 Marika Attili1,2
Marika Attili1,2 Sonia Valentini1,2
Sonia Valentini1,2 Maria Pesavento1,2
Maria Pesavento1,2 Marco Ballarotto3†
Marco Ballarotto3† Viviana Scognamiglio4
Viviana Scognamiglio4 Alfredo Pontecorvi5Antonio Macchiarulo3
Alfredo Pontecorvi5Antonio Macchiarulo3 Fulvio Saccoccia1*‡
Fulvio Saccoccia1*‡ Fabiola Moretti1*‡
Fabiola Moretti1*‡- 1Institute of Biochemistry and Cell Biology, National Research Council of Italy (CNR), Monterotondo, Rome, Italy
- 2Ph.D. Course in Sciences of Nutrition, Metabolism, Aging and Gender-related Pathologies, Catholic University Sacro Cuore, Rome, Italy
- 3Department of Pharmaceutical Sciences, University of Perugia, Perugia, Italy
- 4Institute of Crystallography, National Research Council of Italy (CNR), Rome, Italy
- 5Department of Medicine and Translational Surgery, Catholic University of Roma, Rome, Italy
Introduction: In the era of targeted therapies, molecules for the reactivation of the oncosuppressor p53 in human cancer have not yet reached FDA or EMA approval. Recently, the interaction region of the MDM2/MDM4 heterodimer, the most efficient inhibitor of p53 levels and function, has been successfully targeted. Disruption of the heterodimer activated p53 oncosuppressive function in vitro and in vivo. Despite these encouraging results, further studies on the MDM2/MDM4 interaction region have yet to progress.
Methods: Here, we undertook a detailed bioinformatic and biochemical analysis of this region. Using molecular dynamics simulation followed by umbrella sampling.
Results and discussion: We characterized a short peptide and modified derivatives with increased binding affinity and pharmacodynamics features compared to previous molecules. Our results uncover the intrinsic plasticity of the MDM2 RING domain through different binding clefts and provide evidence of its ability to host different peptides by key residues. This data may guide the development of next-generation therapeutic inhibitors.
1 Introduction
The p53 gene is one of the most relevant oncosuppressors. Numerous clinical and experimental evidences support this notion: the gene is mutated or inactivated in almost 50% of overall human cancer; the p53 knockout mouse develops tumors by 6 months of life; the LiFraumeni syndrome, characterized by the risk of developing multiple invasive cancers (approximately 90% by age 70) is associated with p53 mutation (Hassin and Oren, 2023). The relevance of the oncosuppressive activity of p53 in human cancer has sustained the development of numerous molecules to correct the altered structure of mutant p53 and/or re-establish its function (Hassin and Oren, 2023). Following the discovery of MDM2 in 1992 and its function as a crucial inhibitor of p53, targeting the interaction between p53 and MDM2 has been regarded as a leading approach to therapeutic intervention (Zhu et al., 2022; Hassin and Oren, 2023). Research studies realized the prototype of these molecules, Nutlin-3a, with great excitement and hope (Vassilev et al., 2004). From that point, numerous molecules have been developed. Unfortunately, cancer patients still do not benefit from these efforts.
The reasons for these partial failures are multiple (Haronikova et al., 2021; Zhu et al., 2022). One adverse effect that impairs the advancement of some p53-reactivating molecules is their toxicity to non-malignant cells, due to bone marrow cytopenia and gastrointestinal disorders (Lu et al., 2012). In addition, the discovery of the MDM2 homolog, MDM4, suggested one additional explanation for the lack of success of previous MDM2 inhibitors since those inhibitors showed reduced ability in MDM4 targeting (Yu et al., 2020). Consequently, new molecules have been identified to target MDM4 or both proteins simultaneously (Skalniak et al., 2019; Teveroni et al., 2016; Yu et al., 2020). Given the lower ability of MDM4 to inhibit p53, MDM4 inhibitors have shown low efficiency and did not go beyond Phase I clinical trials (Yu et al., 2020). Conversely, some success was obtained by dual inhibitors, new molecules able to target MDM4 and MDM2 simultaneously (Skalniak et al., 2019; Teveroni et al., 2016).
At the same time, other approaches for the reactivation of p53 emerged, such as those targeting specific activities of MDM2. Indeed, this protein exerts a double control over p53: i) it maintains the p53 protein at low levels through its ubiquitin ligase activity; ii) it interferes with the interaction of p53 with factors of the transcription machinery by binding the transactivation domain of p53 (Karni-Schmidt et al., 2016; Shadfan et al., 2012). Data from animal models expressing knock-in MDM2 genes with impaired E3 ubiquitin ligase activity suggested that the impairment of this activity may represent a favorable approach for p53 reactivation. Inhibition of the sole MDM2 ubiquitin ligase activity allows for maintaining the control of basal p53 transcriptional function in nontumor cells and can avoid the observed toxicities of MDM2-p53 binding disruptors due to p53 overfunction in healthy tissues (Humpton et al., 2021; Tollini et al., 2014).
Moreover, since the ubiquitin ligase MDM2 is provided with oncogenic activities additional to the control of p53 (Coutts et al., 2007; Fu et al., 2009; Yang et al., 2006), inhibiting this activity has opened potential therapeutic approaches in tumors with MDM2 overexpression (Weissman et al., 2008).
Additional detailed studies of p53 degradation evidenced the superior efficiency of the ubiquitin ligase function of the heterodimer MDM2-MDM4 compared to the sole MDM2 in controlling p53 levels (Gu et al., 2002; Linke et al., 2008; Tanimura et al., 1999). Therefore, some approaches were proposed to interfere with the heterodimer to hamper its inhibitory activity (Herman et al., 2011). Since the interaction region of the two proteins is an extended β-sheet flat surface, the search was also directed toward protein-protein interaction inhibitors. In 2015, a peptide able to interfere with the heterodimerization of the two proteins was reported (Pellegrino et al., 2015). This peptide binds MDM2 and activates p53 in vitro and in vivo, inducing an oxidative stress response and a consequent apoptotic collapse of tumor cells. From that study, an in-depth analysis of the chemical structure of the heterodimer region emerged as mandatory to deepen binding features at an atomic level and to design novel, more effective inhibitors. In this work, we performed a detailed bioinformatic and biochemical analysis of the MDM2/MDM4 interaction region and, by in silico screening, characterized two peptides targeting this region that uncovered additional conformations of the MDM2 RING (Really Interesting New Gene) domain.
2 Materials and methods
2.1 Alignment of peptides KVFIA and PWFRW
Starting from the crystal structure of the heterodimer complex (PDB code: 2VJF), a complex of KVFIA with MDM2 was generated in PyMol by truncating the RING domain of MDM4 (chain B). After selecting KVFIA and the amino acid around 4Å, the functions “find any contact” and “find polar contacts” were used to detect the interactions between the peptide and MDM2. MD trajectory files were inspected frame-by-frame to analyse the different binding positions of KVFIA and PWFRW and which regions of MDM2 counteract. We highlight those using the functions described above.
2.2 Alignment of MDM2-MDM4 sequences and WebLogo graphic
The sequences used for the alignments were obtained from the UniProt database using only verified sequences. The sequences were loaded into the online tool Clustal Omega for Multiple Sequence Alignment (MSA) (Madeira et al., 2022). These alignments were created using the online tool Weblogo.berkeley.edu. The total height of the stack indicates the sequence conservation at that position. In contrast, the height of the symbols within the stack indicates the relative abundance of each amino acid at that position.
2.3 Docking and molecular dynamics
KVFIA peptide (ligand) was generated, trimming out the last five amino acids from the MDM4 structure in the PDB 2VJF entry. The datasets generated and/or analysed during the current study are available in the RCSB Protein Data Bank repository, https://www.rcsb.org/structure/2VJF. Before docking, the ligand was fully hydrogenated and was subjected to geometry optimization performed with obminimize subroutine within Open Babel, v. 3.1.0 (O'Boyle et al., 2011) and MMFF94 force field; minimization was left free up to 10,000 minimization steps to reach 1e-8 kcal mol-1 under steepest descent algorithm. The RING/MDM2 (receptor) was extracted from the 2VJF entry, holding a single monomer. All water was removed from the crystal structure. Hence, AutoDock Tools (ADT, v. 1.5.7) (Morris et al., 2009) was used to edit the structure (where needed), add formal charges, and convert the files into .pdbqt format. Docking was performed by AutoDock Vina, v.1.2.3 (Eberhardt et al., 2021; Trott and Olson, 2010): a cubic (8,000 Å3) search box was centered around the MDM4 C-terminal binding site, and an exhaustive search (set at 24) was performed. For the small-scale virtual screening campaign on the 72 peptides, ligands were fully hydrogenated and subjected to geometry optimization performed with obminimize as KVFIA. Docking was performed by limiting the searching grid (20 × 20 × 20 Å) over the binding cleft of KVFIA. For the small-scale virtual screening campaign of the relative’s compounds, a series of 72 peptides (out of the 3,200,000 possible permutations) was screened, which stemmed from KVFIA and then by gradually modifying the original peptide, based on the predicted energy by Autodock Vina after any substitutions. For each compound, hydrogenation and energy minimization steps were performed as above. GROMACS, v. 2023, compiled with CUDA support, was used in all simulation and analysis steps; additional data analysis and associated plots were performed with GNU/Octave (Eaton et al., 2024). Molecular dynamics simulation started from the best (highest scoring) binding pose after the Vina run. The protein and the peptide were initially modeled as a single multi-chain specimen using CHARMM36 (release July 2022). TIP3P water was used to solvate the system. All hydrogens and water were removed from the initial model; proper coordination of the two zinc ions embedded in the RING/MDM2 was assured by setting a harmonic potential between zinc and cysteines S atoms or histidine proximal N atoms, using equilibrium distances of 0.2 nm and 0.24 nm, respectively, and a force constant of 4 × 103 kJ mol-1 nm2 as parameters. The two atoms were also restrained in position (k = 105 kJ mol-1 nm2) during energy minimization and equilibration steps. The Verlet algorithm was used to integrate Newton’s equation of generated motion. Electrostatic interactions were treated using the particle mesh Ewald (PME) method with a cutoff value of 1.2 Å and grid spacing for FFT set at 1.6Ǻ; short-range electrostatic cutoff and Wan der Waals cutoff were set at 16 Å each. A triclinic solvation box of 6.06924, 6.06924, 4.29160 nm axes length was generated and filled with 4,714 water molecules, keeping the distance between the protein and the walls of the box at 1 nm; K+ and Cl− (38 and 52 atoms, respectively) were added to ensure neutrality while keeping salt concentration at 400 mM. Energy minimization was performed with a steep descent method up to 100 kJ mol-1 nm-1 allowed force. Two subsequent steps of proper equilibration were performed under canonical (NVT, 200 ps) and thermobaric-thermostatic (NPT, 500 ps) ensemble. Restraints were removed, and the production phase was performed under NPT conditions, with a time step of 2 fs, up to 200 ns.
