 Christine Stark
Christine Stark Sarah E. Jenison
Sarah E. Jenison Mai T. Ngo
Mai T. Ngo- Department of Chemical and Biological Engineering, University of Wisconsin-Madison, Madison, WI, United States
-Omics technologies have emerged as powerful tools to uncover cellular diversity within tissues, and the role of cell-cell communication in tissue development, function, and disease. In this review, we will discuss recent advancements in -omics technologies that are used to interrogate the biomolecular mechanisms that underly tissue form and function. We will specifically discuss the application of -omics technologies, along with bioinformatic tools, towards identifying new cell types and cell-cell interactions within native tissues. We will then examine how insights from -omics technologies can inform the design of engineered tissues, particularly through the lens of recapitulating native cell-cell interactions. Finally, we will discuss how -omics can be employed to benchmark and analyze engineered tissues for applications that span fundamental science and translation. Overall, the integration of -omics and tissue engineering will improve our understanding of the roles of cellular diversity and cell-cell communication in regulating tissue health and disease and subsequently inform how cell-cell interactions can be leveraged to design therapies for human health applications.
1 Introduction
The term “tissue engineering” was first widely recognized by the scientific community in the late 1980s, following a National Science Foundation meeting under the same name (Vacanti, 2006). At the time, the field focused primarily on the surgical integration of living tissues and prosthetic devices. Initial experiments involved seeding cells onto natural scaffolds present in the body or applying cell sheets to damaged tissues, not yet realizing the potential to design synthetic scaffolds to direct regeneration. Tissue engineering, as we recognize it today—related to the design of new tissues by integrating cells, biologics, and scaffolds—was reportedly first used in a 1991 article titled “Functional Organ Replacement: The New Technology of Tissue Engineering.” Since then, tissue engineering has evolved beyond the design of prosthetic devices and surgical manipulation of native tissues, and now encompasses the development of synthetic tissue platforms for both in vitro and in vivo applications. For example, tissue engineering is now used to generate in vitro tissue models for biological discovery and therapeutic screening, as well as implantable therapies for drug delivery and regenerative medicine.
Modern-day advances in developing and characterizing engineered tissues have been facilitated by the integration of tissue engineering design principles with bioinformatics and -omics technologies. The roots of these -omics technologies can be traced back to the early 2000s, when the publication of the Human Genome Project triggered widespread interest in genomics research (Hood and Rowen, 2013). Rapid progress following this landmark study enabled researchers to explore and characterize biomolecules beyond genomic DNA, leading to the birth of various '-omics’ fields (e.g., epigenomics, transcriptomics, proteomics, metabolomics) (Manzoni et al., 2018). These technologies have unlocked new possibilities for identifying molecular interactions between cells, signaling cues, and the extracellular matrix (ECM) within native tissues. Recent advances in -omics methodologies have also led to single-cell insights, including the identification of cell types previously undetected in heterogenous tissues and the discovery of cell-cell interactions that drive critical tissue-specific functions (Baysoy et al., 2023). In parallel, bioinformatics tools have evolved to aid in interpreting the extensive datasets generated by -omics tools, allowing for insight into biomolecular-scale phenomena that result in cell- or tissue-level function.
Collectively, -omics tools have been applied to study developmental, regenerative, and disease processes across many different tissues and organs. Many of these biological processes arise from interactions between heterotypic cell types (e.g., parenchymal, stromal, immune, vascular) that are present within tissues and organs. -Omics tools have shed light on these complex cell-cell interactions, and how they influence tissue form and function (Armingol et al., 2021). These insights can be leveraged to define cell subpopulations and signaling interactions that must be recapitulated within engineered tissues to accurately mimic specific properties or functions of native tissues. In turn, -omics tools can also be used to characterize cellular heterogeneity and cell-cell interactions within engineered tissues. For example, -omics analysis can be used to derive new biological insight from in vitro model systems, or to assess the integration and performance of transplanted tissues. As -omics and bioinformatics tools continue to improve, further elucidation of the complex cell-cell signaling processes that underly tissue development, regeneration, and disease will enhance our ability to design and validate multicellular engineered tissues that closely mimic native tissues, and to utilize these engineered tissues for applications that span basic science to clinical translation.
In this review, we will first provide an overview of -omics technologies that are commonly used to characterize tissues and provide insight into the biomolecular processes that underly tissue-level behavior and function. Subsequently, we will highlight how -omics have been specifically employed to define cellular diversity of tissues, along with cell-cell interactions that contribute to tissue development, regeneration, and disease. Finally, we will discuss how -omics technologies can inform the design of multicellular engineered tissues and enable the analysis of these living systems for basic science and translational applications.
2 -Omics techniques for characterizing cells and tissues
The advent of -omics technologies has revolutionized our ability to characterize cell states and cell identities, along with the composition and organization of higher-order tissues and organs (Gulati et al., 2025). -Omics technologies leverage advanced experimental, analytical, and computational strategies to characterize the biomolecular landscapes of cell and tissue specimens. Within the field of -omics, scientists have established subcategories of technologies that focus on distinct biomolecules, with the most commonly-used techniques being genomics, transcriptomics, proteomics, and metabolomics (Dai and Shen, 2022). The insights gained from -omics studies have informed our understanding of cell identities and cell states that exist across health and disease, as well as intercellular interactions that result in tissue- and organ-level structure and function. Additionally, while an extensive amount of insight can be gained from using a single -omics technique, holistic characterization of biological specimens and their functions can be achieved by integrating information from complimentary -omics technologies. Such “multi-omics” strategies enable researchers to elucidate systems-level relationships by which genomic, transcriptomic, proteomic, and metabolomic landscapes collectively define biological states and functions (Hasin et al., 2017).
In the following sub-sections, we will examine how the leading -omics technologies (e.g., genomics, transcriptomic, proteomics, metabolomics) contribute to our understanding of biological systems (Figure 1). We will discuss the workflow of the most relevant techniques in each -omics field, followed by a discussion of future trends for these technologies. Additionally, we will highlight the need for advanced bioinformatics tools to derive insights from -omics data using statistical and data science techniques (Uesaka et al., 2022).
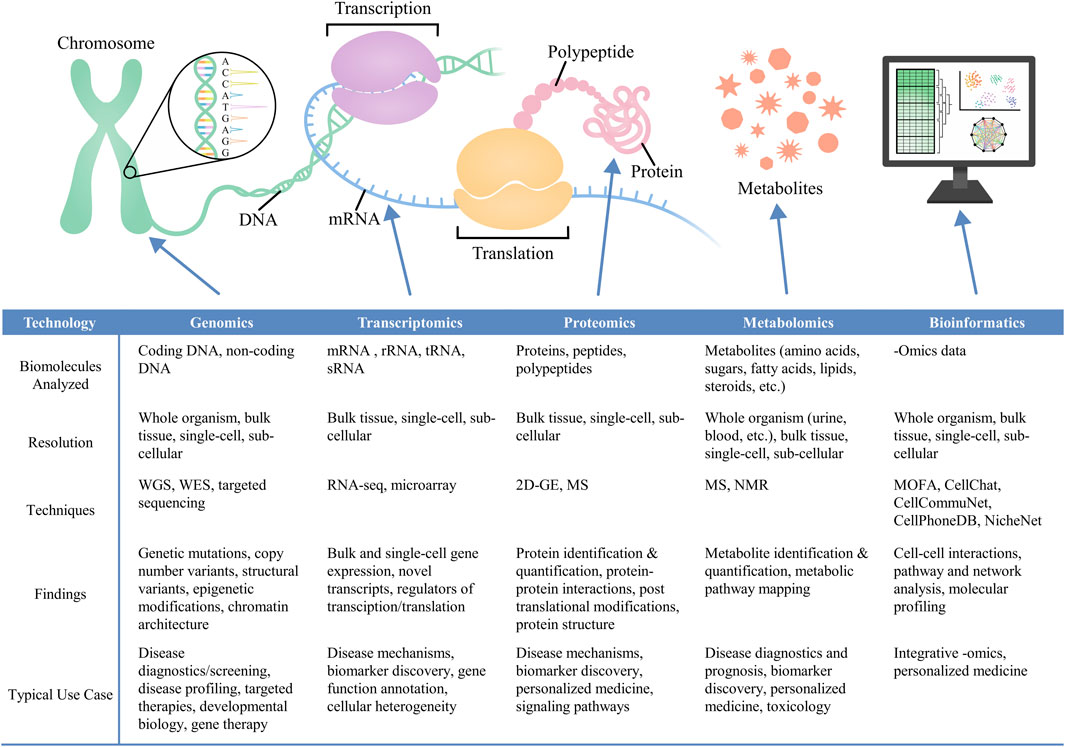
Figure 1. Various–omics technologies—including genomics (Murrell et al., 2005; Hood and Rowen, 2013; Smallwood et al., 2014; Tomczak et al., 2015; Bonev and Cavalli, 2016; Bewicke-Copley et al., 2019; Wong et al., 2021), transcriptomics (Tang et al., 2009; Natarajan et al., 2017; Li and Wang, 2021; Rao et al., 2021; Supplitt et al., 2021; Mubarak and Zahir, 2022; Williams et al., 2022), proteomics (Blagoev et al., 2003; Altelaar et al., 2013; Ragelle et al., 2017; Lundberg and Borner, 2019; Palii et al., 2019; Al-Amrani et al., 2021; Gerritsen and White, 2021), and metabolomics (Wishart, 2005; Dunn et al., 2007; 2011; Pinto and Eileen Dolan, 2011; Levy et al., 2017; Pinu et al., 2019; Wang et al., 2022; Alexandrov, 2023)—combined with bioinformatics approaches (Argelaguet et al., 2018; Browaeys et al., 2020; Efremova et al., 2020; Jin et al., 2021; Uesaka et al., 2022; Ma et al., 2024), can provide biomolecular-level characterization of cell identities, cell-cell interactions, and cell-microenvironment interactions.
2.1 Genomics
The genome is comprised of the complete set of DNA molecules within an organism, providing all genetic instructions for organismal development and function. Within genomic analysis, there are three common approaches: whole-genome sequencing (WGS), which evaluates the complete set of DNA sequences within an organism; whole-exome sequencing (WES), which analyzes the protein-coding regions of the genome; and targeted sequencing, which examines specific sections of the genome (Ng and Kirkness, 2010; Rabbani et al., 2014; Bewicke-Copley et al., 2019). In addition to protein-coding DNA sequences, WGS also captures non-coding sequences and regulatory regions of the genome, which contribute to the regulation of gene activity (Gloss and Dinger, 2018). Changes to non-coding regions can significantly influence gene expression and whole-organism phenotypes. Thus, by studying sequences beyond the exome, scientists have gained a greater understanding of non-coding genetic variations that contribute to disease emergence and progression (Belkadi et al., 2015). In contrast, while the exome comprises a small percentage of an organism’s entire DNA sequence, most known mutations that are linked to disease occur in these regions, and therefore WES by itself can yield significant insights regarding disease-causing genetic variants (Rabbani et al., 2014). Once potential disease-causing genetic variants have been identified, gene editing strategies can be used to generate in vitro tissue models to study the mechanisms by which candidate variants contribute to disease onset and progression (Dobrindt et al., 2021; Longo et al., 2022). Additionally, disease-specific engineered tissues can act as preliminary models to screen potential gene therapies for correcting genetic variants (Kolli et al., 2017; Yang et al., 2021). Lastly, targeted sequencing is utilized to detect specific genetic variations, and represents a more cost-effective strategy to identify mutations specific to certain conditions (Rehm, 2013).
Analogous to genomics, epigenomics is a closely-related field which investigates epigenetic modifications that influence gene expression without modifying the DNA itself (Murrell et al., 2005). Epigenetic modifications typically modulate DNA folding and architecture, which in turn controls transcription factor access to DNA by organizing chromatin into higher-order structures (Wang and Chang, 2018). Epigenetic changes also occur through chemical alterations to DNA, commonly including histone modifications and DNA methylation. Techniques such as Cleavage Under Targets and Tagmentation (CUT&Tag) and Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) have enabled comprehensive profiling of histone modifications and chromatin accessibility (Buenrostro et al., 2013; Bartosovic et al., 2021; Jiang et al., 2022). Intriguingly, epigenetics alone, or in combination with gene expression data, can be used to define cell identities and cell states. Collectively, these tools have allowed for further insight into regulatory elements that mediate gene expression. Techniques for probing epigenomics will continue to be invaluable for characterizing cell state transitions and the processes that govern the emergence of new cell identities during tissue development, regeneration, and disease.
2.1.1 Sample preparation and data analysis for genomics
WGS is the most widely-used genomic analysis technique, providing a precise DNA fingerprint of the biological specimen. To perform WGS, DNA is extracted from cells or tissue via lysis and purified to remove any RNA fragments, proteins, and chemical additives (Linnarsson, 2010). The isolated DNA is then fragmented through mechanical or enzymatic shearing. In the past, standard WGS protocols amplified DNA by polymerase chain reaction (PCR), but modern techniques no longer require this step, thus eliminating bias regarding which sequences are amplified (Zhou et al., 2022). The DNA fragments are then analyzed by a sequencing platform to identify the nucleotide bases that make up of each fragment (Ng and Kirkness, 2010). The resultant data includes millions of DNA reads, which are aligned and assembled through computational pipelines to piece together the DNA fragments into the entire genomic sequence of the biological specimen. Following genome assembly, genetic variations such as single point mutations, structural variants, and single-nucleotide polymorphisms can be detected. The assembled genome sequences are compared to reference sequences using reference-alignment tools and variant-calling algorithms to detect genetic variations (Logsdon et al., 2020). Analysis of genomic data enables the identification of disease-causing variants, and advanced algorithms can even identify abnormal repeats, methylation patterns, and structural variants that influence disease mechanisms.
2.1.2 Genomics trends and future directions
Since the completion of the Human Genome Project, genomic analysis has gained widespread recognition in the field of human health and become a critical biomedical research tool (Lander et al., 2001). In recent years, 3D genomics has emerged as a rapidly-advancing technique. Spatial organization of genes, and how DNA is packaged in the nucleus, dictate how transcriptional and translational machinery and DNA-binding proteins interact with DNA. 3D genomics focuses on analyzing dynamic changes in the spatial organization of the genome, and evaluating how these changes influence gene expression and regulation of essential cellular functions (Zhang and Li, 2020). Standard 3D genomics methods include chromosome conformation capture (3C)-based approaches (Dekker et al., 2002). These methods use proximity ligation to identify chromatin contacts amongst nearby DNA sequences (Han et al., 2018). DNA is first crosslinked to maintain its spatial organization, and then the chromatin is fragmented. The ends of the resulting DNA fragments are ligated and sequenced to detect spatially-proximal DNA. 3D genomics has been employed to study chromatin structure in embryonic stem cells, and changes in 3D chromatin organization were found to correlate with chromatin accessibility and gene activity during early lineage specification (Dixon et al., 2015). Furthermore, spatial genomics techniques allow for spatially-resolved DNA sequencing. Zhao et al. developed slide-DNA-seq, which can analyze DNA sequences from whole tissue samples and therefore map genomic data onto local tissue architecture (Zhao et al., 2022). Future studies that apply 3D and spatial genomic technologies to whole tissues are expected to uncover how genomic architecture and organization influences biological processes across development, homeostasis, and disease.
2.2 Transcriptomics
The transcriptome is the complete set of RNA molecules synthesized by a cell (Wang et al., 2009). Specifically, the transcriptome consists of coding and non-coding RNA. Coding RNA includes messenger RNA (mRNA), which are transcripts for protein-coding genes. When exclusively examining gene expression, transcriptomics techniques like RNA sequencing (RNA-seq) are typically performed by selectively sequencing mRNA. Beyond gene expression, RNA-seq can also identify alternative splicing modifications by revealing multiple mRNA isoforms (Wang et al., 2008), and can also be used to study non-coding RNAs that play a critical role in gene regulation (Atkinson et al., 2012). Non-coding RNA includes ribosomal RNA (rRNA), transfer RNA (tRNA), and small RNA (sRNA), which are not translated into proteins but instead are involved in a variety of functions such as protein synthesis and gene regulation (Palazzo and Lee, 2015). Therefore, transcriptomic analysis not only provides insight into the genes that are translated into proteins to regulate cell phenotypes, but also the molecular machinery that regulates the transcription and translation of these genes.
Much of our current understanding of transcriptomics and its relation to cell or tissue phenotypes is credited to next-generation sequencing (NGS) technologies. NGS was developed to overcome the limitations of DNA microarrays, an earlier technology that first enabled multiplexed analysis of gene expression (Kothapalli et al., 2002). RNA-seq is the current gold standard approach for transcriptomic studies. In the past decade alone, RNA-seq has enabled researchers to identify and quantify gene expression levels, discover novel transcripts, and elucidate differential gene expression dynamics across biological states and specimens (Wang et al., 2009). Initially, the establishment of bulk RNA-seq provided composite gene expression profiles for entire tissues or cell populations. However, recent advances in single-cell RNA sequencing (scRNA-seq) have enabled transcriptomic analysis at the level of an individual cell (Li and Wang, 2021). scRNA-seq data has generated unprecedented insight into the cellular heterogeneity of tissues, allowing researchers to identify and distinguish between different cell types and characterize cell-specific gene expression patterns. In particular, researchers have frequently used transcriptomic approaches to identify cell types and differentially-expressed genes during embryonic development, tissue regeneration, and disease progression (Sleep et al., 2010; Park et al., 2018; Cao et al., 2019; Pijuan-Sala et al., 2019; McKellar et al., 2021; Mascharak et al., 2022; Fabrizio et al., 2025).
