 Shauna Hill
Shauna Hill Kavithalakshmi Sataranatarajan1
Kavithalakshmi Sataranatarajan1- 1Aging and Metabolism Research Program, Oklahoma Medical Research Foundation, Oklahoma City, OK, United States
- 2Department of Cell Systems & Anatomy, University of Texas Health at San Antonio, San Antonio, TX, United States
- 3Department of Pathology, University of Washington, Seattle, WA, United States
- 4Oklahoma City VA Medical Center, Oklahoma City, OK, United States
Mitochondria are established essential regulators of cellular function and metabolism. Mitochondria regulate redox homeostasis, maintain energy (ATP) production through oxidative phosphorylation, buffer calcium levels, and control cell death through apoptosis. In addition to these critical cell functions, recent evidence supports a signaling role for mitochondria. For example, studies over the past few years have established that peptides released from the mitochondria mediate stress responses such as the mitochondrial unfolded protein response (UPRMT) through signaling to the nucleus. Mitochondrial damage or danger associated molecular patterns (DAMPs) provide a link between mitochondria, inflammation and inflammatory disease processes. Additionally, a new class of peptides generated by the mitochondria affords protection against age-related diseases in mammals. In this short review, we highlight the role of mitochondrial signaling and regulation of cellular activities through the mitochondrial UPRMT that signals to the nucleus to affect homeostatic responses, DAMPs, and mitochondrial derived peptides.
Introduction
Mitochondria are essential double membrane cellular organelles that provide the energy to the cell through oxidative phosphorylation. Mitochondria also play a key role in cellular function and homeostasis through adenosine triphosphate (ATP) production, calcium homeostasis, apoptosis signaling, and fatty acid oxidation. In addition, mitochondria modulate redox signaling through the production of reactive oxygen species (ROS). While the role of mitochondrial ROS signaling in modulation of cellular processes has been established, recently several studies have highlighted additional signaling roles for mitochondria that are not initiated by ROS. The idea that mitochondria can regulate cellular metabolism through mitochondrial derived signaling was first described in yeast. Using mitochondrial DNA depleted rho° yeast cells, Ron Butow discovered yeast mitochondria have adapted a mitochondria-to-nucleus signal transduction pathway termed the retrograde response to induce the transcription of nuclear-encoded mitochondrial genes and alleviate mitochondrial stress (Parikh et al., 1987). Higher organisms have adapted a similar retrograde signaling response known as the mitochondrial unfolded protein response (UPRMT). This response is initiated by the accumulation of unfolded proteins in the mitochondria resulting in the induction of UPRMT components (Benedetti et al., 2006). In addition, mitochondria can regulate cell function and metabolism by signaling pathways that involve mitochondrial-derived peptides and other mitochondrial signals such as mitochondrial damage-associated molecular patterns (mito-DAMPs). In this review, we highlight the role of mitochondrial signaling and regulation of cellular activities through the mitochondrial UPRMT that signals to the nucleus to affect homeostatic responses, damage or danger associated molecular patterns (DAMPs), and mitochondrial derived peptides (MDPs) (Figure 1).
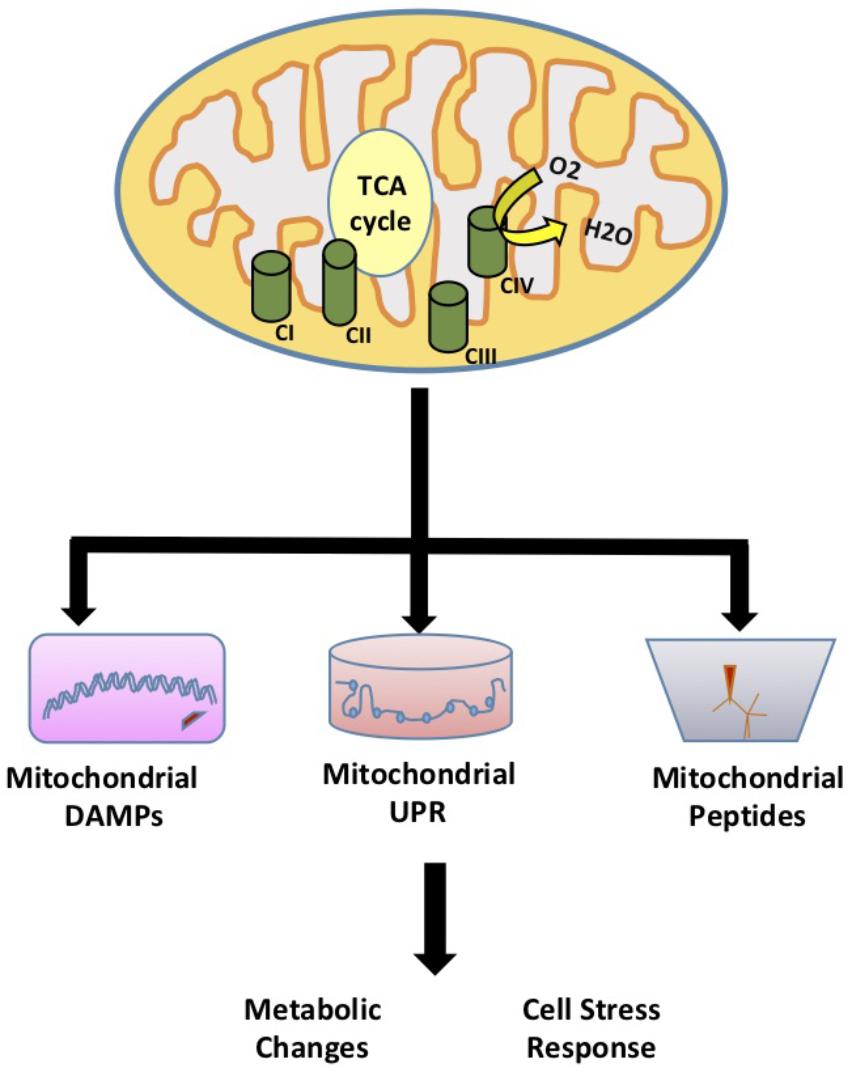
FIGURE 1. Mitochondrial Signaling. The role of mitochondria in signaling and regulation of cellular activities through the mitochondrial unfolded protein response (UPRMT) that signals to the nucleus to affect homeostatic responses, damage or danger associated molecular patterns (DAMPs), and mitochondrial derived peptides (MDPs).
Mitochondrial Unfolded Protein Response (UPRMT)
The UPRMT signaling pathway was first elucidated in C. elegans. Mitochondrial stress generated from misfolded proteins was shown to activate the adenosine triphosphate (ATP)-dependent mitochondrial protease, ClpP, to cleave misfolded proteins (Benedetti et al., 2006; Haynes et al., 2007). The exported peptides in turn activate the nuclear translocation of ATFS-1 (Activating Transcription Factor associated with Stress) where it subsequently activates UBL-5 to form a complex with transcription factor DVE-1 to transcriptionally activate UPRMT genes, heat shock protein 6 (hsp-6) and hsp-10 (Martinus et al., 1996; Haynes et al., 2007, 2010). Subsequent studies found that under normal conditions ATFS-1 accumulates in the mitochondria, but during mitochondrial stress (ETC dysfunction, ROS production, proteotoxic stress, etc.) ATFS-1 accumulates in the cytosol and is transported to the nucleus to induce the expression of mitochondrial stress proteins (Nargund et al., 2012). A recent study in C. elegans showed that the H3K27 demethylases jmjd-1.2 and jmjd-3.1 are required for the UPRMT suggesting that the UPRMT can also be regulated at the epigenetic level (Merkwirth et al., 2016). In another recent report in C. elegans, the UPRMT was proposed to promote the propagation of deleterious mtDNA rather than eliminating deleterious mtDNA (Lin et al., 2016). These findings suggest that the UPRMT could be used as a therapeutic target for mtDNA disorders and associated diseases (further reviewed in Tian et al., 2016).
