371/journal.pone.0201369
48
Hirschhorn T. Stockwell B. R. (2019). The development of the concept of ferroptosis.Free Radic. Biol. Med.133130–143. 10.1016/j.freeradbiomed.2018.09.043
49
Hsieh C. H. Lin Y. J. Chen W. L. Huang Y. C. Chang C. W. Cheng F. C. et al (2017). HIF-1α triggers long-lasting glutamate excitotoxicity via system x(c)(-) in cerebral ischaemia-reperfusion.J. Pathol.241337–349. 10.1002/path.4838
50
Ignowski E. Winter A. N. Duval N. Fleming H. Wallace T. Manning E. et al (2018). The cysteine-rich whey protein supplement, Immunocal®, preserves brain glutathione and improves cognitive, motor, and histopathological indices of traumatic brain injury in a mouse model of controlled cortical impact.Free Radic. Biol. Med.124328–341. 10.1016/j.freeradbiomed.2018.06.026
51
Imai H. Matsuoka M. Kumagai T. Sakamoto T. Koumura T. (2017). Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis.Curr. Top. Microbiol. Immunol.403143–170. 10.1007/82_2016_508
52
Jabaudon D. Scanziani M. Gähwiler B. H. Gerber U. (2000). Acute decrease in net glutamate uptake during energy deprivation.Proc. Natl. Acad. Sci. U.S.A.975610–5615. 10.1073/pnas.97.10.5610
53
Ji C. Kosman D. J. (2015). Molecular mechanisms of non-transferrin-bound and transferring-bound iron uptake in primary hippocampal neurons.J. Neurochem.133668–683. 10.1111/jnc.13040
54
Jin G. Arai K. Murata Y. Wang S. Stins M. F. Lo E. H. et al (2008). Protecting against cerebrovascular injury: contributions of 12/15-lipoxygenase to edema formation after transient focal ischemia.Stroke392538–2543. 10.1161/strokeaha.108.514927
55
Kagan V. E. Mao G. Qu F. Angeli J. P. Doll S. Croix C. S. et al (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis.Nat. Chem. Biol.1381–90. 10.1038/nchembio.2238
56
Karatas H. Eun Jung J. Lo E. H. van Leyen K. (2018). Inhibiting 12/15-lipoxygenase to treat acute stroke in permanent and tPA induced thrombolysis models.Brain Res.1678123–128. 10.1016/j.brainres.2017.10.024
57
Karuppagounder S. S. Alin L. Chen Y. Brand D. Bourassa M. W. Dietrich K. et al (2018). N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E(2) to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice.Ann. Neurol.84854–872. 10.1002/ana.25356
58
Kim J. H. Lewin T. M. Coleman R. A. (2001). Expression and characterization of recombinant rat Acyl-CoA synthetases 1, 4, and 5. Selective inhibition by triacsin C and thiazolidinediones.J. Biol. Chem.27624667–24673. 10.1074/jbc.m010793200
59
Knutson M. D. (2019). Non-transferrin-bound iron transporters.Free Radic. Biol. Med.133101–111. 10.1016/j.freeradbiomed.2018.10.413
60
Kroemer G. Galluzzi L. Vandenabeele P. Abrams J. Alnemri E. S. Baehrecke E. H. et al (2009). Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009.Cell Death Differ.163–11. 10.1038/cdd.2008.150
61
Lachaier E. Louandre C. Godin C. Saidak Z. Baert M. Diouf M. et al (2014). Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors.Anticancer Res.346417–6422.
62
Lan B. Ge J. W. Cheng S. W. Zheng X. L. Liao J. He C. et al (2020). Extract of Naotaifang, a compound Chinese herbal medicine, protects neuron ferroptosis induced by acute cerebral ischemia in rats.J. Integr. Med.18344–350. 10.1016/j.joim.2020.01.008
63
Latunde-Dada G. O. (2017). Ferroptosis: role of lipid peroxidation, iron and ferritinophagy.Biochim. Biophys. Acta Gen. Subj.18611893–1900. 10.1016/j.bbagen.2017.05.019
64
LeBlanc R. H. III Chen R. Selim M. H. Hanafy K. A. (2016). Heme oxygenase-1-mediated neuroprotection in subarachnoid hemorrhage via intracerebroventricular deferoxamine.J. Neuroinflammation13:244.
