 Tiziano D’Andrea
Tiziano D’Andrea Sergio Fucile
Sergio Fucile- 1IRCCS Neuromed, Pozzilli, Italy
- 2Department of Physiology and Pharmacology, Sapienza Rome University, Rome, Italy
Excitotoxic damage is due to an excessive Ca2+ entry in cells following overactivation of Ca2+-permeable ion channels. In neurons, Ca2+-dependent excitotoxicity is linked to the prominent activation of N-Methyl-d-Aspartate receptors (NMDARs), exhibiting a high permeability to Ca2+. Different neurodegenerative diseases share glutamate-and NMDAR-dependent excitotoxicity as a pathogenic mechanism, but also different ligand-gated ion channels (LGICs) may be involved in excitotoxic-related pathologies, such as muscle nicotinic acetylcholine receptor in some forms of congenital myasthenic syndrome. We posit that excitotoxicity due to the overactivation of Ca2+-permeable LGICs may be counteracted by using molecules able to reduce selectively the Ca2+ entry, without blocking Na+ influx, thus reducing the adverse effects induced by channel blockers. In this review, we recapitulate: (i) the techniques used to quantify the Ca2+ permeability of LGICs, with a particular focus on the fractional Ca2+ current (Pf, i.e., the percentage of the total current carried by Ca2+); (ii) the known Pf values of the main LGICs; (iii) the modulation of the LGIC Pf values induced by drugs and measured to date. These data support the possibility of fighting excitotoxicity-related pathologies with a new therapeutic approach.
Introduction
Ca2+ is the most widespread and versatile intracellular second messenger, used by all cells as a signal to control their activities in response to extrinsic and intrinsic stimuli (Berridge et al., 2000; Bootman and Bultynck, 2020). In particular, the intracellular free Ca2+ concentration ([Ca2+]i) is fundamental in several processes in neuronal physiology: it regulates the release of neurotransmitters from the presynaptic terminals, influencing long-term potentiation (LTP; Grover and Teyler, 1990) and long-term depression (Bolshakov and Siegelbaum, 1994) through N-Methyl-D-Aspartate receptor (NMDAR)-related influx. In neurons, Ca2+ regulates gene expression, membrane excitability, dendrite development, synaptogenesis, and many other processes contributing to the neuronal primary functions of information processing and memory storage (Kawamoto et al., 2012). Most of [Ca2+]i changes result from influx into the cell regulated through the opening of voltage-dependent Ca2+ channels (VDCCs), NMDARs, or other types of ligand-gated ion channels (LGICs) located in the plasma membrane (Kawamoto et al., 2012). Additionally, [Ca2+]i levels can be increased by Ca2+ release from ER intracellular Ca2+ stores via ryanodine receptors (RyRs) and inositol-1,4,5-triphosphate receptors (IP3Rs), or by Na+-dependent Ca2+ efflux (Na+/Ca2+ exchanger) from mitochondria (Kawamoto et al., 2012). The return to basal [Ca2+]i is achieved by Ca2+ extrusion or organelle uptake, which uses proteins like sarcoplasmic–endoplasmic reticulum Ca2+-ATPase and mitochondrial uniporter (Kawamoto et al., 2012). Moreover, given its importance and potential danger, a high variety of proteins and mechanisms are related to [Ca2+]i sensing and buffering. In particular, neurons possess specialized homeostatic mechanisms to ensure a tight command of cytosolic Ca2+ levels (Arundine and Tymianski, 2003), such as specialized Ca2+-permeable ionic channels, Ca2+ pumps, intracellular and intracellular Ca2+ buffering systems (Tymianski and Tator, 1996). Any alteration in cellular Ca2+ signaling and/or Ca2+ homeostasis can lead to serious pathological outcomes and has been linked to the activation of death pathways, which in the Central Nervous System (CNS) cause neuropathologies and neurodegenerative diseases (Sun et al., 2024) sharing excitotoxicity as a fundamental cause, such as amyotrophic lateral sclerosis (ALS; Van Den Bosch et al., 2006), Alzheimer’s disease (AD; Hynd et al., 2004), Parkinson’s disease (PD; Beal, 1998), Huntington’s disease (HD; Sepers and Raymond, 2014), epilepsy (Meldrum, 1993) and schizophrenia (Plitman et al., 2014).
Excitotoxicity
Ca2+-related excitotoxicity is considered one of the key processes of neuropathologies and neurodegenerative diseases (Schlaepfer and Bunge, 1973). Several Ca2+-permeable LGICs are involved in excitotoxicity generation, in particular glutamate receptors (Lau and Tymianski, 2010) and ATP P2X receptors (Sáez-Orellana et al., 2016). On the other hand, several studies reported that the activation of neuronal nAChRs (nicotinic acetylcholine receptors) containing the α7 subunit promotes neuronal survival (Dajas-Bailador et al., 2000; Laudenbach et al., 2002). So, despite its fundamental role, an imbalance in glutamate homeostasis and the consequent overactivation of glutamate receptors can damage and kill neurons (Mattson, 2019). Neuronal excitotoxic damage is mostly due to a massive influx of Ca2+ through NMDAR (Choi et al., 1988) and VDCCs that cause reactive oxygen species (ROS) release and consequent mitochondrial dysfunction (Mattson, 2003, 2019). Another example of excitotoxic damage happens in the muscular endplate due to rare genetic disorders, such as congenital myasthenic syndromes (CMS; Grassi and Fucile, 2020), which involves mutations in the genes of proteins related to neuromuscular transmission, mostly (about 15%) AChE gene (Beeson et al., 2005; Engel, 2012). Endplate overstimulation causes excitotoxic endplate damage (endplate myopathy) due to the sustained Ca2+ influx through ε-nAChR-channels, and the Ca2+ overloading triggers degenerative events of the endplate structure (Grassi and Fucile, 2014).