2.2.1 Sample preparation and data analysis for transcriptomics
Sample preparation for RNA-seq begins with the lysis of cells or tissues to extract their RNA. For scRNA-seq, it is necessary to obtain a clean, single-cell suspension prior to lysis (Haque et al., 2017). Alternative strategies to single-cell preparation include isolation of single nuclei and ‘split-pooling’ approaches, which can offer cost-effective and flexible protocols that require less sophisticated equipment and reduce aberrant transcription (Habib et al., 2016; Lacar et al., 2016). After lysis, poly [T] primers are used to selectively capture polyadenylated mRNAs, which are easier to analyze compared to other RNA molecules (Haque et al., 2017). The polyadenylated mRNAs are subsequently converted to complementary DNA (cDNA) via reverse transcription. Sequencing libraries are then prepared by ligating adapters onto the cDNA fragments, and the libraries are then analyzed by sequencing platforms (Haque et al., 2017). Individual reads are mapped to known genes, exons, or annotated transcripts by aligning the sequencing data to reference datasets (Chen et al., 2023).
RNA-seq data can be analyzed to reveal differentially-expressed genes that are present among cell and tissue specimens. Furthermore, identified genes can be grouped into categories that are based on functions or processes associated with each gene. Here, gene set analysis (GSA) can be performed to test differential expression between these groups of genes, thereby providing insight into biological processes or signaling pathways that may vary between experimental groups (Chen et al., 2023). Additionally, over-representation analysis can be used to determine whether specific gene sets are enriched amongst up- or downregulated differentially-expressed genes. Gene set enrichment analysis (GSEA) is another computational method to determine whether a gene set exhibits statistically-significant differences between experimental groups. Functional enrichment analysis can also be employed to infer biological insight from differentially-expressed genes or gene sets. This process commonly uses bioinformatics tools such as the Database for Annotation, Visualization and Integrated Discovery (DAVID) and Ingenuity Pathway Analysis (IPA) (Huang et al., 2007; Krämer et al., 2014).
2.2.2 Transcriptomics trends and future directions
Future sequencing platforms for transcriptomics analysis are expected to focus on improving spatial transcriptomics, which is capable of measuring gene expression within the spatial context of tissues and cells. This is typically conducted by image-based approaches, which detect mRNA molecules directly within the tissue, or next-generation sequencing (NGS) approaches, where spatial information is recorded with each transcript (Williams et al., 2022). In image-based approaches, researchers commonly use in situ hybridization (ISH) methods, where labelled nucleic acid probes bind to specific sequences (Jin and Lloyd, 1997). Alternatively, in situ sequencing (ISS) directly sequences RNA molecules in the tissue using methods like sequencing by ligation or sequencing by synthesis (Lee et al., 2022). NGS methods retain spatial context through techniques such as microdissection and microfluidics that directly capture location information, or the use of microarrays with barcoded probes that correspond to locations within a tissue (Williams et al., 2022). Spatial transcriptomics enables the characterization of transcriptional patterns in 3D space, such as signaling pathway and protein gradients, revealing how gene expression is influenced by tissue architecture (Garcia-Alonso et al., 2021). These techniques can also detect the local organization of cells within a tissue and provide insights into the local signaling environment, including cell-ECM and cell-cell interactions (Coy et al., 2022).
The immense volume of data that is generated from existing RNA-seq workflows can pose significant challenges in data processing and interpretation. For instance, it can be difficult to distinguish background noise from meaningful observations. Additionally, transcriptomic analysis typically produces high-dimensional data, where data points are sparse and traditional analysis methods are less effective. As a result, researchers have turned to developing machine learning and high-throughput computational tools to improve the accuracy, efficiency, and scalability of transcriptomic analysis (Zeng et al., 2022).
2.3 Proteomics
Proteomics is the study of the entire complement of proteins expressed at the cellular, tissue, or organismal level. The results of proteomic analysis can provide information about the identities of proteins that are present in a biological specimen, as well as modifications to protein structures and protein-protein interactions (Al-Amrani et al., 2021; Cui et al., 2022). Previous studies have shown that transcriptomic sequencing alone is weakly correlated with protein expression, supporting the use of proteomics in tandem with transcriptomics and other -omics technologies to obtain a complete picture of the biomolecular landscape for a biological specimen (Zhang et al., 2014; 2016; Mertins et al., 2016; Sinha et al., 2019). Proteomic analysis can provide insight into dynamic protein expression profiles that change in response to environmental cues, enabling characterization of cell states as they evolve over time (Altelaar et al., 2013). In particular, proteomics has been applied to study disease progression, where analyzing protein expression levels at different disease states can reveal critical disease-driving cellular processes and biomolecular-level responses to treatment (Tremlett et al., 2015; Carlsson et al., 2017; Bai et al., 2020). Additionally, analyzing differential protein expression amongst heterogeneous cell types enhances our understanding of the cellular makeup of tissues, and the roles of individual cells in overall tissue function.
A valuable subfield of proteomics is phosphoproteomics, which focuses on the study of protein phosphorylation, a critical post-translational modification (Thingholm et al., 2009; Riley and Coon, 2016). These transient modifications, which entail the addition and subtraction of phosphate groups onto amino acids, are responsible for regulating signaling pathways essential to numerous cellular functions (e.g., proliferation, migration) (Thingholm et al., 2009). Many signal transduction pathways that regulate cellular response to cell-cell and cell-matrix signaling rely on phosphorylation to elicit specific cell behaviors in response to extracellular stimuli. Furthermore, phosphoproteomics have been widely used to investigate alterations to phosphorylation events that are associated with dysregulated signaling and abnormal cell behavior in disease states (Gerritsen and White, 2021; Morshed et al., 2021). In sum, insights from phosphoproteomics are invaluable for studying cell behaviors that arise from cell-cell signaling, as well as signaling landscapes associated with different cell states.
Early proteomics methods relied on two-dimensional (2D) gel electrophoresis to separate proteins by charge and size, which enabled the initial characterization of proteins extracted from various biological samples (Chandramouli and Qian, 2009). These techniques are now more commonly used for qualitative proteomic analysis, as they fall short in terms of reproducibility, sensitivity, and detecting low-abundance proteins. The current leading method for proteomic analysis is mass spectrometry (MS), a technique that can offer precise characterization of proteins, their isoforms, and post-translational modifications. MS offers advantages over traditional immunoassays by detecting intact proteins and peptides (Glish and Vachet, 2003). Depending on the complexity and composition of the biological sample, different proteomics approaches can be used. Bottom-up proteomic strategies analyze proteolytic peptide mixtures, and represent the most widely-used analytical approaches to study protein expression. Alternative strategies such as top-down approaches analyze intact peptides. Finally, middle-down approaches analyze long but not fully-intact peptides, allowing for additional insights into post-translational modifications and different protein isoforms (Han et al., 2008).
2.3.1 Sample preparation and data analysis for proteomics
All proteomic approaches begin by extracting the proteins from the biological specimen, often by using detergents, organic solvents, lyophilization, and mechanical disruption (Al-Amrani et al., 2021). The extracted proteins are then purified by gel-based and chromatography-based approaches, resulting in a mixture of purified proteins. The isolated proteins are then broken down by digestion with a sequence-specific protease to produce a final solution of purified peptides (Sinha and Mann, 2020).
The downstream process for analyzing samples by MS involves ionizing the protein or peptide molecules, commonly by electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI) (Han et al., 2008). The samples are processed in a mass analyzer which separates the peptides according to mass-to-charge ratios. There are several types of mass analyzers that use different separation and ion detection techniques, but here we highlight three common types. Quadrupoles separate ionic peptides based on radio frequency electrical fields, where the ions are fragmented through collisions with inert gases (Haag, 2016). For time-of-flight (TOF) analyzers, the ions are separated based on velocity, and microchannel plate (MCP) detectors amplify and measure emissions from ion ejected electrons. Lastly, Orbitrap analyzers separate ions based on oscillation frequencies where the ‘image current’, which represents the strength of the ion produced, is measured.
The experimental spectra data is analyzed by software that identifies peptides through comparison with known database-generated spectra, generating qualitative or quantitative findings. Peptide quantification strategies are either label-free, where MS signals are extracted and normalized from raw data, or label-based, where stable isotopes are introduced to tag proteins (Sinha and Mann, 2020). Common analysis software includes MSstats, an R package that uses linear mixed models for label-free and label-based workflows, thereby allowing for data-dependent and data-independent spectral readings (Choi et al., 2014). The Perseus platform is another software that utilizes traditional statistical techniques in combination with machine learning to interpret proteomic data (Tyanova et al., 2016). Additionally, there are specialized databases that can be used to infer biological insight from proteomics data. For example, the Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome, PANTHER, and pathways interaction database (PID) all provide extensive information on protein interactions, pathways, and associated gene functions (Mi et al., 2007; Schaefer et al., 2009; Croft et al., 2011; Kanehisa et al., 2012).
2.3.2 Proteomics trends and future directions
Understanding the spatial arrangement of proteins at the cellular and sub-cellular level offers information regarding signaling networks between and within cells, protein localization, and signaling pathways as they correlate to cell function and dysfunction. Spatial proteomic techniques are typically conducted to investigate one of three different findings: proteomic constituents of individual organelles within a cell, analysis of protein-protein interactions, and imaging of protein location within 3D space (Lundberg and Borner, 2019). MS has been used to characterize the structure and composition of organelles, in order to gain better insight into organelle function (Andersen et al., 2003). MS and fluorescent protein tagging-based imaging has also been applied to map the subcellular spatial arrangement of proteins, thereby identifying proximal proteins which may interact in meaningful ways to induce whole cell responses (Liu et al., 2018).
However, MS-based proteomic techniques vary significantly depending on the MS instrument that is used and the sample processing protocol. Variability amongst proteomics protocols therefore affect the accuracy and reproducibility of proteomic data. Additionally, interpretation of collected data is complicated by highly-varied processing parameters, limited quality assessment, and shortage of standardized data formats (Domon and Aebersold, 2006). Difficulty in ensuring consistency of analysis across different data platforms and computational tools limits the ability to draw reliable conclusions. Future development of proteomic technologies should prioritize the standardization of MS preparation and processing protocols, alongside the optimization of data analysis workflows that can enhance the reproducibility and reliability of proteomic studies.
2.4 Metabolomics
The metabolome is the complete set of metabolites found in a biological sample. Metabolites are the intermediate and end products of metabolic processes and include a wide variety of small molecules that contribute to functional phenotypes in whole tissues and single cells (Zhang et al., 2012). Metabolomics is the comprehensive study of metabolites, providing qualitative and quantitative information on the abundance of metabolites in biological systems. Approaches for characterizing the metabolome include targeted analysis, which identifies the presence and quantity of specific metabolites; metabolite profiling, which provides a global and semi-quantitative view of metabolites within a sample; and metabolic fingerprinting, which characterizes patterns of metabolic activity (Shulaev, 2006). Metabolomics has been used to identify metabolites related to prognosis, diagnosis, and intermediate disease states, allowing for a comprehensive understanding of disease progression (Dunn et al., 2007; Sreekumar et al., 2009; Floegel et al., 2013). Over the past decade, metabolomics has become an increasingly attractive experimental method for studying biological systems in combination with other leading -omics technologies, providing insights into the complex interactions between genes, proteins, and metabolites that drive cellular phenotypes.
2.4.1 Sample preparation and data analysis for metabolomics
A wide variety of biological samples can undergo metabolomic analysis, including blood, urine, cerebrospinal fluid, solid tissues, or cells (Tan et al., 2016). Quenching metabolic activity immediately after sample collection is critical for slowing or stopping metabolic flux. After quenching, metabolite isolation and extraction can be performed to prepare samples for analysis. Analysis is conducted by either untreated or targeted approaches. Untargeted approaches generate a comprehensive metabolic profile of all metabolites within a biological specimen, while targeted approaches focus on specific metabolites or sets of metabolites.
Both untargeted and targeted metabolomics utilize MS and nuclear magnetic resonance (NMR) spectroscopy for analysis. NMR spectroscopy is an analytical technique that characterizes chemical bonding and structure of molecules and atoms based on magnetic properties of certain atomic nuclei (Dunn et al., 2011). Metabolites are identified according to signaling peaks that are generated during MS and NMR. There are two classes of identification: putative and definitive. Putative identification uses one or two molecular properties (e.g., retention time, NMR spectrum) for identification without an authentic chemical standard. Definitive identification is more accurate, as it employs at least two properties along with an authentic chemical standard for identification (Tan et al., 2016). Definitive identification is often used after putative identification, in which selected metabolites are confirmed using authentication standards and compared against metabolomics databases (Dunn et al., 2011). There are several software tools that offer comprehensive analysis of metabolomic data, such as MetaboAnalyst 5.0, which provides both statistical and functional analysis for several data collection methods (Pang et al., 2021). Additionally, many specialized platforms are available to derive specific biological insights from metabolomics data. For instance, IMPaLA and MetPA are software tools that focus on identifying relevant metabolic pathways (Kamburov et al., 2011; Xia et al., 2011).
2.4.2 Metabolomic trends and future directions
Spatial metabolomics is a rapidly advancing technology that interprets the presence and quantity of metabolites while conserving their spatial organization within tissues (Alexandrov, 2023). This approach primarily uses mass spectrometry imaging (MSI), a derivative of MS, which overlays a coordinate grid onto a sample, enabling the mass spectrometer to generate spatially-resolved molecular data at each grid point (Buchberger et al., 2018). MSI performs spatial mapping of metabolic heterogeneity within tissues, and has successfully been employed to identify different cell types (Luo et al., 2023). MSI has also been used to characterize diseased tissues, resulting in the generation of metabolically-defined disease subtypes and the identification of dysregulated metabolic pathways (Scott et al., 2017; Neumann et al., 2022; Wang et al., 2022). Current MSI technologies can proficiently assess the spatial organization of metabolites at single-cell resolution, but subcellular analysis remains challenging. Subcellular spatial metabolomics requires complex protocols and advanced technical expertise, thus highlighting the need for improved methods to achieve simplified spatial mapping methodologies at the subcellular level (Petras et al., 2017).
Metabolomics has been largely used in clinical settings, primarily to assess patient health by measuring the presence of specific metabolites in biological specimens. In recent years, metabolomics has also become a powerful tool in precision medicine. It has led to the identification of specific disease biomarkers, as well as the development of personalized treatments that are based on individual metabolic profiles (Soni et al., 2023). However, challenges in absolute quantification, which would allow for the identification of abnormal metabolite levels within a sample, currently limits the translational potential of metabolomics (Pinu et al., 2019). Most metabolomics data is semi-quantitative and normalized in non-standardized ways, and thus quantitative reference data that can be used to calibrate pathway analysis is lacking. While absolute quantification of targeted metabolomics data is possible, methodologies to obtain absolute quantification from untargeted approaches are still in development.
2.5 Multi-omics
Individual -omics techniques can provide insights into cellular identity and function through the perspective of different biomolecules. These tools become more impactful when used together to capture relationships between different biomolecules, resulting in a comprehensive understanding of the biological sample. Multi-omic approaches primarily focus on the downstream analysis of data from separate -omics technologies. For example, computational tools such Multi-Omics Factor Analysis (MOFA) have been developed to integrate multi-omics datasets (Argelaguet et al., 2018). By employing statistical analysis and machine learning tools, multi-omics methods can infer biologically-relevant patterns that reflect biomolecular landscapes defined by combined data from individual -omics techniques.
2.5.1 Multi-omics trends and future directions
Integrating multiple -omics techniques significantly increases data volume and complexity, making it more challenging to decipher synergistic relationships among different biomolecules. These challenges are already present in analyzing data from single -omics techniques, emphasizing the need for advanced computational methods and integrated datasets to effectively combine the information gathered across multiple -omics technologies. Although there are many tools for single -omics analysis, limited tools currently exist for multi-omic analysis. In particular, strategies to integrate data from more than two -omics techniques are lacking.
One comprehensive database, called the Single Cell Atlas (SCA), provides a detailed multi-omics map of human tissues (Pan et al., 2024). This atlas includes data from 125 healthy adult and fetal tissues across eight -omics modalities, including single-cell RNA sequencing, ATAC sequencing, immune profiling, mass cytometry, flow cytometry, spatial transcriptomics, bulk RNA sequencing, and whole-genome sequencing. The Omics Discovery Index (OmicsDI) is another open-source platform that synthesizes -omics datasets, including proteomics, genomics, metabolomics, and transcriptomics (Perez-Riverol et al., 2017). Additional single-omics databases have agreed upon standardizing data structures to contribute to OmicsDI. One of the most notable disease-focused databases is The Cancer Genome Atlas (TCGA), a project launched by the National Institutes of Health to identify and record major cancer-causing genome alterations (Weinstein et al., 2013; Tomczak et al., 2015). TCGA incorporates genomic, epigenomic, transcriptomic, and proteomic data for tumors of over 30 different cancer types. Compared to single -omics databases, these collective databases provide extensive depth of information, though they are limited by smaller sample sizes due to the difficulty of acquiring and combining different datasets. Future efforts will focus on developing more integrative databases designed to meet the computational demands required to extract meaningful insights from multi-omics analysis.
3 Application of -omics technologies to characterize cellular diversity and cell-cell interactions
Cell diversity is essential for tissue structure and function. Advances in -omics technologies have revolutionized the ability to identify new cell types and uncover their specialized functions. These technologies also provide insight into cell-cell interactions, thereby enhancing our understanding of the roles of different cell subpopulations in tissue development, maintaining homeostasis, and responding to physiological demands and stressors (Armingol et al., 2021). Identifying new cell types and understanding their interactions are essential for deciphering the complex biological processes that underlie tissue development, regeneration, repair, and disease. Such knowledge can then be leveraged to develop next-generation therapies. For instance, insights into neuronal cell diversity have advanced knowledge of brain function and disease, while the discovery of novel immune cell subsets may enable future breakthroughs in immunotherapy (Mukamel and Ngai, 2019; Chen et al., 2020). In the following sections, we will describe how -omics technologies have been specifically employed to identify cell types and subtypes, as well as shed light on cell-cell interactions that underpin tissue structure and function.
