 Mark Zervas
Mark Zervas Allyson Gage
Allyson Gage Magali Haas
Magali Haas- Cohen Veterans Bioscience, Cambridge, MA, United States
Introduction: No novel therapeutic targets for post-traumatic stress disorder (PTSD) have been successfully advanced in over two decades, despite substantial unmet clinical need. High-throughput genomic and transcriptomic studies have generated large pools of candidate targets, yet many lack mechanistic relevance, clinical applicability, or druggability. We developed a systematic, biologically rationalized prioritization framework to identify high-confidence CNS-relevant PTSD targets.
Methods: A three-phase quantitative prioritization strategy was applied to 2,467 initial candidate targets derived from PTSD transcriptomic datasets. Phase 1 identified targets expressed in CNS tissues that replicated in independent cohorts, showed consistent differential expression in PTSD CNS tissues, and had concordant direction of effect. Phase 2 advanced targets with moderate or strong CNS disease associations using DisGeNET scores. Phase 3 ranked targets using a composite pathogenicity score incorporating drug trial data, predicted loss-of-function intolerance, and protein-protein interaction network connectivity.
Results: Phase 1 reduced 2,467 candidates to 177 targets enriched for PTSD-relevant traits such as irritability, emotional symptoms, and insomnia. Phase 2 refinement yielded 55 targets with strong CNS phenotypic associations. Phase 3 prioritization identified 20 top-ranked targets with robust PTSD brain association and high CNS pathogenicity, implicating neurotransmitter systems, neurite structural regulation, and protein homeostasis.
Discussion: This three-phase prioritization framework enables efficient de-risking of PTSD target discovery, focusing resources on the most promising and biologically relevant candidates. The approach is adaptable to other poorly understood CNS disorders and may help overcome decades-long stagnation in PTSD therapeutic innovation.
Introduction
Post-Traumatic Stress Disorder (PTSD) is the fourth most common psychiatric condition in the United States, affecting both military and civilian populations, and contributes to a substantial public health burden. PTSD is currently diagnosed based on complex clinical symptoms, including re-experiencing, avoidance, negative cognitions and mood, and arousal [Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, Text Revision (DSM-5-TR)] (American Psychiatric Association, 2022). Available therapies exist, but are limited to broadly treating PTSD, including only two FDA approved selective serotonin reuptake inhibitors (SSRIs) (FDA, 2001; American Psychological Association, 2025; Henney, 2000), which have limited efficacy, undesirable side effects, and target only the symptoms of PTSD, but not cellular pathology, largely because of the sparsity of specific pathways and mechanisms underlying the disorder. As a result, no additional approvals have been issued for PTSD in over 20 years including a recent FDA complete response letter for MDMA-assisted therapy (Grossie, 2024; Lykos Therapeutics, 2024). The PTSD drug development deadlock is driven by multiple contributing factors including blinding, the lack of understanding of the biological underpinnings of PTSD, the generally poor predictive value of preclinical models (Khodosevich et al., 2024; Steckler et al., 2015) and specifically preclinical in vivo PTSD models such as Single Prolonged Stress (SPS) (Ferland-Beckham et al., 2021), a lack of disease relevant and predictive therapeutic biomarkers, and the challenges of identifying potent molecules within the stringent range of molecular properties necessary for blood-brain barrier (BBB) penetration (Jeromin et al., 2020; Wu et al., 2023). Thus, there is a striking need for rational drug development to identify and advance novel PTSD therapeutic targets.
Based on the premise that the expression of PTSD relies on a complex interaction between severity of exposure to traumatic stress and underlying biological and genetic susceptibility, Cohen Veterans Bioscience (CVB), a nonprofit biomedical research organization, invested in research to improve the probability of drug discovery and biomarker success. Initial investments focused on evaluating genetic susceptibility for PTSD through the establishment of a PTSD Consortium to lead the first analysis of global genome wide association studies (GWAS) (Nievergelt et al., 2019; Gelernter et al., 2019; Stein et al., 2021; Nievergelt et al., 2023; Nievergelt et al., 2024). This program established that PTSD susceptibility is heritable and highly polygenic (Nievergelt et al., 2023; Nievergelt et al., 2024), which reinforces that PTSD has a biological underpinning. However, there are significant challenges in mapping single nucleotide polymorphisms (SNPs) identified in GWAS to definitive, select genes (Li and Ritchie, 2021; Gazal et al., 2022; McManus et al., 2023), which limits the identification of causal pathways and therapeutic targets. To understand how diverse genetic perturbations converge onto functional pathways, CVB funded a project that resulted in the development of a protein-protein interaction (PPI) network encoded by genes strongly associated with PTSD based on SNPs identified by the PTSD Psychiatric Genomics Consortium, an approach that has advanced biological underpinnings of autsim spectrum disorder (Pintacuda et al., 2023).
Alternative approaches for target identification include conducting bulk (tissue) or cell-type specific (single cell, sc) RNA sequencing (RNAseq) studies, which have been applied both broadly to developmental disorders (Duan W et al., 2020) and specifically to PTSD (Girgenti et al., 2021; Seah et al., 2022; Wang et al., 2023; Chatzinkakos et al., 2023). Unsurprisingly, a number of brain regions are implicated in PTSD including the prefrontal cortex (PFC) and the amygdala. Accordingly, studies focused on human RNAseq (aka transcriptomics) have been conducted on specific regions derived from postmortem CNS tissue (e.g., PFC subdomains) and from human iPSC-derived excitatory neurons (iPSC-NeuExc) (Girgenti et al., 2021; Seah et al., 2022; Wang et al., 2023; Chatzinkakos et al., 2023). Importantly, these studies have provided lists containing transcripts that may play a role in PTSD. The utility of RNAseq has been demonstrated for recent drug discovery efforts, yielding actionable targets, in particular in the context of oncology drug development (Yang et al., 2020; Rydzewski et al., 2021; Bell, 2023). With the ultimate goal of facilitating novel therapeutic discovery efforts to ameliorate PTSD, we set out to identify and prioritize PTSD-relevant targets that would further our understanding of neuronal phenotypes underpinning PTSD. Knowing that we could not confidently associate GWAS-identified SNPs with definitive causal genes, we took advantage of the publicly available transcript-based data supplemented with published GWAS (Girgenti et al., 2021; Seah et al., 2022; Wang et al., 2023; Chatzinkakos et al., 2023) to identify target genes with increased confidence.
Notably, transcriptomic studies typically identify large numbers of transcripts that are either up-regulated or down-regulated. Thus, identifying and prioritizing transcripts using large transcriptomic data sets is a challenge due to the number of possible targets and their specific relevance to underlying cellular pathology. Therefore, we developed a novel 3-phase quantitative prioritization strategy to identify and rank PTSD-associated transcripts derived from publicly available sources (Girgenti et al., 2021; Seah et al., 2022; Wang et al., 2023; Chatzinkakos et al., 2023) and using the principle of consilience we ultimately prioritized and selected 20 ‘index’ transcripts and their related proteins to further interrogate and generate PTSD PPI networks. Our central hypothesis was this three-phased approach would advance PTSD disease understanding and therapeutic development by contributing to knowledge of PTSD neuronal phenotypes and disease signatures. Because it is unlikely that any one target will meaningfully treat all clinical phenotypes of such a heterogenous disorder as PTSD, our proposed method has the goal of identifying and prioritizing multiple putative PTSD targets. This could inspire a new investment in untangling cellular disease that underpins the clinical phenotypes. Ideally PPI networks, derived from our selected index proteins, would be functionally validated using a series of experimental assays, as part of future research efforts. Finally, we believe our approach, which we applied to PTSD as a practical example, will be applicable to other complex disorders to aid in target identification.
Methods
The following describes a novel, rationalized, and systematic three-phased bioinformatics prioritization strategy to (i) Nominate independently replicated PTSD-associated targets, (ii) Determine their observed differential expression in PTSD brain tissues, and (iii) Characterize evidence to support CNS-relevant pathogenicity (Figure 1) for eventual advancement to functional assays using induced excitatory neurons (iNeuExc) (Mertens et al., 2015; Nehme et al., 2018), which capture cellular disease phenotypes and are distinct from iPSC-NeuExc, which eliminate phenotypes due to reprogramming to a ground state (Takahashi et al., 2007). We utilized a number of publicly available tools to facilitate our efforts: STRING (Szklarczyk, 2019; Szklarczyk, 2023) is a free PPI database that captures physical and functional interactions, parsed on species (e.g., human), which can be viewed in STRING, but can also be imported, visualized, and further analyzed via a plugin module with Cytoscape (Cline et al., 2007; Doncheva et al., 2019; Singhal et al., 2020). As protein-protein associations may not necessarily indicated physical interactions, we took an additional (first neighbors) step in Cytoscape to build on our core proteins and develop a PTSD protein interaction network emphasizing likely physical interactions. The three phases are:
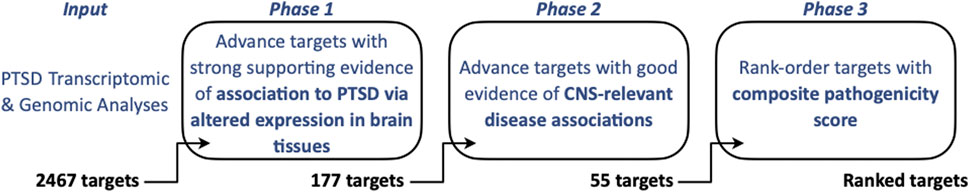
Figure 1. Overview of PTSD Brain Target Prioritization Strategy. Transcriptomic and genomic datasets were assessed to identify targets in brain tissues/iPSC-NeuExc that are confidently associated with PTSD (Phase 1, yielding n = 177 targets) and associated with other CNS phenotypes using DisGeNET (Phase 2, yielding n = 55 targets), and are likely to be pathogenic based on a three-component pathogenicity score (Phase 3) ultimately leading to a prioritized and top-ranked PTSD target cohort (n = 20 targets).
Phase 1
All data and calculations related to Phase 1 are provided in Supplementary Table S1. Candidate PTSD-associated transcriptomic targets and their direction of regulated expression were extracted from 10 studies, encompassing 29 analyses. Across each study, potential targets were annotated as to whether the published findings represented (i) Direct observation of differential transcript expression in brain tissues or in iPSC-NeuExc from PTSD cases versus controls (Girgenti et al., 2021; Logue et al., 2021; Seah et al., 2022; Wang et al., 2023; Chatzinkakos et al., 2023), (ii) Imputation of differential transcript or protein expression from brain tissues in PTSD cases versus controls (Girgenti et al., 2021; Jaffe et al., 2022; Wingo et al., 2022; Zhang et al., 2022) and (iii) Inference of PTSD-associated genes through GWAS (Nievergelt et al., 2019; Gelernter et al., 2019; Stein et al., 2021; Maihofer et al., 2022; Nievergelt et al., 2023). Literature was selected based on the following criteria: for study inclusion both a PTSD and a control cohort were required; No published studies during time of analysis were excluded. Details of analyses that were interrogated relevant to Phase 1 are provided (Supplementary Table S1). For clarity, imputation and related analyses were conducted and described in the cited studies. We utilized the collective information gained from these studies to generate an initial list of targets. Four criteria, listed below, reduced presumptive targets (2,467) to generate an actionable list.
1. Directly observed to be differentially expressed in PTSD brain tissues or iPSC-NeuExc: n = 4 cortical tissues, n = 1 subcortical tissue (amygdala), and n = 1 cell type (excitatory neuron) are represented.
2. Observed in independent cohorts: n = 6 fully independent cohorts represented in the data set.
3. Exhibited consistent (>50%) direction of difference relative to controls across all analysis in which it was identified.
