 José L. Medina-Franco
José L. Medina-Franco Edgar López-López
Edgar López-López Juan F. Avellaneda-Tamayo
Juan F. Avellaneda-Tamayo William J. Zamora
William J. Zamora- 1DIFACQUIM Research Group, Department of Pharmacy, School of Chemistry, Universidad Nacional Autónoma de México, Avenida Universidad 3000, Mexico City, Mexico
- 2Department of Chemistry and Graduate Program in Pharmacology, Center for Research and Advanced Studies of the National Polytechnic Institute, Section 14-740, Mexico City, Mexico
- 3CBio3 Laboratory, School of Chemistry, University of Costa Rica, San José, Costa Rica
- 4Laboratory of Computational Toxicology and Biological Testing Laboratory (LEBi), University of Costa Rica, San José, Costa Rica
1 Introduction
The “chemical space” (CS), “chemical compound space,” or “chemical universe” terms are frequently used in drug discovery and other areas, including chemical synthesis, catalysis, materials science, food chemistry, and agrochemistry, among others (Kim et al., 2024). While the concept is often used intuitively or colloquially, CS is inherently complex, and numerous formal definitions have been proposed and reviewed (Medina-Franco et al., 2022). A commonly accepted notion of CS relates to the number of chemical compounds that could theoretically exist—the “size” of chemical space—which varies greatly depending on the classes of compounds considered (e.g., small organic molecules, peptides, odorants). Another perspective views CS as a multidimensional space in which molecular properties (both structural and functional) define coordinates and relationships between compounds (Virshup et al., 2013; Martinez-Mayorga and Medina-Franco, 2014). These definitions give rise to the concept of chemical subspaces (ChemSpas): subsets of the broader chemical universe distinguished by shared structural or functional features. Within this framework, the biologically relevant chemical space (BioReCS) comprises molecules with biological activity—both beneficial and detrimental. BioReCS spans diverse application areas such as drug discovery, agrochemistry, sensory chemistry (e.g., flavor and odor), food science, and natural product research. It also includes compounds with reactive molecules, including promiscuous and poly-active molecules, as well as those with highly detrimental or undesirable effects, such as toxic and allergic compounds.
Chemical compound databases are key resources for exploring the CS and are central to chemoinformatics (Williams and Richard, 2025). Numerous public databases—varying in size and specialization—target specific regions of BioReCS. Table 1 provides representative examples of freely available libraries across several domains. Comprehensive reviews of chemoinformatic and bioinformatic databases have been published elsewhere (Rigden and Fernández, 2025; de Azevedo et al., 2024).
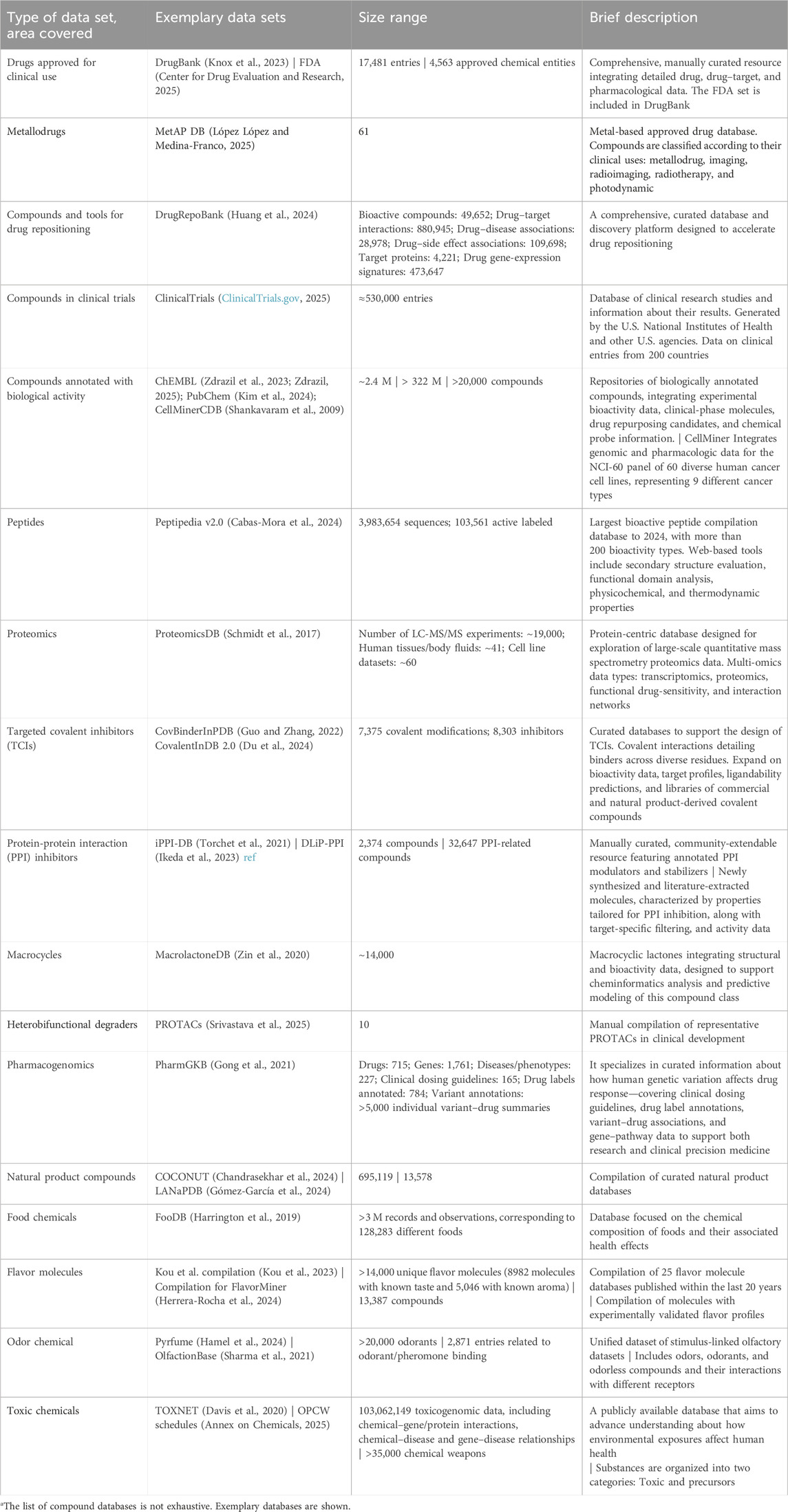
Table 1. Representative public compound data sets covering different regions of the BioReCS.a
A systematic study of CS requires molecular descriptors that define the dimensionality of the space. The choice of descriptors depends on project goals, compound classes (e.g., metal-containing vs purely organic molecules), and the dataset size and diversity. Large and ultra-large chemical libraries that are highly used today in drug discovery projects (Lyu et al., 2019; Corrêa Veríssimo et al., 2024), for example, demand descriptors that strike a balance between computational efficiency and chemical relevance (Warr et al., 2022). The rise of machine learning has led to the development of novel molecular representations (Wigh et al., 2022). Visualization is another critical tool for CS analysis, because these spaces often involve many dimensions; dimensionality-reduction techniques are commonly used to project them into two or three dimensions for interpretation. Recent reviews detail advancements in the visualization of chemical space (Sosnin, 2025).
In this article, we offer an integrative perspective on BioReCS, highlighting common considerations for its consistent and meaningful exploration. We also address its size, historical evolution, and future expansion.