3.1 Identification of cell types and subtypes
Advances in -omics technologies have enabled unprecedented insights into the identification of cell types and their functions. In particular, scRNA-seq has allowed for the analysis of gene expression at the single-cell level, thus revealing distinct transcriptomic profiles for different cell subpopulations. For example, researchers at Massachusetts General Hospital and the Broad Institute utilized scRNA-seq to characterize airway epithelial cells and discovered a novel cell type called the pulmonary ionocyte (Montoro et al., 2018). These cells were found to constitute approximately 1% of airway epithelial cells. Furthermore, they expressed the cystic fibrosis transmembrane conductance regulator gene (CFTR), which has been implicated in cystic fibrosis. Thus, the discovery of this cell type may re-direct therapeutic strategies toward targeting pulmonary ionocytes or their progenitors for cystic fibrosis treatment. In another study performed at Washington University in St. Louis, Lake et al. generated a cellular atlas for healthy and diseased kidneys, which revealed the presence of 51 human kidney cell types and the emergence of altered epithelial, stromal, and vascular cells in diseased kidneys (Lake et al., 2023). This dataset will allow for further investigation into the cell-cell signaling mechanisms that regulate kidney dysfunction.
Spatial transcriptomics has also been employed to identify new cell subpopulations. For example, scientists in Germany integrated spatial transcriptomics with scRNA-seq data to generate a spatial transcriptomics map of the embryonic mouse brain (Marco et al., 2023). The researchers identified six distinct neuronal populations and their spatial organization, which aligned with anatomical regions that have been defined for the embryonic mouse brain. Specifically, the different neuronal populations correspond to the lateral ganglionic eminence (LGE), subventricular zone (SVZ) and mantle zone, the septal areal, the preplate and cortical SVZ, the pial surface, the cortical ventricular zone (VZ), and the LGE VZ. To confirm the identities of the transcriptionally-defined clusters, the scientists cross-referenced their results with known regional and cell-type markers. They visualized gene expression both anatomically and within t-SNE clusters, confirming distinct spatial domains. For instance, Neurog2 and Sox3 were enriched in the cortical VZ, Ascl1 and Ddah1 marked the LGE VZ, and markers such as Neurod6 and Tiam2 highlighted the cortical SVZ, while Zic4 and Onecut2 were enriched in the septal region. Non-neural populations, including fibroblasts and endothelial cells, were identified in the pial surface by markers such as Col1a2 and Cldn5. Moving forward, data from this study can be used to provide spatial context for cell-cell interactions within the developing mouse brain. The ability to study neurogenesis through the lens of tissue organization and architecture will reveal unprecedented insight into the mechanisms that underlie neurodevelopment, and such knowledge can potentially be used to inform strategies for neural regeneration or therapies for neurodevelopmental disorders.
In addition to transcriptomics, multi-omics approaches have also been employed to interrogate cell type diversity within tissues. For example, a research group in Sweden employed multi-omics integration to investigate cellular heterogeneity in the human liver by using a combination of single-nucleus RNA sequencing, proteomics, and chromatin interaction profiling (Cavalli et al., 2020). By analyzing transcriptomic profiles at single-cell resolution alongside mass spectrometry-based proteomic data, the researchers were able to identify seven major liver cell populations, including hepatocytes, cholangiocytes, endothelial cells, Kupffer cells, and hepatic stellate cells, liver progenitor cells, and fibroblasts (Cavalli et al., 2020). Intriguingly, transcriptomic profiling allowed for the identification of four distinct subpopulations of hepatic stellate cells. The integration of transcriptomic and proteomic datasets revealed a modest correlation between RNA and protein expression levels. In addition to cell-type identification, the study incorporated data from HiCap, which is a method for mapping regulatory elements that interact with promoter regions by capturing and sequencing DNA fragments that are in close proximity within the 3D structure of the genome (Zhigulev and Sahlén, 2022). This approach provided an epigenomic perspective on gene regulation, by revealing connections between different regulatory elements. By integrating single-nuclei RNA-seq, proteomics, and HiCap data, the authors identified several enhancer regions for DPYD, which is a gene that encodes for the enzyme dihydropyrimidine dehydrogenase (DPD). DPD deficiency can result in increased chemotoxicity amongst cancer patients, and therefore understanding the causes of DPD deficiency can lead to strategies to alleviate treatment side effects. DYPD expression was associated with both hepatocyte and Kupffer cell subpopulations. The authors also identified several enhancers for SCL2A2, which encodes for a glucose transporter and has been used as a prognostic marker for hepatocellular carcinoma (HCC). SCL2A2 expression was primarily found in hepatocytes. Overexpression of SCL2A2 is associated with cancer cell proliferation and metabolism, and therefore identifying the regulatory elements that influence SCL2A2 expression can reveal biological mechanisms that facilitate HCC progression. Overall, the ability to combine transcriptomic, proteomic, and epigenomic data allowed for a more comprehensive understanding of liver cellular diversity and regulatory elements that influence gene expression in the liver, thereby offering insights into both normal cellular function and disease processes.
3.2 Cell-cell interaction networks
Understanding how cells communicate within tissues is essential for comprehending complex biological processes. Cell-cell communication involves interactions mediated by ligand-receptor pairs and extracellular matrix (ECM) components (Armingol et al., 2021). -Omics technologies have allowed for unprecedented insight into the signaling pathways that cells use to communicate across tissue development, repair, regeneration, and disease. In particular, scRNA-seq has revolutionized the analysis of cell-cell interactions by providing detailed information on ligand-receptor expression patterns, enabling the mapping and understanding of communication pathways between different cell types (Liu et al., 2022). Here, we highlight some of the tools that have been used to infer cell-cell interactions from scRNA-seq data and provide examples of the insights that have been obtained from these tools.
CellChat is a powerful tool that utilizes scRNA-seq data to predict how cells interact and influence each other’s behavior (Jin et al., 2021). This methodology has been instrumental in studying cell-cell interactions across various biological contexts, including immune response, tissue development, and cancer progression. CellChat leverages network-based inference, pattern recognition, and manifold learning to predict dominant cellular signaling inputs and outputs, enabling the classification of signaling pathways and the delineation of conserved versus context-specific communication patterns across diverse transcriptomic datasets. In a recent study investigating the progression of nonalcoholic fatty liver disease (NAFLD) to hepatocellular carcinoma (HCC), CellChat was applied to single-nuclei RNA sequencing data to characterize dynamic signaling networks within the liver microenvironment (Koelsch et al., 2023). The analysis revealed that structural cells, particularly fibroblasts and hepatocytes, served as dominant signal senders, while macrophages, monocytes, and endothelial cells were major recipients of these signals, especially during disease progression. Importantly, CellChat uncovered that signaling molecules such as TGF-β, IL-1β, and TNF-α exhibited context-dependent functions, targeting different cell populations at various disease stages. These shifts reflected a broader reorganization of immune communication, marked by a transition from adaptive to innate immune dominance. Moreover, the study highlighted that certain pro-tumorigenic functions and metabolic alterations were only detectable when considering the collective behavior of the immune network, opposed to analyzing individual cell types in isolation. Through this systems-level approach, CellChat enabled the identification of early signaling changes predictive of carcinogenesis, emphasizing its utility in uncovering emergent immune functions within the complex tissue environments. By understanding these interactions, targeted therapies can be developed to disrupt these communication networks and inhibit tumor progression.
CellCommuNet is a database for scRNA-seq data from human and mouse tissue, with samples that encompass both normal and diseased tissues. This resource aids researchers in deciphering complex cell-cell interactions within tissues, as well as identifying shifts in cell-cell communication that occur with disease (Ma et al., 2024). For example, CellCommuNet has been used to study interactions between macrophages and CD8+T cells in lung adenocarcinoma, revealing enhanced communication mediated by the SPP1-CD44 signaling axis. This interaction suggests a tumor-associated phenotype shift in macrophages, potentially contributing to immune suppression and poor prognosis. Additionally, analysis of clear cell renal cell carcinoma revealed increased cell-cell communication activity, especially involving endothelial cells. CellCommuNet also revealed that VEGF, FN1, and FGF pathways were upregulated in tumor-containing samples compared to healthy controls. These examples underscore CellCommuNet’s utility in uncovering context-specific and disease-relevant signaling dynamics across diverse tissues.
CellPhoneDB and NicheNet are other notable tools that utilize scRNA-seq data to predict cell-cell interactions. CellPhoneDB infers cell-cell communication by integrating a repository of ligands, receptors, and their interactions with scRNA-seq data. It utilizes a statistical framework to predict enriched ligand-receptor interactions between cell types based on gene expression levels and the subunit architecture of protein complexes (Efremova et al., 2020). In one example, CellPhoneDB has been used to explore the role of chemokine receptors in immune cell migration across various immune-mediated diseases (Rahman et al., 2024). The authors were able to identify 39 unique chemokine-based ligand-receptor pairs, many of which occurred only in diseased tissues. Additionally, 30 ligand-receptor pairs associated with immune cell extravasation were identified, and these occurred in both healthy and diseased tissues. Tissue-specific sender-receiver pairs were observed in healthy tissues, while both tissue-specific and generalized sender-receiver pairs were observed in diseased tissues.
NicheNet, on the other hand, predicts how ligands influence gene expression in receiver cells by integrating gene expression data with prior knowledge of ligand-receptor interactions, intracellular signaling pathways, and gene regulatory networks. This computational model uses weighted networks to calculate regulatory potential scores, thus identifying which ligands affect specific target genes (Browaeys et al., 2020). NicheNet was validated against scRNA-seq data from head-and-neck squamous cell carcinomas (Puram et al., 2017), and the results support the hypothesis that cancer-associated fibroblast (CAF) ligands drive a partial epithelial-to-mesenchymal transition program (p-EMT) in nearby malignant cells. NicheNet ranked TGFB3 as one of the strongest CAF ligands, and 18 of the top 20 predicted ligands were also known EMT regulators. Mechanistic tracing connected TGFB3 to downstream genes such as TGFBI, LAMC2, and TNC through SMAD3. These findings demonstrate how NicheNet can be used to uncover functional, ligand-driven regulatory mechanisms in the tumor microenvironment, offering insights into how stromal cells influence cancer progression and identifying potential targets for therapeutic intervention.
4 -Omics-guided design and analysis of engineered tissues
4.1 Design of engineered tissues
The field of tissue engineering can benefit from cellular and molecular insights that are obtained from -omics technologies. By revealing cell-cell and cell-matrix interactions that regulate tissue development, regeneration, and disease, data from -omics studies can inform the design of engineered tissues by highlighting cell types, biologics, and extracellular matrix components that are needed to recapitulate native tissue structure and function (Figure 2). Here, we highlight how -omics can be used to define relevant cell subpopulations for engineered tissues, as well as inform the incorporation of biologics and extracellular matrix components that recapitulate native cell-cell interactions.
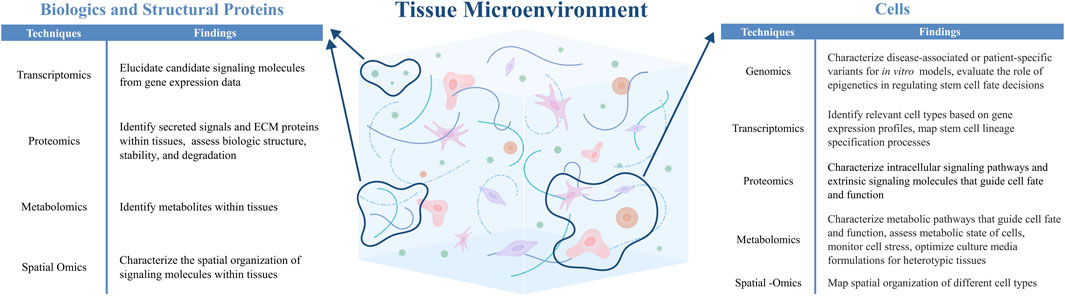
Figure 2. -Omics technologies can be applied to identify and characterize relevant cells, biologics, and structural ECM proteins within the native tissue microenvironment, thereby informing the design of engineered tissues (Hynes and Naba, 2012; Ragelle et al., 2017; Nguyen et al., 2019; Shaik et al., 2019; Bressan et al., 2023; Lammi and Qu, 2024).
4.1.1 Selection of cell types to incorporate into engineered tissues
Cells are key components in establishing the functionality of engineered tissues. Engineered tissues are designed to closely resemble their native tissue counterparts in both cellular composition and organization. Here, we highlight how -omics technologies can inform the cellular make-up of engineered tissues, as well as provide insight into mechanisms for guiding cell fate decisions and cell behavior towards desired functions.
4.1.1.1 Primary cells
A growing body of -omics research has reinforced the view that tissue function is an emergent property of coordinated interactions among multiple cell types, rather than isolated activity of any single population. These insights underscore the importance of cellular diversity, not only in identifying novel or rare subpopulations, but in revealing how established cell types interact to support complex physiological outcomes. Particularly for engineered tissues, the integration of multiple primary cell types, such as parenchymal, vascular, immune, and stromal cells, can be more practical and impactful than focusing solely on rare or transitional populations, which may be difficult to isolate, expand, or maintain in culture.
The selection of appropriate primary cells for incorporation into engineered tissues is increasingly guided by spatial and single-cell omics approaches, which provide high-resolution maps of intercellular communication. These technologies have illuminated how paracrine signaling, extracellular matrix remodeling, and direct cell-cell contact contribute to tissue-specific functions. For instance, in the liver, spatial transcriptomics and scRNA-seq has shown that sinusoidal endothelial cells provide signaling cues that direct proper liver development and zonation, as well as the emergence of functional Kupffer cells (MacParland et al., 2018; Hu et al., 2022; Kent et al., 2024). Engineered liver constructs that include only hepatocytes often fail to achieve mature function, but the addition of non-parenchymal components has been shown to enhance functional outputs such as albumin secretion and urea synthesis, demonstrating the value of recreating the native cellular ecosystem (Li et al., 2014; Stevens et al., 2017; Chhabra et al., 2022). Similar principles apply in cardiac tissue engineering. -Omics studies have revealed extensive crosstalk between cardiomyocytes, endothelial cells, and fibroblasts during development (Skelly et al., 2018; Wu et al., 2022; Hou et al., 2024). Accordingly, cardiomyocyte function is significantly improved when endothelial cells and fibroblasts are included in engineered tissue constructs, as cell-cell communication leads to better electrical conduction, mechanical performance, and resilience under stress (Caspi et al., 2007; Stevens et al., 2009).
4.1.1.2 Stem cells
While primary cells have been used to develop and optimize many engineered tissue platforms, limited availability of primary cell sources, and concerns regarding host-donor compatibility for in vivo applications, have sparked interest in developing alternative cell sourcing strategies. Here, stem cells are considered to be a promising cell source, as they are capable of indefinite expansion, can differentiate into multiple cell types, and can be sourced from the patient to generate personalized therapies (Teo and Vallier, 2010). However, controlling stem cell fate decisions to obtain the appropriate cell types within engineered tissues remains challenging. Advances in -omics technologies have transformed our ability to decode the molecular programs that govern stem cell self-renewal and differentiation, providing a systems-level framework to guide lineage specification. For example, transcriptomics and proteomics have been employed to track gene expression and transcription factor protein expression during lineage specification (Semrau et al., 2017; Palii et al., 2019; Lee et al., 2024). Metabolomics have also demonstrated that stem cells and their progenitors differ into terms of their metabolite composition, and that metabolic signaling can directly regulate stem cell self-renewal and quiescence (Schönberger et al., 2022). Finally, employing -omics technologies to characterize the stem cell niche can identify cell-extrinsic microenvironmental cues that regulate stem cell fate decisions (Kjell et al., 2020). Moving forward, these insights from -omics studies can be combined with cell engineering and synthetic biology tools to encode synthetic molecular programs within stem cells to directly regulate their fate decisions (Tewary et al., 2018). For example, synthetic gene circuits can be designed to control the timing and level of transcription factor activation to guide lineage-specific differentiation, or to directly modulate stem cell quiescence or proliferation. Additionally, incorporating epigenomic profiling, such as chromatin accessibility and histone modification analyses, can provide critical insights into the regulatory landscape of governing stem cell fate, thus enabling more precise engineering of cellular identity. In turn, -omics can be used to analyze engineered stem cells, in order to benchmark their phenotype against their native counterparts and assess the robustness of differentiation protocols (Simmons et al., 2024).
4.1.2 Mimicking cell-cell interactions with biologics and extracellular matrices
Cells communicate through the production and exchange of signaling molecules, which can take the form of growth factors, metabolites, and extracellular matrix proteins. While cells can directly communicate with each other through secreted, soluble factors or ligand-receptor pairs that remain tethered to the cell surface, cell-cell communication can also be achieved through interactions with the surrounding extracellular matrix (ECM) (Rosso et al., 2004). Cells dynamically remodel the ECM by depositing new components and degrading existing scaffolds, which can also regulate the availability of growth factors and other signaling molecules that are sequestered within the ECM. These changes to the ECM are then sensed by surrounding cell types, thereby modulating cellular processes such as migration, growth, and differentiation (Nguyen and D’amore, 2001). Thus, while the incorporation of multiple cell types into an engineered tissue represents one strategy to mimic native cell-cell interactions, a complementary approach involves the inclusion of biologics and ECM cues that represent the signaling molecules that are exchanged between cells in native tissues. By incorporating the correct growth factors, metabolites, or ECM proteins within engineered tissues, it may be possible to reduce the complexity of cell types that are needed to achieve a desired tissue phenotype or function. By analyzing cell–cell interactions in native tissues, -omics technologies can help researchers identify biologics and ECM proteins to design engineered tissues that replicate native cell-cell communication and influence cell function.