The regulation of the UPRMT in mammals is not fully understood, but a few key components have been identified in mammals with the first report by Zhao et al. (2002) showing that UPRMT components are elevated in mitochondrial DNA (mtDNA) depleted mammalian cells. The mammalian transcription factors, CHOP and C/EBPβ were implicated as putative transcription factors for this pathway based on conserved regulatory element in promoters of the UPRMT related genes (Aldridge et al., 2007). In this model, mitochondrial proteotoxic stress mediates CHOP transcriptional activation through the Jnk/c-Jun pathway (Weiss et al., 2003; Jaeschke et al., 2006). This pathway is conserved in mammals as the Jnk/c-Jun pathway is required for transcriptional activation of CHOP and UPRMT induction in mitochondrial ornithine transcarbamylase (OTC) mutant Cos-7 cells under proteotoxic stress (Rath et al., 2012). A recent study by Fiorese et al. (2016) identified the mammalian transcription factor ATF-5 to be the functional ortholog of the C. elegans ATFS-1 transcription factor. The transcription factor, ATF-4 has also been recently shown to be an important mediator of mitochondrial stress response (Quiros et al., 2017). However, ATF-4 was not shown to regulate UPRMT genes but cytoprotective genes that regulate cellular metabolism. Studies in mouse tissue show a consistent correlation between H3K27 demethylases and UPRMT transcripts supporting the notion that UPRMT gene regulation is through demethylases proceeding the retrograde signaling from the mitochondria (Merkwirth et al., 2016).
Studies from our laboratory have shown that mice harboring a null mutation in the SURF1 gene, an electron transport chain complex IV assembly factor, show an induction of the UPRMT in several tissues that is associated with a number of beneficial phenotypes including increased insulin sensitivity, mitochondrial biogenesis, and increased resistance to oxidative stress in cultured fibroblasts (Deepa et al., 2013; Lin et al., 2013; Pulliam et al., 2014; Pharaoh et al., 2016). Shpilka and Haynes (2018) have extensively reviewed the implications of the UPRMT in aging and age-related diseases. These studies suggest a potential link between the UPRMT, metabolism and stress resistance in mammals.
Signaling Peptides Associated With the Mitochondrial Unfolded Protein Response
The mitochondrial UPRMT is of interest as a direct form of communication between the mitochondria and the nucleus. However, a study by Durieux et al. (2011) suggested that mitochondrial stress could be signaled through cell non-autonomous mechanisms. Specifically, studies in C. elegans showed that mitochondrial proteotoxic stress restricted to neuronal tissue induced the mitochondrial UPRMT in a heterologous tissue, the intestine (Durieux et al., 2011). These findings suggest that the UPRMT can be activated in a cell non-autonomous fashion. Interestingly, components of the UPRMT, ATFS-1, and DVE-1 in the neurons are required to induce the UPRMT in the intestine. Further studies have been conducted to determine the signaling factor released by the neurons to induce the UPRMT in the intestine. One study suggested that distal activation of the UPRMT requires UNC-31 mediated secretion of serotonin (Berendzen et al., 2016). However, this study used a general proteotoxic stress model, PolyQ40, and not a mitochondrial specific model. In another study, neuropeptides were investigated to determine if they mediate the UPRMT in a cell non-autonomous fashion using a neuronal specific CRISPR-Cas9 approach to manipulate mitochondrial stress (Shao et al., 2016). Neuropeptides are released from dense core vesicles derived at the synapse and may function as hormones to systemically alter cellular activities. This study showed that deletion of 6 out of the 103 reported neuropeptides blocked the neuronal-specific mitochondrial stress induction of the UPRMT in the intestine. These neuropeptides include INS-17, INS-34, FLP-2, FLP-15, NLP-10, and NLP-28. Subsequently, only constitutive overexpression of flp-2 was shown to induce the UPRMT on its own. These studies suggest that FLP-2 is released by neurons during mitochondrial stress to signal for the induction of the UPRMT in the periphery. Interestingly, overexpression of flp-2 activates the UPRMT but does not extend lifespan. This supports other studies where the UPRMT was shown to not have a casual effect on lifespan (Bennett et al., 2014). The identification of these signaling molecules could have significant implications in the development of therapies to target mitochondrial disease and improve healthspan.
Mitochondrial Damage Associated Molecular Patterns
In recent years, studies have shown that mitochondria can modulate inflammation and the immune response through the release of signaling factors known as Mito-DAMPs (Zhang et al., 2010). Under physiological conditions, damage associated molecular patterns (DAMPs) are not recognized by immune system. During cellular stress or tissue injury, these molecules can be released from dying cells or damaged extracellular matrix in to the extracellular environment (Wenceslau et al., 2014; Land, 2015). During pathological insults, Mito-DAMPs have consequences on the innate immune response and inflammation (Krysko et al., 2011). Mito-DAMPs include mtDNA, TFAM, cardiolipin, ATP, and N-formyl peptides (NFPs) (Nakahira et al., 2015) (Figure 2).
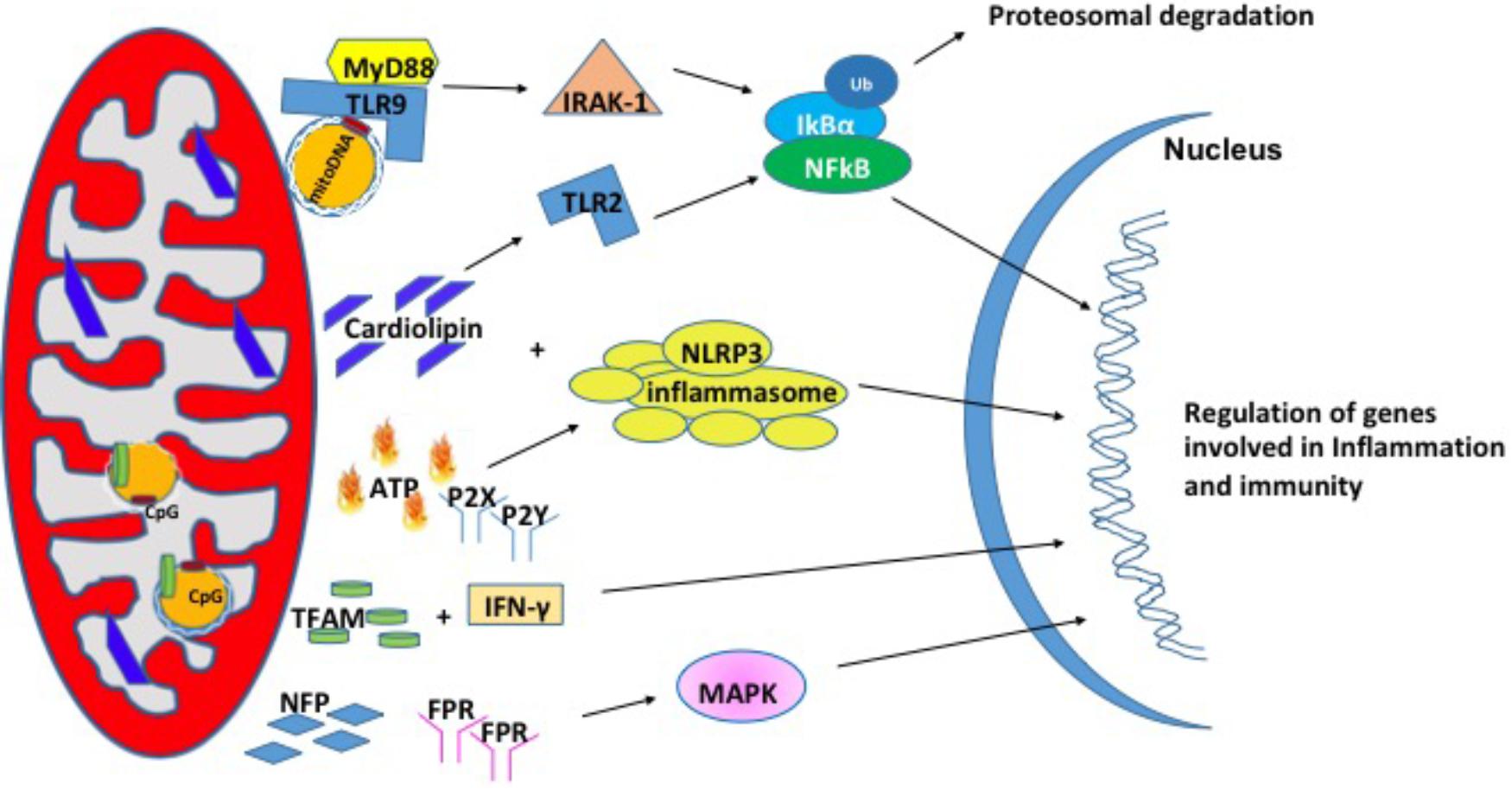
FIGURE 2. Mitochondrial damage-associated molecular patterns (Mito-DAMPs). Mitochondrial damage associated molecular patterns are in the form of mitochondrial DNA (mtDNA), cardiolipin, ATP, TFAM, and NFP. In response to stress, mitochondria release these small molecules to signal for the nuclear transcription of genes involved in inflammation and immunity.
