 Yongli Zhang1
Yongli Zhang1 Hui Song
Hui Song- 1Grassland Agri-Husbandry Research Center, College of Grassland Science, Qingdao Agricultural University, Qingdao, China
- 2College of Agronomy, Henan Agricultural University, Zhengzhou, China
To date, at least eight Arachis genomes have been completely sequenced, including two Arachis duranensis, two Arachis ipaensis, one Arachis monticola, and three Arachis hypogaea. These datasets can provide a powerful starting point to understand the evolution of Arachis species. In addition to a comparison of Arachis species at the whole-genome level, evolutionary masks can be uncovered based on the analysis of Arachis gene families. Although many gene families have been identified and characterized in Arachis, different methods and strategies have been used by different researchers. This paper offers advice on the methods and strategies for identification, nomenclature, and quantitative real-time PCR (qRT-PCR) primer-design based on published datasets of Arachis gene families. The presented analyses provide a theoretical foundation for the improvement of the identification and characterization of gene families in Arachis.
Genome Sequencing and Identification of Gene Families in ARACHIS
The cultivated peanut (A. hypogaea, AABB genome) was formed by the crossing of two wild peanuts: A. duranensis (AA genome) and A. ipaensis (BB genome) (Bertioli et al., 2016, 2019). In 2014, the genome sequences of A. duranensis (V14167) and A. ipaensis (K30076) were released on PeanutBase (https://peanutbase.org); however, their datasets were not usable at the time because the related paper had not been published then. It was not until 2016 when researchers could begin to use the datasets once the paper was finally published in Nature Genetics (Bertioli et al., 2016). In addition to these two lines, researchers sequenced two other lines: A. duranensis (PI 475845) and A. ipaensis (ICG_8206) (Chen et al., 2016; Lu et al., 2018). The genome sequences of three cultivated peanut species, namely A. hypogaea cv. Tifrunner, A. hypogaea cv. Shitouqi, and A. hypogaea cv. Fuhuasheng, were sequenced and released in 2018 (Bertioli et al., 2019; Chen et al., 2019; Zhuang et al., 2019). Simultaneously, the genome of a wild tetraploid peanut, A. monticola, was completely sequenced (Yin et al., 2018, 2019). These eight available genomic datasets provide raw material for the study of Arachis evolution.
Several researchers have focused on genome-wide analyses of the evolution and expression of gene families with canonical domains in Arachis. The WRKY transcription factor, a ~60-residue DNA-binding domain containing a conserved heptapeptide motif WRKYGQK, was first identified after the A. duranensis and A. ipaensis genomes had been released (Song et al., 2016b). Subsequently, aquaporin (AQP), basic/helix-loop-helix (bHLH), basic leucine zipper (bZIP), EXP (expansin), heat shock transcription factor (HSF), lipoxygenase (LOX), mildew resistance locus (MLO), nucleotide-binding sit–leucine-rich repeat (NBS–LRR), and phosphatidyl ethanolamine-binding protein (PEBP) gene families were identified in the A. duranensis (V14167) and A. ipaensis (K30076) genomes (Rispail and Rubiales, 2016; Song et al., 2016a, 2017; Gao et al., 2017; Guimaraes et al., 2017; Wang et al., 2017, 2019; Jin et al., 2019; Shivaraj et al., 2019) (Table S1). Growth-regulating factor (GRF) and NBS–LRR gene families were identified in the A. hypogaea cv. Tifrunner genome (Song et al., 2019; Zhao et al., 2019) (Table S1). However, different methods and strategies were used for the identification of gene families in Arachis.