4. At least one of the following:
• One or more additional observations of differential expression in PTSD brain tissues or neurons.
• Supported by imputation of differential expression in brain tissues from PTSD GWAS data (for review of these methods, see Li and Ritchie, 2021).
• Implicated by a genome-wide significant SNP from a publicly available PTSD GWAS.
Ultimately, the utility of Phase 1 was to generate an actionable list of targets (n = 177), defined as an economically viable set of transcripts/proteins, to be experimentally evaluated by most academic or industry labs while still revealing molecular signatures that provide insight to underlying pathogenic mechanisms.
Phase 2
All data and calculations related to Phase 2 are provided in Supplementary Table S2. DisGeNET (www.disgenet.org) gene disease association (GDA) scores were obtained from DisGeNET (see DisGeNET Supplement) and used to establish orthogonal evidence for target relevance in a CNS disease context. DisGeNET GDA scores (Supplementary Table S2) reflect the strength of evidence for a gene-disease linkage across multiple databases, have previously been used successfully in CNS target prioritization, and include bioinformatic/computational efforts in support of neurodegeneration and Parkinson’s Disease (Deng et al., 2023; Birkenbihl et al., 2023). DisGeNET has also validated prediction efforts using either in vitro or in vivo models of Alzheimer’s Disease (Tun et al., 2023) and ADHD (Li et al., 2023). In a drug development context, a retrospective analysis found that strong DisGeNET GDA scores outclassed all other parameters in increasing chances of success from phase 1 to launch (Mungall et al., 2016). Thus, for our approach, targets with GDA scores <0.3 were deprioritized, which left 55 targets to be advanced to Phase 3 (Figure 1).
Phase 3
Targets (n = 55) advanced to Phase 3 were associated to PTSD through altered brain tissue or neuron expression (Phase 1) and have linkages to CNS-relevant phenotypes (Phase 2). The goal of Phase 3 was to prioritize targets allowing for efficient use of resources during future validation experiments. Therefore, we integrated additional metrics that estimated a target’s pathogenicity and generated a composite pathogenicity score, which was ultimately combined with the Phase 2 GDA score and yielded 20 top tier PTSD targets. All Phase 3 data and calculations are provided in Supplementary Table S3. To benchmark effects of Phase 1 on the candidate target pool, enrichment analysis using the Monarch human phenotype-genotype database (Scannell et al., 2018) was performed with the target pool before (n = 2,467) and after (n = 177) Phase 1-selection, using the whole genome for statistical background (analysis tool is available through www.string-db.org).
Results
PTSD target identification
The goal of phase 1 was to identify and advance targets with strong supporting evidence of association with PTSD via altered expression in brain tissues (Figure 1). We combed through 10 studies and identified 2,467 potential targets. Of these, 177 targets met our four criteria, listed above, and were advanced to Phase 2 (Supplementary Table S4; Figure 2). We conducted phenotype enrichment to validate that our phase 1 approach appropriately refined the target list in accordance with phenotypes that may be associated with PTSD. While candidate targets prior to selection showed some (minor) enrichment for relevant phenotypes such as panic attack (Figure 2A), there was a notable emergence (5-fold increase) of multiple psychiatric and CNS disease phenotypes that resulted following Phase 1 selection (Figure 2B). All terms derived from Phase I were significant at FDR <0.05. Note that “Strength” on the x-axis represents Effect Size, which is defined in the legend of Figure 2, and reflects enrichment of specific phenotypes. These results indicate that advancing putative targets on the basis of a reproducible RNAseq signal derived from PTSD tissues/iPSC-NeuExc enriched the target pool with candidates implicated in neuronal/CNS phenotypes, which we believe have relevance to PTSD. Subsequently, targets that survived Phase 1 were advanced to a second enrichment step to further validate/refine the target list.
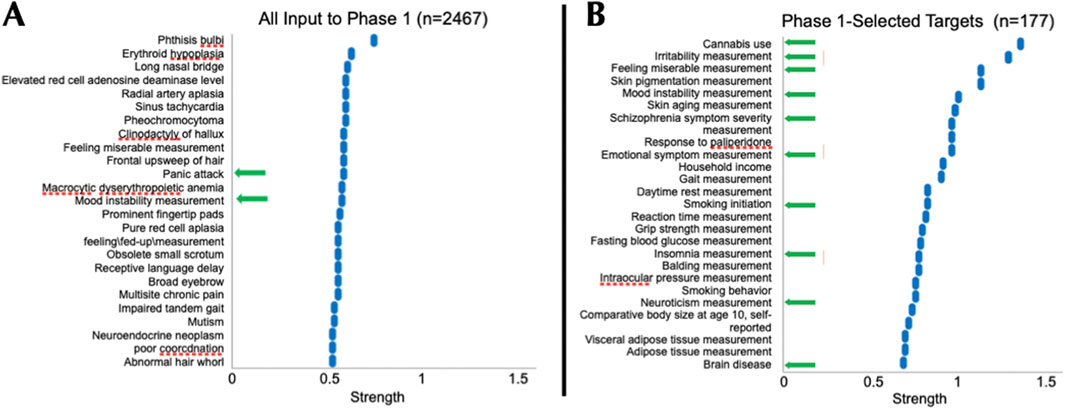
Figure 2. Phase-1 Selected Targets were Enriched for CNS Phenotypes. Panels represent the top 25 terms resulting from Monarch human phenotype enrichment analysis (via STRING) using all PTSD candidate targets (n = 2,467) identified in source publications (A) and phase 1-selected targets (n = 177, (B). Targets selected in Phase 1, reflecting confidence of association to PTSD in brain tissues, were enriched 5-fold for multiple PTSD-relevant phenotypes (from 2 to 10), including irritability, emotional symptom measurement, insomnia, and brain disease (B). All analyses were conducted using the whole genome as the background. “Strength” on the x-axis represents Effect Size, which equals log10 [(Observed number of proteins in the analyzed network annotated to phenotype ‘X':Total number of all proteins annotated to phenotype ‘X') (Expected number of proteins annotated to Phenotype ‘X' in a random network of the same size)]. For perspective, an effect size of 0.3 would be equivalent to a 2-fold enrichment. Phase 1 selected targets (n = 177) were advanced to Phase 2.
The goal of phase 2 was to advance Phase 1-nominated PTSD-associated targets that have orthogonal evidence of association to CNS pathological (disease) phenotypes (Figure 1). CNS-relevant GDA scores were defined by disease terms containing “Mental Disorder”, “Nervous System Disease”, or “Behavioral Mechanism” and were first annotated to their respective Phase 1 PTSD relevant targets (Figure 3). Each target was categorized by its highest GDA score as having “weak” (<0.3), “moderate” (0.3–0.39), or “strong” (>0.4) effect size. Among the 177 Phase 1 targets, 135 (77.4%) had at least one CNS-relevant GDA of any strength, with 55 (31.07%) possessing at least one moderate or strong CNS-relevant GDA (Figures 2, 3A). Moderate and strong gene-disease associations, which included linkages to schizophrenia, autism, depression, neurodegeneration (and more) were archived in a PTSD gene-disease linkage map for future reference; this can be found in Supplementary Table S2. The 55 targets that had at least one moderate or strong CNS-relevant GDA were advanced (Figure 3B) from Phase 2: For simplicity, GDA scores were assigned a “Phase 2 Score” of 3 (i.e., with strong GDA, n = 34) or “Phase 2 Score” of 2 (i.e., with moderate GDA, n = 21) (Table 1, Supplementary Table S5). To validate that the Phase 2 selection procedure, based on strength of DisGeNET GDA score, enriched the target list for disease an enrichment analysis was performed using Monarch human phenotype-gene associations (Mungall et al., 2016). Specifically, Phase 2 targets were tested for enrichment against a background of Phase 1 targets, revealing significant enrichment for CNS phenotypic abnormalities (Figure 4). This analysis supports that selecting targets in Phase 2, on the basis of CNS-relevant DisGeNET GDA scores, had the intended effect of enriching the network for CNS-relevant pathology.
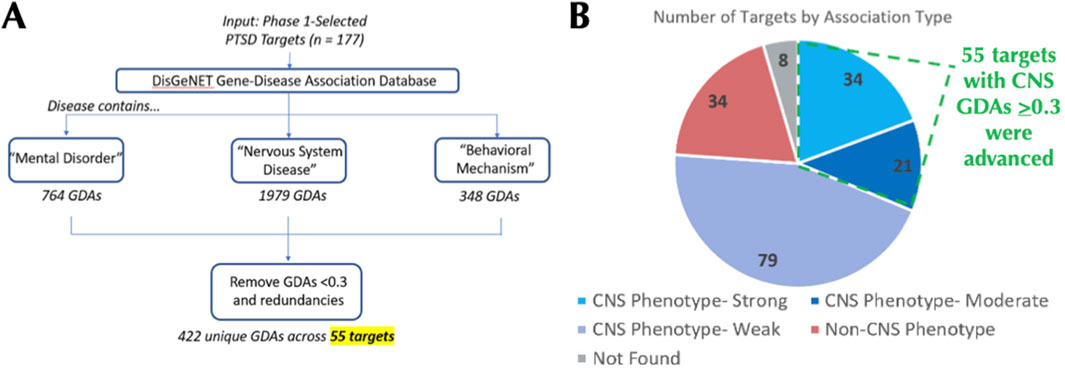
Figure 3. Phase 2 Leverages DisGeNET GDA Scores to Identify PTSD Targets with CNS Phenotype Linkages. For each Phase 1-advanced target (n = 177), DisGeNET gene-disease association (GDA) scores for CNS-relevant disorders were exported and filtered to retain only GDA scores of >0.3 (A). 135 of the 177 phase 1-advanced targets had a CNS-relevant GDA score of any strength, while 55 targets had at least one moderate (0.3–0.39) or strong (>0.4) CNS-relevant GDA score (B).
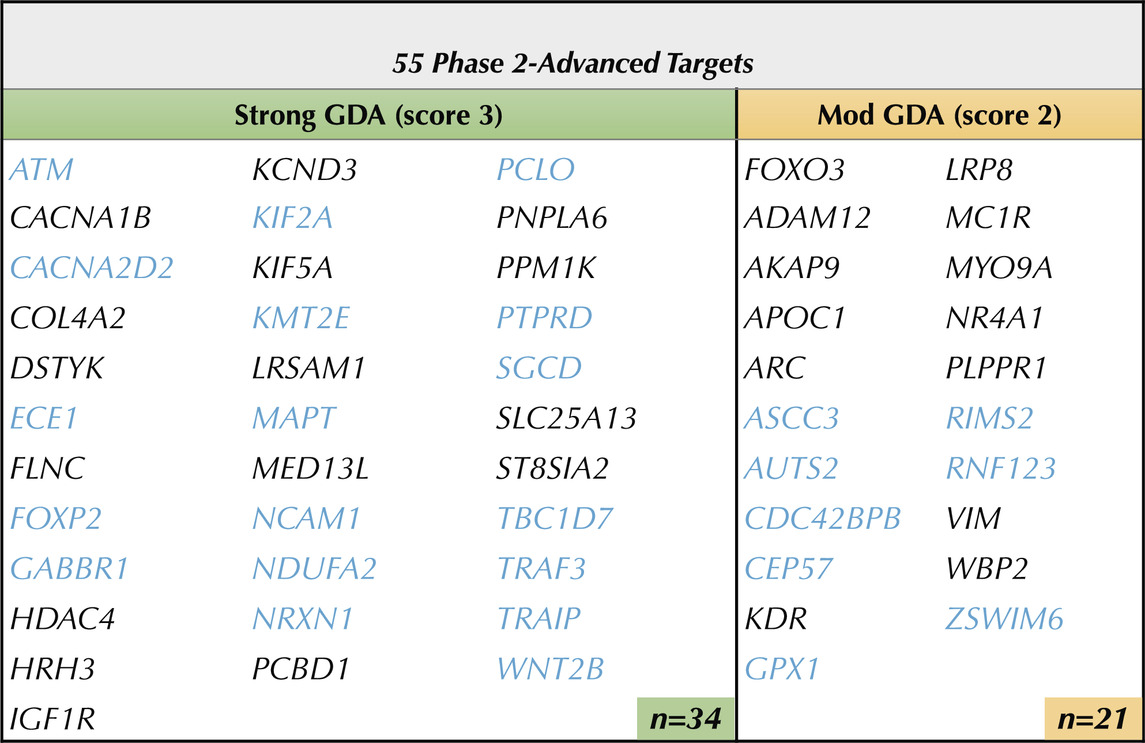
Table 1. Phase 2 Advanced 55 Brain Targets with Evidence of CNS Disease Linkages. 55 of the Phase 1 targets were advanced through phase 2 on the basis of having at least one DisGeNET CNS-relevant disease association score with an Effect Size ≥ 0.3. Targets in blue (n=26) are supported by both primary transcriptomic and supporting genomic evidence of association to PTSD. Strong GDA Effect Size (≥ 0.4) —> score 3. Moderate (Mod) GDA Effect Size (0.3-0.39) —> score 2. Note: see Supplemental Table 2 for GDA score calculations.
