 Martin P. Helley
Martin P. Helley Jennifer Pinnell
Jennifer Pinnell Carolina Sportelli
Carolina Sportelli Kim Tieu
Kim Tieu- 1Department of Environmental Health Sciences, Florida International University, Miami, FL, United States
- 2Peninsula Schools of Medicine and Dentistry, Plymouth University, Plymouth, United Kingdom
Parkinson’s disease (PD) is a devastating neurological movement disorder. Since its first discovery 200 years ago, genetic and environmental factors have been identified to play a role in PD development and progression. Although genetic studies have been the predominant driving force in PD research over the last few decades, currently only a small fraction of PD cases can be directly linked to monogenic mutations. The remaining cases have been attributed to other risk associated genes, environmental exposures and gene–environment interactions, making PD a multifactorial disorder with a complex etiology. However, enormous efforts from global research have yielded significant insights into pathogenic mechanisms and potential therapeutic targets for PD. This review will highlight mitochondrial dysfunction as a common pathway involved in both genetic mutations and environmental toxicants linked to PD.
Introduction
Parkinson’s disease (PD), characterized by degeneration of the nigrostriatal dopaminergic pathway, is the second most prevalent neurodegenerative disorder after Alzheimer’s disease. Pathological hallmarks of PD include the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the presence of cytoplasmic protein aggregates known as Lewy bodies in remaining dopaminergic neurons (Przedborski, 2017). When degeneration in these neurons results in a threshold reduction of approximately 80% of striatal dopamine, motor symptoms of PD such as rigidity, resting tremor, bradykinesia and postural instability start to emerge (Dauer and Przedborski, 2003). PD is a complex and multifactorial brain disorder. Approximately 30% of the familial and 3–5% of sporadic PD cases are caused by monogenic mutations, while the other remaining cases are classified as idiopathic and sporadic with unknown etiology (Klein and Westenberger, 2012; Hernandez et al., 2016). Environmental factors and gene–environment interactions have been implicated in idiopathic PD. This neurological disorder is therefore a polygenic disease with various genetic and environmental contributors cumulatively directing its pathological development.
Since the identification of the first missense substitution in SNCA (Polymeropoulos et al., 1997), the gene encoding α-synuclein, the understanding of the genetic contribution to PD has progressed rapidly and has become rather complex. In addition to the emergence of multiple loci as causative factors in familial PD (Table 1), genome wide association studies (GWAS) and Genome-wide complex trait analysis (GCTA) have uncovered a significant genetic component to the idiopathic disease (Hernandez et al., 2016; Przedborski, 2017). Although mutations in these genes alone may not be considered as causative, they significantly increase the risk of developing idiopathic PD. This is, at least in part, due to incomplete and variable penetrance of the majority of these mutations. The emergence of genetic risk factors for cases of idiopathic PD raises an important question: What is causing the increased susceptibility to the disease in individuals that carry these mutations? One possible and particularly pertinent explanation is that risk factor mutations render the individual more sensitive to the pathological influence of environmental factors thus bringing the idea of gene–environment interactions to the forefront. This review will discuss the impact of genetic mutations, environmental toxicants and gene–environment interactions on PD pathogenesis. Furthermore, we will highlight mitochondria as a common target for both genes and environmental toxicants linked to PD.
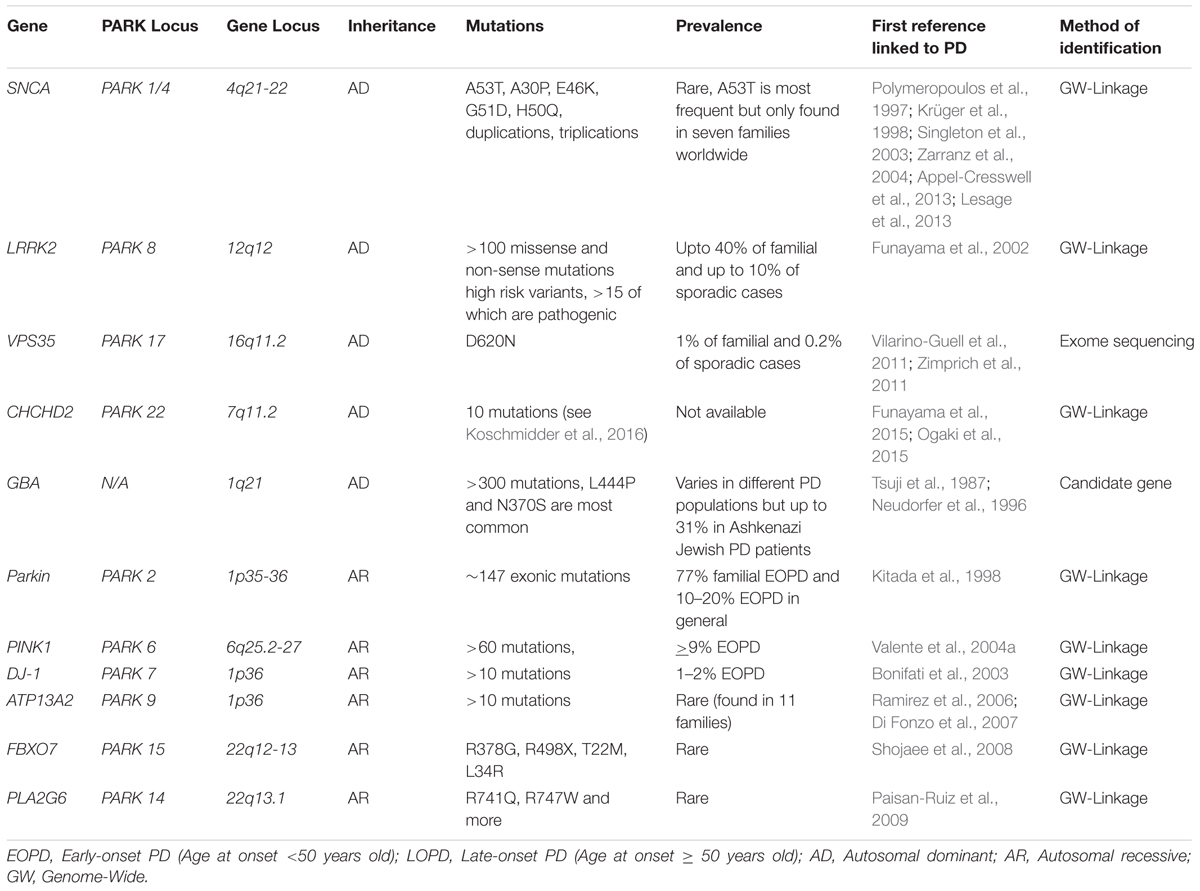
TABLE 1. Major monogenic mutations and associated risk genes in PD.
Genetic Mutations In PD
Autosomal-Dominant Genes
SNCA
Perhaps the most researched PD-linked gene to date is SNCA, which encodes α-synuclein. While mutations in this gene are rare, the SNCA locus is appreciated to be a significant variant factor for PD development (Lill et al., 2012). The missense mutation α-synuclein-A53T (Polymeropoulos et al., 1997) was the first gene to be attributed to disease development and its discovery represents a defining moment in PD research, not only preceding the identification of many other genetic determinants of PD but also mechanisms of sporadic disease. Since then, other point mutations in α-synuclein (A30P, E46K, G51D, and H50Q), as well as gene duplications and triplications, have been identified in familial PD (Krüger et al., 1998; Singleton et al., 2003; Zarranz et al., 2004; Appel-Cresswell et al., 2013; Lesage et al., 2013). The fact that duplications and triplications of SNCA can also cause PD (Singleton et al., 2003) is significant because it indicates that elevated wild type α-synuclein alone is sufficient to cause disease. So far, A53T has been found in seven families worldwide and only one family for each other four missense mutations. Duplications are more common than triplications, which has been found in several families (Hernandez et al., 2016). α-synuclein is also understood to be a key factor of sporadic PD and present in Lewy bodies, which are abnormal proteins commonly observed in PD (Spillantini et al., 1998). α-synuclein-associated mechanisms have therefore been at the forefront of the PD research and multiple high profile discoveries have greatly contributed to the understanding of disease pathology. For example, as discussed below, the discovery that α-synuclein pathology can spread from one cell to another in a prion-like fashion has provided a key insight into how PD may develop and provide novel therapeutic strategies.
α-synuclein is a pre-synaptic protein that plays a role in SNARE complex assembly and the exocytosis of neurotransmitters (Burré et al., 2010; Garcia-Reitböck et al., 2010; Bendor et al., 2013). The pathology of α-synuclein is largely due to its propensity to aggregate; gradually transitioning from small soluble oligomers to larger insoluble fibrils, ultimately forming Lewy bodies. Although the majority of α-synuclein is present in the cytosol, the protein also localizes to mitochondria and induces dysfunction (Devi et al., 2008; Parihar et al., 2008; Liu G. et al., 2009; Nakamura et al., 2011; Subramaniam et al., 2014; Chen et al., 2015; Di Maio et al., 2016). Multiple mitochondrial targeting domains have been identified within the N-terminal domain of the protein and confer its ability to bind to components of the mitochondrial membrane (Devi et al., 2008). Specifically, α-synuclein can bind to cardiolipin (Nakamura et al., 2008), TOM20 (Di Maio et al., 2016), TOM40 (Devi et al., 2008) and VDAC (Rostovtseva et al., 2015) either to directly promote dysfunction at the membrane level or to allow import into other mitochondrial compartments. The importance of the N-terminal domain in mitochondrial targeting is emphasized by the fact that α-synuclein-A53T, a mutant with a substitution in the N-terminal region, has a greater binding affinity for mitochondria than the wild type protein (Devi et al., 2008). However, more recently it has also been argued that, rather than localizing to the mitochondria, α-synuclein specifically targets the mitochondria-associated endoplasmic reticulum membrane (MAM) where it plays a role in regulating mitochondrial morphology (Guardia-Laguarta et al., 2014).
Recent studies have demonstrated that α-synuclein induces mitochondrial dysfunction in vivo. Mice with global overexpression of human wild type α-synuclein in the brain using the Thy1 promoter exhibited age dependent accumulation of α-synuclein in mitochondria of the nigrostriatal dopaminergic pathway, impaired electron transport chain function and enhanced oxidative stress (Subramaniam et al., 2014). Mice with inducible α-synuclein-A53T exhibited severe mitochondrial fragmentation that preceded dopaminergic neurodegeneration and other pathologies (Chen et al., 2015). These detrimental effects may be caused by the direct and specific binding between α-synuclein and outer mitochondrial membrane components such as TOM20 and VDAC (Rostovtseva et al., 2015; Di Maio et al., 2016). Di Maio et al. (2016) demonstrated that oligomeric, dopamine-modified and phosphorylated species of α-synuclein bound to TOM20 and prevented the import of multiple mitochondrial proteins leading to mitochondrial dysfunction. Similarly, binding of wild-type α-synuclein to VDAC was sufficient to block the channel preventing bi-directional metabolite transfer thus promoting dysfunction (Rostovtseva et al., 2015). In addition to blocking the channel, α-synuclein was able to translocate through VDAC to gain access to inner mitochondrial compartments, many of which being direct pathological targets, including respiratory complex components. A similar relationship has been reported for TOM40 as antibodies against this channel prevented the mitochondrial import of α-synuclein (Devi et al., 2008). In addition to these detrimental effects on mitochondrial function, α-synuclein also interferes with normal mitochondrial dynamics (fission, fusion, and transport). Overexpression of both wild type and mutated forms of α-synuclein is associated with severe fragmentation and reduced motility in vitro and in vivo through direct and indirect mechanisms (Kamp et al., 2010; Nakamura et al., 2011; Gui et al., 2012; Guardia-Laguarta et al., 2014; Chen et al., 2015; Bido et al., 2017). Another negative impact of α-synuclein on mitochondria is its ability to reduce mitochondrial biogenesis (Zheng et al., 2010; Siddiqui et al., 2012). Following the onset of oxidative stress, α-synuclein can bind to the promoter region of Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α), a key component of mitochondrial biogenesis, and inhibit the expression of downstream genes (Siddiqui et al., 2012). Lastly, multiple α-synuclein species may also promote the formation of dysfunctional mitochondria by increasing the prevalence of mitochondrial DNA mutations (Martin et al., 2006; Bendor et al., 2013).
In addition to these direct effects, α-synuclein can also affect mitochondrial function through indirect mechanisms. For example, it is increasingly evident that mitochondrial function and autophagy are tightly linked. A reduced autophagic capacity has been identified in many neurodegenerative diseases, including PD. Many of the genes implicated in familial PD, including SNCA, PINK1, Parkin, LRRK2, DJ-1 and glucocerebrosidase (GBA), have functions related to autophagy and mitophagy, emphasizing the importance of this pathway in the disease. Mitochondrial dysfunction may be able to inhibit the autophagy pathway, perhaps by a reduction in ATP availability and increased reactive oxygen species (ROS). Equally, impaired autophagy can promote mitochondrial dysfunction by the reduced clearance of damaged, dysfunctional organelles. The complex relationship between these two cellular pathways is not fully understood, particularly regarding the question of whether mitochondrial dysfunction precedes impaired autophagy or vice versa. Regardless, the interaction between these two fundamental processes represents a critical area of future research in the neurodegenerative field.
As mentioned above, α-synuclein has a direct interaction with the autophagy pathway. Overexpression of wild type α-synuclein was shown to directly modulate autophagy via inhibition of Rab1a protein, which is important for autophagosome formation (Winslow et al., 2010). Impaired autophagy has been demonstrated in vivo using mice overexpressing wild-type α-synuclein (Ebrahimi-Fakhari et al., 2011) or A53T α-synuclein (Yu et al., 2009; Chen et al., 2015). Conversely, an impairment in autophagy pathways promotes the formation and accumulation of higher order α-synuclein species, thus creating a cycle of neurotoxicity (Xilouri et al., 2016). In summary, α-synuclein can impair mitochondrial function at multiple levels: inhibition of the electron transport chain, imbalanced mitochondrial dynamics, reduced mitochondrial biogenesis and autophagy blockade.
Emerging evidence indicates that α-synuclein can spread inter-cellularly through multiple mechanisms, including non-classical exocytosis, exosomal release and nanotubes that directly connect the two cells (Guo and Lee, 2014). According to the Braak staging hypothesis (Hawkes et al., 2007), the olfactory bulb and the gut are the initial spreading sites of misfolded α-synuclein. Consistent with this hypothesis, accumulation of α-synuclein has been identified in enteric neurons of patients with early-stage PD (Hilton et al., 2014). Mitochondria have been implicated in the pathological spread in neurons of both the enteric and central nervous systems. For example, structural mitochondrial abnormalities have been identified in enteric neurons of post-mortem tissue from PD patients (Baumuratov et al., 2016). In primary human fetal enteric neurons, exposure to recombinant α-synuclein induces mitochondrial dysfunction involving a reduction in complex I activity and impairment of mitochondrial respiration (Braidy et al., 2014). Exposure of enteric neurons to the environmental neurotoxin, rotenone, promotes the release of α-synuclein species which are subsequently taken up by and form aggregates in second-order neurons (Pan-Montojo et al., 2012). Exposure of rat ventral midbrain neurons to exogenous, pre-formed fibrillar structures of α-synuclein, often used to study the transmission of the protein, also produce mitochondrial dysfunction (Tapias et al., 2017). These studies, and others, suggest that mitochondria may play a central role in the spread of α-synuclein from one neuron to another. That said, however, the topic of whether α-synuclein is transmitted trans-synaptically in PD is still a topic of debate (Brundin and Melki, 2017; Surmeier et al., 2017), which is beyond the scope of this article.
LRRK2
First identified in a family of Japanese origin (Funayama et al., 2002) and subsequently independently confirmed in several other families from different countries, leucine-rich repeat kinase 2 (LRRK2) mutations are the most common group of mutations in PD cases (Cookson, 2015). Although highly dependent on ethnicity, LRRK2 mutations are estimated to account for up to 40% of familial cases, depending on the ethnic background, and up to 10% of sporadic PD cases worldwide (Hernandez et al., 2016), producing a disease phenotype that is similar to that of classical late-onset idiopathic PD. The most common and well characterized LRRK2 mutation, G2019S, alone is suggested to account for 4% of familial and 1% of sporadic cases. All described LRRK2 mutations, to date, affect a central enzymatic region of the protein consisting of a GTPase and kinase domain and confer a gain of function (West et al., 2007). Although more than 100 LRRK2 mutations have been reported, only a few such as (N1437H, R1441C, R1441G, R1441H, Y1699C, G2019S, and I2020T0) have been proven to cause PD and two have been nominated as risk factors (R1628P and G2385R) (Dächsel and Farrer, 2010; Paisan-Ruiz et al., 2013). The incomplete penetrance of multiple mutations has led to LRRK2 being identified as a risk factor for sporadic PD (Nalls et al., 2014; Hernandez et al., 2016). Sporadic PD patient derived cells also display a slightly increased basal activity of LRRK2, however, it is not known whether this is causative or a consequence of pre-existing pathology (Esteves et al., 2015). Interestingly, a recent study demonstrated that variability within the DNM3 gene influences the age of onset in G2019S carriers (Trinh et al., 2016). The presence of other genetic modifiers may therefore influence disease manifestation in the case of LRRK2 mutations.
The complex physiological roles of LRRK2 are not fully understood; however, it is associated with many cellular functions, particularly those involving membrane dynamics, including autophagy, cytoskeletal dynamics, vesicle dynamics and mitochondrial function. Attempts to better understand how LRRK2 mutations contribute toward PD pathology have been restricted partly due to the difficulty in creating a LRRK2 transgenic model that recapitulates important features of PD. The majority of mutant LRRK2 transgenic models do not display appreciable dopaminergic neurodegeneration, but do exhibit multiple synaptic deficits including reduced vesicle endocytosis (Arranz et al., 2015) and altered dopamine release, uptake and signaling (Li et al., 2009; Tong et al., 2009; Melrose et al., 2010; Walker et al., 2014; Beccano-Kelly et al., 2015; Yue et al., 2015). LRRK2 has been shown to impair mitochondrial dynamics by physically interacting with Drp1 and promoting mitochondrial fragmentation associated with enhanced mitochondrial fission, increased ROS generation and impaired autophagy (Niu et al., 2012; Wang et al., 2012; Stafa et al., 2014; Esteves et al., 2015). In addition to increased Drp1 expression, a concomitant increase in Drp1-S616 phosphorylation has been reported. This specific modification is a marker for increased Drp1 activity (Wang et al., 2012; Stafa et al., 2014; Esteves et al., 2015). Overexpression of the gain of function mutations G2019S and R1441C resulted in a reduction of mitochondrial membrane potential, reduced complex IV activity, increased uncoupling, altered mitochondrial motility, and an imbalance in calcium signaling (Cooper et al., 2012; Papkovskaia et al., 2012; Cherra et al., 2013; Godena et al., 2014). Ultrastructural studies show an accumulation of condensed, disorganized and damaged mitochondria consistent with deficits in mitophagy and mitochondrial dynamics (Ramonet et al., 2011; Yue et al., 2015). Finally, increased mtDNA mutations have been identified in cerebrospinal fluid (CSF) and iPSCs derived from patients with the G2019S mutation (Sanders et al., 2014; Podlesniy et al., 2016). Taken together, although the mechanism is not entirely clear, a toxic gain of kinase function in LRRK2 mutations negatively affects mitochondrial function.
Mitochondrial dysfunction induced by LRRK2 also provides a strong link to the impaired autophagy pathway in multiple models. For example, an accumulation in autophagic and lysosomal structures occur both in vitro and in vivo caused by overexpression of PD-associated mutations (MacLeod et al., 2006; Plowey et al., 2008; Gomez-Suaga et al., 2012). A block in autophagy flux, increase in p62 levels and an accumulation of autophagosomes have been demonstrated in dopaminergic neurons generated from PD patient-derived induced pluripotent stem cells (iPSCs) expressing LRRK2-G2019S at endogenous levels (Sánchez-Danés et al., 2012). Importantly, the inhibition of mitochondrial fission, using the Drp1 peptide inhibitor P110, was shown to improve autophagy and reduce detrimental changes in cell morphology caused by the G2019S mutation in PD patient fibroblasts and dopaminergic neurons derived from iPSCs (Su and Qi, 2013). Conversely, another study showed that disrupted mitochondrial motility and an increased vulnerability to multiple stressors induced by G2019S and R1441C mutations in PD patient iPSCs was rescued by the autophagy promoter rapamycin (Cooper et al., 2012) Together, these studies suggest that autophagy and mitochondrial function are bi-directionally regulating one another.