Identification Method of Gene Families in ARACHIS
At least three methods can be used to identify the members of a gene family. The first method identifies members based on gene annotations. The gene annotation that was generated based on reference genomes was added to the gene name. A gene family was identified using each gene name. This method requires more time when the larger genome is used. In addition, if the gene annotation is wrong, false-positive sequences emerge. The second method identifies members based on local BLAST (PSI-BLAST) or searches tool data from a public database (i.e., PeanutBase). Query sequences always originate from Arabidopsis thaliana, Medicago truncatula, and Glycine max. This method may lose particular gene family members because of species-specific genes. However, this method plays an important role for the identification of gene families with non-canonical domains. The third method identifies members based on a hidden Markov model (HMM) using the HMMER program (Finn et al., 2011). The HMM file was generated by a gene family from various organisms. HMM-based methods can provide an even better representation of gene families and allow the identification of more distant family members.
A total of 12 gene families with canonical domains have been identified in Arachis (Rispail and Rubiales, 2016; Song et al., 2016a, 2017, 2019; Gao et al., 2017; Guimaraes et al., 2017; Wang et al., 2017, 2019; Jin et al., 2019; Shivaraj et al., 2019; Zhao et al., 2019). However, researchers used different methods to identify members among these gene families, specifically BLAST-based (four gene families) and HMM-based (eight gene families) methods (Table S1). Previous studies have demonstrated that more WRKY gene family members could be identified using the HMM-based method than the BLAST-based method in legumes (Song et al., 2018). To evaluate this result in various Arachis gene families, four gene families (AQP, EXP, MLO, and GRF) that were detected using the BLAST-based method in previous studies were re-identified using a HMM-based method. Previous studies identified gene families using different E-value thresholds (Table S2). If a smaller E-value was set, a smaller number of gene family members was obtained in the BLAST-based and HMM-based methods. For the PSI-BLAST and HMM programs, the default E-value parameter was 10. To compare the number of identified gene family members that used BLAST-based and HMM-based methods, this study used an E-value of 10 to re-identify the above-mentioned gene family members in Arachis. To verify the gene family domain, the obtained sequences were submitted to the Pfam database. The sequence was considered a gene family member if it contained a gene family domain. The obtained results showed that more members were identified using the HMM-based and BLAST-based method with an E-value of 10 than previous studies that used the BLAST-based method with an E-value below 10 among the above-mentioned four gene families (Figure 1 and Table S2). All members from the BLAST-based method were found in the HMM-based methods (Figure 1 and Table S3). In addition to this, compared with the BLAST-based method, the HMM-based method can identify a stable number of gene family members under an E-value of 10 in Arachis. Using A. thaliana, Orazy sativa, and G. max AQP and GRF gene family members to query against the Arachis genome for identification of a corresponding gene family in BLAST-based method, the same number of gene family members were detected using both the HMM-based and BLAST-based methods in AQP. However, a larger number of gene family members was detected using the HMM-based method than that when the BLAST-based method was used in GRF. In MLO, A. thaliana MLO was used as query sequence to identify gene family members in A. duranensis and A. ipaensis. The results showed that the same number of gene family members was detected using both HMM-based and BLAST-based methods. Nevertheless, more false positive sequences were found in BLAST-based method rather than HMM-based method (Table S4). To obtain more gene family members, multiple queries from different plants were considered when the BLAST-based method was used to identify gene families. However, if using the HMM-based method to identify gene families, the query sequence only selected the HMM file. Therefore, the HMM-based method is rapid and accurate. In summary, this study proposes that the best way to identify gene families in Arachis is the HMM-based method.
