 Mohammad Sobhan Karvandi
Mohammad Sobhan Karvandi Farzam Sheikhzadeh Hesari2
Farzam Sheikhzadeh Hesari2 Amir Reza Aref
Amir Reza Aref Majid Mahdavi
Majid Mahdavi- 1Department of Cell and Molecular Sciences, Faculty of Biological Sciences, Kharazmi University, Tehran, Iran
- 2Department of Animal Biology, Faculty of Natural Sciences, University of Tabriz, Tabriz, Iran
- 3Department of Medical Oncology, Belfer Center for Applied Cancer Science, Dana-Farber Cancer Institute and Harvard Medical School, Boston, MA, United States
- 4Institute of Biochemistry and Biophysics, University of Tehran, Tehran, Iran
Neuronal loss is one of the striking causes of various central nervous system (CNS) disorders, including major neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and Amyotrophic lateral sclerosis (ALS). Although these diseases have different features and clinical manifestations, they share some common mechanisms of disease pathology. Progressive regional loss of neurons in patients is responsible for motor, memory, and cognitive dysfunctions, leading to disabilities and death. Neuronal cell death in neurodegenerative diseases is linked to various pathways and conditions. Protein misfolding and aggregation, mitochondrial dysfunction, generation of reactive oxygen species (ROS), and activation of the innate immune response are the most critical hallmarks of most common neurodegenerative diseases. Thus, endoplasmic reticulum (ER) stress, oxidative stress, and neuroinflammation are the major pathological factors of neuronal cell death. Even though the exact mechanisms are not fully discovered, the notable role of mentioned factors in neuronal loss is well known. On this basis, researchers have been prompted to investigate the neuroprotective effects of targeting underlying pathways to determine a promising therapeutic approach to disease treatment. This review provides an overview of the role of ER stress, oxidative stress, and neuroinflammation in neuronal cell death, mainly discussing the neuroprotective effects of targeting pathways or molecules involved in these pathological factors.
Introduction
Neurodegenerative diseases are nervous system disorders in which millions of people, especially the elderly, are being affected worldwide. The rising prevalence of these diseases has put the world with a serious challenge (Bloem et al., 2021; Milošević et al., 2021). Despite the developments in this field of study and advancements in pharmacological aspects, there is not a promising drug to consummately cure neurodegenerative diseases yet (Pohl and Kong Thoo Lin, 2018). However, there are still so many studies to alleviate disease symptoms and extend life span (Breijyeh and Karaman, 2020). Neurodegenerative diseases are mostly characterized by toxic protein aggregates with abnormal conformation within neurons or neuroglia, leading to memory, cognitive, and/or movement disorders (Dugger and Dickson, 2017). These diseases include a wide range of neurological disorders, but the major types are Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS; Lezi and Swerdlow, 2012). Protein misfolding and accumulation of amyloid-β (Aβ) and phosphorylated Tau is the major pathological feature in AD, as well as α-synuclein in PD, and mutant superoxide dismutase 1 (mSOD1) in ALS (Ghemrawi and Khair, 2020). Clinical manifestations in these kinds of diseases mainly occur as a consequence of neuron dysfunction or neuronal cell death (Andreone et al., 2020). Besides apoptosis, depending on the conditions, other different types of cell deaths, such as ferroptosis, necroptosis, and parthanatos are also possible to affect cell fidelity (Wang et al., 2018; Ferrada et al., 2020; Reichert et al., 2020; David et al., 2022; Mangalmurti and Lukens, 2022). The mitochondria and endoplasmic reticulum (ER) play crucial role in the occurrence of neuronal cell death among many other organelles (Gorman et al., 2012; Johnson et al., 2021; Markovinovic et al., 2022). Mitochondria are dynamic organelles that participate in producing energy and maintaining cellular redox balance, among many other functions (Johri and Beal, 2012). Therefore, mitochondrial dysfunction, including excessive reactive oxygen species (ROS) production, mitochondrial calcium overload, loss of the mitochondrial membrane potential leading to release of apoptosis-inducing factor (AIF), and other pro-apoptotic factors could lead to caspase activation and cell death (Culmsee and Plesnila, 2006; Kaminskyy and Zhivotovsky, 2014; Hoffmann et al., 2021). Evidence also reveals that mitochondrial DNA (mtDNA) mutations are present in patients with neurodegeneration (Johri and Beal, 2012). Also, aberrant ROS production and imbalance in antioxidant activity could influence mitochondria and impair mitochondria’s function, leading cells to death (Angelova and Abramov, 2018; Doroudian et al., 2021), which is explained in the following sections. Of note, ROS can also contribute to the production of protein aggregates and exacerbate disease pathology (Van Dam and Dansen, 2020).
On the other hand, the ER is a large and dynamic organelle responsible for protein folding and maturation. Once a protein folds with an abnormal conformation, the misfolded protein enters the ER-associated degradation (ERAD) pathway to prevent the following plausible detrimental effects of the protein (Schwarz and Blower, 2016). Aberrant misfolded proteins or aggregates can potentially trigger the process “Unfolded Protein Response” (UPR) to attenuate ER stress or initiate apoptosis pathways (Schwarz and Blower, 2016). UPR has three signaling arms, including IRE1-α, PERK, and ATF6, which are highly conserved pathways (Shi et al., 2022). However, toxic protein aggregates may also undergo degradation by lysosomes (i.e., autophagy), to ameliorate disease progression (Djajadikerta et al., 2020). Autophagy is able to activate or inhibit the apoptosis signaling to maintain intracellular balance or induce neuronal cell death (Gupta R. et al., 2021). All three UPR arms, Ca2+ release, and oxidative stress can directly or indirectly activate autophagy induction (Andhavarapu et al., 2019; Ramirez-Moreno et al., 2019; Ren et al., 2021). Although protein aggregates are the key reasons for the pathology of neurodegenerative diseases, other factors, including activation of glutamate ionotropic receptors, excitotoxicity from dysregulation of neuronal calcium homeostasis, dysfunction of lysosomes, aberrant cell-cycle re-entry, and impairments in axonal transport and synaptic function can also contribute to neuronal injury or death in various neurodegenerative diseases such as AD and PD (Emerit et al., 2004; Fricker et al., 2018; Sushma and Mondal, 2019; Behl et al., 2021; Hoffmann et al., 2021). In addition, increased levels of inflammatory factors in the serum and brain tissue, known as neuroinflammation, participates in the pathophysiology of neurodegenerative diseases (Calsolaro and Edison, 2016). Emerging evidence indicates that neuroinflammation can be the cause and consequence of both ER stress and oxidative stress (Salminen et al., 2009; Sochocka et al., 2013; Pintado et al., 2017). A neurotoxic microenvironment caused by the activation of microglial cells and release of cytotoxic inflammatory factors in the CNS can affect cell fidelity and induce neuronal cell death (Behl et al., 2021; Wu and Zou, 2022). This can be carried out by triggering pyroptosis, an inflammasome-mediated type of cell death (Kovacs and Miao, 2017). However, the undeniable contribution of age, genetics, and environmental factors in the disruption of neuronal homeostasis and subsequently neuronal cell death cannot be discounted (Bejanin et al., 2017).
There have been clinical trials targeting neuropathological hallmarks of neurodegenerative diseases, investigating glucagon-like peptide-1 receptor (GLP-1R) agonists, monoclonal antibodies against toxic protein aggregates, antioxidant agents, beta-secretase (BACE1) inhibitors and other receptor inhibitors such as 5HT-6 serotonin receptor inhibitor (Table 1, Hung and Fu, 2017). The results were controversial, as there was no evidence of beneficial effect on patients’ cognitive and functional status in most trials; while in some cases the condition of patients who received drugs worsened, compared with those who received placebo (Table 1, Egan et al., 2019). These results indicate that novel agents with different features must be studied and trialled. Given the complex interplay of ER stress, oxidative stress, and neuroinflammation in the pathology of most neurodegenerative diseases, developments in the knowledge of underlying mechanisms may be crucial for researchers to propose a promising therapeutic strategy to achieve a more efficient treatment for neurodegenerative diseases.
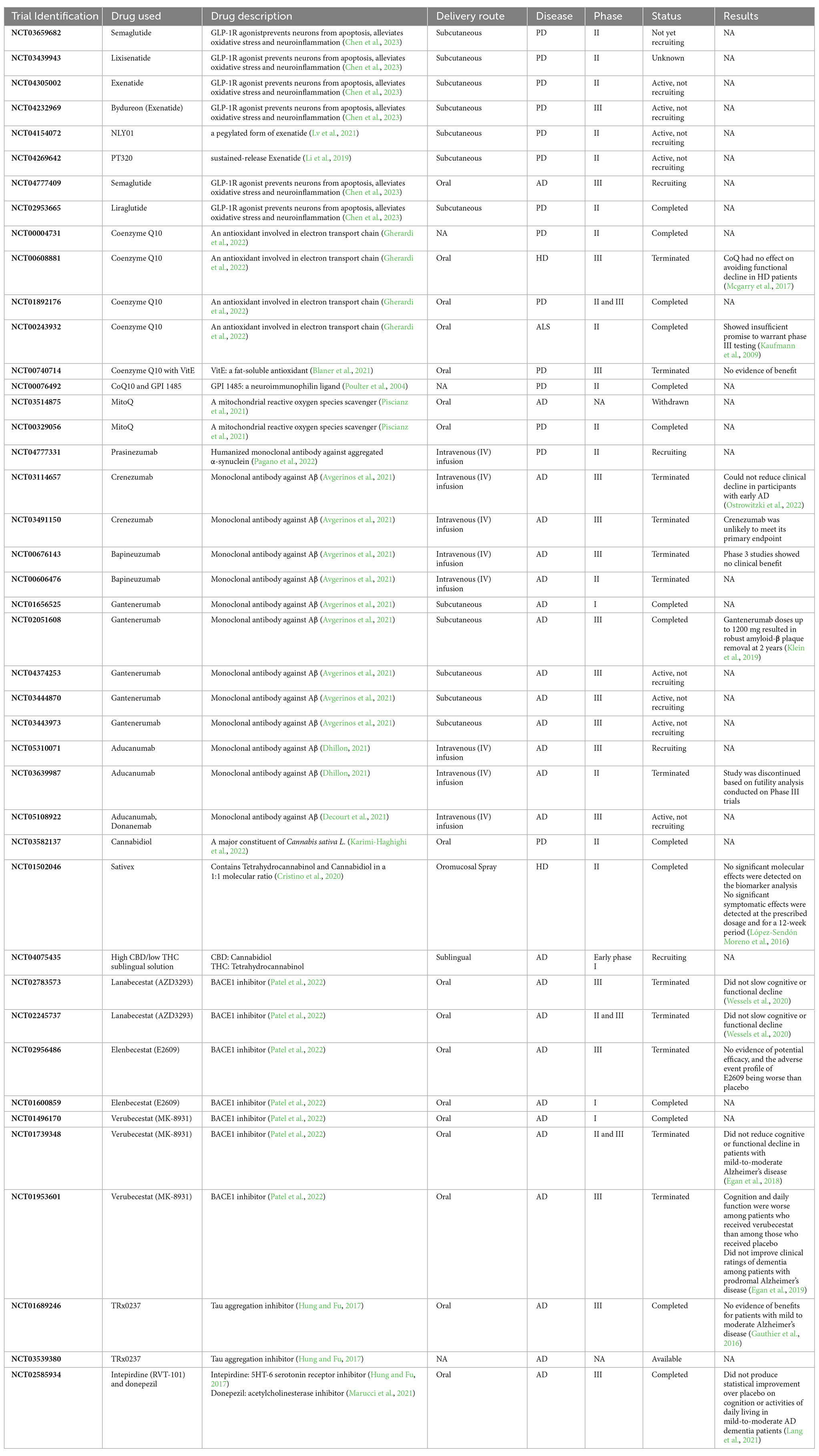
Table 1. Clinical trials associated with neurodegenerative diseases.
ER stress-induced cell death in neurodegenerative diseases
Mechanism of ER stress-induced apoptotic cell death
The ER is known as an organelle involved in protein maturation and folding (Read and Schröder, 2021). Toxic protein aggregates in neurodegenerative diseases, pathogen-associated molecular patterns (PAMPs), danger-associated molecular patterns (DAMPs), ROS, and reactive nitrogen species (RNS), can disrupt protein folding processes in the ER lumen, leading to ER stress (Zhang and Kaufman, 2008). In the condition of ER stress, the aggregation of unfolded or misfolded proteins within the ER lumen of neurons and neuroglia leads to failure of ER in maintaining protein homeostasis through UPR and ERAD. For instance, accumulation of tau protein in AD can affect essential components of ERAD and block this pathway, leading to the accumulation of more misfolded proteins in the ER lumen (Hetz and Saxena, 2017; Ghemrawi and Khair, 2020). Subsequently, UPR-dependent inflammation and apoptotic pathways are induced, resulting in neuronal cell death (Sprenkle et al., 2017; Ghemrawi and Khair, 2020). The ER stress can also be induced by ER Ca2+ dysregulation, impairments in vesicular trafficking, or any defects in UPR components (Cooper et al., 2006; Sprenkle et al., 2017). PKR-like ER kinase (PERK), inositol-requiring transmembrane kinase/endoribonuclease 1 α (IRE1α), and activating transcription factor 6 (ATF6) are three vital sensor proteins that are involved in UPR regulation (Ghemrawi and Khair, 2020). Under normal conditions, these proteins are inactive due to association with ER chaperone proteins such as Immunoglobulin binding protein (BiP) or 78 kDa glucose-regulated protein (GRP78), which are members of heat shock protein families (Halperin et al., 2014).
Under ER stress conditions, the misfolded proteins interact with the substrate binding domain of BiP. Consequently, BiP is released and leads to dimerization and auto-phosphorylation of PERK, as well as intramembrane proteolysis of ATF6 and phosphorylation of IRE1α. Subsequently, the UPR cascade activates to maintain protein homeostasis (Ghemrawi and Khair, 2020). To elaborate, the phosphorylation of the alpha subunit of eukaryotic translation initiation factor (eIF2α) followed by activation of PERK occurs through BiP dissociation. This process inhibits protein synthesis to prevent overload of proteins in the ER lumen (Hetz and Saxena, 2017; Almeida et al., 2022), therefore attempting to restore protein homeostasis (Da Silva et al., 2020). Besides, under prolonged ER stress conditions and failure in the UPR mechanism, p-eIF2α promotes activating transcription factor 4 (ATF4) translation, which enhances up-regulation of pro-apoptotic factors, including CHOP (also known as GADD153; Ghemrawi and Khair, 2020). Eventually, down-regulation of anti-apoptotic Bcl-2 family makes neurons more susceptible to death (Doyle et al., 2011; Hetz and Saxena, 2017; Da Silva et al., 2020; Figure 1A). Moreover, it has been claimed that TRB3 genes, GADD34, death receptor 5 (DR5), ER oxidase 1 (ERO1), and other apoptotic molecules can potentially receive apoptosis signals from CHOP and induce cell death (Taalab et al., 2018; Da Silva et al., 2020). ATF4 also induces transcription of the p53-upregulated modulator of apoptosis (PUMA), which results in ER-stress-induced neuronal apoptosis (Galehdar et al., 2010). Interestingly, experiments have indicated that CHOP could not induce apoptosis in PUMA-deficient neurons, demonstrating the key role of PUMA in CHOP-induced neuronal apoptosis (Galehdar et al., 2010). Moreover, in IRE1-α signaling pathway, the second arm of UPR, after the release of BiP by aggregated proteins, IRE1-α undergoes oligomerization and auto-phosphorylation. p-IRE1α facilitates neuronal death by activation of the apoptotic-signaling kinase-1 (ASK1) and other apoptotic factors as a result of c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38 MAPK) pathways activation (Ghemrawi and Khair, 2020). This pathway may induce p53 activation and lead to the upregulation of Bcl-2 associated X (BAX) protein, which triggers the release of cytochrome C (Cyt C) from the mitochondria to the cytosol and cause apoptotic neuronal cell death (Stefani et al., 2012). Furthermore, the RNase activity of IRE1α plays a critical role in splicing the mRNA coding for X-box binding protein 1 (XBP1) and increases the expression of genes involved in ER machinery, such as BiP (Lee et al., 2003; Hirota et al., 2006; Chen et al., 2022). Besides outlined functions, IRE1α participates in the degradation of some mRNAs and microRNAs, known as “regulated IRE1α-dependent decay” (RIDD; Hetz and Saxena, 2017; Figure 1B). Mutations in Presenilin 1 and 2 (PS1 and PS2), which are frequently involved in AD, can inhibit IRE1 and impair UPR, leading to AD pathology and neuronal cell death (Doyle et al., 2011). ATF6 is the third sensor protein of UPR cascades which is embedded in the ER membrane. By interaction of aggregated proteins with ATF6 in the ER lumen and release of BiP, ATF6 translocates to the Golgi apparatus and undergoes proteolysis. Subsequently, cleaved ATF6 induces transcription of ER chaperones and XBP1 in the nucleus and participates in protein homeostasis (Da Silva et al., 2020; Figure 1C).