Based on the pathogenic concept of overactivation of the excitatory pathways, NMDA receptors have been a longstanding therapeutic target for designing drugs that could be used against these otherwise heterogeneous and complex pathologies (Villmann and Becker, 2007). NMDAR-mediated excitotoxicity seems to start from a signaling mechanism regulated by extrasynaptic NMDARs (Sattler et al., 1999a,b). Conditions that specifically attenuated synaptic NMDAR function did not affect the toxicity caused by exogenous NMDA or glutamate, meaning that extrasynaptic NMDARs must be linked to a molecular pathway triggering neuronal damage (Sattler et al., 1999a,b). Extrasynaptic NMDARs are enriched with the subunit GluN2B, forming GluN2B-containing heterodimers, supporting the hypothesis that extrasynaptic NMDARs constitute a distinct population, serving a specific function; moreover, GluN2B-containing NMDARs have a higher affinity for glutamate, whose concentration is lower in the extrasynaptic space (Papouin and Oliet, 2014). The prevailing theory suggests that synaptic and extrasynaptic NMDAR activation have opposing effects on cell fate: the activity of synaptic NMDARs is thought to favor neuronal survival, via the phosphorylation of intracellular factors such as CREB or Erk1/2; in contrast, cell death is mainly mediated by the activation of extrasynaptic NMDARs, which inhibits the above pathways and promotes the expression of caspase-3, a pro-apoptotic signal (Hardingham et al., 2002; Papouin and Oliet, 2014). To date, the intense research into the mechanisms of excitotoxicity has not enlightened the key intracellular steps responsible for neuronal death in glutamate-dependent excitotoxic processes, mostly because of the heterogeneity of the neurodegeneration that follows glutamate application (Lau and Tymianski, 2010). First, neuronal death in cellular cultures can happen through both apoptosis and necrosis, depending on the severity of NMDA insult: necrotic-like damage predominates in response to acute, relatively intense excitotoxicity (2 mM NMDA), while apoptotic-like neuronal death may develop over many hours after less severe insults, using 300 μM NMDA (Bonfoco et al., 1995). The first to recognize the central role of [Ca2+]i in glutamate excitotoxicity-mediated neuronal damage and death was Choi in 1985, demonstrating that excitotoxicity in neuronal cultures was potentiated in a Ca2+-rich extracellular solution, while neurodegeneration was markedly reduced in a Ca2+-free extracellular solution (Choi, 1985). Furthermore, intracellular Ca2+ chelation by cell-permeant molecules such as BAPTA-AM and EGTA-AM protects neurons against excitotoxic injury both in vitro and in vivo (Tymianski et al., 1994a, 1994b). However, it seems that neuronal mortality is not directly connected to [Ca2+]i: higher lethality is associated with lower Ca2+ influxes via NMDARs compared to higher Ca2+ influxes via other Ca2+-permeant channels, probably due to the spatial link NMDARs share with neuronal nitric oxide synthase (nNOS), which can produce toxic levels of nitric oxide (NO), through postsynaptic density protein of 95 kDa, the PSD-95 (Chariton and Hafner, 1998; Sattler et al., 1999a,b). Some evidence also supports the idea of free radical generation in mitochondria after the Ca2+ influx via NMDARs; radicals, especially superoxide, can interact with other radicals, such as nitric oxide, to form powerful oxidants (Lau and Tymianski, 2010). In fact, the increasing [Ca2+]i accompanying NMDAR activation is partially buffered by the intracellular mitochondria, but this Ca2+ accumulation causes several dysfunctions, with resultant effects on mitochondrial membrane potential, ATP synthesis and glycolysis (key processes in a cell that consumes a lot of energy, like a neuron), reactive oxygen species generation (ROS) and ultimately failure of cytoplasmic Ca2+ homeostasis (Nicholls and Budd, 1998). Moreover, this mitochondrial impairment leads to the release (from the mitochondrial membrane) of Cytochrome C, which causes not only a defect in mitochondrial electron transport (Luetjens et al., 2000) but also caspase-9-dependent caspase-3 activation, which is an “executioner” caspase that leads to neuronal death through the activation of the apoptotic signaling (Seo et al., 2009). More recently, a physical interaction between NMDAR and TRPM4 (a Ca2+-impermeant Ca2+-activated cationic channel; Rajamanickam et al., 2025) has been shown to be required to observe glutamate-induced excitotoxicity (Yan et al., 2020), introducing a new chapter in the large field of excitotoxicity. All these excitotoxic mechanisms are linked to Ca2+ overload, suggesting that avoiding an excessive Ca2+ influx could exert neuroprotection.
Techniques for the quantitative measure of the Ca2+ permeability of ion channels
The high relevance of Ca2+ influx in physiological processes and its key role in the onset of several neuropathologies and neurodegenerative diseases makes it crucial to understand how to measure and modulate Ca2+ entry. The Ca2+ permeability of ion channels has been calculated for decades by measuring the channel reversal potential alterations induced by different [Ca2+]o (Lewis, 1979), but estimating the Ca2+ entry at non-zero electrochemical driving forces, functionally more important, rested on the assumption that there is no interaction among ions passing through the channel (Zhou and Neher, 1993). Then, a new method to quantify the Ca2+ flowing through a cationic ion channel was introduced in 1992, involving the simultaneous use of both electrophysiological and Ca2+ imaging techniques (Augustine and Neher, 1992): a Ca2+ indicator (e.g., Fura-2) is loaded into the cells using a patch micropipette (Zhou and Neher, 1993), and the cell response to an agonist is measured both as a current and a fluorescence emission variation. This mixed approach has been used in the late 90s to measure the Pf (the fractional Ca2+ current, which represents the percentage of the total current carried by Ca2+ ions, at a given membrane potential) of several Ca2+ permeable cationic channels: nAChRs (2.5–11.4%, based on species and subunits composition; Zhou and Neher, 1993; Fucile et al., 2003; Fucile, 2004), human AMPAR (from 0.5 to 3.9%, based on subunits composition; (Burnashev et al., 1995) and human NMDAR (from 8.2 to 11%, based on subunits composition). The NMDARs Pf is the highest among all the glutamate-gated ion channels currently known (Burnashev et al., 1995). More recently, our group introduced a new way to evaluate the ion influxes through the human NMDARs (and other Ca2+-permeable cationic channel) and demonstrate the possibility of modulating the ion selectivity of these receptors, pharmacologically reducing their fractional Ca2+ current, causing the consequent reduction of NMDA-mediated Ca2+ entry in human neurons (D’Andrea et al., 2024). This approach allows us to record simultaneously the intracellular free Ca2+ and Na+ concentrations ([Ca2+]i and [Na+]i, respectively D’Andrea et al., 2024). Our innovative approach is no longer centered on directly measuring ionic fluxes, but on the influx-related changes of [Ca2+]i and [Na+]i, measured simultaneously by fluoresce microscopy. This technique, already described in different contexts (Miyazaki et al., 2019), was used by us for the first time to evaluate the LGICs Ca2+ permeability.
Ca2+-permeable LGICs and related pathologies
Ca2+ ions enter the cells, following their strong concentration gradient, through many kinds of ion channels, exhibiting a large spectrum of functional properties, such as open kinetics, conductance, and ionic selectivity. This wide range of differently regulated pathways, together with the possibility to mobilize Ca2+ from intracellular stores, allow cells to strictly regulate the intracellular free Ca2+ concentration ([Ca2+]i), which in turn drives the function of Ca2+-dependent proteins and related mechanisms (Berridge et al., 2000). Many Ca2+ permeable channels are driven by changes in membrane potential, transducing electrical signals in cellular responses (Zamponi et al., 2015). These VDCCs are highly selective for Ca2+ (Tsien et al., 1987), and their activation and inactivation kinetics are crucial to allow Ca2+ entry avoiding excitotoxic damage (Christel and Lee, 2012). By contrast, cationic LGICs exhibit a much lower Ca2+ selectivity, with Na+ being the ion conducting most of the inward current (Burnashev, 1998; Pankratov and Lalo, 2014). Nevertheless, Ca2+ flowing through LGICs has a paramount physiological function, in processes such as synaptogenesis (Pagano et al., 2021), synaptic plasticity (Gnegy, 2000), and neuromodulation (Kunz et al., 2013), while excessive activation of these channels has been causally related to relevant pathologies (Sattler and Tymianski, 2001; Spalloni et al., 2013; Guo and Ma, 2021). Obviously, the higher the Ca2+ permeability of a channel, the bigger the risk that channel overactivation may lead to excitotoxicity. For these reasons in the last decades many different cationic LGICs have been characterized in terms of their fractional Ca2+ current, which is the percentage of the total current carried by Ca2+ at a certain membrane potential (Zhou and Neher, 1993; Neher, 1995). These experiments explained how different subunits determine the Ca2+ permeability of channels of the same subfamily (Fucile, 2004), and the crucial role of the number and position of electrically charged aminoacidic residues in building the Ca2+ selectivity filter (Watanabe et al., 2002; Fucile, 2017). In Table 1 we summarize the known fractional Ca2+ currents of different kinds of LGICs, as well as the same parameter measured in mutated channels, some of them resulting in Ca2+-related channelopathies. We included in Table 1 only mutated channels with known Pf values, but other mutated LGICs with increased Ca2+ permeability have been described in pathologies (Lemke et al., 2014; Amin et al., 2018).
