 Abdulelah S. Alshehri
Abdulelah S. Alshehri Fengqi You
Fengqi You- 1Robert Frederick Smith School of Chemical and Biomolecular Engineering, Cornell University, Ithaca, NY, United States
- 2Department of Chemical Engineering, College of Engineering, King Saud University, Riyadh, Saudi Arabia
The application of deep learning to a diverse array of research problems has accelerated progress across many fields, bringing conventional paradigms to a new intelligent era. Just as the roles of instrumentation in the old chemical revolutions, we reinforce the necessity for integrating deep learning in molecular systems engineering and design as a transformative catalyst towards the next chemical revolution. To meet such research needs, we summarize advances and progress across several key elements of molecular systems: molecular representation, property estimation, representation learning, and synthesis planning. We further spotlight recent advances and promising directions for several deep learning architectures, methods, and optimization platforms. Our perspective is of interest to both computational and experimental researchers as it aims to chart a path forward for cross-disciplinary collaborations on synthesizing knowledge from available chemical data and guiding experimental efforts.
Introduction
Chemicals play a central role not only in finding solutions to many pressing issues, but also in sustainably developing the global economy (Miodownik, 2015). Several chemical inventions have had profound impacts on our lives, like the pivotal roles of synthetic fertilizers and industrial catalysis in our food-energy-water nexus (Hagen, 2015; Garcia and You, 2016; Garcia and You, 2017). Many past developments, however, have arisen from accidental or heuristic rule-based experiments while being restricted by time and resources, and to a small class of molecular structures (Gani, 2004; Austin et al., 2016). From a chemical engineering viewpoint, another complexity dimension to the development of new chemicals lies in the multi-scale nature of chemical products and processes (Alshehri et al., 2020; Zhang et al., 2020). As chemicals offer hope for addressing urgent needs, persisting to perceive current design difficulties to be solely a result of molecular complexities can lead global environmental efforts to fall far short of 2,030 targets (Miodownik, 2015). As seen, a shift from the current paradigm is needed to allow new drivers of innovations in chemical-based products and processes to take root.
Deep learning has emerged to transform many elements of today’s services and technologies, from search engines to robots and recommendation systems (LeCun et al., 2015). Deep neural network architectures have outperformed many hand-crafted and traditional AI methods and remain to hold state-of-the-art performances on many complex learning tasks. As deep learning algorithms become increasingly sophisticated in processing complex information, their applications have bourgeoned across chemical engineering, including in molecular design (Alshehri et al., 2020), computational chemistry (Cova and Pais, 2019), and control and optimization (Ning and You, 2019; Pappas et al., 2021). Notably, deep learning has made a breakthrough in biological research by beating decades of theoretical and experimental work on predicting the 3D structure of a protein from its genetic sequence (Senior et al., 2020). As seen, the potential for a transformative paradigm shift in chemical engineering with deep learning has never been as accessible and urgent as it is today.
Within the ever-developing deep learning architectures and algorithms, remarkable progress across several learning tasks has been attributed to attention-based neural network architectures (Transformers) (Vaswani et al., 2017). Since their debut in 2017 for language tasks, applications of transformers have moved to vision tasks (Dosovitskiy et al., 2020), reinforcement learning (RL) (Parisotto et al., 2020), and sequencing proteins (Rao et al., 2021), matching or exceeding the performance of conventional neural networks (Lu et al., 2021). Besides, developments in deep learning have transcended the Euclidian domains to approach problems operating in the graph and manifold domains, such as molecules, with graph neural networks (GNNs) (Monti et al., 2017; Zhou et al., 2018). While such developments arose in the supervised learning framework of deep learning, gleaning deeper knowledge across different domains has recently been demonstrated by the self-supervised learning framework (Baevski et al., 2020; LeCun and Misra, 2021). Advances in deep learning also extend to integrated platforms as in self-driving laboratories (Schwaller et al., 2018; MacLeod et al., 2020), and the interpretability of models as a result of medical and scientific applications (Gunning et al., 2019; Tjoa and Guan, 2020). We envisage that recent advances in deep learning offer opportunities for rapid and exciting developments in the design of new chemical products and processes. Such developments can be realized more efficiently through integrating scientific and engineering domain knowledge to decide on molecular representation, modeling, and physical constraints.
Herein, we first present the current status of deep learning applications to chemicals design, namely, molecular representation, property estimation, representation learning, and synthesis planning as shown in Figure 1. Then, we discuss and describe the new trends and open research lines related to deep learning-based chemical products and process design. Finally, the article concludes with an outlook on future directions and recommendations.
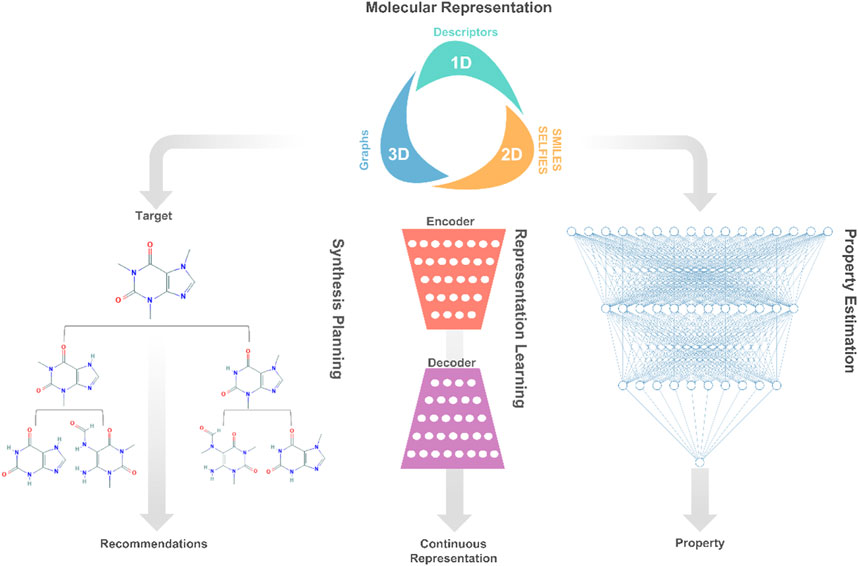
FIGURE 1. A high-level description of prominent deep learning applications involved in molecular systems engineering and design.