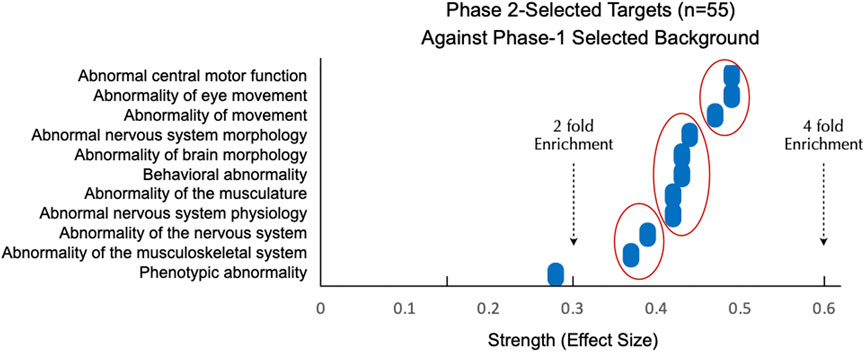
Figure 4. Phase 2-Selected Targets are Enriched for CNS Disease Above and Beyond Phase 1-Selected Targets. Panels represent all significantly enriched (FDR<0.05) terms resulting from Monarch human phenotype enrichment analysis via STRING. Phase 2 targets were enriched for CNS phenotypic abnormalities when tested against a stringent background of Phase 1-selected targets. “Strength” on the x-axis represents the effect size = log10 [(Observed number or proteins in the analyzed network annotated to phenotype ‘X':Total number of all proteins annotated to phenotype ‘X') (Expected number of proteins annotated to phenotype ‘X' in a random network of the same size)]. For perspective, a 2-fold enrichment would be equivalent to an effect size of 0.3. Three clusters of targets with between 2 and 4 fold enrichment are shown by red ovals.
The goal of phase 3 was to rank order targets with a composite pathogenicity score (Figure 1), which was comprised of three components termed “Metrics” (Figure 5), which are described below. Each Metric was weighted equally to minimize biases in determining how the three metric values influenced target prioritization and thus each had a maximum score of 1, which yielded a maximum pathogenicity score of 3 that was used along with GDA scores to rank order PTSD targets (described below). The goal of assigning the following metrics was to provide quantitative values to properties of each target using established criteria and to minimizing biases when prioritizing targets.
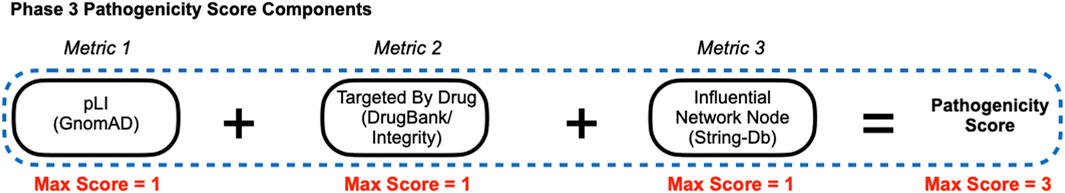
Figure 5. Phase 3 Pathogenicity Score Components. Phase 3 derived scores for equally weighted components (metrics) were used to calculate a pathogenicity score for each target. Metric 1: Predicted loss-of-function intolerance (pLI), Metric 2: Targeted by drug, and Metric 3: Influential network node.
[Metric 1] Predicted loss-of-function intolerance (pLI)
Predicted loss-of-function intolerance (pLI) is a continuous metric with a range of 0–1 that quantifies the selection pressure against (i.e., the relative rarity of) loss-of-function (LOF) variants for a particular gene (Karczewski et al., 2020). A pLI score >0.9 is considered to be extremely loss-of-function intolerant and likely to be haploinsufficient; that is when one functional copy of a gene is not capable of rescuing a phenotype (Karczewski et al., 2020). Importantly, the reason certain genes are loss-of-function intolerant is that these genes tend to be essential for normal cellular function, such that LOF results in severe, if not fatal phenotypes (Karczewski et al., 2020). Therefore, pLI provides strong positive evidence that a putative target is pathogenic. Additionally, a high pLI score is often associated with a potential drug target (Fabre and Mancini, 2022) and thus targets with higher pLI scores may contribute to PTSD. However, it is important to stress that a low a pLI score does not effectively rule out a target as benign (Karczewski et al., 2020). For each Phase 2-advanced target pLI, reported by GnomAD/DisGeNET, was used with no additional transformation (Figure 6; Supplementary Table S3).
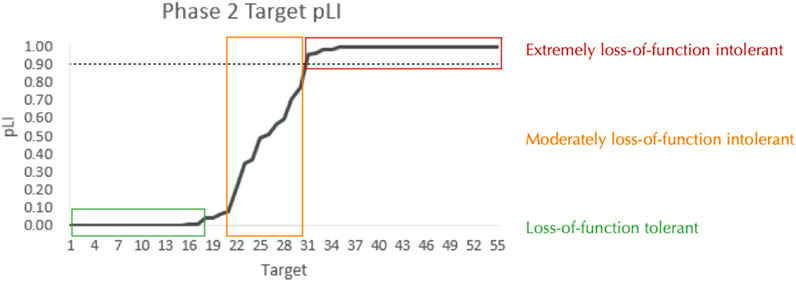
Figure 6. pLI among Phase 2-Advanced Targets. The predicted loss-of-function intolerance (pLI: 0-1) for each of the 55 Phase 2-advanced targets. See Supplementary Table S5 for detailed information on target-specific pLI. The dashed line indicates the standard threshold for a gene to be considered extremely loss-of-function intolerant (0.9).
[Metric 2] Targeted by drug
Drug information can inform estimates of a given target’s pathogenicity by providing positive evidence that modifying the drug target alleviates (portions of) the disease state. To generate a “targeted by drug” score for each target, drug information was mined from two sources: DrugBank (www.drugbank.com) and a legacy version of the Integrity Discovery Database. To estimate confidence that manipulating a target could be therapeutically effective, each target was scored on whether it has been targeted by a drug with a CNS indication and how advanced in development the drug was. Among the 55 targets that we advanced from Phase 2, twelve targets had therapeutic drugs developed for CNS indications; the full scoring of these twelve targets is shown in Table 3.

Table 2. “Targeted by Drug” Scoring Schema. Based on current drug information, each target was evaluated to determine if a drug that modifies it is in development for a CNS indication and how far advanced in the development process it is.
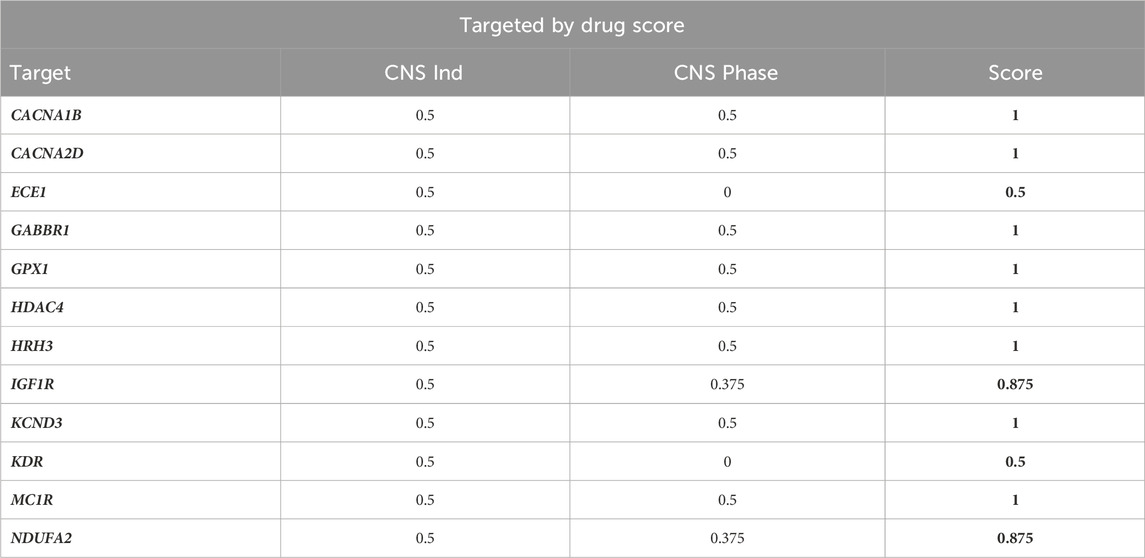
Table 3. “Targeted by Drug” Scores of Phase 2-Advanced Targets. The 12 (out of 55) targets that had at least one targeting compound in-development for a CNS indication were scored based on the criteria described in Table 2. The remaining 43 targets received a score of 0.
[Metric 3] Influential network node
Network analysis considers physical and biochemical interactions among selected targets and can help identify targets that reside in an ‘influential’ position by quantifying target connectivity within a disease-relevant network. To establish network nodes, the STRING database analysis tool (Szklarczyk, 2019) was used to first generate a PTSD-relevant interaction network using the 177 targets advanced from Phase 1 (Figure 7). Second, each of the 55 targets advanced from Phase 2 (represented as green nodes in Figure 7) were annotated based on connectivity, where node degree is the number of proteins in the network with which a target interacts within the PTSD interaction network. The network node scores were assigned as a function of node degree with 0° = 0, 1–3° = 0.5, 4–5° = 0.75, and >5° = 1 (Supplementary Figure S1).