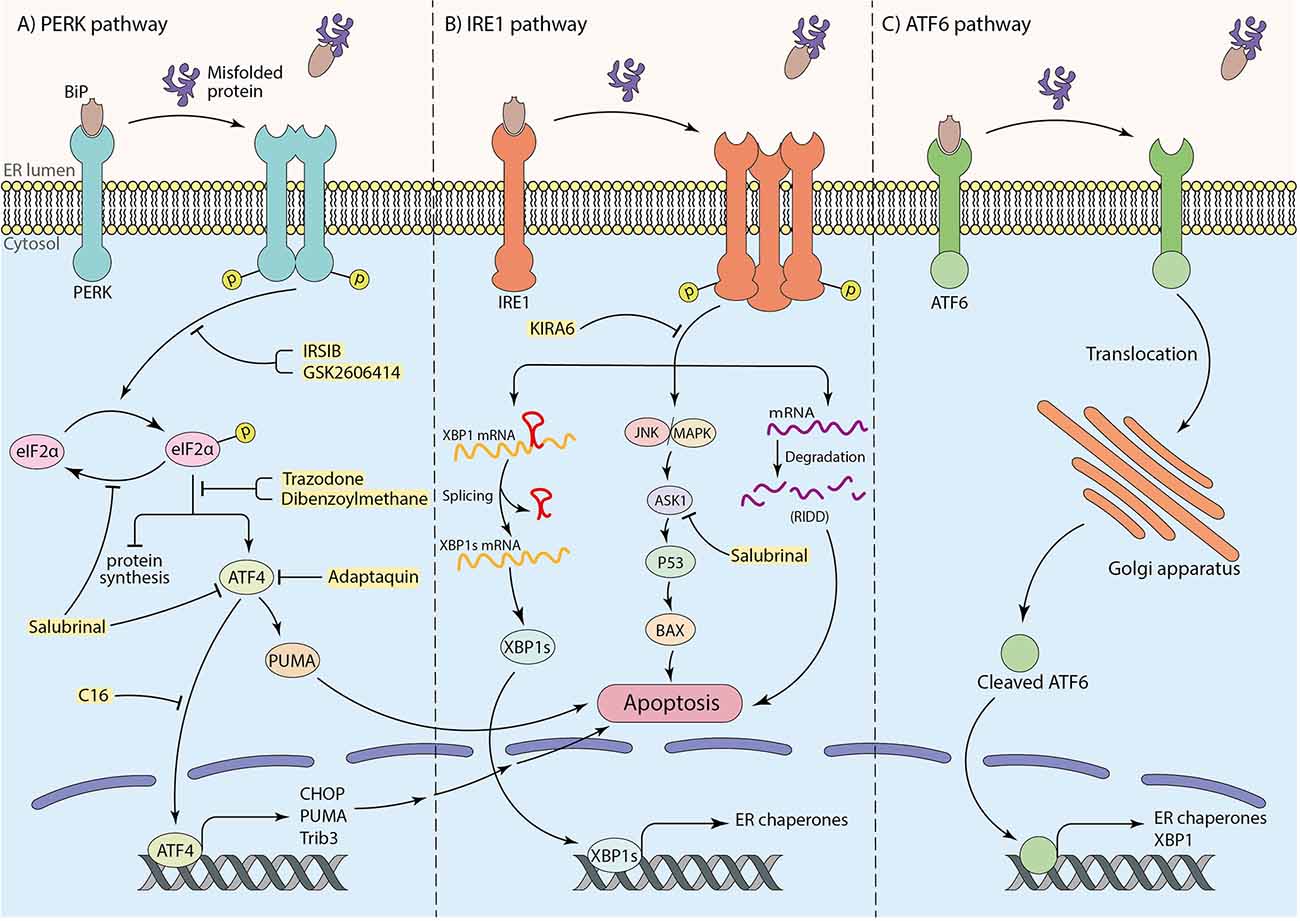
Figure 1. The role of three arms of UPR in inducing apoptosis and the neuroprotective effects of particular inhibitors (shown in yellow box). (A) The PERK pathway: interaction of substrate binding domain of BiP with misfolded or aggregated proteins leads to BiP dissociation, dimerization, and autophosphorylation of PERK, which further causes eIF2α phosphorylation. Phosphorylated eIF2α induces cell death by transcription of apoptotic factors by means of ATF4 transcription factor as well as inhibition of protein synthesis. (B) The IRE1 pathway: after dissociation of BiP from IRE1 receptor by misfolded or aggregated proteins in the ER lumen, IRE1 undergoes oligomerization and autophosphorylation. This results in mRNA degradation termed “regulated IRE1alpha-dependent decay” (RIDD) and inducing apoptotic factors by initiating JNK/MAPK cascade. To mitigate ER stress, the IRE1 pathway also leads to XBP1 mRNA splicing to transcript ER chaperones to improve ER machinery. (C) The ATF6 pathway: translocation of ATF6 to the Golgi apparatus as a result of BiP dissociation, and the proteolysis of ATF6 in Golgi brings out an activated ATF6 transcription factor to transcript ER chaperones and XBP1 for ER machinery.
ER stress-associated alterations of apoptotic factors
In AD, Aβ can trigger ER stress, mitochondrial fragmentation, and neuronal death through ER Ca2+ release by ryanodine receptors (RyRs) and inositol triphosphate receptors (IP3R; Chami and Checler, 2020). Based on studies, overexpression of RyRs contributed to Ca2+ dysregulation in AD mouse models and cell lines. In addition, increase in IP3 receptor-mediated Ca2+ signaling was indicated in AD patients’ fibroblast cells (Callens et al., 2021). Aβ oligomer-dependent ER stress responses can subsequently activate different kinases which phosphorylate specific epitopes on tau leading to the development of neurofibrillary tangles (NFTs) and propagating AD pathology (Sprenkle et al., 2017). Aβ peptides can activate ASK1 and JNK pathways, which can subsequently mediate ER stress-induced apoptosis (Ghemrawi and Khair, 2020). Both ASK1 and JNK were reported to be upregulated in transgenic mouse brains and post-mortem AD samples, respectively (Galvan et al., 2007; Sbodio et al., 2019). It has been revealed that CHOP activation plays a crucial role in the triggering and progression of pathological hallmarks of AD. In agreement, CHOP and its downstream effectors, including caspase-12 and GADD34, are markedly upregulated in the brains of AD patients (Ghemrawi and Khair, 2020). In addition, phosphorylated forms of PERK and eIF2α were significantly increased in the hippocampal pyramidal cells and frontal cortex of AD patients (Stutzbach et al., 2013). The evidence also shows that ER chaperones, including BiP, are also upregulated in the cerebrospinal fluid (CSF) and AD brains (Ghemrawi and Khair, 2020).
Mutations in PARK7, a gene involved in familial PD, might activate ASK1-induced neuronal death in PD. This can be due to the dysfunction in protecting against the Daxx-ASK1 cell death axis, which plays a key role in the completion of signaling pathways from cell surface death receptors (Chang et al., 1998; Homma et al., 2009). In addition, upregulation of ER stress markers, such as GRP78, p-PERK, and p-eIF2α in dopaminergic (DA) neurons of post-mortem PD samples (Shi et al., 2022), demonstrate their function in initiating apoptosis pathways, which could cause serious clinical implications. According to evidence, upregulation in ER stress markers, including BiP and CHOP in post-mortem HD brains, may be associated with neuronal death in HD (Shi et al., 2022). Mutation in genes such as SOD1, a gene encoding Superoxide dismutase 1 (SOD1), can also induce ER stress in neurons in ALS and cause neuronal damages (Sprenkle et al., 2017). ALS-associated mutations in vesicle-associated membrane protein-associated protein B (VAPB) can physically interact with ATF6 and disturb its natural function (Hetz and Saxena, 2017). Patients with ALS-associated VAPB mutations indicated malfunctions in Ca2+ signaling and storage, excessive ER stress, and neuronal death as a result of inhibition of ATF6 (Ghemrawi and Khair, 2020). Furthermore, upregulation of PERK, IRE1α, and ATF6 was found in the ALS mouse models (Ghemrawi and Khair, 2020; Zhao et al., 2022).
Targeting ER stress-induced apoptotic cell death
According to the critical role of ER stress in the occurrence of neuronal cell death in neurodegenerative diseases, targeting associated pathways seem to have hopeful effects on protecting neurons from death (Figure 1). Among three arms of UPR in ER stress conditions, the PERK pathway is the most well-studied in the neuroprotective effects of inhibition of ER stress. In parallel with this, Salubrinal, an anti-ER stress compound, has been well investigated in neurodegenerative disease pathology and treatment (Gupta S. et al., 2021; Ajoolabady et al., 2022). Salubrinal is an activator of UPR, which raises ER chaperone levels, including BiP. It inhibits eIF2α dephosphorylation which can attenuate neuronal death by interfering with death-related signaling pathways, including ATF4 or ASK1 (Figure 1, Table 2; Niso-Santano et al., 2011; Wu et al., 2014; Sprenkle et al., 2017). Accumulating evidence indicates that Salubrinal reduced ER accumulation of α-synuclein and significantly protected against α-synuclein-mediated dopaminergic (DA) neuronal death in transgenic mouse models (Colla et al., 2012). Also, Salubrinal reduced the accumulation of mutant huntingtin (mHTT) by upregulation of BiP and p-eIF2α, and prevent neuronal cell death (Maity et al., 2022). In addition, the drug Adaptaquin blocks Tribbles pseudokinase 3 (Trib3) induction by inhibiting ATF4 and CHOP activity probably through an eIF2α-independent mechanism, leading to neuronal protection in mouse models of PD (Figure 1, Table 2). More investigation is required for the neuroprotective effects of Adaptaquin in ER stress-induced neuronal cell death (Karuppagounder et al., 2016; Aime et al., 2020). Moreover, the PKR inhibitor “C16” can reduce transcriptional induction of pro-apoptotic target genes of ATF4, such as CHOP, Trib3, and PUMA (Figure 1, Table 2). This could significantly reduce MPP+ and 6-OHDA neurotoxin-induced neuronal cell death in PD models (Demmings et al., 2021). According to the experimental study, PUMA expression can be downregulated by directly targeting CHOP to decrease ER stress-induced neuronal apoptosis (Galehdar et al., 2010). Notably, pharmacological inhibition of ATF4, using imidazole-oxindole PKR inhibitor, indicated neuroprotection against neurotoxin-induced cell death in PD models (Demmings et al., 2021). Comparing motor neuron death in ATF4-ablated transgenic ALS mouse models with those expressing normal levels of ATF4 demonstrated the possible role of ATF4 ablation in neuroprotection against ALS by reducing apoptosis components, including CHOP (Matus et al., 2013). Likewise, another study revealed an increase in neuronal death in PD rat models by overexpression of ATF4 using recombinant Adeno-Associated Virus (rAAV; Gully et al., 2016). Halliday et al. (2017) revealed the inhibition of UPR-induced p-eIF2α signaling and neuronal survival by two chemical compounds termed “Trazodone” and “dibenzoylmethane” (DBM) in prion-infected mice (Figure 1, Table 2), presumably by reversing translational attenuation and lowering levels of ATF4 and CHOP which needs to be more inquired in other neurodegenerative diseases including AD and PD. Interestingly, it has been demonstrated that PERK inhibitor GSK2606414 (Figure 1, Table 2), despite its pancreatic toxicity (Halliday et al., 2015), inhibits and reduces PERK expression, which has a neuroprotective effect on DA neurons in Substantia Nigra pars compacta (SNpc) of PD mouse models, and improves the motor performance and neuronal excitability of PD mice (Mercado et al., 2018). In addition, inhibition of PERK signaling with IRSIB has been investigated in ALS rodent models, and a reduction in ATF4 and CHOP levels has been indicated (Figure 1, Table 2), which results in significant neuronal survival. In the same study, a reduction in IRE1-dependent signaling has also been indicated (Halliday et al., 2015, 2017).
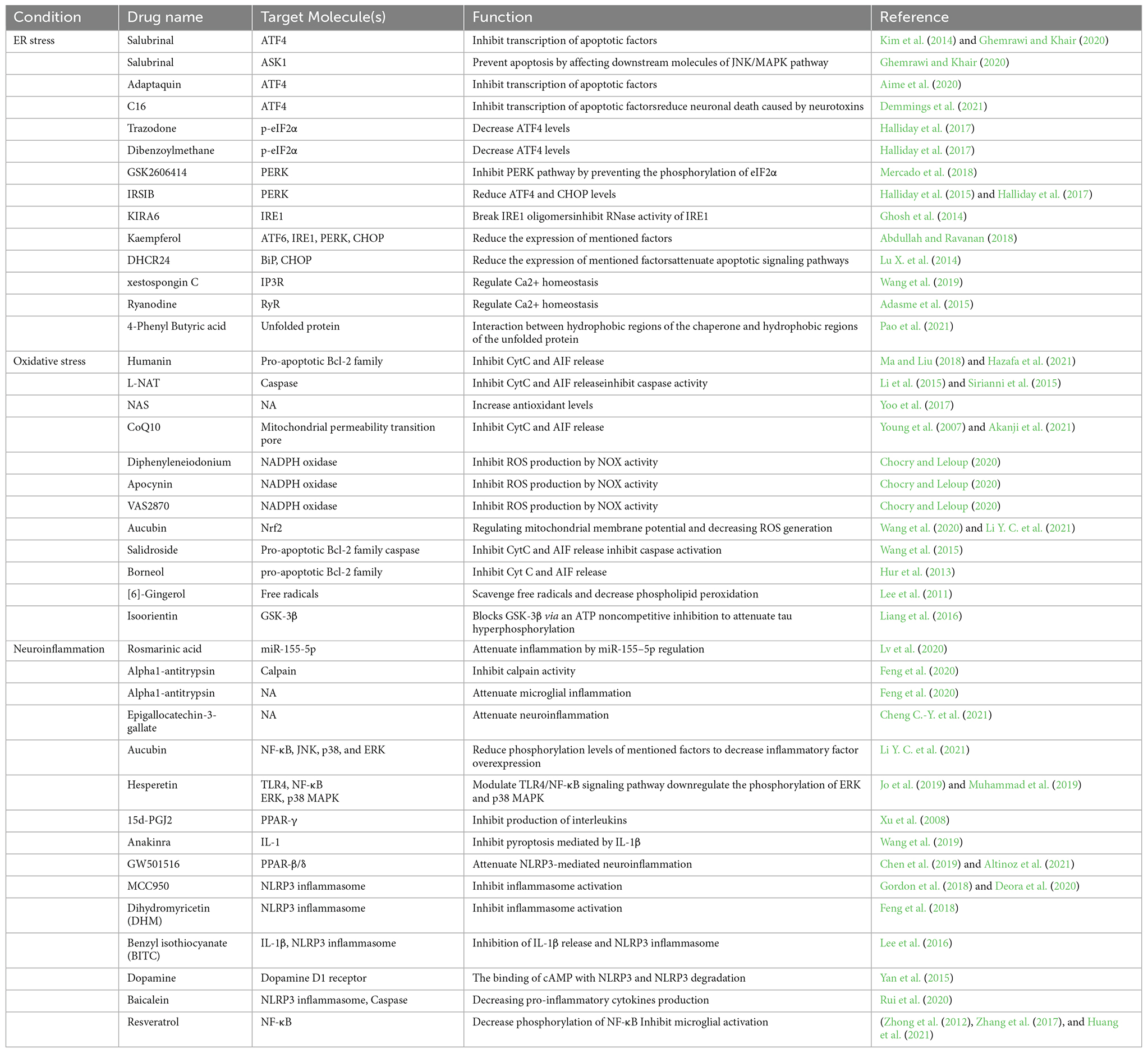
Table 2. The function and target molecules of drugs tested in neurodegenerative disease models in different cellular stress conditions.