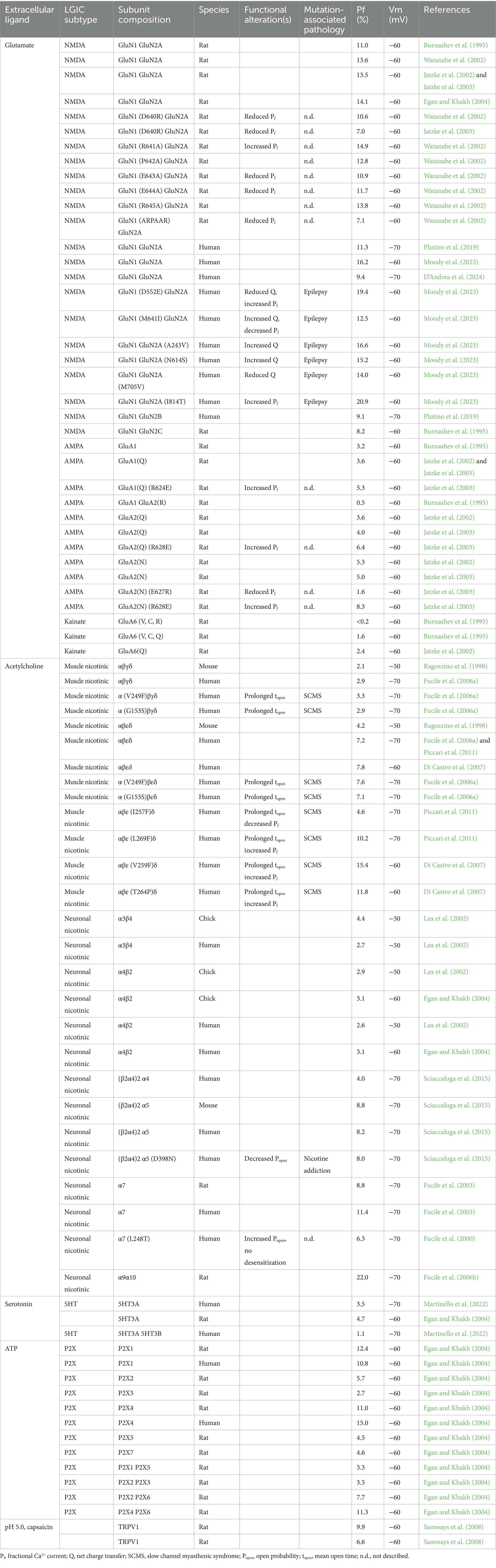
Table 1. Fractional Ca2+ current of cation-selective LGICs.
Physiological modulation of the Ca2+ permeability of LGICs
The Ca2+ permeability of NMDARs can be modulated by physiological processes. Subunits composition of the NMDAR greatly alters several physical and pharmacological properties of the channel: the expression and insertion of the GluN3B subunit, which can co-assemble with endogenous GluN1 and GluN2A and play an important role in modulating synaptic plasticity and neuronal death, reduces the Ca2+ permeability of the NMDAR (Matsuda et al., 2003). PKA signaling cascade controls NMDAR Ca2+ permeability and NMDAR-mediated [Ca2+]i rise in dendritic spines, and in this way PKA directly modulates the induction of NMDAR-dependent LTP at hippocampal Schaffer collateral-CA1 synapses (Skeberdis et al., 2006). NMDAR-mediated Ca2+ influx can also be greatly potentiated by the EphB receptor, a member of the Ephrin receptors family, a large family of receptor tyrosine kinases (Takasu et al., 2002).
Recently, GluA2-containing AMPARs, the most abundant in the central nervous system, have been shown to possess a spectrum of different Ca2+ permeabilities depending on the subunit composition of AMPAR tetramers, as well as the spatial orientation of transmembrane AMPAR and regulatory proteins and cornichon auxiliary subunits (Miguez-Cabello et al., 2025). The extreme complexity in the physiological Ca2+ permeability modulation of Ca2+ permeable LGICs demonstrates the importance of those processes and the possibility to alter and modulate the influx of Ca2+ through ion channels.
Pharmacological modulation of the Ca2+-permeability of LGICs
Given the high number of Ca2+ permeable LGICs, and their causative role in Ca2+-related pathologies (calciumopathies; Stutzmann, 2007), we posit the possibility to reduce the noxious impact of their activation by the selective reduction of their Ca2+ permeation. We started to investigate this strategy more than a decade ago, analyzing the effect on the Pf of the human muscle nicotinic acetylcholine receptor (nAChR) exerted by molecules shown to beneficially act in patients with the slow channel myasthenic syndrome (SCMS; Sadeh et al., 2011). SCMS is a genetic muscle pathology due to mutations of the endplate nAChR dramatically increasing the channel open time and causing a Ca2+-dependent degeneration of the endplate (Engel et al., 1982; Sine et al., 1995; Zhu et al., 2015). The same mutations cause a more severe phenotype in human than in rodent animal models, due to the higher Ca2+ permeability of the human adult nAChR subtype (containing the e subunit; Gomez et al., 2002; Fucile et al., 2006a). Our data explained why nifedipine, salbutamol and verapamil were able to reduce SCMS symptoms in patients, highlighting their capacity to reduce the fractional Ca2+ current through human muscle nAChRs (Piccari et al., 2011). This approach has a crucial aspect: the reduction of Ca2+ entry without blocking the channel. This point is extremely relevant, in particular for pathologies due to channel overactivation, in which it is not possible to completely eliminate channel openings. This is true for the endplate, whose proper function requires nAChR opening with precise kinetics: even fast channel mutations produce myasthenia, although without endplate degeneration (Engel et al., 1993). Similar consideration may be done for excessive activation of synaptic and extrasynaptic glutamate-gated ion channels, functionally linked to Ca2+-related excitotoxicity and to several forms of neurodegeneration: overactivation is harmful, but block is not possible, leading to severe adverse effects (Hargreaves et al., 1994; Parsons et al., 1999). For this reason, a partial block of NMDAR is one of the few therapeutic options for Alzheimer’s disease, in particular using memantine (partial open channel blocker; Kornhuber et al., 1994; Rammes et al., 2008; Karimi Tari et al., 2024). Despite its neuroprotective potential, memantine is not sufficient to significantly ameliorate AD course (Matsunaga et al., 2015), and other therapeutic approaches must be found. In the last few years, we have focused our attention on the possibility to reduce the Pf value of the human NMDAR (Plutino et al., 2019; D’Andrea et al., 2024). We have shown that mild acidosis (pH 6.8–6.5) is not only able to reduce the open probability of NMDARs (as already known; Traynelis and Cull-Candy, 1990), but also to decrease significantly their Ca2+ permeability, with a one-third reduction of the Pf value (Plutino et al., 2019), further explain the known neuroprotective role of mild acidosis (Tombaugh and Sapolsky, 1990). More recently, we have quantified in terms of Pf the observations of Stephen Traynelis group, which investigated the effects of newly developed negative allosteric modulators (NAMs) of NMDARs (Katzman et al., 2015; Hansen et al., 2018). Some of these NAMs were found to reduce the Ca2+ permeability of the human NMDAR, measured by the shift of the reversal potential and calculated in terms of PCa/PNa (Perszyk et al., 2021). We measured the Pf value of human GluN1/GluN2A NMDARs in the presence of one of these compounds, EU1794-4, showing a significant 40% reduction (D’Andrea et al., 2024). We included in Table 2 all the Pf values measured to date, to our knowledge, in the presence of modulators able to reduce the Ca2+ permeability of LGICs. This list is rather short.