Mitochondrial DNA (mtDNA)
Mitochondrial DNA encodes essential protein subunits of the oxidative phosphorylation system that play primary role in respiration and ATP production. Oxidized or fragmented mtDNA is released from damaged mitochondria and evokes an immune response (Garcia and Chavez, 2007; Nakahira et al., 2011). The unmethylated CpG site of the mtDNA binds to TLR9 and activates a downstream cascade of reactions by recruiting the adaptor proteins MyD88; IL-1R-associated kinase (IRAK4), interferon regulatory factor-7 (IRF7), tumor-necrosis factor-alpha receptor activated factor-6 (TRAF6) (Hacker et al., 2000; Huang and Yang, 2010). These events are followed by activation of ERK, p38, and IkB pathways that finally culminate in the transcription of inflammatory genes (Yi and Krieg, 1998; Deguine and Barton, 2014). mtDNA levels in the plasma of the elderly (age 90 years) correlated with pro-inflammatory cytokines like TNF-α, IL-1β, IL-6, IL-1Rα, and RANTES (Pinti et al., 2014) in glomerular kidney disease (Bao et al., 2016), in heart (Bliksoen et al., 2016), and in sporadic ALS (Vielhaber et al., 2000). In addition, circulating mtDNA levels are increased in acute liver injury (Marques et al., 2012), hypertension (McCarthy et al., 2015), acute kidney injury (Tsuji et al., 2016). Apart from the increased levels in the circulation, mtDNA copy number has also been shown to be increased in aging skeletal muscle (Masser et al., 2016), lung tissue (Lee et al., 2000), in cancer (Cormio et al., 2012) and the level of mtDNA in the synovial fluid was shown to correlate with the severity of rheumatoid arthritis (Hajizadeh et al., 2003). A recent study from Sandler et al. (2018) suggests that mtDNA may be an early biomarker in post-operative complications such as cardiopulmonary bypass surgery (Sandler et al., 2018) and in traumatic brain injury (Walko et al., 2014). Thus, mtDNA is associated with a number of diseases and may also play a role in pathologies of aging.
TFAM
Mitochondrial transcription factor, TFAM is an abundant protein that plays an important role in regulating mtDNA content. TFAM is tightly bound to mtDNA and amplifies the mtDNA induced TLR9 immune response (Julian et al., 2012). TFAM can act directly as a DAMP. For example, treatment of THP1 monocytic cells with TFAM resulted in increased expression IL-1β, IL-6, and IL-8 (Little et al., 2014). In another study, serum TFAM levels were doubled after hemorrhagic shock and resuscitation in Sprague-Dawley rats (Chaung et al., 2012), which in turn induces inflammatory responses. The impact of TFAM in neurodegeneration diseases has been reviewed briefly in Kang et al. (2018).
Cardiolipin
Cardiolipin is a phospholipid present in the inner mitochondrial membrane that is a central player in various processes like mitochondrial calcium uniporter, mitochondrial protein kinase C signaling, mitophagy, cytochrome c release during apoptosis and inflammasome activation (Dudek, 2017). Upon translocation to the outer membrane, cardiolipin activates NLRP3 promoting inflammation (Iyer et al., 2013). During cell injury, cardiolipin undergoes oxidation, and released into the extracellular environment where it acts as a mito-DAMPs (Claypool and Koehler, 2012). In support of this, patients with pneumonia show high concentration of cardiolipin in lung fluid (Ray et al., 2010) and patients with mitochondrial disease showed higher cardiolipin content in the skeletal muscle (Schlame et al., 1999). There are several reports elucidating the mechanism by which cardiolipin acts as mito-DAMP under different pathological conditions. In a study with autoimmune disease patients, cardiolipin signals through TLR2-PI3K-PKN1-AKT-p38MAPK-NFkB pathway to activate antigen presenting cells (Cho et al., 2018). In another study involving pneumonia, cardiolipin inhibits interleukin (IL)-10 production by inducing the SUMOylation of the nuclear receptor PPAR gamma. PPAR gamma SUMOylation results in binding of the repressive complex NCOR/HDAC3 to IL-10 promotor but not the TNF promoter thereby efficiently inhibiting IL-10 production (Chakraborty et al., 2017).
Adenosine Triphosphate (ATP)
Adenosine triphosphate is the currency of intracellular energy. ATP plays an important role in glycolysis, TCA cycle, and beta oxidation. Apart from being used as source of energy, it plays a crucial role in biochemical signaling pathways including DNA and RNA synthesis and protein synthesis. Under physiological conditions, there remains a balance between ATP secretion and its extracellular concentration. When this balance is lost, extracellular ATP (eATP) plays a toxic role. eATP is upregulated in diabetic nephropathy (Chen et al., 2013), hypertension (Ji et al., 2012), induces vascular inflammation, atherosclerosis (Stachon et al., 2016), and lung inflammation (Riteau et al., 2010). eATP can be fueled by stimuli such as membrane damage, mechanical stress, excitation of neural tissue (Fields, 2011). eATP binds to the purinergic receptor subtype P2X or P2Y receptor (Surprenant and North, 2009; Erlinge, 2011) and can play a key role in regulation of vascular endothelium, pain and inflammatory responses. Ectonucleotidases CD39 and CD73 can degrade eATP to ADP, AMP and adenosine and each of these molecules can bind to P2 receptors and activate responses related to tissue damage and inflammation (Wilkin et al., 2001; Bours et al., 2006). eATP has been shown to activate the NLRP3 inflammasome that results in the release of IL-18 (Amores-Iniesta et al., 2017) through P2X7 in allograft rejection (Amores-Iniesta et al., 2017). Importantly, P2X7 -receptor inhibitors CE224, AZD9056, and GSK1482160 are available in clinical use as immunomodulatory agents (Vergani et al., 2014).
N-Formyl Peptides (NFPs)
N-Formyl peptides are a class of peptides that are produced by bacterial cells and mitochondria suggesting their involvement in host defense against bacterial infection and the clearance of damaged cells. NFPs have a high affinity binding site for FPR, NFP (Boulay et al., 1990; Rabiet et al., 2005). FPRs are a class of transmembrane G protein-coupled receptors that have three isoforms (FPR1, FPR2, and FPR3) and are highly expressed on monocytes and neutrophils suggesting their involvement in immune cell response (Waller et al., 2004). NFPs have been shown to drive neutrophil activation through MAPK and ERK1/2 signaling pathways (Hazeldine et al., 2015). In cell culture models, the addition of mito-DAMPs initiated chemotaxis, production of TNFα as well as a rapid release of ROS (Rabiet et al., 2005; Friedenberg et al., 2016). The induction of chemotaxis via NFPs is dependent on calcium influx through FPRs (Le et al., 2002).
A number of studies have shown that NFPs are elevated in response to post-traumatic injury and diseased models (Zhang et al., 2010; Dorward et al., 2017). These elevated levels lead to systemic inflammatory response syndrome (SIRS) (Hazeldine et al., 2015). For example, elevated NFPs lead to airway contraction and lung inflammation (Wenceslau et al., 2016). On the other hand, the loss of NFP binding receptor, FPR increases the susceptibility to L. monocytogenes in mice (Liu et al., 2012). Inflammation is positively correlated with aging and this relationship is coined ‘inflamm-aging’ (Franceschi et al., 2000). Therefore, NFP signaling may be a therapeutic target to reduce systemic inflammation and increase healthspan in humans.