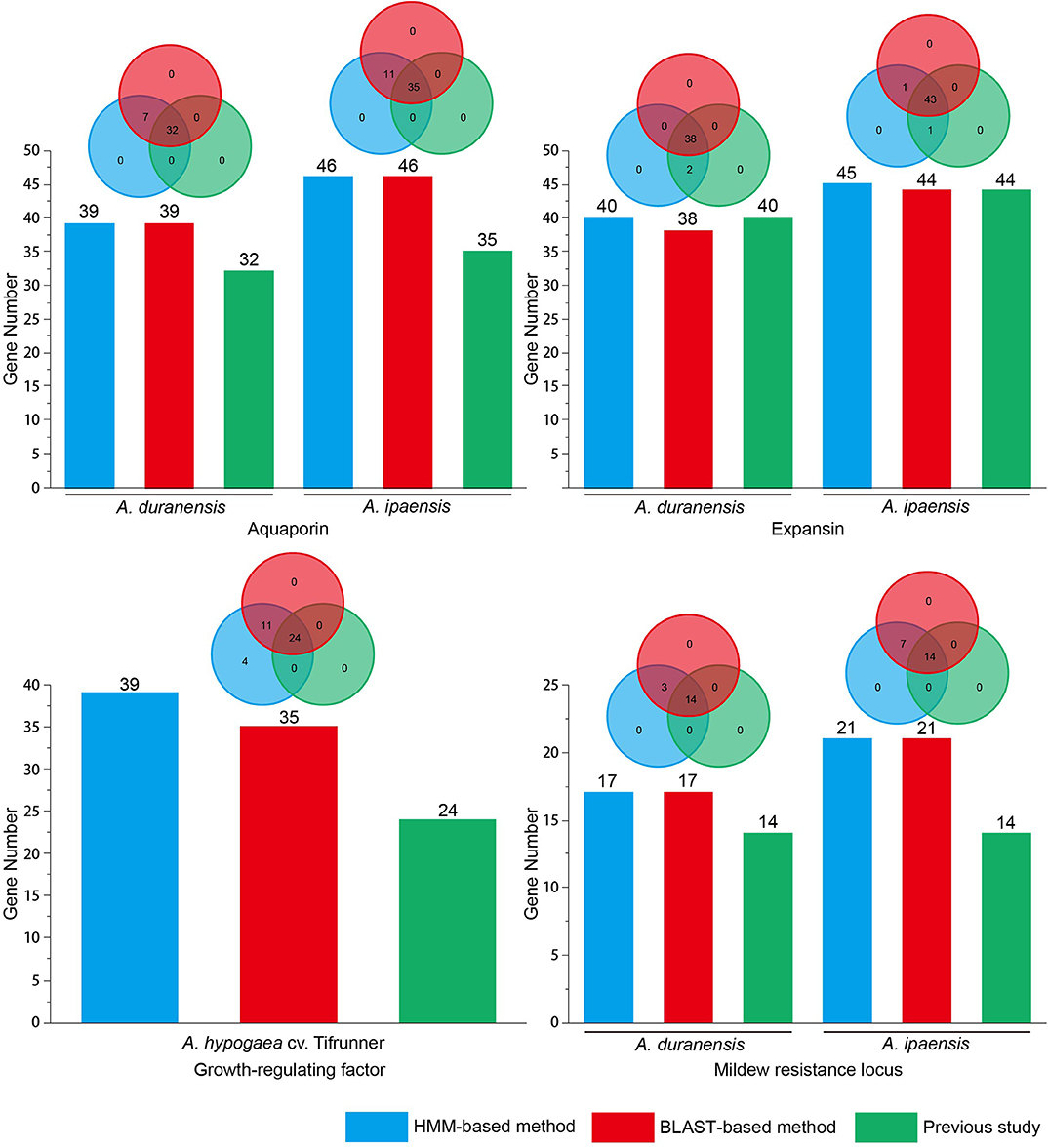
Figure 1. Identification of gene families in Arachis using HMM-based and BLAST-based method. Previous aquaporin data from Shivaraj et al. (2019); Previous expansin data from Guimaraes et al. (2017); Previous growth-regulating factor data from Zhao et al. (2019); Previous mildew resistance locus data from Rispail and Rubiales (2016).