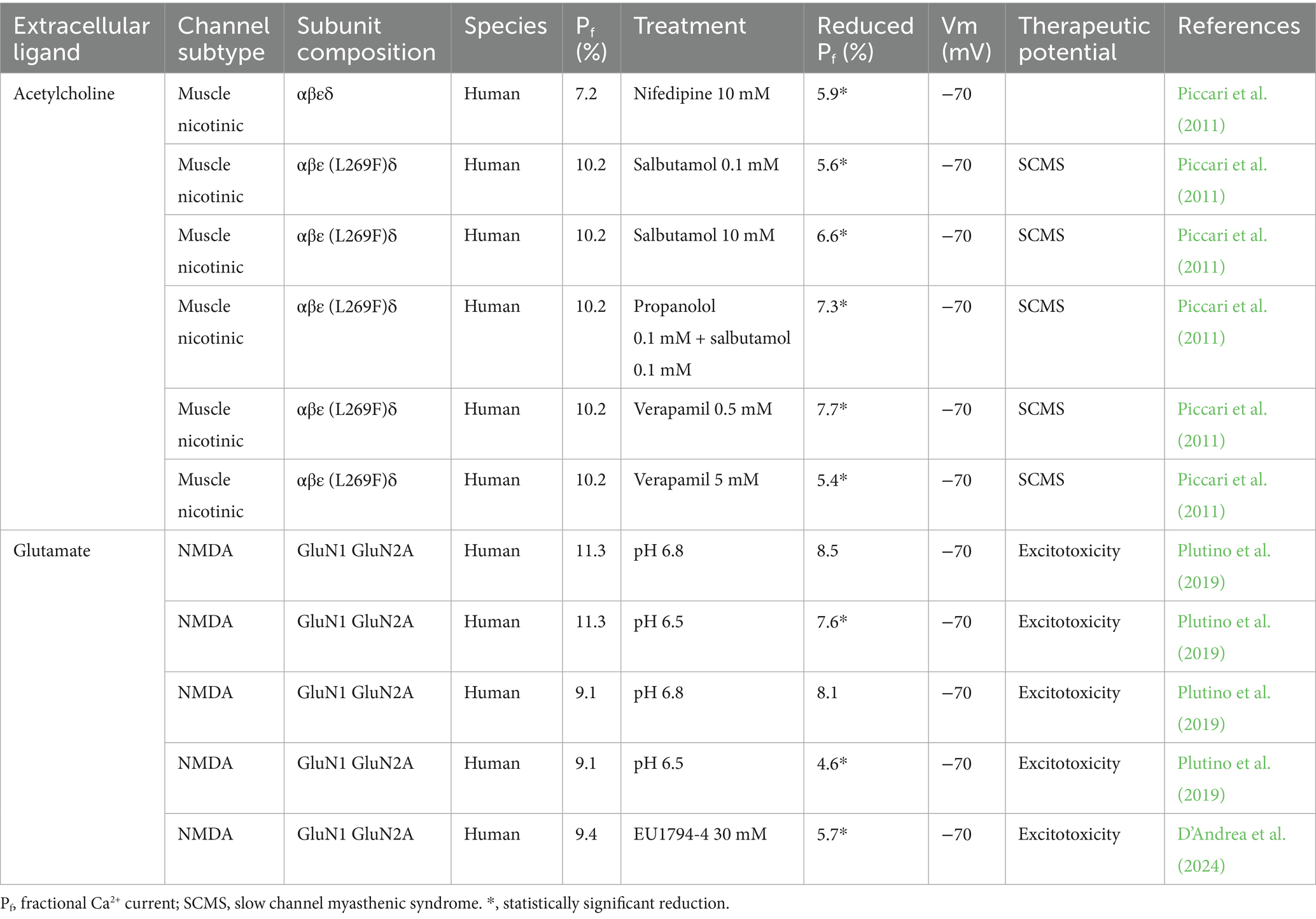
Table 2. Reduction of the fractional Ca2+ current of cationic-selective LGICs.
Discussion
Different therapeutic strategies have been investigated to fight neurodegeneration (see Table 3 for references), but with little impact on clinical applications.
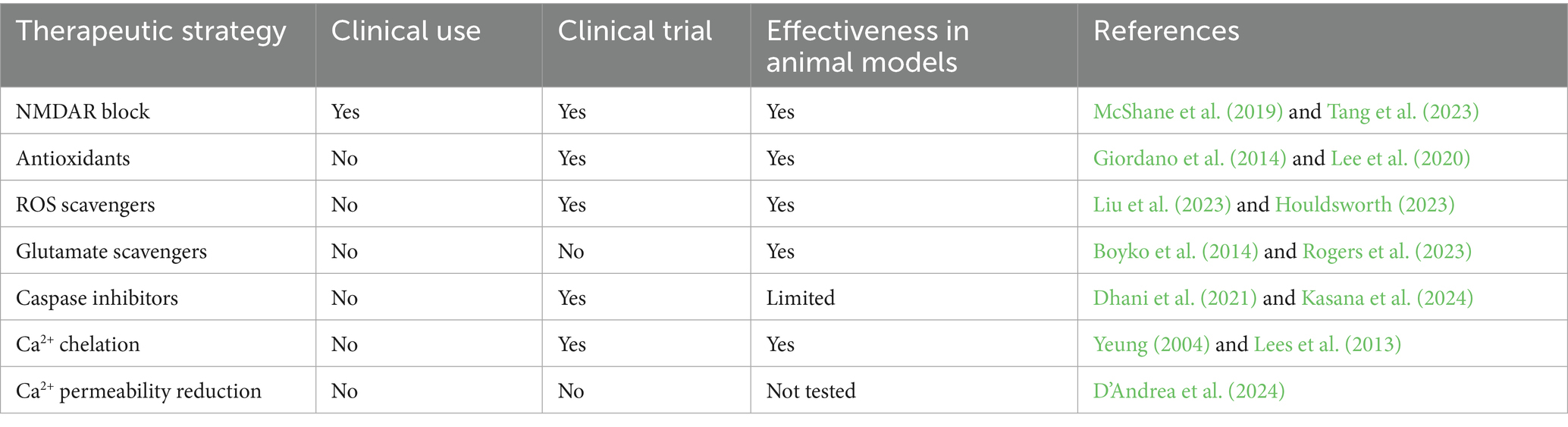
Table 3. Investigated therapeutic strategies against neurodegeneration.
Our data demonstrate that the selective reduction of Ca2+ entry through LGICs is possible, in principle. Reducing Ca2+ influx could also lead to adverse effects. Reduced NMDAR-mediated Ca2+ signaling can alter physiological Ca2+-dependent mechanisms, potentially leading to impaired learning and memory. While excessive NMDAR-mediated Ca2+ influx can trigger pathways leading to neuronal death, insufficient Ca2+ signaling due to reduced NMDAR activity can also be detrimental: this reduction may contribute to the pathophysiology of neurological diseases, as it can hinder the normal signaling required for neuronal health and function (Painuli et al., 2023). Understanding the processes of NMDAR-mediated Ca2+ signaling and its implications for neuronal health will help the design of new drugs, and to maintain a good intracellular Ca2+ homeostasis. In this context, different Ca2+-targeting approaches may be possible. A chelation-based therapy against stroke, head trauma and neurological damage has been proposed, by using DP-b99, a Ca2+/Zn2+ chelator derived from BAPTA (Yeung, 2004). Despite a good tolerability in healthy young and elderly volunteers within the dose range evaluated (Rosenberg et al., 2005), DP-b99 showed no evidence of efficacy in treating human ischemic stroke (Lees et al., 2013). In our opinion, a neuroprotective strategy aiming to reduce Ca2+ entry through LGIC is worth of experimental attention, being in principle applicable to a wide range of Ca2+-related pathologies, especially to the diseases caused by excessive Ca2+ entry through LGICs. Many molecules with described neuroprotective effects may indeed be tried for their ability to selectively reduce Ca2+ entry, with a particular focus on positively charged compounds already known to reduce the channel (sub)conductance levels. For instance, EU1794-4 reduced the single-channel conductance of NMDAR (Perszyk et al., 2021). With this hypothesis in mind, we measured the Pf value of human NMDAR in the presence of memantine, spermine (an intracellular polyamine known to block cation fluxes through LGICs; Tombaugh and Sapolsky, 1990), IEM 1754 and IEM 1460 (open channel blockers; Antonov and Johnson, 1996). None of these molecules was able to reduce the Pf of GluN1/GluN2A NMDAR (Plutino et al., 2019), and to date, besides protons, to our knowledge only NAMs developed by the Traynelis groups exhibit this property.