2 BioReCS
2.1 Current view
In many research projects, the chemical universe—and by extension, BioReCS—is explored through distinct sections of chemical subspaces (ChemSpas). For instance, CS analyses may focus specifically on small-molecule drug candidates, peptides (Orsi and Reymond, 2024), or proteolysis-targeting chimeras (PROTACs) (Danishuddin et al., 2023; Sincere et al., 2023). Other studies target agrochemicals, odorants, natural products, or metal-containing compounds. Some research initiatives are at the intersection of multiple ChemSpas, such as investigating bioactive compounds that straddle both natural product and food chemical domains (Avellaneda-Tamayo et al., 2024) or studying the overlap between flavor and odor chemicals (Cui et al., 2025). Analyzing these intersecting regions of chemical space often requires integrating methodologies from diverse disciplines. In this section, we highlight both heavily explored and underexplored regions of BioReCS.
2.2 Heavily explored chemical subspaces
In drug discovery, widely used public databases such as ChEMBL (Zdrazil, 2025) and PubChem (Kim et al., 2024) serve as major sources of biologically active small molecules, primarily organic compounds. Owing to their extensive biological activity annotations, these databases are major sources of poly-active compounds and promiscuous structures. Table 1 summarizes these and other key databases that cover different regions of BioReCS. The chemical space of drug-like molecules, particularly small organic compounds and natural products, has been extensively studied. Closely related areas, such as small peptides and other beyond Rule of 5 (bRo5) entities, are also well-characterized using computational approaches (Price et al., 2024; Capecchi and Reymond, 2021; López-López et al., 2023). Importantly, to fully chart the boundaries of BioReCS, it is crucial to include negative biological data—that is, compounds known to lack bioactivity (Williams et al., 2016; López-López et al., 2022). These data help define the non-biologically relevant portions of chemical space. A notable example is dark chemical matter, a large-scale dataset comprising small molecules from corporate compound collections that have repeatedly failed to show activity in high-throughput screening assays (Wassermann et al., 2015). Also, a recent development is the generation of InertDB, a compound collection with 3,205 curated inactive compounds obtained from PubChem (An et al., 2025). The database also includes 64,368 putative inactive molecules generated with a deep generative artificial intelligence (AI) model trained on the experimentally determined inactive molecules (An et al., 2025).
2.3 Underexplored chemical subspaces
Certain types of chemical structures remain underrepresented in chemoinformatics due to modeling challenges. A prominent example is metal-containing molecules, which are often excluded during data curation because most chemoinformatics tools are optimized for small organic compounds (Fourches et al., 2016; Bento et al., 2020; Valle-Núñez et al., 2025). Metallodrugs, therefore, represent a structurally and functionally important class that is commonly filtered out by default. However, the difficulty of modeling a region of BioReCS should not justify its exclusion. Similarly, various compound classes are rarely targeted in drug discovery efforts, including large and complex natural products, macrocycles (compounds containing rings of ≥12 atoms), protein-protein interaction (PPI) modulators or inhibitors, PROTACs, and mid-sized peptides. Many of these molecules fall into the beyond Rule of 5 (bRo5) category (Price et al., 2024; Whitty and Zhou, 2015; Schaub et al., 2021) (Table 1). Despite their complexity, interest in characterizing these regions of chemical space is growing. Recent studies have addressed the CS of peptides (Orsi and Reymond, 2024; Capecchi et al., 2019), agrochemicals (Zhang et al., 2018), metallodrugs (Meggers, 2007; López López and Medina-Franco, 2025), macrocycles (Viarengo-Baker et al., 2021; Kim et al., 2025), and PPIs (Zhang et al., 2014; Choi et al., 2021).