65
Lee J. Kosaras B. Del Signore S. J. Cormier K. McKee A. Ratan R. R. et al (2011). Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice.Acta Neuropathol.121487–498. 10.1007/s00401-010-0788-5
66
Lewerenz J. Ates G. Methner A. Conrad M. Maher P. (2018). Oxytosis/Ferroptosis-(Re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system.Front. Neurosci.12:214. 10.3389/fnins.2018.00214
67
Li Q. Han X. Lan X. Gao Y. Wan J. Durham F. et al (2017). Inhibition of neuronal ferroptosis protects hemorrhagic brain.JCI Insight2:e90777.
68
Li Q. Weiland A. Chen X. Lan X. Han X. Durham F. et al (2018). Ultrastructural characteristics of neuronal death and white matter injury in mouse brain tissues after intracerebral hemorrhage: coexistence of ferroptosis, autophagy, and necrosis.Front. Neurol.9:581. 10.3389/fneur.2018.00581
69
Li Y. Feng D. Wang Z. Zhao Y. Sun R. Tian D. et al (2019). Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion.Cell Death Differ.262284–2299. 10.1038/s41418-019-0299-4
70
Li Y. Liu Y. Wu P. Tian Y. Liu B. Wang J. et al (2021). Inhibition of ferroptosis alleviates early brain injury after subarachnoid hemorrhage in vitro and in vivo via reduction of lipid peroxidation.Cell. Mol. Neurobiol.41263–278. 10.1007/s10571-020-00850-1
71
Liang C. Zhang X. Yang M. Dong X. (2019). Recent progress in ferroptosis inducers for cancer therapy.Adv. Mater.31:e1904197.
72
Lipscomb D. C. Gorman L. G. Traystman R. J. Hurn P. D. (1998). Low molecular weight iron in cerebral ischemic acidosis in vivo.Stroke29487–492. 10.1161/01.str.29.2.487
73
Liu H. Hua Y. Keep R. F. Xi G. (2019). Brain ceruloplasmin expression after experimental intracerebral hemorrhage and protection against iron-induced brain injury.Transl. Stroke Res.10112–119. 10.1007/s12975-018-0669-0
74
Liu J. Guo Z. N. Yan X. L. Huang S. Ren J. X. Luo Y. et al (2020). Crosstalk between autophagy and ferroptosis and its putative role in ischemic stroke.Front. Cell. Neurosci.14:577403. 10.3389/fncel.2020.577403
75
Liu J. J. Green P. John Mann J. Rapoport S. I. Sublette M. E. (2015). Pathways of polyunsaturated fatty acid utilization: implications for brain function in neuropsychiatric health and disease.Brain Res.1597220–246. 10.1016/j.brainres.2014.11.059
76
Liu L. Cao Y. Chen C. Zhang X. McNabola A. Wilkie D. et al (2006). Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5.Cancer Res.6611851–11858. 10.1158/0008-5472.can-06-1377
77
Lou M. Lieb K. Selim M. (2009). The relationship between hematoma iron content and perihematoma edema: an MRI study.Cerebrovasc. Dis.27266–271. 10.1159/000199464
78
Lu S. C. (2013). Glutathione synthesis.Biochim. Biophys. Acta18303143–3153.