The search for new molecules able to reduce the Pf of excitotoxicity-related Ca2+-permeable LGICs may produce highly relevant results to improve our pharmacological tools for neuroprotection.
Author contributions
TD’A: Writing – review & editing, Writing – original draft. SF: Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Italian Ministry of Health (Ricerca Corrente) to SF.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Amin, J. B., Leng, X., Gochman, A., Zhou, H.-X., and Wollmuth, L. P. (2018). A conserved glycine harboring disease-associated mutations permits NMDA receptor slow deactivation and high Ca2+ permeability. Nat. Commun. 9:3748. doi: 10.1038/s41467-018-06145-w
Antonov, S. M., and Johnson, J. W. (1996). Voltage-dependent interaction of open-channel blocking molecules with gating of NMDA receptors in rat cortical neurons. J. Physiol. 493, 425–445. doi: 10.1113/jphysiol.1996.sp021394
Arundine, M., and Tymianski, M. (2003). Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 34, 325–337. doi: 10.1016/S0143-4160(03)00141-6
Augustine, G. J., and Neher, E. (1992). Calcium requirements for secretion in bovine chromaffin cells. J. Physiol. 450, 247–271. doi: 10.1113/jphysiol.1992.sp019126
Beal, M. F. (1998). Excitotoxicity and nitric oxide in Parkinson’s disease pathogenesis. Ann. Neurol. 44, S110–S114. doi: 10.1002/ana.410440716
Beeson, D., Hantaï, D., Lochmüller, H., and Engel, A. G. (2005). 126th International Workshop: Congenital Myasthenic Syndromes, 24–26 September 2004, Naarden, The Netherlands. Neuromuscul. Disord. 15, 498–512. doi: 10.1016/j.nmd.2005.05.001
Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21. doi: 10.1038/35036035
Bolshakov, V. Y., and Siegelbaum, S. A. (1994). Postsynaptic induction and presynaptic expression of hippocampal Long-Term Depression. Science 264, 1148–1152. doi: 10.1126/science.7909958
Bonfoco, E., Krainc, D., Ankarcronat, M., Nicoterat, P., and Lipton, S. A. (1995). Apoptosis and necrosis: Two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Neurobiology 92, 7162–7166. doi: 10.1073/pnas.92.16.7162
Bootman, M. D., and Bultynck, G. (2020). Fundamentals of cellular calcium signaling: a primer. Cold Spring Harb. Perspect. Biol. 12:a038802. doi: 10.1101/cshperspect.a038802
Boyko, M., Gruenbaum, S. E., Gruenbaum, B. F., Shapira, Y., and Zlotnik, A. (2014). Brain to blood glutamate scavenging as a novel therapeutic modality: a review. J. Neural Transm. 121, 971–979. doi: 10.1007/s00702-014-1181-7
Burnashev, N. (1998). Calcium permeability of ligand-gated channels. Cell Calcium 24, 325–332. doi: 10.1016/S0143-4160(98)90056-2
Burnashev, N., Zhou, Z., Nehert, E., and Sakmann, B. (1995). Fractional calcium currents through recombinant GluA channels of the NMDA, AMPA and kainate receptor subtypes. J. Physiol. 485, 403–418. doi: 10.1113/jphysiol.1995.sp020738
Chariton, M., and Hafner, M. (1998). Distinct influx pathways, not calcium load, determine neuronal vulnerability to calcium. J. Neurochem. 71, 2349–2364. doi: 10.1046/j.1471-4159.1998.71062349.x
Choi, D. W. (1985). Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci. Lett. 58, 293–297. doi: 10.1016/0304-3940(85)90069-2
Choi, D. W., Koh, J.-Y., and Peters, S. (1988). Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J. Neurosci. 8, 185–196. doi: 10.1523/JNEUROSCI.08-01-00185.1988
Christel, C., and Lee, A. (2012). Ca2+-dependent modulation of voltage-gated Ca2+ channels. Biochim. Biophys. Acta Gen. Subj. 1820, 1243–1252. doi: 10.1016/j.bbagen.2011.12.012
D’Andrea, T., Benedetti, M. C., Monaco, L., Rosa, A., and Fucile, S. (2024). Selective reduction of Ca2+ entry through the human NMDA receptor: a quantitative study by simultaneous Ca2+ and Na+ Imaging. Mol. Neurobiol. 61, 5841–5850. doi: 10.1007/s12035-024-03944-9
Dajas-Bailador, F. A., Lima, P. A., and Wonnacott, S. (2000). The α7 nicotinic acetylcholine receptor subtype mediates nicotine protection against NMDA excitotoxicity in primary hippocampal cultures through a Ca2+ dependent mechanism. Neuropharmacology 39, 2799–2807. doi: 10.1016/S0028-3908(00)00127-1
Dhani, S., Zhao, Y., and Zhivotovsky, B. (2021). A long way to go: caspase inhibitors in clinical use. Cell Death Dis. 12:949. doi: 10.1038/s41419-021-04240-3
Di Castro, A., Martinello, K., Grassi, F., Eusebi, F., and Engel, A. G. (2007). Pathogenic point mutations in a transmembrane domain of the ε subunit increase the Ca2+ permeability of the human endplate ACh receptor. J. Physiol. 579, 671–677. doi: 10.1113/jphysiol.2007.127977
Egan, T. M., and Khakh, B. S. (2004). Contribution of calcium ions to P2X channel responses. J. Neurosci. 24, 3413–3420. doi: 10.1523/JNEUROSCI.5429-03.2004
Engel, A. G. (2012). Current status of the congenital myasthenic syndromes. Neuromuscul. Disord. 22, 99–111. doi: 10.1016/j.nmd.2011.10.009
Engel, A. G., Lambert, E. H., Mulder, D. M., Torres, C. F., Sahashi, K., Bertorini, T. E., et al. (1982). A newly recognized congenital myasthenic syndrome attributed to a prolonged open time of the acetylcholine-induced ion channel. Ann. Neurol. 11, 553–569. doi: 10.1002/ana.410110603
Engel, A. G., Uchitel, O. D., Walls, T. J., Nagel, A., Harper, C. M., and Bodensteiner, J. (1993). Newly recognized congenital myasthenic syndrome associated with high conductance and fast closure of the acetylcholine receptor channel. Ann. Neurol. 34, 38–47. doi: 10.1002/ana.410340109
Fucile, S. (2004). Ca2+ permeability of nicotinic acetylcholine receptors. Cell Calcium 35, 1–8. doi: 10.1016/j.ceca.2003.08.006
Fucile, S. (2017). The distribution of charged amino acid residues and the Ca2+ permeability of nicotinic acetylcholine receptors: a predictive model. Front. Mol. Neurosci. 10:155. doi: 10.3389/fnmol.2017.00155
Fucile, S., Palma, E., Mileo, A. M., Miledi, R., and Eusebi, F. (2000). Human neuronal threonine-for-leucine-248 α7 mutant nicotinic acetylcholine receptors are highly Ca2+ permeable. Proc. Natl. Acad. Sci. 97, 3643–3648. doi: 10.1073/pnas.97.7.3643
Fucile, S., Renzi, M., Lax, P., and Eusebi, F. (2003). Fractional Ca2+ current through human neuronal α7 nicotinic acetylcholine receptors. Cell Calcium 34, 205–209. doi: 10.1016/S0143-4160(03)00071-X
Fucile, S., Sucapane, A., and Eusebi, F. (2006a). Ca2+ permeability through rat cloned α9-containing nicotinic acetylcholine receptors. Cell Calcium 39, 349–355. doi: 10.1016/j.ceca.2005.12.002
Fucile, S., Sucapane, A., Grassi, F., Eusebi, F., and Engel, A. G. (2006b). The human adult subtype ACh receptor channel has high Ca2+ permeability and predisposes to endplate Ca2+ overloading. J. Physiol. 573, 35–43. doi: 10.1113/jphysiol.2006.108092
Giordano, S., Darley-Usmar, V., and Zhang, J. (2014). Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2, 82–90. doi: 10.1016/j.redox.2013.12.013
Gnegy, M. E. (2000). Ca2+/calmodulin signaling in NMDA-induced synaptic plasticity. Crit. Rev. Neurobiol. 14, 91–129. doi: 10.1615/CritRevNeurobiol.v14.i2.10
Gomez, C. M., Maselli, R. A., Groshong, J., Zayas, R., Wollmann, R. L., Cens, T., et al. (2002). Active calcium accumulation underlies severe weakness in a panel of mice with slow-channel syndrome. J. Neurosci. 22, 6447–6457. doi: 10.1523/JNEUROSCI.22-15-06447.2002
Grassi, F., and Fucile, S. (2014). “Nicotinic AChR in Congenital Myasthenic Syndromes” in Pathologies of Calcium Channels (Berlin, Heidelberg: Springer Berlin Heidelberg), 695–711.