2.3.1 Dark regions of the underexplored BioReCS
Beyond beneficial regions, BioReCS also encompasses gray-to-dark areas—zones that include compounds with undesirable biological effects, such as toxic chemicals (Tihányi et al., 2025; (Annex on Chemicals, 2025). Understandably, these regions have received less attention than areas linked to therapeutic or beneficial activity. Nonetheless, distinguishing the characteristics that separate harmful compounds from beneficial ones is vital for the design of safer, human-beneficial, and ecologically responsible molecules.
3 Common considerations to explore BioReCS
In this section, we highlight common challenges associated with exploring BioReCS, along with possible workarounds and emerging directions. While not exhaustive, these topics are meant to illustrate recurring issues and encourage a holistic consideration of the BioReCS.
3.1 Towards universal descriptors
The structural diversity across underexplored regions of BioReCS presents a major challenge to define a consistent chemical space using molecular descriptors. Traditional descriptors, tailored to specific ChemSpas such as small molecules, peptides, or metallodrugs, lack universality. However, there are ongoing efforts to develop structure-inclusive, general-purpose descriptors. Notable examples include molecular quantum numbers (Nguyen et al., 2009) and the MAP4 fingerprint (Capecchi et al., 2020 ref), which is designed to accommodate entities ranging from small molecules to biomolecules and even metabolomic data. More recently, neural network embeddings derived from chemical language models have shown promise in encoding chemically meaningful representations that can reconstruct molecular structures or predict properties (Lžičař and Gamouh, 2024). However, there is still a pressing need to develop systematic molecular fingerprints for the study of biomaterials and inorganic molecules.
3.2 pH-dependent chemical space
Many bioactive compounds, especially drugs, are weak bases, acids, or ampholytes that can ionize depending on the pH of their environment. Pioneering studies have reported that 62.9% of compounds in the World Drug Index (n = 582) are ionizable, with the majority being bases, fewer acids, and some ampholytes (Manallack, 2007), however, chemogenomic analyses on contemporary drugs (n = 3766) have shown that this percentage can reach 80% (Manallack et al., 2013). In consequence, the ionization state—charged or neutral—of a bioactive compound profoundly impacts its solubility, permeability, absorption, distribution, toxicity, and binding, making this distinction essential in drug development and computational modeling. However, CS analyses typically assume molecular structures with neutral charge, which may not reflect the actual bioactive species of compounds under physiological or environmental conditions. Even when the structural representation of an ionizable compound is accurate, chemoinformatics tools often calculate molecular descriptors such as lipophilicity (logP) based solely on the neutral species, overlooking the dominant ionic forms. Computing lipophilicity using logD at physiological pH is much more relevant than using logP for small molecules (Bhal et al., 2007; Zamora et al., 2017), including standard amino acid residues (Zamora et al., 2019) to non-standard residues (Viayna et al., 2024). Those limitations underscore the need for implementing chemoinformatics tools capable of calculating molecular properties contingent on the ionization state of bioactive compounds as a function of environmental pH in CS research (Bertsch et al., 2023; Bertsch-Aguilar et al., 2024). This highlights that neglecting the pH-dependent behavior of bioactive compounds could limit the biological relevance of BioReCS. Consequently, future efforts should aim to incorporate protonation state dynamics to enhance their representativeness in pH-dependent CS analysis.
3.3 De novo generated libraries: expanding the BioReCS
In drug discovery and beyond, there is growing interest in creating on-demand, synthetically accessible virtual libraries for high-throughput screening (Perebyinis and Rognan, 2022; Grygorenko et al., 2020; Chávez-Hernández et al., 2023). Advances in generative models have accelerated the enumeration of the large and ultra-large chemical libraries, expanding the known chemical space and enabling the design of extensive libraries guided by structure or property constraints (Ye, 2024). However, evaluating the usefulness of such libraries requires more than sheer size; chemical diversity, as assessed through fingerprints, scaffolds, and physicochemical descriptors, is equally critical. Notably, a recent historical analysis of ChEMBL, PubChem, and DrugBank revealed that newer libraries are not necessarily more diverse (Lopez Perez et al., 2025). A similar trend could be observed for the continuously enumerated ultra-large chemical libraries, highlighting the need to quantify their chemical diversity using multiple structural representations. For BioReCS, we must consider not only the scale and diversity of expansion but also its direction—whether new molecules occupy unexplored regions or merely populate existing subspaces. Depending on the application area (e.g., drug discovery), the bioactivity profile should also be considered to avoid populating regions of BioReCS with promiscuous compounds associated with undesirable clinical effects.
