 Sandra Lorena Ament-Velásquez1†
Sandra Lorena Ament-Velásquez1† Veera Tuovinen1,2†
Veera Tuovinen1,2† Linnea Bergström1Toby Spribille3
Linnea Bergström1Toby Spribille3 Dan Vanderpool4
Dan Vanderpool4 Juri Nascimbene5Yoshikazu Yamamoto6
Juri Nascimbene5Yoshikazu Yamamoto6 Göran Thor2
Göran Thor2 Hanna Johannesson1*
Hanna Johannesson1*- 1Department of Organismal Biology, Uppsala University, Uppsala, Sweden
- 2Department of Ecology, Swedish University of Agricultural Sciences, Uppsala, Sweden
- 3Biological Sciences CW 405, University of Alberta, Edmonton, AB, Canada
- 4Department of Biology, Indiana University, Bloomington, IN, United States
- 5Department of Biological, Geological and Environmental Sciences, University of Bologna, Bologna, Italy
- 6Department of Bioproduction Science, Faculty of Bioresource Sciences, Akita Prefectural University, Akita, Japan
The study of the reproductive biology of lichen fungal symbionts has been traditionally challenging due to their complex lifestyles. Against the common belief of haploidy, a recent genomic study found a triploid-like signal in Letharia. Here, we infer the genome organization and reproduction in Letharia by analyzing genomic data from a pure culture and from thalli, and performing a PCR survey of the MAT locus in natural populations. We found that the read count variation in the four Letharia specimens, including the pure culture derived from a single sexual spore of L. lupina, is consistent with haploidy. By contrast, the L. lupina read counts from a thallus' metagenome are triploid-like. Characterization of the mating-type locus revealed a conserved heterothallic configuration across the genus, along with auxiliary genes that we identified. We found that the mating-type distributions are balanced in North America for L. vulpina and L. lupina, suggesting widespread sexual reproduction, but highly skewed in Europe for L. vulpina, consistent with predominant asexuality. Taken together, we propose that Letharia fungi are heterothallic and typically haploid, and provide evidence that triploid-like individuals are hybrids between L. lupina and an unknown Letharia lineage, reconciling classic systematic and genetic studies with recent genomic observations.
Introduction
The way an organism reproduces is one of its most important traits, since patterns of inheritance drastically affect a species ability to adapt and survive over evolutionary time (Bell, 1982; Whittle et al., 2011). In Fungi, mating types are key players in reproduction, as they define the sexual identity of an individual at the haploid stage, making it able to recognize and mate with compatible cell types (Kronstad and Staben, 1997). Researchers can use the structure of the mating-type (MAT) locus, and the frequency of mating types in natural populations to shed light on the reproductive biology of a species. In particular, the configuration of the MAT locus can inform us about the species' sexual system.
The most basic MAT configuration occurs in heterothallic ascomycetes, where either of two allelic variants (also referred to as idiomorphs as they represent highly dissimilar sequences that encode different transcription factors) are found at the MAT locus (Butler, 2007). One idiomorph contains a gene encoding a transcriptional activator protein with an α-box domain (referred to herein as MAT1-1-1, see Turgeon and Yoder, 2000), and the other contains a gene encoding a high-mobility group (HMG) protein (MAT1-2-1). In heterothallic species, successful mating is only possible between haploid individuals of opposite mating types (Glass et al., 1988). By contrast, both idiomorphs are often present in homothallic species, allowing them to mate with itself, and in many cases with any other individual (e.g., Billiard et al., 2012; Gioti et al., 2012). In addition, the idiomorphic regions of many ascomycetes harbor additional MAT genes, which may contribute to the sexual development of the species (e.g., Ferreira et al., 1998; Mandel et al., 2007; Tsui et al., 2013; Dyer et al., 2016; Hutchinson et al., 2016). Moreover, the relative frequency of allelic variants at the MAT-locus in natural populations can be used to investigate the reproductive mode of the species (e.g., Mandel et al., 2007), as an even distribution indicates frequent sexual reproduction while an uneven distribution points toward a relatively high frequency of asexual propagation. Within the class Lecanoromycetes, which includes many lichen symbionts, multiple studies have found a single mating type per thallus, indicating a widespread heterothallic sexual system (Singh et al., 2012; Ludwig et al., 2017; Allen et al., 2018; Dal Grande et al., 2018; Pizarro et al., 2019). However, homothallic Lecanoromycetes do exist, such as some members of the genus Xanthoria s. lat. (Honegger et al., 2004; Scherrer et al., 2005), emphasizing that taxa must be investigated in a case-by-case manner.
Ascomycete lichen symbionts are usually assumed to grow vegetatively as haploids, as the majority of Ascomycota (Honegger and Scherrer, 2008). However, there have been suggestions of alternative genetic compositions present in lichens. Examples include different nuclear organizations, like dikaryosis (n + n), diploidy, and higher ploidy levels (Tripp et al., 2017; Tripp and Lendemer, 2018; Pizarro et al., 2019), as well as intrathallus variation, in which multiple individuals (genotypes) of the lichen fungal symbiont come together into a single thallus but without cell fusion (Jahns and Ott, 1997). Cases of the latter phenomenon are also known as “chimeras” or “mechanical hybrids” (Murtagh et al., 2000; Dyer et al., 2001; Altermann, 2004). Commonly, authors make no distinction between different nuclear organizations and the “chimera” scenarios (Honegger et al., 2004; Mansournia et al., 2012). This is partly due to the fact that very little is known about the genetics and composition of thalli, as few species have been successfully grown in isolation from their symbiotic partners (Crittenden et al., 1995; Honegger et al., 2004; Honegger and Zippler, 2007). Historically, it has remained extremely difficult to artificially reconstruct the full lichen symbiosis and to mate different individuals in the lab (Bubrick and Galunj, 1986; Jahns, 1993; Stocker-Wörgötter, 2001). However, the advent of next generation sequencing technologies has opened the door for a re-evaluation of many of our assumptions on lichen biology, including the genetic composition of the dominant ascomycete partner (Tripp et al., 2017; Dal Grande et al., 2018; Armaleo et al., 2019; McKenzie et al., 2020). In particular, Tripp et al. (2017) inferred the ploidy level of seven unrelated fungal lichen symbionts using sequencing data from cultures derived from multiple spores, as well as from thalli containing all symbiotic partners (i.e., metagenomic data). They found evidence of some lineages deviating from haploidy. As compelling as such results may initially seem, such a pattern could have multiple explanations, not all of which involve ploidy changes. Multispore cultures could represent a collection of siblings that would naturally look different from haploid samples. Moreover, it is unclear if lichen fungi have a vegetative incompatibility system in place as other ascomycetes (Dyer et al., 2001; Sanders, 2014), which could influence the fusion or lack thereof between germinating spores. Thus, there is no clear expectation as to what would happen in a multispore culture in which different genotypes might meet and undergo cell fusion. Furthermore, the metagenomes studied might include sexual fruiting bodies (apothecia) or nearby tissue, which presumably contains fertilized (dikaryotic or diploid) cells. Hence, general ploidy levels in lichen thalli remain unclear.
In a recent study, McKenzie et al. (2020) used the Oxford Nanopore MinION technology to produce long-read data from whole thalli of two wolf lichens: Letharia lupina and Letharia columbiana. Unexpectedly, they discovered the presence of three Letharia genotypes in the metagenomic data of both samples, suggesting that Letharia fungal symbionts are triploid-like. They took advantage of the fact that one of the three genotypes within the L. lupina sample was markedly different from the other two, allowing them to phase the assembly graph and to produce a highly continuous haploid-like genome assembly. While methodologically convenient, their findings are nothing short of puzzling, since Letharia is a genus that belongs to the diverse family Parmeliaceae, whose members are heterothallic and in general assumed to be haploid (Honegger and Zippler, 2007; Pizarro et al., 2019). In plant and animal taxa, triploid individuals often have difficulties during meiosis as the chromosome numbers are unbalanced (Ramsey and Schemske, 1998; Tiwary et al., 2004; Köhler et al., 2010), and many higher ploidy populations often persist via asexual reproduction or selfing (Levin, 1975; Mable, 2003; Bicknell and Koltunow, 2004). The sexual system of Letharia has not been studied, but it can be hypothesized that it follows the heterothallic, self-infertile ancestral state. How, then, do triploid or triploid-like fungal symbionts deal with sexual reproduction under heterothallism?
In this study, we explored the reproductive biology of Letharia. In their seminal paper, Kroken and Taylor (2001a) studied the diversity within this genus and defined six cryptic, closely related species based on the analyses of 12 loci and their coalescent patterns. That and several subsequent studies have further refined these species hypotheses based on morphological characters, photosynthetic partner lineage, and their assumed main reproductive mode—sexual or asexual (Kroken and Taylor, 2001a; Altermann et al., 2014, 2016). Sexual Letharia lineages (referred to as “apotheciate”) produce multiple apothecia that release sexual fungal spores, while asexual lineages (“sorediate”) produce abundant asexual symbiotic propagules also known as soredia. The distinction, however, is not clear-cut as apotheciate species might have isidia, another type of symbiotic asexual propagule, and sorediate species occasionally produce apothecia (Kroken and Taylor, 2001a). The global species diversity center of Letharia is found in western North America, where all six lineages of the genus are present. In contrast, only the sorediate species L. lupina and L. vulpina are known in parts of Europe, Asia and Africa. Letharia vulpina, the only representative of the genus in Norway and Sweden, is red-listed in these countries, where it produces apothecia extremely rarely and the genetic variation is low (Högberg et al., 2002; Arnerup et al., 2004; Henriksen and Hilmo, 2015; SLU ArtDatabanken, 2020).
Here we aimed at collecting Letharia samples to represent the diversity of species as previously defined. We gathered genomic data from thalli of four taxa (as metagenomes), as well as from a pure culture derived from a single spore of the species L. lupina, and performed PCR surveys of the mating type in additional specimens. With this data in hand, we re-evaluated the ploidy level and verified the sexual system in Letharia. We also contrasted patterns of mating type frequencies in different populations of the sorediate species to gain insight into the occurrence of sexual vs. asexual reproduction in the species of conservation concern.
Materials and Methods
Lichen Material Used in the Study
We sequenced the genome and the transcriptome of the pure-cultured L. lupina (culture number: 0602M; Supplementary Table 1), originating from a single spore from Canada and maintained in the culture collection at Akita Prefectural University (Yamamoto et al., 1998). Likewise, we sequenced both the metagenome and the metatranscriptome of one lichen thallus from each of four Letharia taxa: L. columbiana, L. lupina, L. “rugosa,” and L. vulpina, all collected from Montana, U.S.A. (Supplementary Table 1, SRA: SRP149293 from Tuovinen et al., 2019; Figure 1A). In addition, we used 317 Letharia thalli for PCR analyses of the MAT locus, sampled from Sweden (N = 98; six localities with 1–20 thalli/locality), Switzerland (N = 18; one locality), Italy (N = 60; six localities with 10 thalli/locality) and the U.S.A. (N = 138; 14 localities with 1–31 thalli/locality; Supplementary Table 1). All lichen specimens were collected from different trees in order to avoid collecting clones of the individual thalli dispersed on the same tree trunk, with the exception of eight putative L. gracilis thalli which we received from B. McCune and that were all collected from the same tree. In Sweden, only a small part of each thallus (<3 cm in length) was collected due to the red-listed status of L. vulpina. After collection, the specimens were air-dried and stored at −20°C until DNA was extracted. The thalli used for RNA extraction were snap frozen in the field in liquid nitrogen, transferred to the lab and stored at −80°C until further processing. Letharia species were identified based on a combination of morphology and internal transcribed spacer (ITS) sequence, as the combination of these characteristics should distinguish the previously described cryptic species (Kroken and Taylor, 2001a). In addition, the algal ITS identity was used to assign two sorediate specimens to species following Altermann et al. (2016) (data not shown). We aligned the ITS sequences from all specimens included in this study (GenBank accessions MG645014–MG645052 from Tuovinen et al., 2019) with the previously published Letharia ITS sequences from Kroken and Taylor (2001a, TreeBASE ID # 1126), Altermann et al. (2014, TreeBASE ID # 15485), and Altermann et al. (2016, NCBI PopSet 1072900244) using MUSCLE in AliView v.1.18 (Larsson, 2014). Detailed information about the specimens, their collection sites and ITS variants is given in Supplementary Table 1, and the specimens are deposited in the Uppsala University Herbarium (UPS).