Inhibition of the IRE1 pathway is also a possible way to attenuate neuronal cell death. For instance, Kinase-Inhibiting RNase Attenuator 6 (KIRA6) inhibits apoptosis by breaking IRE1 oligomers and inhibiting RNase activity of IRE1α (Figure 1, Table 2; Ghosh et al., 2014). Given the vital role of ASK1 in IRE1-mediated UPR and inducing apoptosis, targeting and deletion of ASK1 in mutant SOD1-transgenic mice have been indicated to mitigate motor neuronal death (Homma et al., 2009). Additionally, evidence shows that overexpression of XBP1 protects DA neurons against neurotoxin-induced ER Stress-associated cell death (Valdes et al., 2014; Shi et al., 2022). Furthermore, upregulation of autophagy by targeting XBP1 in ALS and HD models is known to be another way of protection from neuronal cell death (Remondelli and Renna, 2017). The experiments have been demonstrated that ablation of ATF6 facilitates DA neuronal death caused by neurotoxins, including 6-hydroxydopamine (6-OHDA) and 1-methyl-4-phenyl-pyridinium (MPP+; Shi et al., 2022). This indicates the plausible role of the third arm of UPR pathways in inducing neuronal death. Kaempferol (Table 2), a plant-derived ER stress-induced cell death inhibitor, has also reduced the expression of ATF6, PERK, IRE1α, as well as CHOP in Brefeldin A (BFA)-induced ER stress in IMR32 cell lines. More investigations is needed to determine whether it is effective in animal and human neurodegenerative models (Abdullah and Ravanan, 2018). It is also claimed that 3β-Hydroxysteroid-Δ24 reductase (DHCR24) can protect neuronal cells by reducing BiP and CHOP levels and attenuating ER stress-specific apoptotic signaling pathways (Table 2; Lu X. et al., 2014). Targeting other indirect factors involved in ER stress, such as IP3 receptors and Ryanodine receptors, has also been examined. Remarkably, the first research confirming blocking Inositole triphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) to decrease ER stress-induced Ca2+ dyshomeostasis in DA neurons revealed that a RyRs blocker (RY) markedly reduced 6-OHDA-induced cytosolic Ca2+ increases. In contrast, an IP3Rs blocker (Xes) had no considerable effect on cytosolic Ca2+ levels and neuronal cell death (Table 2). Moreover, pre-treatment with an ER stress inhibitor 4-phenyl butyric acid (4-PBA) had a neuroprotective effect on DA neurons from 6-OHDA-induced apoptosis (Table 2; Huang et al., 2017).
Oxidative stress-induced apoptotic cell death in neurodegenerative diseases
Mechanism of oxidative stress-induced apoptotic cell death
Healthy mitochondria produce ROS as a byproduct of oxidative phosphorylation mainly as signaling messengers (Hajam et al., 2022; Trushina et al., 2022), while defective mitochondria generate aberrant amounts of ROS and cause oxidative stress and suspend cellular homeostasis due to the disruption of the balance between ROS generation and antioxidant function (Figure 2; Höhn et al., 2020; Holubiec et al., 2022). Neurons are susceptible to produce free radicals due to being metabolically very active. Evidently, any pathological situation or dysfunction in neurons can generate excess ROS leading to oxidative stress (Bhat et al., 2015). Given that the metabolism rate of neurons is very high, the brain has a high oxygen consumption rate (20%–25% of the total body oxygen consumption). Furthermore, the high content of easily peroxidizable unsaturated fatty acids (PUFA) and the relative paucity of antioxidant enzymes compared with other organs makes the brain vulnerable to free radical damage (Nunomura et al., 2007; Rocha et al., 2018). Therefore, the excessive production of ROS and RNS resulting from various factors, including calcium influx and mitochondrial dysfunction, can compromise cell fidelity and exacerbate disease progression. Hydrogen peroxide (H2O2), superoxide anion (O2−), and highly reactive hydroxyl radical (HO•) are the ROS involved in neurodegeneration. The RNS, such as nitric oxide (NO), are also found to have a deleterious effect on neurons (Singh et al., 2019; Korovesis et al., 2023).
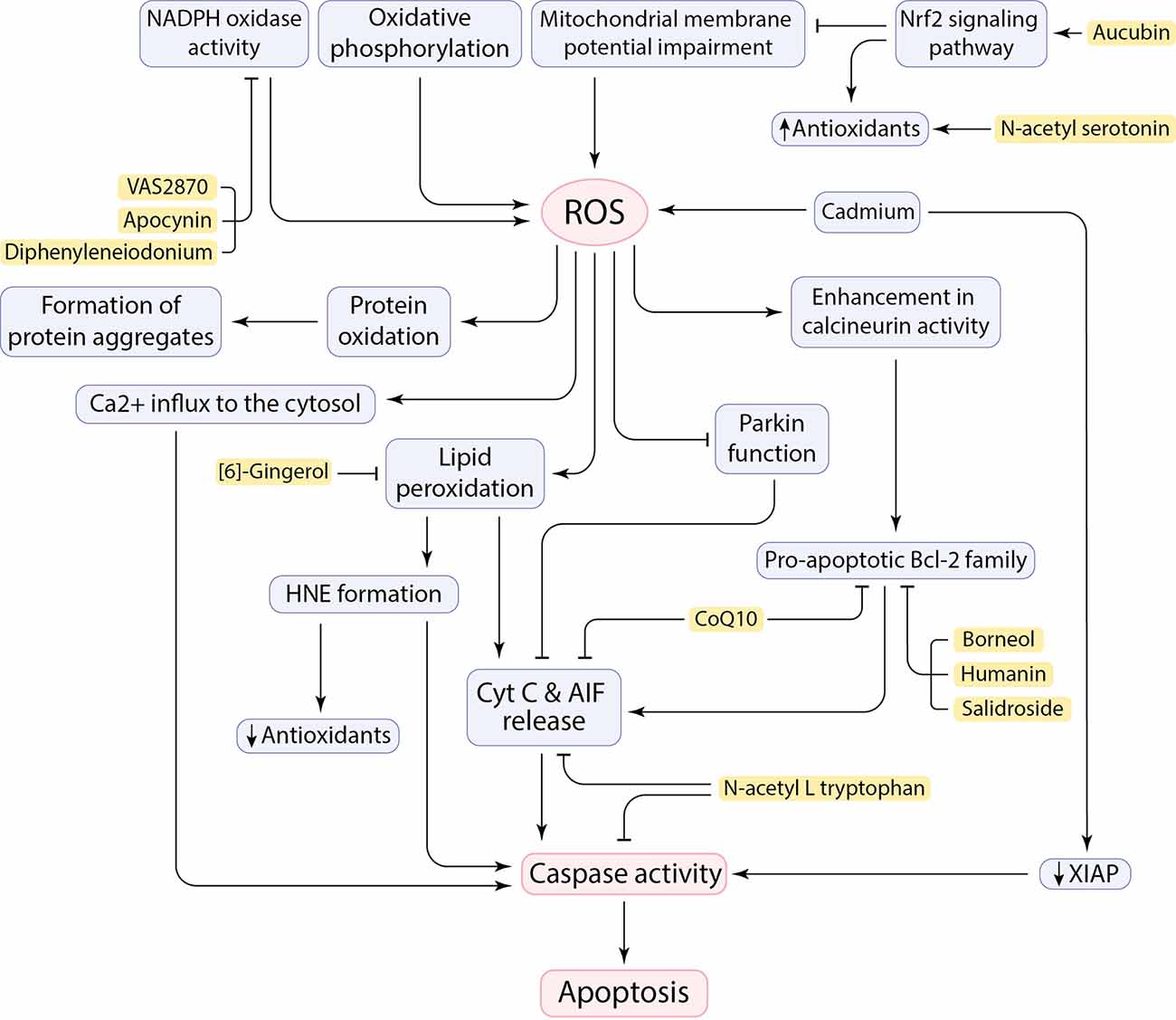
Figure 2. Cause and consequences of ROS production in the CNS and the neuroprotective effects of inhibitory factors (shown in yellow box).
ROS adversely affects the oxidation or peroxidation of specific macromolecules such as lipid peroxidation to malondialdehyde (MDA), protein carbonylation, and oxidation of specific nucleic acids (Singh et al., 2019). It has been claimed that ROS allows Cyt C and AIF to be released from the inner mitochondrial membrane (IMM) and initiate an apoptotic cascade (Figure 2; Bhat et al., 2015). The neural brain cells are enriched in PUFA, such as docosahexaenoic acid, arachidonic acid, and cardiolipin, which makes cells susceptible to lipid peroxidation and subsequent outcomes (Höhn et al., 2020; Falabella et al., 2021). For example, Cardiolipin (CL), a specific phospholipid of IMM, located in the sites of ROS production in the mitochondrial electron transport chain, can potentially be a target for ROS due to its high composition of unsaturated acyl chains. After peroxidation by ROS, CL is supposed to be involved in the conformational changes in IMM and the release of pro-apoptotic proteins, including Cyt C (Bhat et al., 2015; Falabella et al., 2021). However, a serine protease called HTRA2 takes part in the inhibition of pro-apoptotic protein release from mitochondria, but its function may not be sufficient, or it may be disturbed (Bhat et al., 2015). Moreover, the brain is also enriched in redox-active metals (copper and iron) that involve in generating free radicals and peroxidation of lipids (Sbodio et al., 2019; Falabella et al., 2021). These metals could be reduced by proteins such as Aβ, which leads to the formation of H2O2 and pro-apoptotic lipid peroxidation (LPO) products, such as 4-hydroxy-2-nonenal (HNE; Opazo et al., 2002; Jiang et al., 2009). In addition to the role of HNE in decreasing antioxidant levels by reacting with sulfhydryl groups (Taso et al., 2019), HNE forms stable adducts with amine or thiol groups in proteins and may eventually anomalously activate caspases and triggers neuronal cell death (Figure 2; Gaschler and Stockwell, 2017; Barrera et al., 2018). It has been demonstrated that Cadmium (Cd) could easily penetrate the blood-brain barrier and contribute to neurotoxicity. Cd can induce mitochondrial ROS production in neurons as well as downregulation of x-linked inhibitor of apoptosis (XIAP), leading to an increase in mouse double minute 2 (MDM2). Consequently, decrease in p53 facilitates neuronal apoptosis cell death (Figure 2; Zhao et al., 2020).
ROS can influence protein oxidation and contribute to the formation of insoluble protein aggregates (Figure 2), including Aβ peptides and NFTs, α-synuclein, and mSOD1. Oxidative stress can enhance expression of gamma-secretase and beta-secretase (BACE1) through activation of MAPK pathway and involves in Aβ production in neurons and amyloidogenic processing of amyloid precursor protein (APP; Tamagno et al., 2005; Lin and Beal, 2006; Höhn et al., 2020). Oxidative stress also increases tau phosphorylation by activation of glycogen synthase kinase 3 (GSK3; Lin and Beal, 2006). ROS also mediate JNK/stress-activated protein kinase pathways, which subsequently contributes to hyper-phosphorylation of tau proteins, formation of intracellular NFTs, and Aβ-induced neuronal death (Liu et al., 2017). Indeed, hydrogen peroxide and deficiency of mitochondrial antioxidant enzymes has been tested in animal models, which led to increase in Aβ levels (Gerakis and Hetz, 2019) and neuronal cell death (Lin and Beal, 2006). Interestingly, it has been revealed that neurons close to Aβ-plaques seem to be more at risk of cell death due to more severe toxicity of the microenvironment caused by oxidative stress in AD (Xie et al., 2013). Aβ-mediated oxidative stress can enhance the activity of a serine/threonine phosphatase, known as calcineurin, and promotes neuronal death by associating with caspases and/or triggering pro-apoptotic Bcl-2 proteins (Figure 2; Awasthi et al., 2005; Akanji et al., 2021). Moreover, ROS can have noxious effects by affecting Ca2+ cation channels on the ER and plasma membrane. Impaired Ca2+ channels can, in turn, lead to Ca2+ influx to the cytosol, as well as impairment in pumping intracellular Ca2+ out of the cell to maintain homeostasis (Brini et al., 2014). Toxic levels of calcium can trigger cell death through activation of apoptotic factors, including calcium-dependent proteases calpain and caspases (Figure 2; Fairless et al., 2014). Importantly, ROS has also a deleterious effect on nuclear factor erythroid 2-related factor 2 (Nrf2) regulation. Nrf2 is a transcription factor that has an essential role in regulating cellular redox homeostasis (Kovac et al., 2015). Reduced levels of Nrf2 can subsequently result in mitochondrial dysfunction and apoptosis. Therefore, upregulation of Nrf2 reduces oxidative stress by promoting the expression of antioxidant enzymes (Figure 2; Li Y. C. et al., 2021). Besides, the Repressor element 1-silencing transcription factor (REST) regulates cell death-associated genes, including BAX, BH3 interacting domain death agonist (BID), and also PUMA, and maintains resistance to stress conditions. REST-depleted neurons are more susceptible to oxidative stress and anomalously express apoptosis-inducing genes which facilitates neuronal death in AD (Lu T. et al., 2014). Remarkably, upregulation of the transient receptor potential melastatin-2 (TRPM2) in the SNpc of human PD brains agrees with the role of TRPM2 in ROS-induced cell death in PD pathogenesis (Malko et al., 2021). The function of Parkin can be affected by mitochondrial dysfunction and oxidative stress. This will promote Cyt C release and caspase-9 activation, which leads to neuronal cell death and facilitating PD pathogenesis (Figure 2; Lin and Beal, 2006). Defect in Complex I of the mitochondrial electron transport chain by aggregation of α-synuclein and PTEN-induced putative kinase 1 (PINK1) mutations can be also involved in PD pathogenesis by inducing neuronal apoptosis and failure in maintaining mitochondrial membrane potential, respectively (Liu et al., 2017; Morales-Martínez et al., 2022).
Oxidative stress-associated alterations of apoptotic factors
Besides the oxidation of macromolecules is elevated in the brain of patients, decreased levels of antioxidants, including uric acid, vitamin C and E, superoxide dismutase (SOD), catalase, and especially the antioxidant glutathione (GSH), lead to decreased detoxification of ROS in the brain cells which has been discovered in various AD, PD, and other neurodegenerative disease patients (Singh et al., 2019). Oxidative damage occurs before the onset of significant plaque pathology in the AD by triggering glycogen synthase kinase 3 (Lin and Beal, 2006; Wu et al., 2019). Overproduction of ROS and RNS in AD patients has been detected. Additionally, 8-hydroxydeoxyguanosine (8-OHdG), a biomarker of oxidative damage, was elevated in AD ventricular CSF (Niedzielska et al., 2016). However, alterations in plasma levels of antioxidants in AD patients are paradoxical in experimental reports (Niedzielska et al., 2016). Aβ plaques can cause Ca2+ dyshomeostasis in ER leading to Ca2+ influx in the cytosol. Consequently, endogenous GSH levels are reduced, and ROS can cause neurotoxic effects (Liu et al., 2017). The alterations of transition metals, including Cu2+, Zn2+, Fe3+, have been assessed in AD samples. The results indicated that transition metals seem to be imbalanced in AD brains which contributed to oxidative damage and subsequent neuronal death (Bhat et al., 2015). Evidence also shows reduction in nuclear REST levels in neurons of degenerated regions in AD brains, such as prefrontal cortical and hippocampal neurons, which transcriptional dysregulation in apoptotic genes and vulnerability to oxidative stress has made them susceptible to apoptosis cell death (Lu T. et al., 2014). Elevated levels of activated JNK have been reported in post-mortem AD samples, which is probably associated with Aβ formation. JNK implicates in the upregulation of BACE1 and promotes the formation of Aβ, leading to oxidative stress and neuronal apoptosis cell death (Yao et al., 2005; Guglielmotto et al., 2011; Sbodio et al., 2019).
HNE levels were significantly high in the CSF of AD and PD patients, which can be considered as an important reason for neuronal demise and behavioral symptoms in neurodegenerative diseases (Taso et al., 2019). Decreased level of GSH in the Substantia Nigra (SN) of PD patients is one of the earliest biochemical alterations that facilitate the neurotoxic effects of ROS (Niedzielska et al., 2016). Moreover, overexpression of α-synuclein in transgenic mice results in mitochondrial dysfunction and increased oxidative stress (Song et al., 2004). It has been observed that aberrant activity of mutant human SOD1 in ALS patients leads to increase in the level of free radicals in CSF, serum, and urine samples of ALS patients, which exacerbates neuronal damage (Liu and Wang, 2017). Indeed, mSOD1 accumulation in outer mitochondrial membrane (OMM) can result in mitochondrial dysfunction and promotes aberrant ROS production. In an experimental study, mice expressing mSOD1 showed more oxidative damage to mitochondrial lipids and molecules (Mattiazzi et al., 2002; Liu et al., 2004). P53 can regulate genes involved in oxidative stress and mitochondrial function. Environmental toxicants such as bisphenol A (BPA) play a role in inducing neurotoxicity by significantly increased oxidative stress. BPA subsequently leads to upregulation in apoptotic inducing factors, including p53, PUMA, and Drp-1 (Ishtiaq et al., 2021). Additionally, in Huntington’s disease pathology, mHTT can interact with p53 and increase p53 levels, and eventually upregulates apoptotic factors BAX and PUMA, which leads to apoptosis (Bae et al., 2005; Lin and Beal, 2006). mHTT also interacts with mitochondrial membranes, causing mitochondrial abnormalities and an increase in ROS generation, which potentially leads to neuronal degeneration and cell death (Ross and Tabrizi, 2011; Liu et al., 2017). Elevated levels of lipid peroxidation and decreased levels of GSH content have been indicated in the plasma of HD patients (Klepac et al., 2007). In addition, increased levels of 8-OHdG have been observed in the serum of HD patients and post-mortem HD samples (Sbodio et al., 2019).