3.4 Developing novel computational approaches
As the concept and application of chemical space evolve, so too must the computational tools used to explore it (Reymond, 2025). Novel or less conventional regions of drug-like space, such as bRo5 compounds discussed in Section 2.2, demand innovative methodologies or adaptations of existing ones. For instance, a recently developed hybrid fingerprint was designed specifically to accommodate metal-containing molecules, extending traditional organic-focused fingerprints by incorporating metal-specific features (López López and Medina-Franco, 2025). Looking ahead, we anticipate increasing use of hybrid computational workflows, which combine descriptor-based, rule-based, and AI-driven methods (Medina-Franco et al., 2024). In parallel, new methods for analyzing multiple dimensions and types of information—such as chemical multiverse analysis and the creation of consensus chemical spaces (Medina-Franco et al., 2022; Medina-Franco et al., 2019; López-López and Medina-Franco, 2023) —will enable more efficient use and integration of available data. Finally, machine learning models trained in known regions of BioReCS will play a pivotal role in navigating uncharted subspaces and improving coverage of BioReCS.
4 Summary
In this opinion article, we offered a holistic perspective on the biologically relevant chemical space (BioReCS) as a subset of the broader chemical universe. Effective navigation of BioReCS requires not only cataloging active compounds but also systematically reporting biologically inactive molecules, which help define the limits of relevance. While most of the explored regions focus on human-beneficial activities—such as therapeutic development, agriculture, and food sciences—BioReCS also includes dark regions populated by undesirable or toxic compounds. Recognizing and learning from these contrasts is essential for safer, ecologically responsible, and more targeted molecular design. The exploration of understudied ChemSpas may drive the development or refinement of computational tools, especially in cases where current methods fall short. Broadening the scope of BioReCS analysis—from both a structural and functional standpoint—could reveal hidden subspaces containing compounds with novel or unexpected biological activities. Importantly, training machine learning models on known BioReCS data will enhance our capacity to identify uncharted regions and optimize exploration strategies. As chemical databases continue to grow, it is important to emphasize that expansion alone does not equate to increased chemical diversity or biological relevance. Future research should consider not only the scale of these libraries but also their directionality, structural diversity, and applicability to real-world biological contexts.
Author contributions
JM-F: Conceptualization, Funding acquisition, Resources, Writing – review and editing, Writing – original draft, Project administration, Supervision, Formal Analysis. EL-L: Formal Analysis, Investigation, Writing – review and editing. JA-T: Formal Analysis, Investigation, Writing – review and editing. WZ: Funding acquisition, Formal Analysis, Writing – review and editing, Investigation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. We thank the Dirección General de Cómputo y de Tecnologías de la Información y Comunicación (DGTIC), Universidad Nacional Autónoma de México, for the computational resources to use Miztli under project LANCAD-UNAM-DGTIC-335. WZR thanks the Vice Chancellor for Research of the University of Costa Rica for its support via the research project 908-C3-610.
Acknowledgments
Insights and rich discussions with Karina Martinez-Mayorga and Gerald M. Maggiora are highly acknowledged. EL-L and JFA-T thank the Consejo Nacional de Humanidades, Ciencias y Tecnología (CONAHCyT) for the PhD scholarships 762342 (No. CVU: 894234), and 1270553, respectively.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AI, artificial intelligence; bRo5, beyond Rule of 5; ChemSpa, chemical subspace; CS, chemical space; BioReCS, biological-relevant chemical space; PROTACs, proteolysis-targeting chimeras; PPI, protein-protein interaction.
References
An, S., Lee, Y., Gong, J., Hwang, S., Park, I. G., Cho, J., et al. (2025). InertDB as a generative AI-expanded resource of biologically inactive small molecules from PubChem. J. Cheminformatics 17 (1), 49–14. doi:10.1186/s13321-025-00999-1
Annex on Chemicals (2025). OPCW. Available online at: https://www.opcw.org/chemical-weapons-convention/annexes/annex-chemicals/annex-chemicals (Accessed July 25, 2025).