4.1.2.1 Identification of signaling molecules that recapitulate cell-cell communication
The identification of soluble growth factors and metabolites that are produced through cell-cell communication has been enabled by transcriptomics, proteomics, and metabolomics analyses. Analyzing the gene expression of related ligands and receptors, and their localization to different cell subpopulations, can reveal paracrine and juxtacrine mediators of cell-cell communication (Pavličev et al., 2017; Parikh et al., 2022). In addition, proteomics analysis has enabled the characterization of growth factors and the molecular mechanisms that underly their function and interaction with cellular receptors (Soskic et al., 1999; Blagoev et al., 2003; Tsai et al., 2008). These insights help to identify key domains within growth factor proteins that are responsible for receptor binding and downstream signaling. This knowledge can then inform the rational selection of ligands for incorporation into engineered tissues to mimic native cell-cell signaling, as well as the design of peptide motifs that enhance receptor engagement and signal transduction.
Proteomics has also played a key role in analyzing ECM proteins and ECM-sequestered signaling molecules. Here, high-resolution proteomic mapping of native tissues can identify essential matrix proteins such as collagens, laminins, fibronectins, and proteoglycans, along with ECM-associated proteins, regulators, and secreted factors (Hynes and Naba, 2012; Ragelle et al., 2017). In a study by Asthana et al., proteomic analysis using a combination of MS and multiplex ELISA enabled more detailed profiling of the human pancreatic proteome. The researchers identified proteins in pancreatic tissue that are often underrepresented, enabling the design of engineered tissues that incorporate these previously-unidentified components to create more physiologically-relevant scaffolds (Asthana et al., 2021). Ultimately, proteomic datasets can guide the design of biomaterials by informing the inclusion and organization of protein components that mimic the composition and structure of the native ECM. In addition to characterizing composition and structure, it is also important to understand how cells interact with their surrounding ECM. Here, proteomics and transcriptomics have been valuable in studying how different cell types bind to and remodel the ECM, and in identifying the downstream signaling pathways by which the ECM regulates cell behavior (Nguyen et al., 2019; Shaik et al., 2019). Collectively, this information can be used to engineer both cells and biomaterials to elicit appropriate cell-matrix interactions to guide tissue formation and function (Ragelle et al., 2017; Lammi and Qu, 2024).
4.1.2.2 Engineering tissues to mimic and modulate cell-cell communication
Over the past few decades, the field of tissue engineering has developed several strategies to mimic paracrine, juxtacrine, and ECM-mediated signaling within engineered tissues. With regards to paracrine signaling, many research groups have devised approaches to incorporate growth factors into engineered tissues to direct cell behavior towards tissue repair and regeneration (Lee et al., 2011). Efforts have focused on engineering the growth factor itself to enhance long-term stability and bioactivity (Jones et al., 2011). In parallel, innovations in biomaterials design have resulted in different strategies to control the temporal release of growth factors. These strategies include passive sorption of the growth factor onto the biomaterial backbone, covalent conjugation of the growth factor directly onto the biomaterial backbone, and sequestration strategies that leverage reversible electrostatic interactions to transiently bind and release growth factors (Teixeira et al., 2020).
In contrast to paracrine signaling, in which soluble signaling cues are exchanged between cells, juxtracrine signaling relies on direct contact between neighboring cells. To replicate juxtacrine signaling in engineered tissues, researchers have explored strategies to immobilize juxtracrine ligands into scaffolds (Deng et al., 2022). In particular covalent tethering chemistries offer strong, irreversible bonding between the ligand and scaffold (O’Grady et al., 2020). By tethering juxtacrine signals to scaffold backbones, we can mimic the spatial environments of native cell–cell contact and direct cellular responses.
To recapitulate ECM-mediated signals, efforts have focused on synthesizing biomaterials from natural or synthetic biopolymers that mimic the native ECM. Natural proteins that are commonly used in biomaterials include collagen, hyaluronic acid (HA), and fibrin. These components offer key advantages such as biocompatibility and biodegradability, tissue-specific biochemical cues, and enhanced cell adhesion, making them valuable for replicating aspects of the native ECM (Aazmi et al., 2024). Hybrid materials that utilize multiple biopolymers can also be designed to better mimic the heterogeneous composition of native ECM, and to enable better control over the microscale-properties of the materials. However, these biomaterial platforms often fail to capture the full complexity of the native ECM, resulting in growing interest in decellularization techniques, which aim to maximize retention of native ECM constituents. In recent years, analysis of decellularized tissues for different organs and disease states has gained significant popularity (Sackett et al., 2018; J. Xu et al., 2021; Yang et al., 2008). For example, one study employed liquid chromatography (LC)-MS/MS to analyze homogenized pericardium matrices (HPMs) and identify tissue-specific structural proteins present in native heart tissues that could be integrated into hybrid scaffolds as vascular tissue grafts (Bracaglia et al., 2017). The researchers hypothesized that the hybrid scaffold would offer both structural support and enhanced biochemical function, thereby improving cell adhesion, enabling controlled growth factor release and reducing inflammatory response. Ultimately, decellularized tissue models can offer a physiologically-relevant framework for studying cell–cell interactions within native-like tissue environments. Applying–omics techniques to characterize decellularized tissue platforms additionally enables a deeper understanding of how specific matrix components influence tissue form and function, thereby guiding the design of synthetic biomaterials to replicate key aspects of native ECMs and overcome translational challenges associated with animal-derived materials.
4.1.3 Mimicking spatial organization of native tissues
Tissue function arises not only from the presence of appropriate cell types, but also from the proper spatial organization of these cells. For instance, the liver arranges its hepatocytes in hexagonal lobules to streamline metabolism and filtration, while the brain places neurons and glial cells in precise layers to conduct complex signaling. For decades, it has been known that these spatial patterns are essential for regulating tissue function, but there was a lack of tools to mechanistically understand how spatial organization impacts cell behavior and higher-level tissue function. That has since changed with the emergence of spatial -omics technologies, which now allow for the mapping of individual genes, proteins, and metabolites expressed across intact tissues (Bressan et al., 2023). Spatial -omics data can be paired with biofabrication strategies to recreate the cellular organization and architecture of native tissues. For example, bioprinting is a technique that can build tissues in a layer-by-layer fashion with astounding precision. Much like an inkjet printer that deposits pigment on paper, bioprinters deposit cells, ECM components, and growth factors into 3D structures that mirror the architecture of native tissues. Guided by -omics data, bioprinted constructs can now recreate the zonation patterns of the liver, or the layered cellular organization of skin and cartilage (Madiedo-Podvrsan et al., 2021; Janani et al., 2022; Jorgensen et al., 2023).
Tissue-on-chip platforms offer another method to mimic the organization of native tissues. Here, microfluidic devices can be designed with different compartments to segregate different cell subpopulations (Adjei-Sowah et al., 2022; Humayun et al., 2022). Multiple cell types can be co-cultured and arranged according to spatial-omics maps to observe how the cells interact under physiologically-relevant conditions. Due to the advancements in both spatial mapping and microfabrication, these microfluidic platforms can now be engineered with precise spatial heterogeneity, which allows them to model complex tissue interfaces such as the blood-brain barrier (BBB) or intestinal lining (Adriani et al., 2017; De Hoyos-Vega et al., 2023; Kaiser et al., 2024). Ultimately, the ability to more accurately recapitulate native tissue architecture is necessary for engineering tissue constructs that not only reflect the composition and complexity of native tissues, but also support functional outcomes that mimic their physiological behavior.
4.1.4 Mimicking temporal and dynamic changes in engineered tissues
Tissues are inherently dynamic systems that evolve over time, through processes such as cell fate transitions, changes in ECM composition and stiffness, and the emergence of metabolic gradients within the tissue microenvironment. Capturing these temporal changes is critical for understanding and recapitulating tissue development and function. Longitudinal -omics technologies, which characterize samples that are obtained at different time points, offer a powerful means to dissect these dynamic biological processes with high resolution. In particular, time-resolved scRNA-seq has revealed how heterogeneous cell populations transition through lineage trajectories during development. For instance, Trapnell et al. used pseudotime analysis to model gene expression changes during myoblast differentiation, identifying regulatory cascades and transient states that inform cell fate decisions (Trapnell et al., 2014). Similarly, Weinreb et al. applied scRNA-seq with lineage tracing to hematopoiesis, uncovering how transcriptional landscapes evolve as progenitors commit to specific blood lineages (Weinreb et al., 2020). These insights into dynamic transcriptional changes that regulate cell states can inform the development of cell engineering strategies to directly control stem cell fate decisions in vitro.
To recapitulate time-dependent phenomena in engineered tissues, researchers are developing strategies that integrate temporal control over both cellular and material components. Cell engineering approaches that employ inducible circuits or tunable promoters can program time-dependent transcriptional responses to mimic natural gene expression dynamics (Tewary et al., 2018). Advances in materials science have enabled the development of dynamic biomaterials, such as hydrogels with tunable stiffness or degradation profiles that can evolve over time to reflect native ECM remodeling (Xie et al., 2023). Furthermore, hydrogels have been developed with oxygen-dependent chemistries to mimic hypoxic microenvironments (Lewis et al., 2017), while gradient materials can be developed to recapitulate spatial differences in biochemical and biophysical properties that recapitulate the heterogeneity of native tissues (Lowen and Leach, 2020; Ngo et al., 2021).
Beyond static environments, dynamic culture systems such as perfusion bioreactors and mechanically actuated platforms further improve the physiological-relevance of engineered tissues. For example, vascularized microfluidic platforms have been used to stimulate blood flow and nutrient delivery in engineered tissues, enabling more accurate modeling of tissue perfusion. -Omics approaches can be applied pre- and post-perfusion to assess how shear stress regulates vascular cell phenotypes, vascular inflammatory response, or ECM remodeling. Mechanical actuation is another critical cue in many tissues, and recent work has shown that phosphoproteomics can identify signaling pathways activated in response to cyclic strain. For instance, phosphoproteomics analysis revealed that dynamic actuation of engineered muscle grafts increases phosphorylation of proteins related to muscle contractility and repair, angiogenesis, and innervation (Rousseau et al., 2023) Together, these integrated -omics and dynamic culture strategies provide a powerful framework to guide the design and optimization of bioactive and functional tissue constructs.
4.2 Analysis and applications of engineered tissues
Comprehensive analysis of engineered tissues is essential for validating their structure and function, which can subsequently inform their physiological relevance and translational potential. -Omics tools enable the precise characterization of cell composition, cell-cell interactions, and resulting biological processes within engineered tissues. -Omics profiling can therefore be used to benchmark engineered tissues against their native counterparts, allowing for iterative tissue design to achieve desired phenotypes or functions. Once an engineered tissue construct has been validated, -omics can be used in conjunction with both in vivo and in vitro applications (Figure 3). Here, we will describe how -omics can be applied to evaluate the in vivo performance of engineered tissues for transplantation and regenerative medicine, gain new biological insights from in vitro tissue models, and perform pre-clinical therapeutic screening with engineered tissue testbeds.
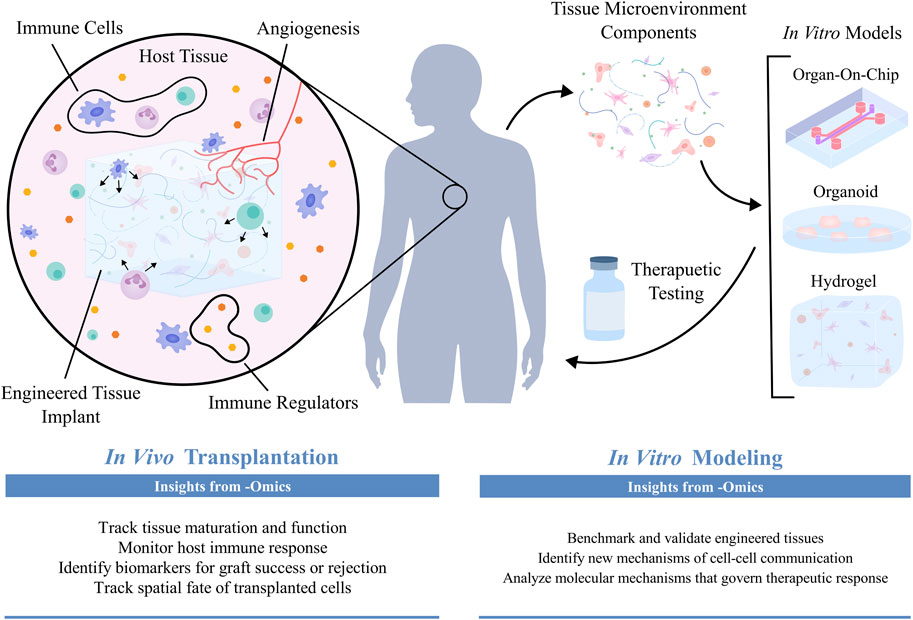
Figure 3. Once engineered tissues have been transplanted into in vivo environments, -omics technologies can be used to assess patient-transplant interactions and validate functional integration (Kumar et al., 2011; Malone and Humphreys, 2019; Balboa et al., 2022; Parikh et al., 2023; Sirolli et al., 2023; Geo et al., 2024). When modeling tissue microenvironments in vitro, -omics technologies are employed to evaluate how well engineered tissues recapitulate their native counterparts, thereby determining their physiological relevance as biomimetic models capable of uncovering novel biological insight and assessing patient-specific therapeutic responses (Finkbeiner et al., 2015; Lassé et al., 2023).
4.2.1 In vivo analysis of engineered issues
In vivo assessment of engineered tissues has been greatly enhanced by the application of -omics technologies, which offer detailed insights into how these constructs function and integrate within living systems. -Omics analysis can help to reveal how engineered tissues interact with surrounding host environments, adapt to physiological conditions, and respond to external stimuli. These approaches are also invaluable for characterizing host immune responses to engineered tissues, and for examining how engineered tissues influence or respond to host-pathogen dynamics.
4.2.1.1 Tissue transplantation: Donor-recipient compatibility and post-transplantation outcomes
Engineered tissues hold significant promise as transplantable therapies to replace damaged or failing tissues. A critical consideration for tissue transplantation is ensuring the compatibility between the patient and donor tissues. In recent years, genomics and transcriptomics have been employed to assess donor-patient compatibility by enabling the characterization of human leukocyte antigen (HLA) genes (Geo et al., 2024).
Beyond initial compatibility assessments, longitudinal monitoring of transplanted tissues is critical for evaluating whether the tissue can successfully integrate into the patient or becomes at risk for rejection. Transcriptomic profiling has been used to analyze gene expression profiles in transplanted tissues, offering insights into cell-cell interactions and signaling mechanisms involved in graft acceptance or rejection (Malone and Humphreys, 2019). Such insights may lead to the development of therapies that can shift cell-cell signaling towards mechanisms that promote transplant acceptance and integration. Proteomics and metabolomics offer additional tools for monitoring immune responses to the transplanted tissue over time. By monitoring fluctuations in protein expression and metabolic profiles, these technologies can provide early warnings of immune activation. For instance, proteomic analysis can detect the upregulation of inflammatory cytokines, chemokines, and other immune mediators, which may be indicative of transplant rejection (Kumar et al., 2011; Sirolli et al., 2023). Simultaneously, metabolomics can identify shifts in key metabolic pathways, such as glycolysis or oxidative stress markers, that may signal aberrant immune activity (Reikvam et al., 2018). These changes typically precede symptom arrival and can therefore act as diagnostic biomarkers that give physicians a critical window for intervention. Thus, the use of -omics can enable timely adjustments to immunosuppressive therapies that maintain immune tolerance while minimizing the side effects associated with generalized immunosuppression.
In addition to interactions between the patient’s immune system and the transplanted tissue, graft performance can also be influenced by pathogens or host microbiota. -Omics technologies provide a molecular-level view of how engineered tissues interact with microbial communities, enabling targeted strategies to improve integration and reduce complications. For instance, proteomic and metabolomic profiling can uncover how pathogens disrupt host defenses or alter tissue metabolism, which are factors that can affect immunocompromised or post-transplant patients (Jean Beltran et al., 2017; Khan et al., 2019; Lehmann et al., 2024). These findings inform the design of anti-microbial materials or pre-conditioning regimens that protect engineered tissues from infection-induced failure. Similarly, the host microbiome plays a critical role in shaping immune response and tissue homeostasis. Metabolomic analyses have shown how microbiome-derived metabolites influence systemic inflammation, immune tolerance, and even wound healing (Holmes et al., 2015; Levy et al., 2017). In tissue engineering, this data can be used to predict graft outcomes and design interventions that support tissue integration. For example, modulating the microbiome through probiotics or tailored diets could enhance compatibility between engineered constructs and host tissues, especially in gut, skin, or mucosal applications. Integrating these omics-driven insights into tissue engineering pipelines aids in creating more resilient, adaptive, and clinically-effective therapies.
4.2.1.2 Validation of engineered tissue performance post-transplantation
Once an engineered tissue is transplanted, it must survive, integrate with surrounding tissue, function, and respond to the physiological demands of its new environment. -Omics technologies allow for molecular-level analyses of engineered tissues after transplantation. Transcriptomics and proteomics can benchmark the maturation of engineered tissues in vivo, which can be characterized through the expression of mature cell markers, proteins, and signaling pathways that are relevant for tissue-specific function (Balboa et al., 2022; Parikh et al., 2023). For example, in engineered liver tissues, -omics data can confirm whether key metabolic enzymes are expressed (Stevens et al., 2017). Metabolomics can be used to profile metabolites that serve as indicators of tissue function or graft recovery after ischemia-reperfusion injury (Wishart, 2005). Finally, -omics techniques can be employed for longitudinal monitoring, which enables dynamic tracking of graft health and can capture early signs of graft failure due to aging, metabolic stress, or overuse, even in the absence of overt immune rejection (Vasaikar et al., 2023). By providing insight on graft performance, -omics technologies enable continual refinement of engineered tissue design for effective transplantation outcomes.