Background
In this section, we provide brief descriptions of key areas in molecular systems engineering and design. Their respective advances are also discussed with respect to promising applications, current state-of-the-art performances, and major limitations. For more extensive discussions on the elements, we point the interested reader to reviews on machine learning techniques for chemical systems (Butler et al., 2018), molecular design (Alshehri et al., 2020), and synthesis planning (Coley et al., 2018).
Molecular Design
Traditionally, molecular design involves the development of molecular representation for predicting physicochemical properties, and the application of optimization for finding molecules that satisfy specific property targets (Gani, 2004). Essentially, the molecular design task can be decomposed into separate components, each representing a decision or learning task. The first decision is concerned with molecular representations, which fall into different categories, such as dimensionality (1D, 2D, or 3D) or data type (numeric, text, or graphs) (Alshehri et al., 2020). This decision is followed by the development of rule-based (Gani, 2019) or learning-based (Gomez-Bombarelli et al., 2018) representations of molecules, which encode/decode a given molecular representation into/from discrete or continuous numerical data. Last, the learned representations of molecules are exploited to learn property models relating molecular structures to their physicochemical properties. These several components are then embedded into a computational optimization framework that seeks to generate promising structures for experimental validation.
Molecular Representation
The digital encoding of an expressive molecular structure representation is at the core of the molecular design learning task. Popular representations in molecular design and discovery are divided into two categories: two-dimensional and three-dimensional. The proper choice of a molecular representation is reliant on the domain knowledge of the problem at hand as well as the deep learning architecture that will be used. Yet, the selection of the best-performing representation for a learning task is not always clear, persisting as an open research avenue in cheminformatics (Butler et al., 2018).
In deep learning applications, two-dimensional representations have enjoyed remarkable success even though their use entails the loss of conformational and bond distance information (Goh et al., 2017). The most popular 2D representation by far is the string-based Simplified Molecular Input Line Entry System (SMILES) representation (Weininger, 1988). Motivated by the limitations of SMILES, SELF-referencIng Embedded Strings (SELFIES) was also developed to create robust representations, ensuring any random combination of characters corresponds to a valid molecule (Krenn et al., 2020). On the other hand, the translational, rotational, and permutation variances of 3D representations have long rendered describing molecules in the 3D space challenging in deep generative models (Goh et al., 2017). Molecular representations in generative modeling remain predominantly two-dimensional. However, recent advances in GNNs and their variants (Schütt et al., 2017; Gao et al., 2018) have addressed many limitations encountered in applying 3D representations, opening unexplored avenues of research (Sourek et al., 2020).
Representation Learning
The choice of molecular representation (input) on which deep learning methods are implemented has a significant impact on their performance. As such, the design of generative models that support successful deep learning applications consumes a large portion of the efforts in creating frameworks for molecular design. Typically, the original molecular representation is transformed across several neural networks into a numerical representation, from which the original molecular representation can also be reconstructed. During the optimization process of these networks, higher and lower-level features of the molecular representations are encoded into the so-called latent or hidden space. In the space, each molecular structure corresponds to a latent representation, which is often a vector of real values. The goal of such generative models is to learn expressive continuous representations that are extended to enhance the optimization of properties and generation of novel promising molecules (Goodfellow et al., 2016; Sanchez-Lengeling and Aspuru-Guzik, 2018).
A diverse array of generative models and frameworks have been applied to not only developing representations, but also controlling the generative task towards objectives. Some have observed that variational autoencoders (VAE) achieve higher generalizability in learning molecular representations by optimizing the latent space over a fixed vector size (Gomez-Bombarelli et al., 2018). Other methods such as generative adversarial networks and recurrent neural networks have also shown promising results in molecular design by controlling the generation process with RL or property objectives (Popova et al., 2018; Jin et al., 2019). Given the growing interest in generative modeling from the deep learning community, applications of more complex and hybrid approaches have also been proposed (Griffiths and Hernandez-Lobato, 2020; Maziarka et al., 2020). Two benchmarking platforms have been developed for evaluating distinct elements of the representations learned by generative models, including validity, novelty, diversity, and uniqueness (Brown et al., 2019; Polykovskiy et al., 2020). The capability to learn a domain-invariant molecular representation on different scales of complexity to generative valid and novel molecules remains a key limitation.
Property Estimation
Property estimation models are instrumental in guiding the design of molecular solutions as such models aim to capture the underlying behavior of the molecular system governed by thermodynamics (Gani, 2019). The purpose of such models is to reduce the time and cost associated with experimental screening while significantly expanding the size of the design space. When learning quantitative structure-property relationship functions, deep learning algorithms systematically search the hypothesis space and uncover complex relationships that would otherwise be too complex to conceptualize by experts (LeCun et al., 2015). As such, deep learning applications have brought a ground-breaking leap in building accurate property estimation models in terms of accuracy and applicability using a wide array of molecular representations (Walters and Barzilay, 2020).