Avellaneda-Tamayo, J. F., Chávez-Hernández, A. L., Prado-Romero, D. L., and Medina-Franco, J. L. (2024). Chemical multiverse and diversity of food chemicals. J. Chem. Inf. Model 64 (4), 1229–1244. doi:10.1021/acs.jcim.3c01617
Bento, A. P., Hersey, A., Félix, E., Landrum, G., Gaulton, A., Atkinson, F., et al. (2020). An open source chemical structure curation pipeline using RDKit. J. Cheminformatics 12 (1), 51. doi:10.1186/s13321-020-00456-1
Bertsch, E., Suñer, S., Pinheiro, S., and Zamora, W. J. (2023). Critical assessment of ph-dependent lipophilicity profiles of small molecules: which one should we use and in which cases? ChemPhysChem 24 (24), e202300548. doi:10.1002/cphc.202300548
Bertsch-Aguilar, E., Piedra, A., Acuña, D., Suñer, S., Pinheiro, S., and Zamora Ramírez, W. J. (2024). LiProS: FAIR simulation workflow to predict accurate lipophilicity profiles for small molecules. Am. Chem. Soc. (ACS). doi:10.26434/chemrxiv-2024-znppb-v2
Bhal, S. K., Kassam, K., Peirson, I. G., and Pearl, G. M. (2007). The rule of five revisited: applying log D in place of log P in drug-likeness filters. Mol. Pharm. 4 (4), 556–560. doi:10.1021/mp0700209
Cabas-Mora, G., Daza, A., Soto-García, N., Garrido, V., Alvarez, D., Navarrete, M., et al. (2024). Peptipedia v2.0: a peptide sequence database and user-friendly web platform. A major update. Database 2024, baae113. doi:10.1093/database/baae113
Capecchi, A., and Reymond, J.-L. (2021). Peptides in chemical space. Med. Drug Discov. 9, 100081. doi:10.1016/j.medidd.2021.100081
Capecchi, A., Zhang, A., and Reymond, J.-L. (2019). Populating chemical space with peptides using a genetic algorithm. J. Chem. Inf. Model. 60 (1), 121–132. doi:10.1021/acs.jcim.9b01014
Capecchi, A., Probst, D., and Reymond, J.-L. (2020). One molecular fingerprint to rule them all: drugs, biomolecules, and the metabolome. J. Cheminformatics 12 (1), 43. doi:10.1186/s13321-020-00445-4
Center for Drug Evaluation and Research (2025). Drugs@FDA data files. U.S. Food Drug Adm. Available online at: https://www.fda.gov/drugs/drug-approvals-and-databases/drugsfda-data-files (Accessed July 25, 2025).
Chandrasekhar, V., Rajan, K., Kanakam, S. R. S., Sharma, N., Weißenborn, V., Schaub, J., et al. (2024). COCONUT 2.0: a comprehensive overhaul and curation of the collection of open natural products database. Nucleic Acids Res. 53 (D1), D634–D643. doi:10.1093/nar/gkae1063
Chávez-Hernández, A. L., López-López, E., and Medina-Franco, J. L. (2023). Yin-yang in drug discovery: rethinking de novo design and development of predictive models. Front. Drug Discov. 3, 1222655. doi:10.3389/fddsv.2023.1222655
Choi, J., Yun, J. S., Song, H., Kim, N. H., Kim, H. S., and Yook, J. I. (2021). Exploring the chemical space of protein–protein interaction inhibitors through machine learning. Sci. Rep. 11 (1), 13369. doi:10.1038/s41598-021-92825-5
ClinicalTrials.gov (2025). ClinicalTrials.gov. Available online at: https://clinicaltrials.gov/(Accessed July 25, 2025).
Corrêa Veríssimo, G., Salgado Ferreira, R., and Gonçalves Maltarollo, V. (2024). Ultra-large virtual screening: definition, recent advances, and challenges in drug design. Mol. Inf. 44 (1), e202400305. doi:10.1002/minf.202400305
Cui, Z., Qi, C., Zhou, T., Yu, Y., Wang, Y., Zhang, Z., et al. (2025). Artificial intelligence and food flavor: how AI models are shaping the future and revolutionary technologies for flavor food development. Compr. Rev. Food Sci. Food Saf. 24 (1), e70068. doi:10.1111/1541-4337.70068
Danishuddin, M., Jamal, M. S., Song, K.-S., Lee, K.-W., Kim, J.-J., and Park, Y.-M. (2023). Revolutionizing drug targeting strategies: integrating artificial intelligence and structure-based methods in PROTAC development. Pharmaceuticals 16 (12), 1649. doi:10.3390/ph16121649
Davis, A. P., Grondin, C. J., Johnson, R. J., Sciaky, D., Wiegers, J., Wiegers, T. C., et al. (2020). Comparative toxicogenomics database (CTD): update 2021. Nucleic Acids Res. 49 (D1), D1138–D1143. doi:10.1093/nar/gkaa891
de Azevedo, D. Q., Campioni, B. M., Pedroz Lima, F. A., Medina-Franco, J. L., Castilho, R. O., and Maltarollo, V. G. (2024). A critical assessment of bioactive compounds databases. Fut. Med. Chem. 16 (10), 1029–1051. doi:10.1080/17568919.2024.2342203
Du, H., Zhang, X., Wu, Z., Zhang, O., Gu, S., Wang, M., et al. (2024). CovalentInDB 2.0: an updated comprehensive database for structure-based and ligand-based covalent inhibitor design and screening. Nucleic Acids Res. 53 (D1), D1322–D1327. doi:10.1093/nar/gkae946