GBA
An interest in GBA as a causative factor of PD followed the clinical observations that some Gaucher’s disease patients developed Parkinsonian symptoms (Neudorfer et al., 1996) and that relatives of these patients had an increased risk of developing PD (Halperin et al., 2006). Gaucher’s disease (GD, Tsuji et al., 1987) is the most common lysosomal storage disorder and is caused by homozygous and compound heterozygous mutations in the GBA gene. GBA is a lysosomal hydrolase, specifically a GBA which acts in the lysosomal membrane to convert the sphingolipid glucosylceramide to glucose and ceramide. Approximately 300 different mutations in GBA have been identified with many shown to have reduced GBA activity. Such reduction has indeed been identified in cases of PD with GBA mutations but, importantly, it has also been identified in the substantia nigra of sporadic PD brains (Gegg et al., 2012). More severe mutations, such as L444P, increase the risk of developing PD and promote an earlier onset as well as greater cognitive decline when compared to milder mutations, such as N370S. However, the overall mortality rate between these two mutations is not significantly different (Gan-Or et al., 2015; Arkadir et al., 2016; Cilia et al., 2016); therefore suggesting that even a partial loss of function is sufficient to cause the disease. The prevalence of GBA mutations varies in different PD populations but is accountable for up to 31% of PD patients with Ashkenazi Jewish ancestry (Sidransky and Lopez, 2012).
A reduction in GBA activity has been associated with mitochondrial dysfunction. GBA inhibition, either pharmacologically or genetically result in a reduction of membrane potential and ATP production, as well as an increase in oxidative stress and mitochondrial fragmentation in vitro (Cleeter et al., 2013). These findings have been confirmed using both fibroblasts isolated from GD patients and mouse models of GD. Fibroblasts from GD patients, with an L444P mutation, exhibited a reduction in complex I-III activity, CoQ10 expression, membrane potential and ATP production as well as an increase in oxidative stress (de la Mata et al., 2015). Mouse models demonstrate a reduction in membrane potential, complex I-III activity, oxygen consumption and an increase in mitochondrial fragmentation in neurons and glia (Osellame et al., 2013; Xu et al., 2014). Further, ultrastructural analysis of mitochondrial morphology showed impaired cristae organization, with mitochondria appearing more rounded and electron dense (Xu et al., 2014).
While the mechanisms of mitochondrial dysfunction associated with GBA mutations are unclear, an impairment of mitophagy is suggested to play an important role. This is perhaps not surprising given the role of GBA in the cell and this observation further strengthens the link between autophagy and mitochondria. In mouse models and primary fibroblasts, decreased mitophagy flux accompanied the observed mitochondrial dysfunction (de la Mata et al., 2015). However, whether an impaired mitophagy response causes the observed mitochondrial dysfunction in these models or whether it is a consequence of an upstream effect is not understood. Another contributing factor to mitochondrial dysfunction in these models is neuroinflammation. In GBA knockout mouse models, activation of both astrocytes and microglia precedes disease manifestation (Enquist et al., 2007; Farfel-Becker et al., 2011). Astrogliosis has also been identified in some GD patients that exhibit parkinsonian symptoms (Wong et al., 2004). It is important to note that the majority of the studies related to GBA discussed in this review have been performed in the setting of GD rather than PD. Therefore, the conclusions made from this work, although relevant, may not be directly transferrable to cases of PD with GBA mutations.
GBA activity appears to be linked to the expression and aggregation of α-synuclein. An accumulation of aggregated α-synuclein, β-amyloid and amyloid precursor protein (APP) in the cortex, hippocampus, striatum and substantia nigra of GBA mutant mice was observed (Xu et al., 2014). Further, inhibiting GBA, using zinc finger techniques, promotes the intercellular transmission of α-synuclein species and therefore contributes toward the spread of PD pathology (Bae et al., 2014). These observations are consistent with the role of GBA in autophagy function. An increase in α-synuclein expression has also been shown to inhibit GBA activity (Mazzulli et al., 2011; Gegg et al., 2012; Murphy et al., 2014), thus further impairing the clearance of α-synuclein mediated by autophagy.
VPS35
A single mutation, D620N, in the gene encoding vacuolar protein sorting 35 (VPS35) causes the development of an autosomal dominant, late-onset PD (Vilarino-Guell et al., 2011; Zimprich et al., 2011; Sharma et al., 2012). A number of other mutations in the gene have been identified; however, their pathological relevance is yet to be demonstrated. VPS35 mutations are rare and only account for approximately 1% of familial and 0.2% of sporadic PD cases.
VPS35 is a core subunit of a heteropentameric complex known as the retromer, which plays an important role in the transport of endosomes to the golgi apparatus and the plasma membrane, as well as in recycling of transmembrane protein cargo (Williams et al., 2017). In recent years VPS35 has also been reported to mediate the shuttling of cargo from mitochondria to peroxisomes and lysosomes through mitochondria-derived vesicles (Braschi et al., 2010; Sugiura et al., 2014). Models of VPS35 mutations successfully demonstrate dopaminergic neurodegeneration (Tsika et al., 2014; Wang et al., 2014, 2016; Tang et al., 2015). The pathogenic mechanism by which VPS35 induces PD is not clear, however, recent studies show that its mutation caused mitochondrial fragmentation, impaired mitochondrial function, increased ROS, α-synuclein accumulation and cell death (Tang et al., 2015; Wang et al., 2016). The D260N mutant has an increased binding interaction with Drp1, an effect that is further promoted by oxidative stress, resulting in an increased turnover of mitochondrial Drp1 complexes via their mitochondria derived vesicular trafficking to the lysosome for degradation (Wang et al., 2016). In addition to increasing mitochondrial fission, VPS35 deficiency or mutation induces mitochondrial fragmentation by impairing mitochondrial fusion (Tang et al., 2015) via mitochondrial E3 ubiquitin ligase 1 (MUL1) upregulation, leading to increased ubiquitination and proteasomal degradation of the fusion protein MFN2. Together, these studies strongly support the role of VPS35 in imbalanced mitochondrial dynamics.
CHCHD2
Although little is known about the physiological roles of Coiled-Coil-Helix-Coiled-Coil-Helix Domain Containing 2 (CHCHD2) or its role in PD pathology, several mutations in the CHCHD2 gene cause an autosomal dominant form of late-onset PD and dementia with Lewy bodies (Funayama et al., 2015; Ogaki et al., 2015; Koschmidder et al., 2016). CHCHD2 contains a mitochondrial targeting sequence in the N-terminal domain and localizes to intermembrane space of mitochondria, where it binds to the complex IV (cytochrome c oxidase, COX). This binding is required for COX activity (Aras et al., 2015). CHCHD2 is therefore necessary for oxidative phosphorylation (Baughman et al., 2009). Indeed, reduced CHCHD2 expression results in reduced COX activity, collapsed membrane potential and an increase in ROS and mitochondrial fragmentation (Aras et al., 2015), resulting in cell death in Drosophila (Meng et al., 2017). PD-related mutant flies exhibited a chronic state of oxidative stress, reduced oxygen consumption, ATP production, increased cytochrome c release and displayed mitochondria with abnormal ultrastructure leading to degeneration of dopaminergic neurons. This study also suggests that CHCHD2 may be an important mitochondrial stress response protein and can modulate cell death signaling by interacting with cytochrome c and MICS1, a member of the Bax inhibitor-1 superfamily (Meng et al., 2017). Although CHCHD2 predominantly localizes in mitochondria and directly binds to complex IV, CHCHD2 is also a transcription factor of the complex IV subunit 4 isoform COX4I2 (Aras et al., 2015). Because of this unique feature, loss of CHCHD2 function negatively affects complex IV both functionally and structurally.
Autosomal-Recessive Genes
PINK1 and Parkin
Mutations in PTEN-induced kinase 1 (PINK1) and Parkin both cause an almost identical form of autosomal recessive, early onset familial PD which can present with or without Lewy Bodies (Kitada et al., 1998; Sasaki et al., 2004; Valente et al., 2004a; Samaranch et al., 2010). A mutation in Parkin was first identified in a Japanese family (Matsumine et al., 1997; Kitada et al., 1998) and are now collectively shown to encompass a wide range of variations spanning the entire length of the gene conferring a loss of function (Klein and Westenberger, 2012). Parkin mutations are considered as the most prevalent autosomal recessive mutations in PD accounting for approximately up to 77% of familial early-onset PD and 10–20% of early-onset PD in general (Klein and Lohmann-Hedrich, 2007; Klein and Westenberger, 2012; Kilarski et al., 2012). Mutations in PINK1, first identified in an Italian family (Valente et al., 2001), are the second most common cause of early-onset autosomal recessive PD accounting for up to 9% of cases (Klein and Westenberger, 2012). Mutations span the length of PINK1, with the majority being missense or non-sense mutations and, similarly to Parkin, confer a loss of function. Heterozygous PINK1 and Parkin mutations may also be considered as a risk factor for developing PD (Khan et al., 2005; Criscuolo et al., 2006; Hedrich et al., 2006; Hiller et al., 2007).
PINK1 is a Ser/Thr kinase that is localized to the mitochondria (Hatano et al., 2004; Valente et al., 2004a,b; Zhou et al., 2008). In a normal, healthy mitochondrion PINK1 binds to the outer mitochondrial membrane (OMM), and is then imported into the inner mitochondrial membrane (IMM) via the translocase of the outer membrane (TOM) complex and translocase of the inner membrane 23 (TIM23) in a membrane potential-dependent manner. Full length PINK1 is then rapidly cleaved by mitochondrial processing peptidase (MPP) and presenilin-associated rhomboid-like protease (PARL). Cleaved PINK1 is released into the cytosol for other non-mitochondrial functions (Dagda et al., 2014) and can be degraded by the proteasome (Meissner et al., 2011; Yamano and Youle, 2013). Hence, PINK1 is maintained at a low level in healthy mitochondria. However, upon mitochondrial damage and the loss of membrane potential, PINK1 import is blocked causing it to accumulate at the OMM (Jin et al., 2010; Lazarou et al., 2012; Okatsu et al., 2013). At the OMM, the kinase domain of PINK1 faces toward the cytosol, enabling it to phosphorylate both mitochondrial and cytosolic proteins (Zhou et al., 2008). Although not consistently observed, PINK1 has been reported to phosphorylate ubiquitin and ubiquitin-like (Ubl) domain of Parkin at a conserved Ser65 residue. This phosphorylation initiates a cascade that consequently activates and recruits Parkin, a ubiquitin E3 ligase, to mitochondria (Durcan and Fon, 2015; Roberts et al., 2016). Activated Parkin promotes ubiquitination of multiple OMM proteins targeting them for degradation via the proteasomal pathway (Gegg et al., 2010; Chan et al., 2011; Yoshii et al., 2011; Kazlauskaite et al., 2014) as well as the recruitment of autophagy adaptor proteins (Roberts et al., 2016). The PINK1/Parkin axis is therefore an important pathway for mitochondrial quality control and the selective removal of the damaged organelle by mitophagy (Geisler et al., 2010; Narendra et al., 2010, 2012; Vives-Bauza et al., 2010).
Mitochondrial abnormalities have been reported in multiple in vivo and in vitro models of PINK1 and Parkin. PINK1 deficient and PD-associated mutant models display reduced mitochondrial membrane potential, ATP levels, reduced respiratory capacity via complex I and IV activity, increased mitochondrial calcium levels, sensitized mitochondrial permeability transition pore opening and increased ROS production (Gautier et al., 2008, 2012; Piccoli et al., 2008; Dagda et al., 2009; Gandhi et al., 2009; Gegg et al., 2009; Liu W. et al., 2009; Morais et al., 2009, 2014; Cui et al., 2010; Heeman et al., 2011). Isolated mitochondria from brains of aged PINK1 null mice display complex I deficits (Gautier et al., 2008; Morais et al., 2014) and reduced calcium buffering capacity (Akundi et al., 2011). Aged Parkin null brains show alterations in the expression of a wide range of mitochondrial proteins, with many relating to a reduced respiratory capacity and increased oxidative damage (Palacino et al., 2004; Periquet et al., 2005; Stichel et al., 2007). PINK1 and Parkin also play a regulatory role in mitochondrial dynamics by mechanisms involving the turnover of fusion and fission associated proteins such as Mfn1/2 and Drp1 (Clark et al., 2006; Deng et al., 2008; Poole et al., 2008; Yang et al., 2008; Lutz et al., 2009; Cui et al., 2010; Gegg et al., 2010; Tanaka et al., 2010; Ziviani et al., 2010; Exner et al., 2012; Buhlman et al., 2014; Pryde et al., 2016). However, whether PINK1 and Parkin are pro-fission or pro-fusion has been a topic of debate (Exner et al., 2012; Pilsl and Winklhofer, 2012).
In addition to the above effects on mitochondrial function and dynamics, PINK1 and Parkin mediate biogenesis through an indirect interaction with PGC-1α (Shin et al., 2011; Stevens et al., 2015; Lee et al., 2017). The loss of PINK1/Parkin activity leads to a reduced clearance, and consequent build-up, of the Parkin Interacting Substrate (PARIS/ZNF746) which is a transcriptional repressor of PGC-1α. An increase in PARIS/ZNF746 is identified in the striatum and substantia nigra of patients with autosomal recessive and sporadic PD, as well as in Parkin knockout mice (Shin et al., 2011).
DJ-1
Homozygosity mapping in two consanguineous families from Italy and the Netherlands with early-onset PD identified DJ-1 mutations (Bonifati et al., 2003). Collectively, mutations in DJ-1 account for approximately 1–2% of early onset recessive cases of PD and encompass deletions, homozygous and heterozygous point mutations and truncations (Bonifati et al., 2003). DJ-1 is a small protein of 189 residues localized to the cytoplasm, the nucleus and associated with mitochondria (Canet-Aviles et al., 2004; Li et al., 2005; Junn et al., 2009). Among its functions, DJ-1 is involved in regulating mitochondrial activity (Hayashi et al., 2009; Junn et al., 2009) and protecting against oxidative stress (Taira et al., 2004).
DJ-1 can interact with mitochondrial complex I subunits and is translocated to the mitochondria under stress conditions (Hayashi et al., 2009). DJ-1 and its mutant forms could associate with the chaperone protein Hsp70, a link that was further increased under oxidative stress conditions. These results suggest that translocation of DJ-1 to the mitochondria may occur with the aid of mitochondrial chaperone proteins (Li et al., 2005). It is believed that the dimeric form of DJ-1 is required for antioxidative stress reactions, while monomeric mutant forms of DJ-1 are toxic for cells (Zhang et al., 2005; Maita et al., 2013). Further studies also demonstrated mitochondrial dysfunction and reduced membrane potential in DJ-1 knockout mice and flies (Hao et al., 2010; Giaime et al., 2012). Consistent with its protective role in mitochondria, DJ-1 knockout mice exhibit enhanced vulnerability to the mitochondrial toxicant 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Kim et al., 2005; Manning-Boğ et al., 2007).
DJ-1 is required for the proper balance of mitochondrial dynamics; primary cortical neurons and embryonic fibroblasts cultured from DJ-1 knockout mice and human dopaminergic neuronal cells with DJ-1 knockdown exhibit mitochondrial fragmentation and reduced Mfn2 levels (Irrcher et al., 2010; Krebiehl et al., 2010; Thomas et al., 2011). Blocking mitochondrial fission or promoting mitochondrial fusion rescues this abnormal mitochondrial morphology (Irrcher et al., 2010; Thomas et al., 2011). DJ-1 has also been suggested to participate in the mitophagy pathway. In fact, loss of DJ-1 leads to mitochondrial impairment and dysfunctional autophagy (Irrcher et al., 2010; Krebiehl et al., 2010; Thomas et al., 2011) and overexpression of PINK1 and Parkin is protective (Irrcher et al., 2010; Thomas et al., 2011). These results are consistent with the proposal that DJ-1 may be aiding PINK1/Parkin in the clearance of misfolded proteins and damaged mitochondria (Xiong et al., 2009; Thomas et al., 2011). Taken together, DJ-1 has a critical role in the oxidative stress response, mitochondrial function and basal autophagy.
PLA2G6
Mutations in the PLA2G6 gene have been described in patients with autosomal recessive early-onset dystonia-parkinsonism with widespread accumulation of Lewy bodies and hyperphosphorylated tau (Paisan-Ruiz et al., 2009; Miki et al., 2017). Brain iron accumulation is found in most affected individuals (Paisan-Ruiz et al., 2009). The PLA2G6 gene encodes a 85-kDa group VI calcium-independent phospholipase A2β (PLA2G6) and is involved in the hydrolysis of glycerophospholipids to release free fatty acids and lysophospholipids (Ma and Turk, 2001). PLA2G6 is confined to the mitochondrial compartment where it remodels membrane phospholipids, protects mitochondria from oxidative damage during apoptotic induction and takes part in calcium signaling (Gadd et al., 2006; Seleznev et al., 2006; Strokin et al., 2012). Although the pathological mechanism of PLA2G6 remains elusive, it has been suggested to affect lipid homeostasis and metabolism as well as impact mitochondrial membranes (Beck et al., 2011). Further, PLA2G6 knockout mice displayed degenerated mitochondrial membranes and elevated levels of synuclein (Sumi-Akamaru et al., 2016). PLA2G6 has been reported to be immunopositive in the Lewy bodies of idiopathic PD, suggesting its pathogenic role extends beyond familial PD (Miki et al., 2017).
ATP13A2
ATP13A2 encodes ATPase type 13A, which is usually localized to lysosomes. Mutations in ATP13A2 were initially identified in Kufor–Rakeb syndrome (Ramirez et al., 2006), which is a recessively inherited disease with multiple brain regions are affected. Fibroblasts derived from these patients display reduced ATP synthesis, increased mtDNA mutations, impaired oxygen consumption and mitochondrial fragmentation (Grunewald et al., 2012). Mutations in ATP13A2 are also found in early onset parkinsonism (Di Fonzo et al., 2007), suggesting that ATP13A2 may also be relevant to PD. ATP13A2 is a multifunctional protein that has been linked to endosome-lysosome dynamics (Dehay et al., 2012; Usenovic et al., 2012), mitochondrial function (Grunewald et al., 2012; Gusdon et al., 2012) and toxicity induced by divalent cation metals (Mn2+ and Zn2+) (Gitler et al., 2009; Kong et al., 2014). Consistent with these roles of ATP13A2, loss of function in this protein has been reported to reduce the ability of various vesicular structures such as lysosomes to regulate divalent metal cations such as Mn2+ and therefore sensitize cells to metal toxicity, accumulation of α-synuclein and mitochondrial dysfunction (Gitler et al., 2009; Kong et al., 2014). These effects are most likely secondary to autophagy deficits, causing an accumulation of misfolded proteins and damaged mitochondria. Collectively, ATP13A2 may exert an indirect, protective role on mitochondria and the cell as a whole. Indeed, overexpression of ATP13A2 is protective against α-synuclein-induced toxicity (Gitler et al., 2009; Kong et al., 2014).
FBXO7
Mutations within the FBXO7 (F-box only protein 7) gene have been shown to promote either a typical PD phenotype (Lohmann et al., 2015) or an early onset form of parkinsonian-pyramidal syndrome (Shojaee et al., 2008; Di Fonzo et al., 2009). FBX07 has been linked to mitochondrial function by acting on the same pathway as PINK1 and Parkin. It directly interacts with both PINK1 and Parkin and therefore plays a role in mitochondrial maintenance and mitophagy (Burchell et al., 2013; Zhou et al., 2015). These studies demonstrate that FBXO7 assists in the translocation of Parkin to the mitochondria in response to cell stress and PD-associated mutations leading to a mislocalization of FBXO7 to the cytosol where it can form toxic aggregates that are deleterious to the cell (Zhou et al., 2015).
Environmental Exposures As Potential Links to PD
Prior to the discovery of the first genetic mutation linked to PD (Polymeropoulos et al., 1997) environmental factors were the major focus of research aiming to understand PD pathogenesis and etiology. The rationale for this dominant environmental theory was largely based on observations such as post-encephalitic infection, manganese (Mn) and MPTP exposure causing parkinsonism (Rail et al., 1981; Langston et al., 1983; Calne et al., 1994; Kwakye et al., 2015). Multiple epidemiological studies and meta-analyses have implicated a number of environmental factors, including pesticides and heavy metals, with increasing PD risk (Tanner et al., 2011; Wang et al., 2011; Kamel, 2013). These studies support the proposition that chronic exposure to environmental toxicants plays a major role in sporadic PD pathogenesis. Pesticides and heavy metals are the most widely researched environmental toxicants implicated in PD, and the ability of some of them to persist in the environment long after their application increases the likelihood of human exposure to these contaminants. Following are some environmental exposures that have been demonstrated to be toxic to the nigrostriatal pathway.