Targeting oxidative stress-induced apoptosis cell death
Undoubtedly, oxidative stress has neurotoxic effects in the pathogenesis of neurodegenerative diseases. However, the exact molecular pathways remain unclear and need to be more inquired about finding a promising therapeutic strategy to decrease neuronal death in neurodegenerative diseases and extend the lifespan of patients. In this regard, the antioxidant properties of many candidate compounds have been reported. In addition, many other molecules that mediate oxidative stress-induced apoptosis have been targeted to prevent neuronal cell death and disease progression (Figure 2). A cytoprotective polypeptide called Humanin (HN), which is encoded by mtDNA, has neuroprotective activity against cellular stress conditions, such as oxidative stress. HN regulates mitochondrial function by targeting apoptotic factors and inhibits apoptosis by upregulation of Bcl-2 and downregulation of Bid and Bax (Figure 1, Table 2; Hazafa et al., 2021). Also, some amino acid derivatives have shown anti-apoptotic effects in ALS models in vitro (Sirianni et al., 2015). N-acetyl-L-tryptophan (L-NAT) and N-acetyl-DL-tryptophan (DL-NAT) have inhibited neuronal cell death in H2O2-induced NSC-34 motor neurons (Figure 1, Table 2). L-NAT inhibits the release of Cyt C/Smac/AIF from mitochondria, as well as inhibition of caspase activity, thereby preventing neuronal apoptosis cell death (Sirianni et al., 2015). Another study by Yoo et al. (2017) showed that N-acetyl serotonin (NAS) has anti-apoptotic properties by activating neurotrophic signaling TrkB/CREB/BDNF pathways. NAS induces and activates the expression of antioxidant enzymes to reduce the level of ROS (Figure 1, Table 2). It also regulates anti- and pro-apoptotic factors and restores mitochondrial membrane potential to prevent neuronal cell death in neurodegenerative disease models (Yoo et al., 2017). According to evidence, mitochondrial permeability transition pore (mPTP) can increase mitochondrial calcium retention and cause cell death. CoQ10 is considered as an inhibitor of mitochondrial permeability transition pore and protects neurons from oxidative stress and apoptosis. The exact protective mechanism of CoQ10 is still indistinct and needs more experiments. CoQ10 may decrease apoptosis by maintaining the integrity of the mitochondrial membrane and inhibiting Cyt C release. CoQ10 may also decrease the Bcl-2 protein level and prevent caspase activation (Figure 1, Table 2; Akanji et al., 2021). As previously mentioned, protein aggregates can potentially induce oxidative stress in neuronal cells. For instance, Aβ can interact and bind to Aβ-binding alcohol dehydrogenase (ABAD), a mitochondrial-matrix protein, and induce apoptosis and free-radical generation. Blocking the interaction of Aβ and ABAD with a “decoy peptide” suppress oxidative stress and neuronal death. In contrast overexpression of ABAD in mouse models contribute to exaggerating cellular stress and further complications (Lustbader et al., 2004). Moreover, NADPH oxidase (NOX) catalyzes the formation of O2− and participates in elevating neurotoxicity and increasing cell death in HD. Treatment of HD models with NOX inhibitors, including diphenyleneiodonium, apocynin, and VAS2870, prevented neurons from cell death (Figure 1, Table 2; Sbodio et al., 2019).
In several studies, it has been reported that plant iridoids have therapeutic applications in several neurodegenerative diseases by regulating apoptotic factors and neuroprotective proteins (Dinda et al., 2019). Aucubin (AU) is an iridoid glycoside with neuroprotective properties, which significantly increases cell viability in neurons via oxidative stress reduction. AU enhances the antioxidant capacity of cells through the Nrf2 signaling pathway and decreases ROS-induced neuronal apoptosis by regulating mitochondrial membrane potential and reducing ROS generation (Figure 1, Table 2; Li Y. C. et al., 2021). The pharmacological effects of other herbal compounds have also been investigated. Salidroside (Sald), is a Chinese plant-derivative compound that could detoxify neurons by suppressing the elevation of the intracellular ROS level and induction of antioxidant enzymes. Sald also participates in the downregulation of pro-apoptotic protein Bax and upregulation of anti-apoptotic protein Bcl-xl, and prevents neuronal cell death (Figure 1, Table 2; Zhang et al., 2010). Some plant-derived organic oils, including a bicyclic monoterpene termed “Borneol”, indicated neuroprotective effects against H2O2-induced apoptosis in vitro. Borneol alleviates neuronal apoptosis by inhibiting Cyt C and AIF release through increase in the expression of anti-apoptotic protein Bcl-2 and decrease in expression of pro-apoptotic protein Bax (Figure 1, Table 2; Hur et al., 2013). -gingerol also attenuates Aβ-induced oxidative stress. Studies revealed that -gingerol scavenges free radicals and decreases phospholipid peroxidation, as well as improves cellular redox balance (Figure 1, Table 2; Lee et al., 2011). Increase in the activity of glycogen synthase kinase-3β (GSK-3β) under oxidative stress condition leads to Nrf2 dysregulation (Kumar et al., 2012). Thus, GSK-3β inhibitors, including an anti-oxidative phytochemical known as Isoorientin, can have neuroprotection against oxidative damage by regulating Nrf2 antioxidant activity (Figure 1, Table 2; Lim et al., 2007; Gianferrara et al., 2022).
Neuroinflammation-induced cell death in neurodegenerative diseases
Mechanism of neuroinflammation-induced cell death
Research findings have indicated that several neurodegenerative diseases are associated with inflammation (Kwon and Koh, 2020). Activated microglia and T lymphocytes have been detected in the SN of PD patients (Dias et al., 2013). In parallel with this, high expression levels of chemokines, interleukins, interferons, and tumor necrosis factor-α (TNF-α) have been discovered in the striatum and substantia nigra of PD post-mortem brain samples and CSF of AD patients (Hirsch and Hunot, 2009; Llano et al., 2012; Gelders et al., 2018). A neurotoxic microenvironment can be promoted by the continuous secretion of inflammatory mediators from microglia and astrocytes, thus facilitating neural degeneration, and glial cell death (Pardillo-Díaz et al., 2022; Song et al., 2022). In addition to apoptosis cell death, pyroptosis, a non-apoptotic programed cell death, can also occur in the CNS, which is mainly mediated by inflammatory processes. Pyroptosis is characterized by cell swelling, formation of pores in the plasma membrane carried out by cleaved Gasdermin D, and the release of pro-inflammatory cytosolic contents into the extracellular space (Figure 3; Walle and Lamkanfi, 2016; Man et al., 2017; Wang et al., 2019). Besides, some specific caspases, including caspase-1, 4, 5, 11, are called “inflammatory caspases”, can mediate pyroptosis (Taylor et al., 2008; Gaidt and Hornung, 2016). Some factors are associated with initiation of inflammatory cascades and promotion of disease pathology. For instance, ER stress can cause inflammation in neurodegenerative diseases. In other words, inducing ER stress in neurons mostly initiates apoptosis, whereas intense ER stress in glial cells can potentially trigger inflammation in neurodegenerative diseases (Sprenkle et al., 2017). The UPR can increase the production and the release of inflammatory factors, such as transcription factor “nuclear factor kappa-light-chain-enhancer of activated B cells” (NF-κB), interleukin 1 (IL-1), IL-6, IL-8, and TNF-α (Feng et al., 2017). Furthermore, in ER stress condition, p-IRE1 can bind to the TRAF-2 protein and forms the TRAF2-IRE1 complex. The complex may bind to ASK-1 and activate the JNK signaling pathway that enhances inflammation (Vukic et al., 2009; Mohammed-Ali et al., 2015). In addition, p-PERK also facilitates neuroinflammation by inducing the JAK1/STAT3 signaling pathway in glial cells (Meares et al., 2014). Moreover, neuroinflammation is a cause and a consequence of chronic oxidative stress. Studies indicate that the production of free radicals (such as ROS) are elevated in neurodegenerative diseases, which can be due to neuroinflammation (Dias et al., 2013; González-reyes et al., 2017). On the other hand, increased levels of ROS can contribute to pro-inflammatory gene transcription and release of cytokines, including IL-1, IL-6, and TNF-α (Sochocka et al., 2013; Teleanu et al., 2022). Debris of dead neurons may trigger glia-mediated neuroinflammation and initiate a pro-inflammatory cascade that can exacerbate disease progression (Wang et al., 2015; Joshi et al., 2019). Recently, it was revealed that microglial pro-inflammatory cytokines are associated with increased α-synuclein aggregation (Guo et al., 2020). Thereby, protein aggregates can be involved in inducing neuroinflammation in the CNS parenchyma.
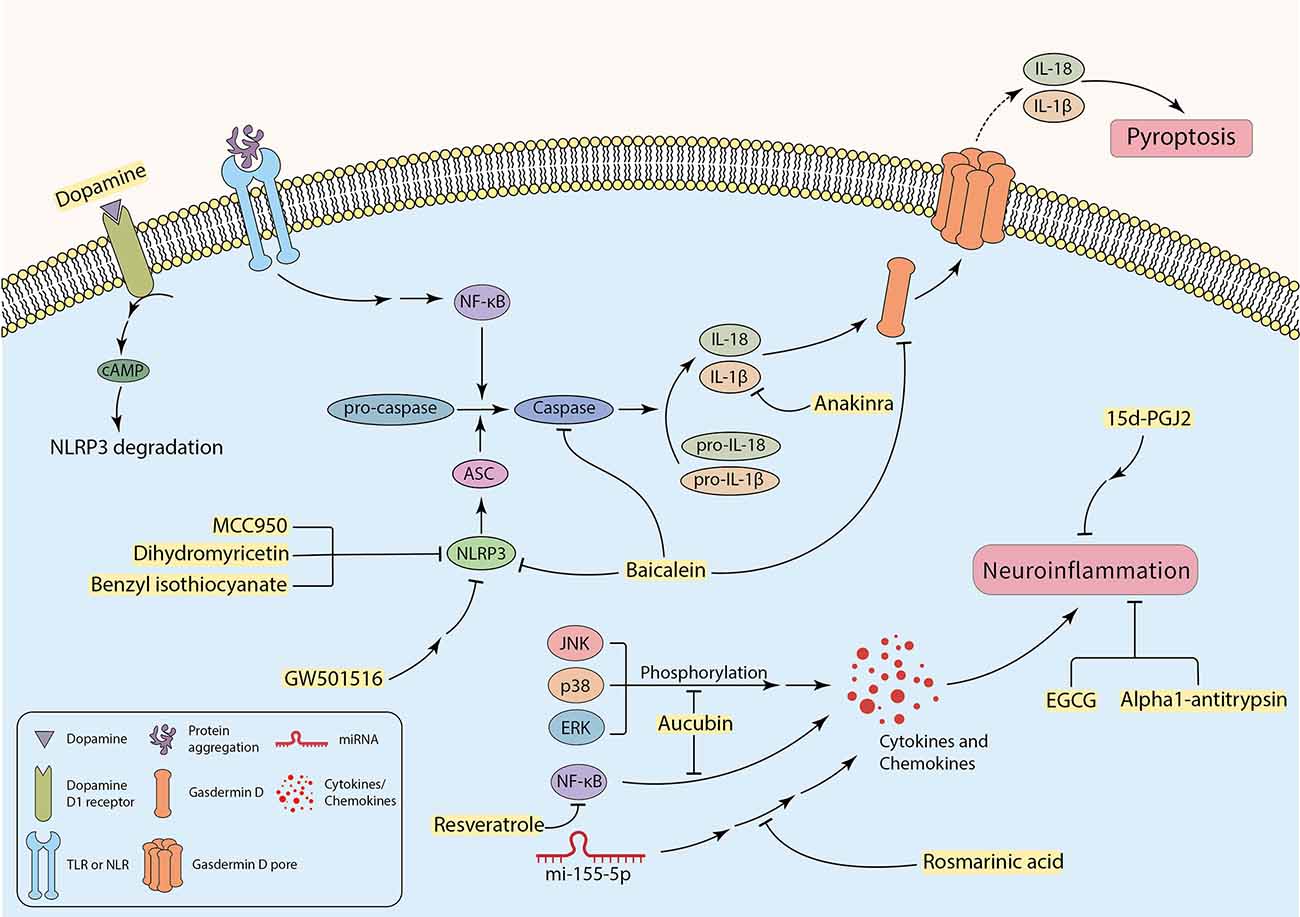
The possible role of inhibiting inflammatory factors to attenuate neuroinflammation-induced neuronal cell death.
It has been investigated that the failure to clear apoptotic cells and activation of glial cells by DAMPs and PAMPs can enhance inflammation response and subsequent neuronal damage and loss (Imbeault et al., 2014; Salter and Stevens, 2017; Voet et al., 2019; Cheng Y. et al., 2021). Activation of complex signaling cascades such as the NLR family pyrin domain containing 3 (NLRP3) inflammasome can be triggered by a wide range of factors including cellular stress, infection (Bader and Winklhofer, 2020; Mahboubi Mehrabani et al., 2022), protein aggregates, and activated microglia (Nichols et al., 2019; Bader and Winklhofer, 2020; Tansey et al., 2022). This phenomenon contributes to producing more neurotoxic cytokines and chemokines such as IL-1β, IL-6, TNF-α, and CCL2 (also known as monocyte chemo-attractant protein-1 or MCP-1), that cause enhancement in neurotoxicity and cell death (Sprenkle et al., 2017; Rocha et al., 2018; Joshi et al., 2019; Nichols et al., 2019). The exact mechanism of neuronal death through activation of NLRP3 inflammasome in microglia has not been perfectly discovered yet (Lee et al., 2019). In fact, NLRP3 inflammasome induces heteromer formation or aggregation of apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), which subsequently activates caspase-1. It results in the maturation of IL-1β and induction of pyroptosis using cleaved Gasdermin D and membrane pores (Figure 3; Stancu et al., 2019; Wang et al., 2019; Bader and Winklhofer, 2020; Feng et al., 2020; Onyango et al., 2021).
Specific receptors, including TLRs and NLRs expressing on neurons or neuroglia, can recognize extracellular neurotoxic protein aggregates (Nichols et al., 2019; Leng and Edison, 2021; Heidari et al., 2022). This may facilitate neuronal cell death (Li Y. et al., 2021) by involvement in caspase activation and secretion of pro-inflammatory factors through NF-κB activation (Figure 3; Rocha et al., 2018; Leng and Edison, 2021). The expression of the receptors for cytokines was indicated in DA neurons, making neurons more susceptible to damage and death (Hirsch and Hunot, 2009). Complement receptors and Fc receptors on microglia can also mediate pro-inflammatory responses independent from extracellular protein aggregates (Leng and Edison, 2021). Activation of microglia can potentially contribute to activating astrocytes, which rapidly upregulates inflammatory signaling molecules (Sims et al., 2022), and can potentially initiate or enhance nitrosative stress due to producing NO (Rocha et al., 2018). In support of this claim, the presence of activated astrocytes is confirmed in post-mortem brain samples of various neurodegenerative disease patients (Hashioka et al., 2021). DAMPs released by dying neurons may also activate microglia through the ionotropic P2X and metabotropic P2Y purinergic receptors and initiate an inflammatory response. For example, under pathological conditions, P2X7 receptors can be overexpressed in the CNS. ATP acts as a DAMP and activates P2X7 receptors, and promotes chronic inflammatory neurological disorders (Thawkar and Kaur, 2019). Post-mortem brain samples of AD patients showed overexpression of P2X7 receptors, which can be associated with disease pathology and progression. It is also suggested that P2X4 receptor overstimulation may result in neuronal cell death (Thawkar and Kaur, 2019). Interestingly, upregulation of P2X and P2Y receptors in ALS patients can subsequently lead to overproduction of TNF-α and cyclooxygenase-2 (COX2), which facilitates neurotoxicity (Liu and Wang, 2017).