Fourches, D., Muratov, E., and Tropsha, A. (2016). Trust, but verify II: a practical guide to chemogenomics data curation. J. Chem. Inf. Model. 56 (7), 1243–1252. doi:10.1021/acs.jcim.6b00129
Gómez-García, A., Acuña Jiménez, D. A., Zamora, W. J., Barazorda-Ccahuana, H. L., Chávez-Fumagalli, M. Á., Valli, M., et al. (2024). Latin American natural product database (LANaPDB): an update. J. Chem. Inf. Model. 64 (22), 8495–8509. doi:10.1021/acs.jcim.4c01560
Gong, L., Whirl-Carrillo, M., and Klein, T. E. (2021). PharmGKB, an integrated resource of pharmacogenomic knowledge. Curr. Protoc. 1 (8), e226. doi:10.1002/cpz1.226
Grygorenko, O. O., Radchenko, D. S., Dziuba, I., Chuprina, A., Gubina, K. E., and Moroz, Y. S. (2020). Generating multibillion chemical space of readily accessible screening compounds. iScience 23 (11), 101681. doi:10.1016/j.isci.2020.101681
Guo, X.-K., and Zhang, Y. (2022). CovBinderInPDB: a structure-based covalent binder database. J. Chem. Inf. Model. 62 (23), 6057–6068. doi:10.1021/acs.jcim.2c01216
Hamel, E. A., Castro, J. B., Gould, T. J., Pellegrino, R., Liang, Z., Coleman, L. A., et al. (2024). Pyrfume: a window to the world’s olfactory data. Sci. Data 11 (1), 1220. doi:10.1038/s41597-024-04051-z
Harrington, R. A., Adhikari, V., Rayner, M., and Scarborough, P. (2019). Nutrient composition databases in the age of big data: FoodDB, a comprehensive, real-time database infrastructure. BMJ Open 9 (6), e026652. doi:10.1136/bmjopen-2018-026652
Herrera-Rocha, F., Fernández-Niño, M., Duitama, J., Cala, M. P., Chica, M. J., Wessjohann, L. A., et al. (2024). FlavorMiner: a machine learning platform for extracting molecular flavor profiles from structural data. J. Cheminformatics 16 (1), 1–12. doi:10.1186/s13321-024-00935-9
Huang, Y., Dong, D., Zhang, W., Wang, R., Lin, Y.-C.-D., Zuo, H., et al. (2024). DrugRepoBank: a comprehensive database and discovery platform for accelerating drug repositioning. Database 2024, baae051. doi:10.1093/database/baae051
Ikeda, K., Maezawa, Y., Yonezawa, T., Shimizu, Y., Tashiro, T., Kanai, S., et al. (2023). DLiP-PPI library: an integrated chemical database of small-to-medium-sized molecules targeting protein–protein interactions. Front. Chem. 10, 1090643. doi:10.3389/fchem.2022.1090643
Kim, S., Chen, J., Cheng, T., Gindulyte, A., He, J., He, S., et al. (2024). PubChem 2025 update. Nucleic Acids Res. 53 (D1), D1516–D1525. doi:10.1093/nar/gkae1059
Kim, T., Baek, E., and Kim, J. (2025). Exploring macrocyclic chemical space: strategies and technologies for drug discovery. Pharmaceuticals 18 (5), 617. doi:10.3390/ph18050617
Knox, C., Wilson, M., Klinger, C. M., Franklin, M., Oler, E., Wilson, A., et al. (2023). DrugBank 6.0: the DrugBank knowledgebase for 2024. Nucleic Acids Res. 52 (D1), D1265–D1275. doi:10.1093/nar/gkad976
Kou, X., Shi, P., Gao, C., Ma, P., Xing, H., Ke, Q., et al. (2023). Data-driven elucidation of flavor chemistry. J. Agric. Food Chem. 71 (18), 6789–6802. doi:10.1021/acs.jafc.3c00909
López López, E., and Medina-Franco, J. L. (2025). Metal-FP: a hybrid molecular fingerprint to encode metal-based approved drugs. ChemRxiv 2025. doi:10.26434/chemrxiv-2025-6zh2h
Lopez Perez, K., López-López, E., Soulage, F., Felix, E., Medina-Franco, J. L., and Miranda-Quintana, R. A. (2025). Growth vs diversity: a time-evolution analysis of the chemical space. J. Chem. Inf. Model. 65 (13), 6788–6796. doi:10.1021/acs.jcim.5c00347
López-López, E., and Medina-Franco, J. L. (2023). Towards decoding hepatotoxicity of approved drugs through navigation of multiverse and consensus chemical spaces. Biomolecules 13 (1), 176. doi:10.3390/biom13010176
López-López, E., Fernández-de Gortari, E., and Medina-Franco, J. L. (2022). Yes SIR! on the structure–inactivity relationships in drug discovery. Drug disc. Today 27 (8), 2353–2362. doi:10.1016/j.drudis.2022.05.005
López-López, E., Robles, O., Plisson, F., and Medina-Franco, J. L. (2023). Mapping the structure–activity landscape of non-canonical peptides with MAP4 fingerprinting. Digit. Disc. 2 (5), 1494–1505. doi:10.1039/d3dd00098b
Lyu, J., Wang, S., Balius, T. E., Singh, I., Levit, A., Moroz, Y. S., et al. (2019). Ultra-large library docking for discovering new chemotypes. Nature 566 (7743), 224–229. doi:10.1038/s41586-019-0917-9
Lžičař, M., and Gamouh, H. (2024). CHEESE: 3D shape and electrostatic virtual screening in a vector space. ChemRxiv 2024. doi:10.26434/chemrxiv-2024-cswth