4.2.2 Biological insights from in vitro engineered tissue platforms
While the field of tissue engineering has traditionally focused on translational applications, many research groups are now employing tissue engineering design principles to generate versatile in vitro models of both healthy and diseased tissues. In vitro models provide a unique advantage by capturing cell and tissue functions under controlled and reproducible conditions, which has been critical for understanding biological phenomena that are often difficult to study native contexts. Such models can be employed to uncover new biological mechanisms underlying development, regeneration, and disease, or may be applied as testbeds to screen new therapies or develop personalized medicine protocols. -Omics can not only inform the design of in vitro models but also enable the characterization of cell-cell interactions within these platforms. Here, we highlight how the integration of -omics and in vitro models have provided new insight into tissue development and disease, and the application of these technologies for therapeutic screening.
4.2.2.1 Developmental biology
Engineered tissues have significantly advanced our understanding of developmental biology, particularly through the use of organoids. Organoids have been traditionally defined as three-dimensional in vitro cultures derived from primary tissues or stem cells and are designed to mimic the self-assembly of tissues during embryonic development (Fatehullah et al., 2016). The first organoid cultures were established by encapsulating primary tissues or stem cells in Matrigel, and culturing the cells in defined media conditions to mimic morphogenetic signaling. In recent years, however, tissue engineering tools such as synthetic biomaterials and biofabrication have been incorporated into organoid protocols to better mimic the spatial organization of native tissues (Garreta et al., 2021). Furthermore, establishing organoids that incorporate stromal, vascular, and immune cell types along with parenchymal cells are an area of ongoing innovation (Takebe and Wells, 2019).
-Omics analyses can be employed to benchmark organoid cultures against native tissues, and to gain insight into new biological mechanisms that regulate tissue development. For example, transcriptomics has been employed to characterize the developmental age of organoid cultures. Finkbeiner et al. used RNA-seq to compare global expression profiles between human intestinal organoids (HIOs), fetal intestinal tissues, and adult intestinal tissues (Finkbeiner et al., 2015). They found that the organoids more closely resembled fetal tissues, and that both the HIOs and fetal tissues had low expression of genes associated with digestive function and host-defense functions. However, once transplanted into an in vivo environment, the HIOs developed adult-like characteristics, suggesting that the in vivo microenvironment contains specific maturation cues that direct intestinal development. Single-cell transcriptomics have also played a key role in identifying cell subpopulations that emerge during organoid culture, along with their corresponding developmental trajectories. In a recent study, scRNA-seq was used to characterize the development of ventral midbrain organoids (Fiorenzano et al., 2021). Here, the authors were able to trace the emergence of different cell populations (e.g., vascular leptomeningeal cells, astrocytes, oligodendrocyte progenitors) over time, as well as identify different dopaminergic neuron subpopulations at different levels of maturity.
In addition to transcriptomics, proteomics analyses can be employed to profile cell type markers, deposited growth factors and extracellular matrix proteins, metabolites, and lipids that are expressed throughout organoid development (Nezvedová et al., 2023). Furthermore, genomic screens have been applied to organoids to understand how individual genes influence tissue development, which not only enhances our biological understanding but also can provide opportunities to refine organoid culture protocols. In one study, CRISPR-based loss-of-function screening resulted in the identification of genes and signaling pathways that are important for kidney development (Ungricht et al., 2022). In particular, BMP, Wnt, Notch, and Sonic Hedgehog signaling were all implicated in early kidney development, and inhibition of ROCK signaling was also found to improve mesoderm induction for the generation of nephron progenitor cells. Taken together, -omics tools will continue to be invaluable for characterizing organoid cultures and deriving biological insight from these model systems.
4.2.2.2 Disease mechanisms
In vitro platforms can be used to model diseases such as cancer and neurological disorders, and to specifically investigate the contributions of cell-cell communication in regulating disease progression. In vitro disease models can be assembled in a bottoms-up fashion, by co-culturing relevant cell types within ECM-mimetic biomaterials or microfabricated platforms. Alternatively, organoids have also been used to model disease; here, diseased tissue can be isolated and cultured, or stem cells can be genetically-edited to mimic mutations that have been implicated in a given disease.
Researchers have turned towards -omics technologies for benchmarking in vitro disease models against native tissues, and for uncovering mechanisms of cell–cell communication in disease contexts. For instance, transcriptomic profiling has been used to compare gene expression patterns between disease-mimetic organoids and patient-derived tissues, helping to validate whether the in vitro models faithfully recapitulate key pathological features. One study by Lassé et al. developed a kidney organoid as a model for TNFα-associated kidney disease (Lassé et al., 2023). Using proteomics and transcriptomics, the authors characterized the organoids at different culture durations, which revealed changes in cellular composition and ECM deposition over time. Upon stimulation with TNFα, the organoids expressed pro-inflammatory markers and increased their secretion of cytokines, cell adhesion proteins, and apoptotic regulators. Transcriptomic signatures that were observed with TNFα stimulation were subsequently confirmed in tissue specimens from kidney disease patients, thereby supporting the physiological relevance of the organoid platform. -Omics techniques can therefore be used in engineered platforms to reveal whether disease-relevant pathways are appropriately activated in the model system.
Once an in vitro platform has been validated, -omics can subsequently be applied to elucidate cell-cell signaling mechanisms that contribute to disease phenotype and progression (Ngo and Harley, 2019; Truong et al., 2019; Ewoldt et al., 2024). Technologies such as scRNA-seq have been particularly useful for the analysis of in vitro platforms. Since cell identities can be inferred from gene expression profiles, the use of scRNA-seq can eliminate the need to physically isolate disparate cell subpopulations from in vitro cultures for downstream analysis. In a recent study, Adjei-Sowah et al. employed scRNA-seq to analyze an in vitro model of the glioblastoma perivascular niche (Adjei-Sowah et al., 2022). Based on marker genes, the authors were able to distinguish endothelial cell, astrocyte, and glioma stem cell subpopulations. Further analysis of copy number variants allowed for additional subtyping of the glioma stem cell population. By characterizing ligand-receptor expression across different cell subpopulations, the authors identified angiocrine SAA1-FPR1 signaling as a novel mediator of glioblastoma invasion. This finding was experimentally verified in their in vitro platform, demonstrating how -omics and engineered tissue models can be used in tandem to derive and test new biological hypotheses.
In addition to scRNA-seq, phosphoproteomics is a powerful tool for identifying dysregulated signaling pathways that are associated with disease states. Within the past decade, phosphoproteomics has been increasingly employed to reveal abnormal phosphorylation-mediated signaling networks that are associated with dysregulated cellular function and disease progression. For example, a recent study identified Rab GTPases as direct substrates of the kinase LRRK2, which is activated in Parkinson’s disease due to mutations in the gene Park8. Phosphorylation of Rab GTPases results in dysregulated intracellular trafficking of Rab and impacts its interactions with regulatory proteins, which may contribute to the development of Parkinson’s disease (Steger et al., 2016). This work highlights how phosphoproteomics can identify precise molecular events, such as kinase-substrate interactions, that contribute to complex disease phenotypes and inform therapeutic strategies. Overall, integrating insights gained from phosphoproteomics with engineered tissue platforms can not only inform design of in vitro models that capture dysregulated cellular communication relevant to different pathologies, but also result in the identification of new signaling mechanisms that are dysregulated during disease progression.
4.2.2.3 In vitro models as therapeutic testbeds
In vitro platforms can not only serve as biological models for basic science, but also as testbeds for preclinical drug evaluation and precision medicine. Advancements in constructing three-dimensional culture systems have significantly enhanced the drug discovery process by offering models that better mimic in vivo environments, thereby improving the accuracy of drug candidate screening compared to traditional 2D tissue cultures (Ingber, 2022). Designing in vitro systems to evaluate therapeutic responses requires careful consideration of how drugs are absorbed and distributed within host tissues. To address this issue, microfluidic technologies have been employed to generate organ-on-a-chip platforms that simulate the structure and function of human organs. These three-dimensional models offer robust tunability, allowing researchers to replicate a wide range of complex organ systems in a reliable and reproducible manner (Shirure et al., 2018). To further enhance the physiological relevance of these models, transport across biological barriers—such as the blood-brain barrier and intestinal epithelium—also need to be modeled (van der Helm et al., 2016; Yu et al., 2018; Arik et al., 2021). By characterizing these barriers and incorporating them into in vitro models, researchers can better assess drug transport into and within tissues. One recent study developed a placenta-on-chip model, offering a new way to study drug transport from mother to fetus and assess safety profiles of drugs administered to pregnant patients (Blundell et al., 2018). Many studies have also focused on modeling the blood–brain barrier, due to its role in limiting drug delivery to the central nervous system (Partyka et al., 2017; Park et al., 2019; Lee et al., 2020; Neumaier et al., 2021). In addition to mimicking individual organs, body-on-chip platforms that link multiple organ chips together have also been developed to model systemic drug transport within the human body (Ronaldson-Bouchard et al., 2022). Due to renewed interest in replacing animal models within the drug development pipeline (Zushin et al., 2023), it is anticipated that continued innovations in fabricating in vitro models will advance the ability for engineered tissue testbeds to accurately recapitulate drug pharmacokinetics and pharmacodynamics.
In vitro testbeds can be developed with patient-derived cells to act as personalized models of disease. When coupled with -omics, these patient-specific platforms can be used to model and study individualized drug responses (Soldatow et al., 2013; Tsamandouras et al., 2017; Vlachogiannis et al., 2018). Genomics has become popular in the field of pharmacogenetics to study how genetic variations among patients affect the metabolism and efficacy of drug treatments (Ma and Lu, 2011). In the past, studies focusing on drug-metabolizing gene variants have accurately predicted patient responses to therapeutics and identified adverse reactions (Pinto and Eileen Dolan, 2011). This approach has revealed key genetic markers that govern differential drug responses, paving the way for more precise and personalized therapeutic interventions. In the future, continued integration of -omics technologies with patient-specific tissue models will enable an in-depth understanding of the molecular mechanisms that govern drug response, allowing for improvements to therapeutic regimens and real-time refinement of treatment protocols.
5 Discussion
-Omics technologies have profoundly transformed the landscape of tissue engineering by enabling a comprehensive understanding of cell diversity and cell-cell communication across tissue development, regeneration, and disease. Insights gleaned from -omics studies have been used to inform the design of engineered tissues, both for fundamental science and translational applications. In turn, -omics can be used to provide molecular-level characterization of engineered tissues, in order to benchmark their composition, structure, and function against their native counterparts. In recent years, -omics technologies have been applied in particular to the design and utilization of in vitro tissue models, in order to validate their physiological relevance and derive new biological insight.
Moving forward, the advent of single-cell and spatial -omics technologies promises to provide even greater insight into tissue organization and cellular heterogeneity. Coupled with innovations in biofabrication, cell engineering, and mammalian synthetic biology, it is expected that single-cell and spatial insights will further enable the fabrication of engineered tissue platforms with precisely-defined architecture and cell composition, thereby moving the field of tissue engineering closer to the development of functional artificial tissues and organs. Integration of -omics technologies with pre-clinical transplantation studies will allow for improved refinement of regenerative tissue therapies, by providing valuable molecular insight into the processes that govern tissue integration, patient-tissue interactions, and tissue maturation and function post-transplantation. Finally, the integration of artificial intelligence and machine learning tools with multi-omics data holds great potential for uncovering new biological mechanisms by which cell-cell communication governs tissue development, function, and dysregulation. Such insights will further inform the development and analysis of engineered tissues as biological models, regenerative therapies, and therapeutic testbeds.
Author contributions
CS: Conceptualization, Writing – original draft, Writing – review and editing. SJ: Conceptualization, Writing – original draft, Writing – review and editing. MN: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Funding for this manuscript was generously provided by the Department of Chemical and Biological Engineering and the College of Engineering at the University of Wisconsin-Madison, as well as the Wisconsin Alumni Research Foundation.
Acknowledgments
The authors would like to acknowledge Tiana Bish for helpful discussions regarding the manuscript. Inkscape was used to generate all figures.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aazmi, A., Zhang, D., Mazzaglia, C., Yu, M., Wang, Z., Yang, H., et al. (2024). Biofabrication methods for reconstructing extracellular matrix mimetics. Bioact. Mater 31, 475–496. doi:10.1016/j.bioactmat.2023.08.018
Adjei-Sowah, E. A., O’Connor, S. A., Veldhuizen, J., Lo Cascio, C., Plaisier, C., Mehta, S., et al. (2022). Investigating the interactions of glioma stem cells in the perivascular niche at single-cell resolution using a microfluidic tumor microenvironment model. Adv. Sci. 9, 2201436. doi:10.1002/advs.202201436
Adriani, G., Ma, D., Pavesi, A., Kamm, R. D., and Goh, E. L. K. (2017). A 3D neurovascular microfluidic model consisting of neurons, astrocytes and cerebral endothelial cells as a blood–brain barrier. Lab. Chip 17, 448–459. doi:10.1039/C6LC00638H
Al-Amrani, S., Al-Jabri, Z., Al-Zaabi, A., Alshekaili, J., and Al-Khabori, M. (2021). Proteomics: concepts and applications in human medicine. World J. Biol. Chem. 12, 57–69. doi:10.4331/wjbc.v12.i5.57