Mitochondrial Derived Peptides (MDPs)
Mitochondrial derived peptides are a novel class of mitochondrial signaling peptides that are encoded by short open reading frames (sORFs) in the mitochondrial genome. MDPs regulate a wide range of cellular signaling pathways and have an implicated role in aging. In this section, we review known MDPs and their role in cellular activities.
Humanin
Humanin was the first discovered MDP using a functional expression screen for peptides that could suppress neuronal cell death induced by Aβ (Hashimoto et al., 2001). Humanin is a 24-amino acid peptide that’s encoded by the mitochondrial 16S rRNA. Humanin has gained significant recognition for its cytoprotective cellular activities and protection against a wide range of pathologies. A number of studies implicate humanin as a potential therapeutic target for a range of diseases. For example, synthetic humanin improves memory deficits and reduced Aβ plaques in rodent models of Alzheimer’s disease (Niikura et al., 2011; Zhang et al., 2012; Chai et al., 2014). Studies have shown that humanin protects against stroke in mice (Xu et al., 2006), ameliorates atherosclerotic plaque formation (Oh et al., 2011), improves insulin sensitivity in rodent models of diabetes (Hoang et al., 2010), and affords cardioprotection against myocardial ischemia (Thummasorn et al., 2016). Additionally, humanin restores cellular ATP levels in cells isolated from human patients affected by the mitochondrial disease, mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (Kariya et al., 2005). The cytoprotective activities of humanin provide this protection in a number of disease models.
Humanin elicits cytoprotection inter- and intracellular via anti-apoptotic effects and binding to two plasma membrane receptors. Humanin interacts via binding with IGFBP-3, Bax, and tBid (Guo et al., 2003; Ikonen et al., 2003; Zhai et al., 2005). In cell culture studies, humanin shows BAX dependent anti-apoptotic effects against serum deprivation and tumor necrosis factor (Guo et al., 2003; Zhai et al., 2005). Humanin also exhibits anti-apoptotic effects by binding to IGFBP-3 to block nuclear translocation required to induce apoptosis (Ikonen et al., 2003). More recently, humanin is shown to prevent endoplasmic reticulum (ER)-stress induced apoptosis by mediating ER-mitochondrial cross talk for cell survival (Matsunaga et al., 2016; Sreekumar et al., 2017). As a result, humanin suppresses apoptosis and promotes cell survival during oxidative and ER stress. Humanin also protects from apoptosis through the preservation of mitochondrial homeostasis in responses to cellular stress (Klein et al., 2013; Thummasorn et al., 2016; Cui et al., 2017). In addition, circulating humanin binds to two plasma membrane receptors to initiate a number of cytoprotective activities. One of the two receptors is the trimeric CNTFR/gp130/WSX-1 receptor (Hashimoto et al., 2001, 2009; Ying et al., 2004). Humanin induces the hetero-oligomerization of CNTFR, gp130, and WSX-1 and subsequently binds to the CNTFR/gp130/WSX-1 receptor to activate the JAK-STAT pathway (Kim et al., 2016). Humanin-induced neuroprotection requires the activation of Stat3 via the CNTFR/gp130/WSX-1 receptor (Hashimoto et al., 2005). Activation of Stat3 by humanin also improves diabetes in a mouse model by inhibiting pancreatic β-cell apoptosis and improving glucose tolerance (Hoang et al., 2010). Humanin protects against damage from Aβ42 by binding to the formyl peptide receptor-like 1/2 (FRPL1/2) receptor (Ying et al., 2004). It’s hypothesized that humanin exerts its neuroprotection by competing with Aβ42 for the binding to FRPL1/2 since Aβ42 accumulation is linked to FRPL1/2 binding.
Due to the universal cytoprotective activities of humanin, it has been proposed to have therapeutic potential to enhance human healthspan. In fact, humanin has been linked to aging. Humanin levels significantly decline with age in humans and rodent models (Muzumdar et al., 2009; Bachar et al., 2010; Hoang et al., 2010). Offspring of centenarians have been shown to have higher levels of humanin compared to the rest of the aging population (Muzumdar et al., 2009). Together, these studies suggest that retaining humanin levels with age may promote healthy aging.
MOTS-c (Mitochondrial Open Reading Frame of the 12S rRNA-c)
MOTS-c is a 16-amino-acid peptide encoded by 12S rRNA sORF in the mtDNA that has been proposed to play a role in regulation of metabolism (Lee et al., 2015). The MOTS-c transcript is polyadenylated and subsequently exported to the cytoplasm for translation. Phylogenetic studies show that MOTS-c is highly conserved across species (Lee et al., 2015). In mice, MOTS-c is present in a number of high-energy demanding tissues with the main targets being skeletal muscle and adipose tissue. MOTS-c is also present in the circulation of humans and mice (Lee et al., 2015).
Microarray analysis in two different mammalian cell culture lines demonstrated that MOTS-c regulates global gene expression specifically affecting metabolic and inflammatory related genes (Lee et al., 2015). The alterations in global metabolic gene expression translate to changes in the metabolite profile. Metabolomic studies showed that MOTS-c reduces metabolites involved in purine and dipeptide metabolism and increases metabolites involved in acylcarnitine and methionine metabolism. MOTS-c also promotes the biosynthesis of the AMPK activator, AICAR (Lee et al., 2015). These alterations in the metabolomic profile induced by MOTS-c suggest a potential target for metabolic disease as well as aging.
Studies have shown that MOTS-c can protect against a number of pathologies. MOTS-c treated mice have reduced body weight, food intake, and blood glucose. MOTS-c promotes insulin sensitivity and protects from high-fat diet induced insulin resistance and obesity in mice (Lee et al., 2015). Specifically, MOTS-c promotes insulin sensitivity in the skeletal muscle by stimulating glucose clearance as measured with the hyperinsulinemic-euglycemic clamp technique (Lee et al., 2015). MOTS-c also has a protective role in ovariectomy-induced bone loss. MOTS-c treatment inhibited receptor activator of nuclear factor-κB ligand (RANK) induced osteoclast differentiation (Ming et al., 2016). The protective effects of MOTS-c on bone loss are partially dependent on MOTS-C mediated AMPK activation since an AMPK inhibitor partially reversed these effects (Ming et al., 2016). Additionally, MOTS-c administration reduced basal circulating levels of IL-6 and TNFα (Lee et al., 2015).
Analyses of the mitochondrial genome from an exceptionally long-lived Japanese population suggest a role for MOTS-c in human longevity (Fuku et al., 2015a). Mitochondrial genome analysis from a Northeast Asian population identified a m.1382A > C polymorphism located in the MOTS-c encoding mtDNA (Fuku et al., 2015a,b). This single nucleotide polymorphism results in a single amino acid substitution predicted to have function consequences on the small peptide. This alteration in MOTS-C could contribute to the exceptional longevity observed in the Northeast Asian population. Together, these data suggest that MOTS-c may be a potential therapeutic target to improve healthspan.
Small Humanin-Like Peptides (SHLPs)
An in silico prediction analyses of sORFs identified six novel peptides named small humanin-like peptides (SHLPs) 1–6 (Cobb et al., 2016). The existence of these novel SHLPs were validated by mRNA and peptide expression levels in cells, tissues, and plasma. The origin of SHLPs 1–6 has been determined by RT-PCR. SHLPs 1, 4, 5, and 6 were confirmed to be mitochondrial in origin. However, SHLPs 2 and 3 were amplified from both mitochondrial and nuclear cDNA, which leaves the possibility that SHLPs 2 and 3 are not exclusively mitochondrial in origin.