79
Ma S. Dielschneider R. F. Henson E. S. Xiao W. Choquette T. R. Blankstein A. R. et al (2017). Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells.PLoS One12:e0182921. 10.1371/journal.pone.0182921
80
MacKenzie E. L. Iwasaki K. Tsuji Y. (2008). Intracellular iron transport and storage: from molecular mechanisms to health implications.Antioxid. Redox Signal.10997–1030. 10.1089/ars.2007.1893
81
Maher P. van Leyen K. Dey P. N. Honrath B. Dolga A. Methner A. (2018). The role of Ca(2+) in cell death caused by oxidative glutamate toxicity and ferroptosis.Cell Calcium7047–55. 10.1016/j.ceca.2017.05.007
82
Maiorino M. Conrad M. Ursini F. (2018). GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues.Antioxid. Redox Signal.2961–74. 10.1089/ars.2017.7115
83
Masaldan S. Bush A. I. Devos D. Rolland A. S. Moreau C. (2019). Striking while the iron is hot: iron metabolism and ferroptosis in neurodegeneration.Free Radic. Biol. Med.133221–233. 10.1016/j.freeradbiomed.2018.09.033
84
Massie A. Boillée S. Hewett S. Knackstedt L. Lewerenz J. (2015). Main path and byways: non-vesicular glutamate release by system xc(-) as an important modifier of glutamatergic neurotransmission.J. Neurochem.1351062–1079. 10.1111/jnc.13348
85
Mleczko-Sanecka K. da Silva A. R. Call D. Neves J. Schmeer N. Damm G. et al (2017). Imatinib and spironolactone suppress hepcidin expression.Haematologica1021173–1184. 10.3324/haematol.2016.162917
86
Mou Y. Wang J. Wu J. He D. Zhang C. Duan C. et al (2019). Ferroptosis, a new form of cell death: opportunities and challenges in cancer.J. Hematol. Oncol.12:34.
87
Pampliega O. Domercq M. Soria F. N. Villoslada P. Rodríguez-Antigüedad A. Matute C. (2011). Increased expression of cystine/glutamate antiporter in multiple sclerosis.J. Neuroinflammation8:63. 10.1186/1742-2094-8-63
88
Park J. Lee D. G. Kim B. Park S. J. Kim J. H. Lee S. R. et al (2015). Iron overload triggers mitochondrial fragmentation via calcineurin-sensitive signals in HT-22 hippocampal neuron cells.Toxicology33739–46. 10.1016/j.tox.2015.08.009
89
Patel S. A. Warren B. A. Rhoderick J. F. Bridges R. J. (2004). Differentiation of substrate and non-substrate inhibitors of transport system xc(-): an obligate exchanger of L-glutamate and L-cystine.Neuropharmacology46273–284. 10.1016/j.neuropharm.2003.08.006
90
Ramos E. Kautz L. Rodriguez R. Hansen M. Gabayan V. Ginzburg Y. et al (2011). Evidence for distinct pathways of hepcidin regulation by acute and chronic iron loading in mice.Hepatology531333–1341. 10.1002/hep.24178
91
Ranard K. M. Erdman J. W. Jr. (2018). Effects of dietary RRR α-tocopherol vs all-racemic α-tocopherol on health outcomes.Nutr. Rev.76141–153. 10.1093/nutrit/nux067
92
Reichert C. O. da Cunha J. Levy D. Maselli L. M. F. Bydlowski S. P. Spada C. (2017). Hepcidin: homeostasis and diseases related to iron metabolism.Acta Haematol.137220–236. 10.1159/000471838
93
Sayan-Ozacmak H. Ozacmak V. H. Barut F. Jakubowska-Dogru E. (2012). Rosiglitazone treatment reduces hippocampal neuronal damage possibly through alleviating oxidative stress in chronic cerebral hypoperfusion.Neurochem. Int.61287–290. 10.1016/j.neuint.2012.05.011
94
Seki Y. Feustel P. J. Keller R. W. Jr. Tranmer B. I. Kimelberg H. K. (1999). Inhibition of ischemia-induced glutamate release in rat striatum by dihydrokinate and an anion channel blocker.Stroke30433–440. 10.1161/01.str.30.2.433
95
Selim M. Foster L. D. Moy C. S. Xi G. Hill M. D. Morgenstern L. B. et al (2019). Deferoxamine mesylate in patients with intracerebral haemorrhage (i-DEF): a multicentre, randomised, placebo-controlled, double-blind phase 2 trial.Lancet Neurol.18428–438.