Advances in property estimation models are twofold: involving the use of more sophisticated molecular representations and advanced deep learning architectures. Earlier models employed simple molecular representations, such as fingerprints, descriptors, and functional groups (Pyzer-Knapp et al., 2015; Zhang et al., 2018). Propelled by progress in computer vision and language models, recent applications have exploited more informative and invertible graph representations and string-based embedding (Su et al., 2019; Ma et al., 2020a). On the other hand, algorithmic and architectural advances have led to remarkable accuracy improvements across different datasets and properties. For example, transfer learning has closed the gap between predictive models and quantum chemistry calculations for estimating the formation energy (Jha et al., 2019), and Bayesian networks were applied to provide uncertainty-calibrated estimates (Zhang, 2019). To the best of our knowledge, a self-supervised graph transformer with transfer learning holds the current state-of-the-art performance on 11 challenging property classification and regression benchmarks (Rong et al., 2020). Despite such a ground-breaking leap, the paucity of property data stands as a major limitation to establishing confidence in deep predictive models as they are data-intensive. The complexity of such an issue is compounded for multiscale problems when products and processes are considered (Alshehri et al., 2020).
Synthesis Planning
Synthesis planning (retrosynthesis) is the process of identifying a sequence of chemical reactions to produce a target molecule from available precursors. The combinatorial problem can also be approached from the backward route by the recursive selection of precursors of a target molecule. Efforts in molecular design have long been devoted to measuring the accessibility of candidate molecules from a synthesis standpoint through the conventional use of synthetic accessibility scores, and expert-crafted rules. An alternative route, deep learning, has brought a successful paradigm shift, surpassing 6 decades of conventional efforts through using deep learning-based models for learning to rank potential precursors. These models mimic expert’s synthesis decision-making to identify promising routes, avoid reactivity conflicts, and estimate reaction mechanisms (Coley et al., 2018; Segler et al., 2018a).
Inspired by advances in recommender systems, several data-driven models have been devised by exploiting large reaction databases to approach the forward and backward synthesis problem. As synthesis planning is a multistep process, complexities are circumvented through preprocessing and simplification steps, such as classifying reaction feasibility (Segler et al., 2018b), predicting reactions from the manual applications of mechanistic rules (Fooshee et al., 2018), and ranking relevant templates (Segler and Waller, 2017). Notable advances in synthesis planning came from hybrid approaches combining template-based forward enumeration and deep learning-based candidate ranking to predict the product(s) of reactions (Coley et al., 2018). Transformer architectures have resulted in several effective models, pushing the state-of-the-art performances on predicting both single-step reactions (Tetko et al., 2020), and multistep retrosynthesis (Lin et al., 2020; Schwaller et al., 2020). Further, handling retrosynthesis planning as a set of logic rules and a conditional graphical model using GNNs has shown significant improvements on common benchmarks (Dai et al., 2020). However, major limitations in data-driven synthesis planning include the absence of detailed final product distribution (concentrations), and expanding the predictive capabilities to cover side reactions.
Perspectives
In this section, we offer our perspective on the most promising directions in deep learning relative to the gaps in the current applications of deep learning to molecular systems engineering and design. To envision the path forward, we distill relevant trends and advances in deep learning that have resulted in recent breakthroughs and state-of-the-art results with connections to molecular systems. On the molecular representation task, we describe advances in transformers and GNNs as ideal future directions for dealing with sequential 2D data and 3D molecular representations, respectively. As a step towards moving beyond fitting data to functions, we highlight the exploratory application of deep learning with self-supervised learning (SSL). Design platforms are also discussed to offer our views on the systematic multiscale modeling and optimization of molecular systems. The first three approaches are depicted in Figure 2.
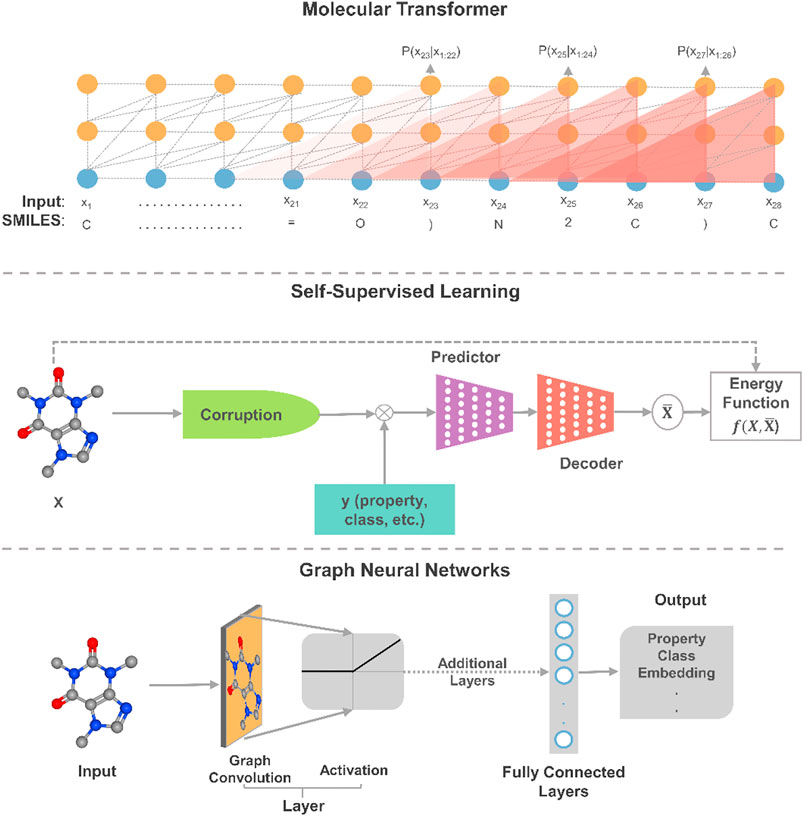
FIGURE 2. An illustrative schematic of three promising applications of deep learning to molecular design. In self-supervised learning, this is a masked property-structure model, which is an instance of contrastive self-supervised learning. In the model, parts of some molecules are masked and properties are used to reconstruct the molecule from its masked/corrupted version.