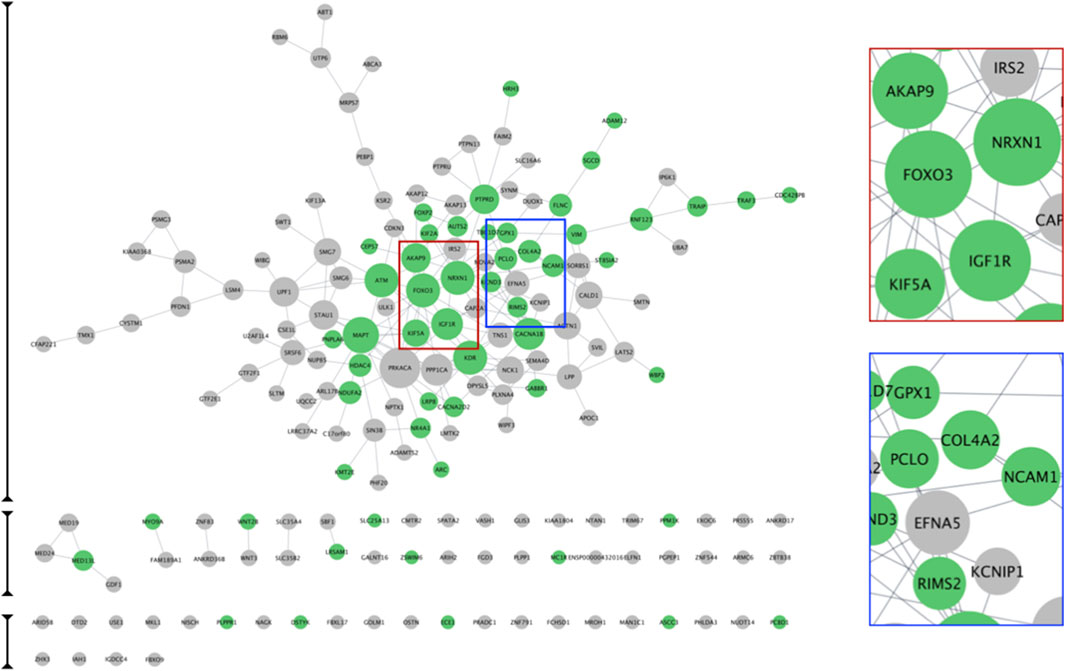
Figure 7. PTSD Target Interaction Network. STRING was used to generate a PTSD interaction network based on the 177 high confidence PTSD targets advanced from phase 1. The subset of 55 targets advanced by phase 2 are indicated as green nodes. Node size reflects the node degree, such that larger node size indicates greater node degree and hence more connections with other nodes. Two examples show examples of larger (red marque) and smaller (blue marque) nodes.
Integrating phase 2 and phase 3: Prioritizing top tier and secondary (ancillary) PTSD targets
To generate a rank-ordered target priority list, target scores from Phase 2 (GDA Score) and Phase 3 (Pathogenicity Score) were summed to yield a novel Target Advancement Prioritization (TAP) Score (Table 4; Supplementary Table S5). Both the directionality of target changes in PTSD and the TAP score are valuable considerations for designing experiments that encompass our proposed Decision Matrix (Supplementary Figure S2). The top transcriptomics-based targets we identified include receptors (e.g., GABBR1, HRH3, IGF1R), ion channels (e.g., CACNA1B, CACNA2D, KCND3), epigenetic regulators (e.g., HDAC4, KMT2E), transcriptional regulators (e.g., MED13L, FOXP2, ARC), neurite regulators (NRXN1, PCLO, MAPT, NCAM1), and mediators of proteolysis (e.g., ECE1, TRAF3). Notably, five of the top ten targets were supported by both transcriptomic and GWAS-derived evidence (Supplementary Table S5).
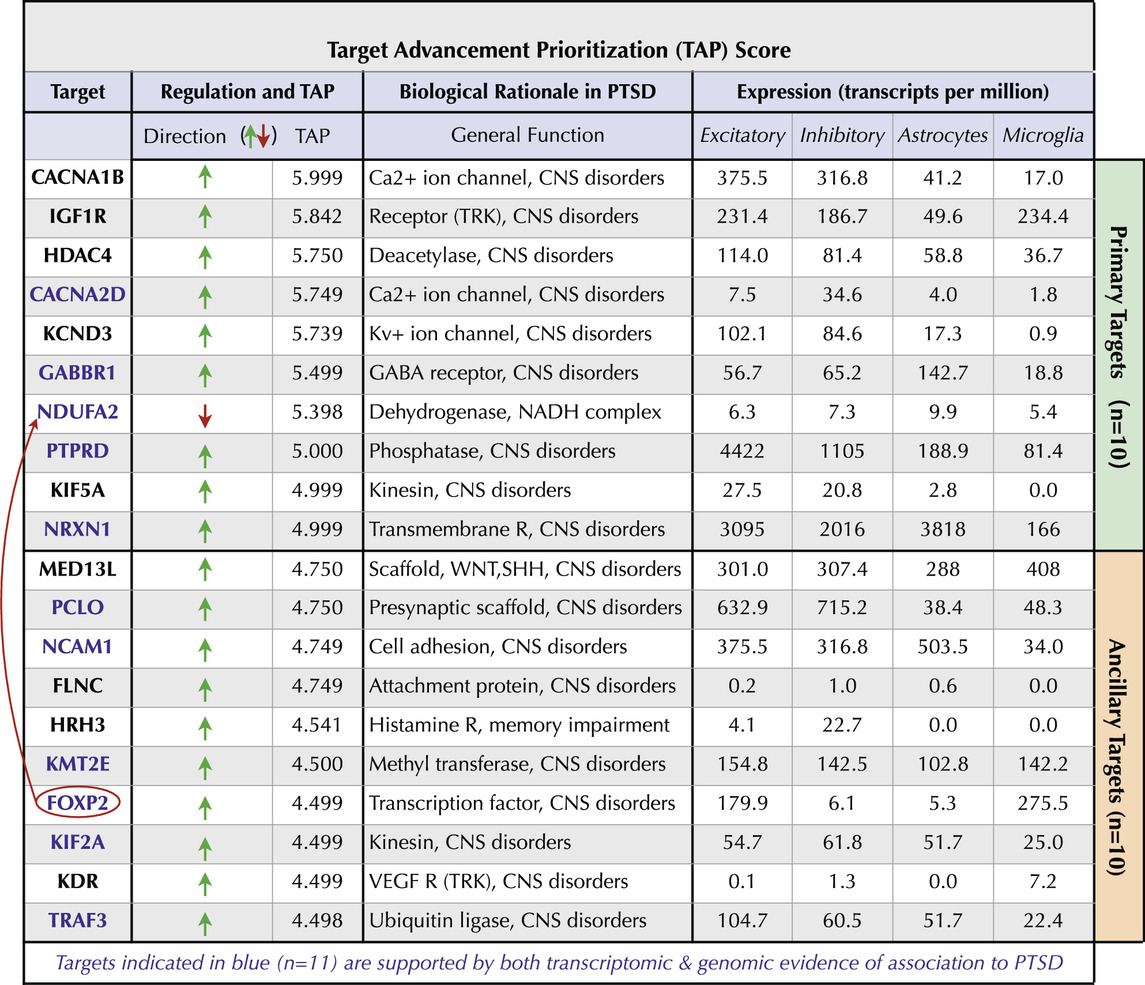
Table 4. PTSD Target Advancement Prioritization (TAP) Score. The top ten targets (green bar) and ten ancillary targets (orange bar) are indicated along with directionality changes in PTSD. The TAP score has a maximal potential value of 6.000. The biological rationale of each target relevant to PTSD and expression in Excitatory and Inhibitory neurons as well as in Astrocytes and Microglia are shown.
PTSD target interaction network
As a next step we explored the interactions of our top 20 PTSD index proteins, which serve as nodes in our network. Nodes, which are connected by ‘edges’ reveal relations to each other and were used to essentially build a PTSD PPI network. To accomplish this, all 177 Phase 1 proteins were entered into STRING using the multiple proteins search tool and interrogated using STRING functions including the Markov Cluster (MCL) algorithm (Supplementary Figure S3), which revealed unique clusters. Subsequently, we imported our proteins into Cytoscape 3.10.2 to visualize, interrogate, and expand the PTSD PPI network using the Cytoscape ‘First Neighbor’ tool (Figure 8). Our top target based on a TAP score of 5.999 (out of 6) was CACNA1B, which is a Ca2+ ion channel involved in CNS disorders (Table 4). CACNA1B had four ‘first neighbors’ (CACNA2D2, NRXN1, GABR1, and KCND3), all of which are on our Top 10 primary list and are involved in brain function. The second ranked target, which had a TAP score of 5.842 was IGF1R, which is a receptor tyrosine kinase (RTK) that is also involved in CNS disorders (Table 4). IGF1R had eight ‘first neighbors’ (Figure 9) all of which were on our Phase-1 Advanced Target list (Supplementary Table S4), but only one appeared on our refined list of Top 20 targets (Table 1). Some of the IGF1R first neighbor proteins are involved in brain function (e.g., axon guidance) as well as other functions including regulating phosphorylation and RAS signaling (Figure 9). A third target (ranked 10) from our top 10 primary target list was NRXN1, which is a transmembrane receptor involved in CNS disorders (Table 4). First neighbor analysis revealed eight first neighbor proteins (NCAM1, RIMS2, FBXL17, FOXP2, PTPRD, AUTS2, CACNA1B, and MAPT) (Figures 10A–C), all of which have brain-specific functions. We also assessed NRXN1 edges in detail, which showed that edges (connections between nodes) were supported by both experimental and co-expression interactions (Figure 10D). Cytoscape also revealed that NRXN1 interacts with numerous synaptic cleft proteins (Rudenko, 2019) (Figure 10E).
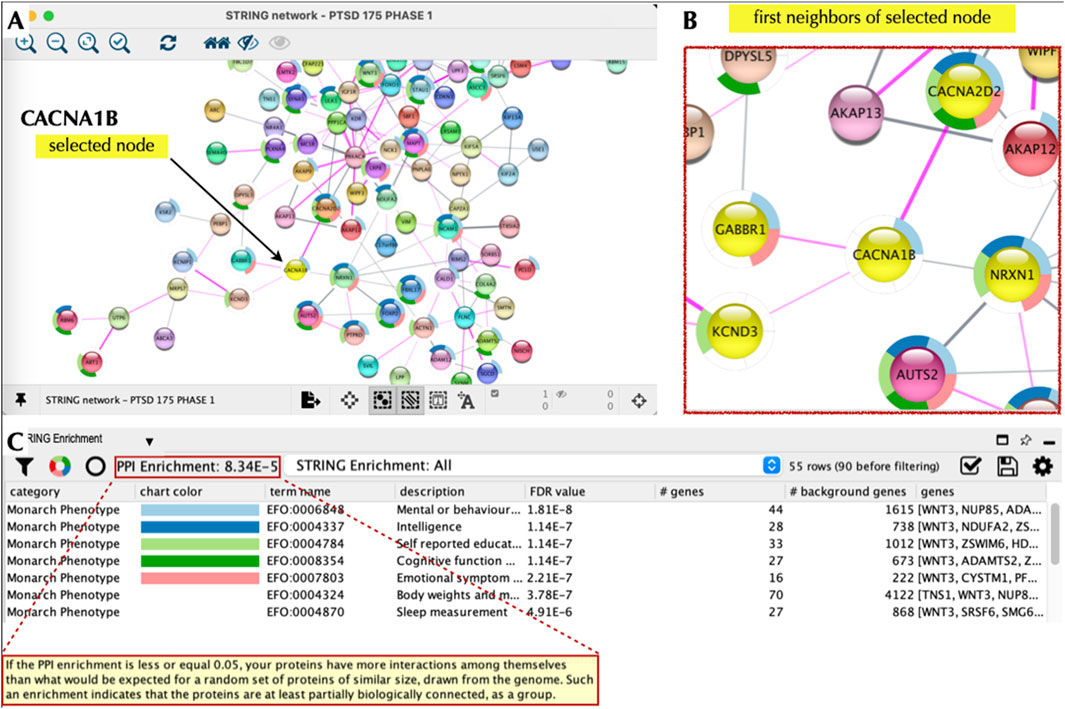
Figure 8. CACNA1B Node and Local Interaction Network. Cytoscape 3.10.2 was used to interrogate the PTSD interaction network based on CACNA1B as the node of interest (A). We then used the ‘first neighbors of selected node’ tool to generate a local interaction network, which revealed the presence of multiple interacting proteins, which were also part of our top 20 list (B). The ‘STRING enrichment’ tool showed that these proteins had a PPI enrichment of 8 × 10−5 and a role in brain health/function with a FDR <10-7 (C).