Grassi, F., and Fucile, S. (2020). Calcium influx through muscle nAChR-channels: One route, multiple roles. Neuroscience 439, 117–124. doi: 10.1016/j.neuroscience.2019.04.011
Grover, L. M., and Teyler, T. J. (1990). Two components of long-term potentiation induced by different patterns of afferent activation. Nature 347, 477–479. doi: 10.1038/347477a0
Guo, C., and Ma, Y.-Y. (2021). Calcium permeable-AMPA receptors and excitotoxicity in neurological disorders. Front. Neural Circuits 15:711564. doi: 10.3389/fncir.2021.711564
Hansen, K. B., Yi, F., Perszyk, R. E., Furukawa, H., Wollmuth, L. P., Gibb, A. J., et al. (2018). Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 150, 1081–1105. doi: 10.1085/jgp.201812032
Hardingham, G. E., Fukunaga, Y., and Bading, H. (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 5, 405–414. doi: 10.1038/nn835
Hargreaves, R. J., Hill, R. G., and Iversen, L. L. (1994). “Neuroprotective NMDA antagonists: the controversy over their potential for adverse effects on cortical neuronal morphology” in Brain Edema IX (Vienna: Springer Vienna), 15–19.
Houldsworth, A. (2023). Role of oxidative stress in neurodegenerative disorders: a review of reactive oxygen species and prevention by antioxidants. Brain Commun. 6:fcad356. doi: 10.1093/braincomms/fcad356
Hynd, M., Scott, H. L., and Dodd, P. R. (2004). Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 45, 583–595. doi: 10.1016/j.neuint.2004.03.007
Jatzke, C., Hernandez, M., and Wollmuth, L. P. (2003). Extracellular vestibule determinants of Ca2+ influx in Ca2+-permeable AMPA receptor channels. J. Physiol. 549, 439–452. doi: 10.1113/jphysiol.2002.034413
Jatzke, C., Watanabe, J., and Wollmuth, L. P. (2002). Voltage and concentration dependence of Ca2+ permeability in recombinant glutamate receptor subtypes. J. Physiol. 538, 25–39. doi: 10.1113/jphysiol.2001.012897
Karimi Tari, P., Parsons, C. G., Collingridge, G. L., and Rammes, G. (2024). Memantine: Updating a rare success story in pro-cognitive therapeutics. Neuropharmacology 244:109737. doi: 10.1016/j.neuropharm.2023.109737
Kasana, S., Kumar, S., Patel, P., Kurmi, B. D., Jain, S., Sahu, S., et al. (2024). Caspase inhibitors: a review on recently patented compounds (2016–2023). Expert Opin. Ther. Pat. 34, 1047–1072. doi: 10.1080/13543776.2024.239773
Katzman, B. M., Perszyk, R. E., Yuan, H., Tahirovic, Y. A., Sotimehin, A. E., Traynelis, S. F., et al. (2015). A novel class of negative allosteric modulators of NMDA receptor function. Bioorg. Med. Chem. Lett. 25, 5583–5588. doi: 10.1016/j.bmcl.2015.10.046
Kawamoto, E. M., Vivar, C., and Camandola, S. (2012). Physiology and pathology of calcium signaling in the brain. Front. Pharmacol. 3, 3–61. doi: 10.3389/fphar.2012.00061
Kornhuber, J., Weller, M., Schoppmeyer, K., and Riederer, P. (1994). Amantadine and memantine are NMDA receptor antagonists with neuroprotective properties. J. Neural Transm. Suppl. 43, 91–104
Kunz, P. A., Roberts, A. C., and Philpot, B. D. (2013). Presynaptic NMDA receptor mechanisms for enhancing spontaneous neurotransmitter release. J. Neurosci. 33, 7762–7769. doi: 10.1523/JNEUROSCI.2482-12.2013
Lau, A., and Tymianski, M. (2010). Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 460, 525–542. doi: 10.1007/s00424-010-0809-1
Laudenbach, V., Medja, F., Zoli, M., Rossi, F. M., Evrard, P., Changeux, J.-P., et al. (2002). Selective activation of central subtypes of the nicotinic acetylcholine receptor has opposite effects on neonatal excitotoxic brain injuries. FASEB J. 16, 423–425. doi: 10.1096/fj.01-0532fje
Lax, P., Fucile, S., and Eusebi, F. (2002). Ca2+ permeability of human heteromeric nAChRs expressed by transfection in human cells. Cell Calcium 32, 53–58. doi: 10.1016/S0143-4160(02)00076-3
Lee, K. H., Cha, M., and Lee, B. H. (2020). Neuroprotective effect of antioxidants in the brain. Int. J. Mol. Sci. 21:7152. doi: 10.3390/ijms21197152
Lees, K. R., Bornstein, N., Diener, H. C., Gorelick, P. B., Rosenberg, G., Shuaib, A., et al. (2013). Results of Membrane-Activated Chelator Stroke Intervention randomized trial of DP-b99 in acute ischemic stroke. Stroke 44, 580–584. doi: 10.1161/STROKEAHA.111.000013
Lemke, J. R., Hendrickx, R., Geider, K., Laube, B., Schwake, M., Harvey, R. J., et al. (2014). GRIN2B mutations in west syndrome and intellectual disability with focal epilepsy. Ann. Neurol. 75, 147–154. doi: 10.1002/ana.24073
Lewis, C. A. (1979). Ion-concentration dependence of the reversal potential and the single channel conductance of ion channels at the frog neuromuscular junction. J. Phyaiol. 286, 417–445. doi: 10.1113/jphysiol.1979.sp012629
Liu, J., Han, X., Zhang, T., Tian, K., Li, Z., and Luo, F. (2023). Reactive oxygen species (ROS) scavenging biomaterials for anti-inflammatory diseases: from mechanism to therapy. J. Hematol. Oncol. 16:116. doi: 10.1186/s13045-023-01512-7
Luetjens, C. M., Bui, N. T., Sengpiel, B., Münstermann, G., Poppe, M., Krohn, A. J., et al. (2000). Delayed mitochondrial dysfunction in excitotoxic neuron death: cytochrome C release and a secondary increase in superoxide production. J. Neurosci. 20, 5715–5723. doi: 10.1523/JNEUROSCI.20-15-05715.2000
Martinello, K., Sucapane, A., and Fucile, S. (2022). 5-HT3 receptors in rat dorsal root ganglion neurons: Ca2+ entry and modulation of neurotransmitter release. Life 12:1178. doi: 10.3390/life12081178
Matsuda, K., Fletcher, M., Kamiya, Y., and Yuzaki, M. (2003). Specific assembly with the NMDA receptor 3b subunit controls surface expression and calcium permeability of NMDA receptors. J. Neurosci. 23:10064-73, 10064–10073. doi: 10.1523/JNEUROSCI.23-31-10064.2003
Matsunaga, S., Kishi, T., and Iwata, N. (2015). Memantine monotherapy for Alzheimer’s disease: a systematic review and meta-analysis. PLoS One 10:e0123289. doi: 10.1371/journal.pone.0123289
Mattson, M. P. (2003). Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. NeuroMolecular Med. 3, 65–94. doi: 10.1385/NMM:3:2:65
Mattson, M. P. (2019). “Excitotoxicity” in Stress: Physiology, Biochemistry, and Pathology Handbook of Stress Series, vol. 3 (London, UK: Academic Press), 125–134.