Alexandrov, T. (2023). Spatial metabolomics: from a niche field towards a driver of innovation. Nat. Metab. 5, 1443–1445. doi:10.1038/s42255-023-00881-0
Altelaar, A. F. M., Munoz, J., and Heck, A. J. R. (2013). Next-generation proteomics: towards an integrative view of proteome dynamics. Nat. Rev. Genet. 14, 35–48. doi:10.1038/nrg3356
Andersen, J. S., Wilkinson, C. J., Mayor, T., Mortensen, P., Nigg, E. A., and Mann, M. (2003). Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426, 570–574. doi:10.1038/nature02166
Argelaguet, R., Velten, B., Arnol, D., Dietrich, S., Zenz, T., Marioni, J. C., et al. (2018). Multi-omics Factor Analysis—A framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 14, e8124. doi:10.15252/msb.20178124
Arik, Y. B., Buijsman, W., Loessberg-Zahl, J., Cuartas-Vélez, C., Veenstra, C., Logtenberg, S., et al. (2021). Microfluidic organ-on-a-chip model of the outer blood-retinal barrier with clinically relevant read-outs for tissue permeability and vascular structure. Lab. Chip 21, 272–283. doi:10.1039/d0lc00639d
Armingol, E., Officer, A., Harismendy, O., and Lewis, N. E. (2021). Deciphering cell–cell interactions and communication from gene expression. Nat. Rev. Genet. 22, 71–88. doi:10.1038/s41576-020-00292-x
Asthana, A., Tamburrini, R., Chaimov, D., Gazia, C., Walker, S. J., Van Dyke, M., et al. (2021). Comprehensive characterization of the human pancreatic proteome for bioengineering applications. Biomaterials 270, 120613. doi:10.1016/j.biomaterials.2020.120613
Atkinson, S. R., Marguerat, S., and Bähler, J. (2012). Exploring long non-coding RNAs through sequencing. Semin. Cell. Dev. Biol. 23, 200–205. doi:10.1016/j.semcdb.2011.12.003
Bai, B., Wang, X., Li, Y., Chen, P. C., Yu, K., Dey, K. K., et al. (2020). Deep Multilayer brain proteomics identifies molecular networks in alzheimer’s disease progression. Neuron 105, 975–991.e7. doi:10.1016/j.neuron.2019.12.015
Balboa, D., Barsby, T., Lithovius, V., Saarimäki-Vire, J., Omar-Hmeadi, M., Dyachok, O., et al. (2022). Functional, metabolic and transcriptional maturation of human pancreatic islets derived from stem cells. Nat. Biotechnol. 40, 1042–1055. doi:10.1038/s41587-022-01219-z
Bartosovic, M., Kabbe, M., and Castelo-Branco, G. (2021). Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat. Biotechnol. 39, 825–835. doi:10.1038/s41587-021-00869-9
Baysoy, A., Bai, Z., Satija, R., and Fan, R. (2023). The technological landscape and applications of single-cell multi-omics. Nat. Rev. Mol. Cell. Biol. 24, 695–713. doi:10.1038/s41580-023-00615-w
Belkadi, A., Bolze, A., Itan, Y., Cobat, A., Vincent, Q. B., Antipenko, A., et al. (2015). Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. U. S. A. 112, 5473–5478. doi:10.1073/pnas.1418631112
Bewicke-Copley, F., Arjun Kumar, E., Palladino, G., Korfi, K., and Wang, J. (2019). Applications and analysis of targeted genomic sequencing in cancer studies. Comput. Struct. Biotechnol. J. 17, 1348–1359. doi:10.1016/j.csbj.2019.10.004
Blagoev, B., Kratchmarova, I., Ong, S.-E., Nielsen, M., Foster, L. J., and Mann, M. (2003). A proteomics strategy to elucidate functional protein-protein interactions applied to EGF signaling. Nat. Biotechnol. 21, 315–318. doi:10.1038/nbt790
Blundell, C., Yi, Y. S., Ma, L., Tess, E. R., Farrell, M. J., Georgescu, A., et al. (2018). Placental Drug Transport-on-a-Chip: a microengineered in vitro model of transporter-mediated Drug efflux in the human placental barrier. Adv. Healthc. Mater 7, 1700786. doi:10.1002/adhm.201700786
Bonev, B., and Cavalli, G. (2016). Organization and function of the 3D genome. Nat. Rev. Genet. 17, 661–678. doi:10.1038/nrg.2016.112
Bracaglia, L. G., Messina, M., Winston, S., Kuo, C. Y., Lerman, M., and Fisher, J. P. (2017). 3D printed pericardium hydrogels to promote wound healing in vascular applications. Biomacromolecules 18, 3802–3811. doi:10.1021/acs.biomac.7b01165
Bressan, D., Battistoni, G., and Hannon, G. J. (2023). The dawn of spatial omics. Science 1979, eabq4964. doi:10.1126/science.abq4964
Browaeys, R., Saelens, W., and Saeys, Y. (2020). NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162. doi:10.1038/s41592-019-0667-5
Buchberger, A. R., DeLaney, K., Johnson, J., and Li, L. (2018). Mass spectrometry imaging: a review of emerging advancements and future insights. Anal. Chem. 90, 240–265. doi:10.1021/acs.analchem.7b04733
Cao, J., Spielmann, M., Qiu, X., Huang, X., Ibrahim, D. M., Hill, A. J., et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. doi:10.1038/s41586-019-0969-x
Carlsson, A. C., Ingelsson, E., Sundström, J., Carrero, J. J., Gustafsson, S., Feldreich, T., et al. (2017). Use of proteomics to investigate kidney function decline over 5 years. Clin. J. Am. Soc. Nephrol. 12, 1226–1235. doi:10.2215/CJN.08780816
Caspi, O., Lesman, A., Basevitch, Y., Gepstein, A., Arbel, G., Habib, I. H. M., et al. (2007). Tissue engineering of vascularized cardiac muscle from Human embryonic stem cells. Circ. Res. 100, 263–272. doi:10.1161/01.RES.0000257776.05673.ff
Cavalli, M., Diamanti, K., Pan, G., Spalinskas, R., Kumar, C., Deshmukh, A. S., et al. (2020). A multi-omics approach to liver diseases: integration of single nuclei transcriptomics with proteomics and HiCap bulk data in human liver, OMICS 24. 180, 194. doi:10.1089/omi.2019.0215
Chandramouli, K., and Qian, P.-Y. (2009). Proteomics: challenges, techniques and possibilities to overcome biological sample complexity. Hum. Genomics Proteomics 1, 239204. doi:10.4061/2009/239204
Chen, Y.-P., Yin, J.-H., Li, W.-F., Li, H.-J., Chen, D.-P., Zhang, C.-J., et al. (2020). Single-cell transcriptomics reveals regulators underlying immune cell diversity and immune subtypes associated with prognosis in nasopharyngeal carcinoma. Cell. Res. 30, 1024–1042. doi:10.1038/s41422-020-0374-x
Chen, J. W., Shrestha, L., Green, G., Leier, A., and Marquez-Lago, T. T. (2023). The hitchhikers’ guide to RNA sequencing and functional analysis. Brief. Bioinform 24, bbac529. doi:10.1093/bib/bbac529
Chhabra, A., Song, H.-H. G., Grzelak, K. A., Polacheck, W. J., Fleming, H. E., Chen, C. S., et al. (2022). A vascularized model of the human liver mimics regenerative responses. Proc. Natl. Acad. Sci. 119, e2115867119. doi:10.1073/pnas.2115867119
Choi, M., Chang, C. Y., Clough, T., Broudy, D., Killeen, T., MacLean, B., et al. (2014). MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526. doi:10.1093/bioinformatics/btu305
Coy, S., Wang, S., Stopka, S. A., Lin, J. R., Yapp, C., Ritch, C. C., et al. (2022). Single cell spatial analysis reveals the topology of immunomodulatory purinergic signaling in glioblastoma. Nat. Commun. 13, 4814. doi:10.1038/s41467-022-32430-w
Croft, D., O’Kelly, G., Wu, G., Haw, R., Gillespie, M., Matthews, L., et al. (2011). Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 39, D691–D697. doi:10.1093/nar/gkq1018
Cui, M., Cheng, C., and Zhang, L. (2022). High-throughput proteomics: a methodological mini-review, Lab. Investig. 102. 1170, 1181. doi:10.1038/s41374-022-00830-7
Dai, X., and Shen, L. (2022). Advances and trends in omics technology development. Front. Med. (Lausanne) 9, 911861. doi:10.3389/fmed.2022.911861
de Hoyos-Vega, J. M., Yu, X., Gonzalez-Suarez, A. M., Chen, S., Mercado-Perez, A., Krueger, E., et al. (2023). Modeling gut neuro-epithelial connections in a novel microfluidic device. Microsyst. Nanoeng. 9, 144. doi:10.1038/s41378-023-00615-y
Dekker, J., Rippe, K., Dekker, M., and Kleckner, N. (2002). Capturing chromosome conformation. Science 1979, 1306–1311. doi:10.1126/science.1067799
Deng, Y., Li, R., Wang, H., Yang, B., Shi, P., Zhang, Y., et al. (2022). Biomaterial-Mediated presentation of Jagged-1 mimetic ligand enhances cellular activation of notch signaling and bone regeneration. ACS Nano 16, 1051–1062. doi:10.1021/acsnano.1c08728
Dixon, J. R., Jung, I., Selvaraj, S., Shen, Y., Antosiewicz-Bourget, J. E., Lee, A. Y., et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. doi:10.1038/nature14222
Dobrindt, K., Hoagland, D. A., Seah, C., Kassim, B., O’Shea, C. P., Murphy, A., et al. (2021). Common genetic variation in humans impacts in vitro susceptibility to SARS-CoV-2 infection. Stem Cell. Rep. 16, 505–518. doi:10.1016/j.stemcr.2021.02.010
Domon, B., and Aebersold, R. (2006). Challenges and opportunities in proteomics data analysis. Mol. and Cell. Proteomics 5, 1921–1926. doi:10.1074/mcp.R600012-MCP200
Dunn, W. B., Broadhurst, D. I., Deepak, S. M., Buch, M. H., McDowell, G., Spasic, I., et al. (2007). Serum metabolomics reveals many novel metabolic markers of heart failure, including pseudouridine and 2-oxoglutarate. Metabolomics 3, 413–426. doi:10.1007/s11306-007-0063-5
Dunn, W. B., Broadhurst, D. I., Atherton, H. J., Goodacre, R., and Griffin, J. L. (2011). Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem. Soc. Rev. 40, 387–426. doi:10.1039/b906712b
Efremova, M., Vento-Tormo, M., Teichmann, S. A., and Vento-Tormo, R. (2020). CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 15, 1484–1506. doi:10.1038/s41596-020-0292-x
Ewoldt, J. K., Wang, M. C., McLellan, M. A., Cloonan, P. E., Chopra, A., Gorham, J., et al. (2024). Hypertrophic cardiomyopathy–associated mutations drive stromal activation via EGFR-mediated paracrine signaling. Sci. Adv. 10, eadi6927. doi:10.1126/sciadv.adi6927
Fabrizio, C., Termine, A., and Caltagirone, C. (2025). Transcriptomics profiling of Parkinson’s disease progression subtypes reveals distinctive patterns of gene expression. J. Cent. Nerv. Syst. Dis. 17, 11795735241286821. doi:10.1177/11795735241286821
Fatehullah, A., Tan, S. H., and Barker, N. (2016). Organoids as an in vitro model of human development and disease. Nat. Cell. Biol. 18, 246–254. doi:10.1038/ncb3312
Finkbeiner, S. R., Hill, D. R., Altheim, C. H., Dedhia, P. H., Taylor, M. J., Tsai, Y. H., et al. (2015). Transcriptome-wide analysis reveals hallmarks of Human Intestine development and maturation in vitro and in vivo. Stem Cell. Rep. 4, 1140–1155. doi:10.1016/j.stemcr.2015.04.010
Fiorenzano, A., Sozzi, E., Birtele, M., Kajtez, J., Giacomoni, J., Nilsson, F., et al. (2021). Single-cell transcriptomics captures features of human midbrain development and dopamine neuron diversity in brain organoids. Nat. Commun. 12, 7302. doi:10.1038/s41467-021-27464-5
Floegel, A., Stefan, N., Yu, Z., Mühlenbruch, K., Drogan, D., Joost, H. G., et al. (2013). Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes 62, 639–648. doi:10.2337/db12-0495
Garcia-Alonso, L., Handfield, L. F., Roberts, K., Nikolakopoulou, K., Fernando, R. C., Gardner, L., et al. (2021). Mapping the temporal and spatial dynamics of the human endometrium in vivo and in vitro. Nat. Genet. 53, 1698–1711. doi:10.1038/s41588-021-00972-2
Garreta, E., Kamm, R. D., Chuva de Sousa Lopes, S. M., Lancaster, M. A., Weiss, R., Trepat, X., et al. (2021). Rethinking organoid technology through bioengineering. Nat. Mater 20, 145–155. doi:10.1038/s41563-020-00804-4
Geo, J. A., Ameen, R., Al Shemmari, S., and Thomas, J. (2024). Advancements in HLA typing techniques and their impact on transplantation medicine. Med. Princ. Pract. 33, 215–231. doi:10.1159/000538176
Gerritsen, J. S., and White, F. M. (2021). Phosphoproteomics: a valuable tool for uncovering molecular signaling in cancer cells. Expert Rev. Proteomics 18, 661–674. doi:10.1080/14789450.2021.1976152
Glish, G. L., and Vachet, R. W. (2003). The basics of mass spectrometry in the twenty-first century. Nat. Rev. Drug Discov. 2, 140–150. doi:10.1038/nrd1011
Gloss, B. S., and Dinger, M. E. (2018). Realizing the significance of noncoding functionality in clinical genomics. Exp. Mol. Med. 50, 1–8. doi:10.1038/s12276-018-0087-0
Gulati, G. S., D’Silva, J. P., Liu, Y., Wang, L., and Newman, A. M. (2025). Profiling cell identity and tissue architecture with single-cell and spatial transcriptomics. Nat. Rev. Mol. Cell. Biol. 26, 11–31. doi:10.1038/s41580-024-00768-2
Haag, A. M. (2016). Mass analyzers and mass spectrometers. Adv. Exp. Med. Biol. 919, 157–169. doi:10.1007/978-3-319-41448-5_7
Habib, N., Li, Y., Heidenreich, M., Swiech, L., Avraham-Davidi, I., Trombetta, J. J., et al. (2016). Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science 1979, 925–928. doi:10.1126/science.aad7038
Han, X., Aslanian, A., and Yates, J. R. (2008). Mass spectrometry for proteomics. Curr. Opin. Chem. Biol. 12, 483–490. doi:10.1016/j.cbpa.2008.07.024
Han, J., Zhang, Z., and Wang, K. (2018). 3C and 3C-based techniques: the powerful tools for spatial genome organization deciphering. Mol. Cytogenet 11, 21. doi:10.1186/s13039-018-0368-2
Haque, A., Engel, J., Teichmann, S. A., and Lönnberg, T. (2017). A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 9, 75. doi:10.1186/s13073-017-0467-4
Hasin, Y., Seldin, M., and Lusis, A. (2017). Multi-omics approaches to disease. Genome Biol. 18, 83. doi:10.1186/s13059-017-1215-1
Holmes, C. J., Plichta, J. K., Gamelli, R. L., and Radek, K. A. (2015). Dynamic role of host stress responses in modulating the cutaneous microbiome: implications for wound healing and infection. Adv. Wound Care (New Rochelle) 4, 24–37. doi:10.1089/wound.2014.0546
Hood, L., and Rowen, L. (2013). The human genome project: big science transforms biology and medicine. Genome Med. 5, 79. doi:10.1186/gm483
Hou, X., Si, X., Xu, J., Chen, X., Tang, Y., Dai, Y., et al. (2024). Single-cell RNA sequencing reveals the gene expression profile and cellular communication in human fetal heart development. Dev. Biol. 514, 87–98. doi:10.1016/j.ydbio.2024.06.004
Hu, S., Liu, S., Bian, Y., Poddar, M., Singh, S., Cao, C., et al. (2022). Single-cell spatial transcriptomics reveals a dynamic control of metabolic zonation and liver regeneration by endothelial cell Wnt2 and Wnt9b. Cell. Rep. Med. 3, 100754. doi:10.1016/j.xcrm.2022.100754
Huang, D. W., Sherman, B. T., Tan, Q., Collins, J. R., Alvord, W. G., Roayaei, J., et al. (2007). The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 8, R183. doi:10.1186/gb-2007-8-9-r183
Humayun, M., Ayuso, J. M., Park, K. Y., Martorelli Di Genova, B., Skala, M. C., Kerr, S. C., et al. (2022). Innate immune cell response to host-parasite interaction in a human intestinal tissue microphysiological system. Sci. Adv. 8, eabm8012. doi:10.1126/sciadv.abm8012
Hynes, R. O., and Naba, A. (2012). Overview of the matrisome-An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 4, a004903. doi:10.1101/cshperspect.a004903
Ingber, D. E. (2022). Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 23, 467–491. doi:10.1038/s41576-022-00466-9
Janani, G., Priya, S., Dey, S., and Mandal, B. B. (2022). Mimicking native liver lobule microarchitecture in vitro with parenchymal and non-parenchymal cells using 3D bioprinting for drug toxicity and drug screening applications. ACS Appl. Mater Interfaces 14, 10167–10186. doi:10.1021/acsami.2c00312
Jean Beltran, P. M., Federspiel, J. D., Sheng, X., and Cristea, I. M. (2017). Proteomics and integrative omic approaches for understanding host–pathogen interactions and infectious diseases. Mol. Syst. Biol. 13, 922. doi:10.15252/msb.20167062
Jiang, P., Zhang, Z., Hu, Y., Liang, Z., Han, Y., Li, X., et al. (2022). Single-cell ATAC-seq maps the comprehensive and dynamic chromatin accessibility landscape of CAR-T cell dysfunction. Leukemia 36, 2656–2668. doi:10.1038/s41375-022-01676-0
Jin, L., and Lloyd, R. V. (1997). In situ hybridization: methods and applications. J. Clin. Lab. Anal. 11, 2–9. doi:10.1002/(SICI)1098-2825(1997)11:1<2::AID-JCLA2>3.0.CO;2-F
Jin, S., Guerrero-Juarez, C. F., Zhang, L., Chang, I., Ramos, R., Kuan, C. H., et al. (2021). Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088. doi:10.1038/s41467-021-21246-9
Jones, D. S., Tsai, P. C., and Cochran, J. R. (2011). Engineering hepatocyte growth factor fragments with high stability and activity as Met receptor agonists and antagonists. Proc. Natl. Acad. Sci. U. S. A. 108, 13035–13040. doi:10.1073/pnas.1102561108
Jorgensen, A. M., Gorkun, A., Mahajan, N., Willson, K., Clouse, C., Jeong, C. G., et al. (2023). Multicellular bioprinted skin facilitates human-like skin architecture in vivo. Sci. Transl. Med. 15, eadf7547. doi:10.1126/scitranslmed.adf7547
Kaiser, K. N., Snyder, J. R., Koppes, R. A., and Koppes, A. N. (2024). A pumpless, high-throughput microphysiological System to mimic enteric innervation of duodenal epithelium and the impact on barrier function. Adv. Funct. Mater 34, 2409718. doi:10.1002/adfm.202409718