Targeting neuroinflammation-induced cell death
Even though the neuroinflammatory cascades exacerbate neurodegenerative disease progression and participate in neuronal death, the exact mechanism of this involvement and therapeutic strategies regarding targeting neuroinflammation is not well studied. According to some investigations, targeting neuroinflammation can be a promising therapeutic approach to decrease inflammation and its further complications in neurodegenerative diseases (Figure 3). Therefore, it can improve patients’ neuronal function in mental and physical activities. Incipiently, some specific microRNAs, such as miR-155-5p, can be key regulators of inflammatory cascades in neurodegenerative diseases. Overexpression of miR-155-5p has been reported in the CSF of AD and MS patients (Lv et al., 2020). It has been recently found that Rosmarinic acid (RA) can inhibit neuroinflammation in neurodegenerative disease samples by regulating miR-155-5p, leading to attenuation in inflammation-associated neuronal damage and loss (Figure 3, Table 2; Lv et al., 2020). Calpains as non-caspase proteases participate in the execution of neuronal cell death and cooperate with key factors of neuronal cell death. Thus, targeting these proteases may result in neuroprotection. Studies have demonstrated that Alpha1-antitrypsin (A1AT) can attenuate microglial neuroinflammation as well as inhibition of calpain activity (Figure 3, Table 2; Feng et al., 2020). Other novel strategies have also been recruited with a focus on neuroinflammation. A recent study by Cheng C.-Y. et al. (2021) targeted inflammation in the substantia nigra of lipopolysaccharide (LPS)-treated rats by liposomes carrying Epigallocatechin-3-gallate (EGCG), a natural antioxidant in green tea. It demonstrated neuroprotection by inhibiting neuroinflammation (Figure 3, Table 2; Cheng C.-Y. et al., 2021). Aucubin, which showed neuroprotective effects in oxidative stress-induced neurotoxicity, can also reduce phosphorylation levels of NF-κB, JNK, p38, and ERK, leading to a decrease in the level of inflammatory factors (Figure 3, Table 2; Li Y. C. et al., 2021). Additionally, a polyphenol named Resveratrol indicated a similar effect by down-regulation of the transcription factor NF-κB in vitro (Figure 3, Table 2; Zhong et al., 2012; Zhang et al., 2017). Targeting TLR4/NF-κB signaling pathway by Hesperetin, a Citrus flavonoid, protected neurons from neuroinflammation and apoptosis (Table 2; Muhammad et al., 2019). Many other similar signaling pathways can be inhibited, aiming to alleviate neuroinflammation in the CNS (Hou et al., 2021). Furthermore, targeting inflammation-associated receptors expressed on neurons can also be an approach. For example, 15d-PGJ2 is a peroxisome proliferator-activated receptor-gamma (PPAR-γ) agonist that inhibits the production of some interleukins and suppresses inflammation in microglial cells in vitro (Figure 3, Table 2; Xu et al., 2008). Suppressing neuroinflammation by targeting P2X7R has been studied, but blood-brain barrier (BBB) permeability limits candidate drugs, so more studies are needed in this case (Thawkar and Kaur, 2019). Intriguingly, Anakinra, an IL-1 receptor antagonist (Mahboubi Mehrabani et al., 2022), reaches CNS easily and inhibits the activity of IL-1β by binding to its receptor and mitigate pyroptosis in neurons (Figure 3, Table 2; Wang et al., 2019).
As mentioned in the former section, since the NLRP3 inflammasome plays a significant role in the enhancement of neurotoxicity, inhibition of NLRP3 and its subsequent pathways might be an effective method to decrease neuroinflammation-induced cell death. MCC950 is a small-molecule NLRP3 inhibitor that has inhibited inflammasome activation in rodent PD models leading to substantial neuroprotection, mitigation in motor deficits, and accumulation of α-synuclein aggregates (Figure 3, Table 2; Gordon et al., 2018). Noteworthy, the neurotransmitter dopamine can bind to the dopamine D1 receptor, which results in ubiquitination and degradation of NLRP3 via the binding of cAMP with NLRP3, leading to the restriction of NLRP3 activation (Figure 3, Table 2; Yan et al., 2015). Some experiments revealed attenuation of NLRP3-mediated neuroinflammation in PD mouse models using peroxisome proliferator-activated receptor beta/delta (PPAR-β/δ) agonist GW501516 (Figure 3, Table 2). There were some limitations with this drug, such as the resistance of the BBB to pass the drug to reach the brain parenchyma. Hence, this compound cannot be considered a candidate for PD treatment until the problem is not solved (Chen et al., 2019). A flavonoid derived from the roots of Scutellaria baicalensis Georgi, termed “Baicalein”, indicated anti-inflammatory and anti-pyroptosis properties in animal models of PD. Experiments suggest that Baicalein may play a role in preventing the loss of DA neurons by reducing the production of various pro-inflammatory cytokines. It can also inhibit NLRP3 and caspase-1 activation, and simultaneously suppress pyroptosis by targeting Gasdermin D in 1-methyl-4phenyl-1,2,3,6-tetrahydropyridine (MPTP) Induced Mice Model of PD (Figure 3, Table 2; Rui et al., 2020). Benzyl isothiocyanate (BITC) and dihydromyricetin (DHM) are other plant-derived compounds with anti-inflammation properties (Lee et al., 2016; Feng et al., 2018). BITC seems to have the neuroprotective effects by inhibition of IL-1β release and NLRP3 inflammasome inhibition in the BV2 microglial cells (Lee et al., 2016). Treatment of APP/PS1 transgenic mice with DHM improved neuroinflammation and memory function, as a result of decreased NLRP3 inflammasome activation (Figure 3, Table 2; Feng et al., 2018). Taken together, the detrimental effects of neuroinflammation in induced neuronal cell death must not be underestimated, and more research is required to provide a better understanding of mechanisms and underlying therapeutic strategies.
Conclusion
As described elaborately, neuronal cell death plays a key role in demonstrating neurodegenerative disease manifestations. Understanding the exact mechanisms and pathways leading to cell death would provide the opportunity for researchers to recommend high-efficiency neuroprotective agents. Until now, many pre-clinical studies have been done in an attempt to cure neurodegenerative diseases, targeting crucial agents involved in well-known pathways leading to neuronal cell death. The field of targeting ER stress and UPR as a therapeutic approach to treat neurodegeneration is growing and has revealed considerable results. On the other hand, ROS damage to mitochondria and homeostasis of the neuron is prominent in neurodegenerative diseases. Hence, this has led to therapeutic approaches using agents with antioxidant properties or inducing the antioxidant activity of the neuron, resulting in inhibition of ROS-mediated neuronal injury. Activation of neuroglia and initiation of neuroinflammation could also lead to a neurotoxic microenvironment for neurons. Unfortunately, the exact mechanism of neuroinflammation-induced cell death is still under debate. Thus, there are not sufficient experimental results of targeting key components of neuroinflammation to decrease neuronal loss in neurodegenerative diseases directly. However, there is strong evidence implicating the role of inhibiting neuroinflammation in attenuating ROS- and ER stress-induced neuronal cell death. Nowadays, the focus on the neuroprotective effects of phytochemicals has significantly increased; nevertheless, there is still much to research and discover to approve phytochemicals as a promising therapeutic agent. Taken together, despite advances in the field of targeting cell death to treat neurodegenerative diseases, there is not an approved compound to directly inhibit cell death yet, so it needs intensive research to find a novel therapeutic strategy for treatment of neurodegenerative diseases.
Author contributions
MK, FSH, and MM provided the idea and mainly wrote the manuscript. MK and AA contributed to the search and assessment of the available literature. MK designed and illustrated the figures. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdullah, A., and Ravanan, P. (2018). Kaempferol mitigates Endoplasmic Reticulum Stress Induced Cell Death by targeting caspase 3/7. Sci. Rep. 8:2189. doi: 10.1038/s41598-018-20499-7
Adasme, T., Paula-Lima, A., and Hidalgo, C. (2015). Inhibitory ryanodine prevents ryanodine receptor-mediated Ca2+ release without affecting endoplasmic reticulum Ca2+ content in primary hippocampal neurons. Biochem. Biophys. Res. Commun. 458, 57–62. doi: 10.1016/j.bbrc.2015.01.065
Aime, P., Karuppagounder, S. S., Rao, A., Chen, Y., Burke, R. E., Ratan, R. R., et al. (2020). The drug adaptaquin blocks ATF4/CHOP-dependent pro-death Trib3 induction and protects in cellular and mouse models of Parkinson’s disease. Neurobiol. Dis. 136:104725. doi: 10.1016/j.nbd.2019.104725
Ajoolabady, A., Lindholm, D., Ren, J., and Pratico, D. (2022). ER stress and UPR in Alzheimer’s disease: mechanisms, pathogenesis, treatments. Cell Death Dis. 13:706. doi: 10.1038/s41419-022-05153-5
Akanji, M. A., Rotimi, D. E., Elebiyo, T. C., Awakan, O. J., and Adeyemi, O. S. (2021). Redox homeostasis and prospects for therapeutic targeting in neurodegenerative disorders. Oxid. Med. Cell. Longev. 2021:9971885. doi: 10.1155/2021/9971885
Almeida, L. M., Pinho, B. R., Duchen, M. R., and Oliveira, J. M. A. (2022). The PERKs of mitochondria protection during stress: insights for PERK modulation in neurodegenerative and metabolic diseases. Biol. Rev. Camb. Philos. Soc. 97, 1737–1748. doi: 10.1111/brv.12860
Altinoz, M. A., Elmaci, İ., Hacimuftuoglu, A., Ozpinar, A., Hacker, E., and Ozpinar, A. (2021). PPARδ and its ligand erucic acid may act anti-tumoral, neuroprotective, and myelin protective in neuroblastoma, glioblastoma, and Parkinson’s disease. Mol. Aspects Med. 78:100871. doi: 10.1016/j.mam.2020.100871
Andhavarapu, S., Mubariz, F., Arvas, M., Bever, C., Jr., and Makar, T. K. (2019). Interplay between ER stress and autophagy: a possible mechanism in multiple sclerosis pathology. Exp. Mol. Pathol. 108, 183–190. doi: 10.1016/j.yexmp.2019.04.016
Andreone, B. J., Larhammar, M., and Lewcock, J. W. (2020). Cell Death and neurodegeneration. Cold Spring Harb. Perspect. Biol. 12:a036434. doi: 10.1101/cshperspect.a036434
Angelova, P. R., and Abramov, A. Y. (2018). Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett. 592, 692–702. doi: 10.1002/1873-3468.12964
Avgerinos, K. I., Ferrucci, L., and Kapogiannis, D. (2021). Effects of monoclonal antibodies against amyloid-β on clinical and biomarker outcomes and adverse event risks: a systematic review and meta-analysis of phase III RCTs in Alzheimer’s disease. Ageing Res. Rev. 68:101339. doi: 10.1016/j.arr.2021.101339
Awasthi, A., Matsunaga, Y., and Yamada, T. (2005). Amyloid-beta causes apoptosis of neuronal cells via caspase cascade, which can be prevented by amyloid-beta-derived short peptides. Exp. Neurol. 196, 282–289. doi: 10.1016/j.expneurol.2005.08.001
Bader, V., and Winklhofer, K. F. (2020). Mitochondria at the interface between neurodegeneration and neuroinflammation. Semin. Cell Dev. Biol. 99, 163–171. doi: 10.1016/j.semcdb.2019.05.028
Bae, B. I., Xu, H., Igarashi, S., Fujimuro, M., Agrawal, N., Taya, Y., et al. (2005). p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 47, 29–41. doi: 10.1016/j.neuron.2005.06.005
Barrera, G., Pizzimenti, S., Daga, M., Dianzani, C., Arcaro, A., Cetrangolo, G. P., et al. (2018). Lipid peroxidation-derived aldehydes, 4-hydroxynonenal and malondialdehyde in aging-related disorders. Antioxidants (Basel) 7:102. doi: 10.3390/antiox7080102
Behl, T., Makkar, R., Sehgal, A., Singh, S., Sharma, N., Zengin, G., et al. (2021). Current trends in neurodegeneration: cross talks between oxidative stress, cell death and inflammation. Int. J. Mol. Sci. 22:7432. doi: 10.3390/ijms22147432
Bejanin, A., Schonhaut, D. R., La Joie, R., Kramer, J. H., Baker, S. L., Sosa, N., et al. (2017). Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 140, 3286–3300. doi: 10.1093/brain/awx243
Bhat, A. H., Dar, K. B., Anees, S., Zargar, M. A., Masood, A., Sofi, M. A., et al. (2015). Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 74, 101–110. doi: 10.1016/j.biopha.2015.07.025
Blaner, W. S., Shmarakov, I. O., and Traber, M. G. (2021). Vitamin A and vitamin E: will the real antioxidant please stand up? Annu. Rev. Nutr. 41, 105–131. doi: 10.1146/annurev-nutr-082018-124228
Bloem, B. R., Okun, M. S., and Klein, C. (2021). Parkinson’s disease. Lancet 397, 2284–2303. doi: 10.1016/S0140-6736(21)00218-X
Breijyeh, Z., and Karaman, R. (2020). Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules 25:5789. doi: 10.3390/molecules25245789
Brini, M., Calì, T., Ottolini, D., and Carafoli, E. (2014). Neuronal calcium signaling: function and dysfunction. Cell. Mol. Life Sci. 71, 2787–2814. doi: 10.1007/s00018-013-1550-7
Callens, M., Kraskovskaya, N., Derevtsova, K., Annaert, W., Bultynck, G., Bezprozvanny, I., et al. (2021). The role of Bcl-2 proteins in modulating neuronal Ca2+ signaling in health and in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 1868:118997. doi: 10.1016/j.bbamcr.2021.118997
Calsolaro, V., and Edison, P. (2016). Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement. 12, 719–732. doi: 10.1016/j.jalz.2016.02.010
Chami, M., and Checler, F. (2020). Alterations of the endoplasmic reticulum (ER) calcium signaling molecular components in Alzheimer’s disease. Cells 9:2577. doi: 10.3390/cells9122577
Chang, H. Y., Nishitoh, H., Yang, X., Ichijo, H., and Baltimore, D. (1998). Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science 281, 1860–1863. doi: 10.1126/science.281.5384.1860
Chen, L., Bi, M., Zhang, Z., Du, X., Chen, X., Jiao, Q., et al. (2022). The functions of IRE1α in neurodegenerative diseases: beyond ER stress. Ageing Res. Rev. 82:101774. doi: 10.1016/j.arr.2022.10177
Chen, S. D., Chuang, Y. C., Lin, T. K., and Yang, J. L. (2023). Alternative role of glucagon-like Peptide-1 receptor agonists in neurodegenerative diseases. Eur. J. Pharmacol. 938:175439. doi: 10.1016/j.ejphar.2022.175439
Chen, L., Xue, L., Zheng, J., Tian, X., Zhang, Y., and Tong, Q. (2019). PPARß/δ agonist alleviates NLRP3 inflammasome-mediated neuroinflammation in the MPTP mouse model of Parkinson’s disease. Behav. Brain Res. 356, 483–489. doi: 10.1016/j.bbr.2018.06.005