In the umbrella sampling simulation of KVFIA, an orthorhombic solvation box of 14 × 8 × 8 nm axes in length was generated; the protein was oriented to align the longest axes of the peptide perpendicular to the x-axes, which had been chosen as the pulling coordinate (the reaction coordinate, ζ, in subsequent WHAM analysis), and keeping the distance between the protein and the wall of the box at 1 nm. The new box was filled with 28,791 water molecules; K+ and Cl− (216 and 230 atoms, respectively) were added to ensure neutrality while keeping salt concentration at 400 mM. Energy minimization and NPT equilibration were performed as above. Then, the pulling force of 500 kJ mol-1 nm-2 was applied to the center of mass (COM) of KVFIA, whereas the COM of the RING/MDM2 was kept in place, applying proper restraints to the whole molecule. The pulling rate was kept fixed at 0.005 nm ps-1. A series of configurations along the reaction coordinate, ζ, was extracted from the pulling simulation to conduct umbrella sampling. Hence, this served as the starting configurations for the umbrella sampling windows, which were run as independent simulations, each comprising an NPT ensemble equilibration (200 ps) and a run of productive step (10 ns). In both cases, restraints were applied to keep RING/MDM2 in place, whereas a harmonic potential with a spring constant equal to 1,500 kJ mol-1 nm-2 was added to the sample peptide conformational space. The force on the spring over time is visible in Supplementary Figure S2. Electrostatic interactions were treated using the particle mesh Ewald (PME) method with a cutoff value of 1.2 Å and grid spacing for FFT set at 1.2 Å. A total of 63 windows, which ultimately recapitulate the simulated detachment of the peptide from its target, were generated, sampled, and analysed to compute the one-dimensional potential of mean force (PMF). The spacing between adjacent windows was chosen to ensure a good overlap of sampled windows in the final histogram, and the distance ranged from 0.1 to 0.5 nm. All the 63 generated windows were used to compute the PMF using the WHAM algorithm implemented in GROMACS. Thus, 642.6 ns simulation time was used to calculate the single protein-peptide interaction PMF. Bootstrap analysis of over 200 replicates was performed to get statistical significance.
Molecular dynamics for the RING/MDM2/PWFRW complex was performed starting from the best (first) binding pose after the Vina run and modeled as already done for KVFIA. Parameters were set as reported above for KVFIA. A triclinic solvation box of 6.06924, 6.06924, 4.29160 nm axis length was generated and filled with 4,701 water molecules, keeping the distance between the protein and the walls of the box at 1 nm; K+ and Cl− (38 and 52 atoms, respectively) were added to ensure neutrality while keeping salt concentration at 400 mM. Energy minimization was performed with a steep descent method up to 100 kJ mol-1 nm-1 allowed force, followed by an additional step with a conjugate gradient method up to 10 kJ mol-1 nm-1 allowed force. Two subsequent steps of proper equilibration were performed under canonical (NVT, 200ps in length) and thermobaric-thermostatic (NPT, 500 ps) ensemble. Restraints were removed, and the production phase was performed under NPT conditions, with a time step of 2 fs, up to 200 ns. The configuration obtained after 160 ns, being in a minimum, was used as starting material to perform the pulling/United States protocol. In the umbrella sampling simulation of PWFRW, an orthorhombic solvation box of 6, 14, 4 nm axis length was generated; the box was chosen to keep it as small as possible to limit computational costs; the protein was oriented to align the longest axes of the peptide perpendicular to the y-axes which had been chosen as the pulling coordinate (the reaction coordinate, ζ, in subsequent WHAM analysis), and keeping the distance between the protein and the walls of the box at 1 nm. The new box was filled with 10,504 water molecules; K+ and Cl− (81 and 95 atoms, respectively) were added to ensure neutrality while keeping salt concentration at 400 mM. Energy minimization and NPT equilibration were performed as above. Then, the pulling procedure and the umbrella sampling were performed as previously described for KVFIA, but the productive step was limited to 5 nsec. Sixty-eight windows were used to compute the PMF using the WHAM algorithm implemented in GROMACS. Thus, 353.6 ns simulation time was used to calculate the single protein-peptide interaction PMF.
MM/GBSA was performed on the central frames obtained after the first production run, avoiding the initial burst and latest (detachment of KVFIA) phases, for a total of 20,050 frames; sampling was performed every 10 frames; oldff/leaprc. ff99SB, leaprc. gaff were used as force fields to build up topologies; PDBradii was set to three; internal and external dielectric constants were set to 1 and 78.5, respectively; salt concentration was set to 0.41 M; solvent probe radius for surface area calculation was set to 1.4 Å.
2.4 Clustering analysis
Clustering analysis was performed by GROMACS on data belonging to both simulations, ranging from 10ns to 150 ns, to get an equal number of frames to be integrated and to avoid both the equilibration phase and the latest period corresponding to the detachment of KVFIA and PWFRW from MDM2. Distances between structures have been determined by the Gromos algorithm. The RMSD cut-off (nm) for two structures to be considered as neighbourhoods was set at 0.085. This value was chosen after several trials to get populations of comparable size in the two systems while trying to avoid too striking shrinkage of the original data. Only clusters containing more than 3% of the population were considered representative of stable conformational states.
2.5 Free energy landscape analysis
Free energy landscapes were constructed using the sham module in Gromacs from RMSD and Rg data obtained from MD simulation ranging from 10 ns to 150 ns; this window ensured an equal number of frames to be integrated and avoid both the equilibration phase and the latest period corresponding to the detachment of KVFIA from MDM2. The final 2D and 3D plots were generated by GNU/Octave.
2.6 GST-MDM2 production
BL21 competent cells were transfected with pGEX4T-1-Flag-HsMdm2 construct by heat shock transformation protocol. Engineered BL21 cells were grown in LB Broth (Luria) with 100 μg/mL ampicillin (Sigma-Merck) and incubated at 37°C on a shaker. When the bacterial culture reached OD600 = 0.7, 0.5 mM Isopropil-β-D-1-tiogalattopiranoside, IPTG (Sigma-Merck), and 200 µM ZnCl2 (Sigma-Merck) were added. After 2 h at 37°C, the culture was pelleted at 3,000 rpm and the pellet was lysed, by sonication, with NETN buffer (50 mM Tris pH 8.3 and 150 mM NaCl) supplemented with 100 μg/mL DNase, 1mM PMSF, 0.2 mg/mL lysozyme and 10% glycerol. The solution was centrifugated at 16,000 rpm for 2 h at 4°C. The salt precipitation protocol was then performed using ammonium sulfate ((NH4)2SO4). Different fractions obtained by precipitation with increasing concentrations of (NH4)2SO4 were controlled by Coomassie gel. The fraction enriched by MDM2 fusion protein (60% and 90%) was collected and purified by binding to glutathione agarose resin (Sigma-Merck) at 4°C for 2 h and then eluted using 2 mM ATP and 10 mM MgCl2 to enhance purification from contaminating E. coli Heat-Shock Proteins. After elution, 60U thrombin (Sigma-Merck) were added to the solution. The cleavage step was left overnight at 4°C under rotation. The next day, ion exchange with HiTrap QFF column (Sigma-Merck) was performed and the fraction containing MDM2 was concentrated with Amicon® Ultra 4 mL (30 kDa MWCO). Every step of the entire procedure was controlled by Coomassie gel, and a BSA standard curve was used for quantitation. Quantitation was also confirmed by Bradford assay.
2.7 Binding studies
The binding of KVFIA to the recombinant form of the human full-length MDM2 was assessed by quenching of intrinsic fluorescence at 340 nm after excitation at 280 nm. Spectra were recorded at 25°C on a Jasco FP-8200 spectrofluorometer equipped with a thermostat. Five traces were collected to get an averaged emission signal. Peptides’ concentration ranged from 30 pM up to 1 nM. After adding each peptide concentration, the system was left free to equilibrate for a few minutes under stirring. The dissociation constant, KD, was obtained from non-linear regression fit to the general binding curve (Langmuir isotherm), assuming the formation of a 1:1 complex, as follows:
where Fi is the measured fluorescence after ith peptide addition, Fmax is the intrinsic MDM2 fluorescence in the absence of any peptide, Fmin is the fluorescence of the fully complexed protein, KD the dissociation constant for the peptide under investigation, and [pep]i the concentration of added peptide at ith step.