McShane, R., Westby, M. J., Roberts, E., Minakaran, N., Schneider, L., Farrimond, L. E., et al. (2019). Memantine for dementia. Cochrane Database Syst. Rev. 3:CD003154. doi: 10.1002/14651858.CD003154.pub6
Meldrum, B. S. (1993). Excitotoxicity and selective neuronal loss in epilepsy. Brain Pathol. 3, 405–412. doi: 10.1111/j.1750-3639.1993.tb00768.x
Miguez-Cabello, F., Wang, X., Yan, Y., Brake, N., Alexander, R. P. D., Perozzo, A. M., et al. (2025). GluA2-containing AMPA receptors form a continuum of Ca2+-permeable channels. Nature 641, 537–544. doi: 10.1038/s41586-025-08736-2
Miyazaki, K., Lisman, J. E., and Ross, W. N. (2019). Improvements in simultaneous sodium and calcium imaging. Front. Cell. Neurosci. 12:514. doi: 10.3389/fncel.2018.00514
Moody, G., Musco, A., Bennett, J., and Wollmuth, L. P. (2023). An integrated approach to evaluate the functional effects of disease-associated NMDA receptor variants. Neuropharmacology 240:109703. doi: 10.1016/j.neuropharm.2023.109703
Neher, E. (1995). The use of Fura-2 for estimating Ca buffer and Ca Fluxes. Neuropharmacology 34, 1423–1442. doi: 10.1016/0028-3908(95)00144-u
Nicholls, D. G., and Budd, S. L. (1998). Neuronal excitotoxicity: the role of mitochondria. Bio Factors 8, 287–299. doi: 10.1002/biof.5520080317
Pagano, J., Giona, F., Beretta, S., Verpelli, C., and Sala, C. (2021). N-methyl-d-aspartate receptor function in neuronal and synaptic development and signaling. Curr. Opin. Pharmacol. 56, 93–101. doi: 10.1016/j.coph.2020.12.006
Painuli, S., Semwal, P., Zam, W., Taheri, Y., Ezzat, S. M., Zuo, P., et al. (2023). NMDA inhibitors: a potential contrivance to assist in management of Alzheimer’s Disease. Comb. Chem. High Throughput Screen. 26, 2099–2112. doi: 10.2174/1386207325666220428112541
Pankratov, Y., and Lalo, U. (2014). Calcium permeability of ligand-gated Ca2+ channels. Eur. J. Pharmacol. 739, 60–73. doi: 10.1016/j.ejphar.2013.11.017
Papouin, T., and Oliet, S. H. R. (2014). Organization, control and function of extrasynaptic NMDA receptors. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 369:20130601. doi: 10.1098/rstb.2013.0601
Parsons, C. G., Danysz, W., and Quack, G. (1999). Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist-a review of preclinical data. Neuropharmacology 38, 735–767. doi: 10.1016/s0028-3908(99)00019-2
Perszyk, R. E., Zheng, Z., Banke, T. G., Zhang, J., Xie, L., McDaniel, M. J., et al. (2021). The negative allosteric modulator EU1794-4 reduces single-channel conductance and Ca2+ permeability of GluN1/GluN2A N-methyl-d-aspartate receptors. Mol. Pharmacol. 99, 399–411. doi: 10.1124/MOLPHARM.120.000218
Piccari, V., Deflorio, C., Bigi, R., Grassi, F., and Fucile, S. (2011). Modulation of the Ca2+ permeability of human endplate acetylcholine receptor-channel. Cell Calcium 49, 272–278. doi: 10.1016/j.ceca.2011.03.002
Plitman, E., Nakajima, S., de la Fuente-Sandoval, C., Gerretsen, P., Chakravarty, M. M., Kobylianskii, J., et al. (2014). Glutamate-mediated excitotoxicity in schizophrenia: a review. Eur. Neuropsychopharmacol. 24, 1591–1605. doi: 10.1016/j.euroneuro.2014.07.015
Plutino, S., Sciaccaluga, M., and Fucile, S. (2019). Extracellular mild acidosis decreases the Ca2+ permeability of the human NMDA receptors. Cell Calcium 80, 63–70. doi: 10.1016/j.ceca.2019.04.001
Ragozzino, D., Barabino, B., Fucile, S., and Eusebi, F. (1998). Ca2+ permeability of mouse and chick nicotinic acetylcholine receptors expressed in transiently transfected human cells. J. Physiol. 507, 749–758. doi: 10.1111/j.1469-7793.1998.749bs.x
Rajamanickam, G., Hu, Z., and Liao, P. (2025). Targeting the TRPM4 channel for neurologic diseases: opportunity and challenge. Neuroscientist. 26:10738584251318979. doi: 10.1177/10738584251318979
Rammes, G., Danysz, W., and Parsons, C. G. (2008). Pharmacodynamics of memantine: an update. Curr Neuropharmacol. 6, 55–78. doi: 10.2174/157015908783769671
Rogers, C. Q., Ramirez, M., Landon, C. S., DeBlasi, J. M., Koutnik, A. P., Ari, C., et al. (2023). A glutamate scavenging protocol combined with Deanna Protocol in SOD1-G93A mouse model of ALS. Nutrients 15:1821. doi: 10.3390/nu15081821
Rosenberg, G., Angel, I., and Kozak, A. (2005). Clinical pharmacology of DP-b99 in healthy volunteers: first administration to humans. Br. J. Clin. Pharmacol. 60, 7–16. doi: 10.1111/j.1365-2125.2005.02378.x
Sadeh, M., Shen, X., and Engel, A. G. (2011). Beneficial effect of albuterol in congenital myasthenic syndrome with epsilon-subunit mutations. Muscle Nerve 44, 289–291. doi: 10.1002/mus.22153
Sáez-Orellana, F., Godoy, P. A., Bastidas, C. Y., Silva-Grecchi, T., Guzmán, L., Aguayo, L. G., et al. (2016). ATP leakage induces P2XR activation and contributes to acute synaptic excitotoxicity induced by soluble oligomers of β-amyloid peptide in hippocampal neurons. Neuropharmacology 100, 116–123. doi: 10.1016/j.neuropharm.2015.04.005
Samways, D. S. K., Khakh, B. S., and Egan, T. M. (2008). Tunable calcium current through TRPV1 receptor channels. J. Biol. Chem. 283, 31274–31278. doi: 10.1074/jbc.C800131200
Sattler, R., and Tymianski, M. (2001). Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol. Neurobiol. 24, 107–130. doi: 10.1385/MN:24:1-3:107
Sattler, R., Xiong, Z., Lu, W.-Y., Hafner, M., MacDonald, J. F., and Tymianski, M. (1999a). Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 284, 1845–1848. doi: 10.1126/science.284.5421.1845
Sattler, R., Xiong, Z., Lu, W.-Y., Macdonald, J. F., and Tymianski, M. (1999b). Distinct roles of synaptic and extrasynaptic NMDA receptors in excitotoxicity. J. Neurosci. 20, 22–33. doi: 10.1523/JNEUROSCI.20-01-00022.2000