Pesticides
Rotenone
A potent non-competitive mitochondrial complex I inhibitor derived from plants of the Leguminosa family (Soloway, 1976), rotenone is a pesticide/piscicide associated with increased risk of developing PD (Tanner et al., 2011; Kamel, 2013). Because the half-life of rotenone is relatively short (∼3 days) (Hisata, 2002), consumption of contaminated food products is an unlikely exposure route. The role of rotenone in PD is therefore most likely through low chronic exposure rather than acute toxicity. Epidemiological studies have demonstrated increased PD risk of ∼1.5–3-fold in individuals who utilized rotenone agriculturally or who lived in close proximity to its use (Tanner et al., 2011). As a highly lipophilic chemical, rotenone passes freely through the blood-brain-barrier (BBB) and cellular membranes and once inside cells it can directly alter mitochondrial function. Rotenone impairs oxidative phosphorylation by inhibiting NADH-ubiquinone reductive activity via binding to the PSST subunit of complex I of the electron transport chain (Schuler and Casida, 2001). This mechanism of rotenone is relevant to the observations that mitochondrial complex I activity is reduced in PD patients (Parker et al., 1989; Schapira et al., 1989). Rotenone has also been shown to enhance mitochondrial fission. In primary cortical neurons, rotenone induces rapid mitochondrial fragmentation within 2 h of treatment, prior to any cellular changes indicative of cytotoxicity (Barsoum et al., 2006). Altering mitochondrial dynamics, by promoting mitochondrial fusion with Mfn2 and blocking Drp1 with Drp1-K38A averts this neurotoxicity (Barsoum et al., 2006). Although the neuropathology is variable, rotenone treatment has been shown to induce motor impairment, neuroinflammation, nigrostriatal degeneration, and accumulation and phosphorylation of α-synuclein (Betarbet et al., 2000; Cannon et al., 2009) in rodent models of PD.
In addition to impairing mitochondrial function, rotenone has been demonstrated to alter mitochondrial transport and protein interactions within neurons. For example, rotenone induces microtubule depolymerisation, thereby interfering with the intracellular transport of vesicles and organelles, including mitochondria (Choi et al., 2011). A recent study revealed increased α-synuclein association with mitochondria, specifically outer membrane protein TOM20, in nigrostriatal dopaminergic neurons of rotenone-treated rats relative to controls (Di Maio et al., 2016). This observation suggests that rotenone exposure may induce α-synuclein-mediated changes in mitochondrial function and morphology to further promote PD pathology.
Paraquat
The herbicidal mechanism of paraquat (PQ) involves interference with photosynthesis and oxygen free radical production, resulting in damage of plant membranes (Tanner et al., 2011; Breckenridge et al., 2013). Initially based on its similar structure to MPP+, this widely used herbicide was hypothesized to be a potential environmental parkinsonian toxicant (Snyder and D’Amato, 1985). Epidemiological studies demonstrate that PQ exposure, both alone and in conjunction with other pesticides, increases the risk of developing PD (Costello et al., 2009; Ritz et al., 2009; Tanner et al., 2011). To further understand the mechanism by which this molecule induces dopaminergic neurotoxicity, PQ has been used experimentally to model PD (Tieu, 2011). PQ is unable to diffuse across the BBB and relies on transport by the L-neutral amino acid transporter to enter the brain (Shimizu et al., 2001; McCormack and Di Monte, 2003). Once inside the brain, PQ can enter cells through the dopamine transporter (DAT) and organic cation transporter-3 (Rappold et al., 2011). In the cytosol, PQ induces toxicity primarily through redox cycling with cellular diaphorases such as NADPH oxidase and nitric oxide synthase (Day et al., 1999), leading to the generation of superoxide. Despite its similar structure to MPP+, PQ is not a complex I inhibitor (Richardson et al., 2005), although this is the site where superoxide is generated from PQ redox-cycling in the mitochondria (Cochemé and Murphy, 2008). Other work also implicates complex III in the formation of paraquat-induced ROS (Castello et al., 2007). Consistent with the role of generating mitochondrial ROS, a recent study reported mitochondrial DNA damage in PD patients with prior history of exposure to PQ (Sanders et al., 2017). The mechanism by which PQ enters mitochondria, however, is currently unknown.
PQ has also been reported to increase mitochondrial fission by reducing the levels of key mitochondrial fusion factors, Mfn1/Mfn2 (Tanaka et al., 2010). In rodent models PQ has been shown to cause neuroinflammation, as well as α-synuclein upregulation and aggregation (Manning-Bog et al., 2002; Wu et al., 2005; Fernagut et al., 2007; Purisai et al., 2007). The use of PQ to model PD has been contested by some, partly because studies utilizing wild-type mice demonstrated variable results, with loss of dopaminergic nigral cell bodies but a lack of consistency in the loss of the corresponding striatal terminals (McCormack et al., 2002; Thiruchelvam et al., 2003; Tieu, 2011); however, this lack of striatal damage has been suggested to be the result of compensatory sprouting from surviving dopaminergic neurons and uptake of PQ into non-dopaminergic striatal cells. In contrast to wild-type mice, mutant mice deficient in OCT3 (Slc22a3), a cation transporter which can uptake PQ into cells, display enhanced striatal damage upon exposure to PQ. This increased sensitivity likely results from reduced buffering capacity by non-dopaminergic cells, leading to increased availability of PQ to damage dopaminergic terminals (Rappold et al., 2011). Despite the controversy which still surrounds the use of PQ to model PD, current evidence from epidemiological studies and mechanistic investigations appear to sustain the conclusion that PQ may increase the risk of developing PD.
Organochlorines
Dieldrin is one of the organochlorine pesticides that was first commercially available in 1950, with wide use in the United States until the late 1980’s. Despite decades of research into potential risk of dieldrin, it is still extensively used in many developing countries. Dieldrin has a long half-life (∼25 years) which contributes to a persistent high risk of exposure, even decades after its use has ceased. Human exposure risk may be mediated through consumption of dairy products, meat, and fish (Kanthasamy et al., 2005), although its high lipophilicity also confers risk of dermal exposure, as it can be rapidly absorbed through the skin. In addition to its environmental persistence, the ability of dieldrin to bioaccumulate, with a half-life of ∼300 days in humans, contributes to the risk it confers, as it can readily pass the BBB and amass in the brain. Indeed, dieldrin has been detected in 30% of post-mortem brain tissue of PD patients, with none in age-matched control brains (Fleming et al., 1994). Another study reported dieldrin in both control and patient brain tissue, with significantly higher levels in the caudate nucleus of PD patients (Corrigan et al., 1996). These results support a potential role of this pesticide in promoting nigral cell death and PD progression (Kanthasamy et al., 2005).
Dieldrin has been shown to exert multiple effects on mitochondria, including increased ROS production (Chun et al., 2001) and stimulation of mitochondria-mediated apoptosis (Kitazawa et al., 2003; Kanthasamy et al., 2008). Dopaminergic cells have increased sensitivity to dieldrin when compared to other cell types as demonstrated in vitro (Sanchez-Ramos et al., 1998; Kitazawa et al., 2001) and in vivo (Sharma et al., 1976; Wagner and Greene, 1978; Heinz et al., 1980). Dieldrin (and other pesticides) have also been shown to block the function of mitochondrial aldehyde dehydrogenase (ALDH class 2) (Fitzmaurice et al., 2013, 2014). ALDH2 metabolizes toxic aldehydes including 3,4-dihydroxyphenylacetaldehyde (DOPAL), which is a reactive neurotoxic metabolite of dopamine. Exposure of individuals with genetic variation in ALDH2 to these pesticides was associated with a two–sixfold increase in their risk of developing PD (Fitzmaurice et al., 2014). ALDH2 inhibition therefore has been suggested as a pathogenic mechanism in PD. Furthermore, based on a recent work that investigated the proteomic effects of dieldrin, 18 proteins involved in the respiratory chain were affected by dieldrin treatment (Cowie et al., 2017), further supporting the influence of this pesticide on mitochondrial function.
Dieldrin has also been demonstrated to directly stimulate α-synuclein fibril formation in vitro, by inducing a conformational change which may serve as a precursor or seed for the aggregation of further protein (Uversky et al., 2001b). Prolonged dieldrin exposure has been reported to inhibit the ubiquitin-proteasome protein degradation system (Sun et al., 2004), further promoting protein aggregation. In summary, although the mechanistic understanding remains incomplete, dieldrin has been shown to induce dopaminergic neurotoxicity through mitochondrial dysfunction, decreased protein degradation and apoptosis initiation.
Pyrethroids
Pyrethroids are synthetic derivatives of the naturally occurring pyrethrum from chrysanthemum flowers. These pesticides are widely used both agriculturally and in the household (Elwan et al., 2006), however, despite their classification as safe substances (Casida et al., 1983), pyrethroid intoxication in humans has been observed (Chen et al., 1991). These chemicals, which include permethrin and deltamethrin, readily cross the BBB and may accumulate at considerable concentrations within the brain (Anadón et al., 1996). Permethrin and deltamethrine have been shown to increase DAT-mediated dopamine uptake by up to 30% in the mouse striatum and induce apoptosis in dopaminergic neuroblastoma cells (Elwan et al., 2006). Because the DAT mediates neurotoxicity of some neurotoxicants (Gainetdinov et al., 1997; Donovan et al., 1999; Rappold et al., 2011), pyrethroids may enhance the vulnerability of dopaminergic neurons to toxic compounds. Furthermore, permethrin has been shown to potently inhibit mitochondrial complex I function in rat liver (Gassner et al., 1997), however, a subsequent study demonstrated that apoptosis induced by pyrethroids (Deltamethrin) is mitochondrial independent (Hossain and Weiner, 1993). Rather, deltamethrin induces calcium overload through Na+ channels, leading to endoplasmic reticulum stress.
A recent study investigated the potential use of permethrin to model PD in a progressive model of early life exposure to the pesticide (Nasuti et al., 2017). Treatment of Wistar rat pups with permethrin from post-natal days (PND) 6–21 induced cognitive impairments, reduced striatal dopamine levels and reduced dopaminergic neurons in the substantia nigra; all common neuropathological changes detected in PD patients. These changes were first obvious at PND 60, although no motor changes were recorded until testing at PND 150. This study adds a new potential pesticide model of PD to the repertoire currently utilized for research, and highlights an important consideration as early life exposure to some toxicants may pre-dispose individuals to pathogenic changes later in life, or initiate slow progressive changes which culminate in development of the clinical phenotype. Despite the demonstration of the neurotoxicity of pyrethroids in experimental models, there appears to be insufficient human data to link pyrethroids exposure to PD (Costa, 2015).
Heavy Metals
Copper, iron, zinc, and Mn represent some of the heavy metals involved in the development of neurodegeneration. Collectively, these metals are required for many physiological activities, however, excessive levels may detrimentally affect cell viability through mechanisms such as mitochondrial dysfunction and oxidative stress (Martinez-Finley et al., 2013; Park et al., 2015). They can also promote DNA damage and interact with proteins that are directly involved in neurotoxicity, such as α-synuclein and β-amyloid, by altering their structures (Cervantes-Cervantes et al., 2005; Ha et al., 2007; Salvador et al., 2010; Chen et al., 2016). Taken together, these effects will eventually disrupt a variety of cellular processes and lead to increased risk of neurological damage.
Of the heavy metals implicated in PD, Mn has received the most attention. Mn is an essential metal characterized by its vital role in brain development and homeostasis (Aschner and Aschner, 2005). It has several chemical forms with 11 oxidation states and its metabolism is tightly linked to that of other heavy metals, in particular that of iron due to their analogous redox behavior (Claus Henn et al., 2011; Kim et al., 2013; Kwakye et al., 2015). Mn is essential for many enzymatic reactions; it can act as a co-factor for glutamine synthetase in astrocytes and it participates in the mitochondrial antioxidant defence system as part of the superoxide dismutase (MnSoD) (Borgstahl et al., 1992). Depending on its oxidative state, Mn can be imported to the central nervous system (CNS) through separate membrane importers such as the divalent metal transporter 1 (DMT1) (highly expressed in the basal ganglia), the DAT, calcium channels, choline and citrate transporters, transferrin, and the ZIP (Zrt- and Irt-related protein) family metal transporters (Huang et al., 2004; Gunter et al., 2013; Tuschl et al., 2013). Metal efflux also plays a critical role in maintaining the homeostatic levels of Mn; although the number of proteins involved in exporting Mn are limited compared to importer channels, they include ferroportin (Fpn), SLC30A10 (solute carrier family 30 member 10), secretory pathway Ca2+-ATPase 1 (SPCA1) and ATP13A2 (Tuschl et al., 2013). Chronic exposure to Mn can trigger harmful effects in the brain clinically resulting in Manganism (Couper, 1837), which is characterized by motor impairments, behavioral changes and cognitive alterations. This movement disorder has symptoms that resemble (Aschner et al., 2009), but are distinct to, those of PD (Calne et al., 1994; Guilarte and Gonzales, 2015). However, Mn exposure is believed to contribute to increased PD risk in conjunction with other factors, such as genetic susceptibility. In addition to environmental exposure, mutations in the SLC30A10 gene (the first human Mn transporter identified) cause an accumulation of Mn in both the basal ganglia and the liver, inducing hypermanganesemia, childhood onset dystonia and adult onset parkinsonism (Quadri et al., 2012; Tuschl et al., 2012). Furthermore, in 2016, a novel autosomal recessive Mn transporter defect (SLC39A14) was discovered in children affected by hypermanganesemia and progressive parkinsonism–dystonia. SLC39A14 represents a specific Mn transporter as mutations in this gene impair Mn uptake, without affecting any other metal levels (Tuschl et al., 2016).
Although parkinsonism induced by Mn and PD share some similar features, it is important to note that they are two distinctive disorders (Calne et al., 1994; Guilarte and Gonzales, 2015; Kwakye et al., 2015). In fact, Mn-induced-parkinsonism does not respond to levodopa, the most commonly used and effective drug in PD. Moreover, it is believed that Mn primarily affects gamma aminobutyric acid (GABA) producing neurons in the globus pallidus while the numbers of dopaminergic neurons in the substantia nigra remain relatively unaffected; stereological cell quantification needs to be performed to confirm these results (Guilarte et al., 2006; Guilarte and Gonzales, 2015). Moreover, Guilarte et al. (2008) used Positron Emission Tomography (PET) to show that Mn-exposed animals exhibited reduced dopamine release in the dorsal striatum, demonstrating an impaired nigrostriatal dopaminergic system in Mn-exposed monkeys. Although Mn-induced parkinsonism cannot be considered as idiopathic PD, Mn can still affect PD pathogenesis by interacting with an individual’s genetic makeup as discussed below, and epidemiological study has established an accumulation of Mn levels in PD patients (Fukushima et al., 2010). In addition, chronic exposure to Mn results in significantly increased Mn levels in dopaminergic neurons of rodent brains compared to control animals (Robison et al., 2015). Therefore, it is possible that Mn can enhance the susceptibility of the individual to developing PD or accelerate the disease onset.
The precise pathological mechanism of Mn toxicity is not known, however, evidence suggests a direct interaction with mitochondria and subsequent induction of mitochondrial damage. Mn can enter mitochondria as Mn2+ through the Ca2+ uniporter channel and slow export occurs via the Na+-independent Ca2+ channel (Gavin et al., 1999). High Mn2+ concentrations can accumulate in the mitochondrial matrix and promote Na+-dependent and Na+-independent Ca2+ efflux channel through direct competitive inhibition. This subsequently causes an increase of mitochondrial Ca2+ levels, which interfere with oxidative respiration and induce oxidative stress (Gavin et al., 1990). It has been suggested that ROS induction can originate from superoxide which can then be converted in the mitochondria to hydrogen peroxide by the Mn and Cu/Zn superoxide dismutase. Hydrogen peroxide can be reduced to form hydroxyl radicals in the presence of Mn or other transition metals via the Fenton reaction (Goldstein et al., 1993; Martinez-Finley et al., 2013). The ROS generated by excessive Mn levels promote opening of the mitochondrial permeability transition pore, causing a loss of membrane potential, impaired ATP synthesis and mitochondrial swelling, contributing to cellular apoptosis (Gavin et al., 1990; Gavin et al., 1992). Gunter et al. (2010) measured ATP production to study how Mn2+ promotes inhibition of the complex oxidative phosphorylation system in multiple organs (liver, heart, and brain mitochondria). Their results showed that Mn2+ inhibits ATP production in different manners when comparing the different tissues. It appears that Mn2+ inhibits two independent sites in brain mitochondria: the primary site is electron transport chain complex II, while the second is either the glutamate/aspartate exchanger or the aspartate aminotransferase (Gunter et al., 2010).
Although previous studies have focused their attention on the effects of Mn on mitochondria and its capacity to interfere with oxidative phosphorylation, there is little evidence of the involvement of mitochondrial dynamics following Mn neurotoxicity. A recent study, however, showed that Mn induced changes in mitochondrial-shaping proteins (Opa-1, Mfn-2, and Drp-1) by shifting the balance toward mitochondrial fission (Alaimo et al., 2014). Indeed, an increased amount of Drp-1 translocation to the mitochondria was observed, indicative of increased mitochondrial fission. Mn-induced apoptosis was prevented when Drp-1 was inhibited (Alaimo et al., 2014). Furthermore, Mn toxicity could promote inflammatory events by directly modulating mitochondrial dynamics in astrocytes, and addition of mito-apocynin (a mitochondrial targeted antioxidant) significantly reduced Mn-induced inflammation (Sarkar et al., 2017). These studies demonstrate that Mn impairs mitochondrial function in both glia and neurons and that restoring mitochondrial health in either cell type is neuroprotective.
Dietary Exposures
Diets have been documented to have an impact on the risk of developing PD. Strong epidemiological evidence indicates, for example, that cigarette smoking and coffee drinking are inversely correlated to PD development (Hernán et al., 2002). On the other hand, certain dietary exposures have also been linked to parkinsonism. Annonacin is one such example.
Annonacin is a chemical derived from plants of the Annonacae family, such as the soursop fruit, which has been implicated in the development of atypical parkinsonism in Guadeloupe. In the 1990’s it was observed that a high proportion of Guadeloupe’s population exhibited levodopa-resistant parkinsonism, with presentation of bradykinesia, rigidity, postural instability, and in some cases dementia (Caparros-Lefebvre and Elbaz, 1999; Caparros-Lefebvre et al., 2002). Annonacin belongs to the acetogenins, a class of large, structurally homogenous polyketides which act as potent mitochondrial poisons via inhibition of mitochondrial complex I (Tormo et al., 1999; Motoyama et al., 2002). Studies of annonacin have demonstrated its potency for complex I inhibition to be up to 50 times greater than that of MPP+, and at least equal to that of rotenone, consistent with its ability to cause drastic neuronal loss and the development of parkinsonian symptoms in a similar manner to these two PD-linked complex I inhibitors (Lannuzel et al., 2006). Acetogenins are highly toxic and have been shown to cause loss of dopaminergic neurons at nanomolar concentrations in vitro (Lannuzel et al., 2006). Due to its lipophilic nature, annonacin does not require specific transporters for entry into the brain or cells and it inhibits complex I function in both neuronal and glial populations (Degli Esposti, 1998; Lannuzel et al., 2006). Although the effects of annonacins are not specific to dopaminergic neurons, neurodegeneration of the basal ganglia is the most pronounced, with reduced alteration of the hippocampus, cerebellum and cerebral cortex (Lannuzel et al., 2006). Studies have confirmed that annonacin-treated rats exhibit neurodegeneration characteristic of PD, with stereological cell counts detailing significant loss of dopaminergic neurons in the substantia nigra, reduced dopaminergic striatal terminals and significantly increased astrocyte and microglial reactivity (Champy et al., 2004; Lannuzel et al., 2006).
Annonacin treatment of primary embryonic striatal neurons induces retrograde mitochondrial transport, with some mitochondria displaying tau protein association with their outer membranes, suggesting that annonacin may also confer risk for other neurodegenerative disorders, including Alzheimer’s disease (Escobar-Khondiker et al., 2007). The authors surmised that ATP-depletion induces this mitochondrial withdrawal from cellular extremities, which may in turn mediate loss of these axonal terminals due to insufficient fulfillment of their energy demands (Escobar-Khondiker et al., 2007). In humans with chronic exposure from fruit consumption, synergistic effects of annonacin with other acetogenins and alkaloids, which are also present in some annonacin-containing plants, may contribute to the putative toxicity of these fruits and the development of neurodegenerative diseases (Lannuzel et al., 2003; Potts et al., 2012).