3 Results
3.1 Characterization of MDM2-MDM4 interaction interface
The MDM2 and MDM4 proteins interact through their RING (Really Interesting New Gene) domains and form an extended six-strand β-barrel region, contributed by the secondary structure elements belonging to both proteins (Figure 1A). Since the RING domains of MDM2 (RING/MDM2) and MDM4 (RING/MDM4) are highly conserved alongside with their tertiary structures, their interaction generates a nearly symmetrical heterodimer (the root-mean-square deviation - RMSD - among the two proteins being 0.334 Å) (Figure 1A).
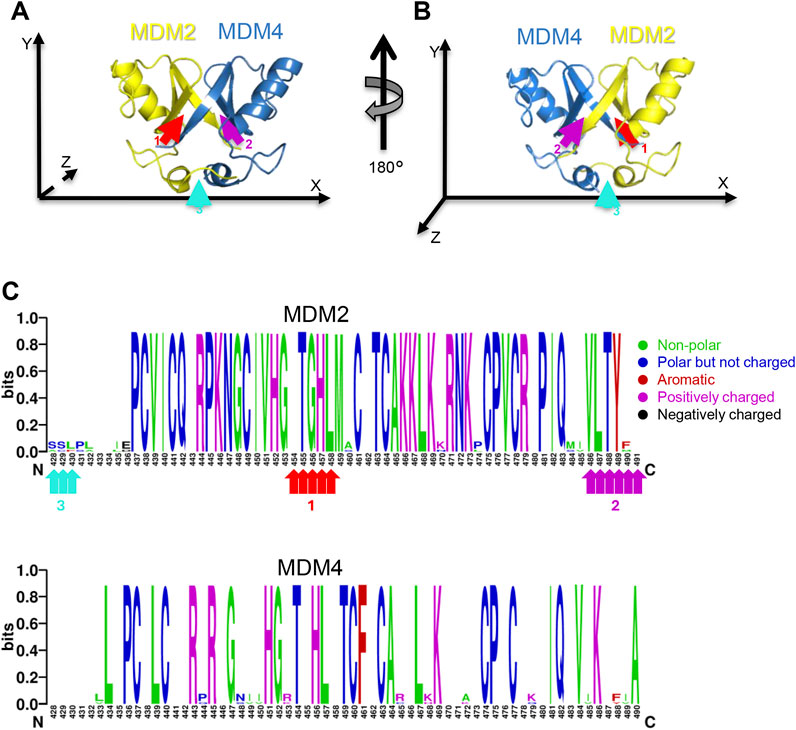
Figure 1. RING-RING interaction region of MDM2/MDM4 heterodimer. (A, B) Different angles of the heterodimer crystal structure. Yellow is the RING domain of MDM2, and blue is the one of MDM4. (A, B) are obtained by 180° rotation around the Y-axis. The arrows indicate the three contact regions of the RING domains. The red arrow (number 1) indicates the interaction between the C-terminus of MDM4 and the second beta-sheet of MDM2, the magenta arrow (number 2) indicates the interaction between the C-terminus of MDM2 and the second beta-sheet of MDM4, the cyan arrow (number 3) indicates the interaction between the two small alpha-helices of MDM2 and MDM4 (picture created by PyMol PDB Code: 2VJF). (C) Visual representation of the MDM2 and MDM4 RING sequence conservation by Web Logo. The image is created by aligning four sequences of Danio Rerio, Xenopus Leavis, Mouse, and Human, representing three out of five classes of chordates: fish, amphibians, and mammals. The bits measure the conservation of the residues. For MDM2 alignment the sequences used are: sp|O42354|MDM2_DANRE, sp|P56273|MDM2_XENLA, sp|P23804|MDM2_MOUSE, sp|Q00987|MDM2_HUMAN. For MDM4 alignment the sequences used are sp|Q7ZUW7|MDM4_DANRE, sp|Q7ZYI3|MDM4_XENLA, sp|O35618|MDM4_MOUSE, sp|O15151|MDM4_HUMAN. The different colours indicate the physico-chemical features of amino acids, as reported in the legend on the right. The arrows indicate the regions of interaction described in (A, B).
By setting an arbitrary reference system, 180° rotation of the structure along the y-axis shows the presence of three interaction regions that contribute to the binding of the two RING domains: i) the first one involves the C-terminus of MDM4 with the second alpha-helix of MDM2 (Figures 1A,B, red arrow 1); ii) the second one involves the C-terminus of MDM2 with the second β-sheet of MDM4 (Figures 1A,B, magenta arrow 2); iii) the third one involves the small α-helix at the N-terminus of the two proteins (Figures 1A,B, cyan arrow 3). Previous studies showed that the first interaction region is mandatory for the formation of the heterodimer (Linke et al., 2008). Indeed, analysis of the conserved residues in MDM2 revealed that the amino acid sequences are highly conserved at the interaction region 1 (Figure 1C red arrows; Supplementary Figure S1A), supporting the crucial function of this region. Interaction region 2 also showed high level of conservation, whereas region 3 was poorly conserved. In comparison, the MDM4 sequence is less conserved (Figure 1C; Supplementary Figure S1B). Based on this data, we investigated peptides interacting with MDM2 region 1 as potential inhibitors of the heterodimer formation.
3.2 Chemical features of short peptide Pep3S
The previously identified Peptide 3 (Pep3) mimics the MDM4 C-terminus and interacts with the region 1 of MDM2 encompassing the last C-terminus twelve amino acids of MDM4 (NH2-KEIQLVIKVFIA-COOH). Pep3 competes with endogenous MDM4 for binding to MDM2, interfering with the heterodimer assembly. Based on the available crystal structure of MDM2 and MDM4 heterodimer and the binding data of the dodecamer peptide (Ballarotto et al., 2024; Pellegrino et al., 2015), a shorter peptide was designed, contributed by only the last five amino acids of Pep3 (Pep3S, hereafter also referred to as KVFIA).
Structural analysis of the deposited PDB 2VJF entry (Linke et al., 2008) revealed that this peptide makes multiple interactions with the residues of the interaction region 1 of the RING/MDM2, namely, N433, K446, K454, T455, G456, H457, L458, and M459 (Figure 2; Supplementary Table S1). Besides some non-polar contacts, the binding of KVFIA is fostered by six polar interactions, supporting the hypothesis of high affinity of this peptide (Supplementary Table S1).
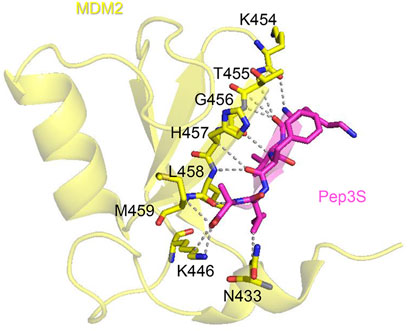
Figure 2. KVFIA/MDM2 interaction. PyMOL cartoon structure depicting the crystal structure of MDM2 RING (yellow) interacting with KVFIA (magenta). The residues of MDM2 interacting with KVFIA are shown in sticks.
3.3 Docking and molecular dynamics simulation of KVFIA peptide
To get a more precise picture of the putative binding of KVFIA to MDM2, we performed docking studies starting from the available crystal structure of the MDM2/MDM4 RING complex, PDB 2VJF. All but the C-terminal tail of MDM4 was trimmed out from the structure, resulting in the Pep3S, NH2-KVFIA-COOH sequence. This peptide was left free to bind the RING/MDM2 region in a targeted docking run by centering the search grid region where the C-terminal tail of MDM4 binds its partner in the crystal structure. Docking was performed to get an initial estimation of binding energy and to generate a starting structure for subsequent molecular dynamics simulation. The best binding poses account for predicted energies from −5.7 to −5.3 kcal/mol (in replicates of docking runs), roughly placing KVFIA binding affinity in the middle micromolar range (68–133 µM) at 300 K.
To validate this docking pose, we investigated the stability of the KVFIA-RING/MDM2 complex by molecular dynamics (MD) simulation performed in explicit water solvent for 200 ns under constant pressure (1 bar) and constant temperature (300K). Initially, MD aimed to get the best stable interaction pose compatible with the size and shape of the RING/MDM2 groove with KVFIA. Root Mean Square Deviation (RMSD) analysis revealed that the system equilibrates after a few nanoseconds (Figure 3A). A subsequent almost flat region could be recognized up to 150 ns. The latest part of the simulation showed the detachment of the peptide from the binding groove (Figure 3A) due to the direct contact with the solvent that made the peptide free to explore a broader range of conformations.