Transformers for Molecules and Beyond
Given the sequential nature of many molecular representations and reactions, the transformer architecture (Vaswani et al., 2017) has proven to be powerful enough to provide state-of-the-art results on many complex tasks through attention mechanisms. Additionally, the architectures have recently shown to possess generalization capabilities that can be transferred from one modality to another (Lu et al., 2021). Such feature allows for transferring learned knowledge from language models to and between, for example, different physicochemical properties at 1% of the required computational power for training. As such, by combining the powers of transformers and transfer learning, we foresee that the application of larger transformer models would catalyze the pace of innovation in molecular engineering and design. Further, although transformer models maintain state-of-the-art performances on property prediction (Rong et al., 2020) and synthesis planning (Tetko et al., 2020), advances around attention-based structured knowledge unlock the potential to explore structural properties and lead to potential improvements. The augmentation of knowledge graphs also presents a clear advantage in reducing the need for complex statistical reasoning as many of the chemical/physical features are encoded within the graphs (Reis et al., 2021). As seen, more complex models and advanced data representations, such as graph theory-based embedding (Yun et al., 2019), are projected to hold remarkable progress in constructing more powerful models.
Self-Supervised Learning for Autonomous Exploration
For deep learning models to bring AI systems closer to chemist-level intelligence, they would need to obtain a nuanced and deep understanding of the phenomena behind data. Experts in the field of deep learning believe that SSL is a major exciting direction towards constructing background knowledge and common sense from deep learning (Ravanelli et al., 2020; LeCun and Misra, 2021). Indeed, it is observed that many models trained using SSL yield considerably higher performance than their supervised counterparts by learning “more” from the data (Liu et al., 2019; Baevski et al., 2020). In SSL, the general technique is to predict a masked or unobserved part of the input. For example, we can predict hidden parts of molecules or unobserved properties from the observed molecule by jointly learning the embedding of molecules and properties. The same masking procedure can also be applied to synthesis planning where parts of the experimental parameters (reactants, reagents, catalysts, temperature, concentrations, time, etc.) are hidden. This technique expresses the observed and unobserved parts as an energy-based model that captures the compatibility of the two parts, and maximizes the compatibility of unseen observations. Given the scale and complexity of current molecular dataset, we anticipate that SSL approaches could lead to performance improvements in training molecular design models. Moreover, insights into chemical properties and reactions can be derived from the exploratory application of SSL to property estimation and synthesis planning.
Graph Neural Networks for the 3D Modeling of Molecules
As a result of the expressive ability of learned molecular representations, GNNs have become quite popular for deep learning on molecules, especially for property estimation. Yet, for example, when property data are limited, the highly dimensional representations of GNNs become more susceptible to overfitting than other molecular representations as molecular fingerprints. It has been shown that such a drawback for molecular data can be alleviated by pretraining and meta-learning (Pappu and Paige, 2020). A promising direction for applying GNNs to chemical structures is the use of graph transformer networks (Yun et al., 2019), which exploits attention mechanisms and the expression of heterogeneous edges and nodes. Moreover, the use of 3D molecular representations offers a window into model interpretability and explainability. Consequently, deep learning models could better understand model reasoning and synthesize chemical knowledge as shown in synthesis planning (Dai et al., 2020). It is worth noting that graph neural networks have also been extended to several chemical engineering applications, such as control (Wang et al., 2018), process scheduling (Ma et al., 2020b), fault diagnosis (Wu and Zhao, 2021), among others. In the realm of molecular systems, applications of GNNs have reaped numerous successes. Yet, such networks remain limited in scalability and depth (Zhou et al., 2018), and future algorithmic advances in GNNs will play a role in expanding their application to chemical engineering data.
Optimization Platforms for Integrated Multiscale Design
The application of deep learning to molecular systems engineering and design is merely a stepping stone toward building multifaceted systems that concurrently integrate nanoscale and macroscale decisions (Pistikopoulos et al., 2021). The development of such systems requires synergetic interactions between the key elements underlying the molecular system as well as the optimization framework exploiting the mathematical structure to find solutions. Currently, there is no published work that considers the simultaneous optimization of molecular design, synthesis routes, and product/process design. As the design space of multiscale molecular systems expands, the need for more efficient optimization strategies controlled by uncertainties present in the data becomes more critical (Alshehri et al., 2020). RL attempts to learn an optimal policy (mapping) of decisions to actions that maximizes the rewards across a single or multiple objective(s). Thereby, the policy extracts patterns from the data to strike a balance between exploration and exploitation in the search for optimality (Nian et al., 2020). A current focus in RL research is centered around creating systems that learn more efficiently with remarkable recent advances in causal discovery (Zhu et al., 2019) and meta-learning (Co-Reyes et al., 2021). Looking ahead, the RL framework holds the potential to improve solutions across many complex chemical engineering problems, such as scheduling (Hubbs et al., 2020), control (Shin et al., 2019), and process optimization (Petsagkourakis et al., 2020). The framework is also a viable alternative to exact optimization-based approaches for large design spaces. Furthermore, the use of RL as an optimization framework is ideal for integrating multiple facets into the design, such as automated robotics in self-driving laboratories for molecular validation (Roch et al., 2018).