Gene–Environment Interactions in Parkinson’s Disease
As comprehension of the role of genetics and environmental exposures in the development of PD increases, it has also become clear that independently these factors cannot account for all cases of sporadic PD. For example, it has been a topic of great interest and debate why a single heterozygous mutation in the recessive PINK1 and Parkin genes would lead to PD in some patients (Klein et al., 2007). It is possible that these heterozygous mutations increase susceptibility to environmental toxicants. Equally interesting is the observation that mutations in LRRK2 have also been detected in sporadic PD (Gilks et al., 2005). Additional familial PD genes have also been found among the risk loci identified for sporadic PD (Nalls et al., 2014). The view of gene–environment interactions that culminate in PD has increasingly been appreciated. However, the specifics of such interactions have not been well documented. Although some recent epidemiological studies have identified a number of specific interactions that might be relevant to PD (Cannon and Greenamyre, 2013), most results are derived from genetically modified experimental models to evaluate the sensitivity of neurons to toxicants as described below.
Rotenone has been shown to interact with or to increase PD risk due to association with multiple genes, including Parkin and VPS-35. When primary neuronal cultures from wild type or Parkin-knockout mice were treated with different rotenone concentrations; the Parkin-knockout cultures were much more sensitive to rotenone toxicity (Casarejos et al., 2006). Because Parkin also functions in the mitophagy pathway, the cumulative effects of Parkin defective cells with rotenone may relate to the reduced clearance of defective mitochondria. Rotenone exposure may therefore increase the likelihood of disease onset in individuals with Parkin mutations, or it may decrease the age of disease onset by potentiating the pathogenic process. Increased vulnerability of primary neuronal cultures to rotenone and VPS35 mutations have also been reported (Tsika et al., 2014), suggesting that increased PD susceptibility due to combinatory effects between rotenone and genetic modifications is not limited to just a single gene.
Paraquat (PQ) also has a number of genetic modifications linked to increased risk and PD susceptibility, including CHCHD2 and PINK1 (Gegg et al., 2009; Meng et al., 2017). Viability of PINK1-/- cells is reduced following treatment with PQ compared to control cells (Gegg et al., 2009). This enhanced vulnerability is likely due to the combination of reduced ATP production and increased intracellular oxidative stress. Increased mitochondrial dysfunction and susceptibility to apoptosis or oxidative stress over time has also been reported in brain cells with loss of PINK1 activity (Gautier et al., 2008; Wood-Kaczmar et al., 2008). CHCHD2 is also shown to increase the sensitivity of Drosophila to PQ-induced oxidative stress (Meng et al., 2017), suggesting that genetic mutations which reduce the ability of cells or organisms to scavenge ROS likely increase their susceptibility to PQ neurotoxicity.
Attention has also been given to the interactions between Mn and PD-linked gene products including α-synuclein, Parkin, PINK1, DJ-1, LRRK2, ATP13A2, and VPS35, as recently discussed in these reviews (Covy and Giasson, 2011; Roth, 2014, Peres et al., 2016). Regarding its interaction with α-synuclein, Mn can bind to this protein via three residues in the C-terminal domain: Asp-121, Asn-122, and Glu-123 (Uversky et al., 2001a), although biophysical studies showed that Mn exhibits a low affinity for α-synuclein (1 mM range) compared to other metals (Binolfi et al., 2006). X-ray fluorescence imaging also indicates an interaction between α-synuclein and Mn in rat primary midbrain neurons (Dućič et al., 2015). This study reported that intracellular content of Mn was higher in cells with α-synuclein overexpression, suggesting that α-synuclein acts as an intracellular storage of Mn (Dućič et al., 2015). Perhaps partly because of this property of α-synuclein, its effects on cell viability has been reported to be both neurotoxic and neuroprotective when combined with Mn. As extensively discussed (Peres et al., 2016), although Mn has been reported by several laboratories to enhance α-synuclein aggregation and neurotoxicity when combined with α-synuclein, some studies also showed that wild type α-synuclein is protective against Mn neurotoxicity in Caenorhabditis elegans (Bornhorst et al., 2014) and rat dopaminergic neuronal cells (Harischandra et al., 2015). For the later study, protection was only observed during the early stages of Mn exposure, suggesting that accumulation of sequestered Mn would eventually induce protein aggregation and neurotoxicity.
Most of the current evidence on ATP13A2 function comes from studies focusing on the interaction between this protein and α-synuclein. Given its role in lysosomal degradation it is not surprising that mutations in this gene can lead to diminished lysosomal-mediated clearance of autophagosomes and accumulation of aggregated α-synuclein (Gitler et al., 2009; Usenovic and Krainc, 2012). On the other hand, ATP13A2 has been shown to protect cells against Mn toxicity (Gitler et al., 2009), however, this protection is lost when mutations in this gene occur. A recent study showed that ATP13A2 mutations could increase the detrimental effects of Mn on motor coordination in humans exposed to the heavy metal (Rentschler et al., 2012).
Mn-exposed animals displayed mitochondrial impairment and loss of tyrosine hydroxylase, a phenotypic marker for dopaminergic neurons (Sriram et al., 2010). The expression of Parkin and DJ-1 were also affected in these cells. Relevant to Mn neurotoxicity, Parkin has been reported to regulate Mn transport (Roth et al., 2010). The DMT1 transporter represents the primary route for Mn uptake in the brain and four isoforms have been identified. Parkin is involved in the ubiquitination and subsequent proteosomal degradation of one of these isoforms (Roth et al., 2010). Therefore, a loss of Parkin function could facilitate Mn transport, promoting Mn accumulation in the basal ganglia and accelerating its toxicity. DJ-1 and Mn have further been implicated in mitochondrial dysfunction. Lee et al. (2012) showed that Mn could reduce both protein and RNA levels of DJ-1 inducing an effect that resembled the neurotoxic activity of mutant forms of DJ-1. Furthermore, the ability of both factors to individually promote oxidative stress, increase opening of the mitochondrial permeability transition pore, decrease membrane potential and impair ATP represent possible exacerbating effects of Mn and DJ-1 on mitochondrial activity. In summary, Mn has been demonstrated to interact with many PD-linked gene products and dysregulation of Mn homeostasis or exposure to Mn may contribute to PD pathogenesis.
Conclusion and Future Directions
First described in 1817, PD is now recognized as a major debilitating neurological disorder. To develop effective disease modifying therapies or a cure, it is critical to understand what causes PD and its associated pathogenic mechanism(s). Until the first PD linked mutation was discovered in 1997, the search for the cause of PD was primarily focused on environmental factors. This approach seems logical given the absence of genetic evidence at the time and the observations that PD/parkinsonism could be caused by post-encephalitic infection and exposure to pesticides, Mn, MPTP and diets as discussed in this review. However, after the first discovery PD-linked mutation, PD research has progressed at a rapid pace with exciting directions. In the last few decades, we have learned tremendously about this disease. However, the more we know, the more we realize how complex this disorder truly is. To date, no other neurodegenerative disorders exhibit so many genetic mutations (and yet they represent only a small fraction of all cases), and with such a strong environmental component as PD. The etiology of PD is complex and multi-factorial.
What have we learned so far about PD etiology and pathogenesis? We have identified several autosomal dominant and recessive genes linked to familial PD, as well as those that increase the risk of PD. The successive and rapid discoveries of these genes have led many PD researchers to shift their focus to genetic studies. However, we have now reached a stage where we realize that even with additional genes to be discovered in the future, genetics alone cannot account for all PD cases. Data from epidemiological and genome-wide association studies support the role of environmental factors and gene–environment interactions in sporadic PD. Experimental models also reveal that all three factors (genetic, environmental and gene–environment interaction) have an impact on the nigrostriatal pathway. In combination, these studies have provided significant insights into mechanisms of neuronal dysfunction and degeneration in PD. These non-mutually exclusive mechanisms include mitochondrial dysfunction, oxidative stress, neuroinflammation, toxic misfolded proteins, and insufficient protein degradation (as illustrated in Figure 1).
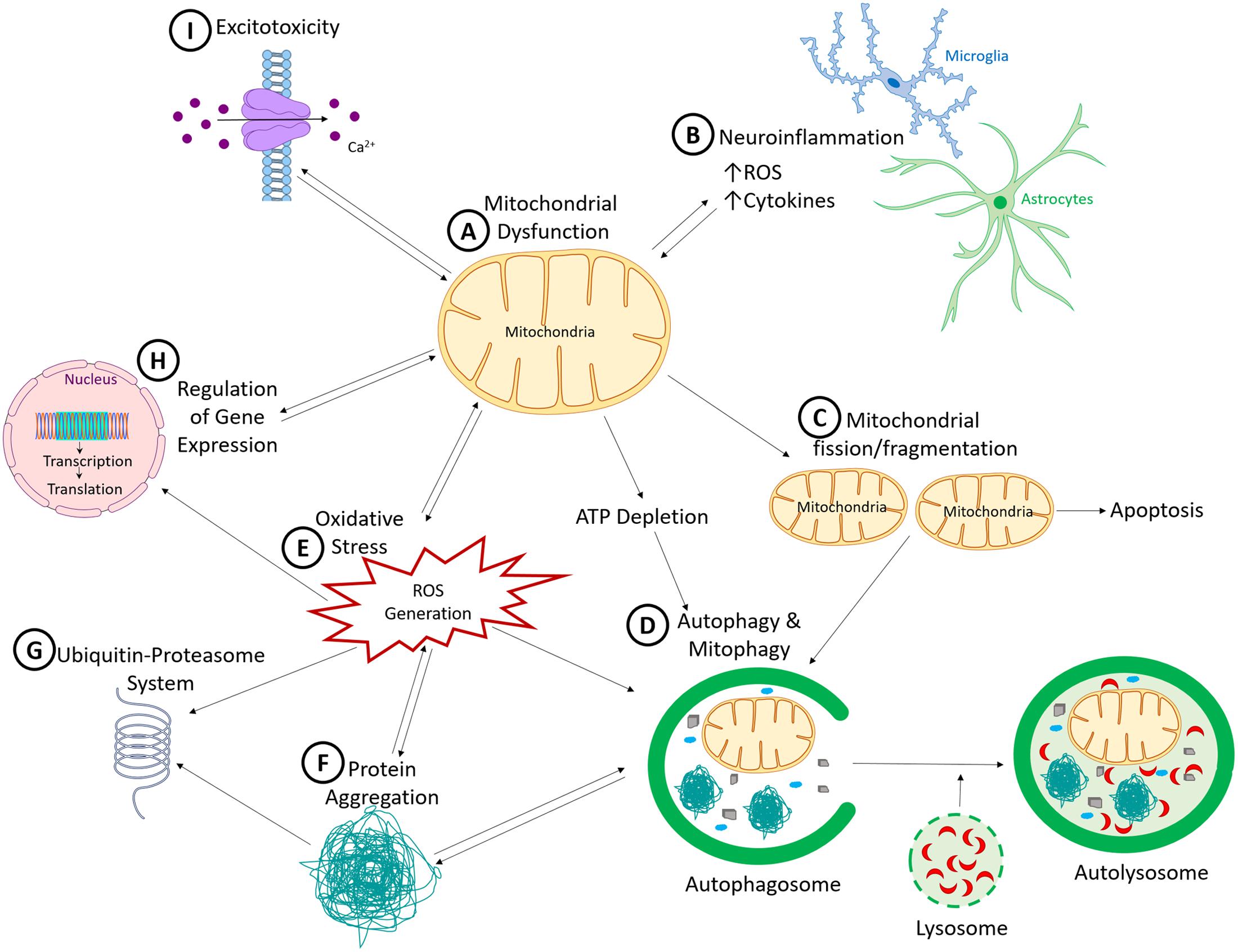
FIGURE 1. Common pathogenic mechanisms in Parkinson’s disease. (A) Mitochondrial dysfunction, a common pathogenic mechanism induced by many of the environmental toxicants and genetic mutations linked to PD, results in a cascade of interconnected cellular dysfunction. (B) Neuroinflammation, facilitated by microglia and to a lesser extent astrocytes, which release neurotoxic factors such as cytokines, interleukins and reactive oxygen species, leading to non-cell autonomous neurotoxicity. (C) Increased mitochondrial fission and fragmentation, which can initiate apoptotic cell death by inducing cytochrome C release. (D) Because autophagy and ubiquitin-proteasome system (UPS) are ATP-dependent processes, ATP reduction would reduces autophagic clearance of damaged proteins and organelles. This process is also sensitive to reactive oxygen species (ROS). (E) Generation of ROS, which has the capacity to promote the formation of toxic oligomers and protein aggregates (F), impair ubiquitin proteasomal system function (G), and induce DNA damage (both nuclear and mitochondrial DNA). (H) Nuclear DNA encodes numerous mitochondrial proteins. DNA damage results in altered nuclear function, genomic instabilities, and mitochondrial dysfunction. (I) Dysregulated cellular Ca2+ is influenced by mitochondrial dysfunction, because mitochondria help to regulate intracellular Ca2+ levels. When damaged, mitochondria release more Ca2+ into the cytosol, thereby increasing cellular excitotoxicity. Over-activation of the excitatory receptors also results in excitotoxicity due to Ca2+ influx which then produces downstream defects such as Ca2+-induced mitochondrial depolarization. As illustrated, all these mechanisms cross-talk and culminate in neurodegenerative processes in PD.
In this review we focus our attention on the role of mitochondrial dysfunction. By examining all the major genetic mutations and environmental toxicants linked to PD, it is evident that mitochondria are a common pathogenic target (Figure 2). Given the extensive and critical role of mitochondria, ranging from the classic “powerhouses” of the cell, to maintaining calcium homeostasis, controlling synaptic activity, initiating apoptosis and the spread of α-synuclein, it is not surprising that impaired mitochondrial function would negatively impact neuronal function and viability. The proposal that mitochondria represent a common and convergent pathway for PD raises some questions that might be worthwhile for future research:
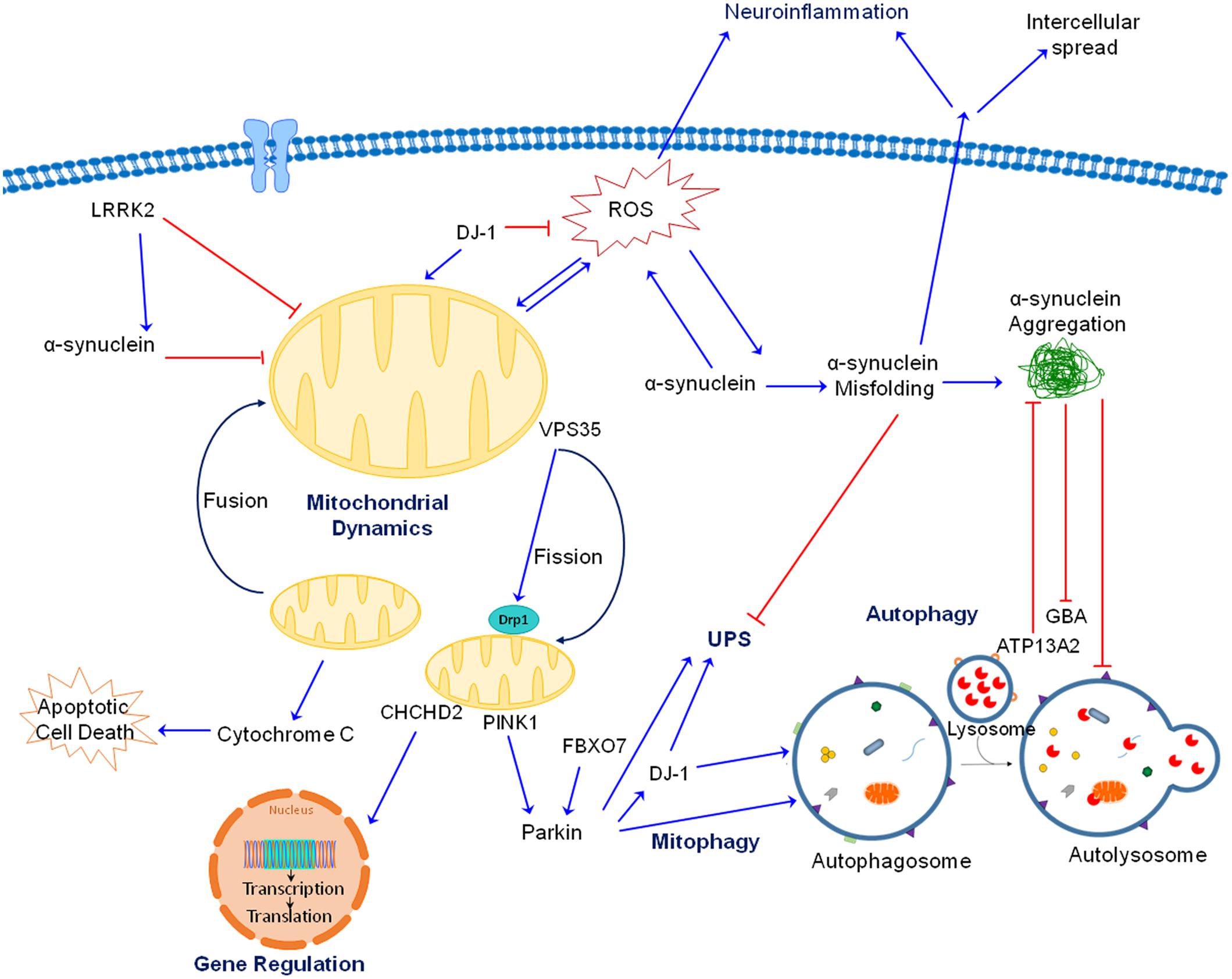
FIGURE 2. Interactions between gene products linked to PD. As discussed in the text, both genetic mutations and neurotoxicants linked to PD impair mitochondrial function, whether directly or indirectly by interacting with each other. For the autosomal recessive gene products, PINK1 is accumulated at outer mitochondrial membrane (OMM) of damaged mitochondria, where it recruits Parkin which ubiquitinates proteins and target them for degradation. Together, PINK1 and Parkin play an important role in maintaining mitochondrial quality through mitophagy. As an antioxidant protein, DJ-1 is translocated to dysfunctional mitochondria when ROS is generated. DJ-1 also aids PINK1 and Parkin in removing damaged mitochondria. Additionally, FBX07 is involved in the recruitment of Parkin to damaged mitochondria and subsequent mitophagy. ATP13A2 is localized in lysosomes and is important for protein degradation. For the autosomal dominant gene products, accumulation of misfolded and aggregated α-synuclein can be induced by a variety of neurotoxicants and genetic mutations. Toxic α-synuclein can impair mitochondrial function, generate ROS and block autophagy/lysosomal function. Mutation in LRRK2 results in gain of toxic kinase function which impairs mitochondrial and autophagic function as well as promotes α-synuclein aggregation. VPS35 mutations increase binding to Drp1 resulting in mitochondrial fragmentation. Of note, mitochondrial fragmentation is a common observation induced by genes and toxicants linked to PD. CHCHD2 is rather unique because it is both a mitochondrial protein and transcription factor. Its mutation impairs complex IV function and expression of a complex IV subunit. Mutations in GBA, a lysosomal protein, impairs protein degradation and mitochondrial function. Although not shown for simplicity, neurotoxicants such as Mn also interact with these proteins (such as α-synuclein) to induce neurotoxicity. Together, there are extensive interactions between these proteins and neurotoxicants leading to neurodegeneration.