96
Silva B. Faustino P. (2015). An overview of molecular basis of iron metabolism regulation and the associated pathologies.Biochim. Biophys. Acta18521347–1359. 10.1016/j.bbadis.2015.03.011
97
Singh N. K. Rao G. N. (2019). Emerging role of 12/15-Lipoxygenase (ALOX15) in human pathologies.Prog. Lipid Res.7328–45. 10.1016/j.plipres.2018.11.001
98
Śliwińska A. Luty J. Aleksandrowicz-Wrona E. Małgorzewicz S. (2018). Iron status and dietary iron intake in vegetarians.Adv. Clin. Exp. Med.271383–1389. 10.17219/acem/70527
99
Słomka A. Świtoñska M. Żekanowska E. (2015). Hepcidin levels are increased in patients with acute ischemic stroke: preliminary report.J. Stroke Cerebrovasc. Dis.241570–1576. 10.1016/j.jstrokecerebrovasdis.2015.03.031
100
Sonnweber T. Nachbaur D. Schroll A. Nairz M. Seifert M. Demetz E. et al (2014). Hypoxia induced downregulation of hepcidin is mediated by platelet derived growth factor BB.Gut631951–1959. 10.1136/gutjnl-2013-305317
101
Steliga A. Kowiañski P. Czuba E. Waśkow M. Moryś J. Lietzau G. (2020). Neurovascular unit as a source of ischemic stroke biomarkers-limitations of experimental studies and perspectives for clinical application.Transl. Stroke Res.11553–579. 10.1007/s12975-019-00744-5
102
Stockwell B. R. Friedmann Angeli J. P. Bayir H. Bush A. I. Conrad M. Dixon S. J. et al (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease.Cell171273–285. 10.1016/j.cell.2017.09.021
103
Sun Y. Zheng Y. Wang C. Liu Y. (2018). Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells.Cell Death Dis.9:753.
104
Tan G. Liu L. He Z. Sun J. Xing W. Sun X. (2016). Role of hepcidin and its downstream proteins in early brain injury after experimental subarachnoid hemorrhage in rats.Mol. Cell. Biochem.41831–38. 10.1007/s11010-016-2730-1
105
Tuo Q. Z. Lei P. Jackman K. A. Li X. L. Xiong H. Li X. L. et al (2017). Tau-mediated iron export prevents ferroptotic damage after ischemic stroke.Mol. Psychiatry221520–1530. 10.1038/mp.2017.171
106
Ursini F. Maiorino M. (2020). Lipid peroxidation and ferroptosis: the role of GSH and GPx4.Free Radic. Biol. Med.152175–185. 10.1016/j.freeradbiomed.2020.02.027
107
van Leyen K. Holman T. R. Maloney D. J. (2014). The potential of 12/15-lipoxygenase inhibitors in stroke therapy.Future Med. Chem.61853–1855. 10.4155/fmc.14.129
108
Weiland A. Wang Y. Wu W. Lan X. Han X. Li Q. et al (2019). Ferroptosis and its role in diverse brain diseases.Mol. Neurobiol.564880–4893. 10.1007/s12035-018-1403-3
109
Winterbourn C. C. (1995). Toxicity of iron and hydrogen peroxide: the Fenton reaction.Toxicol. Lett.82-83969–974. 10.1016/0378-4274(95)03532-x
110
Wrighting D. M. Andrews N. C. (2006). Interleukin-6 induces hepcidin expression through STAT3.Blood1083204–3209. 10.1182/blood-2006-06-027631
111
Wu J. Hua Y. Keep R. F. Nakamura T. Hoff J. T. Xi G. (2003). Iron and iron-handling proteins in the brain after intracerebral hemorrhage.Stroke342964–2969. 10.1161/01.str.0000103140.52838.45
112
Xi G. Keep R. F. Hoff J. T. (2006). Mechanisms of brain injury after intracerebral haemorrhage.Lancet Neurol.553–63. 10.1016/s1474-4422(05)70283-0
113
Xie Y. Hou W. Song X. Yu Y. Huang J. Sun X. et al (2016). Ferroptosis: process and function.Cell Death Differ.23369–379. 10.1038/cdd.2015.158
114
Xiong X. Y. Liu L. Wang F. X. Yang Y. R. Hao J. W. Wang P. F. et al (2016). Toll-Like Receptor 4/MyD88-mediated signaling of hepcidin expression causing brain iron accumulation, oxidative injury, and cognitive impairment after intracerebral hemorrhage.Circulation1341025–1038. 10.1161/circulationaha.116.021881
115
Yagoda N. von Rechenberg M. Zaganjor E. Bauer A. J. Yang W. S. Fridman D. J. et al (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels.Nature447864–868.