Manallack, D. T. (2007). The pKa distribution of drugs: application to drug discovery. Perspect. Med. Chem. 1, 1177391X0700100003. doi:10.1177/1177391x0700100003
Manallack, D. T., Prankerd, R. J., Nassta, G. C., Ursu, O., Oprea, T. I., and Chalmers, D. K. (2013). A chemogenomic analysis of ionization constants—Implications for drug discovery. ChemMedChem 8 (2), 242–255. doi:10.1002/cmdc.201200507
Martinez-Mayorga, K., and Medina-Franco, J. L. (2014). Foodinformatics: applications of chemical information to food chemistry. Springer. doi:10.1007/978-3-319-10226-9
Medina-Franco, J. L., Naveja, J. J., and López-López, E. (2019). Reaching for the bright StARs in chemical space. Drug disc. Today 24 (11), 2162–2169. doi:10.1016/j.drudis.2019.09.013
Medina-Franco, J. L., Rodríguez-Pérez, J. R., Cortés-Hernández, H. F., and López-López, E. (2024). Rethinking the “best method” paradigm: the effectiveness of hybrid and multidisciplinary approaches in chemoinformatics. Artif. Intell. Life Sci. 6, 100117. doi:10.1016/j.ailsci.2024.100117
Medina-Franco, J. L., Chávez-Hernández, A. L., López-López, E., and Saldívar-González, F. I. (2022). Chemical multiverse: an expanded view of chemical space. Mol. Inf. 41 (11), 2200116. doi:10.1002/minf.202200116
Meggers, E. (2007). Exploring biologically relevant chemical space with metal complexes. Curr. Op. Chem. Biol. 11 (3), 287–292. doi:10.1016/j.cbpa.2007.05.013
Nguyen, K. T., Blum, L. C., van Deursen, R., and Reymond, J. (2009). Classification of organic molecules by molecular quantum numbers. ChemMedChem 4 (11), 1803–1805. doi:10.1002/cmdc.200900317
Orsi, M., and Reymond, J. (2024). Navigating a 1E+60 chemical space of peptide/peptoid oligomers. Mol. Inf. 44 (1), e202400186. doi:10.1002/minf.202400186
Perebyinis, M., and Rognan, D. (2022). Overlap of on-demand ultra-large combinatorial spaces with on-the-shelf drug-like libraries. Mol. Inf. 42 (1), 2200163. doi:10.1002/minf.202200163
Price, E., Weinheimer, M., Rivkin, A., Jenkins, G., Nijsen, M., Cox, P. B., et al. (2024). Beyond rule of five and PROTACs in modern drug discovery: polarity reducers, chameleonicity, and the evolving physicochemical landscape. J. Med. Chem. 67 (7), 5683–5698. doi:10.1021/acs.jmedchem.3c02332
Reymond, J.-L. (2025). Chemical space as a unifying theme for chemistry. J. Cheminformatics 17 (1), 6. doi:10.1186/s13321-025-00954-0
Rigden, D. J., and Fernández, X. M. (2025). The 2025 nucleic acids research database issue and the online molecular biology database collection. Nucleic Acids Res. 53 (D1), D1–D9. doi:10.1093/nar/gkae1220
Schaub, J., Zielesny, A., Steinbeck, C., and Sorokina, M. (2021). Description and analysis of glycosidic residues in the largest open natural products database. Biomolecules 11 (4), 486. doi:10.3390/biom11040486
Schmidt, T., Samaras, P., Frejno, M., Gessulat, S., Barnert, M., Kienegger, H., et al. (2017). ProteomicsDB. Nucleic Acids Res. 46 (D1), D1271-D1281–D1281. doi:10.1093/nar/gkx1029
Shankavaram, U. T., Varma, S., Kane, D., Sunshine, M., Chary, K. K., Reinhold, W. C., et al. (2009). CellMiner: a relational database and query tool for the NCI-60 cancer cell lines. BMC Genomics 10 (1), 277. doi:10.1186/1471-2164-10-277
Sharma, A., Saha, B. K., Kumar, R., and Varadwaj, P. K. (2021). OlfactionBase: a repository to explore odors, odorants, olfactory receptors and odorant–receptor interactions. Nucleic Acids Res. 50 (D1), D678–D686. doi:10.1093/nar/gkab763
Sincere, N. I., Anand, K., Ashique, S., Yang, J., and You, C. (2023). PROTACs: emerging targeted protein degradation approaches for advanced druggable strategies. Molecules 28 (10), 4014. doi:10.3390/molecules28104014
Sosnin, S. (2025). Chemical space visual navigation in the era of deep learning and big data. Drug Discov. Today 30 (7), 104392. doi:10.1016/j.drudis.2025.104392
Srivastava, A., Pike, A., Swedrowska, M., Nash, S., and Grime, K. (2025). In vitro ADME profiling of PROTACs: successes, challenges, and lessons learned from analysis of clinical protacs from a diverse physicochemical space. J. Med. Chem. 68 (9), 9584–9593. doi:10.1021/acs.jmedchem.5c00358
Tihányi, J., Horváthová, E., Fábelová, L., Murínová, Ľ. P., Sisto, R., Moleti, A., et al. (2025). Environmental ototoxicants: an update. Environ. Sci. Pollut. Res. 32, 8629–8642. doi:10.1007/s11356-025-36230-9
Torchet, R., Druart, K., Ruano, L. C., Moine-Franel, A., Borges, H., Doppelt-Azeroual, O., et al. (2021). The iPPI-DB initiative: a community-centered database of protein–protein interaction modulators. Bioinformatics 37 (1), 89–96. doi:10.1093/bioinformatics/btaa1091