First, would therapeutically targeting mitochondria alone be sufficiently efficacious? Given that other pathogenic mechanisms (oxidative stress, neuroinflammation, protein aggregation, and kinase activity) also occur in PD and that the mitochondrion is not always the first neurotoxic target, perhaps a “cocktail” therapy, a strategy used for HIV and cancer treatment, is necessary. Second, regardless of whether mitochondrial impairment is induced by environmental toxicants or genetic mutations, multiple cell types in various brain regions would be affected. Yet, dopaminergic neurons are the most vulnerable cell type. Understanding what dictates this selective cell death not only will shed light on disease pathogenesis but also therapeutic strategy. It has been proposed that this process involves a combination of selective expression of L-type Ca2+ channel leading to elevated cytosolic Ca2+, oxidized cytosolic dopamine, α-synuclein and the inherent long, highly branched axons of nigral dopaminergic neurons (Obeso et al., 2017). Although attractive, this hypothesis remains to be validated. Third, in addition to mitochondrial dysfunction, another theme gaining significant momentum is the autophagic impairment pathway. Although limited, evidence exists to support that these mechanisms, mitochondria and autophagy, are not mutually exclusive and they can bi-directionally regulate each other. It is not clear, however, which one would play a more prominent role. Fourth, emerging evidence seems to support the idea of blocking mitochondrial fission as a therapeutic strategy for PD and other neurodegenerative disorders. However, because a balance of mitochondrial fission/fusion is critical to normal physiological process, how to target this pathway to achieve optimal therapeutic with minimal side effects will require more efforts. In summary, mitochondrial dysfunction is a pathogenic mechanism shared by genetic mutations and environmental toxicants linked to PD. However, substantial further research is required before a safe and effective disease-modifying therapy may be translated into the clinical setting.
Author Contributions
MH, JP, CS, and KT wrote the manuscript. KT edited the manuscript.
Funding
This work was supported in part by National Institute of Environmental Health Services (R01-ES022274) and Stempel College of Environmental and Occupational Health, Florida International University.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We apologize to those authors whose work we have not cited due to space constraints.
References
Akundi, R. S., Huang, Z., Eason, J., Pandya, J. D., Zhi, L., Cass, W. A., et al. (2011). Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLOS ONE 6:e16038. doi: 10.1371/journal.pone.0016038
Alaimo, A., Gorojod, R. M., Beauquis, J., Muñoz, M. J., Saravia, F., and Kotler, M. L. (2014). Deregulation of mitochondria-shaping proteins Opa-1 and Drp-1 in manganese-induced apoptosis. PLOS ONE 9:e91848. doi: 10.1371/journal.pone.0091848
Anadón, A., Martinez-Larrañaga, M. R., Fernandez-Cruz, M. L., Diaz, M. J., Fernandez, M. C., and Martinez, M. A. (1996). Toxicokinetics of deltamethrin and its 4’-HO-metabolite in the rat. Toxicol. Appl. Pharmacol. 141, 8–16. doi: 10.1006/taap.1996.0254
Appel-Cresswell, S., Vilarino-Guell, C., Encarnacion, M., Sherman, H., Yu, I., Shah, B., et al. (2013). Alpha-synuclein p.H50q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 28, 811–813. doi: 10.1002/mds.25421
Aras, S., Bai, M., Lee, I., Springett, R., Huttemann, M., and Grossman, L. (2015). MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 20, 43–51. doi: 10.1016/j.mito.2014.10.003
Arkadir, D., Dinur, T., Mullin, S., Mehta, A., Baris, H. N., Alcalay, R. N., et al. (2016). Trio approach reveals higher risk of PD in carriers of severe vs. mild GBA mutations. Blood Cells Mol. Dis. doi: 10.1016/j.bcmd.2016.11.007 [Epub ahead of print].
Arranz, A. M., Delbroek, L., Van Kolen, K., Guimarães, M. R., Mandemakers, W., Daneels, G., et al. (2015). LRRK2 functions in synaptic vesicle endocytosis through a kinase-dependent mechanism. J. Cell Sci. 128, 541–552. doi: 10.1242/jcs.158196
Aschner, J. L., and Aschner, M. (2005). Nutritional aspects of manganese homeostasis. Mol. Aspects Med. 26, 353–362. doi: 10.1016/j.mam.2005.07.003
Aschner, M., Erikson, K. M., Herrero Hernández, E., Hernández, E. H., and Tjalkens, R. (2009). Manganese and its role in Parkinson’s disease: from transport to neuropathology. Neuromol. Med. 11, 252–266. doi: 10.1007/s12017-009-8083-0
Bae, E. J., Yang, N. Y., Song, M., Lee, C. S., Lee, J. S., Jung, B. C., et al. (2014). Glucocerebrosidase depletion enhances cell-to-cell transmission of alpha-synuclein. Nat. Commun. 5:4755. doi: 10.1038/ncomms5755
Barsoum, M. J., Yuan, H., Gerencser, A. A., Liot, G., Kushnareva, Y., Gräber, S., et al. (2006). Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 25, 3900–3911. doi: 10.1038/sj.emboj.7601253
Baughman, J. M., Nilsson, R., Gohil, V. M., Arlow, D. H., Gauhar, Z., and Mootha, V. K. (2009). A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLOS Genet. 5:e1000590. doi: 10.1371/journal.pgen.1000590
Baumuratov, A. S., Antony, P. M. A., Ostaszewski, M., He, F., Salamanca, L., Antunes, L., et al. (2016). Enteric neurons from Parkinson’s disease patients display ex vivo aberrations in mitochondrial structure. Sci. Rep. 6:33117. doi: 10.1038/srep33117
Beccano-Kelly, D. A., Volta, M., Munsie, L. N., Paschall, S. A., Tatarnikov, I., Co, K., et al. (2015). LRRK2 overexpression alters glutamatergic presynaptic plasticity, striatal dopamine tone, postsynaptic signal transduction, motor activity and memory. Hum. Mol. Genet. 24, 1336–1349. doi: 10.1093/hmg/ddu543
Beck, G., Sugiura, Y., Shinzawa, K., Kato, S., Setou, M., Tsujimoto, Y., et al. (2011). Neuroaxonal dystrophy in calcium-independent phospholipase A2β deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci. 31, 11411–11420. doi: 10.1523/jneurosci.0345-11.2011
Bendor, J., Logan, T., and Edwards, R. H. (2013). The function of α-synuclein. Neuron 79, 1044–1066. doi: 10.1016/j.neuron.2013.09.004
Betarbet, R., Sherer, T., MacKenzie, G., Garcia-Osuna, M., Panov, A., and Greenamyre, J. (2000). Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 3, 1301–1306. doi: 10.1038/81834
Bido, S., Soria, F. N., Fan, R. Z., Bezard, E., and Tieu, K. (2017). Mitochondrial division inhibitor-1 is neuroprotective in the A53T-α-synuclein rat model of Parkinson’s disease. Sci. Rep. 7:7495. doi: 10.1038/s41598-017-07181-0
Binolfi, A., Rasia, R. M., Bertoncini, C. W., Ceolin, M., Zweckstetter, M., Griesinger, C., et al. (2006). Interaction of alpha-synuclein with divalent metal ions reveals key differences: a link between structure, binding specificity and fibrillation enhancement. J. Am. Chem. Soc. 128, 9893–9901. doi: 10.1021/ja0618649
Bonifati, V., Rizzu, P., van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E., et al. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. doi: 10.1126/science.1077209
Borgstahl, G. E., Parge, H. E., Hickey, M. J., Beyer, W. F., Hallewell, R. A., and Tainer, J. A. (1992). The structure of human mitochondrial manganese superoxide dismutase reveals a novel tetrameric interface of two 4-helix bundles. Cell 71, 107–118. doi: 10.1016/0092-8674(92)90270-M
Bornhorst, J., Chakraborty, S., Meyer, S., Lohren, H., Brinkhaus, S. G., Knight, A. L., et al. (2014). The effects of pdr1, djr1.1 and pink1 loss in manganese-induced toxicity and the role of α-synuclein in C. elegans. Metallomics 6, 476–490. doi: 10.1039/c3mt00325f
Braidy, N., Gai, W. P., Xu, Y. H., Sachdev, P., Guillemin, G. J., Jiang, X. M., et al. (2014). Alpha-synuclein transmission and mitochondrial toxicity in primary human foetal enteric neurons in vitro. Neurotox. Res. 25, 170–182. doi: 10.1007/s12640-013-9420-5
Braschi, E., Goyon, V., Zunino, R., Mohanty, A., Xu, L., and McBride, H. M. (2010). Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr. Biol. 20, 1310–1315. doi: 10.1016/j.cub.2010.05.066
Breckenridge, C. B., Sturgess, N. C., Butt, M., Wolf, J. C., Zadory, D., Beck, M., et al. (2013). Pharmacokinetic, neurochemical, stereological and neuropathological studies on the potential effects of paraquat in the substantia nigra pars compacta and striatum of male C57BL/6J mice. Neurotoxicology 37, 1–14. doi: 10.1016/j.neuro.2013.03.005
Brundin, P., and Melki, R. (2017). Prying into the prion hypothesis for Parkinson’s disease. J. Neurosci. 37, 9808–9818. doi: 10.1523/JNEUROSCI.1788-16.2017
Buhlman, L., Damiano, M., Bertolin, G., Ferrando-Miguel, R., Lombes, A., Brice, A., et al. (2014). Functional interplay between Parkin and Drp1 in mitochondrial fission and clearance. Biochim. Biophys. Acta 1843, 2012–2026. doi: 10.1016/j.bbamcr.2014.05.012
Burchell, V. S., Nelson, D. E., Sanchez-Martinez, A., Delgado-Camprubi, M., Ivatt, R. M., Pogson, J. H., et al. (2013). The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 16, 1257–1265. doi: 10.1038/nn.3489
Burré, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Südhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Calne, D. B., Chu, N. S., Huang, C. C., Lu, C. S., and Olanow, W. (1994). Manganism and idiopathic parkinsonism: similarities and differences. Neurology 44, 1583–1586. doi: 10.1212/WNL.44.9.1583
Canet-Aviles, R. M., Wilson, M. A., Miller, D. W., Ahmad, R., McLendon, C., Bandyopadhyay, S., et al. (2004). The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. U.S.A. 101, 9103–9108. doi: 10.1073/pnas.0402959101
Cannon, J. R., and Greenamyre, J. T. (2013). Gene-environment interactions in Parkinson’s disease: specific evidence in humans and mammalian models. Neurobiol. Dis. 57, 38–46. doi: 10.1016/j.nbd.2012.06.025
Cannon, J. R., Tapias, V. M., Na, H. M., Honick, A. S., Drolet, R. E., and Greenamyre, J. T. (2009). A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 34, 279–290. doi: 10.1016/j.nbd.2009.01.016
Caparros-Lefebvre, D., and Elbaz, A. (1999). Possible relation of atypical parkinsonism in the French West Indies with consumption of tropical plants: a case-control study. Caribbean Parkinsonism study group. Lancet 354, 281–286. doi: 10.1016/S0140-6736(98)10166-6
Caparros-Lefebvre, D., Sergeant, N., Lees, A., Camuzat, A., Daniel, S., Lannuzel, A., et al. (2002). Guadeloupean parkinsonism: a cluster of progressive supranuclear palsy-like tauopathy. Brain 125(Pt 4), 801–811. doi: 10.1093/brain/awf086
Casarejos, M. J., Menéndez, J., Solano, R. M., Rodríguez-Navarro, J. A., García de Yébenes, J., and Mena, M. A. (2006). Susceptibility to rotenone is increased in neurons from Parkin null mice and is reduced by minocycline. J. Neurochem. 97, 934–946. doi: 10.1111/j.1471-4159.2006.03777.x
Casida, J. E., Gammon, D. W., Glickman, A. H., and Lawrence, L. J. (1983). Mechanisms of selective action of pyrethroid insecticides. Annu. Rev. Pharmacol. Toxicol. 23, 413–438. doi: 10.1146/annurev.pa.23.040183.002213
Castello, P. R., Drechsel, D. A., and Patel, M. (2007). Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. J. Biol. Chem. 282, 14186–14193. doi: 10.1074/jbc.M700827200
Cervantes-Cervantes, M. P., Calderón-Salinas, J. V., Albores, A., and Muñoz-Sánchez, J. L. (2005). Copper increases the damage to DNA and proteins caused by reactive oxygen species. Biol. Trace Elem. Res. 103, 229–248. doi: 10.1385/bter
Champy, P., Höglinger, G. U., Féger, J., Gleye, C., Hocquemiller, R., Laurens, A., et al. (2004). Annonacin, a lipophilic inhibitor of mitochondrial complex I, induces nigral and striatal neurodegeneration in rats: possible relevance for atypical parkinsonism in Guadeloupe. J. Neurochem. 88, 63–69. doi: 10.1046/j.1471-4159.2003.02138.x
Chan, N. C., Salazar, A. M., Pham, A. H., Sweredoski, M. J., Kolawa, N. J., Graham, R. L., et al. (2011). Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737. doi: 10.1093/hmg/ddr048
Chen, L., Xie, Z., Turkson, S., and Zhuang, X. (2015). A53T human alpha-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J. Neurosci. 35, 890–905. doi: 10.1523/jneurosci.0089-14.2015
Chen, P., Miah, M. R., and Aschner, M. (2016). Metals and Neurodegeneration. F1000Res 5:F1000FacultyRev–366. doi: 10.12688/f1000research.7431.1
Chen, S. Y., Zhang, Z. W., He, F. S., Yao, P. P., Wu, Y. Q., Sun, J. X., et al. (1991). An epidemiological study on occupational acute pyrethroid poisoning in cotton farmers. Br. J. Ind. Med. 48, 77–81. doi: 10.1136/oem.48.2.77
Cherra, S. J., Steer, E., Gusdon, A. M., Kiselyov, K., and Chu, C. T. (2013). Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am. J. Pathol. 182, 474–484. doi: 10.1016/j.ajpath.2012.10.027
Choi, W.-S., Palmiter, R. D., and Xia, Z. (2011). Loss of mitochondrial complex I activity potentiates dopamine neuron death induced by microtubule dysfunction in a Parkinson’s disease model. J. Cell Biol. 1992, 873–882. doi: 10.1083/jcb.201009132
Chun, H. S., Gibson, G. E., DeGiorgio, L. A., Zhang, H., Kidd, V. J., and Son, J. H. (2001). Dopaminergic cell death induced by MPP(+), oxidant and specific neurotoxicants shares the common molecular mechanism. J. Neurochem. 76, 1010–1021. doi: 10.1046/j.1471-4159.2001.00096.x
Cilia, R., Tunesi, S., Marotta, G., Cereda, E., Siri, C., Tesei, S., et al. (2016). Survival and dementia in GBA-associated Parkinson’s disease: the mutation matters. Ann. Neurol. 80, 662–673. doi: 10.1002/ana.24777
Clark, I. E., Dodson, M. W., Jiang, C., Cao, J. H., Huh, J. R., Seol, J. H., et al. (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with Parkin. Nature 441, 1162–1166. doi: 10.1038/nature04779
Claus Henn, B., Kim, J., Wessling-Resnick, M., Téllez-Rojo, M. M., Jayawardene, I., Ettinger, A. S., et al. (2011). Associations of iron metabolism genes with blood manganese levels: a population-based study with validation data from animal models. Environ. Health 10:97. doi: 10.1186/1476-069x-10-97
Cleeter, M. W., Chau, K. Y., Gluck, C., Mehta, A., Hughes, D. A., Duchen, M., et al. (2013). Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 62, 1–7. doi: 10.1016/j.neuint.2012.10.010
Cochemé, H. M., and Murphy, M. P. (2008). Complex I is the major site of mitochondrial superoxide production by paraquat. J. Biol. Chem. 283, 1786–1798. doi: 10.1074/jbc.M708597200
Cookson, M. R. (2015). LRRK2 pathways leading to neurodegeneration. Curr. Neurol. Neurosci. Rep. 15:42. doi: 10.1007/s11910-015-0564-y
Cooper, O., Seo, H., Andrabi, S., Guardia-Laguarta, C., Graziotto, J., Sundberg, M., et al. (2012). Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 4, 141ra190. doi: 10.1126/scitranslmed.3003985
Corrigan, F. M., French, M., and Murray, L. (1996). Organochlorine compounds in human brain. Hum. Exp. Toxicol. 15, 262–264. doi: 10.1177/096032719601500314
Costa, L. G. (2015). The neurotoxicity of organochlorine and pyrethroid pesticides. Handb. Clin. Neurol. 131, 135–148. doi: 10.1016/B978-0-444-62627-1.00009-3
Costello, S., Cockburn, M., Bronstein, J., Zhang, X., and Ritz, B. (2009). Parkinson’s disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am. J. Epidemiol. 169, 919–926. doi: 10.1093/aje/kwp006
Couper, J. (1837). On the effects of black oxide of manganese which inhaled into the lungs. Br. Ann. Med. Pharm. Vital Stat. Gen. Sci. 1, 41–42. doi: 10.1002/jcp.22692
Covy, J. P., and Giasson, B. I. (2011). α-Synuclein, leucine-rich repeat kinase-2, and manganese in the pathogenesis of Parkinson disease. Neurotoxicology 32, 622–629. doi: 10.1016/j.neuro.2011.01.003
Cowie, A. M., Sarty, K. I., Mercer, A., Koh, J., Kidd, K. A., and Martyniuk, C. J. (2017). The pesticide dieldrin disrupts proteins related to oxidative respiration and mitochondrial stress in the central nervous system. Data Brief 11, 628–633. doi: 10.1016/j.dib.2017.03.008
Criscuolo, C., Volpe, G., De Rosa, A., Varrone, A., Marongiu, R., Mancini, P., et al. (2006). PINK1 homozygous W437X mutation in a patient with apparent dominant transmission of parkinsonism. Mov. Disord. 21, 1265–1267. doi: 10.1002/mds.20933
Cui, M., Tang, X., Christian, W. V., Yoon, Y., and Tieu, K. (2010). Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. J. Biol. Chem. 285, 11740–11752. doi: 10.1074/jbc.M109.066662
Dächsel, J. C., and Farrer, M. J. (2010). LRRK2 and Parkinson disease. Arch. Neurol. 67, 542–547. doi: 10.1001/archneurol.2010.79
Dagda, R. K., Cherra, S. J., Kulich, S. M., Tandon, A., Park, D., and Chu, C. T. (2009). Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 284, 13843–13855. doi: 10.1074/jbc.M808515200
Dagda, R. K., Pien, I., Wang, R., Zhu, J., Wang, K. Z., Callio, J., et al. (2014). Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J. Neurochem. 128, 864–877. doi: 10.1111/jnc.12494
Dauer, W., and Przedborski, S. (2003). Parkinson’s disease: mechanisms and models. Neuron 39, 889–909. doi: 10.1016/S0896-6273(03)00568-3
Day, B. J., Patel, M., Calavetta, L., Chang, L. Y., and Stamler, J. S. (1999). A mechanism of paraquat toxicity involving nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 96, 12760–12765. doi: 10.1073/pnas.96.22.12760
de la Mata, M., Cotan, D., Oropesa-Avila, M., Garrido-Maraver, J., Cordero, M. D., Villanueva Paz, M., et al. (2015). Pharmacological chaperones and coenzyme Q10 treatment improves mutant beta-glucocerebrosidase activity and mitochondrial function in neuronopathic forms of gaucher disease. Sci. Rep. 5:10903. doi: 10.1038/srep10903
Degli Esposti, M. (1998). Inhibitors of NADH-ubiquinone reductase: an overview. Biochim. Biophys. Acta 1364, 222–235. doi: 10.1016/S0005-2728(98)00029-2
Dehay, B., Ramirez, A., Martinez-Vicente, M., Perier, C., Canron, M. H., Doudnikoff, E., et al. (2012). Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 109, 9611–9616. doi: 10.1073/pnas.1112368109
Deng, H., Dodson, M. W., Huang, H., and Guo, M. (2008). The Parkinson’s disease genes pink1 and Parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 105, 14503–14508. doi: 10.1073/pnas.0803998105
Devi, L., Raghavendran, V., Prabhu, B., Avadhani, N., and Anandatheerthavarada, H. (2008). Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100. doi: 10.1074/jbc.M710012200
Di Fonzo, A., Chien, H. F., Socal, M., Giraudo, S., Tassorelli, C., Iliceto, G., et al. (2007). ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 68, 1557–1562. doi: 10.1212/01.wnl.0000260963.08711.08
Di Fonzo, A., Dekker, M. C., Montagna, P., Baruzzi, A., Yonova, E. H., Correia Guedes, L., et al. (2009). FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 72, 240–245. doi: 10.1212/01.wnl.0000338144.10967.2b
Di Maio, R., Barrett, P. J., Hoffman, E. K., Barrett, C. W., Zharikov, A., Borah, A., et al. (2016). a-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 8:342ra78. doi: 10.1126/scitranslmed.aaf3634
Donovan, D. M., Miner, L. L., Perry, M. P., Revay, R. S., Sharpe, L. G., Przedborski, S., et al. (1999). Cocaine reward and MPTP toxicity: alteration by regional variant dopamine transporter overexpression. Brain Res. Mol. Brain Res. 73, 37–49. doi: 10.1016/S0169-328X(99)00235-1
Dućič, T., Carboni, E., Lai, B., Chen, S., Michalke, B., Lázaro, D. F., et al. (2015). Alpha-synuclein regulates neuronal levels of manganese and calcium. ACS Chem. Neurosci. 6, 1769–1779. doi: 10.1021/acschemneuro.5b00093
Durcan, T. M., and Fon, E. A. (2015). The three ’P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 29, 989–999. doi: 10.1101/gad.262758.115
Ebrahimi-Fakhari, D., Cantuti-Castelvetri, I., Fan, Z., Rockenstein, E., Masliah, E., Hyman, B. T., et al. (2011). Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J. Neurosci. 31, 14508–14520. doi: 10.1523/jneurosci.1560-11.2011
Elwan, M. A., Richardson, J. R., Guillot, T. S., Caudle, W. M., and Miller, G. W. (2006). Pyrethroid pesticide-induced alterations in dopamine transporter function. Toxicol. Appl. Pharmacol. 211, 188–197. doi: 10.1016/j.taap.2005.06.003
Enquist, I. B., Lo Bianco, C., Ooka, A., Nilsson, E., Mansson, J. E., Ehinger, M., et al. (2007). Murine models of acute neuronopathic Gaucher disease. Proc. Natl. Acad. Sci. U.S.A. 104, 17483–17488. doi: 10.1073/pnas.0708086104
Escobar-Khondiker, M., Höllerhage, M., Muriel, M. P., Champy, P., Bach, A., Depienne, C., et al. (2007). Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. J. Neurosci. 27, 7827–7837. doi: 10.1523/jneurosci.1644-07.2007
Esteves, A. R., Fernandes, M. G., Santos, D., Januario, C., and Cardoso, S. (2015). The upshot of LRRK2 inhibition to Parkinson’s disease paradigm. Mol. Neurobiol. 52, 1804–1820. doi: 10.1007/s12035-014-8980-6
Exner, N., Lutz, A. K., Haass, C., and Winklhofer, K. F. (2012). Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. EMBO J. 31, 3038–3062. doi: 10.1038/emboj.2012.170
Farfel-Becker, T., Vitner, E. B., Pressey, S. N., Eilam, R., Cooper, J. D., and Futerman, A. H. (2011). Spatial and temporal correlation between neuron loss and neuroinflammation in a mouse model of neuronopathic Gaucher disease. Hum. Mol. Genet. 20, 1375–1386. doi: 10.1093/hmg/ddr019
Fernagut, P. O., Hutson, C. B., Fleming, S. M., Tetreaut, N. A., Salcedo, J., Masliah, E., et al. (2007). Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha-synuclein over-expression. Synapse 61, 991–1001. doi: 10.1002/syn.20456
Fitzmaurice, A. G., Rhodes, S. L., Cockburn, M., Ritz, B., and Bronstein, J. M. (2014). Aldehyde dehydrogenase variation enhances effect of pesticides associated with Parkinson disease. Neurology 82, 419–426. doi: 10.1212/wnl.0000000000000083
Fitzmaurice, A. G., Rhodes, S. L., Lulla, A., Murphy, N. P., Lam, H. A., O’Donnell, K. C., et al. (2013). Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 110, 636–641. doi: 10.1073/pnas.1220399110
Fleming, L., Mann, J. B., Bean, J., Briggle, T., and Sanchez-Ramos, J. R. (1994). Parkinson’s disease and brain levels of organochlorine pesticides. Ann. Neurol. 36, 100–103. doi: 10.1002/ana.410360119
Fukushima, T., Tan, X., Luo, Y., and Kanda, H. (2010). Relationship between blood levels of heavy metals and Parkinson’s disease in China. Neuroepidemiology 34, 18–24. doi: 10.1159/000255462
Funayama, M., Hasegawa, K., Kowa, H., Saito, M., Tsuji, S., and Obata, F. (2002). A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2–q13.1. Ann. Neurol. 51, 296–301. doi: 10.1002/ana.10113
Funayama, M., Ohe, K., Amo, T., Furuya, N., Yamaguchi, J., Saiki, S., et al. (2015). CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: a genome-wide linkage and sequencing study. Lancet Neurol. 14, 274–282. doi: 10.1016/s1474-4422(14)70266-2
Gadd, M. E., Broekemeier, K. M., Crouser, E. D., Kumar, J., Graff, G., and Pfeiffer, D. R. (2006). Mitochondrial iPLA2 activity modulates the release of cytochrome c from mitochondria and influences the permeability transition. J. Biol. Chem. 281, 6931–6939. doi: 10.1074/jbc.M510845200
Gainetdinov, R. R., Fumagalli, F., Jones, S. R., and Caron, M. G. (1997). Dopamine transporter is required for in vivo MPTP neurotoxicity: evidence from mice lacking the transporter. J. Neurochem. 69, 1322–1325. doi: 10.1046/j.1471-4159.1997.69031322.x
Gandhi, S., Wood-Kaczmar, A., Yao, Z., Plun-Favreau, H., Deas, E., Klupsch, K., et al. (2009). PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell. 33, 627–638. doi: 10.1016/j.molcel.2009.02.013
Gan-Or, Z., Amshalom, I., Kilarski, L. L., Bar-Shira, A., Gana-Weisz, M., Mirelman, A., et al. (2015). Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 84, 880–887. doi: 10.1212/wnl.0000000000001315
Garcia-Reitböck, P., Anichtchik, O., Bellucci, A., Iovino, M., Ballini, C., Fineberg, E., et al. (2010). SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain 133(Pt 7), 2032–2044. doi: 10.1093/brain/awq132
Gassner, B., Wüthrich, A., Scholtysik, G., and Solioz, M. (1997). The pyrethroids permethrin and cyhalothrin are potent inhibitors of the mitochondrial complex I. J. Pharmacol. Exp. Ther. 281, 855–860.