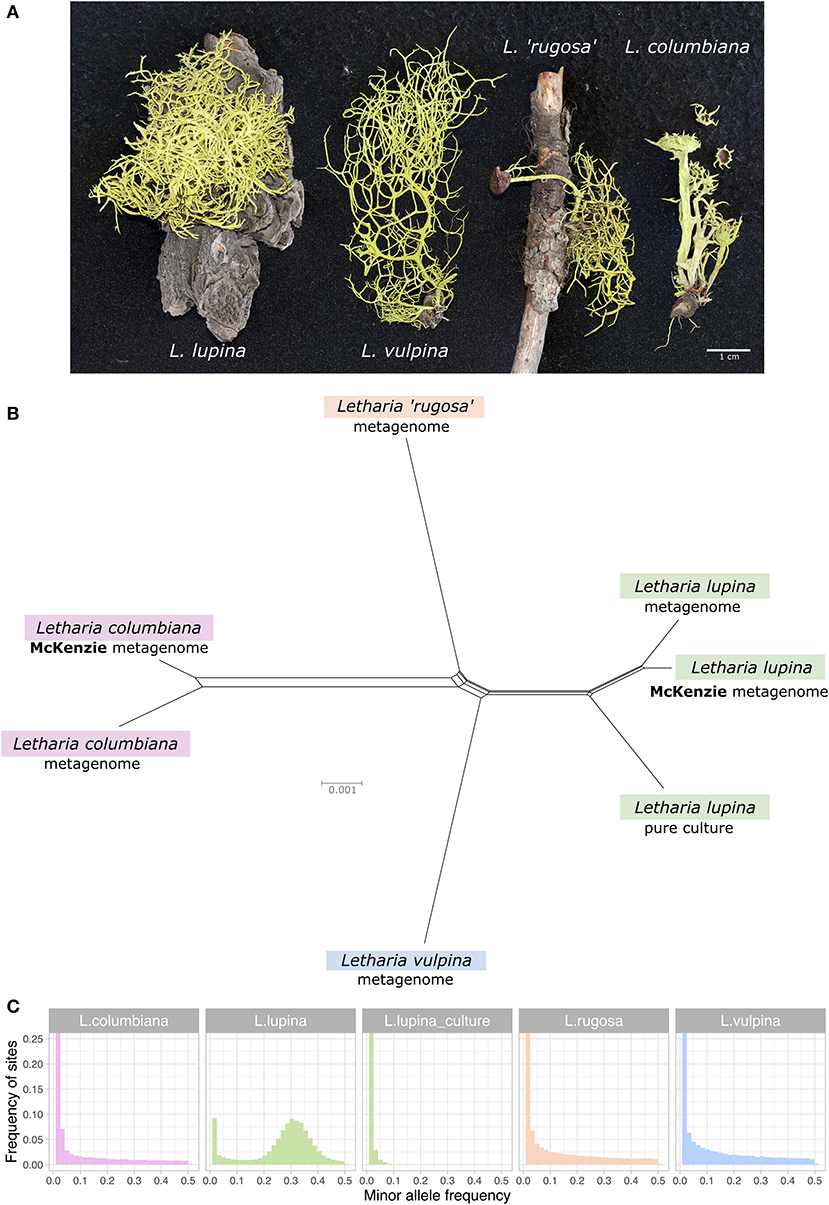
Figure 1. Relatedness and minor allele frequency (MAF) distributions of Letharia specimens. (A) Thalli used for metagenome sequencing in this study. (B) Unrooted NeighborNet split network based on 680 genes from our samples with genomic or metagenomic data available and those from McKenzie et al. (2020). (C) Folded MAF distributions of the metagenomes from Letharia specimens in (A), as well as the whole genome derived from the pure culture of a single L. lupina spore. All samples show distributions concordant with haploidy, with the exception of the triploid-like metagenome of L. lupina.
DNA and RNA Extraction
For the reference genome and transcriptome sequencing, mycelia of the pure culture of L. lupina was placed in a 2 ml safe lock tube with two sterile 3 mm tungsten carbide beads, frozen in a liquid nitrogen and pulverized in a TissueLyzer II (Qiagen) with 25 r/sec for 1 min. In addition, pieces of the vegetative thallus part, carefully avoiding any apothecia or their close proximity, of four Letharia used for metagenome and metatranscriptome sequencing and 317 specimens used for PCR screening were physically disrupted as described in Tuovinen et al. (2019). DNA was extracted from both mycelia and lichen tissue by using the DNeasy Plant Mini kit, following the manufacturer's instructions with a prolonged incubation at 65°C for 1 h. RNA of the mycelia from the pure culture was extracted with RNeasy Plant Mini Kit (Qiagen) with RLT buffer. RNA from the thalli was extracted with the Ambion Ribopure Yeast Kit (Life Technologies). For detailed description of DNA and RNA extraction of the lichen thalli, see Tuovinen et al. (2019).
Library Preparation and NGS Sequencing
Libraries for genome sequencing were prepared from 100 ng of DNA using the TruSeq Nano DNA Sample Preparation Kit (#15041110, rev B). The pure culture was sequenced twice on a split lane of Illumina MiSeq v3 with 300 bp paired-end sequencing. The DNA from each thallus was sequenced on 1/5 lane of Illumina HiSeq 2500 with 100 bp paired-end sequencing by the SNP&SEQ Technology Platform, Science for Life Laboratory at Uppsala University (Tuovinen et al., 2019).
Libraries for the mRNA sequencing were made by using the Illumina TrueSeq Stranded mRNA Kit (#15031047, rev E) and sequenced with Illumina HiSeq 2500. The transcriptome of the pure culture was sequenced on 1/5 of a lane at the SNP&SEQ Technology Platform, and the metatranscriptomes from the thalli on 1/6 of a lane each at the Huntsman Cancer Institute, University of Utah sequencing core (Tuovinen et al., 2019).
Quality Control and Genome and Transcriptome Assembly
Both the DNA and RNA raw reads were trimmed and quality controlled using Trimmomatic 0.32 (Bolger et al., 2014) (with options ILLUMINACLIP:/adapters.fa:2:20:10:4:true LEADING:10 TRAILING:10 SLIDINGWINDOW:4:15 MINLEN:25 HEADCROP:0). Data quality was inspected using FastQC (Patel and Jain, 2012). The draft genome of the pure cultured L. lupina was de novo assembled from the trimmed reads using SPAdes v. 3.5.0 (Bankevich et al., 2012) with k-mers 21,33,55,77,97,127 and the --careful flag. The raw reads of the metagenomes were assembled with SPAdes using k-mers 55, 75, 85, 95, as preliminary analyses using trimmed reads performed worse (not shown). Likewise, downstream analyses of the L. lupina pure culture were done with trimmed reads while the raw reads were used for the metagenomes. A summary of the genome assembly statistics is presented in Supplementary Table 2 as calculated with QUAST 3.2 (Gurevich et al., 2013). The filtered RNA reads of the pure cultured L. lupina, excluding the orphan reads produced after trimming, were assembled using Trinity 2.0.6 (Grabherr et al., 2011; Haas et al., 2013).
Genome Characterization of L. lupina
As the long-read L. lupina assembly from McKenzie et al. (2020), hereafter referred to as the McKenzie L. lupina assembly, was derived from an entire lichen thallus, we compared it to the SPAdes assembly of the pure-cultured L. lupina. We produced whole genome alignments with the NUCmer program from the MUMmer package v. 4.0.0beta2 (Kurtz et al., 2004) with options -b 2000 -c 200 --maxmatch. This analysis revealed only small alignments (<11 kbp) between the contig 1 of the McKenzie L. lupina assembly and multiple scaffolds from the SPAdes assembly of the pure-cultured L. lupina. To evaluate if the contig 1 of the McKenzie L. lupina assembly really is part of the genome of the ascomycete and not other symbiotic partners, we explored the repetitive content of both assemblies with RepeatMasker v. 4.0.8 (http://www.repeatmasker.org/) and the repeat library from McKenzie et al. (2020) with parameters -a -xsmall -gccalc -gff -excln.
Phylogenetic Analyses of Marker Genes
Alignments were produced with MAFFT version 7.245 (Katoh and Standley, 2013) and options --maxiterate 1000 --retree 1 --localpair. We used the program RAxML v. 8.0.20 (Stamatakis, 2014) to infer a Maximum Likelihood (ML) tree of the previously published Letharia ITS variants along with variants discovered in our data. Likewise, we produced ML trees from the idiomorph sequences (MAT1-1 or MAT1-2) as above but partitioning by gene and intergenic regions. Branch support was assessed with 1000 bootstrap pseudo-replicates. The specimens with unique ITS variants were putatively assigned to species based on the ML tree, previously published informative SNPs that can separate between L. vulpina and L. lupina, and previously described morphological characters (Kroken and Taylor, 2001a; McCune and Altermann, 2009; Altermann et al., 2014, 2016).
In order to place our genomic samples within the diversity of the genus, we extracted the sequence from 11 markers used in previous Letharia studies (ribosomal ITS, the 18S intron, chitin synthase I, and the anonymous loci 11, 2, 13, DO, CT, BA, 4, and CS) from the haploid individuals (see below) and fused them with the concatenated alignment from Altermann et al. (2016) (TreeBASE study ID S18729). We then inferred a ML topology with IQ-TREE v. 1.6.8 (Nguyen et al., 2015). Branch support was estimated with 100 standard bootstraps. Rooting was arbitrarily done with the well-supported L. columbiana (L. “lucida”) lineage.
Network Analysis of Genomic Data
Previous population studies using few markers have shown that the Letharia species are very closely related (Kroken and Taylor, 2001a; Altermann et al., 2014, 2016). In order to approximate the genetic distance and relationships between the Letharia individuals for which we had whole-genome data available, we used as reference the genome annotation of the McKenzie L. lupina. We randomly selected 1000 gene features, extracted their corresponding sequences from the McKenzie L. lupina assembly (including introns), and used them as queries in BLAST searches performed on the SPAdes assembly of our four metagenomes and the L. lupina pure culture. We also included the long-read assembly of the L. columbiana individual from McKenzie et al. (2020), hereafter referred to as the McKenzie L. columbiana. We filtered the BLAST output to include only hits that had at least a 250 bp-long hit alignment and 90% identity to the query. We extracted the sequence corresponding to the surviving hits and aligned them using MAFFT version 7.458 (Katoh and Standley, 2013) with options –adjustdirection --maxiterate 1000 --retree 1 --localpair. With the aim of approximating single-copy orthologs, only those alignments that contained a single hit for all samples were retained. The resulting 680 genes were concatenated into a matrix of 1,184,497 bp from which 35,458 bp were variable. We used the matrix as input for SplitsTree v. 4.14.16, build 26 Sep 2017 (Huson and Bryant, 2006) to produce an unrooted split network with a NeighborNet (Bryant and Moulton, 2004) distance transformation (uncorrected distances), an Equal Angle splits transformation and excluding gap sites.
Inferring Ploidy Levels Based on the Minor Allele Frequency (MAF) Distribution
As the McKenzie samples were inferred to be triploid-like (McKenzie et al., 2020), we determined the ploidy levels of our metagenomes and the pure culture by calculating the distribution of the minor (i.e., less common) allele in read counts of biallelic SNPs (Yoshida et al., 2013; Ament-Velásquez et al., 2016). Normally, a diploid individual should have a minor allele frequency (MAF) distribution around 0.5, while in a triploid individual the mode should be around 1/3. A tetraploid individual is expected to have two overlapping curves, one centered around 0.25 and another around 0.5. The width of the curves is proportional to factors like coverage variance, mapping biases, and hidden paralogy (Heinrich et al., 2011). In addition, any given sample is expected to have a large number of very low frequency alleles due to sequencing errors. As a result, a haploid sample should only exhibit a curve corresponding to the sequencing errors. Note that a caveat of this approach is that the haploid signal cannot be distinguished from an extremely homozygous higher-ploidy level individual, as these will show few heterozygous sites. Moreover, a diploid or higher ploidy signal in MAF distributions could correspond to a heterokaryon condition (n + n, n + n + n, etc.), which strictly speaking is composed of haploid nuclei. Hence, “ploidy” is used here in a broad sense to also include genetically different heterokaryons.
To produce MAF distributions, we mapped the Illumina reads of all of our metagenomes to the pure-cultured L. lupina, which should exclude most reads from other symbionts, using BWA v. 0.7.17 (Li and Durbin, 2010) with PCR duplicates marked by Picard v. 2.23.0 (http://broadinstitute.github.io/picard/). To call variants we used VarScan2 v. 2.4.4 (Koboldt et al., 2012) with parameters --p-value 0.1 --min-var-freq 0.005 to ensure the recovery of low frequency alleles and no further influence of the variant caller on the shape of the MAF distribution. Only biallelic SNPs from contigs larger than 100 kbp and with no missing data were used. We further filtered out all SNPs overlapping with repeated elements annotated above with RepeatMasker, and modified the output of VarScan2 to be compliant with the vcf format (https://samtools.github.io/hts-specs/). Based on the general coverage levels of all samples, we excluded variants with coverage above 300x or below 50x. These high quality SNPs were processed with the R packages vcfR v. 1.10.0 (Knaus and Grünwald, 2017), dplyr v. 0.8.0.1 (Wickham et al., 2018), ggplot2 v. 3.1.1 (Wickham, 2016), and cowplot v. 1.0.0 (Wilke, 2019). MAF distributions were produce from sites with MAF > 0 and scaled such that the area of all bars amount to 1. A full Snakemake v. 5.19.2 (Köster and Rahmann, 2012) pipeline is available at https://github.com/johannessonlab/Letharia.