116
Yan H. F. Tuo Q. Z. Yin Q. Z. Lei P. (2020). The pathological role of ferroptosis in ischemia/reperfusion-related injury.Zool. Res.41220–230. 10.24272/j.issn.2095-8137.2020.042
117
Yang W. S. SriRamaratnam R. Welsch M. E. Shimada K. Skouta R. Viswanathan V. S. et al (2014). Regulation of ferroptotic cancer cell death by GPX4.Cell156317–331. 10.1016/j.cell.2013.12.010
118
Yang W. S. Stockwell B. R. (2016). Ferroptosis: death by lipid peroxidation.Trends Cell Biol.26165–176. 10.1016/j.tcb.2015.10.014
119
Yang X. Park S. H. Chang H. C. Shapiro J. S. Vassilopoulos A. Sawicki K. T. (2017). Sirtuin 2 regulates cellular iron homeostasis via deacetylation of transcription factor NRF2.J. Clin. Invest.1271505–1516. 10.1172/jci88574
120
Yigitkanli K. Pekcec A. Karatas H. Pallast S. Mandeville E. Joshi N. et al (2013). Inhibition of 12/15-lipoxygenase as therapeutic strategy to treat stroke.Ann. Neurol.73129–135. 10.1002/ana.23734
121
Yuan H. Li X. Zhang X. Kang R. Tang D. (2016). Identification of ACSL4 as a biomarker and contributor of ferroptosis.Biochem. Biophys. Res. Commun.4781338–1343. 10.1016/j.bbrc.2016.08.124
122
Zhang Z. Wu Y. Yuan S. Zhang P. Zhang J. Li H. et al (2018). Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage.Brain Res.1701112–125. 10.1016/j.brainres.2018.09.012
123
Zhao H. Chen Y. Feng H. (2018). P2X7 receptor-associated programmed cell death in the pathophysiology of hemorrhagic stroke.Curr. Neuropharmacol.161282–1295. 10.2174/1570159x16666180516094500
124
Zille M. Karuppagounder S. S. Chen Y. Gough P. J. Bertin J. Finger J. et al (2017). Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis.Stroke481033–1043. 10.1161/strokeaha.116.015609
Summary
Keywords
ferroptosis, programmed cell death, iron overload, lipid peroxidation, ischemic stroke, hemorrhagic stroke, glutathione inhibition
Citation
Zhang Y, Lu X, Tai B, Li W and Li T (2021) Ferroptosis and Its Multifaceted Roles in Cerebral Stroke. Front. Cell. Neurosci. 15:615372. doi: 10.3389/fncel.2021.615372
Received
08 October 2020
Accepted
09 April 2021
Published
03 June 2021
Volume
15 - 2021
Edited by
Zhen-Ni Guo, First Affiliated Hospital of Jilin University, China
Reviewed by
Hulya Karatas, Hacettepe University, Turkey; Jian Liu, Capital Medical University, China
Updates
Copyright
© 2021 Zhang, Lu, Tai, Li and Li.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Li, taoli_kh@163.comWeijia Li, 11943953@qq.com
†These authors have contributed equally to this work and share first authorship
This article was submitted to Cellular Neuropathology, a section of the journal Frontiers in Cellular Neuroscience
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.