Cell viability and apoptosis studies revealed that SHLPs 2 and 3 promotes cell viability and protection against cellular apoptosis in NIT-1 and 22Rv1, mouse beta and human prostate cancer cells, respectively (Cobb et al., 2016). One method in which SHLPs 2 and 3 promote cell viability is through the reduction of ROS production. The peptides, SHLPs 2 and 3 increase basal oxygen consumption and ATP production (Cobb et al., 2016). Furthermore, SHLPs 2 and 3 promotes adipocyte differentiation of 3T3L pre-adipocytes. SHLPs 2 and 3 activate ERK and STAT-3 signaling (Cobb et al., 2016).
Similar to humanin and MOTS-c, SHLP 2 circulating levels decline with age making it a potential therapeutic target for age-related diseases (Cobb et al., 2016). In fact, SHLP 2 has similar effects on insulin sensitivity as humanin and MOTS-c. In Sprague-Dawley rats, SHLP 2 enhanced the exogenous glucose infusion rate by 50% and promoted glucose uptake by peripheral tissues measured with the hyperinsulinemic-euglycemic clamp technique (Cobb et al., 2016). These findings suggest that SHLP 2 is an insulin sensitizer and implicates a therapeutic potential for SHLP 2 for diabetes. Additionally, SHLP 2 supplementation prevented Aβ-induced neuronal cell death (Cobb et al., 2016). SHLP 2 may exert neuroprotection similar to humanin via the activation of STAT-3. Future studies should investigate the effects of SHLP 2 in Alzheimer’s disease and diabetic mammalian models. Furthermore, it is important to establish the role of SHLP 2 in modulating healthspan as well as characterize the biological and physiological effects of the other SHLPs.
Summary
Since the initial finding of mitochondrial nuclear signaling in yeast, our understanding of the potential for mitochondrial to act in a cell signaling capacity has expanded significantly. A similar mitochondrial-nuclear signaling pathway, the UPRMT, was demonstrated in C. elegans and more recently studies have demonstrated that mitochondrial stress restricted to neuronal tissue induced the UPRMT in a heterologous tissue, the intestine. These findings suggest that there is not only intracellular signaling resulting in UPRMT activation, but that there is also signaling between/among tissues resulting in UPRMT activation. This is a novel concept that has important implications for extending our understanding of the role of mitochondria in cellular function and resistance to stress. Mitochondrial peptides have also been shown to regulate cytoprotective activities and mitochondrial stress responses in mammals. For example, a recently discovered MDP, humanin, is released during mitochondrial dysfunction resulting in an increase of cytoprotective activities and providing protection against a range of pathologies. Since the discovery of humanin, MOTS-C and a class of six other mitochondrial derived peptides (SHLPs) have been identified. Similar to humanin, these peptides have shown to afford cytoprotection but not much is known. Future work will likely continue to demonstrate exciting new signaling and regulatory functions for the mitochondria.
Author Contributions
SH, KS, and HVR wrote the text. KS prepared the figures.
Funding
HVR is supported by a Senior Research Career Scientist Award from the Department of Veterans Affairs and SH was supported by a Ruth L. Kirschstein National Research Service Award for Individual Predoctoral Fellows (F31AG047764-03).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Van Remmen laboratory for rich discussions and their comments throughout the preparation of this mini-review.
References
Aldridge, J. E., Horibe, T., and Hoogenraad, N. J. (2007). Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One 2:e874. doi: 10.1371/journal.pone.0000874
Amores-Iniesta, J., Barbera-Cremades, M., Martinez, C. M., Pons, J. A., Revilla-Nuin, B., Martinez-Alarcon, L., et al. (2017). Extracellular ATP activates the NLRP3 inflammasome and is an early danger signal of skin allograft rejection. Cell Rep. 21, 3414–3426. doi: 10.1016/j.celrep.2017.11.079
Bachar, A. R., Scheffer, L., Schroeder, A. S., Nakamura, H. K., Cobb, L. J., Oh, Y. K., et al. (2010). Humanin is expressed in human vascular walls and has a cytoprotective effect against oxidized LDL-induced oxidative stress. Cardiovasc. Res. 88, 360–366. doi: 10.1093/cvr/cvq191
Bao, W., Xia, H., Liang, Y., Ye, Y., Lu, Y., Xu, X., et al. (2016). Toll-like receptor 9 can be activated by endogenous mitochondrial DNA to induce podocyte apoptosis. Sci. Rep. 6:22579. doi: 10.1038/srep22579
Benedetti, C., Haynes, C. M., Yang, Y., Harding, H. P., and Ron, D. (2006). Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 174, 229–239. doi: 10.1534/genetics.106.061580
Bennett, C. F., Vander Wende, H., Simko, M., Klum, S., Barfield, S., Choi, H., et al. (2014). Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat. Commun. 5:3483. doi: 10.1038/ncomms4483
Berendzen, K. M., Durieux, J., Shao, L. W., Tian, Y., Kim, H. E., Wolff, S., et al. (2016). Neuroendocrine coordination of mitochondrial stress signaling and proteostasis. Cell 166, 1553.e10–1563.e10. doi: 10.1016/j.cell.2016.08.042
Bliksoen, M., Mariero, L. H., Torp, M. K., Baysa, A., Ytrehus, K., Haugen, F., et al. (2016). Extracellular mtDNA activates NF-kappaB via toll-like receptor 9 and induces cell death in cardiomyocytes. Basic Res. Cardiol. 111:42. doi: 10.1007/s00395-016-0553-6
Boulay, F., Tardif, M., Brouchon, L., and Vignais, P. (1990). The human N-formylpeptide receptor. Characterization of two cDNA isolates and evidence for a new subfamily of G-protein-coupled receptors. Biochemistry 29, 11123–11133. doi: 10.1021/bi00502a016
Bours, M. J., Swennen, E. L., Di Virgilio, F., Cronstein, B. N., and Dagnelie, P. C. (2006). Adenosine 5’-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 112, 358–404. doi: 10.1016/j.pharmthera.2005.04.013
Chai, G. S., Duan, D. X., Ma, R. H., Shen, J. Y., Li, H. L., Ma, Z. W., et al. (2014). Humanin attenuates Alzheimer-like cognitive deficits and pathological changes induced by amyloid beta-peptide in rats. Neurosci. Bull. 30, 923–935. doi: 10.1007/s12264-014-1479-3
Chakraborty, K., Raundhal, M., Chen, B. B., Morse, C., Tyurina, Y. Y., Khare, A., et al. (2017). The mito-DAMP cardiolipin blocks IL-10 production causing persistent inflammation during bacterial pneumonia. Nat. Commun. 8:13944. doi: 10.1038/ncomms13944
Chaung, W. W., Wu, R., Ji, Y., Dong, W., and Wang, P. (2012). Mitochondrial transcription factor A is a proinflammatory mediator in hemorrhagic shock. Int. J. Mol. Med. 30, 199–203. doi: 10.3892/ijmm.2012.959
Chen, K., Zhang, J., Zhang, W., Zhang, J., Yang, J., Li, K., et al. (2013). ATP-P2X4 signaling mediates NLRP3 inflammasome activation: a novel pathway of diabetic nephropathy. Int. J. Biochem. Cell Biol. 45, 932–943. doi: 10.1016/j.biocel.2013.02.009
Cho, J. A., Kim, T. J., Moon, H. J., Kim, Y. J., Yoon, H. K., and Seong, S. Y. (2018). Cardiolipin activates antigen-presenting cells via TLR2-PI3K-PKN1-AKT/p38-NF-kB signaling to prime antigen-specific naive T cells in mice. Eur. J. Immunol. 48, 777–790. doi: 10.1002/eji.201747222
Claypool, S. M., and Koehler, C. M. (2012). The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 37, 32–41. doi: 10.1016/j.tibs.2011.09.003
Cobb, L. J., Lee, C., Xiao, J., Yen, K., Wong, R. G., Nakamura, H. K., et al. (2016). Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging 8, 796–809. doi: 10.18632/aging.100943
Cormio, A., Guerra, F., Cormio, G., Pesce, V., Fracasso, F., Loizzi, V., et al. (2012). Mitochondrial DNA content and mass increase in progression from normal to hyperplastic to cancer endometrium. BMC Res. Notes 5:279. doi: 10.1186/1756-0500-5-279
Cui, A. L., Zhang, Y. H., Li, J. Z., Song, T., Liu, X. M., Wang, H., et al. (2017). Humanin rescues cultured rat cortical neurons from NMDA-induced toxicity through the alleviation of mitochondrial dysfunction. Drug Des. Devel. Ther. 11, 1243–1253. doi: 10.2147/DDDT.S133042