Gautier, C. A., Giaime, E., Caballero, E., Nunez, L., Song, Z., Chan, D., et al. (2012). Regulation of mitochondrial permeability transition pore by PINK1. Mol. Neurodegener. 7:22. doi: 10.1186/1750-1326-7-22
Gautier, C. A., Kitada, T., and Shen, J. (2008). Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 105, 11364–11369. doi: 10.1073/pnas.0802076105
Gavin, C. E., Gunter, K. K., and Gunter, T. E. (1990). Manganese and calcium efflux kinetics in brain mitochondria. Relevance to manganese toxicity. Biochem. J. 266, 329–334. doi: 10.1042/bj2660329
Gavin, C. E., Gunter, K. K., and Gunter, T. E. (1992). Mn2+ sequestration by mitochondria and inhibition of oxidative phosphorylation. Toxicol. Appl. Pharmacol. 115, 1–5. doi: 10.1016/0041-008X(92)90360-5
Gavin, C. E., Gunter, K. K., and Gunter, T. E. (1999). Manganese and calcium transport in mitochondria: implications for manganese toxicity. Neurotoxicology 20, 445–453.
Gegg, M. E., Burke, D., Heales, S. J., Cooper, J. M., Hardy, J., Wood, N. W., et al. (2012). Glucocerebrosidase deficiency in substantia nigra of Parkinson disease brains. Ann. Neurol. 72, 455–463. doi: 10.1002/ana.23614
Gegg, M. E., Cooper, J. M., Chau, K. Y., Rojo, M., Schapira, A. H., and Taanman, J. W. (2010). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/Parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870. doi: 10.1093/hmg/ddq419
Gegg, M. E., Cooper, J. M., Schapira, A. H., and Taanman, J. W. (2009). Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLOS ONE 4:e4756. doi: 10.1371/journal.pone.0004756
Geisler, S., Holmstrom, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131. doi: 10.1038/ncb2012
Giaime, E., Yamaguchi, H., Gautier, C. A., Kitada, T., and Shen, J. (2012). Loss of DJ-1 does not affect mitochondrial respiration but increases ROS production and mitochondrial permeability transition pore opening. PLOS ONE 7:e40501. doi: 10.1371/journal.pone.0040501
Gilks, W. P., Abou-Sleiman, P. M., Gandhi, S., Jain, S., Singleton, A., Lees, A. J., et al. (2005). A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 365, 415–416. doi: 10.1016/s0140-6736(05)17830-1
Gitler, A. D., Chesi, A., Geddie, M. L., Strathearn, K. E., Hamamichi, S., Hill, K. J., et al. (2009). Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 41, 308–315. doi: 10.1038/ng.300
Godena, V. K., Brookes-Hocking, N., Moller, A., Shaw, G., Oswald, M., Sancho, R. M., et al. (2014). Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat. Commun. 5:5245. doi: 10.1038/ncomms6245
Goldstein, S., Meyerstein, D., and Czapski, G. (1993). The Fenton reagents. Free Radic. Biol. Med. 15, 435–445. doi: 10.1016/0891-5849(93)90043-T
Gomez-Suaga, P., Luzon-Toro, B., Churamani, D., Zhang, L., Bloor-Young, D., Patel, S., et al. (2012). Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 21, 511–525. doi: 10.1093/hmg/ddr481
Grunewald, A., Arns, B., Seibler, P., Rakovic, A., Munchau, A., Ramirez, A., et al. (2012). ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol. Aging 33, 1843.e1–7. doi: 10.1016/j.neurobiolaging.2011.12.035
Guardia-Laguarta, C., Area-Gomez, E., Rüb, C., Liu, Y., Magrané, J., Becker, D., et al. (2014). α-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259. doi: 10.1523/jneurosci.2507-13.2014
Gui, Y.-X., Wang, X.-Y., Kang, W.-Y., Zhang, Y.-J., Zhang, Y., Zhou, Y., et al. (2012). Extracellular signal-regulated kinase is involved in alpha-synuclein-induced mitochondrial dynamic disorders by regulating dynamin-like protein 1. Neurobiol. Aging 33, 2841–2854. doi: 10.1016/j.neurobiolaging.2012.02.001
Guilarte, T. R., Burton, N. C., McGlothan, J. L., Verina, T., Zhou, Y., Alexander, M., et al. (2008). Impairment of nigrostriatal dopamine neurotransmission by manganese is mediated by pre-synaptic mechanism(s): implications to manganese-induced parkinsonism. J. Neurochem. 107, 1236–1247. doi: 10.1111/j.1471-4159.2008.05695.x
Guilarte, T. R., Chen, M. K., McGlothan, J. L., Verina, T., Wong, D. F., Zhou, Y., et al. (2006). Nigrostriatal dopamine system dysfunction and subtle motor deficits in manganese-exposed non-human primates. Exp. Neurol. 202, 381–390. doi: 10.1016/j.expneurol.2006.06.015
Guilarte, T. R., and Gonzales, K. K. (2015). Manganese-induced parkinsonism is not idiopathic Parkinson’s disease: environmental and genetic evidence. Toxicol. Sci. 146, 204–212. doi: 10.1093/toxsci/kfv099
Gunter, T. E., Gerstner, B., Gunter, K. K., Malecki, J., Gelein, R., Valentine, W. M., et al. (2013). Manganese transport via the transferrin mechanism. Neurotoxicology 34, 118–127. doi: 10.1016/j.neuro.2012.10.018
Gunter, T. E., Gerstner, B., Lester, T., Wojtovich, A. P., Malecki, J., Swarts, S. G., et al. (2010). An analysis of the effects of Mn2+ on oxidative phosphorylation in liver, brain, and heart mitochondria using state 3 oxidation rate assays. Toxicol. Appl. Pharmacol. 249, 65–75. doi: 10.1016/j.taap.2010.08.018
Guo, J. L., and Lee, V. M. (2014). Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 20, 130–138. doi: 10.1038/nm.3457
Gusdon, A. M., Zhu, J., Van Houten, B., and Chu, C. T. (2012). ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol. Dis. 45, 962–972. doi: 10.1016/j.nbd.2011.12.015
Ha, C., Ryu, J., and Park, C. B. (2007). Metal ions differentially influence the aggregation and deposition of Alzheimer’s beta-amyloid on a solid template. Biochemistry 46, 6118–6125. doi: 10.1021/bi7000032
Halperin, A., Elstein, D., and Zimran, A. (2006). Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells Mol. Dis. 36, 426–428. doi: 10.1016/j.bcmd.2006.02.004
Hao, L. Y., Giasson, B. I., and Bonini, N. M. (2010). DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. U.S.A. 107, 9747–9752. doi: 10.1073/pnas.0911175107
Harischandra, D. S., Jin, H., Anantharam, V., Kanthasamy, A., and Kanthasamy, A. G. (2015). α-Synuclein protects against manganese neurotoxic insult during the early stages of exposure in a dopaminergic cell model of Parkinson’s disease. Toxicol. Sci. 143, 454–468. doi: 10.1093/toxsci/kfu247
Hatano, Y., Li, Y., Sato, K., Asakawa, S., Yamamura, Y., Tomiyama, H., et al. (2004). Novel PINK1 mutations in early-onset parkinsonism. Ann. Neurol. 56, 424–427. doi: 10.1002/ana.20251
Hawkes, C. H., Del Tredici, K., and Braak, H. (2007). Parkinson’s disease: a dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 33, 599–614. doi: 10.1111/j.1365-2990.2007.00874.x
Hayashi, T., Ishimori, C., Takahashi-Niki, K., Taira, T., Kim, Y. C., Maita, H., et al. (2009). DJ-1 binds to mitochondrial complex I and maintains its activity. Biochem. Biophys. Res. Commun. 390, 667–672. doi: 10.1016/j.bbrc.2009.10.025
Hedrich, K., Hagenah, J., Djarmati, A., Hiller, A., Lohnau, T., Lasek, K., et al. (2006). Clinical spectrum of homozygous and heterozygous PINK1 mutations in a large German family with Parkinson disease: role of a single hit? Arch. Neurol. 63, 833–838. doi: 10.1001/archneur.63.6.833
Heeman, B., Van den Haute, C., Aelvoet, S. A., Valsecchi, F., Rodenburg, R. J., Reumers, V., et al. (2011). Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J. Cell Sci. 124(Pt 7), 1115–1125. doi: 10.1242/jcs.078303
Heinz, G. H., Hill, E. F., and Contrera, J. F. (1980). Dopamine and norepinephrine depletion in ring doves fed DDE, dieldrin, and Aroclor 1254. Toxicol. Appl. Pharmacol. 53, 75–82. doi: 10.1016/0041-008X(80)90383-X
Hernán, M. A., Takkouche, B., Caamaño-Isorna, F., and Gestal-Otero, J. J. (2002). A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann. Neurol. 52, 276–284. doi: 10.1002/ana.10277
Hernandez, D. G., Reed, X., and Singleton, A. B. (2016). Genetics in Parkinson disease: mendelian versus non-Mendelian inheritance. J. Neurochem. 139, 59–74. doi: 10.1111/jnc.13593
Hiller, A., Hagenah, J. M., Djarmati, A., Hedrich, K., Reetz, K., Schneider-Gold, C., et al. (2007). Phenotypic spectrum of PINK1-associated parkinsonism in 15 mutation carriers from 1 family. Mov. Disord. 22, 145–147. doi: 10.1002/mds.21059
Hilton, D., Stephens, M., Kirk, L., Edwards, P., Potter, R., Zajicek, J., et al. (2014). Accumulation of alpha-synuclein in the bowel of patients in the pre-clinical phase of Parkinson’s disease. Acta Neuropathol. 127, 235–241. doi: 10.1007/s00401-013-1214-6
Hisata, J. (2002). Final Supplemental Environmental Impact Statement. Lake and Stream Rehabilitation: Rotenone Use and Health Risks. Washington, DC: Washington State Department of Fish and Wildlife.
Hossain, M. A., and Weiner, N. (1993). Dopaminergic functional supersensitivity: effects of chronic L-dopa and carbidopa treatment in an animal model of Parkinson’s disease. J. Pharmacol. Exp. Ther. 267, 1105–1111.