Outlook
Integrating nanoscale decisions (molecules) with macroscale behaviors and properties of chemical systems has been an open challenge to the chemical engineering community (Pistikopoulos et al., 2021). Yet, rapid progress in molecular engineering and design has been enabled by advances in deep learning architectures and algorithms, as well as the availability of large chemical informatics and datasets. Looking ahead, much of the remaining work is perceived as being reliant on innovations around developing expressive representation encoding physics and chemistry theory and building multifaceted frameworks. Applicable developments in such areas are central to allowing data-driven models to assist decisions in molecular systems engineering and design, which is critical to solving pressing problems in the chemical, agriculture, energy, and healthcare industries.
Advances in graph representations and deep attention-based architectures have shown the capacity to encode more relevant graphical data, and meaningful patterns, respectively. As a result, the error gap between experimental data and predictions in the field keeps shrinking, building the momentum to break the barriers for general-purpose molecular design frameworks. Still, many challenges persist, from the quality of generative and multistep synthesis models, to interpretability and expressivity of property estimation models and molecular representations. With the “black-box” nature of deep learning as a common barrier to adoption, SSL, uncertainty estimation (Loquercio et al., 2020), and interpretability (Preuer et al., 2019; Arrieta et al., 2020) offer quantitative descriptions of error and a better in-depth understating of decisions.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Author Contributions
AA and FY conceived the ideas and wrote the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alshehri, A. S., Gani, R., and You, F. (2020). Deep Learning and Knowledge-Based Methods for Computer-Aided Molecular Design-Toward a Unified Approach: State-Of-The-Art and Future Directions. Comput. Chem. Eng. 141, 107005. doi:10.1016/j.compchemeng.2020.107005
Arrieta, A. B., Díaz-Rodríguez, N., Del Ser, J., Bennetot, A., Tabik, S., Barbado, A., et al. (2020). Explainable Artificial Intelligence (XAI): Concepts, Taxonomies, Opportunities and Challenges toward Responsible AI. Inf. Fusion 58, 82–115. doi:10.1016/j.inffus.2019.12.012
Austin, N. D., Sahinidis, N. V., and Trahan, D. W. (2016). Computer-aided Molecular Design: An Introduction and Review of Tools, Applications, and Solution Techniques. Chem. Eng. Res. Des. 116, 2–26. doi:10.1016/j.cherd.2016.10.014
Baevski, A., Zhou, H., Mohamed, A., and Auli, M. (2020). wav2vec 2.0: A Framework for Self-Supervised Learning of Speech Representations. arXiv. doi:10.1109/icassp40776.2020.9054224
Brown, N., Fiscato, M., Segler, M. H. S., and Vaucher, A. C. (2019). GuacaMol: Benchmarking Models for De Novo Molecular Design. J. Chem. Inf. Model. 59 (3), 1096–1108. doi:10.1021/acs.jcim.8b00839
Butler, K. T., Davies, D. W., Cartwright, H., Isayev, O., and Walsh, A. (2018). Machine Learning for Molecular and Materials Science. Nature 559, 547–555. doi:10.1038/s41586-018-0337-2
Coley, C. W., Green, W. H., and Jensen, K. F. (2018). Machine Learning in Computer-Aided Synthesis Planning. Acc. Chem. Res. 51 (5), 1281–1289. doi:10.1021/acs.accounts.8b00087
Co-Reyes, J. D., Miao, Y., Peng, D., Real, E., Levine, S., Le, Q. V., et al. (2021). Evolving Reinforcement Learning Algorithms. ArXiv.
Cova, T. F. G. G., and Pais, A. A. C. C. (2019). Deep Learning for Deep Chemistry: Optimizing the Prediction of Chemical Patterns. Front. Chem. 7 (809). doi:10.3389/fchem.2019.00809
Dai, H., Li, C., Coley, C. W., Dai, B., and Song, L. (2020). Retrosynthesis Prediction with Conditional Graph Logic Network. ArXiv. doi:10.1287/20a2211b-f3ae-4d98-845f-3c68e9f392dc
Dosovitskiy, A., Beyer, L., Kolesnikov, A., Weissenborn, D., Zhai, X., Unterthiner, T., et al. (2020). An Image Is worth 16x16 Words: Transformers for Image Recognition at Scale. arXiv.
Fooshee, D., Mood, A., Gutman, E., Tavakoli, M., Urban, G., Liu, F., et al. (2018). Deep Learning for Chemical Reaction Prediction. Mol. Syst. Des. Eng. 3 (3), 442–452. doi:10.1039/C7ME00107J
Gani, R. (2004). Chemical Product Design: Challenges and Opportunities. Comput. Chem. Eng. 28 (12), 2441–2457. doi:10.1016/j.compchemeng.2004.08.010
Gani, R. (2019). Group Contribution-Based Property Estimation Methods: Advances and Perspectives. Curr. Opin. Chem. Eng. 23, 184–196. doi:10.1016/j.coche.2019.04.007
Gao, H., Wang, Z., and Ji, S. (2018). “Large-scale Learnable Graph Convolutional Networks,” in Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining), London, United Kingdom, 1416–1424.