Deepa, S. S., Pulliam, D., Hill, S., Shi, Y., Walsh, M. E., Salmon, A., et al. (2013). Improved insulin sensitivity associated with reduced mitochondrial complex IV assembly and activity. FASEB J. 27, 1371–1380. doi: 10.1096/fj.12-221879
Deguine, J., and Barton, G. M. (2014). MyD88: a central player in innate immune signaling. F1000Prime Rep. 6:97. doi: 10.12703/P6-97
Dorward, D. A., Lucas, C. D., Doherty, M. K., Chapman, G. B., Scholefield, E. J., Conway Morris, A., et al. (2017). Novel role for endogenous mitochondrial formylated peptide-driven formyl peptide receptor 1 signalling in acute respiratory distress syndrome. Thorax 72, 928–936. doi: 10.1136/thoraxjnl-2017-210030
Dudek, J. (2017). Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell. Dev. Biol. 5:90. doi: 10.3389/fcell.2017.00090
Durieux, J., Wolff, S., and Dillin, A. (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91. doi: 10.1016/j.cell.2010.12.016
Erlinge, D. (2011). P2Y receptors in health and disease. Adv. Pharmacol. 61, 417–439. doi: 10.1016/B978-0-12-385526-8.00013-8
Fields, R. D. (2011). Nonsynaptic and nonvesicular ATP release from neurons and relevance to neuron-glia signaling. Semin. Cell Dev. Biol. 22, 214–219. doi: 10.1016/j.semcdb.2011.02.009
Fiorese, C. J., Schulz, A. M., Lin, Y. F., Rosin, N., Pellegrino, M. W., and Haynes, C. M. (2016). The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 26, 2037–2043. doi: 10.1016/j.cub.2016.06.002
Franceschi, C., Bonafe, M., Valensin, S., Olivieri, F., De Luca, M., Ottaviani, E., et al. (2000). Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 908, 244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x
Friedenberg, S. G., Strange, H. R., Guillaumin, J., Vangundy, Z. C., Crouser, E. D., and Papenfuss, T. L. (2016). Effect of disrupted mitochondria as a source of damage-associated molecular patterns on the production of tumor necrosis factor alpha by splenocytes from dogs. Am. J. Vet. Res. 77, 604–612. doi: 10.2460/ajvr.77.6.604
Fuku, N., He, Z. H., Sanchis-Gomar, F., Pareja-Galeano, H., Tian, Y., Arai, Y., et al. (2015a). Exceptional longevity and muscle and fitness related genotypes: a functional in vitro analysis and case-control association replication study with SNPs THRH rs7832552, IL6 rs1800795, and ACSL1 rs6552828. Front. Aging Neurosci. 7:59. doi: 10.3389/fnagi.2015.00059
Fuku, N., Pareja-Galeano, H., Zempo, H., Alis, R., Arai, Y., Lucia, A., et al. (2015b). The mitochondrial-derived peptide Mots-c: a player in exceptional longevity? Aging Cell 14, 921–923. doi: 10.1111/acel.12389
Garcia, N., and Chavez, E. (2007). Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci. 81, 1160–1166. doi: 10.1016/j.lfs.2007.08.019
Guo, B., Zhai, D., Cabezas, E., Welsh, K., Nouraini, S., Satterthwait, A. C., et al. (2003). Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 423, 456–461. doi: 10.1038/nature01627
Hacker, H., Vabulas, R. M., Takeuchi, O., Hoshino, K., Akira, S., and Wagner, H. (2000). Immune cell activation by bacterial CpG-DNA through myeloid differentiation marker 88 and tumor necrosis factor receptor-associated factor (TRAF)6. J. Exp. Med. 192, 595–600. doi: 10.1084/jem.192.4.595
Hajizadeh, S., Degroot, J., Tekoppele, J. M., Tarkowski, A., and Collins, L. V. (2003). Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res. Ther. 5, R234–R240. doi: 10.1186/ar787
Hashimoto, Y., Kurita, M., and Matsuoka, M. (2009). Identification of soluble WSX-1 not as a dominant-negative but as an alternative functional subunit of a receptor for an anti-Alzheimer’s disease rescue factor Humanin. Biochem. Biophys. Res. Commun. 389, 95–99. doi: 10.1016/j.bbrc.2009.08.095
Hashimoto, Y., Niikura, T., Tajima, H., Yasukawa, T., Sudo, H., Ito, Y., et al. (2001). A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Abeta. Proc. Natl. Acad. Sci. U.S.A. 98, 6336–6341. doi: 10.1073/pnas.101133498
Hashimoto, Y., Suzuki, H., Aiso, S., Niikura, T., Nishimoto, I., and Matsuoka, M. (2005). Involvement of tyrosine kinases and STAT3 in Humanin-mediated neuroprotection. Life Sci. 77, 3092–3104. doi: 10.1016/j.lfs.2005.03.031
Haynes, C. M., Petrova, K., Benedetti, C., Yang, Y., and Ron, D. (2007). ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 13, 467–480. doi: 10.1016/j.devcel.2007.07.016
Haynes, C. M., Yang, Y., Blais, S. P., Neubert, T. A., and Ron, D. (2010). The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 37, 529–540. doi: 10.1016/j.molcel.2010.01.015
Hazeldine, J., Hampson, P., Opoku, F. A., Foster, M., and Lord, J. M. (2015). N-Formyl peptides drive mitochondrial damage associated molecular pattern induced neutrophil activation through ERK1/2 and P38 MAP kinase signalling pathways. Injury 46, 975–984. doi: 10.1016/j.injury.2015.03.028
Hoang, P. T., Park, P., Cobb, L. J., Paharkova-Vatchkova, V., Hakimi, M., Cohen, P., et al. (2010). The neurosurvival factor Humanin inhibits beta-cell apoptosis via signal transducer and activator of transcription 3 activation and delays and ameliorates diabetes in nonobese diabetic mice. Metabolism 59, 343–349. doi: 10.1016/j.metabol.2009.08.001
Huang, X., and Yang, Y. (2010). Targeting the TLR9-MyD88 pathway in the regulation of adaptive immune responses. Expert Opin. Ther. Targets 14, 787–796. doi: 10.1517/14728222.2010.501333
Ikonen, M., Liu, B., Hashimoto, Y., Ma, L., Lee, K. W., Niikura, T., et al. (2003). Interaction between the Alzheimer’s survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proc. Natl. Acad. Sci. U.S.A. 100, 13042–13047. doi: 10.1073/pnas.2135111100
Iyer, S. S., He, Q., Janczy, J. R., Elliott, E. I., Zhong, Z., Olivier, A. K., et al. (2013). Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323. doi: 10.1016/j.immuni.2013.08.001
Jaeschke, A., Karasarides, M., Ventura, J. J., Ehrhardt, A., Zhang, C., Flavell, R. A., et al. (2006). JNK2 is a positive regulator of the cJun transcription factor. Mol. Cell 23, 899–911. doi: 10.1016/j.molcel.2006.07.028
Ji, X., Naito, Y., Weng, H., Endo, K., Ma, X., and Iwai, N. (2012). P2X7 deficiency attenuates hypertension and renal injury in deoxycorticosterone acetate-salt hypertension. Am. J. Physiol. Renal Physiol. 303, F1207–F1215. doi: 10.1152/ajprenal.00051.2012
Julian, M. W., Shao, G., Bao, S., Knoell, D. L., Papenfuss, T. L., Vangundy, Z. C., et al. (2012). Mitochondrial transcription factor A serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. J. Immunol. 189, 433–443. doi: 10.4049/jimmunol.1101375
Kang, I., Chu, C. T., and Kaufman, B. A. (2018). The mitochondrial transcription factor TFAM in neurodegeneration: emerging evidence and mechanisms. FEBS Lett. 592, 793–811. doi: 10.1002/1873-3468.12989
Kariya, S., Hirano, M., Furiya, Y., Sugie, K., and Ueno, S. (2005). Humanin detected in skeletal muscles of MELAS patients: a possible new therapeutic agent. Acta Neuropathol. 109, 367–372. doi: 10.1007/s00401-004-0965-5
Kim, S. J., Guerrero, N., Wassef, G., Xiao, J., Mehta, H. H., Cohen, P., et al. (2016). The mitochondrial-derived peptide humanin activates the ERK1/2, AKT, and STAT3 signaling pathways and has age-dependent signaling differences in the hippocampus. Oncotarget 7, 46899–46912. doi: 10.18632/oncotarget.10380
Klein, L. E., Cui, L., Gong, Z., Su, K., and Muzumdar, R. (2013). A humanin analog decreases oxidative stress and preserves mitochondrial integrity in cardiac myoblasts. Biochem. Biophys. Res. Commun. 440, 197–203. doi: 10.1016/j.bbrc.2013.08.055
Krysko, D. V., Agostinis, P., Krysko, O., Garg, A. D., Bachert, C., Lambrecht, B. N., et al. (2011). Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 32, 157–164. doi: 10.1016/j.it.2011.01.005
Land, W. G. (2015). The role of damage-associated molecular patterns in human diseases: part i - promoting inflammation and immunity. Sultan Qaboos Univ. Med. J. 15, e9–e21.