Cheng, C.-Y., Barro, L., Tsai, S. T., Feng, T. W., Wu, X. Y., Chao, C. W., et al. (2021). Epigallocatechin-3-gallate-loaded liposomes favor anti-inflammation of microglia cells and promote neuroprotection. Int. J. Mol. Sci. 22:3037. doi: 10.3390/ijms22063037
Cheng, Y., Song, Y., Chen, H., Li, Q., Gao, Y., Lu, G., et al. (2021). Ferroptosis mediated by lipid reactive oxygen species: a possible causal link of neuroinflammation to neurological disorders. Oxid. Med. Cell. Longev. 2021:5005136. doi: 10.1155/2021/5005136
Chocry, M., and Leloup, L. (2020). The NADPH oxidase family and its inhibitors. Antioxid. Redox Signal. 33, 332–353. doi: 10.1089/ars.2019.7915
Colla, E., Coune, P., Liu, Y., Pletnikova, O., Troncoso, J. C., Iwatsubo, T., et al. (2012). Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 32, 3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012
Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M., Hill, K. J., Bhullar, B., et al. (2006). α-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313, 324–328. doi: 10.1126/science.1129462
Cristino, L., Bisogno, T., and Di Marzo, V. (2020). Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 16, 9–29. doi: 10.1038/s41582-019-0284-z
Culmsee, C., and Plesnila, N. (2006). Targeting Bid to prevent programmed cell death in neurons. Biochem. Soc. Trans. 34, 1334–1340. doi: 10.1042/BST0341334
Da Silva, D. C., ValentãO, P., Andrade, P. B., and Pereira, D. M. (2020). Endoplasmic reticulum stress signaling in cancer and neurodegenerative disorders: tools and strategies to understand its complexity. Pharmacol. Res. 155:104702. doi: 10.1016/j.phrs.2020.104702
David, S., Jhelum, P., Ryan, F., Jeong, S. Y., and Kroner, A. (2022). Dysregulation of iron homeostasis in the central nervous system and the role of ferroptosis in neurodegenerative disorders. Antioxid. Redox Signal. 37, 150–170. doi: 10.1089/ars.2021.0218
Decourt, B., Boumelhem, F., Pope, E. D., 3rd, Shi, J., Mari, Z., and Sabbagh, M. N. (2021). Critical appraisal of amyloid lowering agents in AD. Curr. Neurol. Neurosci. Rep. 21:39. doi: 10.1007/s11910-021-01125-y
Demmings, M. D., Tennyson, E. C., Petroff, G. N., Tarnowski-Garner, H. E., and Cregan, S. P. (2021). Activating transcription factor-4 promotes neuronal death induced by Parkinson’s disease neurotoxins and α-synuclein aggregates. Cell Death Differ. 28, 1627–1643. doi: 10.1038/s41418-020-00688-6
Deora, V., Lee, J. D., Albornoz, E. A., McAlary, L., Jagaraj, C. J., Robertson, A. A. B., et al. (2020). The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 68, 407–421. doi: 10.1002/glia.23728
Dhillon, S. (2021). Aducanumab: first approval. Drugs 81, 1437–1443. doi: 10.1007/s40265-021-01569-z
Dias, V., Junn, E., and Mouradian, M. M. (2013). The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 3, 461–491. doi: 10.3233/JPD-130230
Dinda, B., Dinda, M., Kulsi, G., Chakraborty, A., and Dinda, S. (2019). Therapeutic potentials of plant iridoids in Alzheimer’s and Parkinson’s diseases: a review. Eur. J. Med. Chem. 169, 185–199. doi: 10.1016/j.ejmech.2019.03.009
Djajadikerta, A., Keshri, S., Pavel, M., Prestil, R., Ryan, L., and Rubinsztein, D. C. (2020). Autophagy induction as a therapeutic strategy for neurodegenerative diseases. J. Mol. Biol. 432, 2799–2821. doi: 10.1016/j.jmb.2019.12.035
Doroudian, M., Azhdari, M. H., Goodarzi, N., O’Sullivan, D., and Donnelly, S. C. (2021). Smart nanotherapeutics and lung cancer. Pharmaceutics 13:1972. doi: 10.3390/pharmaceutics13111972
Doyle, K. M., Kennedy, D., Gorman, A. M., Gupta, S., Healy, S. J., and Samali, A. (2011). Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J. Cell. Mol. Med. 15, 2025–2039. doi: 10.1111/j.1582-4934.2011.01374.x
Dugger, B. N., and Dickson, D. W. (2017). Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 9:a028035. doi: 10.1101/cshperspect.a028035
Egan, M. F., Kost, J., Tariot, P. N., Aisen, P. S., Cummings, J. L., Vellas, B., et al. (2018). Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 378, 1691–1703. doi: 10.1056/NEJMoa1706441
Egan, M. F., Kost, J., Voss, T., Mukai, Y., Aisen, P. S., Cummings, J. L., et al. (2019). Randomized trial of verubecestat for prodromal Alzheimer’s disease. N. Engl. J. Med. 380, 1408–1420. doi: 10.1056/NEJMoa1812840
Emerit, J., Edeas, M., and Bricaire, F. (2004). Neurodegenerative diseases and oxidative stress. Biomed. Pharmacother. 58, 39–46. doi: 10.1016/j.biopha.2003.11.004
Fairless, R., Williams, S. K., and Diem, R. (2014). Dysfunction of neuronal calcium signalling in neuroinflammation and neurodegeneration. Cell Tissue Res. 357, 455–462. doi: 10.1007/s00441-013-1758-8
Falabella, M., Vernon, H. J., Hanna, M. G., Claypool, S. M., and Pitceathly, R. D. S. (2021). Cardiolipin, mitochondria and neurological disease. Trends Endocrinol. Metab. 32, 224–237. doi: 10.1016/j.tem.2021.01.006
Feng, Y., Li, X., Zhou, W., Lou, D., Huang, D., Li, Y., et al. (2017). Regulation of SET gene expression by NFkB. Mol. Neurobiol. 54, 4477–4485. doi: 10.1007/s12035-016-9967-2
Feng, Y. S., Tan, Z. X., Wu, L. Y., Dong, F., and Zhang, F. (2020). The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Res. Rev. 64:101192. doi: 10.1016/j.arr.2020.101192
Feng, J., Wang, J. X., Du, Y. H., Liu, Y., Zhang, W., Chen, J. F., et al. (2018). Dihydromyricetin inhibits microglial activation and neuroinflammation by suppressing NLRP3 inflammasome activation in APP/PS1 transgenic mice. CNS Neurosci. Ther. 24, 1207–1218. doi: 10.1111/cns.12983
Ferrada, L., Barahona, M. J., Salazar, K., Vandenabeele, P., and Nualart, F. (2020). Vitamin C controls neuronal necroptosis under oxidative stress. Redox Biol. 29:101408. doi: 10.1016/j.redox.2019.101408
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., and Brown, G. C. (2018). Neuronal cell death. Physiol. Rev. 98, 813–880. doi: 10.1152/physrev.00011.2017
Gaidt, M. M., and Hornung, V. (2016). Pore formation by GSDMD is the effector mechanism of pyroptosis. EMBO J. 35, 2167–2169. doi: 10.15252/embj.201695415
Galehdar, Z., Swan, P., Fuerth, B., Callaghan, S. M., Park, D. S., and Cregan, S. P. (2010). Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J. Neurosci. 30, 16938–16948. doi: 10.1523/JNEUROSCI.1598-10.2010
Galvan, V., Banwait, S., Spilman, P., Gorostiza, O. F., Peel, A., Ataie, M., et al. (2007). Interaction of ASK1 and the β-amyloid precursor protein in a stress-signaling complex. Neurobiol. Dis. 28, 65–75. doi: 10.1016/j.nbd.2007.06.017
Gaschler, M. M., and Stockwell, B. R. (2017). Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 482, 419–425. doi: 10.1016/j.bbrc.2016.10.086
Gauthier, S., Feldman, H. H., Schneider, L. S., Wilcock, G. K., Frisoni, G. B., Hardlund, J. H., et al. (2016). Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 388, 2873–2884. doi: 10.1016/S0140-6736(16)31275-2
Gelders, G., Baekelandt, V., and Van der Perren, A. (2018). Linking neuroinflammation and neurodegeneration in Parkinson’s disease. J. Immunol. Res. 2018:4784268. doi: 10.1155/2018/4784268
Gerakis, Y., and Hetz, C. (2019). Brain organoids: a next step for humanized Alzheimer’s disease models? Mol. Psychiatry 24, 474–478. doi: 10.1038/s41380-018-0343-7
Ghemrawi, R., and Khair, M. (2020). Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseases. Int. J. Mol. Sci. 21:6127. doi: 10.3390/ijms21176127
Gherardi, G., Corbioli, G., Ruzza, F., and Rizzuto, R. (2022). CoQ10 and resveratrol effects to ameliorate aged-related mitochondrial dysfunctions. Nutrients 14:4326. doi: 10.3390/nu14204326
Ghosh, R., Wang, L., Wang, E. S., Perera, B. G., Igbaria, A., Morita, S., et al. (2014). Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 158, 534–548. doi: 10.1016/j.cell.2014.07.002
Gianferrara, T., Cescon, E., Grieco, I., Spalluto, G., and Federico, S. (2022). Glycogen synthase kinase 3β involvement in neuroinflammation and neurodegenerative diseases. Curr. Med. Chem. 29, 4631–4697. doi: 10.2174/0929867329666220216113517
González-reyes, R. E., Nava-Mesa, M. O., Vargas-Sánchez, K., Ariza-Salamanca, D., and Mora-Muñoz, L. (2017). Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 10:427. doi: 10.3389/fnmol.2017.00427
Gordon, R., Albornoz, E. A., Christie, D. C., Langley, M. R., Kumar, V., Mantovani, S., et al. (2018). Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 10:eaah4066. doi: 10.1126/scitranslmed.aah4066
Gorman, A. M., Healy, S. J., Jäger, R., and Samali, A. (2012). Stress management at the ER: regulators of ER stress-induced apoptosis. Pharmacol. Ther. 134, 306–316. doi: 10.1016/j.pharmthera.2012.02.003
Guglielmotto, M., Monteleone, D., Giliberto, L., Fornaro, M., Borghi, R., Tamagno, E., et al. (2011). Amyloid-β42 activates the expression of BACE1 through the JNK pathway. J. Alzheimers Dis. 27, 871–883. doi: 10.3233/JAD-2011-110884
Gully, J. C., Sergeyev, V. G., Bhootada, Y., Mendez-Gomez, H., Meyers, C. A., Zolotukhin, S., et al. (2016). Up-regulation of activating transcription factor 4 induces severe loss of dopamine nigral neurons in a rat model of Parkinson’s disease. Neurosci. Lett. 627, 36–41. doi: 10.1016/j.neulet.2016.05.039
Guo, M., Wang, J., Zhao, Y., Feng, Y., Han, S., Dong, Q., et al. (2020). Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain 143, 1476–1497. doi: 10.1093/brain/awaa090
Gupta, R., Ambasta, R. K., and Pravir, K. (2021). Autophagy and apoptosis cascade: which is more prominent in neuronal death? Cell. Mol. Life Sci. 78, 8001–8047. doi: 10.1007/s00018-021-04004-4
Gupta, S., Mishra, A., and Singh, S. (2021). Cardinal role of eukaryotic initiation factor 2 (eIF2α) in progressive dopaminergic neuronal death, DNA fragmentation: implication of PERK:IRE1α:ATF6 axis in Parkinson’s pathology. Cell Signal. 81:109922. doi: 10.1016/j.cellsig.2021.109922
Hajam, Y. A., Rani, R., Ganie, S. Y., Sheikh, T. A., Javaid, D., Qadri, S. S., et al. (2022). Oxidative stress in human pathology and aging: molecular mechanisms and perspectives. Cells 11:552. doi: 10.3390/cells11030552
Halliday, M., Radford, H., Sekine, Y., Moreno, J., Verity, N., Le Quesne, J., et al. (2015). Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 6:e1672. doi: 10.1038/cddis.2015.49
Halliday, M., Radford, H., Zents, K. A. M., Molloy, C., Moreno, J. A., Verity, N. C., et al. (2017). Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain 140, 1768–1783. doi: 10.1093/brain/awx074
Halperin, L., Jung, J., and Michalak, M. (2014). The many functions of the endoplasmic reticulum chaperones and folding enzymes. IUBMB Life 66, 318–326. doi: 10.1002/iub.1272
Hashioka, S., Wu, Z., and Klegeris, A. (2021). Glia-driven neuroinflammation and systemic inflammation in Alzheimer’s disease. Curr. Neuropharmacol. 19, 908–924. doi: 10.2174/1570159X18666201111104509
Hazafa, A., Batool, A., Ahmad, S., Amjad, M., Chaudhry, S. N., Asad, J., et al. (2021). Humanin: a mitochondrial-derived peptide in the treatment of apoptosis-related diseases. Life Sci. 264:118679. doi: 10.1016/j.lfs.2020.118679
Heidari, A., Yazdanpanah, N., and Rezaei, N. (2022). The role of toll-like receptors and neuroinflammation in Parkinson’s disease. J. Neuroinflammation 19:135. doi: 10.1186/s12974-022-02496-w
Hetz, C., and Saxena, S. (2017). ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 13, 477–491. doi: 10.1038/nrneurol.2017.99
Hirota, M., Kitagaki, M., Itagaki, H., and Aiba, S. (2006). Quantitative measurement of spliced XBP1 mRNA as an indicator of endoplasmic reticulum stress. J. Toxicol. Sci. 31, 149–156. doi: 10.2131/jts.31.149
Hirsch, E. C., and Hunot, S. (2009). Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 8, 382–397. doi: 10.1016/S1474-4422(09)70062-6
Hoffmann, L., Waclawczyk, M. S., Tang, S., Hanschmann, E. M., Gellert, M., Rust, M. B., et al. (2021). Cofilin1 oxidation links oxidative distress to mitochondrial demise and neuronal cell death. Cell Death Dis. 12:953. doi: 10.1038/s41419-021-04242-1
Höhn, A., Tramutola, A., and Cascella, R. (2020). Proteostasis failure in neurodegenerative diseases: focus on oxidative stress. Oxid. Med. Cell. Longev. 2020:5497046. doi: 10.1155/2020/5497046
Holubiec, M. I., Gellert, M., and Hanschmann, E. M. (2022). Redox signaling and metabolism in Alzheimer’s disease. Front. Aging Neurosci. 14:1003721. doi: 10.3389/fnagi.2022.1003721
Homma, K., Katagiri, K., Nishitoh, H., and Ichijo, H. (2009). Targeting ASK1 in ER stress-related neurodegenerative diseases. Expert Opin. Ther. Targets 13, 653–664. doi: 10.1517/14728220902980249
Hou, Y., Wei, Y., Lautrup, S., Yang, B., Wang, Y., Cordonnier, S., et al. (2021). NAD+ supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. U S A 118:e2011226118. doi: 10.1073/pnas.2011226118
Huang, J., Huang, N., Xu, S., Luo, Y., Li, Y., Jin, H., et al. (2021). Signaling mechanisms underlying inhibition of neuroinflammation by resveratrol in neurodegenerative diseases. J. Nutr. Biochem. 88:108552. doi: 10.1016/j.jnutbio.2020.108552
Huang, L., Xue, Y., Feng, D., Yang, R., Nie, T., Zhu, G., et al. (2017). Blockade of RyRs in the ER attenuates 6-OHDA-induced calcium overload, cellular hypo-excitability and apoptosis in dopaminergic neurons. Front. Cell Neurosci. 11:52. doi: 10.3389/fncel.2017.00052
Hung, S. Y., and Fu, W. M. (2017). Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci. 24:47. doi: 10.1186/s12929-017-0355-7
Hur, J., Pak, S. C., Koo, B. S., and Jeon, S. (2013). Borneol alleviates oxidative stress via upregulation of Nrf2 and Bcl-2 in SH-SY5Y cells. Pharm. Biol. 51, 30–35. doi: 10.3109/13880209.2012.700718
Imbeault, E., Mahvelati, T. M., Braun, R., Gris, P., and Gris, D. (2014). Nlrx1 regulates neuronal cell death. Mol. Brain 7:90. doi: 10.1186/s13041-014-0090-x
Ishtiaq, A., Ali, T., Bakhtiar, A., Bibi, R., Bibi, K., Mushtaq, I., et al. (2021). Melatonin abated bisphenol A-induced neurotoxicity via p53/PUMA/Drp-1 signaling. Environ. Sci. Pollut. Res. Int. 28, 17789–17801. doi: 10.1007/s11356-020-12129-5
Jiang, D., Li, X., Williams, R., Patel, S., Men, L., Wang, Y., et al. (2009). Ternary complexes of iron, amyloid-β, and nitrilotriacetic acid: binding affinities, redox properties and relevance to iron-induced oxidative stress in Alzheimer’s disease. Biochemistry 48, 7939–7947. doi: 10.1021/bi900907a
Jo, S. H., Kim, M. E., Cho, J. H., Lee, Y., Lee, J., Park, Y. D., et al. (2019). Hesperetin inhibits neuroinflammation on microglia by suppressing inflammatory cytokines and MAPK pathways. Arch. Pharm. Res. 42, 695–703. doi: 10.1007/s12272-019-01174-5