Garcia, D. J., and You, F. (2016). The Water-Energy-Food Nexus and Process Systems Engineering: A New Focus. Comput. Chem. Eng. 91, 49–67. doi:10.1016/j.compchemeng.2016.03.003
Garcia, D., and You, F. (2017). Systems Engineering Opportunities for Agricultural and Organic Waste Management in the Food-Water-Energy Nexus. Curr. Opin. Chem. Eng. 18, 23–31. doi:10.1016/j.coche.2017.08.004
Goh, G. B., Hodas, N. O., and Vishnu, A. (2017). Deep Learning for Computational Chemistry. J. Comput. Chem. 38 (16), 1291–1307. doi:10.1002/jcc.24764
Gómez-Bombarelli, R., Wei, J. N., Duvenaud, D., Hernández-Lobato, J. M., Sánchez-Lengeling, B., Sheberla, D., et al. (2018). Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 4 (2), 268–276. doi:10.1021/acscentsci.7b00572
Griffiths, R.-R., and Hernández-Lobato, J. M. (2020). Constrained Bayesian Optimization for Automatic Chemical Design Using Variational Autoencoders. Chem. Sci. 11 (2), 577–586. doi:10.1039/c9sc04026a
Gunning, D., Stefik, M., Choi, J., Miller, T., Stumpf, S., and Yang, G.-Z. (2019). XAI-explainable Artificial Intelligence. Sci. Robot. 4 (37), eaay7120. doi:10.1126/scirobotics.aay7120
Hagen, J. (2015). Industrial Catalysis: A Practical Approach. Weinheim,Germany: John Wiley & Sons. doi:10.1002/9783527684625
Hubbs, C. D., Li, C., Sahinidis, N. V., Grossmann, I. E., and Wassick, J. M. (2020). A Deep Reinforcement Learning Approach for Chemical Production Scheduling. Comput. Chem. Eng. 141, 106982. doi:10.1016/j.compchemeng.2020.106982
Jha, D., Choudhary, K., Tavazza, F., Liao, W.-k., Choudhary, A., Campbell, C., et al. (2019). Enhancing Materials Property Prediction by Leveraging Computational and Experimental Data Using Deep Transfer Learning. Nat. Commun. 10 (1), 5316. doi:10.1038/s41467-019-13297-w
Jin, W., Yang, K., Barzilay, R., and Jaakkola, T. (2019). “Learning Multimodal Graph-To-Graph Translation for Molecular Optimization,” in 7th International Conference on Learning Representations, New Orleans, LA (ICLR).
Krenn, M., Häse, F., Nigam, A., Friederich, P., and Aspuru-Guzik, A. (2020). Self-Referencing Embedded Strings (SELFIES): A 100% Robust Molecular String Representation. Machine Learn. Sci. Tech. 1 (4), 045024. doi:10.1088/2632-2153/aba947
LeCun, Y., and Misra, I. (2021). Self-supervised Learning: The Dark Matter of Intelligence. [Online]. Facebook AI. Available at: https://ai.facebook.com/blog/self-supervised-learning-the-dark-matter-of-intelligence/(Accessed).
LeCun, Y., Bengio, Y., and Hinton, G. (2015). Deep Learning. Nature 521 (7553), 436–444. doi:10.1038/nature14539
Lin, K., Xu, Y., Pei, J., and Lai, L. (2020). Automatic Retrosynthetic Route Planning Using Template-free Models. Chem. Sci. 11 (12), 3355–3364. doi:10.1039/C9SC03666K
Liu, Y., Ott, M., Goyal, N., Du, J., Joshi, M., Chen, D., et al. (2019). Roberta: A Robustly Optimized Bert Pretraining Approach. arXiv.
Loquercio, A., Segu, M., and Scaramuzza, D. (2020). A General Framework for Uncertainty Estimation in Deep Learning. IEEE Robot. Autom. Lett. 5 (2), 3153–3160. doi:10.1109/LRA.2020.2974682
Lu, K., Grover, A., Abbeel, P., and Mordatch, I. (2021). Pretrained Transformers as Universal Computation Engines. arXiv.
Ma, H., Bian, Y., Rong, Y., Huang, W., Xu, T., Xie, W., et al. (2020a). Multi-View Graph Neural Networks for Molecular Property Prediction. arXiv.
Ma, T., Ferber, P., Huo, S., Chen, J., and Katz, M. (2020b). “Online Planner Selection with Graph Neural Networks and Adaptive Scheduling,” in Proceedings of the AAAI Conference on Artificial Intelligence), New York, NY, 5077–5084.