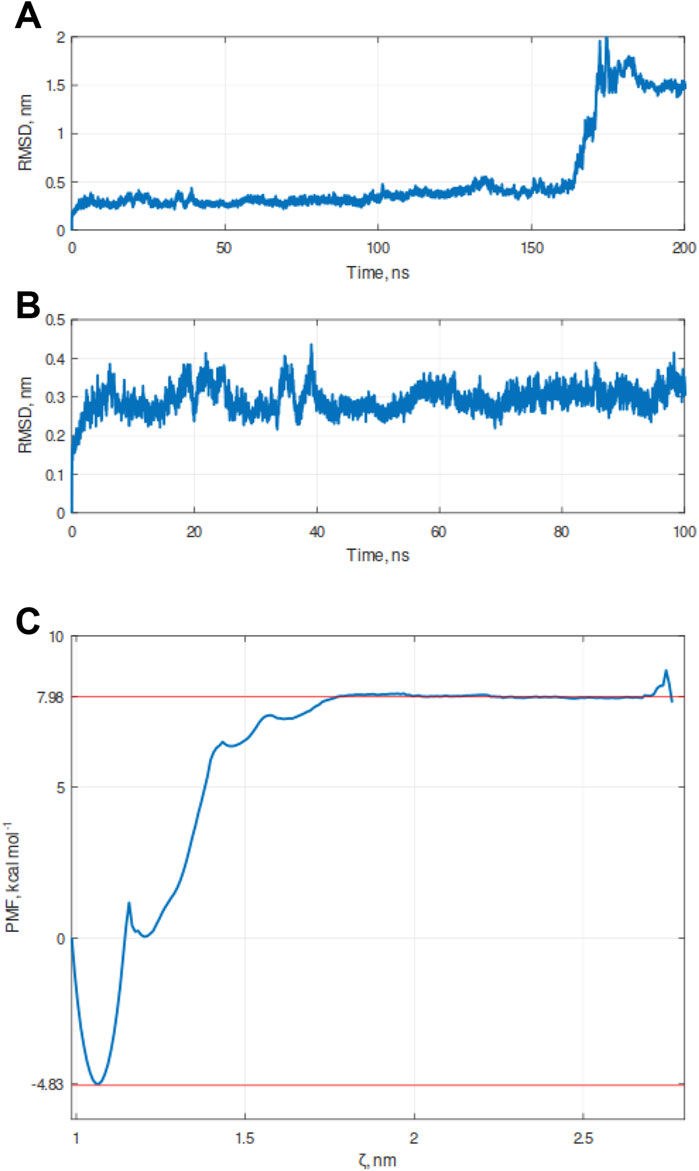
Figure 3. Molecular dynamics (MD) simulation of the KVFIA/RING-MDM2 complex. (A) Graph of Root Mean Square Deviation (RMSD) calculated over the simulation timeframe of 200 ns under constant pressure (1 bar) and constant temperature (300K). The detachment of KVFIA was observed after 160 ns. (B) Magnification of the previous graph between the time frame 0–100 ns. In the nearly 50 ns, the system was at a local minimum, and this point was chosen as a starting point for the pulling procedure. (C) Free energy landscape of simulated detachment of KVFIA from RING/MDM2, after umbrella simulation (US) procedure. The binding energy (ΔGbind) was evaluated by integrating the potential of mean force (PMF) obtained after a series of umbrella sampling (US) simulations; a total pulling time of 600 ps was performed. The plot was generated by the combination of 68 frames (Supplementary Figure S2). Red dotted lines delimit the lower (LV, −4.83 kcal mol-1) and the upper values (as the average of the flat upper region, UV, 7.98 kcal mol-1), whereby the ΔG was calculated as ΔG = UV-LV. A second local minimum, centered at 1.2 nm, likely accounts for a metastable state generated during the pulling procedure. The cusp in the PMF plot reflects an under-sampled region among the two minima (see Supplementary Figure S2B). Any attempts to get enhanced resolution of the profile in that region was unsuccessful.
This first simulation paved the way for a more accurate estimation of the energy associated with the binding of KVFIA to RING/MDM2 by the umbrella sampling (United States), an in silico procedure well suited for this kind of system (Broomhead and Soliman, 2017; Pantsar and Poso, 2018). The US procedure permits the calculation of the binding energy between two specimens by using the pulling simulation (Kästner, 2011), a computational method by which an imaginary force is applied to the center of mass (COM) of one molecule belonging to a bimolecular complex along an arbitrary path for the detachment of the molecule while keeping the other COM restrained in its original position. To recapitulate KVFIA dissociation from the target RING/MDM2, we chose the 50 ns time point, at which RMSD reached the minimum value (Figure 3B), as starting point. We applied a pulling force of 500 kJ mol-1 nm-2 with a pulling rate of 0.005 nm ps-1 to the COM of KVFIA while keeping MDM2 restrained in its original place. This procedure resulted in a constant increase of pulling force followed by a steep fall and an oscillating behaviour at a longer distance (Supplementary Figure S2A). The US was then performed by getting a series of initial configurations, taken at increasing COM distance from RING/MDM2. Each distance corresponds to a location where KVFIA was harmonically restrained by applying a biasing potential (set at 1500 kJ mol-1), which keeps the peptide in place while it explores a larger conformational space in that region. This procedure created a series of snapshots of the KVFIA detachment route from MDM2. The Weighted Histogram Analysis Method (WHAM, Supplementary Figure S2B) calculated after pulling and umbrella sampling indicated a predicted binding energy, ΔGbind, equal to −12.81 kcal mol-1 (calculated as the difference between the top flat region and the absolute minimum in the final PMF plot; Figure 3C). The obtained ΔGbind places KVFIA affinity in the sub-nanomolar range, with a calculated value of 430 pM at 300 K (Supplementary Table S2). This value is much higher than that obtained from docking and highlights the importance of further exploring the initial docking poses to get a better estimate of the binding affinity (Broomhead and Soliman, 2017; Pantsar and Poso, 2018; Tam et al., 2022). To further reinforce the umbrella sampling data, we pursued the MM/GBSA approach. In this case, the ΔG was equal to −12.6 ± 4.16. In the MM/GBSA method, the ΔG is obtained from the ΔG = ΔH-TΔS relation, obtaining ΔH = −30.55 ± 2.86 and -TΔS = 19.95 ± 3.02. Thus, the ΔG obtained with this approach is in line with that obtained after the US procedure.
3.4 In vitro analysis of KVFIA affinity
To confirm the high affinity of KVFIA for the RING/MDM2, as indicated by the MD/US simulation, we tested its value in solution. We performed an in vitro binding assay by measuring the intrinsic fluorescence of full-length recombinant MDM2 and its quenching in the presence of KVFIA. The sequence of full-length MDM2 has 11 Phe, 14 Tyr, and 4 Trp, which ultimately contribute to generating a fluorescent signal. We set up a protocol for producing human recombinant full-length MDM2 exploiting the Glutathione S-transferase (GST) Gene Fusion System and salting out by ammonium sulfate precipitation. The dissociation constant (KD) obtained by titrating a fixed amount of MDM2 (0.2 µM), with increasing concentration of Pep3S (from 30 pM to 1 nM), was 0.152 ± 0.007 nM (Table 1), in good agreement with that obtained by molecular dynamics and umbrella sampling simulation (0.430 nM), reinforcing the reliability of such bioinformatic approach. KVFIA affinity was almost half-decreased compared to Pep3 (Table 1), indicating increased efficacy of the short peptide in MDM2 binding. In comparison, a control scramble peptide (VAIKF) showed an affinity value equal to 0.447 ± 0.028 (Valentini et al., 2025).

Table 1. Binding affinity of peptides to full-length MDM2. Reported are the average value of two independent experiments performed in duplicates ±standard deviation.
3.5 In silico studies of KVFIA derivatives
We then focused on the relevance of the peptide sequence in determining the binding affinity. Using in silico screening, we tested a series of small peptides, each with up to four out of five residues altered from the original KVFIA sequence. From 3.2 million possible combinations, we shrank the list by observing that phenylalanine (Phe) at the central position and tryptophan (Trp) at the C-terminal (C-term) position consistently yielded lower docking energies. Fixing these residues to enhance binding affinity, we selected 72 pentapeptides (primarily composed mainly of L-α-amino acids) based on top docking scores (Supplementary Table S3).
These findings align with the physicochemical properties of KVFIA and literature data: Trp, being bulkier than Ala, more effectively fills the MDM2 binding groove, strengthening peptide-target interactions, while Phe is crucial for MDM4 binding to MDM2. (Poyurovsky et al., 2007; Uldrijan et al., 2007).
Among those peptides contributed by only α-amino acids and retaining the invariant Phe and the C-term Trp, PWFRW showed the highest predicted binding energy in docking (−7.4 kcal/mol; Supplementary Table S3), a neat increase over the original KVFIA (−5.7/–5.3 kcal/mol). Interestingly, the docking pose suggested a slightly different binding region compared to KVFIA (Supplementary Figures S3A, S3B). To compare MDM2 in complex with PWFRW and KVFIA, we performed molecular dynamics simulation. The initial MD run showed that PWFRW behaved differently from the original KVFIA: RMSD showed that PWFRW did not detach from RING/MDM2, unlike KVFIA (Figure 4A, blue line). Moreover, by comparing RMSF profiles obtained for alpha carbons of PWFRW and KVFIA, we observed that the former induced the RING/MDM2 to acquire a more rigid structure (Figure 4B). Finally, WHAM analysis provided predicted binding energy of 12.33 kcal mol-1, corresponding to 966 pM affinity (Figure 4C), a value that does not deviate too much from that obtained for KVFIA. These findings foster the idea that the heterodimer can be targeted by peptides whose composition is highly different from the original sequence of the MDM4 C-term while still maintaining high affinity to the RING/MDM2. Moreover, the crucial Phe at position three in both peptides appears to be a pivot for the two interaction regions, 1 and 2 of the MDM4/MDM2 heterodimer.