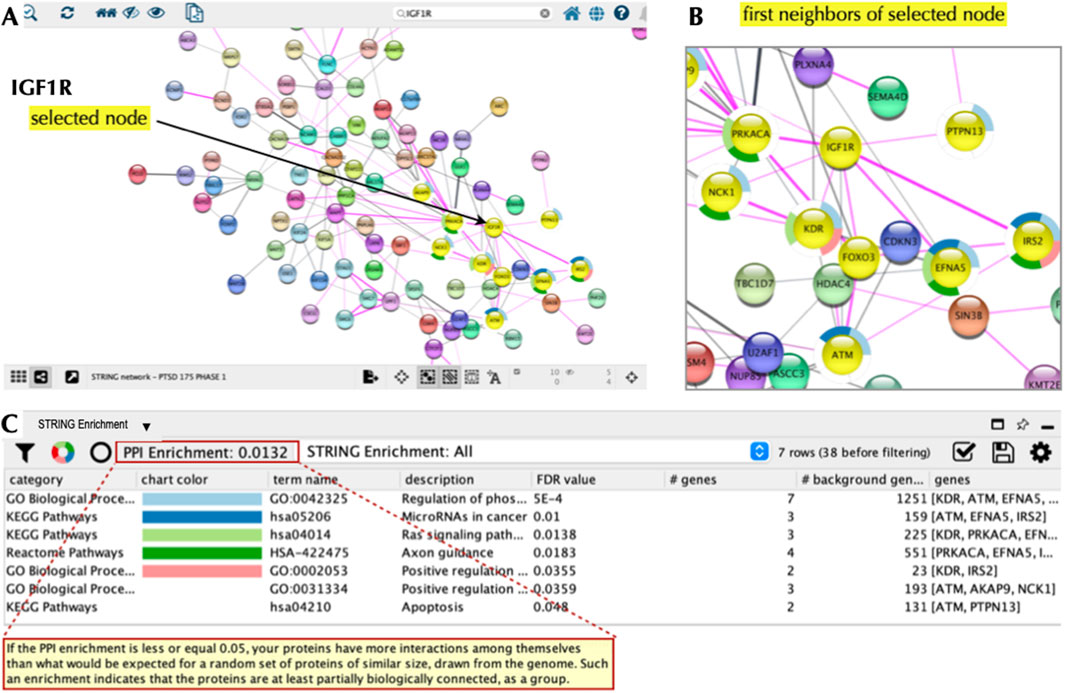
Figure 9. IGF1R Node and Local Interaction Network. Cytoscape 3.10.2 was used to interrogate the PTSD interaction network based on IGF1R as the node of interest (A). We then used the ‘first neighbors of selected node’ tool to generate a local interaction network revealing eight interacting proteins, which were also part of our top 20 list (B). The ‘STRING enrichment’ tool showed that these proteins had PPI enrichment and a role in stem cell differentiation, axonal path finding, and dendritic outgrowth (Deak and Sonntag, 2012) with a FDR<0.05 (C).

Figure 10. NRXN1 Node and Local Interaction Network. Cytoscape 3.10.2 was used to interrogate the PTSD interaction network based on NRXN1 as the node of interest (A). We then used the ‘first neighbors of selected node’ tool to generate a local interaction network revealing eight interacting proteins, which were also part of our top 20 list (B). The ‘STRING enrichment’ tool showed that these proteins had a PPI enrichment of 8 × 10−5 and a role in brain health/function with a FDR <10-7 (C). The panels (A–C) show node assessment while panel (D) shows edge assessment with subscores relevant to supporting evidence based on experiments and co-expression. The panel in the bottom right (E) shows NRXN1 (blue ovals) interacting with numerous distinct partners in the synaptic cleft (Rudenko, 2019).
Clustering of targets
We clustered the 20 top tier targets based on Cytoscape 3.10.2 and placed them in context with each other (Figure 11). Nodes with significant edges, that is connections, with other top 20 targets from our initial list (Table 1) are shown as lines connecting with an oval. Targets that had support from both our primary transcriptomic analysis and support from GWAS are indicated with blue font. Only FOXP2 (bold blue font) was observed in a previous internal (CVB) PTSD SNP-AG assessment and in the current transcriptomic-based identification of PTSD targets. Interestingly, four super (functional)-clusters (Figure 11) bind targets into a functional network although each putative target has a distinct function (Figure 11; Table 4). Cluster III contains three nodes (NCAM1, MAPT, and KIFA5). Although, MAPT was not derived from transcriptomics directly it appeared as a node via edges connected to other nodes in our analysis with Cytoscape. MAPT also appeared in a previous internal SNP-AG identification campaign. Cluster I was connected to Cluster II via connection between the CACNA1B and NRXN1 nodes. We denote Cluster II as a central hub with multiple edges (connections) that bind two nodes within Cluster III. Finally, Cluster IV initially appeared to be independent, but was revealed to be indirectly bound to Cluster III by edges that were connected by NDUFA2 and PRKACA. Notably, PRKACA was not identified via our top 20 list and thus represents an expansion component ascertained using Cytoscape and bridges the two clusters. The interactions of PTSD transcript nodes were partitioned into four distinct super (functional) clusters, which allow for more readily assessing both function and putative underlying pathobiology of PTSD.
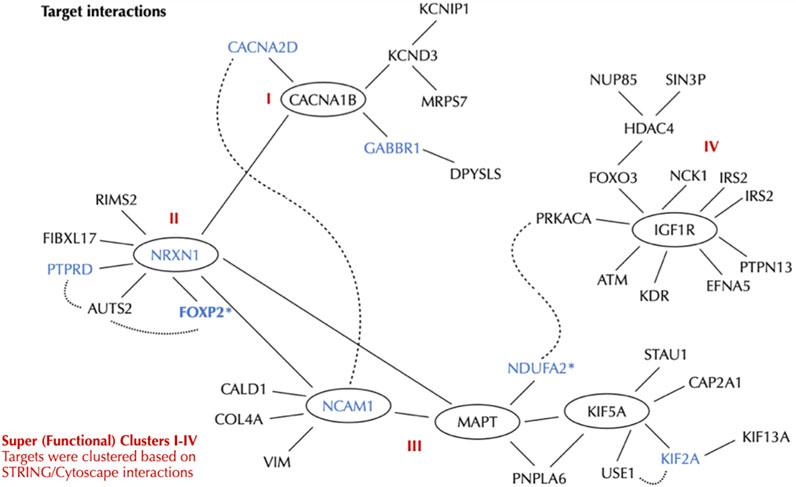
Figure 11. PTSD Targets and Local Interaction Network. Cytoscape 3.10.2 was used to interrogate the PTSD interaction network based on nodes, which were further organized into four super clusters (I-IV). The primary 10 ten targets are: CACNA1B, IGF1R, HDAC4, CACNA2D, KCND3, GABBR1, NDUFA2*, PTPRD, KIF5A, NRXN. The ancillary 10 targets are: MED13L, PCLO, NCAM1, FLNC, HRH3, KMT2E, FOXP2*, KIF2A, KDR, TRAF3. FOXP2* is an ancillary target and was swapped in as a primary target due to NDUFA2 having low expression in neurons (see Table 4). Targets with both transcriptomics and GWAS support are shown by blue font.
Nodes within functional clusters, which represent possible druggable targets, and their associated pathways, may be utilized to ameliorate cellular disease phenotypes. CACNA1B, which is the node of Cluster 1, is closely associated with CACNA2D. Both are voltage gated calcium channels involved in neurotransmitter release and implicated in a number of CNS disorders including seizures, ataxia, and schizophrenia (Sundararajan et al., 2018). CACNA1B is connected to NRXN1, which is the node of Cluster II and is a cell surface receptor. Cell surface receptors represent traditional pharmacologically-druggable targets. NRXN1 regulates Ca2+ dependent processes including neurotransmitter release and synaptic function (Walker et al., 2012; Han et al., 2020). NRXN1 is connected to NCAM1, which is part of Complex III, is a cell adhesion molecule involved in cell-cell interactions and cell-matrix interactions. NRXN1 is also implicated in schizophrenia and bipolar disorder. KIF5A, also part of Complex III, is a kinesin enzyme involved in axonal transport, as well as other functions, and is implicated in neurological disorders (Flippo and Strack, 2020). IGF1R, the node within Cluster IV is a receptor tyrosine kinase, which is another traditional pharmacologically-druggable target involved in cell growth and survival. IGF1R is implicated in both Alzheimer’s disease and Parkinson’s Disease (Deak and Sonntag, 2012).
The ten primary targets identified using our approach (CACNA1B, IGF1R, HDAC4, CACNA2D, KCND3, GABBR1, NDUFA2*, PTPRD, KIF5A, NRXN) were expanded and clustered revealing broadly related functional interactions that at face-value help distinguish underlying biological processes disrupted as a result of PTSD. In addition to the convergence of functionality there are neurological diseases related to the clusters that have strong affiliation with PTSD and at a cellular level suggests overlap between mechanisms contributing to PTSD and other neurological disorders. According to GTeX, NDUFA2 is expressed at very low levels in neurons and was thus swapped with FOXP2 from our ancillary target list. To be consistent with identifying and prioritizing NDUFA2 using our approach we still included this target, but re-positioned it within the ancillary targets. It is critical that all targets be experimentally validated for expression using existing reagents and cells/tissues. However, at this point we considered the evidence at hand and decided to reposition this target. Additionally, FOXP2 is expressed highly in excitatory neurons and is also implicated in schizophrenia, autism, and major depressive disorder.
Discussion
PTSD is a significant and complex disorder with few therapeutic options, which notably have not been updated in over 20 years. GWAS have been employed to identify genetic targets of disease and substantial efforts and resources have been invested in PTSD GWAS, which have revealed that there is a heritable, and thus genetic contribution, to the disorder (Nievergelt et al., 2019; Gelernter et al., 2019; Stein et al., 2021; Nievergelt et al., 2023; Nievergelt et al., 2024). However, it noteworthy that GWAS have typically not led to novel therapeutic targets nor a deeper understanding of neurological disorders (Uffelmann et al., 2021). Although progress has been made to overcome GWAS limitations (Watanabe et al., 2017; Schaid et al., 2018; Wang et al., 2020), aligning GWAS SNPs to specific actionable genes still has significant challenges. Therefore, we developed a streamlined approach that relied on transcripts derived from published RNAseq studies (Girgenti et al., 2021; Seah et al., 2022; Wang et al., 2023; Chatzinkakos et al., 2023) applied to postmortem CNS tissue and iPSC-NeuExc, from both PTSD and control samples allowing us to identify novel targets. We subsequently utilized publicly available tools to prioritize the transcriptome-based targets with relevance to PTSD.