Schlaepfer, W. W., and Bunge, R. P. (1973). Effects of calcium ion concentration on the degeneration of amputated axons in tissue culture. J. Cell Biol. 59, 456–470. doi: 10.1083/jcb.59.2.456
Sciaccaluga, M., Moriconi, C., Martinello, K., Catalano, M., Bermudez, I., Stitzel, J. A., et al. (2015). Crucial role of nicotinic α5 subunit variants for Ca2+ fluxes in ventral midbrain neurons. FASEB J. 29, 3389–3398. doi: 10.1096/fj.14-268102
Seo, D. W., Lopez-Meraz, M. L., Allen, S., Wasterlain, C. G., and Niquet, J. (2009). Contribution of a mitochondrial pathway to excitotoxic neuronal necrosis. J. Neurosci. Res. 87, 2087–2094. doi: 10.1002/jnr.22035
Sepers, M. D., and Raymond, L. A. (2014). Mechanisms of synaptic dysfunction and excitotoxicity in Huntington’s disease. Drug Discov. Today 19, 990–996. doi: 10.1016/j.drudis.2014.02.006
Sine, S. M., Ohno, K., Bouzat, C., Auerbach, A., Milone, M., Pruitt, J. N., et al. (1995). Mutation of the acetylcholine receptor α subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron 15, 229–239. doi: 10.1016/0896-6273(95)90080-2
Skeberdis, V. A., Chevaleyre, V., Lau, C. G., Goldberg, J. H., Pettit, D. L., Suadicani, S. O., et al. (2006). Protein kinase A regulates calcium permeability of NMDA receptors. Nat. Neurosci. 9, 501–510. doi: 10.1038/nn1664
Spalloni, A., Nutini, M., and Longone, P. (2013). Role of the N-methyl-d-aspartate receptors complex in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1832, 312–322. doi: 10.1016/j.bbadis.2012.11.013
Stutzmann, G. E. (2007). The pathogenesis of Alzheimer’s disease—is it a lifelong “calciumopathy”? Neuroscientist 13, 546–559. doi: 10.1177/1073858407299730
Sun, D., Amiri, M., Meng, Q., Unnithan, R. R., and French, C. (2024). Calcium signalling in neurological disorders, with insights from miniature fluorescence microscopy. Cells 14:4. doi: 10.3390/cells14010004
Takasu, M. A., Dalva, M. B., Zigmond, R. E., and Greenberg, M. E. (2002). Modulation of NMDA receptor-dependent calcium influx and gene expression through EphB receptors. Science 295, 491–495. doi: 10.1126/science.1065983
Tang, B., Wang, Y., and Ren, J. (2023). Basic information about memantine and its treatment of Alzheimer’s disease and other clinical applications. Ibrain 9, 340–348. doi: 10.1002/ibra.12098
Tombaugh, G. C., and Sapolsky, R. M. (1990). Mild acidosis protects hippocampal neurons from injury induced by oxygen and glucose deprivation. Brain Res. 506, 343–345. doi: 10.1016/0006-8993(90)91277-N
Traynelis, S. F., and Cull-Candy, S. G. (1990). Proton inhibition of N-methyl-D-aspartate receptors in cerebellar neurons. Nature 345, 347–350. doi: 10.1038/345347a0
Tsien, R. W., Hess, P., McCleskey, E. W., and Rosenberg, R. L. (1987). Calcium channels: mechanisms of selectivity, permeation, and block. Annu. Rev. Biophys. Biophys. Chem. 16, 265–290. doi: 10.1146/annurev.bb.16.060187.001405
Tymianski, M., Charlton, M. P., Carlen, P. L., and Tator, C. H. (1994a). Properties of neuroprotective cell-permeant Ca2+ chelators: effects on [Ca2+]i and glutamate neurotoxicity in vitro. J. Neurophysiol. 72, 1973–1992. doi: 10.1152/jn.1994.72.4.1973
Tymianski, M., Spigelman, I., Zhang, L., Carlen, P. L., Tator, C. H., Charlton, M. P., et al. (1994b). Mechanism of action and persistence of neuroprotection by cell-permeant Ca2+ chelators. J. Cereb. Blood Flow Metab. 14, 911–923. doi: 10.1038/jcbfm.1994.122
Tymianski, M., and Tator, C. H. (1996). Normal and abnormal calcium homeostasis in neurons: a basis for the pathophysiology of traumatic and ischemic central nervous system injury. Neurosurgery 38, 1176–1195. doi: 10.1097/00006123-199606000-00028
Van Den Bosch, L., Van Damme, P., Bogaert, E., and Robberecht, W. (2006). The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 1068–1082. doi: 10.1016/j.bbadis.2006.05.002
Villmann, C., and Becker, C. M. (2007). On the hypes and falls in neuroprotection: Targeting the NMDA receptor. Neuroscientist 13, 594–615. doi: 10.1177/1073858406296259
Watanabe, J., Beck, C., Kuner, T., Premkumar, L. S., and Wollmuth, L. P. (2002). DRPEER: a motif in the extracellular vestibule conferring high Ca2+ flux rates in NMDA receptor channels. J. Neurosci. 22, 10209–10216. doi: 10.1523/JNEUROSCI.22-23-10209.2002
Yan, J., Bengtson, C. P., Buchthal, B., Hagenston, A. M., and Bading, H. (2020). Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science 370:370. doi: 10.1126/science.aay3302
Zamponi, G. W., Striessnig, J., Koschak, A., and Dolphin, A. C. (2015). The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol. Rev. 67, 821–870. doi: 10.1124/pr.114.009654
Zhou, Z., and Neher, E. (1993). Calcium permeability of nicotinic acetylcholine receptor channels in bovine adrenal chromaffin cells. Pflugers Arch. 425, 511–517. doi: 10.1007/BF00374879
Keywords: neuroprotection, neurodegeneration, NMDA receptor, nicotinic acetylcholine receptor, fractional Ca2+ current
Citation: D’Andrea T and Fucile S (2025) Reduction of the Ca2+ permeability of ligand-gated ion channels as a strategy against excitotoxicity. Front. Cell. Neurosci. 19:1617006. doi: 10.3389/fncel.2025.1617006
Edited by:
Richard Anthony DeFazio, University of Michigan, United StatesReviewed by:
Manish Shukla, Penn State Milton S. Hershey Medical Center, United StatesCopyright © 2025 D’Andrea and Fucile. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergio Fucile, c2VyZ2lvLmZ1Y2lsZUB1bmlyb21hMS5pdA==