Exploring the Origin of the Subgenomes Within the L. lupina Metagenome
As the L. lupina metagenome was suspected to be of hybrid origin (see Results), we attempted to determine the identity of the subgenomes by comparing them with the genome of the pure-cultured L. lupina as well as the genomes of the other sampled Letharia lineages (L. columbiana, L. rugosa, and L. vulpina). We assumed that the pure-cultured L. lupina corresponds to the “true” L. lupina genotype, and assigned all sites in the L. lupina metagenome with identical genotype to the pure culture as “lupina,” while all alternative alleles were assigned to “other.” We created an artificial haplotype of the “other” subgenome within the L. lupina metagenome by concatenating the “other” allele at every variable site in the set of high quality SNPs. Only sites with “other” allele frequency >0.2 were accepted as true “other.” Sites with frequency <0.1 were considered sequencing errors, and were assigned to the “lupina” allele instead. Remaining sites were treated as missing data to be conservative against potential paralogs. Equivalent sequences were created for all the haploid metagenomes and the pure culture, excluding sites with MAF <0.05. As a result, we obtained an alignment of variable sites, from which only sites genotyped for all samples were retained. We then divided the alignment of each contig by non-overlapping windows of 50 SNPs and calculated a Neighbor Joining tree of uncorrected distances per window using the R package phangorn v. 2.5.5 (Schliep, 2011), inspired by the logic of the program Twisst (Martin and Van Belleghem, 2017). We examined the topology of each window and recorded if the “lupina” and “other” haplotypes were sister to each other or to another taxon using the python library ETE 3 v. 3.1.1 (Huerta-Cepas et al., 2016). This analysis was done by rooting all genealogies with L. columbiana, since this is the best defined and less mixed of all Letharia lineages based on previous population and phylogenetic studies (Altermann et al., 2014, 2016). Windows of physical distance longer than 10 Kb were discarded as they often contain long tracks of missing data.
Identifying Scaffolds of the Genomes Containing the MAT Locus
We performed searches with the BLAST 2.2.31+ suit (Camacho et al., 2009) of the MAT-gene domains (HMG and α; see Martin et al., 2010) in the assembly of the L. lupina pure culture. As queries we used the corresponding gene sequences of Polycauliona polycarpa (Xanthoria polycarpa; Lecanoromycetes, Genbank accession numbers AJ884598 and AJ884599). Furthermore, we searched for APN2 (encoding a basic endonuclease/DNA lyase) and SLA2 (encoding a protein that binds to cortical patch actin) since these genes are typically flanking the MAT genes in Pezizomycotina (Heitman et al., 2007). As queries we used the ortholog clusters (OG5 126768 and OG5 129034 for APN2 and SLA2, respectively) retrieved from OrthoMCL-DB (Chen et al., 2006) and restricted to Pezizomycotina. Finally, we used the MAT locus retrieved from the L. lupina pure culture assembly as a query to perform BLAST searches against the assemblies of the metagenomes (L. lupina, L. vulpina, L. columbiana, and L. “rugosa”).
Annotating the MAT locus
We used three ab initio gene predictors to annotate Letharia scaffolds containing the MAT locus: SNAP release 2013-02-16 (Korf, 2004), Augustus v. 3.2.2 (Stanke and Waack, 2003), and GeneMark-ES v. 4.32 (Lomsadze et al., 2005; Ter-Hovhannisyan et al., 2008). All gene predictors were trained for the Letharia genomes as described in the Supplementary Materials.
We used STAR v. 2.5.1b (Dobin et al., 2013) to map filtered RNAseq reads to the entire genome assembly of the pure-cultured L. lupina, or to the relevant scaffold extracted from the metagenomes. Cufflinks v. 2.2.1 (Trapnell et al., 2010) was used to create transcript models from which ORFs were inferred using TransDecoder v. 3.0.1 (Haas et al., 2013). The ortholog sequences of APN2 and SLA2 of Aspergillus oryzae (OrthoMCL-DB: RIB40_aory) and the MAT genes of Polycauliona polycarpa were aligned to the Letharia scaffolds using Exonerate v. 2.2.0 (Slater and Birney, 2005) with the options —exhaustive TRUE —maxintron 1000. Using all these sources of evidence, we created weighted consensus gene structures using EVidenceModeler (EVM) v. 1.1.1 (Haas et al., 2008) that were subject to manual curation with Artemis release 16.0.0 (Rutherford et al., 2000).
To determine the identity of auxiliary MAT genes found in Letharia, we used the protein sequences of L. rugosa (MAT1-1) and L. columbiana (MAT1-2) to conduct BLASTp and DELTA-BLAST searches in the NCBI Genbank database (Boratyn et al., 2012) (searches conducted the 29th of April of 2020), as well as HMMER v. 3.3 (www.hmmer.org; Eddy, 2011) with the uniprotrefprot database, version 2019_09. We also compared the sequences with the protein models provided by Pizarro et al. (2019) for multiple lecanoromycete species. In addition, we re-annotated the scaffold with the MAT locus of the species Leptogium austroamericanum and Dibaeis baeomyces (Pizarro et al., 2019), as well as the MAT1-2 idiomorph of Cladonia grayi (accession number MH795990; Armaleo et al., 2019) using the pipeline above in order to confirm their gene order and composition. We found that the protein model of the MAT1-1 auxiliary gene as provided by Pizarro et al. (2019) is relatively variable at the C' terminus, which could be explained by inconsistencies in the gene annotation process in the absence of sources of evidence additional to the ab initio prediction (e.g., RNAseq data), or by true biological differences. Hence, only the first exon was analyzed further with IQ-TREE. Likewise, we only used the first and second exon of the MAT1-2 auxiliary gene for an IQ-TREE analysis.
PCR Screening of the Mating Type and Additional Annotation of the Idiomorphs
We designed idiomorph specific primer pairs based on the annotation of the MAT locus (MAT1-1: MAT11L6F and MAT11L6kR, MAT1-2: MAT12LF1 and MAT12LR1; Supplementary Table 3) to screen for the mating-type ratios in natural populations. Each sample was amplified with primer pairs for both idiomorphs and the presence/absence of the PCR-product of each idiomorph was determined by using agarose gel electrophoresis. A subsample of PCR products from each idiomorph was sequenced to ensure the amplification of the correct target sequence with the PCR primers. We tested whether the distribution of mating types in the sorediate species (L. lupina and L. vulpina) in U.S.A., the Alps and Sweden follows the 1:1 ratio expected for a population that regularly reproduces sexually by using an exact binomial test with 95% confidence interval.
In order to sequence and annotate both idiomorphs for each Letharia species, we selected specimens of the same species (based on ITS sequence information) as the individual from which the genomic data originated, but of the opposite mating type, and amplified a circa 3,800 bp long part of the MAT locus using primers LMATF2 and LMATR1 and APN2LF and SLA2LR (Supplementary Table 3). The same procedure was done for L. “barbata” and the putative L. gracilis specimens. The entire MAT locus was then sequenced with primers designed for Letharia based on the genome annotations (Supplementary Table 3). The PCR-obtained sequences of the idiomorphs were annotated using the trained ab initio gene predictors and the alignments (with Exonerate) of the final gene models. We define the idiomorphic part of the MAT locus as the whole region between APN2 and SLA2 that lack sequence similarity between individuals of different mating types.
Results
Letharia Species in the Study
Based on morphology and ITS sequence variation, we assigned the collected 322 specimens to five of the six previously recognized Letharia species (Kroken and Taylor, 2001a; Altermann, 2009; Altermann et al., 2016), including those used for metagenome sequencing (see Supplementary Materials). While traditionally-used markers generally provide little resolution to the Letharia species complex (Supplementary Figures 1, 2; Altermann et al., 2014, 2016), a NeighborNet split network of 680 genes extracted from all (meta)genome assemblies confirmed clustering by species for these samples (Figure 1B).
High Correspondence Between Our Pure Culture Assembly and the McKenzie Assembly of L. lupina
The McKenzie L. lupina assembly was produced by manual curation of metagenomic long-read data and is one of the best genome assemblies for an ascomycete lichen symbiont currently available (McKenzie et al., 2020). However, it could still contain sequences from other symbiotic partners. To investigate this possibility, we compared the L. lupina pure culture assembly generated by us to the McKenzie L. lupina assembly and found that all contigs, except contig 1, have good correspondence between the two. There are very few obvious translocations or inversions, which might be either assembly artifacts or biological differences between samples (Supplementary Figure 3). Conversely, contig 1 only produced very small (<11 kbp) alignments to multiple scaffolds of the L. lupina pure culture assembly (Supplementary Figure 4). This contig is the largest in the McKenzie L. lupina assembly (>3 Mbp), with a size comparable to the smallest chromosomes of well-studied ascomycetes like Podospora anserina (Espagne et al., 2008). Remarkably, contig 1 is composed of 87% repetitive elements, in stark contrast with most other contigs that harbor around 30% repetitive elements (Supplementary Figure 5). We reasoned that if the repetitive elements present in contig 1 also occur in the other contigs of either the McKenzie L. lupina or the pure-cultured L. lupina, then this large contig is indeed part of the L. lupina genome and not of another symbiont. Accordingly, we found that the five most abundant repeats of contig 1 are present elsewhere in both assemblies (Supplementary Figures 6, 7), and were probably not recovered as such in the pure culture assembly due to the limitations of short-read technology. Hence, we conclude that the McKenzie L. lupina assembly does reflect the genome of the ascomycete L. lupina and the observation of a triploid-like signal is not an artifact of additional unrelated fungal sequences in their assembly.
Genomic and Metagenomic Datasets Show Haploid-Like Patterns Except for the Metagenome of L. lupina
As the results of McKenzie et al. (2020) showed a triploid-like signal for L. lupina and L. columbiana, we inferred the ploidy levels in our genomic and metagenomic samples by producing MAF distributions using the L. lupina pure culture as reference for variant calling. As expected under haploidy, all but one sample show an exponential decay of low frequency variants produced by sequencing errors, including the L. lupina pure culture (Figure 1C). The exception to the haploid pattern is our L. lupina metagenome, which has a clear peak centered around 1/3, consistent with triploidy. Note that the exponential decay of the variants of the L. lupina pure culture is steeper than for the other samples. This difference results from higher coverage and longer reads, as the L. lupina pure culture was sequenced with MiSeq, as opposed to HiSeq for other samples, leading to better read mapping. In addition, its own genome was used as reference for mapping, minimizing the variance further. We obtained similar results using the McKenzie L. lupina as reference (data not shown).
Letharia Has a Heterothallic MAT-Locus Architecture
Having established that most metagenomes and the pure-cultured L. lupina are most likely haploid, we proceeded to characterize the MAT locus to give us an indication of the sexual system in Letharia. The MAT locus with the flanking genes APN2 and SLA2 was found within a single scaffold for the pure-cultured L. lupina (submitted to GenBank under the accession number MK521630) and from the metagenomes of L. columbiana, L. “rugosa,” and L. vulpina (GenBank numbers MK521629, MK521632 and MK521631, respectively). Only one MAT idiomorph was recovered in each assembled genome, suggesting that Letharia is heterothallic.
The genome of the pure-cultured L. lupina and the metagenomes of L. vulpina, and L. “rugosa” contained the MAT1-1 idiomorph (with the α domain of the MAT1-1-1 gene), while the MAT1-2 idiomorph (with the HMG domain of the MAT1-2-1 gene) was found in the metagenome of L. columbiana. Figure 2 shows the gene content of the MAT1-1 idiomorph as represented by L. “rugosa,” and the MAT1-2 as represented by L. columbiana. Notably, alignments with Exonerate revealed the remnants of the last exon of the MAT1-1-1 gene in the idiomorph MAT1-2 (the * in Figure 2B). Albeit homologous, this piece of exon is highly diverged from the MAT1-1-1 in Letharia MAT1-1, as revealed by the percentage of pairwise identity along the idiomorphs (Figure 2C). This same pattern was observed in the species of the genus Cladonia (family Cladoniaceae) by Armaleo et al. (2019), suggesting that the pseudogenized exon might be ancestral to both families. In addition, based on transcriptomic data of L. columbiana, a smaller ORF was weakly predicted downstream of APN2 in both idiomorphs (as lorf, from Letharia open reading frame, in Figure 2; see also Supplementary Figures 8–11). Likewise, the transcriptomic data revealed an antisense transcript for the MAT1-1-1 gene in all species (Figure 2 and Supplementary Figures 8–11). See Supplementary Materials for more details.