Our primary goal was to identify novel PTSD targets and concomitantly uncover mechanisms that contribute to the disorder. To identify targets we took advantage of publicly available transcriptomic data sets (Supplementary Tables S1–S3) and developed a three-phase strategy that produced a list of twenty putative targets involved in PTSD. Initially we started with 2,467 targets, which were reduced to 177 (Phase 1), and then further validated/reduced using enrichment to 55 targets (Phase 2). The 55 targets were rank ordered (prioritized) by summing target scores from Phase 2 (GDA Score) and Phase 3 (Pathogenicity Score), which yielded a TAP Score producing twenty prioritized targets for PTSD (Table 4). These findings are an important early step in developing novel PTSD therapeutics, which have not had a new therapy developed in greater than 2 decades. Interestingly five of our prioritized targets (CACNA1B, GABBR1, HDAC4, IGF1R, KDR) have a combined ten compounds at various stages of clinical development (Supplementary Table S8). These compounds are either modulators or inhibitors suggesting that they would be appropriate for testing against targets that displayed an upward directionality in PTSD (Table 4) and were localized within Functional Clusters I and IV (Figure 11). We suggest testing these molecules following cell-based assays as described in the Decision Matrix (Supplementary Figure S2). A limitation with our strategy is that we cannot confidently associate any given target with specific PTSD symptom domains. Therefore, this limits us in the ability to address how identified targets contribute to the heterogenous clinical phenotypes associated with PTSD. However, integrating both identification and prioritization provides a cohesive initial approach, which we believe may confidently advance targets for complex neurological disorders such as PTSD. Our target prioritization strategy may also be applied to a wide array of diseases to accommodate the need for additional or new therapeutic drug targets and to facilitate the procurement of cellular disease phenotypes prior to initiating drug discovery efforts. Of course, other cell types and phenotypic classes of interest (e.g., immune or cardiovascular disorders) may be emphasized through choice of input datasets and disease class filtering in DisGeNET. Importantly, as cell-type specific datasets and datasets from additional brain regions become more available, it is anticipated that cell/tissue type of observations will become a more significant variable for Phase I stage prioritization. Other types of biological data may also be incorporated as supporting evidence, such as DNA methylation. In addition, genotype-phenotype linkage information generated as part of a prioritization process can be mined for hypothesis generation and functional interpretation. For example, recently published papers describe several related approaches that leverage gene-disease association maps similar to what was generated in our Phase 2 strategy (described above) to make mechanistic predictions and stratify complex diseases (Torres, 2021; Bermperidis et al., 2022; Bugrim, 2023).
The rationalized and systematic prioritization strategy described and applied here nominated 55 (i) Independently replicated PTSD-associated targets with (ii) Observed differential expression in PTSD brain tissue/excitatory neurons and (iii) Evidence supporting CNS-relevant pathogenicity. The PTSD targets we prioritized may be advanced to in vitro experimental functional validation by following our rationalized Decision Matrix (Supplementary Figure S2), which is anticipated to improve confidence in observing phenotypic effects in specific subsets of neurons. There are alternative methods describing the establishment of PPI networks (Pintacuda et al., 2023), which may result in more robust networks, but the tools we utilized are free, generally easy to use, and allowed us to establish a PTSD network for functional interrogation. We specifically undertook this task because for PTSD there have been no new therapies since the existing class of SSRI therapeutics, which were developed 2 decades ago. This is particularly relevant given that a promising ‘psychedelic’ molecule (MDMA) recently did not receive approval for PTSD (Reardon, 2024). This suggests a need to continue to identify novel targets to both understand this complex disorder and develop therapeutic alternatives to complement or supplement SSRIs or psychedelics, provided the latter is approved for treating PTSD. To provide pathological context for the targets, we next discuss roles of the associated networks in contributing to PTSD.
Functional cluster I
CACNA1B (L-type) and CACNA2D (non-L-type) (Figure 11), belong to voltage gated calcium channels, which modulate diverse functions in the CNS. CACNA1B and GABBR1 are enriched in the GABAergic synapse pathway and are upregulated following propofol (anesthetic) exposures (Li et al., 2018). CACNA1B and GABBR1 are also involved in biological pathways and mechanisms associated with genes implicated in schizophrenia (Sundararajan et al., 2018). CACNA2D is a risk gene for another complex disorder, Autism (Yang and Shcheglovitov, 2019). Because both calcium channel types converge on phenotypic associations with CNS disorders (Table 4), this provides reinforcing evidence that these channels contribute broadly to complex neurological disorders.
Functional cluster II
It has been suggested that FOXP2, DISC1, and the NRXN family are linked in a molecular network that contributes to neurodevelopmental disorders (Walker et al., 2012). However, this is the first report that we are aware of that provides supporting evidence that NRXN1 and FOXP2 are interacting partners (Figure 11). It appears that FOXP2 and PTPRD are both involved in maintaining synaptic architecture, which emerged only after complex mutagenesis assessment (Südhof, 2017; Sclip and Südhof, 2023). In addition, both proteins appear to form both pre-synaptic and post-synaptic structural complexes (Han et al., 2020).
Functional cluster III
NDUFA2 has previously been shown to be related to the avoidance subdomain of the PTSD symptom cluster (Pérez-González et al., 2024). NDUFA2 is an element of the mitochondrial complex 1 and is dysregulated in neurological disorders such as AD (Peng et al., 2020). We could not find a direct link between MAPT and NDUFA2 (Figure 11) in the literature so the interaction between them is considered tenuous at this time. Kinesins (also called KIFs) are molecular motors and implicated in both fast (50–400 mm/day) and slow (less than 8 mm/day) axonal transport (Maday et al., 2014). KIF2A appears to be an essential regulator of neuronal connectivity and for the establishment of precise postnatal hippocampal wiring (Guillaud et al., 2020). Thus, a plausible scenario is that perturbations in KIF2A and KIF5A result in altered interactions with TAU (encoded by MAPT) resulting in disrupted axonal transport as a contributing mechanism to PTSD.
Functional cluster IV
IGF-1 (Figure 11) has a major role in neuronal development as it supports neuronal stem cell differentiation, axonal path finding, and dendritic outgrowth (Deak and Sonntag, 2012). Supportingly, studies on the role of the IGF-1 receptor elicit very similar phenotypes (Deak and Sonntag, 2012). IGF-1 acts locally via IGF1R to augment synaptic connections based on olfactory nervous system assessment in mice (Deak and Sonntag, 2012).
Collectively, the four nodes we identified play a role in synaptic architecture and also function in axonal transport, which suggests that alterations in these processes contributes to PTSD and indicates cellular interactions to probe using discovery model systems. Our prioritized target list (Table 4) coupled with the proposed interaction network (Figures 7, 11) confers a significant advancement by identifying PTSD cellular pathology, specific pathways, and mechanisms that may contribute to the disorder, and may be targeted with existing molecules (Supplementary Table S8). Limitations to our study include the inability to partition targets based on the diverse array of distinct cell types including neuronal subtypes, astrocytes, and microglia that may differentially contribute to PTSD. An additional limitation is the absence of non-cortical tissue representation including the striatum, midbrain, hindbrain, and cerebellum - all off which have been implicated in psychiatric disorders.
A path ahead
Given the poor translational validity of preclinical models of PTSD (e.g., SPS, Ferland-Beckham et al., 2021), which were evaluated through various programs and methods developed to improve their utility such as EQIPD and GOT-IT (Emmerich et al., 2021), we propose, as a future direction to validate these PTSD targets and interrogate their involvement in specific pathways using human iPSC models (Takahashi et al., 2007; Mertens et al., 2015; Nehme et al., 2018). While this section is hypothetical, we wanted to propose a logical strategic example for efficiently and cost-effectively validating targets (Supplementary Figure S2). Initially, proposed targets would be ascertained for their expression and distribution at the RNA/protein level using induced iPSC-derived excitatory neurons (iNeuExc). Traditional iPSC-derived neurons are generated by taking cells with a somatic cell fate (e.g., fibroblasts) and reprogramming them to obtain an iPSC (stem cell) identity (Takahashi and Yamanaka, 2006; Takahashi et al., 2007). However, this process essentially eliminates disease phenotypes. Subsequently, stem cells are programed to obtain a unique somatic fate such as a subtype-specific neuron (e.g., cortical excitatory neuron). In contrast, a process of direct conversion from fibroblast to excitatory neuron (iNeuExc) (Nehme et al., 2018) preserves disease phenotypes making them better suited to understand cellular disease pathology (Mertens et al., 2015). This is an important premise for using advanced iPSC technology to interrogate disease pathology occurring in adult neurons. The protocol for generating iNeuExc has been publicly available for years making them a suitable substrate to begin studies although it would also be valuable to follow the same process to produce induced Inhibitory neurons (iNeuInh). To experimentally validate targets and ascertain phenotypes associated with PTSD targets we propose an experimental paradigm (Supplementary Figure S2), which is described next. The determination of expression and distribution encompass the first proposed decision point with targets having validated expression subsequently being genetically manipulated in accordance of directionality (see Table 4). CRISPR modification is proposed to be coupled with subjecting iNeuExc to a stressor (Supplementary Figure S2A) such as oxytocin or hydrocortisone (Li and Ritchie, 2021; Duan et al., 2020). For PTSD, the goal is to mimic in cellular assays the convergence of transcriptomic perturbation plus an environmental stressor.
We complement our target identification and prioritization strategy with a Decision Matrix (Supplementary Figure S2) that describes a logical methodology of functionally interrogating prioritized PTSD targets using relevant assays for PTSD target validation. Morphometric and electrophysiological assays, respectively, are informative or decision points with altered electrophysiology being viewed as a particularly important phenotype based our top tier target properties (Table 4). Targets demonstrating altered electrophysiological properties would be advanced through the Decision Matrix to gather valuable information to facilitate understanding of the biological alterations that underpin PTSD clinical phenotypes.
The next critical step would be to validate one (or ideally more) of these targets. Validation of our proposed methodology would provide potential industry partners with a tool to make rational drug development decisions that would increase their likelihood of success, decrease unnecessary testing of patients in negative clinical trials, and speed the time to important therapeutic options. Among the 20 putative PTSD targets we identified, four of the top 10 targets (CACNA1B, GABBR1, HDAC4, IGF1R) and one ancillary target (KDR) have a combined ten compounds at various stages of clinical development (Supplementary Table S8). Notably all five of these targets had an upward directionality in PTSD (Table 4), which makes them good candidates to be tested with modulator/inhibitor-based therapeutics that are currently in development (Supplementary Table S8). Interestingly, all ten compounds are being tested for, or are already approved for, one of two CNS indications, epilepsy and neurodegeneration. Two are approved for use in epilepsy and four other compounds of the same targets (CACN1B and HDAC) are in development for epilepsy. Two other targets (GABBR1 and KDR) have one compound each being developed for epilepsy while the compounds targeting the fifth target, IGF1R, are being tested in neurodegeneration. An important next step in validating this methodology would be to use one (or more) target in the Decision Matrix (Supplementary Figure S2) and test a given compound’s ability to ameliorate electrophysiological phenotypes elicited by CRISPR/stress modification. If applicable, a subset of targets could then be validated and considered for repurposing (Fajgenbaum et al., 2025) to treating PTSD.
It is important to acknowledge alternative target identification strategies to place our approach in context. There is currently rapidly increased utilization of computational biology, artificial intelligence, and large language model based approaches for target identification, which are beyond the scope of this discussion. A number of recent reviews nicely summarize advances in drug target identification. For example, network-based approaches (Koutsandreas et al., 2025), database utilization (Liu et al., 2025), and Mendelian randomization on expression quantitative trait loci (Baird et al., 2021) have been described. In addition, experimental-based approaches (Tabana et al., 2023) including phenotypic screening (Chan et al., 2010) and CRISPR screening and multidisciplinary approaches (Jia et al., 2025; Lomenick et al., 2009) have also been utilized. However, often the aforementioned described approaches have not been applied to a specific use case scenario to yield actionable target identification and prioritization. We describe a novel approach that is complementary to these previous approaches and apply it to PTSD, which has been without a novel therapeutic development in more than 2 decades.
In summary, we developed a novel strategy to identify and prioritize PTSD targets and place them in biologically meaningful pathways. In addition, we propose a strategic framework for validating biological properties. Our goal was to make these approaches publicly available to benefit PTSD patients and researchers pursuing therapeutic development for the disorder, but we also propose that the tools may be utilized in other disease discovery contexts.