Le, Y., Murphy, P. M., and Wang, J. M. (2002). Formyl-peptide receptors revisited. Trends Immunol. 23, 541–548. doi: 10.1016/S1471-4906(02)02316-5
Lee, C., Zeng, J., Drew, B. G., Sallam, T., Martin-Montalvo, A., Wan, J., et al. (2015). The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 21, 443–454. doi: 10.1016/j.cmet.2015.02.009
Lee, H. C., Yin, P. H., Lu, C. Y., Chi, C. W., and Wei, Y. H. (2000). Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochem. J. 348(Pt 2), 425–432. doi: 10.1042/bj3480425
Lin, A. L., Pulliam, D. A., Deepa, S. S., Halloran, J. J., Hussong, S. A., Burbank, R. R., et al. (2013). Decreased in vitro mitochondrial function is associated with enhanced brain metabolism, blood flow, and memory in Surf1-deficient mice. J. Cereb. Blood Flow Metab. 33, 1605–1611. doi: 10.1038/jcbfm.2013.116
Lin, Y.-F., Schulz, A. M., Pellegrino, M. W., Lu, Y., Shaham, S., and Haynes, C. M. (2016). Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial upr. Nature 533, 416–419. doi: 10.1038/nature17989
Little, J. P., Simtchouk, S., Schindler, S. M., Villanueva, E. B., Gill, N. E., Walker, D. G., et al. (2014). Mitochondrial transcription factor A (Tfam) is a pro-inflammatory extracellular signaling molecule recognized by brain microglia. Mol. Cell. Neurosci. 60, 88–96. doi: 10.1016/j.mcn.2014.04.003
Liu, M., Chen, K., Yoshimura, T., Liu, Y., Gong, W., Wang, A., et al. (2012). Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci. Rep. 2:786. doi: 10.1038/srep00786
Marques, P. E., Amaral, S. S., Pires, D. A., Nogueira, L. L., Soriani, F. M., Lima, B. H., et al. (2012). Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 56, 1971–1982. doi: 10.1002/hep.25801
Martinus, R. D., Garth, G. P., Webster, T. L., Cartwright, P., Naylor, D. J., Hoj, P. B., et al. (1996). Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem. 240, 98–103. doi: 10.1111/j.1432-1033.1996.0098h.x
Masser, D. R., Clark, N. W., Van Remmen, H., and Freeman, W. M. (2016). Loss of the antioxidant enzyme CuZnSOD (Sod1) mimics an age-related increase in absolute mitochondrial DNA copy number in the skeletal muscle. Age 38, 323–333. doi: 10.1007/s11357-016-9930-1
Matsunaga, D., Sreekumar, P. G., Ishikawa, K., Terasaki, H., Barron, E., Cohen, P., et al. (2016). Humanin protects RPE cells from endoplasmic reticulum stress-induced apoptosis by upregulation of mitochondrial glutathione. PLoS One 11:e0165150. doi: 10.1371/journal.pone.0165150
McCarthy, C. G., Wenceslau, C. F., Goulopoulou, S., Ogbi, S., Baban, B., Sullivan, J. C., et al. (2015). Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc. Res. 107, 119–130. doi: 10.1093/cvr/cvv137
Merkwirth, C., Jovaisaite, V., Durieux, J., Matilainen, O., Jordan, S. D., Quiros, P. M., et al. (2016). Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 165, 1209–1223. doi: 10.1016/j.cell.2016.04.012
Ming, W., Lu, G., Xin, S., Huanyu, L., Yinghao, J., Xiaoying, L., et al. (2016). Mitochondria related peptide MOTS-c suppresses ovariectomy-induced bone loss via AMPK activation. Biochem. Biophys. Res. Commun. 476, 412–419. doi: 10.1016/j.bbrc.2016.05.135
Muzumdar, R. H., Huffman, D. M., Atzmon, G., Buettner, C., Cobb, L. J., Fishman, S., et al. (2009). Humanin: a novel central regulator of peripheral insulin action. PLoS One 4:e6334. doi: 10.1371/journal.pone.0006334
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230. doi: 10.1038/ni.1980
Nakahira, K., Hisata, S., and Choi, A. M. (2015). The roles of mitochondrial damage-associated molecular patterns in diseases. Antioxid. Redox Signal. 23, 1329–1350. doi: 10.1089/ars.2015.6407
Nargund, A. M., Pellegrino, M. W., Fiorese, C. J., Baker, B. M., and Haynes, C. M. (2012). Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590. doi: 10.1126/science.1223560
Niikura, T., Sidahmed, E., Hirata-Fukae, C., Aisen, P. S., and Matsuoka, Y. (2011). A humanin derivative reduces amyloid beta accumulation and ameliorates memory deficit in triple transgenic mice. PLoS One 6:e16259. doi: 10.1371/journal.pone.0016259
Oh, Y. K., Bachar, A. R., Zacharias, D. G., Kim, S. G., Wan, J., Cobb, L. J., et al. (2011). Humanin preserves endothelial function and prevents atherosclerotic plaque progression in hypercholesterolemic ApoE deficient mice. Atherosclerosis 219, 65–73. doi: 10.1016/j.atherosclerosis.2011.06.038
Parikh, V. S., Morgan, M. M., Scott, R., Clements, L. S., and Butow, R. A. (1987). The mitochondrial genotype can influence nuclear gene expression in yeast. Science 235, 576–580. doi: 10.1126/science.3027892
Pharaoh, G., Pulliam, D., Hill, S., Sataranatarajan, K., and Van Remmen, H. (2016). Ablation of the mitochondrial complex IV assembly protein Surf1 leads to increased expression of the UPR(MT) and increased resistance to oxidative stress in primary cultures of fibroblasts. Redox Biol. 8, 430–438. doi: 10.1016/j.redox.2016.05.001
Pinti, M., Cevenini, E., Nasi, M., De Biasi, S., Salvioli, S., Monti, D., et al. (2014). Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 44, 1552–1562. doi: 10.1002/eji.201343921
Pulliam, D. A., Deepa, S. S., Liu, Y., Hill, S., Lin, A. L., Bhattacharya, A., et al. (2014). Complex IV-deficient Surf1(-/-) mice initiate mitochondrial stress responses. Biochem. J. 462, 359–371. doi: 10.1042/BJ20140291
Quiros, P. M., Prado, M. A., Zamboni, N., D’Amico, D., Williams, R. W., Finley, D., et al. (2017). Multi-omics analysis identifies Atf4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 216, 2027–2045. doi: 10.1083/jcb.201702058
Rabiet, M. J., Huet, E., and Boulay, F. (2005). Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. Eur. J. Immunol. 35, 2486–2495. doi: 10.1002/eji.200526338
Rath, E., Berger, E., Messlik, A., Nunes, T., Liu, B., Kim, S. C., et al. (2012). Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut 61, 1269–1278. doi: 10.1136/gutjnl-2011-300767
Ray, N. B., Durairaj, L., Chen, B. B., Mcverry, B. J., Ryan, A. J., Donahoe, M., et al. (2010). Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat. Med. 16, 1120–1127. doi: 10.1038/nm.2213
Riteau, N., Gasse, P., Fauconnier, L., Gombault, A., Couegnat, M., Fick, L., et al. (2010). Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 182, 774–783. doi: 10.1164/rccm.201003-0359OC
Sandler, N., Kaczmarek, E., Itagaki, K., Zheng, Y., Otterbein, L., Khabbaz, K., et al. (2018). Mitochondrial DAMPs are released during cardiopulmonary bypass surgery and are associated with postoperative atrial fibrillation. Heart Lung Circ. 27, 122–129. doi: 10.1016/j.hlc.2017.02.014
Schlame, M., Shanske, S., Doty, S., Konig, T., Sculco, T., Dimauro, S., et al. (1999). Microanalysis of cardiolipin in small biopsies including skeletal muscle from patients with mitochondrial disease. J. Lipid Res. 40, 1585–1592.