Huang, E., Ong, W. Y., and Connor, J. R. (2004). Distribution of divalent metal transporter-1 in the monkey basal ganglia. Neuroscience 128, 487–496. doi: 10.1016/j.neuroscience.2004.06.055
Irrcher, I., Aleyasin, H., Seifert, E. L., Hewitt, S. J., Chhabra, S., Phillips, M., et al. (2010). Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 19, 3734–3746. doi: 10.1093/hmg/ddq288
Jin, S. M., Lazarou, M., Wang, C., Kane, L. A., Narendra, D. P., and Youle, R. J. (2010). Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942. doi: 10.1083/jcb.201008084
Junn, E., Jang, W. H., Zhao, X., Jeong, B. S., and Mouradian, M. M. (2009). Mitochondrial localization of DJ-1 leads to enhanced neuroprotection. J. Neurosci. Res. 87, 123–129. doi: 10.1002/jnr.21831
Kamel, F. (2013). Paths from pesticides to Parkinson’s. Science 341, 722–723. doi: 10.1126/science.1243619
Kamp, F., Exner, N., Lutz, A. K., Wender, N., Hegermann, J., Brunner, B., et al. (2010). Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 29, 3571–3589. doi: 10.1038/emboj.2010.223
Kanthasamy, A. G., Kitazawa, M., Kanthasamy, A., and Anantharam, V. (2005). Dieldrin-induced neurotoxicity: relevance to Parkinson’s disease pathogenesis. Neurotoxicology 26, 701–719. doi: 10.1016/j.neuro.2004.07.010
Kanthasamy, A. G., Kitazawa, M., Yang, Y., Anantharam, V., and Kanthasamy, A. (2008). Environmental neurotoxin dieldrin induces apoptosis via caspase-3-dependent proteolytic activation of protein kinase C delta (PKCdelta): implications for neurodegeneration in Parkinson’s disease. Mol. Brain 1:12. doi: 10.1186/1756-6606-1-12
Kazlauskaite, A., Kondapalli, C., Gourlay, R., Campbell, D. G., Ritorto, M. S., Hofmann, K., et al. (2014). Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 460, 127–139. doi: 10.1042/bj20140334
Khan, N. L., Horta, W., Eunson, L., Graham, E., Johnson, J. O., Chang, S., et al. (2005). Parkin disease in a Brazilian kindred: manifesting heterozygotes and clinical follow-up over 10 years. Mov. Disord. 20, 479–484. doi: 10.1002/mds.20335
Kilarski, L. L., Pearson, J. P., Newsway, V., Majounie, E., Knipe, M. D., Misbahuddin, A., et al. (2012). Systematic review and UK-based study of PARK2 (Parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson’s disease. Mov. Disord. 27, 1522–1529. doi: 10.1002/mds.25132
Kim, J., Buckett, P. D., and Wessling-Resnick, M. (2013). Absorption of manganese and iron in a mouse model of hemochromatosis. PLOS ONE 8:e64944. doi: 10.1371/journal.pone.0064944
Kim, R. H., Smith, P. D., Aleyasin, H., Hayley, S., Mount, M. P., Pownall, S., et al. (2005). Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 102, 5215–5220. doi: 10.1073/pnas.0501282102
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the Parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Kitazawa, M., Anantharam, V., and Kanthasamy, A. G. (2001). Dieldrin-induced oxidative stress and neurochemical changes contribute to apoptopic cell death in dopaminergic cells. Free Radic. Biol. Med. 31, 1473–1485. doi: 10.1016/S0891-5849(01)00726-2
Kitazawa, M., Anantharam, V., and Kanthasamy, A. G. (2003). Dieldrin induces apoptosis by promoting caspase-3-dependent proteolytic cleavage of protein kinase Cdelta in dopaminergic cells: relevance to oxidative stress and dopaminergic degeneration. Neuroscience 119, 945–964. doi: 10.1016/S0306-4522(03)00226-4
Klein, C., and Lohmann-Hedrich, K. (2007). Impact of recent genetic findings in Parkinson’s disease. Curr. Opin. Neurol. 20, 453–464. doi: 10.1097/WCO.0b013e3281e6692b
Klein, C., Lohmann-Hedrich, K., Rogaeva, E., Schlossmacher, M. G., and Lang, A. E. (2007). Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 6, 652–662. doi: 10.1016/s1474-4422(07)70174-6
Klein, C., and Westenberger, A. (2012). Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a008888. doi: 10.1101/cshperspect.a008888
Kong, S. M., Chan, B. K., Park, J. S., Hill, K. J., Aitken, J. B., Cottle, L., et al. (2014). Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alpha-Synuclein externalization via exosomes. Hum. Mol. Genet. 23, 2816–2833. doi: 10.1093/hmg/ddu099
Koschmidder, E., Weissbach, A., Bruggemann, N., Kasten, M., Klein, C., and Lohmann, K. (2016). A nonsense mutation in CHCHD2 in a patient with Parkinson disease. Neurology 86, 577–579. doi: 10.1212/WNL.0000000000002361
Krebiehl, G., Ruckerbauer, S., Burbulla, L. F., Kieper, N., Maurer, B., Waak, J., et al. (2010). Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLOS ONE 5:e9367. doi: 10.1371/journal.pone.0009367
Krüger, R., Kuhn, W., Müller, T., Woitalla, D., Graeber, M., Kösel, S., et al. (1998). Ala30pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108. doi: 10.1038/ng0298-106
Kwakye, G. F., Paoliello, M. M., Mukhopadhyay, S., Bowman, A. B., and Aschner, M. (2015). Manganese-induced parkinsonism and Parkinson’s disease: shared and distinguishable features. Int. J. Environ. Res. Public Health 12, 7519–7540. doi: 10.3390/ijerph120707519
Langston, J., Ballard, P., Tetrad, J., and Irwin, I. (1983). Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science 25, 979–980. doi: 10.1126/science.6823561
Lannuzel, A., Höglinger, G. U., Champy, P., Michel, P. P., Hirsch, E. C., and Ruberg, M. (2006). Is atypical parkinsonism in the Caribbean caused by the consumption of Annonacae? J. Neural Transm. Suppl. 70, 153–157. doi: 10.1007/978-3-211-45295-0_24
Lannuzel, A., Michel, P. P., Höglinger, G. U., Champy, P., Jousset, A., Medja, F., et al. (2003). The mitochondrial complex I inhibitor annonacin is toxic to mesencephalic dopaminergic neurons by impairment of energy metabolism. Neuroscience 121, 287–296. doi: 10.1016/S0306-4522(03)00441-X
Lazarou, M., Jin, S. M., Kane, L. A., and Youle, R. J. (2012). Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 22, 320–333. doi: 10.1016/j.devcel.2011.12.014
Lee, E., Yin, Z., Sidoryk-Wêgrzynowicz, M., Jiang, H., and Aschner, M. (2012). 15-Deoxy-Δ12,14-prostaglandin J2 modulates manganese-induced activation of the NF-κB, Nrf2, and PI3K pathways in astrocytes. Free Radic. Biol. Med. 52, 1067–1074. doi: 10.1016/j.freeradbiomed.2011.12.016
Lee, Y., Stevens, D. A., Kang, S. U., Jiang, H., Lee, Y. I., Ko, H. S., et al. (2017). PINK1 primes Parkin-mediated ubiquitination of PARIS in dopaminergic neuronal survival. Cell Rep. 18, 918–932. doi: 10.1016/j.celrep.2016.12.090
Lesage, S., Anheim, M., Letournel, F., Bousset, L., Honoré, A., Rozas, N., et al. (2013). G51d α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 73, 459–471. doi: 10.1002/ana.23894
Li, H. M., Niki, T., Taira, T., Iguchi-Ariga, S. M., and Ariga, H. (2005). Association of DJ-1 with chaperones and enhanced association and colocalization with mitochondrial Hsp70 by oxidative stress. Free Radic. Res. 39, 1091–1099. doi: 10.1080/10715760500260348
Li, Y., Liu, W., Oo, T. F., Wang, L., Tang, Y., Jackson-Lewis, V., et al. (2009). Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat. Neurosci. 12, 826–828. doi: 10.1038/nn.2349
Lill, C. M., Roehr, J. T., McQueen, M. B., Kavvoura, F. K., Bagade, S., Schjeide, B. M., et al. (2012). Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: the PDGene database. PLOS Genet. 8:e1002548. doi: 10.1371/journal.pgen.1002548
Liu, G., Zhang, C., Yin, J., Li, X., Cheng, F., Li, Y., et al. (2009). alpha-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci. Lett. 454, 187–192. doi: 10.1016/j.neulet.2009.02.056
Liu, W., Vives-Bauza, C., Acin-Perez, R., Yamamoto, A., Tan, Y., Li, Y., et al. (2009). PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and alpha-synuclein aggregation in cell culture models of Parkinson’s disease. PLOS ONE 4:e4597. doi: 10.1371/journal.pone.0004597
Lohmann, E., Coquel, A. S., Honore, A., Gurvit, H., Hanagasi, H., Emre, M., et al. (2015). A new F-box protein 7 gene mutation causing typical Parkinson’s disease. Mov. Disord. 30, 1130–1133. doi: 10.1002/mds.26266
Lutz, A. K., Exner, N., Fett, M. E., Schlehe, J. S., Kloos, K., Lammermann, K., et al. (2009). Loss of Parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 284, 22938–22951. doi: 10.1074/jbc.M109.035774
Ma, Z., and Turk, J. (2001). The molecular biology of the group VIA Ca2+-independent phospholipase A2. Prog. Nucleic Acid Res. Mol. Biol. 67, 1–33. doi: 10.1016/S0079-6603(01)67023-5
MacLeod, D., Dowman, J., Hammond, R., Leete, T., Inoue, K., and Abeliovich, A. (2006). The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52, 587–593. doi: 10.1016/j.neuron.2006.10.008
Maita, C., Maita, H., Iguchi-Ariga, S. M., and Ariga, H. (2013). Monomer DJ-1 and its N-terminal sequence are necessary for mitochondrial localization of DJ-1 mutants. PLOS ONE 8:e54087. doi: 10.1371/journal.pone.0054087
Manning-Boğ, A. B., Caudle, W. M., Perez, X. A., Reaney, S. H., Paletzki, R., Isla, M. Z., et al. (2007). Increased vulnerability of nigrostriatal terminals in DJ-1-deficient mice is mediated by the dopamine transporter. Neurobiol. Dis. 27, 141–150. doi: 10.1016/j.nbd.2007.03.014
Manning-Bog, A. B., McCormack, A. L., Li, J., Uversky, V. N., Fink, A. L., and Di Monte, D. A. (2002). The herbicide paraquat causes up-regulation and aggregation of a-synuclein in mice. J. Biol. Chem. 277, 1641–1644. doi: 10.1074/jbc.C100560200
Martin, L. J., Pan, Y., Price, A. C., Sterling, W., Copeland, N. G., Jenkins, N. A., et al. (2006). Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 26, 41–50. doi: 10.1523/jneurosci.4308-05.2006
Martinez-Finley, E. J., Gavin, C. E., Aschner, M., and Gunter, T. E. (2013). Manganese neurotoxicity and the role of reactive oxygen species. Free Radic. Biol. Med. 62, 65–75. doi: 10.1016/j.freeradbiomed.2013.01.032
Matsumine, H., Saito, M., Shimoda-Matsubayashi, S., Tanaka, H., Ishikawa, A., Nakagawa-Hattori, Y., et al. (1997). Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27. Am. J. Hum. Genet. 60, 588–596.
Mazzulli, J. R., Xu, Y. H., Sun, Y., Knight, A. L., McLean, P. J., Caldwell, G. A., et al. (2011). Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. doi: 10.1016/j.cell.2011.06.001
McCormack, A. L., and Di Monte, D. A. (2003). Effects of L-dopa and other animo acids against paraquat-induced nigrostriatal degeneration. J. Neurochem. 85, 82–86. doi: 10.1046/j.1471-4159.2003.01621.x
McCormack, A. L., Thiruchelvam, M., Manning-Bog, A. B., Thiffault, C., Langston, W. J., Cory-Slechta, D. A., et al. (2002). Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol. Dis. 10, 119–127. doi: 10.1006/nbdi.2002.0507
Meissner, C., Lorenz, H., Weihofen, A., Selkoe, D. J., and Lemberg, M. K. (2011). The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 117, 856–867. doi: 10.1111/j.1471-4159.2011.07253.x
Melrose, H. L., Dachsel, J. C., Behrouz, B., Lincoln, S. J., Yue, M., Hinkle, K. M., et al. (2010). Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol. Dis. 40, 503–517. doi: 10.1016/j.nbd.2010.07.010
Meng, H., Yamashita, C., Shiba-Fukushima, K., Inoshita, T., Funayama, M., Sato, S., et al. (2017). Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun. 8:15500. doi: 10.1038/ncomms15500
Miki, Y., Tanji, K., Mori, F., Kakita, A., Takahashi, H., and Wakabayashi, K. (2017). PLA2G6 accumulates in Lewy bodies in PARK14 and idiopathic Parkinson’s disease. Neurosci. Lett. 645, 40–45. doi: 10.1016/j.neulet.2017.02.027
Morais, V. A., Haddad, D., Craessaerts, K., De Bock, P. J., Swerts, J., Vilain, S., et al. (2014). PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science 344, 203–207. doi: 10.1126/science.1249161
Morais, V. A., Verstreken, P., Roethig, A., Smet, J., Snellinx, A., Vanbrabant, M., et al. (2009). Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 1, 99–111. doi: 10.1002/emmm.200900006
Motoyama, T., Yabunaka, H., and Miyoshi, H. (2002). Essential structural factors of acetogenins, potent inhibitors of mitochondrial complex I. Bioorg. Med. Chem. Lett. 12, 2089–2092. doi: 10.1016/S0960-894X(02)00374-8
Murphy, K. E., Gysbers, A. M., Abbott, S. K., Tayebi, N., Kim, W. S., Sidransky, E., et al. (2014). Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 137(Pt 3), 834–848. doi: 10.1093/brain/awt367
Nakamura, K., Nemani, V., Azarbal, F., Skibinski, G., Levy, J., Egami, K., et al. (2011). Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 286, 20701–20726. doi: 10.1074/jbc.M110.213538
Nakamura, K., Nemani, V. M., Wallender, E. K., Kaehlcke, K., Ott, M., and Edwards, R. H. (2008). Optical reporters for the conformation of alpha-synuclein reveal a specific interaction with mitochondria. J. Neurosci. 28, 12305–12317. doi: 10.1523/jneurosci.3088-08.2008
Nalls, M. A., Pankratz, N., Lill, C. M., Do, C. B., Hernandez, D. G., Saad, M., et al. (2014). Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989–993. doi: 10.1038/ng.3043
Narendra, D., Walker, J. E., and Youle, R. (2012). Mitochondrial Quality Control Mediated by PINK1 and Parkin: Links to Parkinsonism. Cold Spring Harb. Perspect. Biol. 4:a011338. doi: 10.1101/cshperspect.a011338
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., et al. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLOS Biol. 8:e1000298. doi: 10.1371/journal.pbio.1000298
Nasuti, C., Brunori, G., Eusepi, P., Marinelli, L., Ciccocioppo, R., and Gabbianelli, R. (2017). Early life exposure to permethrin: a progressive animal model of Parkinson’s disease. J. Pharmacol. Toxicol. Methods 83, 80–86. doi: 10.1016/j.vascn.2016.10.003
Neudorfer, O., Giladi, N., Elstein, D., Abrahamov, A., Turezkite, T., Aghai, E., et al. (1996). Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 89, 691–694. doi: 10.1093/qjmed/89.9.691
Niu, J., Yu, M., Wang, C., and Xu, Z. (2012). Leucine-rich repeat kinase 2 disturbs mitochondrial dynamics via Dynamin-like protein. J. Neurochem. 122, 650–658. doi: 10.1111/j.1471-4159.2012.07809.x
Obeso, J. A., Stamelou, M., Goetz, C. G., Poewe, W., Lang, A. E., Weintraub, D., et al. (2017). Past, present, and future of Parkinson’s disease: a special essay on the 200th anniversary of the shaking palsy. Mov. Disord. 32, 1264–1310. doi: 10.1002/mds.27115
Ogaki, K., Koga, S., Heckman, M. G., Fiesel, F. C., Ando, M., Labbe, C., et al. (2015). Mitochondrial targeting sequence variants of the CHCHD2 gene are a risk for Lewy body disorders. Neurology 85, 2016–2025. doi: 10.1212/WNL.0000000000002170
Okatsu, K., Uno, M., Koyano, F., Go, E., Kimura, M., Oka, T., et al. (2013). A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J. Biol. Chem. 288, 36372–36384. doi: 10.1074/jbc.M113.509653
Osellame, L. D., Rahim, A. A., Hargreaves, I. P., Gegg, M. E., Richard-Londt, A., Brandner, S., et al. (2013). Mitochondria and quality control defects in a mouse model of Gaucher disease–links to Parkinson’s disease. Cell Metab. 17, 941–953. doi: 10.1016/j.cmet.2013.04.014
Paisan-Ruiz, C., Bhatia, K. P., Li, A., Hernandez, D., Davis, M., Wood, N. W., et al. (2009). Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann. Neurol. 65, 19–23. doi: 10.1002/ana.21415
Paisan-Ruiz, C., Lewis, P. A., and Singleton, A. B. (2013). LRRK2: cause, risk, and mechanism. J. Parkinsons Dis. 3, 85–103. doi: 10.3233/jpd-130192
Palacino, J. J., Sagi, D., Goldberg, M. S., Krauss, S., Motz, C., Wacker, M., et al. (2004). Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 279, 18614–18622. doi: 10.1074/jbc.M401135200
Pan-Montojo, F., Schwarz, M., Winkler, C., Arnhold, M., O’Sullivan, G. A., Pal, A., et al. (2012). Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci. Rep. 2, 898. doi: 10.1038/srep00898
Papkovskaia, T. D., Chau, K. Y., Inesta-Vaquera, F., Papkovsky, D. B., Healy, D. G., Nishio, K., et al. (2012). G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum. Mol. Genet. 21, 4201–4213. doi: 10.1093/hmg/dds244
Parihar, M. S., Parihar, A., Fujita, M., Hashimoto, M., and Ghafourifar, P. (2008). Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol. Life Sci. 65, 1272–1284. doi: 10.1007/s00018-008-7589-1
Park, J., Lee, D. G., Kim, B., Park, S. J., Kim, J. H., Lee, S. R., et al. (2015). Iron overload triggers mitochondrial fragmentation via calcineurin-sensitive signals in HT-22 hippocampal neuron cells. Toxicology 337, 39–46. doi: 10.1016/j.tox.2015.08.009
Parker, W. D., Boyson, S. J., and Parks, J. K. (1989). Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann. Neurol. 26, 719–723. doi: 10.1002/ana.410260606
Peres, T. V., Parmalee, N. L., Martinez-Finley, E. J., and Aschner, M. (2016). Untangling the manganese-α-synuclein web. Front. Neurosci. 10:364. doi: 10.3389/fnins.2016.00364
Periquet, M., Corti, O., Jacquier, S., and Brice, A. (2005). Proteomic analysis of Parkin knockout mice: alterations in energy metabolism, protein handling and synaptic function. J. Neurochem. 95, 1259–1276. doi: 10.1111/j.1471-4159.2005.03442.x
Piccoli, C., Ripoli, M., Quarato, G., Scrima, R., D’Aprile, A., Boffoli, D., et al. (2008). Coexistence of mutations in PINK1 and mitochondrial DNA in early onset parkinsonism. J. Med. Genet. 45, 596–602. doi: 10.1136/jmg.2008.058628
Pilsl, A., and Winklhofer, K. F. (2012). Parkin, PINK1 and mitochondrial integrity: emerging concepts of mitochondrial dysfunction in Parkinson’s disease. Acta Neuropathol. 123, 173–188. doi: 10.1007/s00401-011-0902-3
Plowey, E. D., Cherra, S. J. III, Liu, Y. J., and Chu, C. T. (2008). Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 105, 1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x
Podlesniy, P., Vilas, D., Taylor, P., Shaw, L. M., Tolosa, E., and Trullas, R. (2016). Mitochondrial DNA in CSF distinguishes LRRK2 from idiopathic Parkinson’s disease. Neurobiol. Dis. 94, 10–17. doi: 10.1016/j.nbd.2016.05.019
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Poole, A. C., Thomas, R. E., Andrews, L. A., McBride, H. M., Whitworth, A. J., and Pallanck, L. J. (2008). The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. U.S.A. 105, 1638–1643. doi: 10.1073/pnas.0709336105
Potts, L. F., Luzzio, F. A., Smith, S. C., Hetman, M., Champy, P., and Litvan, I. (2012). Annonacin in Asimina triloba fruit: implication for neurotoxicity. Neurotoxicology 33, 53–58. doi: 10.1016/j.neuro.2011.10.009
Pryde, K. R., Smith, H. L., Chau, K. Y., and Schapira, A. H. (2016). PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J. Cell Biol. 213, 163–171. doi: 10.1083/jcb.201509003
Przedborski, S. (2017). The two-century journey of Parkinson disease research. Nat. Rev. Neurosci. 18, 251–259. doi: 10.1038/nrn.2017.25
Purisai, M. G., McCormack, A. L., Cumine, S., Li, J., Isla, M. Z., and Di Monte, D. A. (2007). Microglial activation as a priming event leading to paraquat-induced dopaminergic cell degeneration. Neurobiol. Dis. 25, 392–400. doi: 10.1016/j.nbd.2006.10.008
Quadri, M., Federico, A., Zhao, T., Breedveld, G. J., Battisti, C., Delnooz, C., et al. (2012). Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am. J. Hum. Genet. 90, 467–477. doi: 10.1016/j.ajhg.2012.01.017
Rail, D., Scholtz, C., and Swash, M. (1981). Post-encephalitic Parkinsonism: current experience. J. Neurol. Neurosurg. Psychiatry 44, 670–676. doi: 10.1136/jnnp.44.8.670
Ramirez, A., Heimbach, A., Grundemann, J., Stiller, B., Hampshire, D., Cid, L. P., et al. (2006). Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191. doi: 10.1038/ng1884
Ramonet, D., Daher, J. P., Lin, B. M., Stafa, K., Kim, J., Banerjee, R., et al. (2011). Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLOS ONE 6:e18568. doi: 10.1371/journal.pone.0018568
Rappold, P. M., Cui, M., Chesser, A. S., Tibbett, J., Grima, J. C., Duan, L., et al. (2011). Paraquat neurotoxicity is mediated by the dopamine transporter and organic cation transporter-3. Proc. Natl. Acad. Sci. U.S.A. 108, 20766–20771. doi: 10.1073/pnas.1115141108
Rentschler, G., Covolo, L., Haddad, A. A., Lucchini, R. G., Zoni, S., and Broberg, K. (2012). ATP13A2 (PARK9) polymorphisms influence the neurotoxic effects of manganese. Neurotoxicology 33, 697–702. doi: 10.1016/j.neuro.2012.01.007
Richardson, J. R., Quan, Y., Sherer, T. B., Greenamyre, J. T., and Miller, G. W. (2005). Paraquat neurotoxicity is distinct from that of MPTP and rotenone. Toxicol. Sci. 88, 193–201. doi: 10.1093/toxsci/kfi304
Ritz, B. R., Manthripragada, A. D., Costello, S., Lincoln, S. J., Farrer, M. J., Cockburn, M., et al. (2009). Dopamine transporter genetic variants and pesticides in Parkinson’s disease. Environ. Health Perspect. 117, 964–969. doi: 10.1289/ehp.0800277
Roberts, R. F., Tang, M. Y., Fon, E. A., and Durcan, T. M. (2016). Defending the mitochondria: the pathways of mitophagy and mitochondrial-derived vesicles. Int. J. Biochem. Cell Biol. 79, 427–436. doi: 10.1016/j.biocel.2016.07.020
Robison, G., Sullivan, B., Cannon, J. R., and Pushkar, Y. (2015). Identification of dopaminergic neurons of the substantia nigra pars compacta as a target of manganese accumulation. Metallomics 7, 748–755. doi: 10.1039/c5mt00023h
Rostovtseva, T. K., Gurnev, P. A., Protchenko, O., Hoogerheide, D. P., Yap, T. L., Philpott, C. C., et al. (2015). α-synuclein shows high-affinity interaction with voltage-dependent anion channel suggesting mechanisms of mitochondrial regulation and toxicity in Parkinson disease. J. Biol. Chem. 290, 18467–18477. doi: 10.1074/jbc.M115.641746
Roth, J. A. (2014). Correlation between the biochemical pathways altered by mutated Parkinson-related genes and chronic exposure to manganese. Neurotoxicology 44, 314–325. doi: 10.1016/j.neuro.2014.08.006
Roth, J. A., Singleton, S., Feng, J., Garrick, M., and Paradkar, P. N. (2010). Parkin regulates metal transport via proteasomal degradation of the 1B isoforms of divalent metal transporter 1. J. Neurochem. 113, 454–464. doi: 10.1111/j.1471-4159.2010.06607.x
Salvador, G. A., Uranga, R. M., and Giusto, N. M. (2010). Iron and mechanisms of neurotoxicity. Int. J. Alzheimers Dis. 2011:720658. doi: 10.4061/2011/720658
Samaranch, L., Lorenzo-Betancor, O., Arbelo, J. M., Ferrer, I., Lorenzo, E., Irigoyen, J., et al. (2010). PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133(Pt 4), 1128–1142. doi: 10.1093/brain/awq051
Sánchez-Danés, A., Richaud-Patin, Y., Carballo-Carbajal, I., Jiménez-Delgado, S., Caig, C., Mora, S., et al. (2012). Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 4, 380–395. doi: 10.1002/emmm.201200215
Sanchez-Ramos, J., Facca, A., Basit, A., and Song, S. (1998). Toxicity of dieldrin for dopaminergic neurons in mesencephalic cultures. Exp. Neurol. 150, 263–271. doi: 10.1006/exnr.1997.6770
Sanders, L. H., Laganiere, J., Cooper, O., Mak, S. K., Vu, B. J., Huang, Y. A., et al. (2014). LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: reversal by gene correction. Neurobiol. Dis. 62, 381–386. doi: 10.1016/j.nbd.2013.10.013
Sanders, L. H., Paul, K. C., Howlett, E. H., Lawal, H., Boppana, S., Bronstein, J. M., et al. (2017). Editor’s highlight: base excision repair variants and pesticide exposure increase Parkinson’s disease risk. Toxicol. Sci. 158, 188–198. doi: 10.1093/toxsci/kfx086
Sarkar, S., Malovic, E., Harischandra, D. S., Ngwa, H. A., Ghosh, A., Hogan, C., et al. (2017). Manganese exposure induces neuroinflammation by impairing mitochondrial dynamics in astrocytes. Neurotoxicology doi: 10.1016/j.neuro.2017.05.009 [Epub ahead of print].