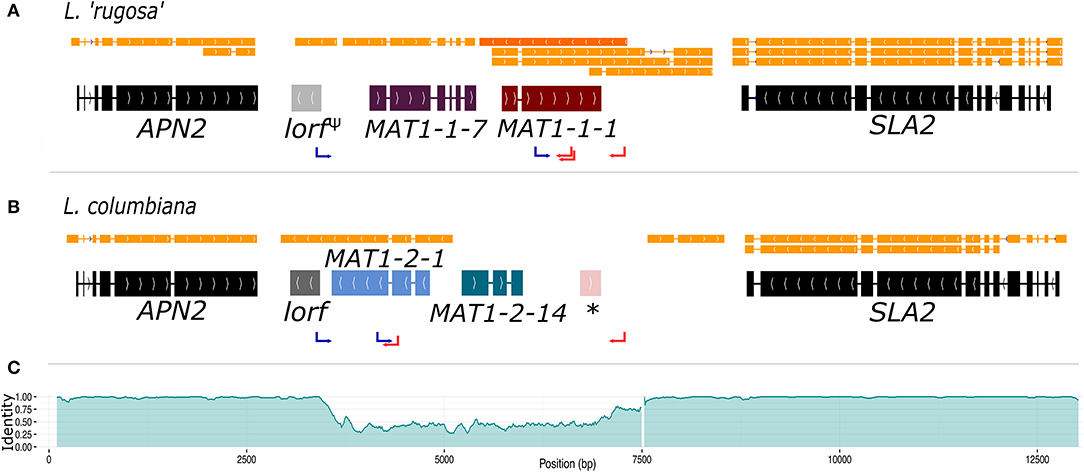
Figure 2. The structure of the MAT idiomorphs in the genus Letharia. The typical gene content of the MAT1-1 idiomorph as exemplified by L. “rugosa” (A) is contrasted to the MAT1-2 idiomorph of L. columbiana (B). The final gene models with intron-exon boundaries are shown along with the transcript models (orange). In (A) the antisense transcript of the MAT1-1-1 gene is marked with a darker orange color. Notice that the lorf gene is pseudogenized in L. “rugosa” (lorfΨ). In (B) a star (*) shows the location of a sequence homologous, but highly divergent, to the last exon of the MAT1-1-1 gene. The pairwise identity between idiomorphs of the MAT locus is plotted in (C) using overlapping windows of 100 bp (incremental steps of 10 bp along the sequence), showing the drop in identity. The white gap toward the 3′ end of the area of low identity corresponds to a track of 129 bp in the MAT1-2 idiomorph of all species. The positions of forward (blue) and reverse (red) primers used for PCR screening are marked with arrows.
We further obtained the sequence of the MAT region from additional 46 thalli by PCR, which corresponded to both the MAT1-1 or MAT1-2 idiomorphs for all species (except L. cf. gracilis, for which all samples had a MAT1-2 idiomorph). As the idiomorphs are very similar between species (indicated by scale bars of MAT gene phylogenies in Supplementary Figure 12), we can use the sequence comparison between L. rugosa and L. columbiana to estimate the size of the non-recombining region in Letharia by examining the level of divergence between genomic sequences of MAT1-1 and MAT1-2 (Figure 2C). Toward APN2, the identity between sequences drops from 98 to 99% (overlapping windows of 100 bp with incremental steps of 10 bp) to on average 48%, marking the beginning of the idiomorphic region. On the 3′ end, toward SLA2, a plateau region of around 70% identity covers approximately 500 bp. The sequence identity reaches this plateau of intermediate values just before the end of the idiomorph, and it is followed by an indel of 129 bp present in MAT1-2, but not in the MAT1-1, for all Letharia species. We consider the 3′-end of this indel the boundary for the 3′-end of the idiomorph. Using these boundaries, we estimate that the size of each idiomorph of Letharia is around 3.8 kbp.
The Auxiliary Genes Present in Letharia MAT Idiomorphs Are Ancestral to the Lecanoromycetes
We found that both idiomorphs of the MAT locus of each Letharia species harbor additional ORFs (one in each idiomorph) designated herein as MAT1-1-7 and MAT1-2-14 (Figure 2). The nomenclature of these additional genes is contentious and discussed extensively in the Supplementary Materials.
Pizarro et al. (2019) reported a number of ORFs associated with the lecanoromycete idiomorphs, but it was not clarified whether these so-called “auxiliary MAT genes” are homologous across lichen symbionts or other fungal taxa, making comparisons difficult. Hence, we used protein sequence and synteny comparisons to define homology of these genes. We found that most, but not all, Lecanoromycetes have the same auxiliary gene in the MAT1-1 idiomorph as Letharia (i.e., MAT1-1-7; left panel of Figure 3) and at the same location (data not shown). In addition, this ORF seems to be homologous to a gene predicted for members of the class Eurotiomycetes (left panel of Figure 3). By contrast, the auxiliary genes present in the MAT1-1 idiomorph of Leptogium austroamericanum (Collemataceae, Lecanoromycetes) and Dibaeis baeomyces (Icmadophilaceae, Lecanoromycetes) (Pizarro et al., 2019) are not related to MAT1-1-7, and are located in a different position (between MAT1-1-1 and SLA2).
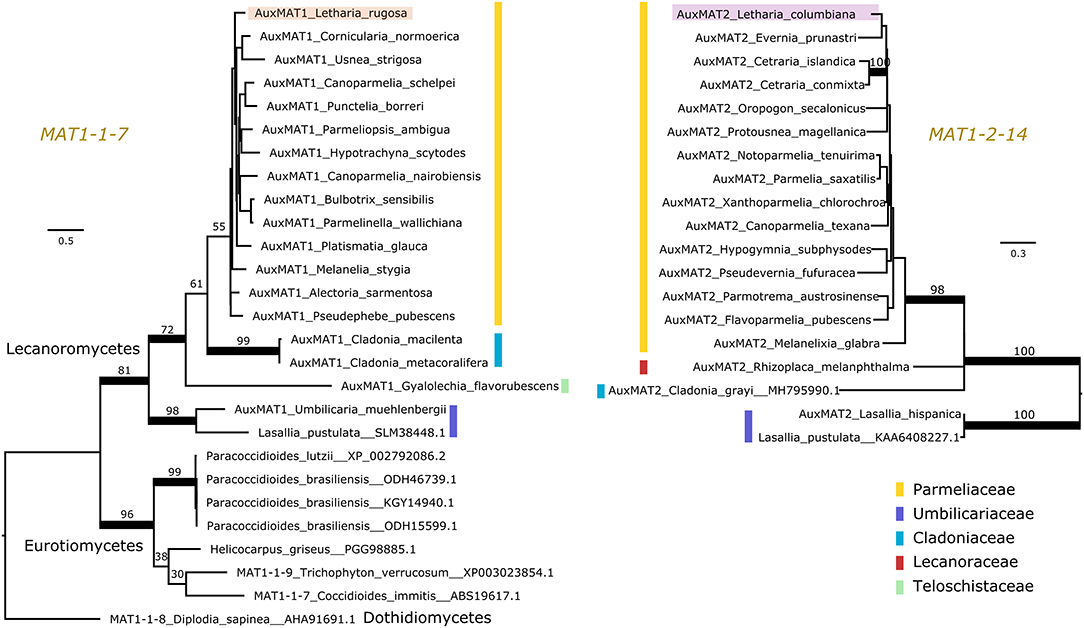
Figure 3. Maximum likelihood trees of the auxiliary genes MAT1-1-7 (Left) and MAT1-2-14 (Right). Colored vertical bars mark the different families within the lichen-forming class Lecanoromycetes, and the Letharia sequences are highlighted. MAT1-1-7 is found outside Lecanoromycetes, while MAT1-2-14 seems unique for this class. Notice that the homologous genes of MAT1-1-7 in Trichophyton verrucosum and Diplodia sapinea have been named differently by other authors. Branches with bootstrap values above 70% are thickened. Branch lengths are proportional to the scale bar (amino acid substitution per site).
For the Letharia auxiliary gene in the MAT1-2 idiomorph (MAT1-2-14 in Figure 2), we also found homologs in other Lecanoromycetes (right panel of Figure 3), but no homolog outside of Lecanoromycetes. This gene was not identified by Armaleo et al. (2019) in Cladonia, but our prediction pipeline recovered it in the Cladonia grayi genome (right panel of Figure 3) in the expected position.
Taken together, our data shows that the general idiomorph architecture of Letharia is conserved at least between Parmeliaceae, Cladoniaceae, and Umbilicariaceae (Figure 3). Given the presence of MAT1-1-7 in a member of Teloschistaceae, and of MAT1-2-14 in a member from Lecanoraceae (Figure 3), potentially the general structure of the idiomorph is the same also in these families.
The Allele Frequencies Along the Genome of the L. lupina Metagenome Suggest Hybridization
To explore the triploid-like signal in our L. lupina metagenome, we mapped its reads to the pure culture assembly. We assigned the SNPs used for the MAF distribution as either matching the pure culture (“lupina” allele) or having an alternative allele (“other”) and produced an unfolded allele frequency distribution (c.f. Bustamante et al., 2001) of the metagenome sample (Figure 4). We found that the “lupina” allele has similar contributions to the 1/3 and 2/3 modes, suggesting the three chromosomal sets in the metagenome are similarly related to the reference. However, plotting the distribution of the “other” allele along the pure culture assembly revealed areas where the “other” allele maintains a single mode (either 1/3 or 2/3) for several consecutive thousands of base pairs (i.e., in blocks), as well as areas of loss of heterozygosity (LOH), mostly in favor of the “lupina” allele (Figure 5). The regions with LOH are not explained by poor mapping of the “other” genotype into the reference pure culture, as their coverage is comparable to that of polymorphic sites (Supplementary Figure 13). This observed genome-wide allele distribution patterns indicates that the three haplotypes within the L. lupina metagenome are not equally related to the reference pure culture across the genome. Such signal is highly suggestive of hybridization between L. lupina and another Letharia lineage, presumably leading to triploidy (i.e., allopolyploidization) or to a triploid-like condition (n + n + n or 2n + n). In addition, the alternating block pattern suggests that there was recombination in the history of the metagenome chromosomes, and/or that the reference pure-culture itself is a hybrid.
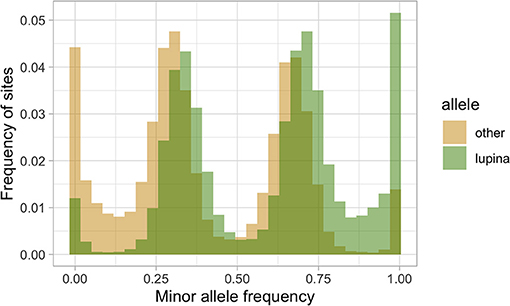
Figure 4. Unfolded allele frequency distribution of the L. lupina metagenome, with alleles classified as either identical to that of the L. lupina pure culture (“lupina”) or an alternative allele (“other”).
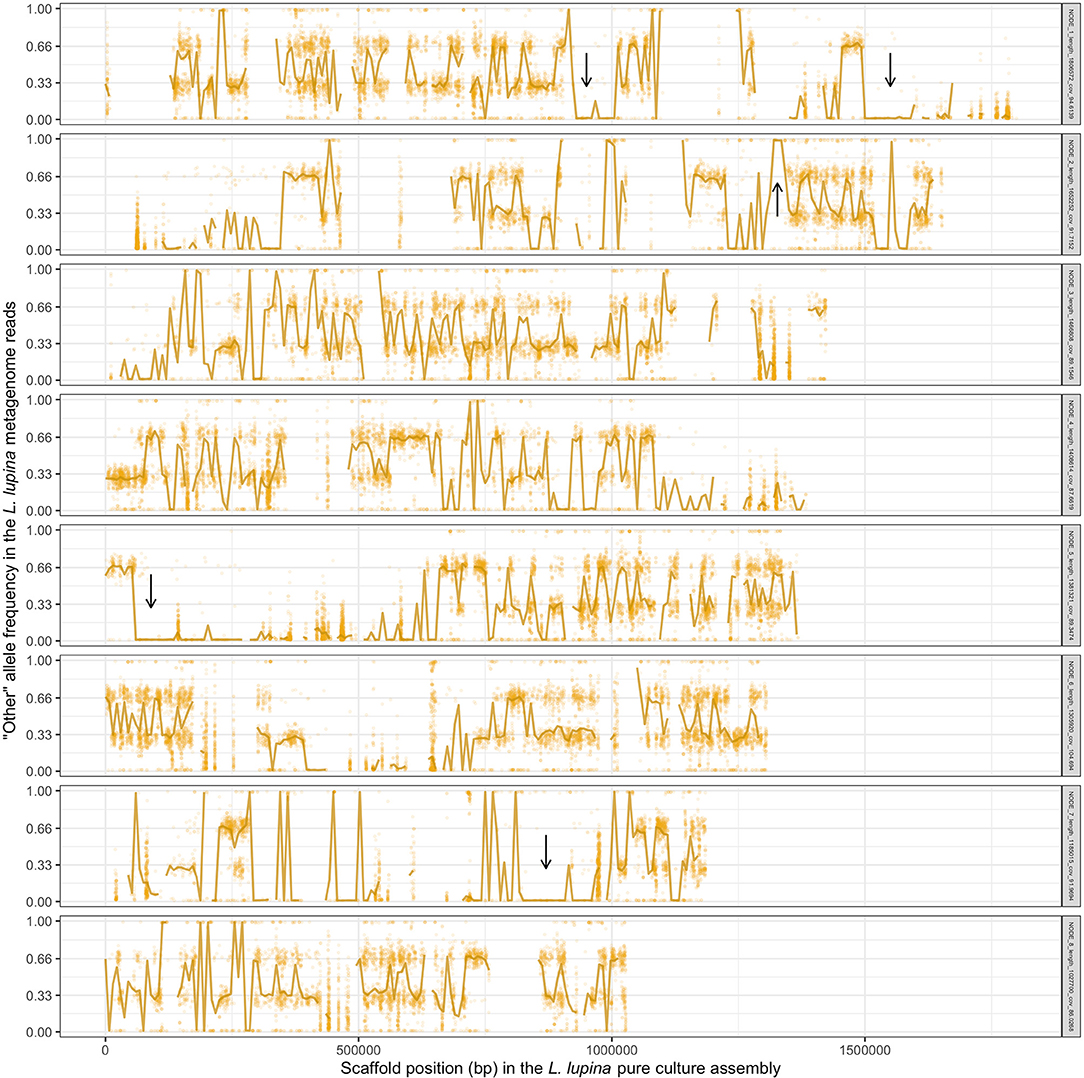
Figure 5. Frequency of the “other” (alternative) allele in the reads of the L. lupina metagenome when mapped to the L. lupina pure culture assembly (only the eight largest scaffolds are shown). Golden points represent the raw allele frequency at each site, while the solid line connects the median allele frequency of non-overlapping 7.5 kb-long windows. Sites and windows overlapping with repetitive elements were discarded (missing data). Representative areas with long tracks of loss of heterozygosity (LOH) are marked with arrows.