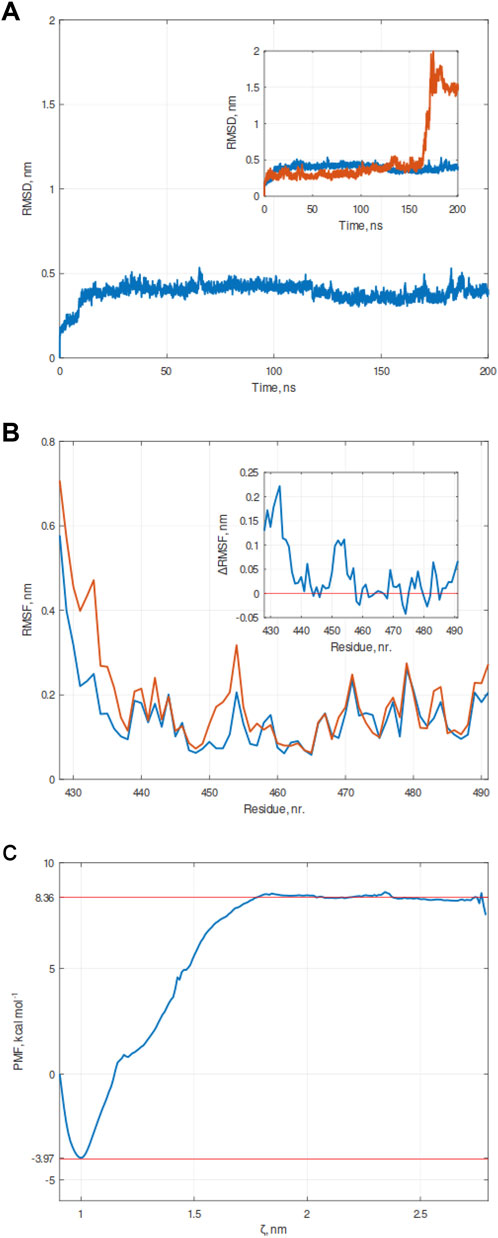
Figure 4. Molecular dynamics (MD) simulation of the PWFRW-RING/MDM2 complex. (A) Graph of Root Mean Square Deviation (RMSD) calculated over the simulation timeframe of 200 ns under constant pressure (1 bar) and constant temperature (300 K). Unlike KVFIA, no detachment was observed. The inset shows the superposition of RMSDs of KVFIA (red line) and PWFRW (blue line). (B) Root-mean-square fluctuation (RMSF) of RING/MDM2 (aa 428–491) in complex with KVFIA (red line) or PWFRW (blue line). A differential RMSF plot is shown in the inset. Any value above the threshold set to zero implies that the corresponding residue of RING/MDM2 (indicated as a number in the x-axis) fluctuates much more in the presence of KVFIA than in PWFRW. (C) Free energy landscape of simulated detachment of PWFRW from RING/MDM2, after Umbrella simulation (US) procedure. The binding energy (ΔGbind) was evaluated by integrating the potential of mean force (PMF) obtained after a series of Umbrella sampling (US) simulations; a total pulling time of 600 ps was performed. The plot was generated by the combination of 68 frames. Red dotted lines enclose the lower (LV, −3.97 kcal mol-1) and upper values (as the average of the flat upper region, UV, 8.36 kcal mol-1), whereby the ΔG was calculated as ΔG = UV-LV.
3.6 Structural comparison and analysis of KVFIA and PWFRW binding to RING/MDM2
Since the two peptides bound two different regions of the RING/MDM2, we wondered whether they experienced different conformational states when bound to RING/MDM2. For this purpose, we used MD trajectories of MDM2 in complex with both peptides to cluster their conformational states. This analysis showed that over 97% of conformational states cluster in three groups for KVFIA or two groups for PWFRW (Figures 5A,B; Table 2). This implies that the binding of KVFIA resulted in a broader number of different conformational states than PWFRW. In addition, clustering confirmed that the two peptides explored different portions of the binding cleft during the simulation (Figure 6A) and that, in turn, the binding was mediated by different interactions (Table 2). Particularly, the polar contacts of KVFIA were retained only by three out of five residues (K1, F3, A5) in all the three most representative clusters (Table 2; Figures 6A–D; Supplementary Tables S4-S6), indicating that the binding of KVFIA to MDM2 is very similar to that of C-terminal of MDM4 in the PDB-2VJF structure. Conversely, PWFRW makes polar contacts by four out of five residues (W2, F3, R4, W5) (Table 2; Figures 6A, C, E; Supplementary Tables S7, S8) in different clusters (Table 2, cluster 15). In addition, although both peptides make non-polar contacts with all their residues (Table 2), PWFRW contacted a higher number of unique residues belonging to RING/MDM2 (Table 2, cyan cells).

Figure 5. Clustering of conformational states of RING/MDM2-peptides’ complexes. Representative structures belonging to the most populated states (≥3%) show the different portions of the binding cleft experienced by the two peptides. (A, B) Representative structures after cluster analysis of the RING/MDM2 in complex with KVFIA - red, panel (A) - and PWFRW - magenta, panel (B). The figure highlights the different portions of the interaction region explored by the two peptides.

Table 2. Contact points of KVFIA and PWFRW with RING/MDM2 in indicated clusters.
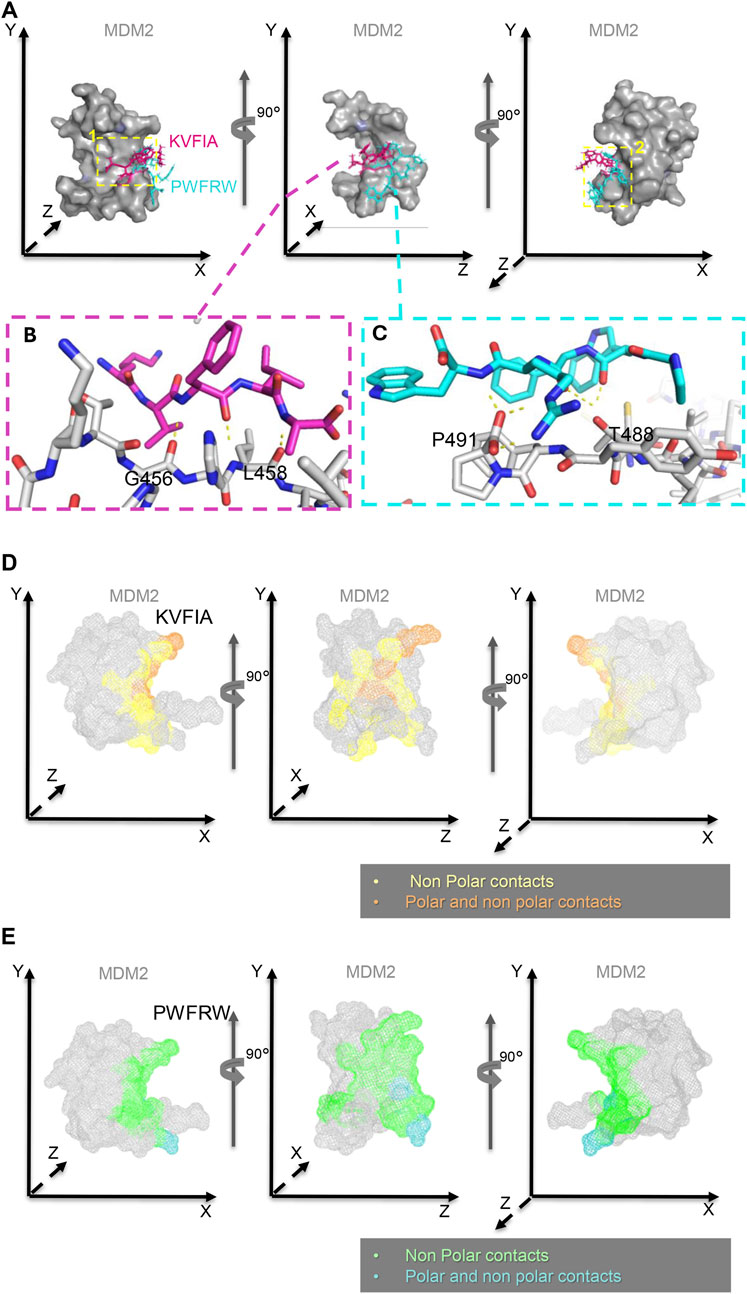
Figure 6. Binding position and interaction surface of KVFIA and PWFRW with MDM2. (A) Superposition of KVFIA (magenta) and PWFRW (blue) on the surface of RING/MDM2 observed by consecutive 90° rotations around the Y-axis. The yellow dotted boxes indicate the groove/cleft occupied by KVFIA (number 1, left panel) or PWFRW (number 2, right panel). PWFRW binds a different groove/cleft in a backward position compared to KVFIA (yellow dotted box number 2, right panel). (B, C) Magnification of the most stable interaction between KVFIA (F3) and G456 (1.8 Å) and (F3 and (A) L458 (1.9 Å) of MDM2 (magenta dotted box, left panel), and between PWFRW (N-term W2 and F) T488 (1.7 Å and 2.5 Å distance, respectively) and (W5 and R) P491 (1.9 Å and 2.0 Å distance, respectively)of MDM2 (blue dotted box, right panel). (D, E) Interaction surface of MDM2 with KVFIA (D) or PWFRW (E) observed by consecutive 90° rotations around the Y-axis. The different colors indicate the chemical nature of contacts, as reported in the relative legends. Structures made by PyMol PDB Code: 2VJF.
Molecular dynamic trajectories proved that PWFRW is more prone to create additional contacts than KVFIA due to marked differences in amino acid composition and moiety (Figures 7A, B). Particularly, PWFRW makes π-interactions with the phenylalanine 490 (F490) of MDM2. Specifically, by R4 via a cation-π interaction (Figure 7C) and by F3 and W5 via π-π interaction (Figures 7D, E) (details on frames whereby observations came from are given in the figure legend). Conversely, no π-interactions were detected in the binding of KVFIA. Interestingly, F3 of PWFRW also contributes to the interaction with Proline 491 (P491) of human MDM2 in the most representative Cluster 15 (Table 2). We speculate that this change of F3 from a purely hydrophobic interaction (with F490 of MDM2 in KVFIA, Table 2) to an extended π-interaction network (F490 and P491 of MDM2 in PWFRW, Table 2) drives the second peptide toward the different binding cleft. These results confirm the pivotal role of F3 and indicate the plasticity of the interaction regions 1 and 2 of the RING/MDM2 in hosting different peptides.