Kamburov, A., Cavill, R., Ebbels, T. M. D., Herwig, R., and Keun, H. C. (2011). Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics 27, 2917–2918. doi:10.1093/bioinformatics/btr499
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M., and Tanabe, M. (2012). KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114. doi:10.1093/nar/gkr988
Kent, G. M., Atkins, M. H., Lung, B., Nikitina, A., Fernandes, I. M., Kwan, J. J., et al. (2024). Human liver sinusoidal endothelial cells support the development of functional human pluripotent stem cell-derived Kupffer cells. Cell. Rep. 43, 114629. doi:10.1016/j.celrep.2024.114629
Khan, M. M., Ernst, O., Manes, N. P., Oyler, B. L., Fraser, I. D. C., Goodlett, D. R., et al. (2019). Multi-Omics strategies uncover host–pathogen interactions. ACS Infect. Dis. 5, 493–505. doi:10.1021/acsinfecdis.9b00080
Kjell, J., Fischer-Sternjak, J., Thompson, A. J., Friess, C., Sticco, M. J., Salinas, F., et al. (2020). Defining the adult neural stem cell niche proteome identifies key regulators of adult neurogenesis. Cell. Stem Cell. 26, 277–293.e8. doi:10.1016/j.stem.2020.01.002
Koelsch, N., Mirshahi, F., Aqbi, H. F., Saneshaw, M., Idowu, M. O., Olex, A. L., et al. (2023). The crosstalking immune cells network creates a collective function beyond the function of each cellular constituent during the progression of hepatocellular carcinoma. Sci. Rep. 13, 12630. doi:10.1038/s41598-023-39020-w
Kolli, N., Lu, M., Maiti, P., Rossignol, J., and Dunbar, G. (2017). CRISPR-Cas9 mediated gene-silencing of the mutant huntingtin gene in an in vitro model of Huntington’s disease. Int. J. Mol. Sci. 18, 754. doi:10.3390/ijms18040754
Kothapalli, R., Yoder, S. J., Mane, S., and Loughran, T. P. (2002). Microarray result: how accurate are they? BMC Bioinforma. 3, 22. doi:10.1186/1471-2105-3-22
Krämer, A., Green, J., Pollard, J., and Tugendreich, S. (2014). Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530. doi:10.1093/bioinformatics/btt703
Kumar, A. R., Li, X., LeBlanc, J. F., Farmer, D. G., Elashoff, D., Braun, J., et al. (2011). Proteomic analysis reveals innate immune activity in intestinal transplant dysfunction. Transplantation 92, 112–119. doi:10.1097/TP.0b013e31821d262b
Lacar, B., Linker, S. B., Jaeger, B. N., Krishnaswami, S., Barron, J., Kelder, M., et al. (2016). Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat. Commun. 7, 11022. doi:10.1038/ncomms11022
Lake, B. B., Menon, R., Winfree, S., Hu, Q., Melo Ferreira, R., Kalhor, K., et al. (2023). An atlas of healthy and injured cell states and niches in the human kidney. Nature 619, 585–594. doi:10.1038/s41586-023-05769-3
Lammi, M. J., and Qu, C. (2024). Spatial transcriptomics, proteomics, and epigenomics as tools in tissue engineering and regenerative medicine. Bioengineering 11, 1235. doi:10.3390/bioengineering11121235
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., et al. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. doi:10.1038/35057062
Lassé, M., El Saghir, J., Berthier, C. C., Eddy, S., Fischer, M., Laufer, S. D., et al. (2023). An integrated organoid omics map extends modeling potential of kidney disease. Nat. Commun. 14, 4903. doi:10.1038/s41467-023-39740-7
Lee, K., Silva, E. A., and Mooney, D. J. (2011). Growth factor delivery-based tissue engineering: general approaches and a review of recent developments. J. R. Soc. Interface 8, 153–170. doi:10.1098/rsif.2010.0223
Lee, S., Chung, M., Lee, S. R., and Jeon, N. L. (2020). 3D brain angiogenesis model to reconstitute functional human blood–brain barrier in vitro. Biotechnol. Bioeng. 117, 748–762. doi:10.1002/bit.27224
Lee, M., Guo, Q., Kim, M., Choi, J., Segura, A., Genceroglu, A., et al. (2024). Systematic mapping of TF-mediated cell fate changes by a pooled induction coupled with scRNA-seq and multi-omics approaches. Genome Res. 34, 484–497. doi:10.1101/gr.277926.123
Lehmann, C. J., Dylla, N. P., Odenwald, M., Nayak, R., Khalid, M., Boissiere, J., et al. (2024). Fecal metabolite profiling identifies liver transplant recipients at risk for postoperative infection. Cell. Host Microbe 32, 117–130.e4. doi:10.1016/j.chom.2023.11.016
Levy, M., Blacher, E., and Elinav, E. (2017). Microbiome, metabolites and host immunity. Curr. Opin. Microbiol. 35, 8–15. doi:10.1016/j.mib.2016.10.003
Lewis, D. M., Blatchley, M. R., Park, K. M., and Gerecht, S. (2017). O2-controllable hydrogels for studying cellular responses to hypoxic gradients in three dimensions in vitro and in vivo. Nat. Protoc. 12, 1620–1638. doi:10.1038/nprot.2017.059
Li, X., and Wang, C. Y. (2021). From bulk, single-cell to spatial RNA sequencing. Int. J. Oral Sci. 13, 36. doi:10.1038/s41368-021-00146-0
Li, C. Y., Stevens, K. R., Schwartz, R. E., Alejandro, B. S., Huang, J. H., and Bhatia, S. N. (2014). Micropatterned cell–cell interactions enable functional encapsulation of primary hepatocytes in Hydrogel microtissues. Tissue Eng. Part A 20, 2200–2212. doi:10.1089/ten.tea.2013.0667
Linnarsson, S. (2010). Recent advances in DNA sequencing methods - general principles of sample preparation. Exp. Cell. Res. 316, 1339–1343. doi:10.1016/j.yexcr.2010.02.036
Liu, X., Salokas, K., Tamene, F., Jiu, Y., Weldatsadik, R. G., Öhman, T., et al. (2018). An AP-MS- and BioID-compatible MAC-tag enables comprehensive mapping of protein interactions and subcellular localizations. Nat. Commun. 9, 1188. doi:10.1038/s41467-018-03523-2
Liu, Z., Sun, D., and Wang, C. (2022). Evaluation of cell-cell interaction methods by integrating single-cell RNA sequencing data with spatial information. Genome Biol. 23, 218. doi:10.1186/s13059-022-02783-y
Logsdon, G. A., Vollger, M. R., and Eichler, E. E. (2020). Long-read human genome sequencing and its applications. Nat. Rev. Genet. 21, 597–614. doi:10.1038/s41576-020-0236-x
Longo, M., Meroni, M., Paolini, E., Erconi, V., Carli, F., Fortunato, F., et al. (2022). TM6SF2/PNPLA3/MBOAT7 loss-of-function genetic variants impact on NAFLD development and progression both in patients and in in vitro models. CMGH 13, 759–788. doi:10.1016/j.jcmgh.2021.11.007
Lowen, J. M., and Leach, J. K. (2020). Functionally graded biomaterials for use as model systems and replacement tissues. Adv. Funct. Mater 30, 1909089. doi:10.1002/adfm.201909089
Lundberg, E., and Borner, G. H. H. (2019). Spatial proteomics: a powerful discovery tool for cell biology. Nat. Rev. Mol. Cell. Biol. 20, 285–302. doi:10.1038/s41580-018-0094-y
Luo, L., Ma, W., Liang, K., Wang, Y., Su, J., Liu, R., et al. (2023). Spatial metabolomics reveals skeletal myofiber subtypes. Sci. Adv. 9, eadd0455. doi:10.1126/sciadv.add0455
Ma, Q., and Lu, A. Y. H. (2011). Pharmacogenetics, pharmacogenomics, and individualized medicine. Pharmacol. Rev. 63, 437–459. doi:10.1124/pr.110.003533
Ma, Q., Li, Q., Zheng, X., and Pan, J. (2024). CellCommuNet: an atlas of cell–cell communication networks from single-cell RNA sequencing of human and mouse tissues in normal and disease states. Nucleic Acids Res. 52, D597–D606. doi:10.1093/nar/gkad906
MacParland, S. A., Liu, J. C., Ma, X.-Z., Innes, B. T., Bartczak, A. M., Gage, B. K., et al. (2018). Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 9, 4383. doi:10.1038/s41467-018-06318-7
Madiedo-Podvrsan, S., Belaïdi, J.-P., Desbouis, S., Simonetti, L., Ben-Khalifa, Y., Collin-Djangone, C., et al. (2021). Utilization of patterned bioprinting for heterogeneous and physiologically representative reconstructed epidermal skin models. Sci. Rep. 11, 6217. doi:10.1038/s41598-021-85553-3
Malone, A. F., and Humphreys, B. D. (2019). Single-cell transcriptomics and solid organ transplantation. Transplantation 103, 1776–1782. doi:10.1097/TP.0000000000002725
Manzoni, C., Kia, D. A., Vandrovcova, J., Hardy, J., Wood, N. W., Lewis, P. A., et al. (2018). Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences. Brief. Bioinform 19, 286–302. doi:10.1093/bib/bbw114
Marco, B. D., Vázquez-Marı́n, J., Monyer, H., Centanin, L., and Alfonso, J. (2023). Spatial transcriptomics map of the embryonic mouse brain – a tool to explore neurogenesis. Biol. Open 12, bio060151. doi:10.1242/bio.060151
Mascharak, S., Talbott, H. E., Januszyk, M., Griffin, M., Chen, K., Davitt, M. F., et al. (2022). Multi-omic analysis reveals divergent molecular events in scarring and regenerative wound healing. Cell. Stem Cell. 29, 315–327.e6. doi:10.1016/j.stem.2021.12.011
McKellar, D. W., Walter, L. D., Song, L. T., Mantri, M., Wang, M. F. Z., De Vlaminck, I., et al. (2021). Large-scale integration of single-cell transcriptomic data captures transitional progenitor states in mouse skeletal muscle regeneration. Commun. Biol. 4, 1280. doi:10.1038/s42003-021-02810-x
Mertins, P., Mani, D. R., Ruggles, K. V., Gillette, M. A., Clauser, K. R., Wang, P., et al. (2016). Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62. doi:10.1038/nature18003
Mi, H., Guo, N., Kejariwal, A., and Thomas, P. D. (2007). PANTHER version 6: protein sequence and function evolution data with expanded representation of biological pathways. Nucleic Acids Res. 35, D247–D252. doi:10.1093/nar/gkl869
Montoro, D. T., Haber, A. L., Biton, M., Vinarsky, V., Lin, B., Birket, S. E., et al. (2018). A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324. doi:10.1038/s41586-018-0393-7
Morshed, N., Lee, M. J., Rodriguez, F. H., Lauffenburger, D. A., Mastroeni, D., and White, F. M. (2021). Quantitative phosphoproteomics uncovers dysregulated kinase networks in Alzheimer’s disease. Nat. Aging 1, 550–565. doi:10.1038/s43587-021-00071-1
Mubarak, G., and Zahir, F. R. (2022). Recent Major transcriptomics and epitranscriptomics contributions toward personalized and precision medicine. J. Pers. Med. 12, 199. doi:10.3390/jpm12020199
Mukamel, E. A., and Ngai, J. (2019). Perspectives on defining cell types in the brain. Curr. Opin. Neurobiol. 56, 61–68. doi:10.1016/j.conb.2018.11.007
Murrell, A., Rakyan, V. K., and Beck, S. (2005). From genome to epigenome. Hum. Mol. Genet. 14, R3–R10. doi:10.1093/hmg/ddi110
Natarajan, K. N., Teichmann, S. A., and Kolodziejczyk, A. A. (2017). Single cell transcriptomics of pluripotent stem cells: reprogramming and differentiation. Curr. Opin. Genet. Dev. 46, 66–76. doi:10.1016/j.gde.2017.06.003
Neumaier, F., Zlatopolskiy, B. D., and Neumaier, B. (2021). Drug penetration into the central nervous System: pharmacokinetic concepts and in vitro model systems. Pharmaceutics 13, 1542. doi:10.3390/pharmaceutics13101542
Neumann, J. M., Freitag, H., Hartmann, J. S., Niehaus, K., Galanis, M., Griesshammer, M., et al. (2022). Subtyping non-small cell lung cancer by histology-guided spatial metabolomics. J. Cancer Res. Clin. Oncol. 148, 351–360. doi:10.1007/s00432-021-03834-w
Nezvedová, M., Jha, D., Váňová, T., Gadara, D., Klímová, H., Raška, J., et al. (2023). Single cerebral Organoid Mass spectrometry of cell-specific protein and glycosphingolipid traits. Anal. Chem. 95, 3160–3167. doi:10.1021/acs.analchem.2c00981
Ng, P. C., and Kirkness, E. F. (2010). “Whole genome sequencing,” in Genetic variation: methods and protocols. Editors M. R. Barnes, and G. Breen (Totowa, NJ: Humana Press), 215–226. doi:10.1007/978-1-60327-367-1_12
Ngo, M. T., and Harley, B. A. C. (2019). Perivascular signals alter global gene expression profile of glioblastoma and response to temozolomide in a gelatin hydrogel. Biomaterials 198, 122–134. doi:10.1016/j.biomaterials.2018.06.013
Ngo, M. T., Barnhouse, V. R., Gilchrist, A. E., Mahadik, B. P., Hunter, C. J., Hensold, J. N., et al. (2021). Hydrogels containing gradients in vascular density reveal dose-dependent role of angiocrine cues on stem cell behavior. Adv. Funct. Mater 31, 2101541. doi:10.1002/adfm.202101541
Nguyen, L. L., and D’amore, P. A. (2001). Cellular interactions in vascular growth and differentiation. Int. Rev. Cytol. 204, 1–48. doi:10.1016/S0074-7696(01)04002-5
Nguyen, E. V., Pereira, B. A., Lawrence, M. G., Ma, X., Rebello, R. J., Chan, H., et al. (2019). Proteomic profiling of human prostate cancer-associated fibroblasts (CAF) reveals LOXL2-dependent regulation of the tumor microenvironment. Mol. Cell. Proteomics 18, 1410–1427. doi:10.1074/mcp.RA119.001496
O’Grady, B. J., Balotin, K. M., Bosworth, A. M., McClatchey, P. M., Weinstein, R. M., Gupta, M., et al. (2020). Development of an N-Cadherin biofunctionalized hydrogel to support the Formation of synaptically connected neural networks. ACS Biomater. Sci. Eng. 6, 5811–5822. doi:10.1021/acsbiomaterials.0c00885
Palazzo, A. F., and Lee, E. S. (2015). Non-coding RNA: what is functional and what is junk? Front. Genet. 5, 2. doi:10.3389/fgene.2015.00002
Palii, C. G., Cheng, Q., Gillespie, M. A., Shannon, P., Mazurczyk, M., Napolitani, G., et al. (2019). Single-Cell proteomics reveal that quantitative changes in Co-expressed lineage-specific transcription factors determine cell fate. Cell. Stem Cell. 24, 812–820.e5. doi:10.1016/j.stem.2019.02.006
Pan, L., Parini, P., Tremmel, R., Loscalzo, J., Lauschke, V. M., Maron, B. A., et al. (2024). Single Cell Atlas: a single-cell multi-omics human cell encyclopedia. Genome Biol. 25, 104. doi:10.1186/s13059-024-03246-2
Pang, Z., Chong, J., Zhou, G., De Lima Morais, D. A., Chang, L., Barrette, M., et al. (2021). MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 49, W388–W396. doi:10.1093/nar/gkab382
Parikh, A. S., Wizel, A., Davis, D., Lefranc-Torres, A., Rodarte-Rascon, A. I., Miller, L. E., et al. (2022). Single-cell RNA sequencing identifies a paracrine interaction that may drive oncogenic notch signaling in human adenoid cystic carcinoma. Cell. Rep. 41, 111743. doi:10.1016/j.celrep.2022.111743
Parikh, B. H., Blakeley, P., Regha, K., Liu, Z., Yang, B., Bhargava, M., et al. (2023). Single-cell transcriptomics reveals maturation of transplanted stem cell–derived retinal pigment epithelial cells toward native state, Proc. Natl. Acad. Sci. 120. e2214842120. doi:10.1073/pnas.2214842120
Park, J., Shrestha, R., Qiu, C., Kondo, A., Huang, S., Werth, M., et al. (2018). Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 1979, 758–763. doi:10.1126/science.aar2131
Park, T. E., Mustafaoglu, N., Herland, A., Hasselkus, R., Mannix, R., FitzGerald, E. A., et al. (2019). Hypoxia-enhanced Blood-Brain Barrier Chip recapitulates human barrier function and shuttling of drugs and antibodies. Nat. Commun. 10, 2621. doi:10.1038/s41467-019-10588-0
Partyka, P. P., Godsey, G. A., Galie, J. R., Kosciuk, M. C., Acharya, N. K., Nagele, R. G., et al. (2017). Mechanical stress regulates transport in a compliant 3D model of the blood-brain barrier. Biomaterials 115, 30–39. doi:10.1016/j.biomaterials.2016.11.012
Pavličev, M., Wagner, G. P., Chavan, A. R., Owens, K., Maziarz, J., Dunn-Fletcher, C., et al. (2017). Single-cell transcriptomics of the human placenta: inferring the cell communication network of the maternal-fetal interface. Genome Res. 27, 349–361. doi:10.1101/gr.207597.116
Perez-Riverol, Y., Bai, M., Da Veiga Leprevost, F., Squizzato, S., Park, Y. M., Haug, K., et al. (2017). Discovering and linking public omics data sets using the Omics discovery Index. Nat. Biotechnol. 35, 406–409. doi:10.1038/nbt.3790
Petras, D., Jarmusch, A. K., and Dorrestein, P. C. (2017). From single cells to our planet—recent advances in using mass spectrometry for spatially resolved metabolomics. Curr. Opin. Chem. Biol. 36, 24–31. doi:10.1016/j.cbpa.2016.12.018
Pijuan-Sala, B., Griffiths, J. A., Guibentif, C., Hiscock, T. W., Jawaid, W., Calero-Nieto, F. J., et al. (2019). A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 566, 490–495. doi:10.1038/s41586-019-0933-9
Pinto, N., and Eileen Dolan, M. (2011). Clinically relevant genetic variations in drug metabolizing enzymes. Curr. Drug Metab. 12, 487–497. doi:10.2174/138920011795495321
Pinu, F. R., Goldansaz, S. A., and Jaine, J. (2019). Translational metabolomics: current challenges and future opportunities. Metabolites 9, 108. doi:10.3390/metabo9060108
Puram, S. V., Tirosh, I., Parikh, A. S., Patel, A. P., Yizhak, K., Gillespie, S., et al. (2017). Single-Cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 171, 1611–1624.e24. doi:10.1016/j.cell.2017.10.044
Rabbani, B., Tekin, M., and Mahdieh, N. (2014). The promise of whole-exome sequencing in medical genetics. J. Hum. Genet. 59, 5–15. doi:10.1038/jhg.2013.114
Ragelle, H., Naba, A., Larson, B. L., Zhou, F., Prijić, M., Whittaker, C. A., et al. (2017). Comprehensive proteomic characterization of stem cell-derived extracellular matrices. Biomaterials 128, 147–159. doi:10.1016/j.biomaterials.2017.03.008