The reads of the L. lupina metagenome that map to the scaffold containing the MAT locus also maintain a 2/3 proportion in favor of the “lupina” allele (Figure 6). BLAST searches revealed that our L. lupina metagenome contains both MAT idiomorphs. As expected, the coverage of the MAT1-1 idiomorph is around 2/3 of the mean (Supplementary Figure 14), confirming that two chromosome sets are MAT1-1 and the other is MAT1-2. In addition, we found both idiomorphs in the raw reads of the McKenzie L. columbiana metagenome, but not in the McKenzie L. lupina one, in which we only found MAT1-1. Thus, the triploid-like Letharia thalli can have either both or a single mating type.
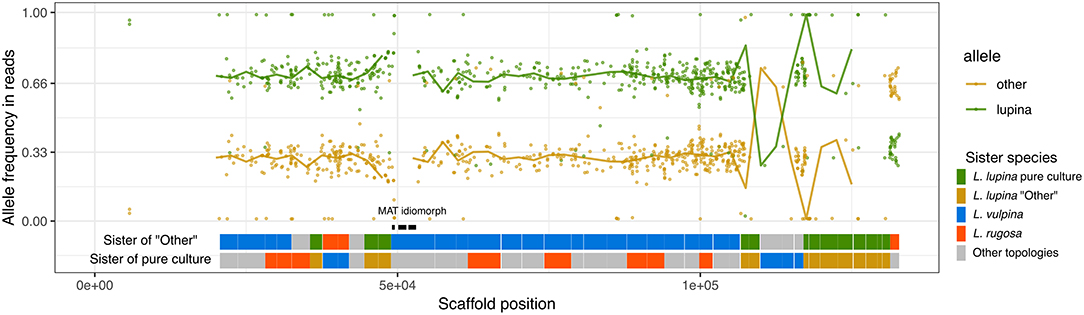
Figure 6. Allele frequencies and ancestry of the L. lupina metagenome variants around the MAT locus. The L. lupina pure culture was used as reference to classify each allele as either “lupina” (reference) or “other” (alternative). Points represent the raw allele frequency at each site, while the solid lines connect the median allele frequency of non-overlapping 2.5 kb-long windows. Sites and windows overlapping with repetitive elements were discarded (missing data). The genes within the MAT1-1 locus are marked with black rectangles. Below the allele frequencies, two horizontal bands represent the inferred sister of the “other” (upper band) and “lupina” (lower band) components across the contig, inferred from genealogies of non-overlapping windows of 50 SNPs each. Genealogies were rooted with L. columbiana.
In an effort to determine the unknown parental lineage, i.e., the origin of the “other” allele, we estimated the phylogenetic relationship between the “other” haplotype, the L. lupina pure culture, and additional sampled species along the genome using non-overlapping windows. For the contig containing the MAT locus in particular, we found that the “other” allele clusters mostly as sister to L. vulpina, while the L. lupina pure culture is occasionally sister to L. “rugosa” (Figure 6). Previous population studies concluded that L. vulpina and L. lupina are not each other's closest relatives (Altermann et al., 2014, 2016). As the similarity of “other” with L. vulpina is continuous for thousands of base pairs, it is likely not due to incomplete lineage sorting. Thus, there is the possibility that L. vulpina (or a closely related lineage) has contributed to the ancestry of the L. lupina metagenome through hybridization. Notably, in the phylogeny of the MAT1-1 idiomorph, some L. vulpina individuals are forming a clade with L. lupina, albeit with moderate support (Supplementary Figure 12). Nonetheless, this signal is less clear in other parts of the genome, where sometimes it is the pure culture that clusters with L. vulpina while the “other” haplotype associates with L. “rugosa,” or simply other topologies are recovered (Supplementary Figure 15). Thus, these results support that the L. lupina pure culture itself is admixed and/or that more complicated gene flow combinations are affecting these Letharia lineages.
Regional Distribution of Mating Types in Sorediate Species Shows Variation in Reproductive Mode
We amplified a single idiomorph in all specimens collected, with two exceptions being one L. vulpina from Italy and one L. lupina from Switzerland. From these two, we amplified both idiomorphs (Supplementary Table 1). With the aim to investigate the primary reproductive mode of the sorediate species (L. lupina and L. vulpina), we characterized the regional occurrence of their mating types. In the U.S.A., even when only a few individuals of each species were collected from the same locality, both mating types were sampled. The mating-type ratio in the U.S.A. (when all American samples of a species were treated as one region) of neither sorediate species deviated from a 1:1 ratio (exact binomial test with 95% confidence interval: N = 85; p = 0.8 for L. lupina and N = 13; p = 1 for L. vulpina). Furthermore, both mating types were present in L. lupina in the Alps and the mating-type ratio in these populations did not deviate from 1:1 (N = 18; p = 0.2). However, the ratio of mating types in L. vulpina is significantly skewed from a 1:1 ratio (N = 59; p = 1.8e-10) in the Alps, and in Sweden only one mating-type was found among 98 samples (Figure 7 and Supplementary Table 1). These findings suggest that sorediate species reproduce sexually in U.S.A, but that L. vulpina reproduces sexually at very low frequency in Europe.
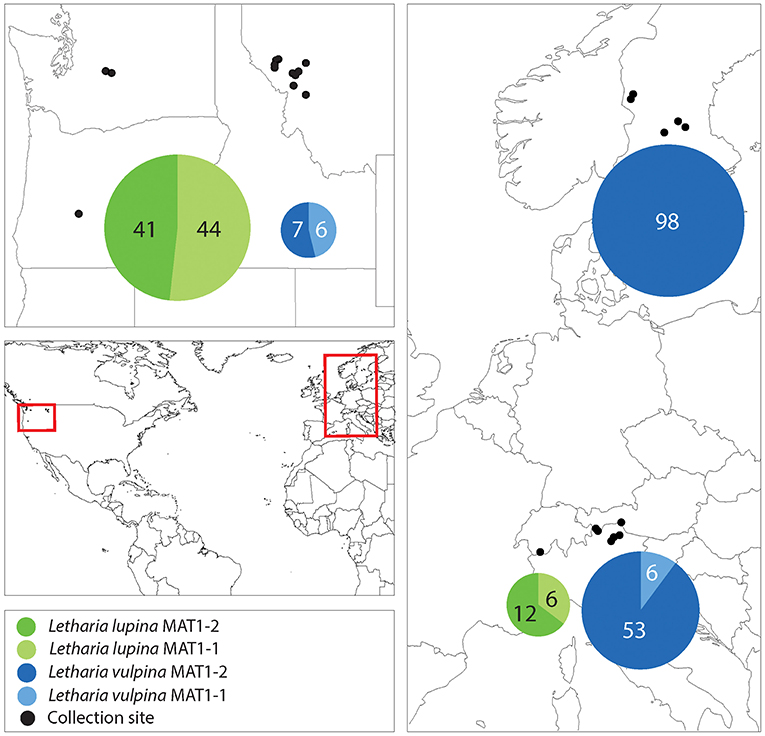
Figure 7. A map showing the collection sites and mating-type distribution in the sorediate species in the U.S.A., the Swiss and Italian Alps and Sweden. The numbers correspond to the number of sampled thalli (n), and the areas of the pie charts are proportional to n.
Discussion
Our combined approach of next-generation sequencing from representative samples, along with regional PCR surveys of the mating types, has allowed us to revise key aspects of the reproductive biology of Letharia. Specifically, we were able to determine haploidy in most genomic samples and a triploid-like signal in one. Furthermore, we revealed conserved heterothallism across the genus, and we uncovered differences in mating type frequencies between North American and European specimens of sorediate species. McKenzie et al. (2020) qualified the genome architecture of Letharia as “enigmatic,” referring to the triploid-like nature of their two samples. Here we further point out the unexpectedly high content of repeated elements in the contig 1 of their L. lupina assembly. Future research should aim at clarifying if this contig is a core feature of the Letharia genome, or if it represents some form of accessory or B chromosome, which typically have a high repeat content (Bertazzoni et al., 2018; Ahmad and Martins, 2019).
In contrast to previous studies addressing ploidy levels (Tripp et al., 2017; McKenzie et al., 2020), we made use of a pure culture derived from a single spore. This approach removes confounding factors related to the presence of other unrelated fungi within the thallus, and to unknown effects of growing alongside siblings. In effect, our pure culture represents the immediate state after sexual reproduction in L. lupina. The fact that the metagenomes of L. columbiana, L. “rugosa,” and L. vulpina are also haploid further suggests that a single spore is enough, with symbionts, for the development of a lichen thallus, and that a Letharia lichen is not formed from a collection of spores as it has been suggested for other species (Jahns and Ott, 1997; Murtagh et al., 2000). Crucially, in their classic study, Kroken and Taylor (2001b) separately genotyped ascomata and maternal thalli from four specimens of L. gracilis and two of L. lupina using restriction enzymes. They consistently found heterozygosity in the fruiting bodies but not in the thalli, which is indicative of outcrossing and concordant with a dominant haploid life stage. Moreover, we detected very few specimens in our regional sampling with both mating types by PCR. Hence, it appears that the majority of Letharia thalli in nature are in fact haploid.
The conspicuous pattern of divergent haplotypes within both our and the McKenzie L. lupina metagenomes is consistent with hybridization. A triploid-like hybrid could be formed in a number of ways, but here we highlight some possible models (Figure 8). It has been proposed before that multiple genotypes could coexist within a single lichen thallus, forming chimeras (Jahns and Ott, 1997; Dyer et al., 2001; Mansournia et al., 2012). In the case of the triploid-like L. lupina thalli, a chimera would be composed of three individuals with mixed ancestry (Figure 8Ai). However, the read count ratios along the genome are perfectly aligned to triploidy in all three, individually prepared metagenome samples. It would be unlikely to recover such strict proportions of hyphal biomass and DNA content in different thalli in the absence of cell fusion. Rather, it is more plausible that the metagenomes are indeed triploid-like in one of three nuclear configurations: 3n, 2n + n, or n + n + n. In plants and animals, hybridization is a common cause of triploidization via fertilization by unreduced gametes, which leads to triploid offspring that are often unstable and aneuploid (Mable, 2003; Köhler et al., 2010). However, the absence of diploid-like variants in our data suggests that there is no obvious aneuploidy, and hence the triploid-like configuration is genome-wide. Moreover, the dominant life stage of fungi is haploidy, and hence the products of meiosis are directly the offspring (spores). It follows that triploidization might occur during meiosis and spore development.