Data availability statement
The datasets presented in this study can be found in the following online repository titled “ZervasFrontiersDrugDiscoverySupplementalTables_2025” which is located at: https://drive.google.com/drive/folders/1FaG0E_5ConmyUvuuV_LznwsHQjoxpoJs.
Ethics statement
Ethical approval was not required for the study involving humans in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements.
Author contributions
MZ: Writing – original draft, Investigation, Formal Analysis, Visualization, Conceptualization, Supervision, Writing – review and editing, Methodology. AG: Methodology, Conceptualization, Supervision, Writing – review and editing, Writing – original draft, Visualization. MH: Resources, Writing – review and editing, Methodology, Supervision, Writing – original draft, Conceptualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Funding for this research project was provided by Cohen Veterans Bioscience.
Acknowledgments
We acknowledge Elena Rotondo Engeldrum, Ph.D. for her significant contribution to developing the transcriptomics strategy and conducting essential analyses. MZ initiated and contributed to the transcriptomics strategy. ERE and MZ developed the local interaction network, wrote the manuscript, and generated figures. ERE, MZ, AG, MH provided equal intellectual contribution. The time and contribution of authors and acknowledged staff occurred exclusively while employees at Cohen Veterans Bioscience. ERE is currently at Regeneron. MZ is currently at Zervas Scientific Consulting. This article was submitted as a preprint to BioRxiv as Zervas et al. 2025.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2025.1613261/full#supplementary-material
References
American Psychiatric Association (2022). Diagnostic and statistical manual of mental disorders. Washington, DC: American Psychiatric Publishing, Inc. 5th ed. doi:10.1176/appi.books.9780890425787
American Psychological Association (2025). APA Clinical Practice Guideline for the Treatment of Posttraumatic Stress Disorder (PTSD) in Adults. Available online at: https://www.apa.org/about/policy/guideline-ptsd-in-adults.pdf.
Baird, D. A., Liu, J. Z., Zheng, J., Sieberts, S. K., Perumal, T., Elsworth, B., et al. (2021). Identifying drug targets for neurological and psychiatric disease via genetics and the brain transcriptome. PLoS Genet. 17 (1), e1009224. doi:10.1371/journal.pgen.1009224
Bell, J. (2023). The value of RNA sequencing in drug discovery. Chicago, Illinois: Tempus. Available online at: www.tempus.com/resources/.
Bermperidis, T., Schafer, S., Gage, F. H., Sejnowki, T., and Torres, E. B. (2022). Dynamic interrogation of stochastic transcriptome trajectories using disease associated genes reveals distinct origins of neurological and neuropsychiatric disorders. bioRxiv Preprint. doi:10.1101/2022.02.26.482124
Birkenbihl, C., Ahmad, A., Massat, N. J., Raschka, T., Avbersek, A., Downey, P., et al. (2023). Artificial intelligence-based clustering and characterization of Parkinson’s disease trajectories. Sci. Rep. 13, 2897. doi:10.1038/s41598-023-30038-8
Bugrim, A. (2023). Identification of disease mechanisms and novel disease genes using clinical concept embedding learned from massive amounts of biomedical data. bioRxiv Preprint. doi:10.1101/2023.04.27.538319
Chan, J. N., Nislow, C., and Emili, A. (2010). Recent advances and method development for drug target identification. Trends Pharmacol. Sci. 31 (2), 82–88. doi:10.1016/j.tips.2009.11.002
Chatzinkakos, C., Pernia, C. D., Morrison, F. G., Iatrou, A., McCullough, K. M., Schuler, H., et al. (2023). Single nucleus transcriptome profiling of dorsolateral prefrontal cortex: mechanistic roles for neuronal gene expression, including the 17q21.31 locus, in PTSD stress response. Am. J. Psychiatry 180, 739–754. doi:10.1176/appi.ajp.20220478
Cline, M. S., Smoot, M., Cerami, E., Kuchinsky, A., Landys, N., Workman, C., et al. (2007). Integration of biological networks and gene expression data using cytoscape. Nat. Protoc. 2 (10), 2366–2382. doi:10.1038/nprot.2007.324
Deak, F., and Sonntag, W. E. (2012). Aging, synaptic dysfunction, and insulin-like growth factor (IGF)-1. Journals Gerontology Ser. A 67A, 611–625. doi:10.1093/gerona/gls118
Deng, L., Fu, P., Ding, L., Duan, X., Feng, S., and Peng, Y. (2023). Virome analysis provides new insights into the association between viruses and Parkinson’s disease. J. Med. Virology 95, e28111. doi:10.1002/jmv.28111
Doncheva, N. T., Morris, J. H., Gorodkin, J., and Jensen, L. J. (2019). Cytoscape StringApp: network analysis and visualization of proteomics data. J. Proteome Res. 18 (2), 623–632. doi:10.1021/acs.jproteome.8b00702
Duan, W., Wang, K., Duan, Y., Chu, X., Ma, R., Hu, P., et al. (2020). Integrated transcriptome analyses revealed key target genes in mouse models of autism. Autism Rese 13, 352–368. doi:10.1002/aur.2240
Emmerich, C. H., Gamboa, L. M., Hofmann, M. C. J., Bonin-Andresen, M., Arbach, O., Schendel, P., et al. (2021). Improving target assessment in biomedical research: the GOT-IT recommendations. Nat. Rev. Drug Disc 20, 64–68. doi:10.1038/s41573-020-0087-33
Fabre, A., and Mancini, J. (2022). No preferential mode of inheritance for highly constrained genes. Intractable Rare Dis. Res. 11 (1), 25–28. doi:10.5582/irdr.2022.01011
Fajgenbaum, D. C., Nijim, S., Mitchell, G., Macak, M., Bizon, C., Tropsha, A., et al. (2025). Pioneering a new field of computational pharmacophenomics to unlock the life-saving potential of existing medicines. Lancet Haematol. 12, e94–e96. doi:10.1016/S2352-3026(24)00278-3
FDA (2001). FDA approved therapeutic drugs (selective serotonin reuptake inhibitors, SSRIs) for PTSD. Silver Spring, Maryland: Food and Drug Administration. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2001/20031s29lbl.pdf.
Ferland-Beckham, C., Chaby, L. E., Daskalakis, N. P., Knox, D., Liberzon, I., Lim, M. M., et al. (2021). Systematic review and methodological considerations for the use of single prolonged stress and fear extinction retention in rodents. Front. Behav. Neurosci. 15, 652636. doi:10.3389/fnbeh.2021.652636
Flippo, K. H., and Strack, S. (2020). An emerging role for mitochondrial dynamics in schizophrenia. Schizophr. Res. 187, 26–32. doi:10.1016/j.schres.2017.05.003
Gazal, S., Weissbrod, O., Hormozdiari, F., Dey, K. K., Nasser, J., Jagadeesh, K. A., et al. (2022). Combining SNP-to-gene linking strategies to identify disease genes and assess disease omnigenicity. Nat. Genet. 2 54 (6), 827–836. doi:10.1038/s41588-022-01087-y
Gelernter, J., Sun, N., Polimanti, R., Pietrzak, R., Levey, D. F., Bryois, J., et al. (2019). Genome-wide association study of post-traumatic stress disorder re-experiencing symptoms in >165,000 US veterans. Nat. Neurosci. 22, 1394–1401. doi:10.1038/s41593-019-0447-7
Girgenti, M. J., Wang, J., Ji, D., Cruz, D. A., Stein, M. B., et al. (2021). Transcriptomic organization of the human brain in post-traumatic stress disorder. Nat. Neurosci. 24, 24–33. doi:10.1038/s41593-020-00748-7
Grossie, G. (2024). MDMA-assisted therapy receives a complete response letter from the FDA. Am. J. Manag. Care. Available online at: https://www.ajmc.com/view/mdma-assisted-therapy-receives-a-complete-response-letter-from-the-fda.
Guillaud, L., El-Agamy, S. E., Otsuki, M., and Teren, M. (2020). Anterograde axonal transport in neuronal homeostasis and disease. Front. Mol. Neurosci. 13, 556175. doi:10.3389/fnmol.2020.556175
Han, K. A., Kim, Y. J., Yoon, T. H., Kim, H., Sungwon Bae, S., Um, J. W., et al. (2020). LAR-RPTPs directly interact with neurexins to coordinate bidirectional assembly of molecular machineries. J. Neurosci. 40, 8438–8462. doi:10.1523/JNEUROSCI.1091-20.2020
Henney, J. E. (2000). Sertraline approved for posttraumatic stress disorder. JAMA 283 (5), 596. doi:10.1001/jama.283.5.596-JFD00000-3-1
Jaffe, A. E., Tao, R., Page, S. C., Maynard, K. R., Pattie, E. A., Nguyen, C. V., et al. (2022). Decoding shared versus divergent transcriptomic signatures across cortico-amygdala circuitry in PTSD and depressive disorders. Am. J. Psychiatry 179 (9), 673–686. doi:10.1176/appi.ajp.21020162
Jeromin, A., Lasseter, H. C., Provost, A. C., Daskalakis, N. P., Etkin, A., Gehrman, P., et al. (2020). Driving progress in posttraumatic stress disorder biomarkers. Biol. Psychiatry 87 (6), e13–e14. doi:10.1016/j.biopsych.2019.07.036
Jia, Z. C., Yang, X., Wu, Y. K., Li, M., Das, D., Chen, M. X., et al. (2025). The art of finding the right drug target: emerging methods and strategies. Pharmacol. Rev. 76 (5), 896–914. doi:10.1124/pharmrev.123.001028
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alfödi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi:10.1038/s41586-020-2308-7
Khodosevich, K., Dragicevic, K., and Howes, O. (2024). Drug targeting in psychiatric disorders - how to overcome the loss in translation? Nat. Rev. Drug Disc 23 (3), 218–231. doi:10.1038/s41573-023-00847-7
Koutsandreas, T., Tsafou, K., Horn, H., Barrett, I., and Petsalaki, E. (2025). Network-based approaches for drug target identification. Annu. Rev. Biomed. Data Sci. 8, 423–446. doi:10.1146/annurev-biodatasci-101424-120950
Li, B., and Ritchie, M. D. (2021). From GWAS to gene: transcriptome-wide association studies and other methods to functionally understand GWAS discoveries. Front. Genet. 12, 713230. doi:10.3389/fgene.2021.713230
Li, Y., Liu, Y., Fan, J., Zhou, Q., Song, X., Peng, Z., et al. (2018). Validation and bioinformatic analysis of propofol-induced differentially expressed microRNAs in primary cultured neural stem cells. Gene 664, 90–100. doi:10.1016/j.gene.2018.04.046
Li, X., Xiao, Z., Pu, W., Jiang, Z., Wang, S., and Zhang, Y. (2023). Network pharmacology, molecular docking, and experimental validation to explore the potential mechanism of long Mu qing xin mixture for the treatment of attention deficit hyperactivity disorder. Front. Pharmacol. 14, 1144907. doi:10.3389/fphar.2023.1144907
Liu, X., Feng, D., Chen, J., Li, T., Wang, X., Zhang, R., et al. (2025). HCDT 2.0: a highly confident DrugTarget database for experimentally validated genes, RNAs, and pathways. Sci. Data 12:695. doi:10.1038/s41597-025-04981-2
Logue, M. W., Zhou, Z., Morrison, F. G., Wolf, E. J., Daskalakis, N. P., Chatzinakos, C., et al. (2021). Gene expression in the dorsolateral and ventromedial prefrontal cortices implicates immune-related gene networks in PTSD. Neurobiol. Stress 15, 100398. doi:10.1016/j.ynstr.2021.100398
Lomenick, B., Hao, R., Jonai, N., Chin, R. M., Aghajan, M., Warburton, S., et al. (2009). Target identification using drug affinity responsive target stability (DARTS). PNAS 106 (51), 21984–21989. doi:10.1073/pnas.0910040106
Lykos Therapeutics (2024). Lykos therapeutics announces complete response letter for midomafetamine capsules for PTSD. San Jose, CA: Lykos Therapeutics. Available online at: https://news.lykospbc.com/2024-08-09-Lykos-Therapeutics-Announces-Complete-Response-Letter-for-Midomafetamine-Capsules-for-PTSD.