Rahman, M. F., Kurlovs, A. H., Vodnala, M., Meibalan, E., Means, T. K., Nouri, N., et al. (2024). Immune disease dialogue of chemokine-based cell communications as revealed by single-cell RNA sequencing meta-analysis. Front. Syst. Biol. 4, 1466368. doi:10.3389/fsysb.2024.1466368
Rao, A., Barkley, D., França, G. S., and Yanai, I. (2021). Exploring tissue architecture using spatial transcriptomics. Nature 596, 211–220. doi:10.1038/s41586-021-03634-9
Rehm, H. L. (2013). Disease-targeted sequencing: a cornerstone in the clinic. Nat. Rev. Genet. 14, 295–300. doi:10.1038/nrg3463
Reikvam, H., Grønningsæter, I.-S., Mosevoll, K. A., Lindås, R., Hatfield, K., and Bruserud, Ø. (2018). Patients with treatment-requiring chronic graft versus host disease after allogeneic stem cell transplantation have altered metabolic profiles due to the disease and immunosuppressive therapy: potential implication for biomarkers. Front. Immunol. 8, 1979. doi:10.3389/fimmu.2017.01979
Riley, N. M., and Coon, J. J. (2016). Phosphoproteomics in the Age of rapid and deep proteome profiling. Anal. Chem. 88, 74–94. doi:10.1021/acs.analchem.5b04123
Ronaldson-Bouchard, K., Teles, D., Yeager, K., Tavakol, D. N., Zhao, Y., Chramiec, A., et al. (2022). A multi-organ chip with matured tissue niches linked by vascular flow. Nat. Biomed. Eng. 6, 351–371. doi:10.1038/s41551-022-00882-6
Rosso, F., Giordano, A., Barbarisi, M., and Barbarisi, A. (2004). From Cell-ECM interactions to tissue engineering. J. Cell. Physiol. 199, 174–180. doi:10.1002/jcp.10471
Rousseau, E., Raman, R., Tamir, T., Bu, A., Srinivasan, S., Lynch, N., et al. (2023). Actuated tissue engineered muscle grafts restore functional mobility after volumetric muscle loss. Biomaterials 302, 122317. doi:10.1016/j.biomaterials.2023.122317
Sackett, S. D., Tremmel, D. M., Ma, F., Feeney, A. K., Maguire, R. M., Brown, M. E., et al. (2018). Extracellular matrix scaffold and hydrogel derived from decellularized and delipidized human pancreas. Sci. Rep. 8, 10452. doi:10.1038/s41598-018-28857-1
Schaefer, C. F., Anthony, K., Krupa, S., Buchoff, J., Day, M., Hannay, T., et al. (2009). PID: the pathway interaction database. Nucleic Acids Res. 37, D674–D679. doi:10.1093/nar/gkn653
Schönberger, K., Obier, N., Romero-Mulero, M. C., Cauchy, P., Mess, J., Pavlovich, P. V., et al. (2022). Multilayer omics analysis reveals a non-classical retinoic acid signaling axis that regulates hematopoietic stem cell identity. Cell. Stem Cell. 29, 131–148.e10. doi:10.1016/j.stem.2021.10.002
Scott, A. J., Post, J. M., Lerner, R., Ellis, S. R., Lieberman, J., Shirey, K. A., et al. (2017). Host-based lipid inflammation drives pathogenesis in Francisella infection. Proc. Natl. Acad. Sci. 114, 12596–12601. doi:10.1073/pnas.1712887114
Semrau, S., Goldmann, J. E., Soumillon, M., Mikkelsen, T. S., Jaenisch, R., and van Oudenaarden, A. (2017). Dynamics of lineage commitment revealed by single-cell transcriptomics of differentiating embryonic stem cells. Nat. Commun. 8, 1096. doi:10.1038/s41467-017-01076-4
Shaik, S., Martin, E. C., Hayes, D. J., Gimble, J. M., and Devireddy, R. V. (2019). Transcriptomic profiling of adipose derived stem cells undergoing osteogenesis by RNA-Seq. Sci. Rep. 9, 11800. doi:10.1038/s41598-019-48089-1
Shirure, V. S., Bi, Y., Curtis, M. B., Lezia, A., Goedegebuure, M. M., Goedegebuure, S. P., et al. (2018). Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids. Lab. Chip 18, 3687–3702. doi:10.1039/c8lc00596f
Shulaev, V. (2006). Metabolomics technology and bioinformatics. Brief. Bioinform 7, 128–139. doi:10.1093/bib/bbl012
Simmons, A. D., Baumann, C., Zhang, X., Kamp, T. J., De La Fuente, R., and Palecek, S. P. (2024). Integrated multi-omics analysis identifies features that predict human pluripotent stem cell-derived progenitor differentiation to cardiomyocytes. J. Mol. Cell. Cardiol. 196, 52–70. doi:10.1016/j.yjmcc.2024.08.007
Sinha, A., and Mann, M. (2020). A beginner’s guide to mass spectrometry–based proteomics. Biochemist 42. 64, 69. doi:10.1042/bio20200057
Sinha, A., Huang, V., Livingstone, J., Wang, J., Fox, N. S., Kurganovs, N., et al. (2019). The proteogenomic landscape of curable prostate cancer. Cancer Cell. 35, 414–427.e6. doi:10.1016/j.ccell.2019.02.005
Sirolli, V., Piscitani, L., and Bonomini, M. (2023). Biomarker-Development proteomics in kidney Transplantation: an updated review. Int. J. Mol. Sci. 24, 5287. doi:10.3390/ijms24065287
Skelly, D. A., Squiers, G. T., McLellan, M. A., Bolisetty, M. T., Robson, P., Rosenthal, N. A., et al. (2018). Single-Cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart. Cell. Rep. 22, 600–610. doi:10.1016/j.celrep.2017.12.072
Sleep, E., Boué, S., Jopling, C., Raya, M., Raya, Á., and Belmonte, J. C. I. (2010). Transcriptomics approach to investigate zebrafish heart regeneration. J. Cardiovasc. Med. 11, 369–380. doi:10.2459/JCM.0b013e3283375900
Smallwood, S. A., Lee, H. J., Angermueller, C., Krueger, F., Saadeh, H., Peat, J., et al. (2014). Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 11, 817–820. doi:10.1038/nmeth.3035
Soldatow, V. Y., Lecluyse, E. L., Griffith, L. G., and Rusyn, I. (2013). In vitro models for liver toxicity testing. Toxicol. Res. (Camb) 2, 23–39. doi:10.1039/c2tx20051a
Soni, V., Bartelo, N., Schweickart, A., Chawla, Y., Dutta, A., and Jain, S. (2023). Future perspectives of metabolomics: gaps, planning, and recommendations. Metabolomics, 479–512. doi:10.1007/978-3-031-39094-4_14
Soskic, V., Görlach, M., Poznanovic, S., Boehmer, F. D., and Godovac-Zimmermann, J. (1999). Functional proteomics analysis of signal transduction pathways of the platelet-derived growth factor β receptor. Biochemistry 38, 1757–1764. doi:10.1021/bi982093r
Sreekumar, A., Poisson, L. M., Rajendiran, T. M., Khan, A. P., Cao, Q., Yu, J., et al. (2009). Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 457, 910–914. doi:10.1038/nature07762
Steger, M., Tonelli, F., Ito, G., Davies, P., Trost, M., Vetter, M., et al. (2016). Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5, e12813. doi:10.7554/elife.12813
Stevens, K. R., Kreutziger, K. L., Dupras, S. K., Korte, F. S., Regnier, M., Muskheli, V., et al. (2009). Physiological function and transplantation of scaffold-free and vascularized human cardiac muscle tissue. Proc. Natl. Acad. Sci. 106, 16568–16573. doi:10.1073/pnas.0908381106
Stevens, K. R., Scull, M. A., Ramanan, V., Fortin, C. L., Chaturvedi, R. R., Knouse, K. A., et al. (2017). In situ expansion of engineered human liver tissue in a mouse model of chronic liver disease. Sci. Transl. Med. 9, eaah5505. doi:10.1126/scitranslmed.aah5505
Supplitt, S., Karpinski, P., Sasiadek, M., and Laczmanska, I. (2021). Current achievements and applications of transcriptomics in personalized cancer medicine. Int. J. Mol. Sci. 22, 1422. doi:10.3390/ijms22031422
Takebe, T., and Wells, J. M. (2019). Organoids by design. Science 364, 956–959. doi:10.1126/science.aaw7567
Tan, S. Z., Begley, P., Mullard, G., Hollywood, K. A., and Bishop, P. N. (2016). Introduction to metabolomics and its applications in ophthalmology. Eye (Basingstoke) 30, 773–783. doi:10.1038/eye.2016.37
Tang, F., Barbacioru, C., Wang, Y., Nordman, E., Lee, C., Xu, N., et al. (2009). mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382. doi:10.1038/nmeth.1315
Teixeira, S. P. B., Domingues, R. M. A., Shevchuk, M., Gomes, M. E., Peppas, N. A., and Reis, R. L. (2020). Biomaterials for sequestration of growth factors and modulation of cell behavior. Adv. Funct. Mater 30, 1909011. doi:10.1002/adfm.201909011
Teo, A. K. K., and Vallier, L. (2010). Emerging use of stem cells in regenerative medicine. Biochem. J. 428, 11–23. doi:10.1042/BJ20100102
Tewary, M., Shakiba, N., and Zandstra, P. W. (2018). Stem cell bioengineering: building from stem cell biology. Nat. Rev. Genet. 19, 595–614. doi:10.1038/s41576-018-0040-z
Thingholm, T. E., Jensen, O. N., and Larsen, M. R. (2009). Analytical strategies for phosphoproteomics. Proteomics 9, 1451–1468. doi:10.1002/pmic.200800454
Tomczak, K., Czerwińska, P., and Wiznerowicz, M. (2015). Review The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Wspolczesna Onkol. 1A, 68–77. doi:10.5114/wo.2014.47136
Trapnell, C., Cacchiarelli, D., Grimsby, J., Pokharel, P., Li, S., Morse, M., et al. (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386. doi:10.1038/nbt.2859
Tremlett, H., Dai, D. L. Y., Hollander, Z., Kapanen, A., Aziz, T., Wilson-McManus, J. E., et al. (2015). Serum proteomics in multiple sclerosis disease progression. J. Proteomics 118, 2–11. doi:10.1016/j.jprot.2015.02.018
Truong, D. D., Kratz, A., Park, J. G., Barrientos, E. S., Saini, H., Nguyen, T., et al. (2019). A human organotypic microfluidic tumor model permits investigation of the interplay between patient-derived fibroblasts and breast cancer cells. Cancer Res. 79, 3139–3151. doi:10.1158/0008-5472.CAN-18-2293
Tsai, M. C., Shen, L. F., Kuo, H. S., Cheng, H., and Chak, K. F. (2008). Involvement of acidic fibroblast growth factor in spinal cord injury repair processes revealed by a proteomics approach. Mol. Cell. Proteomics 7, 1668–1687. doi:10.1074/mcp.M800076-MCP200
Tsamandouras, N., Chen, W. L. K., Edington, C. D., Stokes, C. L., Griffith, L. G., and Cirit, M. (2017). Integrated gut and liver microphysiological systems for quantitative in vitro pharmacokinetic studies. AAPS J. 19, 1499–1512. doi:10.1208/s12248-017-0122-4
Tyanova, S., Temu, T., Sinitcyn, P., Carlson, A., Hein, M. Y., Geiger, T., et al. (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. doi:10.1038/nmeth.3901
Uesaka, K., Oka, H., Kato, R., Kanie, K., Kojima, T., Tsugawa, H., et al. (2022). Bioinformatics in bioscience and bioengineering: recent advances, applications, and perspectives. J. Biosci. Bioeng. 134, 363–373. doi:10.1016/j.jbiosc.2022.08.004
Ungricht, R., Guibbal, L., Lasbennes, M. C., Orsini, V., Beibel, M., Waldt, A., et al. (2022). Genome-wide screening in human kidney organoids identifies developmental and disease-related aspects of nephrogenesis. Cell. Stem Cell. 29, 160–175.e7. doi:10.1016/j.stem.2021.11.001
Vacanti, C. A. (2006). The history of tissue engineering. J. Cell. Mol. Med. 10, 569–576. doi:10.1111/j.1582-4934.2006.tb00421.x
van der Helm, M. W., van der Meer, A. D., Eijkel, J. C. T., van den Berg, A., and Segerink, L. I. (2016). Microfluidic organ-on-chip technology for blood-brain barrier research. Tissue Barriers 4, e1142493. doi:10.1080/21688370.2016.1142493
Vasaikar, S. V., Savage, A. K., Gong, Q., Swanson, E., Talla, A., Lord, C., et al. (2023). A comprehensive platform for analyzing longitudinal multi-omics data. Nat. Commun. 14, 1684. doi:10.1038/s41467-023-37432-w
Vlachogiannis, G., Hedayat, S., Vatsiou, A., Jamin, Y., Fernández-Mateos, J., Khan, K., et al. (2018). Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 1979, 920–926. doi:10.1126/science.aao2774
Wang, K. C., and Chang, H. Y. (2018). Epigenomics technologies and applications. Circ. Res. 122, 1191–1199. doi:10.1161/CIRCRESAHA.118.310998
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., et al. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. doi:10.1038/nature07509
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi:10.1038/nrg2484
Wang, J., Kunzke, T., Prade, V. M., Shen, J., Buck, A., Feuchtinger, A., et al. (2022). Spatial metabolomics identifies distinct tumor-specific subtypes in gastric cancer patients. Clin. Cancer Res. 28, 2865–2877. doi:10.1158/1078-0432.CCR-21-4383
Weinreb, C., Rodriguez-Fraticelli, A., Camargo, F. D., and Klein, A. M. (2020). Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 1979, eaaw3381. doi:10.1126/science.aaw3381
Weinstein, J. N., Collisson, E. A., Mills, G. B., Shaw, K. R. M., Ozenberger, B. A., Ellrott, K., et al. (2013). The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120. doi:10.1038/ng.2764
Williams, C. G., Lee, H. J., Asatsuma, T., Vento-Tormo, R., and Haque, A. (2022). An introduction to spatial transcriptomics for biomedical research. Genome Med. 14, 68. doi:10.1186/s13073-022-01075-1
Wishart, D. S. (2005). Metabolomics: the principles and potential applications to transplantation. Am. J. Transplant. 5, 2814–2820. doi:10.1111/j.1600-6143.2005.01119.x
Wong, A. K., Sealfon, R. S. G., Theesfeld, C. L., and Troyanskaya, O. G. (2021). Decoding disease: from genomes to networks to phenotypes. Nat. Rev. Genet. 22, 774–790. doi:10.1038/s41576-021-00389-x
Wu, L., Li, Y.-F., Shen, J.-W., Zhu, Q., Jiang, J., Ma, S.-H., et al. (2022). Single-cell RNA sequencing of mouse left ventricle reveals cellular diversity and intercommunication. Physiol. Genomics 54, 11–21. doi:10.1152/physiolgenomics.00016.2021
Xia, J., Wishart, D. S., and Valencia, A. (2011). MetPA: a web-based metabolomics tool for pathway analysis and visualization. Bioinformatics. doi:10.1093/bioinformatics/btq418
Xie, W., Wei, X., Kang, H., Jiang, H., Chu, Z., Lin, Y., et al. (2023). Static and dynamic: evolving biomaterial mechanical properties to control cellular mechanotransduction. Adv. Sci. 10, 2204594. doi:10.1002/advs.202204594
Xu, J., Fang, H., Zheng, S., Li, L., Jiao, Z., Wang, H., et al. (2021). A biological functional hybrid scaffold based on decellularized extracellular matrix/gelatin/chitosan with high biocompatibility and antibacterial activity for skin tissue engineering. Int. J. Biol. Macromol. 187, 840–849. doi:10.1016/j.ijbiomac.2021.07.162
Yang, Q., Peng, J., Guo, Q., Huang, J., Zhang, L., Yao, J., et al. (2008). A cartilage ECM-derived 3-D porous acellular matrix scaffold for in vivo cartilage tissue engineering with PKH26-labeled chondrogenic bone marrow-derived mesenchymal stem cells. Biomaterials 29, 2378–2387. doi:10.1016/j.biomaterials.2008.01.037
Yang, Q., Zhou, Y., Chen, J., Huang, N., Wang, Z., and Cheng, Y. (2021). Gene therapy for drug-resistant glioblastoma via lipid-polymer hybrid nanoparticles combined with focused ultrasound. Int. J. Nanomedicine 16, 185–199. doi:10.2147/IJN.S286221
Yu, F., Selva Kumar, N. D., Choudhury, D., Foo, L. C., and Ng, S. H. (2018). Microfluidic platforms for modeling biological barriers in the circulatory system. Drug Discov. Today 23, 815–829. doi:10.1016/j.drudis.2018.01.036
Zeng, Z., Li, Y., Li, Y., and Luo, Y. (2022). Statistical and machine learning methods for spatially resolved transcriptomics data analysis. Genome Biol. 23, 83. doi:10.1186/s13059-022-02653-7
Zhang, Y., and Li, G. (2020). Advances in technologies for 3D genomics research. Sci. China Life Sci. 63, 811–824. doi:10.1007/s11427-019-1704-2
Zhang, A., Sun, H., Wang, P., Han, Y., and Wang, X. (2012). Modern analytical techniques in metabolomics analysis. Analyst 137, 293–300. doi:10.1039/c1an15605e
Zhang, B., Wang, J., Wang, X., Zhu, J., Liu, Q., Shi, Z., et al. (2014). Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387. doi:10.1038/nature13438
Zhang, H., Liu, T., Zhang, Z., Payne, S. H., Zhang, B., McDermott, J. E., et al. (2016). Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 166, 755–765. doi:10.1016/j.cell.2016.05.069
Zhao, T., Chiang, Z. D., Morriss, J. W., LaFave, L. M., Murray, E. M., Del Priore, I., et al. (2022). Spatial genomics enables multi-modal study of clonal heterogeneity in tissues. Nature 601, 85–91. doi:10.1038/s41586-021-04217-4
Zhigulev, A., and Sahlén, P. (2022). Spatial genome Organization. New York, NY: Springer US. doi:10.1007/978-1-0716-2497-5
Zhou, G., Zhou, M., Zeng, F., Zhang, N., Sun, Y., Qiao, Z., et al. (2022). Performance characterization of PCR-free whole genome sequencing for clinical diagnosis. Med. (United States) 101, e28972. doi:10.1097/MD.0000000000028972
Keywords: tissue engineering, cell-cell interactions, cell diversity, genomics, transcriptomics, proteomics, metabolomics
Citation: Stark C, Jenison SE and Ngo MT (2025) -Omics approaches to study and model cell-cell interactions in engineered tissues. Front. Chem. Eng. 7:1629455. doi: 10.3389/fceng.2025.1629455
Received: 15 May 2025; Accepted: 13 August 2025;
Published: 28 August 2025.
Edited by:
Molly Kozminsky, Iowa State University, United StatesReviewed by:
Viviana Monje, University at Buffalo, United StatesErika Y. Wang, Massachusetts Institute of Technology, United States
Copyright © 2025 Stark, Jenison and Ngo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mai T. Ngo, bXRuZ28yQHdpc2MuZWR1
†These authors have contributed equally to this work