Johnson, J., Mercado-Ayon, E., Mercado-Ayon, Y., Dong, Y. N., Halawani, S., Ngaba, L., et al. (2021). Mitochondrial dysfunction in the development and progression of neurodegenerative diseases. Arch. Biochem. Biophys. 702:108698. doi: 10.1016/j.abb.2020.108698
Johri, A., and Beal, M. F. (2012). Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 342, 619–630. doi: 10.1124/jpet.112.192138
Joshi, A. U., Minhas, P. S., Liddelow, S. A., Haileselassie, B., Andreasson, K. I., Dorn, G. W., 2nd, et al. (2019). Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 22, 1635–1648. doi: 10.1038/s41593-019-0486-0
Kaminskyy, V. O., and Zhivotovsky, B. (2014). Free radicals in cross talk between autophagy and apoptosis. Antioxid. Redox Signal. 21, 86–102. doi: 10.1089/ars.2013.5746
Karimi-Haghighi, S., Razavi, Y., Iezzi, D., Scheyer, A. F., Manzoni, O., and Haghparast, A. (2022). Cannabidiol and substance use disorder: dream or reality. Neuropharmacology 207:108948. doi: 10.1016/j.neuropharm.2022.108948
Karuppagounder, S. S., Alim, I., Khim, S. J., Bourassa, M. W., Sleiman, S. F., John, R., et al. (2016). Therapeutic targeting of oxygen-sensing prolyl hydroxylases abrogates ATF4-dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci. Transl. Med. 8:328ra29. doi: 10.1126/scitranslmed.aac6008
Kaufmann, P., Thompson, J. L., Levy, G., Buchsbaum, R., Shefner, J., Krivickas, L. S., et al. (2009). Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann. Neurol. 66, 235–244. doi: 10.1002/ana.21743
Kim, J. S., Heo, R. W., Kim, H., Yi, C. O., Shin, H. J., Han, J. W., et al. (2014). Salubrinal, ER stress inhibitor, attenuates kainic acid-induced hippocampal cell death. J. Neural Transm. (Vienna) 121, 1233–1243. doi: 10.1007/s00702-014-1208-0
Klein, G., Delmar, P., Voyle, N., Rehal, S., Hofmann, C., Abi-Saab, D., et al. (2019). Gantenerumab reduces amyloid-β plaques in patients with prodromal to moderate Alzheimer’s disease: a PET substudy interim analysis. Alzheimers Res. Ther. 11:101. doi: 10.1186/s13195-019-0559-z
Klepac, N., Relja, M., Klepac, R., Hećimović, S., Babić, T., and Trkulja, V. (2007). Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects: a cross-sectional study. J. Neurol. 254, 1676–1683. doi: 10.1007/s00415-007-0611-y
Korovesis, D., Rubio-Tomás, T., and Tavernarakis, N. (2023). Oxidative stress in age-related neurodegenerative diseases: an overview of recent tools and findings. Antioxidants (Basel) 12:131. doi: 10.3390/antiox12010131
Kovac, S., Angelova, P. R., Holmström, K. M., Zhang, Y., Dinkova-Kostova, A. T., and Abramov, A. Y. (2015). Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 1850, 794–801. doi: 10.1016/j.bbagen.2014.11.021
Kovacs, S. B., and Miao, E. A. (2017). Gasdermins: effectors of pyroptosis. Trends Cell. Biol. 27, 673–684. doi: 10.1016/j.tcb.2017.05.005
Kumar, H., Lim, H. W., More, S. V., Kim, B. W., Koppula, S., Kim, I. S., et al. (2012). The role of free radicals in the aging brain and Parkinson’s disease: convergence and parallelism. Int. J. Mol. Sci. 13, 10478–10504. doi: 10.3390/ijms130810478
Kwon, H. S., and Koh, S. H. (2020). Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl. Neurodegener. 9:42. doi: 10.1186/s40035-020-00221-2
Lang, F. M., Mo, Y., Sabbagh, M., Solomon, P., Boada, M., Jones, R. W., et al. (2021). Intepirdine as adjunctive therapy to donepezil for mild-to-moderate Alzheimer’s disease: a randomized, placebo-controlled, phase 3 clinical trial (MINDSET). Alzheimers Dement. (N Y) 7:e12136. doi: 10.1002/trc2.12136
Lee, A.-H., Iwakoshi, N. N., and Glimcher, L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 23, 7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003
Lee, C.-M., Lee, D.-S., Jung, W.-K., Yoo, J. S., Yim, M.-J., Choi, Y. H., et al. (2016). Benzyl isothiocyanate inhibits inflammasome activation in E. coli LPS-stimulated BV2 cells. Int. J. Mol. Med. 38, 912–918. doi: 10.3892/ijmm.2016.2667
Lee, C., Park, G. H., Kim, C.-Y., and Jang, J.-H. (2011). -Gingerol attenuates β-amyloid-induced oxidative cell death via fortifying cellular antioxidant defense system. Food Chem. Toxicol. 49, 1261–1269. doi: 10.1016/j.fct.2011.03.005
Lee, E., Hwang, I., Park, S., Hong, S., Hwang, B., Cho, Y., et al. (2019). MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 26, 213–228. doi: 10.1038/s41418-018-0124-5
Leng, F., and Edison, P. (2021). Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat. Rev. Neurol. 17, 157–172. doi: 10.1038/s41582-020-00435-y
Lezi, E., and Swerdlow, R. H. (2012). Mitochondria in neurodegeneration. Adv. Exp. Med. Biol. 942, 269–286. doi: 10.1007/978-94-007-2869-1_12
Li, W., Fotinos, A., Wu, Q., Chen, Y., Zhu, Y., Baranov, S., et al. (2015). N-acetyl-L-tryptophan delays disease onset and extends survival in an amyotrophic lateral sclerosis transgenic mouse model. Neurobiol. Dis. 80, 93–103. doi: 10.1016/j.nbd.2015.05.002
Li, Y. C., Hao, J. C., Shang, B., Zhao, C., Wang, L. J., Yang, K. L., et al. (2021). Neuroprotective effects of aucubin on hydrogen peroxide-induced toxicity in human neuroblastoma SH-SY5Y cells via the Nrf2/HO-1 pathway. Phytomedicine 87:153577. doi: 10.1016/j.phymed.2021.153577
Li, Y., Vaughan, K. L., Tweedie, D., Jung, J., Kim, H. K., Choi, H. I., et al. (2019). Pharmacokinetics of Exenatide in nonhuman primates following its administration in the form of sustained-release PT320 and Bydureon. Sci. Rep. 9:17208. doi: 10.1038/s41598-019-53356-2
Li, Y., Xia, Y., Yin, S., Wan, F., Hu, J., Kou, L., et al. (2021). Targeting microglial α-synuclein/TLRs/NF-kappaB/NLRP3 inflammasome axis in Parkinson’s disease. Front Immunol. 12:719807. doi: 10.3389/fimmu.2021.719807
Liang, Z., Zhang, B., Su, W. W., Williams, P. G., and Li, Q. X. (2016). C-Glycosylflavones alleviate tau phosphorylation and amyloid neurotoxicity through GSK3β inhibition. ACS Chem. Neurosci. 7, 912–923. doi: 10.1021/acschemneuro.6b00059
Lim, J. H., Park, H. S., Choi, J. K., Lee, I. S., and Choi, H. J. (2007). Isoorientin induces Nrf2 pathway-driven antioxidant response through phosphatidylinositol 3-kinase signaling. Arch. Pharm. Res. 30, 1590–1598. doi: 10.1007/BF02977329
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi: 10.1038/nature05292
Liu, J., Lillo, C., Jonsson, P. A., Vande Velde, C., Ward, C. M., Miller, T. M., et al. (2004). Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 43, 5–17. doi: 10.1016/j.neuron.2004.06.016
Liu, J., and Wang, F. (2017). Role of neuroinflammation in amyotrophic lateral sclerosis: cellular mechanisms and therapeutic implications. Front. Immunol. 8:1005. doi: 10.3389/fimmu.2017.01005
Liu, Z., Zhou, T., Ziegler, A. C., Dimitrion, P., and Zuo, L. (2017). Oxidative stress in neurodegenerative diseases: from molecular mechanisms to clinical applications. Oxid. Med. Cell. Longev. 2017:2525967. doi: 10.1155/2017/2525967
Llano, D. A., Li, J., Waring, J. F., Ellis, T., Devanarayan, V., Witte, D. G., et al. (2012). Cerebrospinal fluid cytokine dynamics differ between Alzheimer disease patients and elderly controls. Alzheimer Dis. Assoc. Disord. 26, 322–328. doi: 10.1097/WAD.0b013e31823b2728
López-Sendón Moreno, J. L., García Caldentey, J., Trigo Cubillo, P., Ruiz Romero, C., García Ribas, G., Alonso Arias, M. A., et al. (2016). A double-blind, randomized, cross-over, placebo-controlled, pilot trial with Sativex in Huntington’s disease. J. Neurol. 263, 1390–1400. doi: 10.1007/s00415-016-8145-9
Lu, T., Aron, L., Zullo, J., Pan, Y., Kim, H., Chen, Y., et al. (2014). REST and stress resistance in ageing and Alzheimer’s disease. Nature 507, 448–454. doi: 10.1038/nature13163
Lu, X., Li, Y., Wang, W., Chen, S., Liu, T., Jia, D., et al. (2014). 3 β-hydroxysteroid-Δ 24 reductase (DHCR24) protects neuronal cells from apoptotic cell death induced by endoplasmic reticulum (ER) stress. PLoS One 9:e86753. doi: 10.1371/journal.pone.0086753
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., et al. (2004). ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452. doi: 10.1126/science.1091230
Lv, R., Du, L., Zhou, F., Yuan, X., Liu, X., and Zhang, L. (2020). Rosmarinic acid alleviates inflammation, apoptosis and oxidative stress through regulating miR-155-5p in a Mice Model of Parkinson’s disease. ACS Chem. Neurosci. 11, 3259–3266. doi: 10.1021/acschemneuro.0c00375
Lv, M., Xue, G., Cheng, H., Meng, P., Lian, X., HöLscher, C., et al. (2021). The GLP-1/GIP dual-receptor agonist DA5-CH inhibits the NF-κB inflammatory pathway in the MPTP mouse model of Parkinson’s disease more effectively than the GLP-1 single-receptor agonist NLY01. Brain Behav. 11:e2231. doi: 10.1002/brb3.2231
Ma, Z. W., and Liu, D. X. (2018). Humanin decreases mitochondrial membrane permeability by inhibiting the membrane association and oligomerization of Bax and Bid proteins. Acta. Pharmacol. Sin. 39, 1012–1021. doi: 10.1038/aps.2017.169
Mahboubi Mehrabani, M., Karvandi, M. S., Maafi, P., and Doroudian, M. (2022). Neurological complications associated with Covid-19; molecular mechanisms and therapeutic approaches. Rev. Med. Virol. 32:e2334. doi: 10.1002/rmv.2334
Maity, S., Komal, P., Kumar, V., Saxena, A., Tungekar, A., and Chandrasekar, V. (2022). Impact of ER stress and ER-mitochondrial crosstalk in Huntington’s disease. Int. J. Mol. Sci. 23:780. doi: 10.3390/ijms23020780
Malko, P., Ding, R., and Jiang, L.-H. (2021). TRPM2 channel in oxidative stress-induced mitochondrial dysfunction and apoptotic cell death. Adv. Protein Chem. Struct. Biol. 125, 51–72. doi: 10.1016/bs.apcsb.2020.12.001
Man, S. M., Karki, R., and Kanneganti, T. D. (2017). Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 277, 61–75. doi: 10.1111/imr.12534
Mangalmurti, A., and Lukens, J. R. (2022). How neurons die in Alzheimer’s disease: implications for neuroinflammation. Curr. Opin. Neurobiol. 75:102575. doi: 10.1016/j.conb.2022.102575
Markovinovic, A., Greig, J., Martín-Guerrero, S. M., Salam, S., and Paillusson, S. (2022). Endoplasmic reticulum-mitochondria signaling in neurons and neurodegenerative diseases. J. Cell Sci. 135:jcs248534. doi: 10.1242/jcs.248534
Marucci, G., Buccioni, M., Ben, D. D., Lambertucci, C., Volpini, R., and Amenta, F. (2021). Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 190:108352. doi: 10.1016/j.neuropharm.2020.108352
Mattiazzi, M., D’aurelio, M., Gajewski, C. D., Martushova, K., Kiaei, M., Beal, M. F., et al. (2002). Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 277, 29626–29633. doi: 10.1074/jbc.M203065200
Matus, S., Lopez, E., Valenzuela, V., Nassif, M., and Hetz, C. (2013). Functional contribution of the transcription factor ATF4 to the pathogenesis of amyotrophic lateral sclerosis. PLoS One 8:e66672. doi: 10.1371/journal.pone.0066672
Mcgarry, A., Mcdermott, M., Kieburtz, K., De Blieck, E. A., Beal, F., Marder, K., et al. (2017). A randomized, double-blind, placebo-controlled trial of coenzyme Q10 in Huntington disease. Neurology 88, 152–159. doi: 10.1212/WNL.0000000000003478
Meares, G. P., Liu, Y., Rajbhandari, R., Qin, H., Nozell, S. E., Mobley, J. A., et al. (2014). PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol. Cell. Biol. 34, 3911–3925. doi: 10.1128/MCB.00980-14
Mercado, G., Castillo, V., Soto, P., López, N., Axten, J. M., Sardi, S. P., et al. (2018). Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson’s disease. Neurobiol. Dis. 112, 136–148. doi: 10.1016/j.nbd.2018.01.004
Milošević, M., Arsić, A., Cvetković, Z., and Vučić, V. (2021). Memorable food: fighting age-related neurodegeneration by precision nutrition. Front. Nutr. 8:688086. doi: 10.3389/fnut.2021.688086
Mohammed-Ali, Z., Cruz, G. L., and Dickhout, J. G. (2015). Crosstalk between the unfolded protein response and NF-κB-mediated inflammation in the progression of chronic kidney disease. J. Immunol. Res. 2015:428508. doi: 10.1155/2015/428508
Morales-Martínez, A., Martínez-Gómez, P. A., Martinez-Fong, D., Villegas-Rojas, M. M., Perez-Severiano, F., Del Toro-Colín, M. A., et al. (2022). Oxidative stress and mitochondrial complex I dysfunction correlate with neurodegeneration in an α-synucleinopathy animal model. Int. J. Mol. Sci. 23:11394. doi: 10.3390/ijms231911394
Muhammad, T., Ikram, M., Ullah, R., Rehman, S. U., and Kim, M. O. (2019). Hesperetin, a citrus flavonoid, attenuates LPS-induced neuroinflammation, apoptosis and memory impairments by modulating TLR4/NF-κB signaling. Nutrients 11:648. doi: 10.3390/nu11030648
Nichols, M. R., St-Pierre, M.-K., Wendeln, A.-C., Makoni, N. J., Gouwens, L. K., Garrad, E. C., et al. (2019). Inflammatory mechanisms in neurodegeneration. J. Neurochem. 149, 562–581. doi: 10.1111/jnc.14674
Niedzielska, E., Smaga, I., Gawlik, M., Moniczewski, A., Stankowicz, P., Pera, J., et al. (2016). Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 53, 4094–4125. doi: 10.1007/s12035-015-9337-5
Niso-Santano, M., Bravo-San Pedro, J. M., Gómez-Sánchez, R., Climent, V., Soler, G., Fuentes, J. M., et al. (2011). ASK1 overexpression accelerates paraquat-induced autophagy via endoplasmic reticulum stress. Toxicol Sci. 119, 156–168. doi: 10.1093/toxsci/kfq313
Nunomura, A., Moreira, P. I., Lee, H. G., Zhu, X., Castellani, R. J., Smith, M. A., et al. (2007). Neuronal death and survival under oxidative stress in Alzheimer and Parkinson diseases. CNS Neurol. Disord. Drug Targets 6, 411–423. doi: 10.2174/187152707783399201
Onyango, I. G., Jauregui, G. V., Čarná, M., Bennett, J. P., Jr., and Stokin, G. B. (2021). Neuroinflammation in Alzheimer’s disease. Biomedicines 9:524. doi: 10.3390/biomedicines9050524
Opazo, C., Huang, X., Cherny, R. A., Moir, R. D., Roher, A. E., White, A. R., et al. (2002). Metalloenzyme-like Activity of Alzheimer’s disease β-amyloid: CU-Dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H2O2*. J. Biol. Chem. 277, 40302–40308. doi: 10.1074/jbc.M206428200
Ostrowitzki, S., Bittner, T., Sink, K. M., Mackey, H., Rabe, C., Honig, L. S., et al. (2022). Evaluating the safety and efficacy of crenezumab vs. placebo in adults with early Alzheimer disease: two phase 3 randomized placebo-controlled trials. JAMA Neurol. 79, 1113–1121. doi: 10.1001/jamaneurol.2022.2909
Pagano, G., Taylor, K. I., Anzures-Cabrera, J., Marchesi, M., Simuni, T., Marek, K., et al. (2022). Trial of prasinezumab in early-stage Parkinson’s disease. N Engl. J. Med. 387, 421–432. doi: 10.1056/NEJMoa2202867