Valle-Núñez, G., Cedillo-González, R., Avellaneda-Tamayo, J. F., Saldívar-González, F. I., Prado-Romero, D. L., and Medina-Franco, J. L. (2025). Machine learning-driven antiviral libraries targeting respiratory viruses. Digit. Discov. 4, 1239–1258. doi:10.1039/d5dd00037h
Viarengo-Baker, L. A., Brown, L. E., Rzepiela, A. A., and Whitty, A. (2021). Defining and navigating macrocycle chemical space. Chem. Sci. 12, 4309–4328. doi:10.1039/d0sc05788f
Viayna, A., Matamoros, P., Blázquez-Ruano, D., and Zamora, W. J. (2024). From canonical to unique: extension of a lipophilicity scale of amino acids to non-standard residues. Explor Drug Sci. 2, 389–407. doi:10.37349/eds.2024.00053
Virshup, A. M., Contreras-García, J., Wipf, P., Yang, W., and Beratan, D. N. (2013). Stochastic voyages into uncharted chemical space produce a representative library of all possible drug-like compounds. J. Am. Chem. Soc. 135 (19), 7296–7303. doi:10.1021/ja401184g
Warr, W. A., Nicklaus, M. C., Nicolaou, C. A., and Rarey, M. (2022). Exploration of ultralarge compound collections for drug discovery. J. Chem. Inf. Model. 62 (9), 2021–2034. doi:10.1021/acs.jcim.2c00224
Wassermann, A. M., Lounkine, E., Hoepfner, D., Le Goff, G., King, F. J., Studer, C., et al. (2015). Dark chemical matter as a promising starting point for drug lead discovery. Nat. Chem. Biol. 11, 958–966. doi:10.1038/nchembio.1936
Whitty, A., and Zhou, L. (2015). Horses for courses: reaching outside drug-like chemical space for inhibitors of challenging drug targets. Fut. Med. Chem. 7 (9), 1093–1095. doi:10.4155/fmc.15.56
Wigh, D. S., Goodman, J. M., and Lapkin, A. A. (2022). A review of molecular representation in the age of machine learning. WIREs Comp. Mol. Sci. 12 (5), e1603. doi:10.1002/wcms.1603
Williams, A. J., and Richard, A. M. (2025). Three pillars for ensuring public access and integrity of chemical databases powering cheminformatics. J. Cheminf. 17, 40. doi:10.1186/s13321-025-00983-9
Williams, R. V., Amberg, A., Brigo, A., Coquin, L., Giddings, A., Glowienke, S., et al. (2016). It’s difficult, but important, to make negative predictions. Regul. Toxicol. Pharmacol. 76, 79–86. doi:10.1016/j.yrtph.2016.01.008
Ye, G. (2024). De novo drug design as GPT language modeling: large chemistry models with supervised and reinforcement learning. J. Comput.-Aided Mol. Des. 38, 20. doi:10.1007/s10822-024-00559-z
Zamora, W. J., Curutchet, C., Campanera, J. M., and Luque, F. J. (2017). Prediction of pH-dependent hydrophobic profiles of small molecules from miertus–scrocco–tomasi continuum solvation calculations. J. Phys. Chem. B 121 (42), 9868–9880. doi:10.1021/acs.jpcb.7b08311
Zamora, W. J., Campanera, J. M., and Luque, F. J. (2019). Development of a structure-based, ph-dependent lipophilicity scale of amino acids from continuum solvation calculations. J. Phys. Chem. Lett. 10 (4), 883–889. doi:10.1021/acs.jpclett.9b00028
Zdrazil, B. (2025). Fifteen years of ChEMBL and its role in cheminformatics and drug discovery. J. Cheminf. 17, 32. doi:10.1186/s13321-025-00963-z
Zdrazil, B., Felix, E., Hunter, F., Manners, E. J., Blackshaw, J., Corbett, S., et al. (2023). The ChEMBL database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nuc. Acids Res. 52 (D1), D1180–D1192. doi:10.1093/nar/gkad1004
Zhang, X., Betzi, S., Morelli, X., and Roche, P. (2014). Focused chemical libraries – design and enrichment: an example of protein–protein interaction chemical space. Fut. Med. Chem. 6 (11), 1291–1307. doi:10.4155/fmc.14.57
Zhang, Y., Lorsbach, B. A., Castetter, S., Lambert, W. T., Kister, J., Wang, N. X., et al. (2018). Physicochemical property guidelines for modern agrochemicals. Pest Manag. Sci. 74, 1979–1991. doi:10.1002/ps.5037
Keywords: chemoinformatics, dark chemical matter, de novo design, food chemicals, metallodrugs, natural products, odor chemicals, peptides
Citation: Medina-Franco JL, López-López E, Avellaneda-Tamayo JF and Zamora WJ (2025) On the biologically relevant chemical space: BioReCS. Front. Drug Discov. 5:1674289. doi: 10.3389/fddsv.2025.1674289
Received: 27 July 2025; Accepted: 12 August 2025;
Published: 25 August 2025.
Edited by:
Rodolpho C. Braga, InsilicAll, BrazilReviewed by:
Ho Leung Ng, Atomwise Inc, United StatesAndrea Trabocchi, University of Florence, Italy
Copyright © 2025 Medina-Franco, López-López, Avellaneda-Tamayo and Zamora. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José L. Medina-Franco, bWVkaW5hamxAdW5hbS5teA==