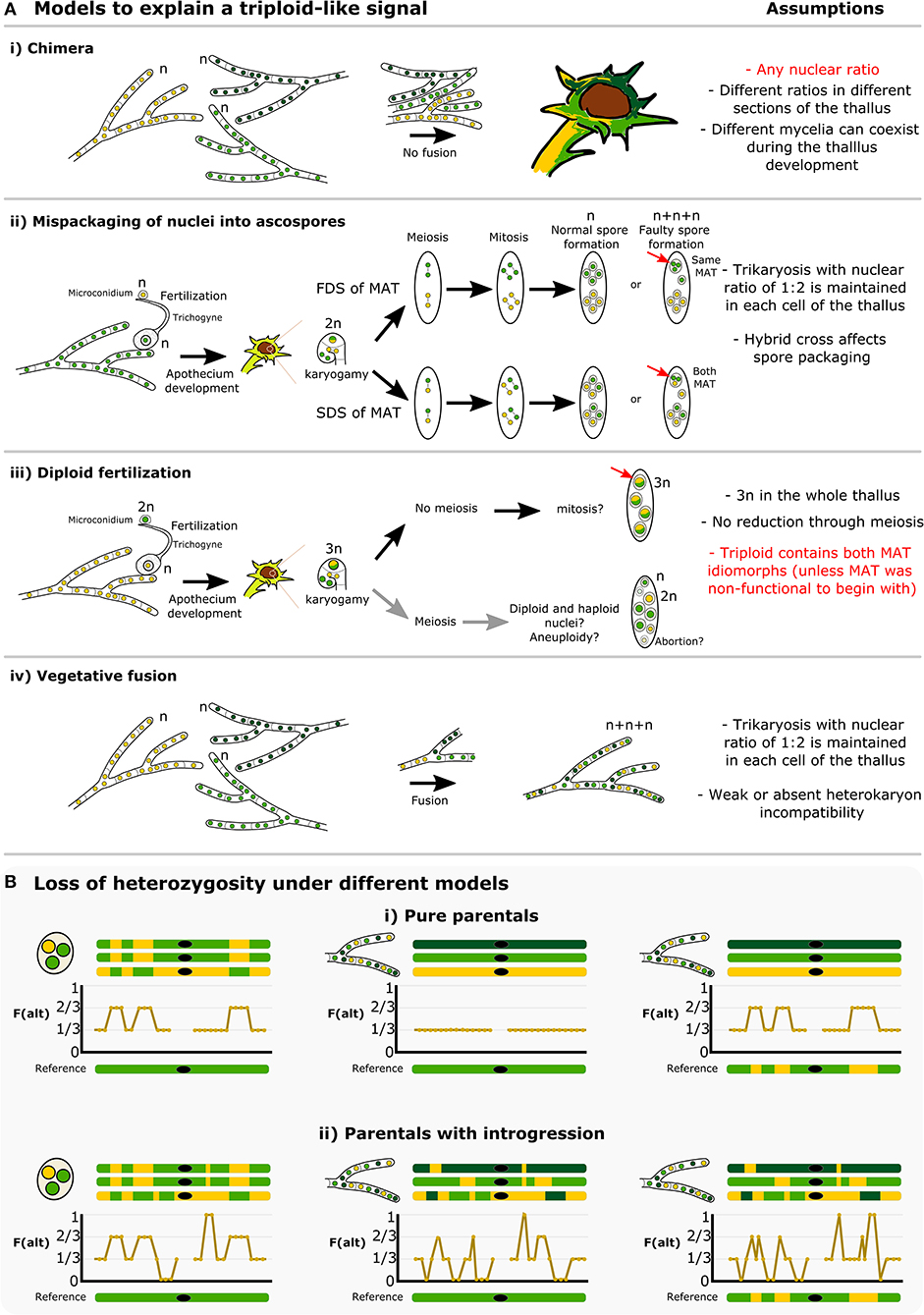
Figure 8. How is a triploid-like hybrid of Letharia formed? (A) We present four main models that can explain a triploid-like pattern where only two out of three (sub)genomes belong to the same species. More complex models or combinations are also possible. The L. lupina allele ancestry is represented by green and the unknown Letharia lineage by yellow. Two shades of green, light and dark, correspond to different variants of the same species. In model (i) chimeric thalli formed from three independent haploid (n) mycelia that do not undergo cell fusion. In model (ii) normal sexual reproduction with fertilization involves haploid parents, followed by a standard meiosis but with mispackaging of nuclei during ascospore formation, creating trikaryotic (n + n + n) spores. These spores can have a single mating type if the MAT locus undergoes first division segregation (FDS), or both mating types if it follows second division segregation (SDS). Model (iii) implies sexual reproduction where one of the parents is diploid (2n), leading to a triploid (3n) zygote during karyogamy, and a failure of meiosis to preserve the triploid condition in the resulting spores. In the last model (iv), vegetative fusion of three individuals leads to trikaryotic mycelia. For each model, a number of testable assumptions is given, highlighting in red those that are unlikely or do not conform to the observed data. Model (i) is unlikely given the triploid-like observation in three independently sequenced lichen thalli (ours and McKenzie's). Model (iii) requires multiple events (e.g., previous diploidization of one of the parentals, no meiotic reduction, a bypass of the heterothallic system to produce spores of the same mating type in case of the McKenzie L. lupina), and given the occurrence of meiosis after karyogamy, aneuploidy is likely to occur in the resulting spores, which is also incompatible with the observed data. Hence, we interpret the models (ii, iv) as the most likely hypotheses, since they can accommodate thalli with a single and two MAT idiomorphs. Still, even under those two models, previous introgression between the two parentals is necessary in order to recover the observed areas of loss of heterozygosity along the genome, as shown by schematic representations in (B). The frequency of the alternative allele is shown along cartoon chromosomes in the absence (i) or presence (ii) of introgression, either in the triploid individual or in the reference. The black dot on the chromosomes represents the centromere or any repetitive area that leads to missing data.
An alternative explanation, to our knowledge absent from the literature, is the faulty packaging of nuclei into ascospores (Figure 8Aii). Normally, the packaging of nuclei during spore development is strictly controlled, but it is known from model ascomycetes like Podospora and Neurospora that occasionally this fails, and spores with an unexpected number of nuclei are accidentally formed at low rates (Ames, 1934; Page, 1936; Raju and Newmeyer, 1977; Raju, 1992). Crosses between different species have been shown to induce or exacerbate irregularities in spore development (Perkins et al., 1976; Perkins, 1994; Jacobson, 1995). Notably, our L. lupina and the McKenzie L. columbiana metagenomes contain both mating types (in a 1:2 ratio), but the McKenzie L. lupina metagenome has only one. These two types of triploid-like cases could be formed by differential segregation of the MAT locus along the ascus: under first division segregation, all nuclei in an accidentally trikaryotic spore (two mitotic sister nuclei plus one non-sister nucleus) can be of the same mating type, while under second division segregation such a spore would have two sister nuclei of one mating type and a non-sister nucleus of the other mating type (Figure 8Aii). Yet another model where fertilization occurs from one diploid parent would require the absence of meiosis to preserve the triploid (3n) configuration in the spores (Figure 8Aiii). However, given a functional heterothallic life-style, triploid spores should always have both mating types, which does not match the observation in the McKenzie L. lupina metagenome. Finally, the triploid-like lichens could be produced via vegetative fusion of different individuals of the ascomycete lichen symbiont (Figure 8Aiv), forming heterokaryons (Tripp and Lendemer, 2018). Nonetheless, fusion of ascomycetes not involved in lichens typically leads to dikaryons (n + n) only. In the absence of knowledge on vegetative incompatibility systems in lichenized fungi, it is unclear how likely heterokaryosis would lead to trikaryons (n + n + n).
Note that these different models lead to specific predictions on the distribution of allele frequency variation along the genome (Figure 8B). For example, in the trikaryotic spore two of the genotypes should be identical, as they are the sister products of mitosis, and there should be perfectly aligned tracks of swapped ancestry between the subgenomes produced by recombination (left side of Figure 8B). This would not be the case in the heterokaryon produced by fusion unless the individuals involved are siblings (center and right side of Figure 8B). Moreover, the trikaryotic spore, the heterokaryon produced by fusion, and even the chimera would not be able to recover the observed distribution of allele frequencies in the absence of introgressed tracks in the parentals and/or in the reference (compare Figure 8Bi with Figure 8Bii). With the available data, it is not possible to distinguish between these different scenarios, but our results suggest that there is extensive hybridization and introgression between L. lupina and another Letharia lineage.
Historically, the taxonomy of the Letharia genus has been chronically plagued by cryptic lineages and blurry species definitions (Kroken and Taylor, 2001a; Altermann, 2009; Altermann et al., 2014, 2016), partially due to their recent diversification (judging by their high genetic similarity), which inevitably leads to high incomplete lineage sorting. It is likely that hybrids and gene flow further complicate matters. Clearly, L. lupina is not the only taxon producing triploid-like thalli. Specifically, while we found that our L. columbiana metagenome is haploid, the sample of McKenzie et al. (2020) turned out to be triploid-like. In addition, one of the L. vulpina samples assayed here with PCR had both mating types. Hence, it is possible that hybridization and formation of triploid-like individuals occurs in other lineages of the genus, contributing further to the taxonomical difficulty in this group.
In a large-scale study, Pizarro et al. (2019) found that a single MAT idiomorph was found in representative samples across Lecanoromycetes, consistent with widespread heterothallism. As expected, all Letharia lineages studied here also follow this pattern. We further characterized both idiomorphs and determined the minimum size of the non-recombining region (3.8 kbp), which has not been studied but for a handful of taxa (Scherrer et al., 2005; Dal Grande et al., 2018; Armaleo et al., 2019; Pizarro et al., 2019). Moreover, we refined the annotation of the auxiliary genes present in Letharia using transcriptomic data and homology searches to other fungal classes. This analysis confirmed that synteny is conserved in both idiomorphs for Parmeliaceae, Cladoniaceae, and Umbilicariaceae, and potentially for other families within Lecanoromycetes, making Letharia an adequate reference for the mating type architecture of this group. However, the MAT locus in Collemataceae and Icmadophilaceae seems to be different, highlighting that the MAT locus architecture is not conserved across all lichen-forming Lecanoromycetes.
Since heterothallism implies sexual self-incompatibility, the relative proportion of mating-types in a population is critical for a population's ability to reproduce sexually (Consolo et al., 2005; Olarte et al., 2012; Saleh et al., 2012; Singh et al., 2012). Here we determined that both sorediate species, L. lupina and L. vulpina, likely reproduce sexually often enough to maintain balanced MAT frequencies in North American populations, despite the fact that apotheciate thalli are relatively rare. In Europe, nonetheless, the MAT frequencies are biased, to the point that only one mating type was found in the Swedish L. vulpina populations. The extreme scarcity of apothecia and low genetic variation in Sweden (Högberg et al., 2002) could be related to the lack of mating partner, as the populations may be pushed to predominantly clonal reproduction. Alternatively, the absence of the MAT1-1 idiomorph might reflect the preferred route of reproduction as mainly clonal propagation can skew the local distribution of mating types even if the populations rarely recombine (Singh et al., 2015). Clonality itself could be a byproduct of the triploid-like condition, as these thalli might be infertile. It must be noted, however, that the two sequenced triploid-like metagenomes of L. lupina and the pure culture came from northwestern North America, and the only indication of possible triploidy in L. vulpina comes from the one Italian specimen with both mating types. Thus, it remains of great interest to determine the geographical extent of non-haploidy in the genus, as well as its relationship with clonality in Europe.
Clonality at the edge of a species' geographical range is commonly observed across different organism groups and can have ecological advantage over short time (Billingham et al., 2003; Kawecki, 2008; Meloni et al., 2013). In the Swedish range edge populations of L. vulpina, clonality may be especially beneficial as it preserves the combination of the compatible symbionts adapted to the local habitat, in addition to reassuring the survival of the individual and the species. However, despite the short-term advantages of clonality, the lack of recombination should eventually lead to limited evolutionary potential of the species and accumulation of deleterious mutations (Kawecki 2008). Currently, there seems to be no, or very low, potential for sexual reproduction of L. vulpina in the studied Swedish populations, which in combination with the low genetic diversity may reduce the fitness and the adaptive potential of these lichens in a changing environment.
Conclusions
Hybridization and polyploidization are emerging as significant influencers of fungal evolution and speciation, based largely on the study of pathogens or human-associated species (Allman et al., 2011; Marcet-Houben and Gabaldón, 2015; Depotter et al., 2016; Steenwyk et al., 2020). The results presented here, along with recent studies (Keuler et al., 2020; McKenzie et al., 2020), suggest that lichen fungal symbionts are also subject to these phenomena. The genus Letharia is a classical model of evolutionary biology amongst lichen-forming species thanks to the work of S. Kroken, J. W. Taylor, and others (Kroken and Taylor, 2001a,b; Högberg et al., 2002; Altermann et al., 2014, 2016). Now, Letharia is elevated into the genomics era, refueling research on lichen reproductive biology. In the process new aspects of its biology are discovered, which are indeed enigmatic.
Data Availability Statement
The datasets presented in this study can be found online as a Dryad repository (https://datadryad.org/stash), doi: 10.5061/dryad.1jwstqjtd, in the BioProject PRJNA523679 of the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra), and in GenBank (https://www.ncbi.nlm.nih.gov/genbank/) under accession numbers MK521629-MK521632.