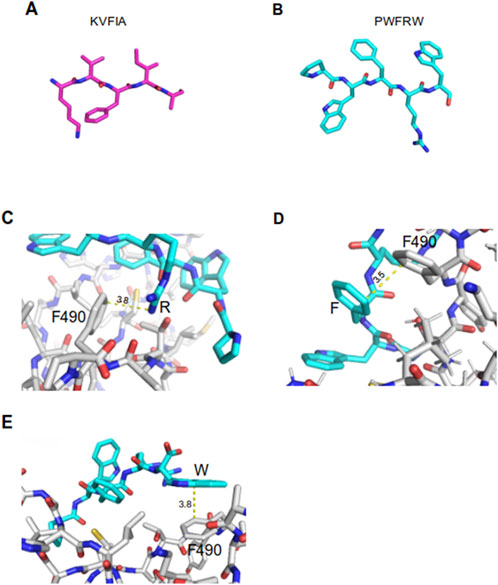
Figure 7. Representation of KVFIA and PWFRW and π-interactions between MDM2 and PWFRW. (A, B) Structural sticks representation of the two peptides created by PyMol. (C) π-cation interaction of F490 of MDM2 with R of PWFRW–mean distance 3.8 Å (dynamic simulation frames: 15,956–16264). (D) π-π interaction of F490 of MDM2 with Phe of PWFRW–mean distance 3.5 Å (dynamic simulation frames: 1892–1973). (E) π- π interaction of F490 of MDM2 with C-term Trp of PWFRW–mean distance 3.8 Å (dynamic simulation frames: 15,269–15,316).
3.7 Conformational state of RING/MDM2
Since the two peptides experience different conformational states, we wondered whether MDM2 would also experience different conformations when bound to one or the other. Combining RMSD and radius of gyration (Rg) data obtained from MD simulation, we generated the free energy profile of the RING/MDM2 in the presence of KVFIA or PWFRW. Although an energy minimum could be recognized in both cases, the plot of KVFIA (Figure 8A) appears more smeared compared to PWFRW (Figure 8B). These data suggest that KVFIA explores many low-populated states with close although different energy from each other. On the contrary, PWFRW showed a more compact plot around the minimum, indicating that many nearly identical conformational states lie within this unique well. These observations agree with the observed differences in the RMSF plots and the number of clusters for the two different systems: in KVFIA, two larger populations account for about 72% and 22% of the total conformational states (Figure 8C; Table 2); in PWFRW, approximately 95% of the population lies in a single deep well (Figure 8D; Table 2). Overall, these data prove that the RING/MDM2 adopts different conformational states by hosting different peptides. This observation could also have important implications for the competitive binding dynamics of MDM4.
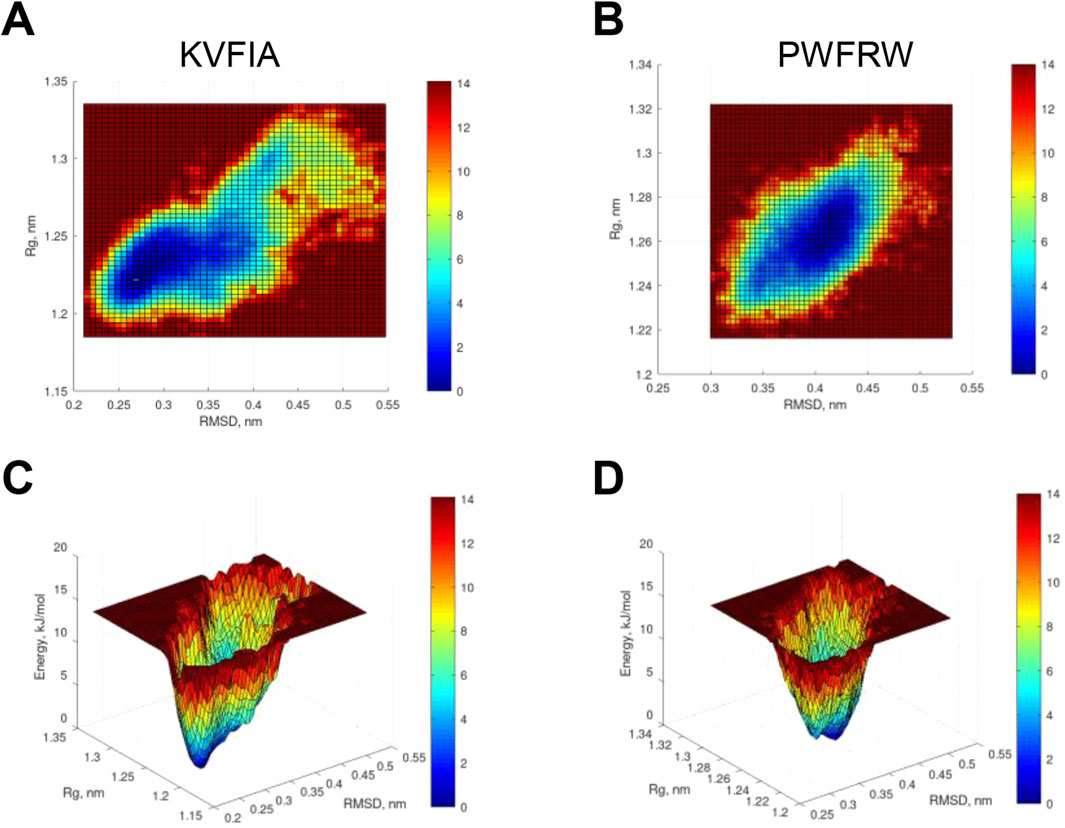
Figure 8. Free energy profile of the MDM2/RING in the presence of KVFIA or PWFRW. (A, B) Heatmap of free energy profile of the MDM2/RING in the presence of KVFIA (A) or PWFRW (B). (C, D) 3D profile of free energy after US procedure.
4 Discussion
In this work, we undertook a detailed bioinformatic and computational analysis of the interaction region of the two homolog proteins MDM2 and MDM4 and characterized novel peptides with different binding properties to this region.
MDM2 and MDM4 are largely recognized as the main inhibitors of p53, one of the most relevant tumor suppressor genes (Hu et al., 2016; Karni-Schmidt et al., 2016). Accordingly, many efforts have been spent on targeting the p53/MDM2 and p53/MDM4 complexes. More recently, targeting the MDM2/MDM4 heterodimer formation has been added to the scope through small molecules and peptides (Herman et al., 2011; Ilic et al., 2022; Merlino et al., 2024; Pellegrino et al., 2015; Wu et al., 2015).
Starting from a detailed analysis of the MDM2/MDM4 interaction region and taking advantage of a previous peptide whose sequence encompasses the C-terminus of MDM4, we demonstrated that the interaction region of MDM2 and MDM4 can host short peptides, five amino acids long, with increased binding affinity to the RING/MDM2/MDM4 compared to the previously published peptide (Pellegrino et al., 2015). For the evaluation of the binding affinity, we have taken a cautious approach, limiting the docking to just obtaining an initial binding pose. We did not rely on predicted affinity via docking and used a more sophisticated and accurate approach, such as umbrella sampling. It is worth noting that results about KVFIA obtained through MD simulation match with those obtained by in vitro fluorescence measurements in the limit of 1 kcal mol-1 as proposed by John Pople to ensure chemical accuracy (Pople, 1999), confirming the reliability of MD prediction. It is worth noting that the ΔG value obtained after MM/GBSA calculation is also within the limit of ±1 kcal mol-1, despite the fact that getting an accurate value for entropy is generally not an easy task, computationally-wise. Nevertheless, there is general compliance to consider as inaccurate any value for entropy whose uncertainty (as standard deviation) is beyond 3.6 (Ekberg and Ryde, 2021). Actually, the uncertainty associated with entropy calculation is within this limit (being -TΔS = 19.95 ± 3.02, as reported in the Results section).
In this regard, it should be mentioned that the KD value determined using the intrinsic fluorescence assay is significantly lower than the KD value previously obtained with microscale thermophoresis (KD = 19.2 µM) (Ballarotto et al., 2024). Such a discrepancy, however, can be ascribed to the different biophysical techniques that make use of the unlabeled target protein (intrinsic fluorescence assay) or the fluorescently labeled target protein (microscale thermophoresis). In this latter case, the presence of a fluorescent tag may have negatively affected the binding affinity of KVFIA to the MDM2 RING domain.
Our work underscores the importance of evaluating the predicted binding affinity and the initial binding poses obtained after virtual screening/docking procedures using molecular dynamics simulation. This finding supported our use of molecular dynamics simulation under fine-tuned conditions for the investigation of other molecules that ultimately resulted in the peptide PWFRW. We regarded the data produced for PWFRW by in silico methods as a starting point in the search of novel MDM2/MDM4 protein-protein interaction disruptors. The data presented here serve as a proof-of-concept to assess the goodness of the implementation of a pipeline encompassing MD followed by umbrella sampling in the pursuit of a new search. Further studies will add value to assess this new peptide, not only in vitro but also in cells.