MacLeod, B. P., Parlane, F. G., Morrissey, T. D., Häse, F., Roch, L. M., Dettelbach, K. E., et al. (2020). Self-driving Laboratory for Accelerated Discovery of Thin-Film Materials. Sci. Adv. 6 (20), eaaz8867. doi:10.1126/sciadv.aaz8867
Maziarka, Ł., Pocha, A., Kaczmarczyk, J., Rataj, K., Danel, T., and Warchoł, M. (2020). Mol-CycleGAN: a Generative Model for Molecular Optimization. J. Cheminform 12 (1). doi:10.1186/s13321-019-0404-1
Miodownik, M. (2015). Materials for the 21st century: What Will We Dream up Next? MRS Bull. 40 (12), 1188–1197. doi:10.1557/mrs.2015.267
Monti, F., Boscaini, D., Masci, J., Rodola, E., Svoboda, J., and Bronstein, M. M. (2017). “Geometric Deep Learning on Graphs and Manifolds Using Mixture Model Cnns”, in: Proceedings of the IEEE conference on computer vision and pattern recognition), Honolulu, HI, 5115–5124.
Nian, R., Liu, J., and Huang, B. (2020). A Review on Reinforcement Learning: Introduction and Applications in Industrial Process Control. Comput. Chem. Eng. 139, 106886. doi:10.1016/j.compchemeng.2020.106886
Ning, C., and You, F. (2019). Optimization under Uncertainty in the Era of Big Data and Deep Learning: When Machine Learning Meets Mathematical Programming. Comput. Chem. Eng. 125, 434–448. doi:10.1016/j.compchemeng.2019.03.034
Pappas, I., Kenefake, D., Burnak, B., Avraamidou, S., Ganesh, H. S., Katz, J., et al. (2021). Multiparametric Programming in Process Systems Engineering: Recent Developments and Path Forward. Front. Chem. Eng. 2 (32). doi:10.3389/fceng.2020.620168
Pappu, A., and Paige, B. (2020). Making Graph Neural Networks Worth it for Low-Data Molecular Machine Learning. arXiv. doi:10.7554/elife.56159.sa1
Parisotto, E., Song, F., Rae, J., Pascanu, R., Gulcehre, C., Jayakumar, S., et al. (2020). “Stabilizing Transformers for Reinforcement Learning,” in International Conference on Machine Learning: PMLR 7487–7498.
Petsagkourakis, P., Sandoval, I. O., Bradford, E., Zhang, D., and del Rio-Chanona, E. A. (2020). Reinforcement Learning for Batch Bioprocess Optimization. Comput. Chem. Eng. 133, 106649. doi:10.1016/j.compchemeng.2019.106649
Pistikopoulos, E. N., Barbosa-Povoa, A., Lee, J. H., Misener, R., Mitsos, A., Reklaitis, G. V., et al. (2021). Process Systems Engineering - the Generation Next? Comput. Chem. Eng. 147, 107252. doi:10.1016/j.compchemeng.2021.107252
Polykovskiy, D., Zhebrak, A., Sanchez-Lengeling, B., Golovanov, S., Tatanov, O., Belyaev, S., et al. (2020). Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models. Front. Pharmacol. 11 (1931). doi:10.3389/fphar.2020.565644
Popova, M., Isayev, O., and Tropsha, A. (2018). Deep Reinforcement Learning for De Novo Drug Design. Sci. Adv. 4 (7), eaap7885. doi:10.1126/sciadv.aap7885
Preuer, K., Klambauer, G., Rippmann, F., Hochreiter, S., and Unterthiner, T. (2019). “Interpretable Deep Learning in Drug Discovery,” in Explainable AI: Interpreting, Explaining and Visualizing Deep Learning (Cham, Switzerland: Springer), 331–345. doi:10.1007/978-3-030-28954-6_18
Pyzer‐Knapp, E. O., Li, K., and Aspuru‐Guzik, A. (2015). Learning from the harvard Clean Energy Project: The Use of Neural Networks to Accelerate Materials Discovery. Adv. Funct. Mater. 25 (41), 6495–6502. doi:10.1002/adfm.201501919
Rao, R., Liu, J., Verkuil, R., Meier, J., Canny, J. F., Abbeel, P., et al. (2021). Msa Transformer. bioRxiv. doi:10.1016/j.surge.2021.02.002
Ravanelli, M., Zhong, J., Pascual, S., Swietojanski, P., Monteiro, J., Trmal, J., et al. (2020). “Multi-task Self-Supervised Learning for Robust Speech Recognition,” in ICASSP 2020-2020 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), Barcelona, Spain (IEEE), 6989–6993.
Reis, E. S. D., Costa, C. A. D., Silveira, D. E. D., Bavaresco, R. S., Righi, R. D. R., Barbosa, J. L. V., et al. (2021). Transformers Aftermath. Commun. ACM 64 (4), 154–163. doi:10.1145/3430937
Roch, L. M., Häse, F., Kreisbeck, C., Tamayo-Mendoza, T., Yunker, L. P. E., Hein, J. E., et al. (2018). ChemOS: Orchestrating Autonomous Experimentation. Sci. Robot. 3 (19), eaat5559. doi:10.1126/scirobotics.aat5559
Rong, Y., Bian, Y., Xu, T., Xie, W., Wei, Y., Huang, W., et al. (2020). Self-Supervised Graph Transformer on Large-Scale Molecular Data. Adv. Neural Inf. Process. Syst. 33.