Shao, L. W., Niu, R., and Liu, Y. (2016). Neuropeptide signals cell non-autonomous mitochondrial unfolded protein response. Cell Res. 26, 1182–1196. doi: 10.1038/cr.2016.118
Shpilka, T., and Haynes, C. M. (2018). The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 19, 109–120. doi: 10.1038/nrm.2017.110
Sreekumar, P. G., Hinton, D. R., and Kannan, R. (2017). Endoplasmic reticulum-mitochondrial crosstalk: a novel role for the mitochondrial peptide humanin. Neural Regen. Res. 12, 35–38. doi: 10.4103/1673-5374.198970
Stachon, P., Geis, S., Peikert, A., Heidenreich, A., Michel, N. A., Unal, F., et al. (2016). Extracellular ATP induces vascular inflammation and atherosclerosis via purinergic receptor Y2 in mice. Arterioscler. Thromb. Vasc. Biol. 36, 1577–1586. doi: 10.1161/ATVBAHA.115.307397
Surprenant, A., and North, R. A. (2009). Signaling at purinergic P2X receptors. Annu. Rev. Physiol. 71, 333–359. doi: 10.1146/annurev.physiol.70.113006.100630
Thummasorn, S., Apaijai, N., Kerdphoo, S., Shinlapawittayatorn, K., Chattipakorn, S. C., and Chattipakorn, N. (2016). Humanin exerts cardioprotection against cardiac ischemia/reperfusion injury through attenuation of mitochondrial dysfunction. Cardiovasc. Ther. 34, 404–414. doi: 10.1111/1755-5922.12210
Tian, Y., Merkwirth, C., and Dillin, A. (2016). Mitochondrial UPR: a double-edged sword. Trends Cell Biol. 26, 563–565. doi: 10.1016/j.tcb.2016.06.006
Tsuji, N., Tsuji, T., Ohashi, N., Kato, A., Fujigaki, Y., and Yasuda, H. (2016). Role of mitochondrial DNA in septic AKI via toll-like receptor 9. J. Am. Soc. Nephrol. 27, 2009–2020. doi: 10.1681/ASN.2015040376
Vergani, A., Tezza, S., Fotino, C., Visner, G., Pileggi, A., Chandraker, A., et al. (2014). The purinergic system in allotransplantation. Am. J. Transplant. 14, 507–514. doi: 10.1111/ajt.12567
Vielhaber, S., Kunz, D., Winkler, K., Wiedemann, F. R., Kirches, E., Feistner, H., et al. (2000). Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain 123(Pt 7), 1339–1348. doi: 10.1093/brain/123.7.1339
Walko, T. D. III, Bola, R. A., Hong, J. D., Au, A. K., Bell, M. J., Kochanek, P. M., et al. (2014). Cerebrospinal fluid mitochondrial DNA: a novel DAMP in pediatric traumatic brain injury. Shock 41, 499–503. doi: 10.1097/SHK.0000000000000160
Waller, A., Sutton, K. L., Kinzer-Ursem, T. L., Absood, A., Traynor, J. R., Linderman, J. J., et al. (2004). Receptor binding kinetics and cellular responses of six N-formyl peptide agonists in human neutrophils. Biochemistry 43, 8204–8216. doi: 10.1021/bi035335i
Weiss, C., Schneider, S., Wagner, E. F., Zhang, X., Seto, E., and Bohmann, D. (2003). JNK phosphorylation relieves Hdac3-dependent suppression of the transcriptional activity of c-Jun. EMBO J. 22, 3686–3695. doi: 10.1093/emboj/cdg364
Wenceslau, C. F., Mccarthy, C. G., Szasz, T., Spitler, K., Goulopoulou, S., Webb, R. C., et al. (2014). Mitochondrial damage-associated molecular patterns and vascular function. Eur. Heart J. 35, 1172–1177. doi: 10.1093/eurheartj/ehu047
Wenceslau, C. F., Szasz, T., Mccarthy, C. G., Baban, B., Nesmith, E., and Webb, R. C. (2016). Mitochondrial N-formyl peptides cause airway contraction and lung neutrophil infiltration via formyl peptide receptor activation. Pulm. Pharmacol. Ther. 37, 49–56. doi: 10.1016/j.pupt.2016.02.005
Wilkin, F., Duhant, X., Bruyns, C., Suarez-Huerta, N., Boeynaems, J. M., and Robaye, B. (2001). The P2Y11 receptor mediates the ATP-induced maturation of human monocyte-derived dendritic cells. J. Immunol. 166, 7172–7177. doi: 10.4049/jimmunol.166.12.7172
Xu, X., Chua, C. C., Gao, J., Hamdy, R. C., and Chua, B. H. (2006). Humanin is a novel neuroprotective agent against stroke. Stroke 37, 2613–2619. doi: 10.1161/01.STR.0000242772.94277.1f
Yi, A. K., and Krieg, A. M. (1998). Rapid induction of mitogen-activated protein kinases by immune stimulatory CpG DNA. J. Immunol. 161, 4493–4497.
Ying, G., Iribarren, P., Zhou, Y., Gong, W., Zhang, N., Yu, Z. X., et al. (2004). Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J. Immunol. 172, 7078–7085. doi: 10.4049/jimmunol.172.11.7078
Zhai, D., Luciano, F., Zhu, X., Guo, B., Satterthwait, A. C., and Reed, J. C. (2005). Humanin binds and nullifies Bid activity by blocking its activation of Bax and Bak. J. Biol. Chem. 280, 15815–15824. doi: 10.1074/jbc.M411902200
Zhang, Q., Raoof, M., Chen, Y., Sumi, Y., Sursal, T., Junger, W., et al. (2010). Circulating mitochondrial Damps cause inflammatory responses to injury. Nature 464, 104–107. doi: 10.1038/nature08780
Zhang, W., Zhang, W., Li, Z., Hao, J., Zhang, Z., Liu, L., et al. (2012). S14G-humanin improves cognitive deficits and reduces amyloid pathology in the middle-aged APPswe/PS1dE9 mice. Pharmacol. Biochem. Behav. 100, 361–369. doi: 10.1016/j.pbb.2011.09.012
Keywords: mitochondria, stress response, longevity, signaling peptides, retrograde response
Citation: Hill S, Sataranatarajan K and Van Remmen H (2018) Role of Signaling Molecules in Mitochondrial Stress Response. Front. Genet. 9:225. doi: 10.3389/fgene.2018.00225
Received: 21 July 2017; Accepted: 07 June 2018;
Published: 10 July 2018.
Edited by:
Aleksandra Trifunovic, Universität zu Köln, GermanyReviewed by:
Paul Kirchman, University of South Florida Sarasota–Manatee, United StatesCarsten Merkwirth, Ferring Research Institute, Inc., United States
Copyright © 2018 Hill, Sataranatarajan and Van Remmen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shauna Hill, aGlsbHM4OEB1dy5lZHU=