The comparison of PWFRW and KVFIA data highlighted striking features and marked differences in the interaction properties with the RING/MDM2: KVFIA is the root in the family of short peptides stemming from the physiological sequence of MDM4, the binding partner of MDM2, and interacts with the central region of the RING domain of MDM2 (the red arrow in Figure 1). PWFRW, although maintaining the phenylalanine (Phe487 of MDM4), an essential point of contact for the heterodimer (Poyurovsky et al., 2007; Uldrijan et al., 2007), binds a different portion of the RING domain while retaining a similar predicted binding affinity to KVFIA. Particularly, PWFRW interacts with the RING/MDM2 C-terminal moiety, corresponding to the second heterodimer interaction region (the magenta arrows in Figure 1). The free energy profile demonstrated that such difference in contacts and composition shifts the relative population of the target MDM2, indicating that PWFRW forces RING/MDM2 in a narrower energy minimum compared to KVFIA (Figure 8). Thus, the RING/MDM2 can adopt different conformations depending on the binding of distinct peptides. The exact mechanism may involve either a change in MDM2 conformation upon initial peptide binding or a shift in the relative populations of its conformers to accommodate the bound molecule at best.
The discovery of a peptide binding the region two of the MDM2/MDM4 interaction domain will help to reveal the functional intersection among the three interaction regions of the heterodimer (synergic/cooperative/antagonistic activity). Indeed, at present, it is not known whether the three interaction regions display different properties or cooperate to strengthen the binding. The biological test of PWFRW could also help to define the specificity of the ubiquitin ligase activity of MDM2 towards other targets (Kosztyu et al., 2019). Indeed, MDM2 develops its ubiquitin ligase activity towards proteins other than p53. However, neither the requirements of the different regions of the RING/MDM2 in the development of this activity have been investigated nor the relevance of the heterodimer MDM4/MDM2. A recent paper reported the crystal structure of MDM2/MDM4 RING-domain heterodimer bound to E2 (UbcH5B) covalently linked to ubiquitin (UbcH5B–Ub) (Nomura et al., 2017). Of note, two residues involved in the binding of the heterodimer with ubiquitin, H457, and N433, are bound by both peptides (Table 2). Therefore, the short peptides here identified could interfere with the general ubiquitin ligase activity of the heterodimer and display additional activities to the reactivation of p53. Similarly, a recent new class of small-molecule inhibitors named MMRi (MDM2-MDM4 Ring Domain Inhibitors) showed activity towards targets other than p53 (Lama et al., 2022), although the authors did not ascertain whether such altered specificity is due to the MMRi binding to other molecules besides MDM2 or to the multiple targets of MDM2.
Finally, considering the increasing interest in the therapeutic use of the ubiquitination and deubiquitination systems in cancer, including PROTAC and glue degraders (Liu et al., 2024), and the inclusion of MDM2 in these studies (Li et al., 2024; Liu et al., 2024), the details provided in this study about the two peptides' interaction may open new application routes.
Peptides may show some improvement over synthetic drugs (Rossino et al., 2023). The peptides' flexible backbone and the multiple contact points with the target allow them to interact with the protein surface efficiently. This favors higher binding affinities between peptide and target, often translating into a higher potency than small compounds. Furthermore, the natural constituents of the peptides translate into low toxicity (Góngora-Benítez et al., 2014). Finally, they can bind to larger surfaces compared to small molecules, which enables the engagement of so-called ‘undruggable’ targets, such as PPIs (Wang et al., 2022). The reduced cost of small peptide production compared to large proteins is also worth mentioning (Góngora-Benítez et al., 2014).
In summary, detailing the MDM2/MDM4 interaction region has revealed increased opportunities for interacting peptides, widening this region’s therapeutic perspective. These results may guide the development of next-generation heterodimer and MDM2 inhibitors.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
MA: Conceptualization, Formal Analysis, Investigation, Methodology, Writing – original draft. SV: Investigation, Methodology, Writing – original draft. MP: Investigation, Writing – review and editing. MB: Investigation, Methodology, Writing – original draft. VS: Methodology, Writing – original draft, Writing – review and editing. AP: Writing – original draft, Funding acquisition, Resources. AM: Writing – original draft, Investigation, Methodology, Writing – review and editing. FS: Investigation, Methodology, Writing – original draft, Writing – review and editing, Conceptualization, Data curation, Formal Analysis. FM: Conceptualization, Data curation, Funding acquisition, Project administration, Resources, Supervision, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was financially supported by the Italian Association for Cancer Research Grant AIRC IG 21814 to FM We acknowledge the CINECA award under the ISCRA initiative (project HP10CVO9TH), for the availability of high-performance computing resources and support.
Acknowledgments
We thank Prof. Giampaolo Barone and Prof. Fabrizio Lo Celso of the University of Palermo for their valuable discussion about molecular dynamics simulation and umbrella sampling. The two peptides (KVFIA and PWFRW) are under PCT evaluation: Application Number: PCT/IB2024/057215N. Submission number: 057215. Date: 25/07/2024 Title: Peptides capable of interfering with the MDM2/MDM4 heterodimer association and their use in cancer treatment. Inventors: F. Moretti, S. Valentini, M. Attili, F. Saccoccia, N. Tirelli, A. Gennari, A. Macchiarulo, A. Temperini, M. Ballarotto.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchbi.2025.1576640/full#supplementary-material
References
Ballarotto, M., Bianconi, E., Valentini, S., Temperini, A., Moretti, F., and Macchiarulo, A. (2024). Rational design, synthesis, and biophysical characterization of a peptidic MDM2-MDM4 interaction inhibitor. Bioorg. & Med. Chem. 113, 117937. doi:10.1016/j.bmc.2024.117937
Broomhead, N. K., and Soliman, M. E. (2017). Can We rely on computational predictions to correctly identify ligand binding sites on novel protein drug targets? Assessment of binding site prediction methods and a protocol for validation of predicted binding sites. Cell Biochem. biophysics 75, 15–23. doi:10.1007/s12013-016-0769-y
Coutts, A. S., Boulahbel, H., Graham, A., and La Thangue, N. B. N. B. (2007). Mdm2 targets the p53 transcription cofactor JMY for degradation. EMBO Rep. 8, 84–90. doi:10.1038/sj.embor.7400855
Eaton, J. W., Bateman, D., and Soren, H. (2024). GNU octave manual version 3: guide books. ACM digital library.
Eberhardt, J., Santos-Martins, D., Tillack, A., and Forli, S. (2021). AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 61, 3891–3898. doi:10.1021/acs.jcim.1c00203
Ekberg, V., and Ryde, U. (2021). On the use of interaction entropy and related methods to estimate binding entropies. J. Chem. theory Comput. 17, 5379–5391. doi:10.1021/acs.jctc.1c00374
Fu, W., Ma, Q., Chen, L., Li, P., Zhang, M., Ramamoorthy, S., et al. (2009). MDM2 acts downstream of p53 as an E3 ligase to promote FOXO ubiquitination and degradation. J. Biol. Chem. 284, 13987–14000. doi:10.1074/jbc.m901758200
Góngora-Benítez, M., Tulla-Puche, J., and Albericio, F. (2014). Multifaceted roles of disulfide bonds. Peptides as therapeutics. Chem. Rev. 114, 901–926. doi:10.1021/cr400031z
Gu, J., Kawai, H., Nie, L., Kitao, H., Wiederschain, D., Jochemsen, A. G., et al. (2002). Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J. Biol. Chem. 277, 19251–19254. doi:10.1074/jbc.c200150200
Haronikova, L., Bonczek, O., Zatloukalova, P., Kokas-Zavadil, F., Kucerikova, M., Coates, P. J., et al. (2021). Resistance mechanisms to inhibitors of p53-MDM2 interactions in cancer therapy: can we overcome them? Cell. & Mol. Biol. Lett. 26, 53. doi:10.1186/s11658-021-00293-6
Hassin, O., and Oren, M. (2023). Drugging p53 in cancer: one protein, many targets. Nat. Rev. Drug Discov. 22, 127–144. doi:10.1038/s41573-022-00571-8
Herman, A. G., Hayano, M., Poyurovsky, M. V., Shimada, K., Skouta, R., Prives, C., et al. (2011). Discovery of Mdm2-MdmX E3 ligase inhibitors using a cell-based ubiquitination assay. Cancer Discov. 1, 312–325. doi:10.1158/2159-8290.cd-11-0104