Author Contributions
VT, LB, and HJ designed the study. VT and LB collected the Swedish specimens. TS collected the American specimens. JN collected the Italian specimens. HJ collected the Swiss specimens. YY provided the pure culture of Letharia. VT, LB, DV, and TS conducted the laboratory work. SLA-V, VT, LB, and DV conducted the data-analyses. GT and HJ provided the research resources. SLA-V, VT, and HJ wrote the paper. TS, DV, JN, and GT commented and contributed to the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This project was supported by a stipend to LB by Leksand municipality, Grants from Stiftelsen Lars Hiertas Minne (FO2013-0370), Stiftelsen Extensus and Lennanders Stiftelse to VT, and by a Grant (DO2011-0022) from Stiftelsen Oscar och Lili Lamms Minne to GT.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge that the sequencing of metagenomes and pure culture was performed by the SNP&SEQ Technology Platform, Science for Life Laboratory at Uppsala University, a national infrastructure supported by the Swedish Research Council (VR-RFI) and the Knut and Alice Wallenberg Foundation. A collecting permit for Letharia vulpina in Sweden was issued by the county administration board of Dalarna (Dnr: 522-2513-2014). Helmut Mayrhofer and John McCutcheon are thanked for supporting metatranscriptome sequencing at University of Graz and University of Montana, respectively. We thank Bruce McCune and Steve Leavitt for providing us with Letharia specimens, Markus Hiltunen for laboratory assistance, and Gulnara Tagirdzhanova, Sergio Tusso, Brendan Furneaux, and Aaron A. Vogan for fruitful discussions about the metagenomes and analyses, and Dr. Markus Wilken about the MAT nomenclature. We also thank Jessica L. Allen and Sean McKenzie for access to their Letharia repeat library and genome annotations. Finally, we thank two reviewers for their useful comments on the original manuscript. This study was published previously as a preprint (Ament-Velásquez et al., 2020).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/ffunb.2021.656386/full#supplementary-material
References
Ahmad, S., and Martins, C. (2019). The modern view of B chromosomes under the impact of high scale omics analyses. Cells 8:156. doi: 10.3390/cells8020156
Allen, J. L., McKenzie, S. K., Sleith, R. S., and Alter, S. E. (2018). First genome-wide analysis of the endangered, endemic lichen Cetradonia linearis reveals isolation by distance and strong population structure. Am. J. Bot. 105, 1556–1567. doi: 10.1002/ajb2.1150
Allman, E. S., Degnan, J. H., and Rhodes, J. A. (2011). Identifying the rooted species tree from the distribution of unrooted gene trees under the coalescent. J. Math. Biol. 62, 833–862. doi: 10.1007/s00285-010-0355-7
Altermann, S. (2004). A second look at Letharia (Th. Fr.) Zahlbr. Bull. Calif. Lichen Soc. 11, 33–36. Available online at: http://californialichens.org/bulletin/cals11_2.pdf
Altermann, S. (2009). Geographic Structure in a Symbiotic Mutualism. Santa Cruz, CA: University of California Santa Cruz. 159 p.
Altermann, S., Leavitt, S. D., and Goward, T. (2016). Tidying up the genus Letharia: Introducing, L. lupina sp. nov. and a new circumscription for L. columbiana. Lichenologist 48, 423–439. doi: 10.1017/S0024282916000396
Altermann, S., Leavitt, S. D., Goward, T., Nelsen, M. P., and Lumbsch, H. T. (2014). How do you solve a problem like Letharia? A new look at cryptic species in lichen-forming fungi using Bayesian clustering and SNPs from multilocus sequence data. PLoS ONE 9:e97556. doi: 10.1371/journal.pone.0097556
Ament-Velásquez, S. L., Figuet, E., Ballenghien, M., Zattara, E. E., Norenburg, J. L., Fernández-Álvarez, F. A., et al. (2016). Population genomics of sexual and asexual lineages in fissiparous ribbon worms (Lineus, Nemertea): hybridization, polyploidy and the Meselson effect. Mol. Ecol. 25, 3356–3369. doi: 10.1111/mec.13717
Ament-Velásquez, S. L. V., Tuovinen, L., Bergström, T., Spribille, D., Vanderpool, J., Nascimbene, Y., et al. (2020). The plot thickens: haploid and triploid-like thalli, hybridization, and biased mating type ratios in Letharia. BioRxiv. doi: 10.1101/2020.12.18.423428
Ames, L. M. (1934). Hermaphroditism involving self-sterility and cross-fertility in the ascomycete Pleurage anserina. Mycologia 26, 392–414. doi: 10.1080/00275514.1934.12020734
Armaleo, D., Müller, O., Lutzoni, F., Andrésson, Ó. S., Blanc, G., Bode, H. B., et al. (2019). The lichen symbiosis re-viewed through the genomes of Cladonia grayi and its algal partner Asterochloris glomerata. BMC Genomics 20:605. doi: 10.1186/s12864-019-5629-x
Arnerup, J., Högberg, N., and Thor, G. (2004). Phylogenetic analysis of multiple loci reveal the population structure within Letharia in the Caucasus and Morocco. Mycol. Res. 108, 311–316. doi: 10.1017/S0953756204009360
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bell, G. (1982). The Masterpiece of Nature: The Evolution and Genetics of Sexuality. Berkeley, CA: University of California Press.
Bertazzoni, S., Williams, A. H., Jones, D. A., Syme, R. A., Tan, K.-C., and Hane, J. K. (2018). Accessories make the outfit: accessory chromosomes and other dispensable DNA regions in plant-pathogenic fungi. Mol. Plant Microbe Interact. 31, 779–788. doi: 10.1094/MPMI-06-17-0135-FI
Bicknell, R. A., and Koltunow, A. M. (2004). Understanding apomixis: recent advances and remaining conundrums. Plant Cell 16(suppl_1), S228–S245. doi: 10.1105/tpc.017921
Billiard, S., López-Villavicencio, M., Hood, M. E., and Giraud, T. (2012). Sex, outcrossing and mating types: unsolved questions in fungi and beyond. J. Evol. Biol. 25, 1020–1038. doi: 10.1111/j.1420-9101.2012.02495.x
Billingham, M. R., Reusch, T. B. H. H., Alberto, F., and Serrão, E. A. (2003). Is asexual reproduction more important at geographical limits? A genetic study of the seagrass Zostera marina in the Ria Formosa, Portugal. Mar. Ecol. Prog. Ser. 265, 77–83. doi: 10.3354/meps265077
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Boratyn, G. M., Schäffer, A. A., Agarwala, R., Altschul, S. F., Lipman, D. J., and Madden, T. L. (2012). Domain enhanced lookup time accelerated BLAST. Biol. Direct 7:12. doi: 10.1186/1745-6150-7-12
Bryant, D., and Moulton, V. (2004). Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol. Biol. Evol. 21, 255–265. doi: 10.1093/molbev/msh018
Bubrick, P., and Galunj, M. (1986). Spore to spore resynthesis of Xanthoria parietina. Lichenologist 18, 47–49. doi: 10.1017/S0024282986000051
Bustamante, C. D., Wakeley, J., Sawyer, S., and Hartl, D. L. (2001). Directional selection and the site-frequency spectrum. Genetics 159, 1779–1788. doi: 10.1093/genetics/159.4.1779
Butler, G. (2007). “The evolution of MAT: the ascomycetes,” in Sex in Fungi: Molecular Determination and Evolutionary Implication, eds J. Heitman, J. W. Kronstad, J. W. Taylor, and L. A. Casselton (Washington, DC: ASM Press), 3–18.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Chen, F., Mackey, A. J., Stoeckert, C. J., and Roos, D. S. (2006). OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 34, D363–D368. doi: 10.1093/nar/gkj123
Consolo, V. F., Cordo, C. A., and Salerno, G. L. (2005). Mating-type distribution and fertility status in Magnaporthe grisea populations from Argentina. Mycopathologia 160, 285–290. doi: 10.1007/s11046-005-4333-3
Crittenden, P. D., David, J. C., Hawksworth, D. L., and Campbell, F. S. (1995). Attempted isolation and success in the culturing of a broad spectrum of lichen-forming and lichenicolous fungi. New Phytol. 130, 267–297. doi: 10.1111/j.1469-8137.1995.tb03048.x
Dal Grande, F., Meiser, A., Greshake Tzovaras, B., Otte, J., Ebersberger, I., and Schmitt, I. (2018). The draft genome of the lichen-forming fungus Lasallia hispanica (Frey) Sancho and A. Crespo. Lichenologist 50, 329–340. doi: 10.1017/S002428291800021X
Depotter, J. R. L., Seidl, M. F., Wood, T. A., and Thomma, B. P. H. J. (2016). Interspecific hybridization impacts host range and pathogenicity of filamentous microbes. Curr. Opin. Microbiol. 32, 7–13. doi: 10.1016/j.mib.2016.04.005
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Dyer, P. S., Inderbitzin, P., and Debuchy, R. (2016). “14 Mating-type structure, function, regulation and evolution in the Pezizomycotina,” in Growth, Differentiation and Sexuality. The Mycota (A Comprehensive Treatise on Fungi as Experimental Systems for Basic and Applied Research), Vol 1, ed J. Wendland (Cham: Springer). doi: 10.1007/978-3-319-25844-7_14
Dyer, P. S., Murtagh, G. J., and Crittenden, P. D. (2001). Use of RAPD-PCR DNA fingerprinting and vegetative incompatibility tests to investigate genetic variation within lichen-forming fungi. Symbiosis 31, 213–229. Available online at: https://hdl.handle.net/10222/77851
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Espagne, E., Lespinet, O., Malagnac, F., Da Silva, C., Jaillon, O., Porcel, B. M., et al. (2008). The genome sequence of the model ascomycete fungus Podospora anserina. Genome Biol. 9:R77. doi: 10.1186/gb-2008-9-5-r77
Ferreira, A. V., An, Z., Metzenberg, R. L., and Glass, N. L. (1998). Characterization of mat A-2, mat A-3 and ▵matA mating-type mutants of Neurospora crassa. Genetics 1067–1079.
Gioti, A., Mushegian, A. A., Strandberg, R., Stajich, J. E., and Johannesson, H. (2012). Unidirectional evolutionary transitions in fungal mating systems and the role of transposable elements. Mol. Biol. Evol. 29, 3215–3226. doi: 10.1093/molbev/mss132
Glass, N. L., Vollmer, S. J., Staben, C., Grotelueschen, J., Metzenberg, R. L., and Yanofsky, C. (1988). DNAs of the two mating-type alleles of Neurospora crassa are highly dissimilar. Science 241, 570–573. doi: 10.1126/science.2840740
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9:R7. doi: 10.1186/gb-2008-9-1-r7
Heinrich, V., Stange, J., Dickhaus, T., Imkeller, P., Krüger, U., Bauer, S., et al. (2011). The allele distribution in next-generation sequencing data sets is accurately described as the result of a stochastic branching process. Nucleic Acids Res. 40, 2426–2431. doi: 10.1093/nar/gkr1073
Heitman, J., Kronstad, J. W., Taylor, J. W., and Casselton, L. A. (2007). Sex in Fungi, 1st Edn. Washington, DC: American Society for Microbiology. doi: 10.1128/9781555815837
Högberg, N., Kroken, S., Thor, G., and Taylor, J. W. (2002). Reproductive mode and genetic variation suggest a North American origin of European Letharia vulpina. Mol. Ecol. 11, 1191–1196. doi: 10.1046/j.1365-294X.2002.01513.x
Honegger, R., and Scherrer, S. (2008). “Sexual reproduction in lichen-forming ascomycete,” in Lichen Biology, 2nd Edn, ed T. H. Nash (New York, NY: Cambridge University Press), 94–103. doi: 10.1017/CBO9780511790478.007
Honegger, R., and Zippler, U. (2007). Mating systems in representatives of Parmeliaceae, Ramalinaceae and Physciaceae (Lecanoromycetes, lichen-forming ascomycetes). Mycol. Res. 111, 424–432. doi: 10.1016/j.mycres.2007.02.005
Honegger, R., Zippler, U., Gansner, H., and Scherrer, S. (2004). Mating systems in the genus Xanthoria (lichen-forming ascomycetes). Mycol. Res. 108, 480–488. doi: 10.1017/S0953756204009682
Huerta-Cepas, J., Serra, F., and Bork, P. (2016). ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 33, 1635–1638. doi: 10.1093/molbev/msw046
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Hutchinson, M. I., Powell, A. J., Tsang, A., O'Toole, N., Berka, R. M., Barry, K., et al. (2016). Genetics of mating in members of the Chaetomiaceae as revealed by experimental and genomic characterization of reproduction in Myceliophthora heterothallica. Fungal Genet. Biol. 86, 9–19. doi: 10.1016/j.fgb.2015.11.007
Jacobson, D. J. (1995). Sexual dysfunction associated with outcrossing in Neurospora tetrasperma, a pseudohomothallic ascomycete. Mycologia 87, 604–617. doi: 10.1080/00275514.1995.12026577
Jahns, H. M. (1993). Culture experiments with lichens. Plant Syst. Evol. 187, 145–174. doi: 10.1007/BF00994097
Jahns, H. M., and Ott, S. (1997). Life strategies in lichens - some general considerations. Bibl. Lichenol. 67, 49–67.