Sanchez-Lengeling, B., and Aspuru-Guzik, A. (2018). Inverse Molecular Design Using Machine Learning: Generative Models for Matter Engineering. Science 361 (6400), 360–365. doi:10.1126/science.aat2663
Schütt, K. T., Arbabzadah, F., Chmiela, S., Müller, K. R., and Tkatchenko, A. (2017). Quantum-chemical Insights from Deep Tensor Neural Networks. Nat. Commun. 8 (1), 1–8. doi:10.1038/ncomms13890
Schwaller, P., Gaudin, T., Lányi, D., Bekas, C., and Laino, T. (2018). “Found in Translation”: Predicting Outcomes of Complex Organic Chemistry Reactions Using Neural Sequence-To-Sequence Models. Chem. Sci. 9 (28), 6091–6098. doi:10.1039/C8SC02339E
Schwaller, P., Petraglia, R., Zullo, V., Nair, V. H., Haeuselmann, R. A., Pisoni, R., et al. (2020). Predicting Retrosynthetic Pathways Using Transformer-Based Models and a Hyper-Graph Exploration Strategy. Chem. Sci. 11 (12), 3316–3325. doi:10.1039/C9SC05704H
Segler, M. H. S., and Waller, M. P. (2017). Neural-Symbolic Machine Learning for Retrosynthesis and Reaction Prediction. Chem. Eur. J. 23 (25), 5966–5971. doi:10.1002/chem.201605499
Segler, M. H. S., Preuss, M., and Waller, M. P. (2018a). Planning Chemical Syntheses with Deep Neural Networks and Symbolic AI. Nature 555 (7698), 604–610. doi:10.1038/nature25978
Segler, M. H. S., Kogej, T., Tyrchan, C., and Waller, M. P. (2018b). Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 4 (1), 120–131. doi:10.1021/acscentsci.7b00512
Senior, A. W., Evans, R., Jumper, J., Kirkpatrick, J., Sifre, L., Green, T., et al. (2020). Improved Protein Structure Prediction Using Potentials from Deep Learning. Nature 577 (7792), 706–710. doi:10.1038/s41586-019-1923-7
Shin, J., Badgwell, T. A., Liu, K.-H., and Lee, J. H. (2019). Reinforcement Learning - Overview of Recent Progress and Implications for Process Control. Comput. Chem. Eng. 127, 282–294. doi:10.1016/j.compchemeng.2019.05.029
Sourek, G., Zelezny, F., and Kuzelka, O. (2020). Learning with Molecules beyond Graph Neural Networks. arXiv.
Su, Y., Wang, Z., Jin, S., Shen, W., Ren, J., and Eden, M. R. (2019). An Architecture of Deep Learning in QSPR Modeling for the Prediction of Critical Properties Using Molecular Signatures. Aiche J. 65 (9), e16678. doi:10.1002/aic.16678
Tetko, I. V., Karpov, P., Van Deursen, R., and Godin, G. (2020). State-of-the-art Augmented NLP Transformer Models for Direct and Single-step Retrosynthesis. Nat. Commun. 11 (1), 1–11. doi:10.1038/s41467-020-19266-y
Tjoa, E., and Guan, C. (2020). A Survey on Explainable Artificial Intelligence (Xai): Toward Medical Xai. IEEE Trans. Neural Netw. Learn. Syst. 1–21. doi:10.1109/TNNLS.2020.3027314
Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A. N., et al. (2017). Attention Is All You Need. Adv. Neural Inf. Process. Syst.
Walters, W. P., and Barzilay, R. (2020). Applications of Deep Learning in Molecule Generation and Molecular Property Prediction. Acc. Chem. Res. 54, 263–270. doi:10.1021/acs.accounts.0c00699
Wang, T., Liao, R., Ba, J., and Fidler, S. (2018). “Nervenet: Learning Structured Policy with Graph Neural Networks,” in International Conference on Learning Representations, Vancouver, Canada.
Weininger, D. (1988). SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Model. 28 (1), 31–36. doi:10.1021/ci00057a005
Wu, D., and Zhao, J. (2021). Process Topology Convolutional Network Model for Chemical Process Fault Diagnosis. Process Saf. Environ. Prot. 150, 93–109. doi:10.1016/j.psep.2021.03.052
Zhang, L., Mao, H., Liu, L., Du, J., Gani, R., and Engineering, C. (2018). A Machine Learning Based Computer-Aided Molecular Design/screening Methodology for Fragrance Molecules. Comput. Chem. Eng. 115, 295–308. doi:10.1016/j.compchemeng.2018.04.018
Zhang, L., Mao, H., Liu, Q., and Gani, R. (2020). Chemical Product Design - Recent Advances and Perspectives. Curr. Opin. Chem. Eng. 27, 22–34. doi:10.1016/j.coche.2019.10.005
Zhang, Y., and Lee, A. A. (2019). Bayesian Semi-supervised Learning for Uncertainty-Calibrated Prediction of Molecular Properties and Active Learning. Chem. Sci. 10 (35), 8154–8163. doi:10.1039/C9SC00616H
Keywords: computational design, molecular design, synthesis planning, deep learning, product design, systems engineering
Citation: Alshehri AS and You F (2021) Paradigm Shift: The Promise of Deep Learning in Molecular Systems Engineering and Design. Front. Chem. Eng. 3:700717. doi: 10.3389/fceng.2021.700717
Received: 26 April 2021; Accepted: 26 May 2021;
Published: 08 June 2021.
Edited by:
José María Ponce-Ortega, Michoacana University of San Nicolás de Hidalgo, MexicoReviewed by:
Rajib Mukherjee, University of Texas of the Permian Basin, United StatesFernando Israel Gómez-Castro, University of Guanajuato, Mexico
Eusiel Rubio-Castro, Autonomous University of Sinaloa, Mexico
Copyright © 2021 Alshehri and You. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengqi You, ZmVuZ3FpLnlvdUBjb3JuZWxsLmVkdQ==