Hu, L., Zhang, H., Bergholz, J., Sun, S., and Xiao, Z. X. (2016). MDM2/MDMX: master negative regulators for p53 and RB. Mol. & Cell. Oncol. 3, e1106635. doi:10.1080/23723556.2015.1106635
Humpton, T. J., Nomura, K., Weber, J., Magnussen, H. M., Hock, A. K., Nixon, C., et al. (2021). Differential requirements for MDM2 E3 activity during embryogenesis and in adult mice. Genes & Dev. 35, 117–132. doi:10.1101/gad.341875.120
Ilic, V., Egorova, O., Tsang, E., Gatto, M., Wen, Y., Zhao, Y., et al. (2022). Hinokiflavone inhibits MDM2 activity by targeting the MDM2-MDMX RING domain. Biomolecules 12, 643. doi:10.3390/biom12050643
Karni-Schmidt, O., Lokshin, M., and Prives, C. (2016). The roles of MDM2 and MDMX in cancer. Annu. Rev. Pathology Mech. Dis. 11, 617–644. doi:10.1146/annurev-pathol-012414-040349
Kosztyu, P., Slaninová, I., Valčíková, B., Verlande, A., Müller, P., Paleček, J. J., et al. (2019). A single conserved amino acid residue as a critical context-specific determinant of the differential ability of Mdm2 and MdmX RING domains to dimerize. Front. physiology 10, 14. doi:10.3389/fphys.2019.00390
Lama, R., Xu, C., Galster, S., Querol-García, J., Portwood, S., Mavis, C., et al. (2022). Small molecule MMRi62 targets MDM4 for degradation and induces leukemic cell apoptosis regardless of p53 status. Front. Oncol. 12, 933446. doi:10.3389/fonc.2022.933446
Li, H., Cai, X., Yang, X., and Zhang, X. (2024). An overview of PROTACs targeting MDM2 as a novel approach for cancer therapy. Eur. J. Med. Chem. 272, 116506. doi:10.1016/j.ejmech.2024.116506
Linke, K., Mace, P. D., Smith, C. A., Vaux, D. L., Silke, J., and Day, C. L. (2008). Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 15, 841–848. doi:10.1038/sj.cdd.4402309
Liu, F., Chen, J., Li, K., Li, H., Zhu, Y., Zhai, Y., et al. (2024). Ubiquitination and deubiquitination in cancer: from mechanisms to novel therapeutic approaches. Mol. Cancer 23, 148–150. doi:10.1186/s12943-024-02046-3
Lu, M., Wang, X., Li, Y., Tripodi, J., Mosoyan, G., Mascarenhas, J., et al. (2012) Combination treatment in vitro with Nutlin, a small-molecule antagonist of MDM2, and pegylated interferon-α 2a specifically targets JAK2V617F-positive polycythemia vera cells. Blood. 120, 3098–3105. doi:10.1182/blood-2012-02-410712
Madeira, F., Pearce, M., Tivey, A., Basutkar, P., Lee, J., Edbali, O., et al. (2022). Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic acids Res. 50, W276–W279. doi:10.1093/nar/gkac240
Merlino, F., Pecoraro, A., Longobardi, G., Donati, G., Di LevaBrignola, F. S. C., Piccarducci, R., et al. (2024). Development and nanoparticle-mediated delivery of novel MDM2/MDM4 heterodimer peptide inhibitors to enhance 5-fluorouracil nucleolar stress in colorectal cancer cells. J. Med. Chem. 67, 1812–1824. doi:10.1021/acs.jmedchem.3c01312
Morris, G., Huey, R., Lindstrom, W., Sanner, M., Belew, R., Goodsell, D., et al. (2009). AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. doi:10.1002/jcc.21256
Nomura, K., Klejnot, M., Kowalczyk, D., Hock, A. K., Sibbet, G. J., Vousden, K. H., et al. (2017). Structural analysis of MDM2 RING separates degradation from regulation of p53 transcription activity. Nat. Struct. & Mol. Biol. 24, 578–587. doi:10.1038/nsmb.3414
O'Boyle, N., Banck, M., James, C., Morley, C., Vandermeersch, T., and Hutchison, G. (2011). Open Babel: an open chemical toolbox. J. cheminformatics 3, 14. doi:10.1186/1758-2946-3-33
Pantsar, T., and Poso, A. (2018). Binding affinity via docking: fact and fiction. Mol. (Basel, Switz) 23, 1899. doi:10.3390/molecules23081899
Pellegrino, M., Mancini, F., Luca, R., Coletti, A., Giacche, N., Manni, I., et al. (2015). Targeting the MDM2/MDM4 interaction interface as a promising approach for p53 reactivation therapy. Cancer Res. 75, 4560–4572. doi:10.1158/0008-5472.can-15-0439
Pople, J. A. (1999). Quantum chemical models (nobel lecture). Angew. Chem. Int. Ed. 38, 1894–1902. doi:10.1002/(sici)1521-3773(19990712)38:13/14<1894::aid-anie1894>3.0.co;2-h
Poyurovsky, M. V., Priest, C., Kentsis, A., Borden, K. L., Pan, Z. Q., Pavletich, N., et al. (2007). The Mdm2 RING domain C-terminus is required for supramolecular assembly and ubiquitin ligase activity. Embo J. 26, 90–101. doi:10.1038/sj.emboj.7601465
Rossino, G., Marchese, E., Galli, G., Verde, F., Finizio, M., Serra, M., et al. (2023). Peptides as therapeutic agents: challenges and opportunities in the green transition era. Mol. (Basel, Switz) 28, 7165. doi:10.3390/molecules28207165
Skalniak, L., Surmiak, E., and Holak, T. A. (2019). A therapeutic patent overview of MDM2/X-targeted therapies (2014-2018). Expert Opin. Ther. Pat. 29, 151–170. doi:10.1080/13543776.2019.1582645
Shadfan, M., Lopez-Pajares, V., and Yuan, Z. M. (2012). MDM2 and MDMX: alone and together in regulation of p53. Transl. Cancer. Res. 1 (2), 88–99. doi:10.3978/j.issn.2218-676X.2012.04.02
Tam, N. M., Nguyen, T. H., Ngan, V. T., Tung, N. T., and Ngo, S. T. (2022). Unbinding ligands from SARS-CoV-2 Mpro via umbrella sampling simulations. R. Soc. Open Sci. 9, 211480. doi:10.1098/rsos.211480
Tanimura, S., Ohtsuka, S., Mitsui, K., Shirouzu, K., Yoshimura, A., and Ohtsubo, M. (1999). MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 447, 5–9. doi:10.1016/s0014-5793(99)00254-9
Teveroni, E., Luca, R., Pellegrino, M., Ciolli, G., Pontecorvi, A., and Moretti, F. (2016). Peptides and peptidomimetics in the p53/MDM2/MDM4 circuitry - a patent review. Expert Opin. Ther. Pat. 26, 1417–1429. doi:10.1080/13543776.2017.1233179
Tollini, L. A., Jin, A., Park, J., and Zhang, Y. (2014). Regulation of p53 by Mdm2 E3 ligase function is dispensable in embryogenesis and development, but essential in response to DNA damage. Cancer Cell 26, 235–247. doi:10.1016/j.ccr.2014.06.006
Trott, O., and Olson, A. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. doi:10.1002/jcc.21334
Uldrijan, S., Pannekoek, W. J., and Vousden, K. H. (2007). An essential function of the extreme C-terminus of MDM2 can be provided by MDMX. Embo J. 26, 102–112. doi:10.1038/sj.emboj.7601469
Valentini, S., Mele, G., Attili, M., Maria Rita, A., Fulvio, S., Francesca, S., et al. (2025). Targeting the MDM2-MDM4 interaction interface reveals an otherwise therapeutically active wild-type p53 in colorectal cancer. Mol. Oncol. 11. doi:10.1002/1878-0261.70006
Vassilev, L. T., Vu, B. T., Graves, B., Carvajal, D., Podlaski, F., Filipovic, Z., et al. (2004). In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848. doi:10.1126/science.1092472
Wang, L., Wang, N., Zhang, W., Cheng, X., Yan, Z., Shao, G., et al. (2022). Therapeutic peptides: current applications and future directions. Signal Transduct. Target. Ther. 7, 48. doi:10.1038/s41392-022-00904-4
Weissman, A. M., Yang, Y., Kitagaki, J., Sasiela, C. A., Beutler, J. A., and O'Keefe, B. R. (2008). Inhibiting Hdm2 and ubiquitin-activating enzyme: targeting the ubiquitin conjugating system in cancer. Ernst Scher. Found. Symp. Proc., 171–190. doi:10.1007/2789_2008_108
Wu, W., Xu, C., Ling, X., Fan, C., Buckley, B. P., Chernov, M. V., et al. (2015). Targeting RING domains of Mdm2-MdmX E3 complex activates apoptotic arm of the p53 pathway in leukemia/lymphoma cells. Cell death & Dis. 6, e2035. doi:10.1038/cddis.2015.358
Yang, J. Y., Zong, C. S., Xia, W., Wei, Y., Ali-Seyed, M., Li, Z., et al. (2006). MDM2 promotes cell motility and invasiveness by regulating E-cadherin degradation. Mol. Cell. Biol. 26, 7269–7282. doi:10.1128/mcb.00172-06
Yu, D. H., Xu, Z. Y., Mo, S., Yuan, L., Cheng, X. D., and Qin, J. J. (2020). Targeting MDMX for cancer therapy: rationale, strategies, and challenges. Front. Oncol. 10, 1389. doi:10.3389/fonc.2020.01389
Keywords: target p53, peptide, MDM2, MDM4 (MDMX), umbrella sampling
Citation: Attili M, Valentini S, Pesavento M, Ballarotto M, Scognamiglio V, Pontecorvi A, Macchiarulo A, Saccoccia F and Moretti F (2025) Exploring the heterodimer MDM2/MDM4 interaction region uncovers high-affinity peptides for therapeutic p53-reactivation. Front. Chem. Biol. 4:1576640. doi: 10.3389/fchbi.2025.1576640
Received: 14 February 2025; Accepted: 18 April 2025;
Published: 12 May 2025.
Edited by:
Adriana Erica Miele, Université Claude Bernard Lyon 1, FranceReviewed by:
Massimiliano Perduca, University of Verona, ItalyNancy Jaiswal, Indiana University, United States
Copyright © 2025 Attili, Valentini, Pesavento, Ballarotto, Scognamiglio, Pontecorvi, Macchiarulo, Saccoccia and Moretti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabiola Moretti, ZmFiaW9sYS5tb3JldHRpQGNuci5pdA==; Fulvio Saccoccia, ZnVsdmlvLnNhY2NvY2NpYUBjbnIuaXQ=
†Present address: Marco Ballarotto, Division of Cancer Therapeutics, Centre for Cancer Drug Discovery, The Institute of Cancer Research, London, SM2 5NG, United Kingdom
‡These authors have contributed equally to this work and share last authorship