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kawecki, T. J. (2008). Adaptation to marginal habitats. Annu. Rev. Ecol. Evol. Syst. 39, 321–342. doi: 10.1146/annurev.ecolsys.38.091206.095622
Keuler, R., Garretson, A., Saunders, T., Erickson, R. J., St. Andre, N., Grewe, F., et al. (2020). Genome-scale data reveal the role of hybridization in lichen-forming fungi. Sci. Rep. 10:1497. doi: 10.1038/s41598-020-58279-x
Knaus, B. J., and Grünwald, N. J. (2017). VcfR: a package to manipulate and visualize variant call format data in R. Mol. Ecol. Res. 17, 44–53. doi: 10.1111/1755-0998.12549
Koboldt, D. C., Zhang, Q., Larson, D. E., Shen, D., McLellan, M. D., Lin, L., et al. (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. doi: 10.1101/gr.129684.111
Köhler, C., Mittelsten, O. S., and Erilova, A. (2010). The impact of the triploid block on the origin and evolution of polyploid plants. Trends Genetics 26, 142–148. doi: 10.1016/j.tig.2009.12.006
Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5:59. doi: 10.1186/1471-2105-5-59
Köster, J., and Rahmann, S. (2012). Snakemake-a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522. doi: 10.1093/bioinformatics/bts480
Kroken, S., and Taylor, J. W. (2001a). A gene genealogical approach to recognize phylogenetic species boundaries in the lichenized fungus Letharia. Mycologia 93, 38–53. doi: 10.1080/00275514.2001.12061278
Kroken, S., and Taylor, J. W. (2001b). Outcrossing and recombination in the lichenized fungus Letharia. Fungal Genet. Biol. 34, 83–92. doi: 10.1006/fgbi.2001.1291
Kronstad, J. W., and Staben, C. (1997). Mating type in filamentous fungi. Annu. Rev. Genet. 31, 245–276. doi: 10.1146/annurev.genet.31.1.245
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Larsson, A. (2014). AliView: a fast and lightweight alignment viewer and editor for large data sets. Bioinformatics 30:btu531. doi: 10.1093/bioinformatics/btu531
Levin, D. A. (1975). Minority cytotype exclusion in local plant populations. Taxon 24, 35–43. doi: 10.2307/1218997
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O., and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506. doi: 10.1093/nar/gki937
Ludwig, L. R., Summerfield, T. C., Lord, J. M., and Singh, G. (2017). Characterization of the mating-type locus (MAT) reveals a heterothallic mating system in Knightiella splachnirima. Lichenologist 49, 373–385. doi: 10.1017/S0024282917000214
Mable, B. K. (2003). Breaking down taxonomic barriers in polyploidy research. Trends Plant Sci. 8, 582–590. doi: 10.1016/j.tplants.2003.10.006
Mandel, M. A., Barker, B. M., Kroken, S., Rounsley, S. D., and Orbach, M. J. (2007). Genomic and population analyses of the mating type loci in coccidioides species reveal evidence for sexual reproduction and gene acquisition. Eukaryot. Cell 6, 1189–1199. doi: 10.1128/EC.00117-07
Mansournia, M. R., Wu, B., Matsushita, N., and Hogetsu, T. (2012). Genotypic analysis of the foliose lichen Parmotrema tinctorum using microsatellite markers: association of mycobiont and photobiont, and their reproductive modes. Lichenologist 44, 419–440. doi: 10.1017/S0024282911000909
Marcet-Houben, M., and Gabaldón, T. (2015). Beyond the whole-genome duplication: phylogenetic evidence for an ancient interspecies hybridization in the baker's yeast lineage. PLoS Biol. 13:e1002220. doi: 10.1371/journal.pbio.1002220
Martin, S. H., and Van Belleghem, S. M. (2017). Exploring evolutionary relationships across the genome using topology weighting. Genetics 206, 429–438. doi: 10.1534/genetics.116.194720
Martin, T., Lu, S., van Tilbeurgh, H., Ripoll, D. R., Dixelius, C., Turgeon, B. G., et al. (2010). Tracing the origin of the fungal α1 domain places its ancestor in the HMG-box superfamily: implication for fungal mating-type evolution. PLoS ONE 5:e15199. doi: 10.1371/journal.pone.0015199
McCune, B., and Altermann, S. (2009). Letharia gracilis (Parmeliaceae), a new species from California and Oregon. Bryologist 112, 375–378. doi: 10.1639/0007-2745-112.2.375
McKenzie, S. K., Walston, R. F., and Allen, J. L. (2020). Complete, high-quality genomes from long-read metagenomic sequencing of two wolf lichen thalli reveals enigmatic genome architecture. Genomics 112, 3150–3156. doi: 10.1016/j.ygeno.2020.06.006
Meloni, M., Reid, A., Caujapé-Castells, J., Marrero, Á., Fernández-Palacios, J. M., Mesa-Coelo, R. A., et al. (2013). Effects of clonality on the genetic variability of rare, insular species: the case of Ruta microcarpa from the Canary Islands. Ecol. Evol. 3, 1569–1579. doi: 10.1002/ece3.571
Murtagh, G. J., Dyer, P. S., and Crittenden, P. D. (2000). Sex and the single lichen. Nature 404:564. doi: 10.1038/35007142
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Olarte, R. A., Horn, B. W., Dorner, J. W., Monacell, J. T., Singh, R., Stone, E. A., et al. (2012). Effect of sexual recombination on population diversity in aflatoxin production by Aspergillus flavus and evidence for cryptic heterokaryosis. Mol. Ecol. 21, 1453–1476. doi: 10.1111/j.1365-294X.2011.05398.x
Page, W. M. (1936). Note on abnormal spores in Podospora minuta. Trans. Br. Mycol. Soc. 20, 186–189. doi: 10.1016/S0007-1536(36)80010-2
Patel, R. K., and Jain, M. (2012). NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 7:e30619. doi: 10.1371/journal.pone.0030619
Perkins, D. D. (1994). How should the infertility of interspecies crosses be designated? Mycologia 86, 758–761. doi: 10.1080/00275514.1994.12026480
Perkins, D. D., Turner, B. C., and Barry, E. G. (1976). Strains of Neurospora collected from nature. Evolution 30, 281–313. doi: 10.1111/j.1558-5646.1976.tb00910.x
Pizarro, D., Dal Grande, F., Leavitt, S. D., Dyer, P. S., Schmitt, I., Crespo, A., et al. (2019). Whole-genome sequence data uncover widespread heterothallism in the largest group of lichen-forming fungi. Genome Biol. Evol. 11, 721–730. doi: 10.1093/gbe/evz027
Raju, N. B. (1992). Functional heterothallism resulting from homokaryotic conidia and ascospores in Neurospora tetrasperma. Mycol. Res. 96, 103–116. doi: 10.1016/S0953-7562(09)80923-4
Raju, N. B., and Newmeyer, D. (1977). Giant ascospores and abnormal croziers in a mutant of Neurospora crassa. Exp. Mycol. 1, 152–165. doi: 10.1016/S0147-5975(77)80040-6
Ramsey, J., and Schemske, D. W. (1998). Pathways, mechanisms, and rates of polyploid formation in flowering plants. Ann. Rev. Ecol. Systemat. 29, 467–501. doi: 10.1146/annurev.ecolsys.29.1.467
Rutherford, K., Parkhill, J., Crook, J., Horsnell, T., Rice, P., Rajandream, M.-A., et al. (2000). Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945. doi: 10.1093/bioinformatics/16.10.944
Saleh, D., Xu, P., Shen, Y., Li, C., Adreit, H., Milazzo, J., et al. (2012). Sex at the origin: an Asian population of the rice blast fungus Magnaporthe oryzae reproduces sexually. Mol. Ecol. 21, 1330–1344. doi: 10.1111/j.1365-294X.2012.05469.x
Sanders, W. B. (2014). Complete life cycle of the lichen fungus Calopadia puiggarii (Pilocarpaceae, Ascomycetes) documented in situ: Propagule dispersal, establishment of symbiosis, thallus development, and formation of sexual and asexual reproductive structures. Am. J. Bot. 101, 1836–1848. doi: 10.3732/ajb.1400272
Scherrer, S., Zippler, U., and Honegger, R. (2005). Characterisation of the mating-type locus in the genus Xanthoria (lichen-forming ascomycetes, Lecanoromycetes). Fungal Genet. Biol. 42, 976–988. doi: 10.1016/j.fgb.2005.09.002
Schliep, K. P. (2011). phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593. doi: 10.1093/bioinformatics/btq706
Singh, G., Dal Grande, F., Cornejo, C., Schmitt, I., and Scheidegger, C. (2012). Genetic basis of self-incompatibility in the lichen-forming fungus Lobaria pulmonaria and skewed frequency distribution of mating-type idiomorphs: implications for conservation. PLoS ONE 7:e51402. doi: 10.1371/journal.pone.0051402
Singh, G., Dal Grande, F., Werth, S., and Scheidegger, C. (2015). Long-term consequences of disturbances on reproductive strategies of the rare epiphytic lichen Lobaria pulmonaria: Clonality a gift and a curse. FEMS Microbiol. Ecol. 91, 1–11. doi: 10.1093/femsec/fiu009
Slater, G. S. C., and Birney, E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6:31. doi: 10.1186/1471-2105-6-31
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., and Waack, S. (2003). Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, ii215–ii225. doi: 10.1093/bioinformatics/btg1080
Steenwyk, J. L., Lind, A. L., Ries, L. N. A., dos Reis, T. F., Silva, L. P., Almeida, F., et al. (2020). Pathogenic allodiploid hybrids of Aspergillus fungi. Curr. Biol. 30, 2495.e7–2507.e7. doi: 10.1016/j.cub.2020.04.071
Stocker-Wörgötter, E. (2001). Experimental lichenology and microbiology of lichens: culture experiments, secondary chemistry of cultured mycobionts, resynthesis, and thallus. Bryologist 104, 576–581. doi: 10.1639/0007-2745(2001)104[0576:ELAMOL]2.0.CO;2
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, Y. O., and Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18, 1979–1990. doi: 10.1101/gr.081612.108
Tiwary, B. K., Kirubagaran, R., and Ray, A. K. (2004). The Biology of triploid fish. Rev. Fish Biol. Fish. 14, 391–402. doi: 10.1007/s11160-004-8361-8
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Tripp, E. A., and Lendemer, J. C. (2018). Twenty-seven modes of reproduction in the obligate lichen symbiosis. Brittonia 70, 1–14. doi: 10.1007/s12228-017-9500-6
Tripp, E. A., Zhuang, Y., and Lendemer, J. C. (2017). A review of existing whole genome data suggests lichen mycelia may be haploid or diploid. Bryologist 120, 302–310. doi: 10.1639/0007-2745-120.3.302
Tsui, C. K.-M., DiGuistini, S., Wang, Y., Feau, N., Dhillon, B., Bohlmann, J., et al. (2013). Unequal recombination and evolution of the mating-type (mat) loci in the pathogenic fungus Grosmannia clavigera and relatives. G33, 465–480. doi: 10.1534/g3.112.004986
Tuovinen, V., Ekman, S., Thor, G., Vanderpool, D., Spribille, T., and Johannesson, H. (2019). Two basidiomycete fungi in the cortex of wolf lichens. Curr. Biol. 29, 476.e5–483.e5. doi: 10.1016/j.cub.2018.12.022
Turgeon, B. G., and Yoder, O. C. (2000). Proposed nomenclature for mating type genes of filamentous ascomycetes. Fungal Genet. Biol. 31, 1–5. doi: 10.1006/fgbi.2000.1227
Whittle, C. A., Nygren, K., and Johannesson, H. (2011). Consequences of reproductive mode on genome evolution in fungi. Fungal Genet. Biol. 48, 661–667. doi: 10.1016/j.fgb.2011.02.005
Wickham, H., François, R., Henry, L., and Müller, K. (2018). dplyr: A Grammar of Data Manipulation. R package version 0.7.6. Available online at: https://CRAN.R-project.org/package=dplyr
Wilke, C. O. (2019). cowplot: Streamlined Plot Theme and Plot Annotations for “ggplot2”. R Package Version 1.0.0. Available online at: https://CRAN.R-project.org/package=cowplot
Yamamoto, Y., Kinoshita, Y., and Yoshimura, I. (1998). Difference of cell-viability and spore discharge capability between two Lichen species, Letharia columbiana (Nutt.) Thoms. and L. vulpina (L.) Hue. Plant Biotechnol. 15, 131–133. doi: 10.5511/plantbiotechnology.15.131
Keywords: lichens, heterothallism, mating type locus, ploidy, hybridization
Citation: Ament-Velásquez SL, Tuovinen V, Bergström L, Spribille T, Vanderpool D, Nascimbene J, Yamamoto Y, Thor G and Johannesson H (2021) The Plot Thickens: Haploid and Triploid-Like Thalli, Hybridization, and Biased Mating Type Ratios in Letharia. Front. Fungal Biol. 2:656386. doi: 10.3389/ffunb.2021.656386
Received: 20 January 2021; Accepted: 24 March 2021;
Published: 26 May 2021.
Edited by:
Toni Gabaldón, Barcelona Supercomputing Center, SpainReviewed by:
Timothy Yong James, University of Michigan, United StatesYen-Ping Hsueh, Academia Sinica, Taiwan
Copyright © 2021 Ament-Velásquez, Tuovinen, Bergström, Spribille, Vanderpool, Nascimbene, Yamamoto, Thor and Johannesson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanna Johannesson, aGFubmEuam9oYW5uZXNzb25AZWJjLnV1LnNl
†These authors have contributed equally to this work and share first authorship