Sasaki, S., Shirata, A., Yamane, K., and Iwata, M. (2004). Parkin-positive autosomal recessive juvenile Parkinsonism with alpha-synuclein-positive inclusions. Neurology 63, 678–682. doi: 10.1212/01.WNL.0000134657.25904.0B
Schapira, A. H., Cooper, J. M., Dexter, D., Jenner, P., Clark, J. B., and Marsden, C. D. (1989). Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1, 1269. doi: 10.1016/S0140-6736(89)92366-0
Schuler, F., and Casida, J. E. (2001). Functional coupling of PSST and ND1 subunits in NADH:ubiquinone oxidoreductase established by photoaffinity labelling. Biochim. Biophys. Acta 1506, 79–87. doi: 10.1016/S0005-2728(01)00183-9
Seleznev, K., Zhao, C., Zhang, X. H., Song, K., and Ma, Z. A. (2006). Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. J. Biol. Chem. 281, 22275–22288. doi: 10.1074/jbc.M604330200
Sharma, M., Ioannidis, J. P., Aasly, J. O., Annesi, G., Brice, A., Bertram, L., et al. (2012). A multi-centre clinico-genetic analysis of the VPS35 gene in Parkinson disease indicates reduced penetrance for disease-associated variants. J. Med. Genet. 49, 721–726. doi: 10.1136/jmedgenet-2012-101155
Sharma, R. P., Winn, D. S., and Low, J. B. (1976). Toxic, neurochemical and behavioral effects of dieldrin exposure in mallard ducks. Arch. Environ. Contam. Toxicol. 5, 43–53. doi: 10.1007/BF02220889
Shimizu, K., Ohtaki, K., Matsubara, K., Aoyama, K., Uezono, T., Saito, O., et al. (2001). Carrier-mediated processes in blood-brain barrier penetration and neural uptake of paraquat. Brain Res. 906, 135–142. doi: 10.1016/S0006-8993(01)02577-X
Shin, J. H., Ko, H. S., Kang, H., Lee, Y., Lee, Y. I., Pletinkova, O., et al. (2011). PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 144, 689–702. doi: 10.1016/j.cell.2011.02.010
Shojaee, S., Sina, F., Banihosseini, S. S., Kazemi, M. H., Kalhor, R., Shahidi, G. A., et al. (2008). Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am. J. Hum. Genet. 82, 1375–1384. doi: 10.1016/j.ajhg.2008.05.005
Siddiqui, A., Chinta, S. J., Mallajosyula, J. K., Rajagopolan, S., Hanson, I., Rane, A., et al. (2012). Selective binding of nuclear alpha-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: implications for Parkinson’s disease. Free Radic Biol. Med. 53, 993–1003. doi: 10.1016/j.freeradbiomed.2012.05.024
Sidransky, E., and Lopez, G. (2012). The link between the GBA gene and parkinsonism. Lancet Neurol. 11, 986–998. doi: 10.1016/s1474-4422(12)70190-4
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). Alpha-synuclein locus triplication causes Parkinson’s disease. Science 302, 841. doi: 10.1126/science.1090278
Snyder, S. H., and D’Amato, R. J. (1985). Predicting Parkinson’s disease. Nature 317, 198–199. doi: 10.1038/317198a0
Soloway, S. (1976). Naturally occuring insecticides. Environ. Health Perspect. 14, 109–117. doi: 10.1289/ehp.7614109
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Sriram, K., Lin, G. X., Jefferson, A. M., Roberts, J. R., Wirth, O., Hayashi, Y., et al. (2010). Mitochondrial dysfunction and loss of Parkinson’s disease-linked proteins contribute to neurotoxicity of manganese-containing welding fumes. FASEB J. 24, 4989–5002. doi: 10.1096/fj.10-163964
Stafa, K., Tsika, E., Moser, R., Musso, A., Glauser, L., Jones, A., et al. (2014). Functional interaction of Parkinson’s disease-associated LRRK2 with members of the dynamin GTPase superfamily. Hum. Mol. Genet. 23, 2055–2077. doi: 10.1093/hmg/ddt600
Stevens, D. A., Lee, Y., Kang, H. C., Lee, B. D., Lee, Y. I., Bower, A., et al. (2015). Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. U.S.A. 112, 11696–11701. doi: 10.1073/pnas.1500624112
Stichel, C. C., Zhu, X. R., Bader, V., Linnartz, B., Schmidt, S., and Lubbert, H. (2007). Mono- and double-mutant mouse models of Parkinson’s disease display severe mitochondrial damage. Hum. Mol. Genet. 16, 2377–2393. doi: 10.1093/hmg/ddm083
Strokin, M., Seburn, K. L., Cox, G. A., Martens, K. A., and Reiser, G. (2012). Severe disturbance in the Ca2+ signaling in astrocytes from mouse models of human infantile neuroaxonal dystrophy with mutated Pla2g6. Hum. Mol. Genet. 21, 2807–2814. doi: 10.1093/hmg/dds108
Su, Y. C., and Qi, X. (2013). Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum. Mol. Genet. 22, 4545–4561. doi: 10.1093/hmg/ddt301
Subramaniam, S. R., Vergnes, L., Franich, N. R., Reue, K., and Chesselet, M. F. (2014). Region specific mitochondrial impairment in mice with widespread overexpression of alpha-synuclein. Neurobiol. Dis. 70, 204–213. doi: 10.1016/j.nbd.2014.06.017
Sugiura, A., McLelland, G. L., Fon, E. A., and McBride, H. M. (2014). A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 33, 2142–2156. doi: 10.15252/embj.201488104
Sumi-Akamaru, H., Beck, G., Shinzawa, K., Kato, S., Riku, Y., Yoshida, M., et al. (2016). High expression of α-synuclein in damaged mitochondria with PLA2G6 dysfunction. Acta Neuropathol. Commun. 4:27. doi: 10.1186/s40478-016-0298-3
Sun, F., Anantharam, V., and Kanthasamy, A. (2004). Dieldrin exposure impairs the ubiquitin-proteosome function and promotes-synuclein aggregation in mesencephalic dopaminergic neuronal cells: relevance of environmental factors in the etiopathogenesis of Parkinson’s disease. Toxicologist 78, 621.
Surmeier, D. J., Obeso, J. A., and Halliday, G. M. (2017). Parkinson’s disease is not simply a prion disorder. J. Neurosci. 37, 9799–9807. doi: 10.1523/JNEUROSCI.1787-16.2017
Taira, T., Saito, Y., Niki, T., Iguchi-Ariga, S. M., Takahashi, K., and Ariga, H. (2004). DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 5, 213–218. doi: 10.1038/sj.embor.7400074
Tanaka, A., Cleland, M. M., Xu, S., Narendra, D. P., Suen, D. F., Karbowski, M., et al. (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380. doi: 10.1083/jcb.201007013
Tang, F. L., Liu, W., Hu, J. X., Erion, J. R., Ye, J., Mei, L., et al. (2015). VPS35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep. 12, 1631–1643. doi: 10.1016/j.celrep.2015.08.001
Tanner, C., Kamel, F., Ross, G., Hoppin, J., Goldman, S., Korell, M., et al. (2011). Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 119, 866–872. doi: 10.1289/ehp.1002839
Tapias, V., Hu, X., Luk, K. C., Sanders, L. H., Lee, V. M., and Greenamyre, J. T. (2017). Synthetic alpha-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell. Mol. Life Sci. 74, 2851–2874. doi: 10.1007/s00018-017-2541-x
Thiruchelvam, M., McCormack, A., Richfield, E. K., Baggs, R. B., Tank, A. W., Di Monte, D. A., et al. (2003). Age-related irreversible progressive nigrostriatal dopaminergic neurotoxicity in the paraquat and maneb model of the Parkinson’s disease phenotype. Eur. J. Neurosci. 18, 589–600. doi: 10.1046/j.1460-9568.2003.02781.x
Thomas, K. J., McCoy, M. K., Blackinton, J., Beilina, A., van der Brug, M., Sandebring, A., et al. (2011). DJ-1 acts in parallel to the PINK1/Parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 20, 40–50. doi: 10.1093/hmg/ddq430
Tieu, K. (2011). A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harbor. Perspect. Med. 1:a009316. doi: 10.1101/cshperspect.a009316
Tong, Y., Pisani, A., Martella, G., Karouani, M., Yamaguchi, H., Pothos, E. N., et al. (2009). R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc. Natl. Acad. Sci. U.S.A. 106, 14622–14627. doi: 10.1073/pnas.0906334106
Tormo, J. R., Gallardo, T., Aragón, R., Cortes, D., and Estornell, E. (1999). Specific interactions of monotetrahydrofuranic annonaceous acetogenins as inhibitors of mitochondrial complex I. Chem. Biol. Interact. 122, 171–183. doi: 10.1016/S0009-2797(99)00120-9
Trinh, J., Gustavsson, E. K., Vilariño-Güell, C., Bortnick, S., Latourelle, J., McKenzie, M. B., et al. (2016). DNM3 and genetic modifiers of age of onset in LRRK2 Gly2019Ser parkinsonism: a genome-wide linkage and association study. Lancet Neurol. 15, 1248–1256. doi: 10.1016/s1474-4422(16)30203-4
Tsika, E., Glauser, L., Moser, R., Fiser, A., Daniel, G., Sheerin, U. M., et al. (2014). Parkinson’s disease-linked mutations in VPS35 induce dopaminergic neurodegeneration. Hum. Mol. Genet. 23, 4621–4638. doi: 10.1093/hmg/ddu178
Tsuji, S., Choudary, P. V., Martin, B. M., Stubblefield, B. K., Mayor, J. A., Barranger, J. A., et al. (1987). A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher’s disease. N. Engl. J. Med. 316, 570–575. doi: 10.1056/NEJM198703053161002
Tuschl, K., Clayton, P. T., Gospe, S. M., Gulab, S., Ibrahim, S., Singhi, P., et al. (2012). Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am. J. Hum. Genet. 90, 457–466. doi: 10.1016/j.ajhg.2012.01.018
Tuschl, K., Meyer, E., Valdivia, L. E., Zhao, N., Dadswell, C., Abdul-Sada, A., et al. (2016). Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat. Commun. 7:11601. doi: 10.1038/ncomms11601
Tuschl, K., Mills, P. B., and Clayton, P. T. (2013). Manganese and the brain. Int. Rev. Neurobiol. 110, 277–312. doi: 10.1016/b978-0-12-410502-7.00013-2
Usenovic, M., and Krainc, D. (2012). Lysosomal dysfunction in neurodegeneration: the role of ATP13A2/PARK9. Autophagy 8, 987–988. doi: 10.4161/auto.20256
Usenovic, M., Tresse, E., Mazzulli, J. R., Taylor, J. P., and Krainc, D. (2012). Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J. Neurosci. 32, 4240–4246. doi: 10.1523/jneurosci.5575-11.2012
Uversky, V. N., Li, J., and Fink, A. L. (2001a). Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 276, 44284–44296. doi: 10.1074/jbc.M105343200
Uversky, V. N., Li, J., and Fink, A. L. (2001b). Pesticides directly accelerate the rate of alpha-synuclein fibril formation: a possible factor in Parkinson’s disease. FEBS Lett. 500, 105–108.
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004a). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
Valente, E. M., Bentivoglio, A. R., Dixon, P. H., Ferraris, A., Ialongo, T., Frontali, M., et al. (2001). Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am. J. Hum. Genet. 68, 895–900. doi: 10.1086/319522
Valente, E. M., Salvi, S., Ialongo, T., Marongiu, R., Elia, A. E., Caputo, V., et al. (2004b). PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann. Neurol. 56, 336–341. doi: 10.1002/ana.20256
Vilarino-Guell, C., Wider, C., Ross, O. A., Dachsel, J. C., Kachergus, J. M., Lincoln, S. J., et al. (2011). VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 89, 162–167. doi: 10.1016/j.ajhg.2011.06.001
Vives-Bauza, C., Zhou, C., Huang, Y., Cui, M., de Vries, R. L., Kim, J., et al. (2010). PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U.S.A. 107, 378–383. doi: 10.1073/pnas.0911187107
Wagner, S. R., and Greene, F. E. (1978). Dieldrin-induced alterations in biogenic amine content of rat brain. Toxicol. Appl. Pharmacol. 43, 45–55. doi: 10.1016/S0041-008X(78)80031-3
Walker, M. D., Volta, M., Cataldi, S., Dinelle, K., Beccano-Kelly, D., Munsie, L., et al. (2014). Behavioral deficits and striatal DA signaling in LRRK2 p.G2019S transgenic rats: a multimodal investigation including PET neuroimaging. J. Parkinsons Dis. 4, 483–498. doi: 10.3233/jpd-140344
Wang, A., Costello, S., Cockburn, M., Zhang, X., Bronstein, J., and Ritz, B. (2011). Parkinson’s disease risk from ambient exposure to pesticides. Eur. J. Epidemiol. 26, 547–555. doi: 10.1007/s10654-011-9574-5
Wang, H. S., Toh, J., Ho, P., Tio, M., Zhao, Y., and Tan, E. K. (2014). In vivo evidence of pathogenicity of VPS35 mutations in the Drosophila. Mol. Brain 7:73. doi: 10.1186/s13041-014-0073-y
Wang, W., Wang, X., Fujioka, H., Hoppel, C., Whone, A. L., Caldwell, M. A., et al. (2016). Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 22, 54–63. doi: 10.1038/nm.3983
Wang, X., Yan, M. H., Fujioka, H., Liu, J., Wilson-Delfosse, A., Chen, S. G., et al. (2012). LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 21, 1931–1944. doi: 10.1093/hmg/dds003
West, A. B., Moore, D. J., Choi, C., Andrabi, S. A., Li, X., Dikeman, D., et al. (2007). Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 16, 223–232. doi: 10.1093/hmg/ddl471
Williams, E. T., Chen, X., and Moore, D. J. (2017). VPS35, the retromer complex and Parkinson’s disease. J. Parkinsons Dis. 7, 219–233. doi: 10.3233/jpd-161020
Winslow, A. R., Chen, C. W., Corrochano, S., Acevedo-Arozena, A., Gordon, D. E., Peden, A. A., et al. (2010). alpha-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J. Cell Biol. 190, 1023–1037. doi: 10.1083/jcb.201003122
Wong, K., Sidransky, E., Verma, A., Mixon, T., Sandberg, G. D., Wakefield, L. K., et al. (2004). Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 82, 192–207. doi: 10.1016/j.ymgme.2004.04.011
Wood-Kaczmar, A., Gandhi, S., Yao, Z., Abramov, A. Y., Abramov, A. S., Miljan, E. A., et al. (2008). PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLOS ONE 3:e2455. doi: 10.1371/journal.pone.0002455
Wu, X. F., Block, M. L., Zhang, W., Qin, L., Wilson, B., Zhang, W. Q., et al. (2005). The role of microglia in paraquat-induced dopaminergic neurotoxicity. Antioxid. Redox. Signal. 7, 654–661. doi: 10.1089/ars.2005.7.654
Xilouri, M., Brekk, O. R., and Stefanis, L. (2016). Autophagy and alpha-synuclein: relevance to Parkinson’s disease and related synucleopathies. Mov. Disord. 31, 178–192. doi: 10.1002/mds.26477
Xiong, H., Wang, D., Chen, L., Choo, Y. S., Ma, H., Tang, C., et al. (2009). Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Invest. 119, 650–660. doi: 10.1172/jci37617
Xu, Y. H., Xu, K., Sun, Y., Liou, B., Quinn, B., Li, R. H., et al. (2014). Multiple pathogenic proteins implicated in neuronopathic Gaucher disease mice. Hum. Mol. Genet. 23, 3943–3957. doi: 10.1093/hmg/ddu105
Yamano, K., and Youle, R. J. (2013). PINK1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769. doi: 10.4161/auto.24633
Yang, Y., Ouyang, Y., Yang, L., Beal, M. F., McQuibban, A., Vogel, H., et al. (2008). Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc. Natl. Acad. Sci. U.S.A. 105, 7070–7075. doi: 10.1073/pnas.0711845105
Yoshii, S. R., Kishi, C., Ishihara, N., and Mizushima, N. (2011). Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 286, 19630–19640. doi: 10.1074/jbc.M110.209338
Yu, W. H., Dorado, B., Figueroa, H. Y., Wang, L., Planel, E., Cookson, M. R., et al. (2009). Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am. J. Pathol. 175, 736–747. doi: 10.2353/ajpath.2009.080928
Yue, M., Hinkle, K. M., Davies, P., Trushina, E., Fiesel, F. C., Christenson, T. A., et al. (2015). Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol. Dis. 78, 172–195. doi: 10.1016/j.nbd.2015.02.031
Zarranz, J. J., Alegre, J., Gómez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., et al. (2004). The new mutation, e46k, of alpha-synuclein causes Parkinson and lewy body dementia. Ann. Neurol. 55, 164–173. doi: 10.1002/ana.107951
Zhang, L., Shimoji, M., Thomas, B., Moore, D. J., Yu, S. W., Marupudi, N. I., et al. (2005). Mitochondrial localization of the Parkinson’s disease related protein DJ-1: implications for pathogenesis. Hum. Mol. Genet. 14, 2063–2073. doi: 10.1093/hmg/ddi211
Zheng, B., Liao, Z., Locascio, J. J., Lesniak, K. A., Roderick, S. S., Watt, M. L., et al. (2010). PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2, 52ra73. doi: 10.1126/scitranslmed.3001059
Zhou, C., Huang, Y., Shao, Y., May, J., Prou, D., Perier, C., et al. (2008). The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc. Natl. Acad. Sci. U.S.A. 105, 12022–12027. doi: 10.1073/pnas.0802814105
Zhou, Z. D., Xie, S. P., Sathiyamoorthy, S., Saw, W. T., Sing, T. Y., Ng, S. H., et al. (2015). F-box protein 7 mutations promote protein aggregation in mitochondria and inhibit mitophagy. Hum. Mol. Genet. 24, 6314–6330. doi: 10.1093/hmg/ddv340
Zimprich, A., Benet-Pages, A., Struhal, W., Graf, E., Eck, S. H., Offman, M. N., et al. (2011). A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 89, 168–175. doi: 10.1016/j.ajhg.2011.06.008
Keywords: Parkinson’s disease, mitochondrial dynamics, mitochondrial dysfunction, neurodegeneration, neurotoxicity, environmental toxins, gene–environment interaction
Citation: Helley MP, Pinnell J, Sportelli C and Tieu K (2017) Mitochondria: A Common Target for Genetic Mutations and Environmental Toxicants in Parkinson’s Disease. Front. Genet. 8:177. doi: 10.3389/fgene.2017.00177
Received: 07 August 2017; Accepted: 01 November 2017;
Published: 17 November 2017.
Edited by:
Joao Batista Teixeira da Rocha, Universidade Federal de Santa Maria, BrazilReviewed by:
Christopher J. Martyniuk, University of Florida, United StatesJason Cannon, Purdue University, United States
Copyright © 2017 Helley, Pinnell, Sportelli and Tieu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kim Tieu, a3RpZXVAZml1LmVkdQ==