Maday, S., Alison, E., Twelvetrees, A. E., Moughamian, A. J., and Holzbaur, E. L. F. (2014). Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 84, 292–309. doi:10.1016/j.neuron.2014.10.019
Maihofer, A. X., Choi, K. W., Coleman, J. R. I., Daskalakis, N. P., Denckla, C. A., Ketema, E., et al. (2022). Enhancing discovery of genetic variants for posttraumatic stress disorder through integration of quantitative phenotypes and trauma exposure information. Biol. Psychiatry 91, 626–636. doi:10.1016/j.biopsych.2021.09.020
McManus, J., Lovelett, R. J., Lowengrub, D., and Christensen, S. (2023). A unifying statistical framework to discover disease genes from GWASs. Cell. Genomics 3 (3), 100264. doi:10.1016/j.xgen.2023.100264
Mertens, J., Paquola, A. C. M., Ku, M., Hatch, E., Bohnke, L., Ladjevardi, S., et al. (2015). Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell. Stem Cell. 17, 705–718. doi:10.1016/j.stem.2015.09.001
Mungall, C. J., McMurry, J. A., Köhler, S., Balhoff, J. P., Borromeo, C., Brush, M., et al. (2016). The monarch initiative: an integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Res. 45, D712–D722. doi:10.1093/nar/gkw1128
Nehme, R., Zuccaro, E., Dia Ghosh, S., Li, C., Sherwood, J. L., Pietilainen, O., et al. (2018). Combining NGN2 programming with developmental patterning generates human excitatory neurons with NMDAR-mediated synaptic transmission. Cell. Rep. 23 (8), 2509–2523. doi:10.1016/j.celrep.2018.04.066
Nievergelt, C. M., Maihofer, A. X., Klengel, T., Atkinson, E. G., Chen, C. Y., Choi, K. W., et al. (2019). International meta-analysis of PTSD genome-wide association studies identifies sex- and ancestry-specific genetic risk loci. Nat. Comm. 10, 4558. doi:10.1038/s41467-019-12576-w
Nievergelt, C. M., Maihofer, A. X., Atkinson, E. G., Chen, C. Y., Choi, K. W., Coleman, J. R., et al. (2023). Discovery of 95 PTSD loci provides insight into genetic architecture and neurobiology of trauma and stress-related disorders. MedRxiv Pre Print, 2023.08.31.23294915. doi:10.1101/2023.08.31.23294915
Nievergelt, C. M., Maihofer, A. X., Atkinson, E. G., Chen, C. Y., Choi, K. W., Coleman, J. R. I., et al. (2024). Genome-wide association analyses identify 95 risk loci and provide insights into the neurobiology of post-traumatic stress disorder. Nat. Genet. 56, 792–808. doi:10.1038/s41588-024-01707-9
Peng, Y. S., Tang, C. W., Peng, Y. Y., Chang, H., Chen, C. H., Guo, S. L., et al. (2020). Comparative functional genomic analysis of Alzheimer’s affected and naturally aging brains. Peer J. 8, e8682. doi:10.7717/peerj.8682
Pérez-González, A. P., García-Kroepfly, A. L., Pérez-Fuentes, K. A., García-Reyes, R. I., Solis-Roldan, F. F., Alba-González, J. A., et al. (2024). The ROSMAP project: aging and neurodegenerative diseases through omic sciences. Front. Neuroinform. 18, 1443865. doi:10.3389/fninf.2024.1443865
Pintacuda, G., Hsu, Y. H., Tsafou, K., Li, K. W., Martín, J. M., Riseman, J., et al. (2023). Protein interaction studies in human induced neurons indicate convergent biology underlying autism spectrum disorders. Cell. Genomics 3, 100250. doi:10.1016/j.xgen.2022.100250
Reardon, S. (2024). MDMA therapy for PTSD rejected by FDA panel. Nat. June 5. doi:10.1038/d41586-024-01622-3
Rudenko, G. (2019). Neurexins - versatile molecular platforms in the synaptic cleft. Curr. Opin. Struct. Biol. 54, 112–121. doi:10.1016/j.sbi.2019.01.009
Rydzewski, N. R., Peterson, E., Lang, J. M., Yu, M., Laura Chang, S., Sjöström, M., et al. (2021). Predicting cancer drug TARGETS - TreAtment response generalized Elastic-neT signatures. NPJ Genomic Med. 6, 76. doi:10.1038/s41525-021-00239-z
Scannell, D., Smietana, K., Fleming, E., and Moller, M. (2018). Precision medicine: opening the aperture. In: Human genetics: the next phase of biopharma R&D. New York, NY: McKinsey and Company. p. 21.
Schaid, D. J., Chen, W., and Larson, N. B. (2018). From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 19, 491–504. doi:10.1038/s41576-018-0016-z
Sclip, A., and Südhof, T. C. (2023). Combinatorial expression of neurexins and LAR-Type phosphotyrosine phosphatase receptors instructs assembly of a cerebellar circuit. Nat. Commun. 14, 4976. doi:10.1038/s41467-023-40526-0
Seah, C., Breen, M. S., Rusielewicz, T., Bader, H. N., Xu, C., Hunter, C. J., et al. (2022). Modeling gene × environment interactions in PTSD using human neurons reveals diagnosis-specific glucocorticoid-induced gene expression. Nat. Neurosci. 25, 1434–1445. doi:10.1038/s41593-022-01161-y
Singhal, A., Cao, S., Churas, C., Pratt, D., Fortunato, S., Zheng, F., et al. (2020). Multiscale community detection in cytoscape. PLoS Comput. Biol. 16 (10), e1008239. doi:10.1371/journal.pcbi.1008239
Steckler, K. B., Haas, M., Kas, M. J., Koustova, E., Bespalov, A., et al. (2015). The preclinical data forum network: a new ECNP initiative to improve data quality and robustness for (preclinical) neuroscience. Eur. Neuropsychopharmacol. 25 (10), 1803–1807. doi:10.1016/j.euroneuro.2015.05.011
Stein, M., Levey, D. F., Cheng, Z., Wendt, F. R., Harrington, K., Pathak, G. A., et al. (2021). Genome-wide association analyses of post-traumatic stress disorder and its symptom subdomains in the million veteran program nat genet. Nat. Genet. 53 (2), 174–184. doi:10.1038/s41588-020-00767-x
Südhof, T. C. (2017). Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell. 171, 745–769. doi:10.1016/j.cell.2017.10.024
Sundararajan, T., Manzardo, A. M., and Butler, M. G. (2018). Functional analysis of schizophrenia genes using GeneAnalytics program and integrated databases. Gene 641, 25–34. doi:10.1016/j.gene.2017.10.035
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47 (D1), D607–D613. doi:10.1093/nar/gky1131
Szklarczyk, D., Kirsch, R., Koutrouli, M., Nastou, K., Mehryary, F., Hachilif, R., et al. (2023). The STRING database in 2023: Protein–Protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646. doi:10.1093/nar/gkac1000
Tabana, Y., Babu, D., Fahlman, R., Siragi, A. G., and Barakat, K. (2023). Target identification of small molecules: an overview of the current applications in drug discovery. BMC Biotechnol. 23, 44. doi:10.1186/s12896-023-00815-4
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 126, 663–676. doi:10.1016/j.cell.2006.07.024
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 131, 861–872. doi:10.1016/j.cell.2007.11.019
Torres, E. B. (2021). Precision autism: genomic stratification of disorders making up the broad spectrum May demystify its “epidemic rates.”. J. Personalized Med. 11, 1119. doi:10.3390/jpm11111119
Tun, X., Wang, E. J., Gao, Z., Lundberg, K., Xu, R., and Hu, D. (2023). Integrin β3-mediated cell senescence associates with gut inflammation and intestinal degeneration in models of Alzheimer’s disease. Int. J. Mol. Sci. 24, 5697. doi:10.3390/ijms24065697
Uffelmann, E., Huang, Q. Q., Munung, N. S., de Vries, J., Okada, Y., Martin, A. R., et al. (2021). Genome-wide association studies. Nat. Rev. Methods Prim. 1, 59. doi:10.1038/s43586-021-00056-9
Walker, R. M., Hill, A. E., Newman, A. C., Hamilton, G., Torrance, H. S., Anderson, S. M., et al. (2012). The DISC1 promoter: characterization and regulation by FOXP2. Hum. Mol. Genet. 21, 2862–2872. doi:10.1093/hmg/dds111
Wang, Q. S., Kelley, D. R., Ulirsch, J., Kanai, M., Sadhuka, S., Cui, R., et al. (2020). Leveraging supervised learning for functionally-informed fine-mapping of cis-eQTLs identifies an additional 20,913 putative causal eQTLs. bioRxiv. doi:10.1101/2020.10.20.347294
Wang, J., Li, H., Wilson, R., Wang, W., Lam, T. T., Lewis, D. A., et al. (2023). A proteome-wide, multi-omics analysis implicates novel protein dysregulation in post-traumatic stress disorder. medRxiv Pre print. doi:10.1101/2023.05.05.23289589
Watanabe, K., Taskesen, E., van Bochoven, A., and Posthuma, D. (2017). Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826. doi:10.1038/s41467-017-01261-5
Wingo, T. S., Gerasimov, E. S., Liu, Y., Duong, D. M., Vattathil, S. M., Lori, A., et al. (2022). Integrating human brain proteomes with genome-wide association data implicates novel proteins in post-traumatic stress disorder. Mol. Psychiatry 27, 3075–3084. doi:10.1038/s41380-022-01544-4
Wu, D., Chen, Q., Chen, X., Han, F., Chen, Z., and Wang, Y. (2023). The blood-brain barrier: Structure, regulation and drug delivery. Signal Transduct Target Ther. 8, 217. doi:10.1038/s41392-023-01481-w
Yang, G., and Shcheglovitov, A. (2019). Probing disrupted neurodevelopment in autism using human stem cell-derived neurons and organoids: an outlook into future diagnostics and drug development. Dev. Dyn. 249, 6–33. doi:10.1002/dvdy.100
Yang, X., Kui, L., Tang, M., Li, D., Wei, K., Chen, W., et al. (2020). High-throughput transcriptome profiling in drug and biomarker discovery. Front. Genet. 11, 19. doi:10.3389/fgene.2020.00019
Zervas, M., Gage, A., and Haas, M. (2025). Bioinformatics-driven identification and prioritization of PTSD targets based on published multi-omic data. doi:10.1101/2025.04.07.647615
Keywords: PTSD, transcriptomics, drug target, prioritization, identification
Citation: Zervas M, Gage A and Haas M (2025) Bioinformatics-driven identification and prioritization of PTSD targets based on published multi-omic data. Front. Drug Discov. 5:1613261. doi: 10.3389/fddsv.2025.1613261
Received: 16 April 2025; Accepted: 25 July 2025;
Published: 10 September 2025.
Edited by:
Florbela Pereira, New University of Lisbon, PortugalCopyright © 2025 Zervas, Gage and Haas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark Zervas, bXplcnZhc0BnbWFpbC5jb20=
†ORCID: Mark Zervas, orcid.org/0000-0002-7027-4299