Pao, H. P., Liao, W. I., Tang, S. E., Wu, S. Y., Huang, K. L., and Chu, S. J. (2021). Suppression of endoplasmic reticulum stress by 4-PBA protects against hyperoxia-induced acute lung injury via up-regulating claudin-4 expression. Front. Immunol. 12:674316. doi: 10.3389/fimmu.2021.674316
Pardillo-Díaz, R., Perez-GarcíA, P., Castro, C., Nunez-Abades, P., and Carrascal, L. (2022). Oxidative stress as a potential mechanism underlying membrane hyperexcitability in neurodegenerative diseases. Antioxidants (Basel) 11:1511. doi: 10.3390/antiox11081511
Patel, S., Bansoad, A. V., Singh, R., and Khatik, G. L. (2022). BACE1: a key regulator in Alzheimer’s disease progression and current development of its inhibitors. Curr. Neuropharmacol. 20, 1174–1193. doi: 10.2174/1570159X19666211201094031
Pintado, C., Macías, S., Domínguez-Martín, H., Castaño, A., and Ruano, D. (2017). Neuroinflammation alters cellular proteostasis by producing endoplasmic reticulum stress, autophagy activation and disrupting ERAD activation. Sci. Rep. 7:8100. doi: 10.1038/s41598-017-08722-3
Piscianz, E., Tesser, A., Rimondi, E., Melloni, E., Celeghini, C., and Marcuzzi, A. (2021). MitoQ is able to modulate apoptosis and inflammation. Int. J. Mol. Sci. 22:4753. doi: 10.3390/ijms22094753
Pohl, F., and Kong Thoo Lin, P. (2018). The potential use of plant natural products and plant extracts with antioxidant properties for the prevention/treatment of neurodegenerative diseases: in vitro, in vivo and clinical trials. Molecules 23:3283. doi: 10.3390/molecules23123283
Poulter, M. O., Payne, K. B., and Steiner, J. P. (2004). Neuroimmunophilins: a novel drug therapy for the reversal of neurodegenerative disease? Neuroscience 128, 1–6. doi: 10.1016/j.neuroscience.2004.06.016
Ramirez-Moreno, M. J., Duarte-Jurado, A. P., Gopar-Cuevas, Y., Gonzalez-Alcocer, A., Loera-Arias, M. J., Saucedo-Cardenas, O., et al. (2019). Autophagy stimulation decreases dopaminergic neuronal death mediated by oxidative stress. Mol. Neurobiol. 56, 8136–8156. doi: 10.1007/s12035-019-01654-1
Read, A., and Schröder, M. (2021). The unfolded protein response: an overview. Biology (Basel) 10:384. doi: 10.3390/biology10050384
Reichert, C. O., De Freitas, F. A., Sampaio-Silva, J., Rokita-Rosa, L., Barros, P. L., Levy, D. L., et al. (2020). Ferroptosis mechanisms involved in neurodegenerative diseases. Int. J. Mol. Sci. 21:8765. doi: 10.3390/ijms21228765
Remondelli, P., and Renna, M. (2017). The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Front. Mol. Neurosci. 10:187. doi: 10.3389/fnmol.2017.00187
Ren, H., Zhai, W., Lu, X., and Wang, G. (2021). The cross-links of endoplasmic reticulum stress, autophagy and neurodegeneration in Parkinson’s disease. Front. Aging Neurosci. 13:691881. doi: 10.3389/fnagi.2021.691881
Rocha, E. M., De Miranda, B., and Sanders, L. H. (2018). Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 109, 249–257. doi: 10.1016/j.nbd.2017.04.004
Ross, C. A., and Tabrizi, S. J. (2011). Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–98. doi: 10.1016/S1474-4422(10)70245-3
Rui, W., Li, S., Xiao, H., Xiao, M., and Shi, J. (2020). Baicalein attenuates neuroinflammation by inhibiting NLRP3/caspase-1/GSDMD pathway in MPTP induced mice model of Parkinson’s disease. Int. J. Neuropsychopharmacol. 23, 762–773. doi: 10.1093/ijnp/pyaa060
Salminen, A., Kauppinen, A., Suuronen, T., Kaarniranta, K., and Ojala, J. (2009). ER stress in Alzheimer’s disease: a novel neuronal trigger for inflammation and Alzheimer’s pathology. J. Neuroinflammation 6:41. doi: 10.1186/1742-2094-6-41
Salter, M. W., and Stevens, B. (2017). Microglia emerge as central players in brain disease. Nat. Med. 23, 1018–1027. doi: 10.1038/nm.4397
Sbodio, J. I., Snyder, S. H., and Paul, B. D. (2019). Redox mechanisms in neurodegeneration: from disease outcomes to therapeutic opportunities. Antioxid. Redox. Signal. 30, 1450–1499. doi: 10.1089/ars.2017.7321
Schwarz, D. S., and Blower, M. D. (2016). The endoplasmic reticulum: structure, function and response to cellular signaling. Cell. Mol. Life Sci. 73, 79–94. doi: 10.1007/s00018-015-2052-6
Shi, M., Chai, Y., Zhang, J., and Chen, X. (2022). Endoplasmic reticulum stress-associated neuronal death and innate immune response in neurological diseases. Front. Immunol. 12:794580. doi: 10.3389/fimmu.2021.794580
Sims, S. G., Cisney, R. N., Lipscomb, M. M., and Meares, G. P. (2022). The role of endoplasmic reticulum stress in astrocytes. Glia 70, 5–19. doi: 10.1002/glia.24082
Singh, A., Kukreti, R., Saso, L., and Kukreti, S. (2019). Oxidative stress: a key modulator in neurodegenerative diseases. Molecules 24:1583. doi: 10.3390/molecules24081583
Sirianni, A. C., Jiang, J., Zeng, J., Mao, L. L., Zhou, S., Sugarbaker, P., et al. (2015). N-acetyl-l-tryptophan, but not N-acetyl-d-tryptophan, rescues neuronal cell death in models of amyotrophic lateral sclerosis. J. Neurochem. 134, 956–968. doi: 10.1111/jnc.13190
Sochocka, M., Koutsouraki, E. S., Gasiorowski, K., and Leszek, J. (2013). Vascular oxidative stress and mitochondrial failure in the pathobiology of Alzheimer’s disease: a new approach to therapy. CNS Neurol. Disord. Drug Targets 12, 870–881. doi: 10.2174/18715273113129990072
Song, J., Han, K., Wang, Y., Qu, R., Liu, Y., Wang, S., et al. (2022). Microglial activation and oxidative stress in PM2.5-induced neurodegenerative disorders. Antioxidants (Basel) 11:1482. doi: 10.3390/antiox11081482
Song, D. D., Shults, C. W., Sisk, A., Rockenstein, E., and Masliah, E. (2004). Enhanced substantia nigra mitochondrial pathology in human α-synuclein transgenic mice after treatment with MPTP. Exp. Neurol. 186, 158–172. doi: 10.1016/S0014-4886(03)00342-X
Sprenkle, N. T., Sims, S. G., Sánchez, C. L., and Meares, G. P. (2017). Endoplasmic reticulum stress and inflammation in the central nervous system. Mol. Neurodegener. 12:42. doi: 10.1186/s13024-017-0183-y
Stancu, I. C., Cremers, N., Vanrusselt, H., Couturier, J., Vanoosthuyse, A., Kessels, S., et al. (2019). Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 137, 599–617. doi: 10.1007/s00401-018-01957-y
Stefani, I. C., Wright, D., Polizzi, K. M., and Kontoravdi, C. (2012). The role of ER stress-induced apoptosis in neurodegeneration. Curr. Alzheimer Res. 9, 373–387. doi: 10.2174/156720512800107618
Stutzbach, L. D., Xie, S. X., Naj, A. C., Albin, R., Gilman, S., Lee, V. M., et al. (2013). The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol. Commun. 1:31. doi: 10.1186/2051-5960-1-31
Sushma, , and Mondal, A. C. (2019). Role of GPCR signaling and calcium dysregulation in Alzheimer’s disease. Mol. Cell Neurosci. 101:103414. doi: 10.1016/j.mcn.2019.103414
Taalab, Y. M., Ibrahim, N., Maher, A., Hassan, M., Mohamed, W., Moustafa, A. A., et al. (2018). Mechanisms of disordered neurodegenerative function: concepts and facts about the different roles of the protein kinase RNA-like endoplasmic reticulum kinase (PERK). Rev. Neurosci. 29, 387–415. doi: 10.1515/revneuro-2017-0071
Tamagno, E., Parola, M., Bardini, P., Piccini, A., Borghi, R., Guglielmotto, M., et al. (2005). β-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J. Neurochem. 92, 628–636. doi: 10.1111/j.1471-4159.2004.02895.x
Tansey, M. G., Wallings, R. L., Houser, M. C., Herrick, M. K., Keating, C. E., and Joers, V. (2022). Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 22, 657–673. doi: 10.1038/s41577-022-00684-6
Taso, O. V., Philippou, A., Moustogiannis, A., Zevolis, E., and Koutsilieris, M. (2019). Lipid peroxidation products and their role in neurodegenerative diseases. Ann. Res. Hosp. 3:2. doi: 10.21037/arh.2018.12.02
Taylor, R. C., Cullen, S. P., and Martin, S. J. (2008). Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 9, 231–241. doi: 10.1038/nrm2312
Teleanu, D. M., Niculescu, A.-G., Lungu, I. I., Radu, C. I., Vladacenco, O., Roza, E., et al. (2022). An overview of oxidative stress, neuroinflammation and neurodegenerative diseases. Int. J. Mol. Sci. 23:5938. doi: 10.3390/ijms23115938
Thawkar, B. S., and Kaur, G. (2019). Inhibitors of NF-κB and P2X7/NLRP3/caspase 1 pathway in microglia: novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmunol. 326, 62–74. doi: 10.1016/j.jneuroim.2018.11.010
Trushina, E., Trushin, S., and Hasan, M. F. (2022). Mitochondrial complex I as a therapeutic target for Alzheimer’s disease. Acta Pharm. Sin. B. 12, 483–495. doi: 10.1016/j.apsb.2021.11.003
Valdes, P., Mercado, G., Vidal, R. L., Molina, C., Parsons, G., Court, F. A., et al. (2014). Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. U S A 111, 6804–6809. doi: 10.1073/pnas.1321845111
Van Dam, L., and Dansen, T. B. (2020). Cross-talk between redox signalling and protein aggregation. Biochem. Soc. Trans. 48, 379–397. doi: 10.1042/BST20190054
Voet, S., Srinivasan, S., Lamkanfi, M., and Van Loo, G. (2019). Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 11:e10248. doi: 10.15252/emmm.201810248
Vukic, V., Callaghan, D., Walker, D., Lue, L. F., Liu, Q. Y., Couraud, P. O., et al. (2009). Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol. Dis. 34, 95–106. doi: 10.1016/j.nbd.2008.12.007
Walle, L. V., and Lamkanfi, M. (2016). Pyroptosis. Curr. Biol. 26, R568–R572. doi: 10.1016/j.cub.2016.02.019
Wang, W.-Y., Tan, M.-S., Yu, J. T., and Tan, L. (2015). Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 3:136. doi: 10.3978/j.issn.2305-5839.2015.03.49
Wang, H. F., Wang, Z. Q., Ding, Y., Piao, M. H., Feng, C. S., Chi, G. F., et al. (2018). Endoplasmic reticulum stress regulates oxygen-glucose deprivation-induced parthanatos in human SH-SY5Y cells via improvement of intracellular ROS. CNS Neurosci. Ther. 24, 29–38. doi: 10.1111/cns.12771
Wang, S., Yuan, Y. H., Chen, N. H., and Wang, H. B. (2019). The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int. Immunopharmacol. 67, 458–464. doi: 10.1016/j.intimp.2018.12.019
Wang, H., Zhou, X. M., Wu, L. Y., Liu, G. J., Xu, W. D., Zhang, X. S., et al. (2020). Aucubin alleviates oxidative stress and inflammation via Nrf2-mediated signaling activity in experimental traumatic brain injury. J. Neuroinflammation 17:188. doi: 10.1186/s12974-020-01863-9
Wessels, A. M., Tariot, P. N., Zimmer, J. A., Selzler, K. J., Bragg, S. M., Andersen, S. W., et al. (2020). Efficacy and safety of lanabecestat for treatment of early and mild Alzheimer disease: the AMARANTH and DAYBREAK-ALZ randomized clinical trials. JAMA Neurol. 77, 199–209. doi: 10.1001/jamaneurol.2019.3988
Wu, L., Luo, N., Zhao, H. R., Gao, Q., Lu, J., Pan, Y., et al. (2014). Salubrinal protects against rotenone-induced SH-SY5Y cell death via ATF4-parkin pathway. Brain Res. 1549, 52–62. doi: 10.1016/j.brainres.2014.01.003
Wu, Q., and Zou, C. (2022). Microglial dysfunction in neurodegenerative diseases via RIPK1 and ROS. Antioxidants (Basel) 11:2201. doi: 10.3390/antiox11112201
Wu, Y., Chen, M., and Jiang, J. (2019). Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 49, 35–45. doi: 10.1016/j.mito.2019.07.003
Xie, H., Hou, S., Jiang, J., Sekutowicz, M., Kelly, J., and Bacskai, B. J. (2013). Rapid cell death is preceded by amyloid plaque-mediated oxidative stress. Proc. Natl. Acad. Sci. U S A 110, 7904–7909. doi: 10.1073/pnas.1217938110
Xu, J., Barger, S. W., and Drew, P. D. (2008). The PPAR-γ agonist 15-deoxy-Δ-prostaglandin J2 attenuates microglial production of IL-12 family cytokines: potential relevance to Alzheimer’s disease. PPAR Res. 2008:349185. doi: 10.1155/2008/349185
Yan, Y., Jiang, W., Liu, L., Wang, X., Ding, C., Tian, Z., et al. (2015). Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160, 62–73. doi: 10.1016/j.cell.2014.11.047
Yao, M., Nguyen, T. V., and Pike, C. J. (2005). β-amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. J. Neurosci. 25, 1149–1158. doi: 10.1523/JNEUROSCI.4736-04.2005
Yoo, J. M., Lee, B. D., Sok, D. E., Ma, J. Y., and Kim, M. R. (2017). Neuroprotective action of N-acetyl serotonin in oxidative stress-induced apoptosis through the activation of both TrkB/CREB/BDNF pathway and Akt/Nrf2/Antioxidant enzyme in neuronal cells. Redox Biol. 11, 592–599. doi: 10.1016/j.redox.2016.12.034
Young, A. J., Johnson, S., Steffens, D. C., and Doraiswamy, P. M. (2007). Coenzyme Q10: a review of its promise as a neuroprotectant. CNS Spectr. 12, 62–68. doi: 10.1017/s1092852900020538
Zhang, K., and Kaufman, R. J. (2008). From endoplasmic-reticulum stress to the inflammatory response. Nature 454, 455–462. doi: 10.1038/nature07203
Zhang, L., Yu, H., Zhao, X., Lin, X., Tan, C., Cao, G., et al. (2010). Neuroprotective effects of salidroside against beta-amyloid-induced oxidative stress in SH-SY5Y human neuroblastoma cells. Neurochem. Int. 57, 547–555. doi: 10.1016/j.neuint.2010.06.021
Zhang, S., Gao, L., Liu, X., Lu, T., Xie, C., and Jia, J. (2017). Resveratrol attenuates microglial activation via SIRT1-SOCS1 pathway. Evid. Based Complement. Alternat. Med. 2017:8791832. doi: 10.1155/2017/8791832
Zhao, C., Liao, Y., Rahaman, A., and Kumar, V. (2022). Towards understanding the relationship between ER stress and unfolded protein response in amyotrophic lateral sclerosis. Front. Aging Neurosci. 14:892518. doi: 10.3389/fnagi.2022.892518
Zhao, R., Yu, Q., Hou, L., Dong, X., Zhang, H., Chen, X., et al. (2020). Cadmium induces mitochondrial ROS inactivation of XIAP pathway leading to apoptosis in neuronal cells. Int. J. Biochem. Cell Biol. 121:105715. doi: 10.1016/j.biocel.2020.105715
Keywords: neurodegenerative diseases, cell death, ER stress, UPR – unfolded protein response, oxidative stress, ROS, neuroinflammation
Citation: Karvandi MS, Sheikhzadeh Hesari F, Aref AR and Mahdavi M (2023) The neuroprotective effects of targeting key factors of neuronal cell death in neurodegenerative diseases: The role of ER stress, oxidative stress, and neuroinflammation. Front. Cell. Neurosci. 17:1105247. doi: 10.3389/fncel.2023.1105247
Received: 22 November 2022; Accepted: 07 February 2023;
Published: 06 March 2023
Edited by:
Danuta Jantas, Maj Institute of Pharmacology (PAN), PolandReviewed by:
Andrii Domanskyi, Orion Corporation (Finland), FinlandA. Raquel Esteves, University of Coimbra, Portugal
Katarzyna Z. Kuter, Maj Institute of Pharmacology (PAN), Poland
Copyright © 2023 Karvandi, Sheikhzadeh Hesari, Aref and Mahdavi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Majid Mahdavi, bWFqaWQubWFoZGF2aUB0YWJyaXp1LmFjLmly; bWFqaWRtYWhkYXZpQHV0LmFjLmly
Glossary