Nomenclature for ARACHIS Gene Family Members
The nomenclature for Arachis gene family members could be classified into three types (Table S1). In the Arachis expansin gene family, A. thaliana expansin was used as reference (Guimaraes et al., 2017). In brief, the nomenclature for A. thaliana expansin was completed based on a chronological order of their discovery and phylogenetic tree (Kende et al., 2004). Synteny was constructed between Arachis and A. thaliana expansin. In four gene families (bHLH, LOX, and PEBP in A. duranensis and A. ipaensis; and NBS–LRR in A. hypogaea cv. Tifrunner), no nomenclature was allotted for members of gene families. The sequencing ID was used as gene name. In seven gene families (AQP, bZIP, HSF, NBS–LRR, MLO, and WRKY in A. duranensis and A. ipaensis; and GRF in A. hypogaea cv. Tifrunner), the nomenclature for members was defined by their chromosomal order. Arachis duranensis, A. ipaensis, and A. hypogaea cv. Tifrunner were referred to as Ad, Ai, and Ah, respectively. Following this procedure, the gene family name was listed and the number was then assigned based on the gene location in chromosomal order (e.g., AdWRKY1 and AdWRKY2). However, if a new member was found after the nomenclature had been assigned to a given gene family, the gene order of the new member should come after the last number of the legacy version.
Identification of Duplicated Genes in ARACHIS Gene Families
Gene duplication is one of the driving forces of evolution and is a potential strategy for the adaptation to environmental change (Panchy et al., 2016; Van de Peer et al., 2017). To date, nine gene families were used to conduct homolog (paralog and ortholog) relationship analysis (Table S1). However, different methods were used to identify homology in Arachis, including phylogenetic tree, BLAST-based methods, and synteny relationship methods (Rispail and Rubiales, 2016; Song et al., 2016a,b, 2017, 2019; Guimaraes et al., 2017; Wang et al., 2017, 2019; Jin et al., 2019). Although these methods have been used to identify homologs in many studies, detailed parameters need to be listed. For example, which model was used and which bootstrap was credible for clades in the phylogenetic tree? Which threshold value was set for the synteny analyses? This paper recommends that researchers should consider using the BLAST-based homolog identification method in Arachis because this method has been verified for the identification of homologs in the cultivated peanut (Clevenger et al., 2016; Bertioli et al., 2019; Chen et al., 2019; Zhuang et al., 2019). The following evaluation criteria were used as thresholds to determine homology: (1) alignment coverage exceeding 80% of the two sequences; (2) identity > 80%; and (3) E-value ≤ 1E−10.
Gene completeness is a crucial factor that affects evolutionary analysis. Confusing results can be obtained when partial sequences are used in gene structure analysis because of the potential loss of introns and exons. In addition, selection pressure cannot be identified when partial homolog sequences are used. Therefore, it is suggested that full-length sequences of Arachis gene family members should be used for the evolutionary analyses. In addition to this, it is also worth noting that pseudogenes were identified during analysis of gene families. Although pseudogenes may play a crucial role in plant development and response to stress, most pseudogenes cannot code for proteins or loss of the original function. Therefore, pseudogenes were excluded when the selective pressures were estimated. In A. duranensis and A. ipaensis, CDSs with premature codons were reported in MLO, NBS–LRR, and WRKY gene families, which have been considered pseudogenes (Rispail and Rubiales, 2016; Song et al., 2016b, 2017).
qRT-PCR Primer Design for ARACHIS Gene Families
The cultivated peanut is allotetraploid and contains many homologs. In addition, the members of gene families contain conserved sequences. Therefore, qRT-PCR primers are difficult to design because of non-specific amplification. Before the cultivated peanut genome was released, qRT-PCR primers were designed using the sum of A. duranensis and A. ipaensis sequences as cultivated peanut genome (Song et al., 2016a, 2017). Researchers focused on a problem to avoid the amplification of homologous sequences when designing the qRT-PCR primers in Arachis NBS–LRR and LOX gene families (Song et al., 2016a, 2017). Until now, the cultivated peanut genome can be used to study the expression of gene families. Future study has to carefully design the qRT-PCR primers to avoid non-specific amplification. The qRT-PCR primers are designed using the CDS with untranslated region (UTR) sequence because the UTR contained non-conserved sequences. Non-conserved regions are identified using multiple sequence alignment before designing the qRT-PCR primers. The Beacon Designer program was used for designing qRT-PCR primers. Beacon Designer can upload the genome sequence as a database. When a pair of qRT-PCR primers is designed, the program searches the database and lists the amplified fragment. This function can help researchers to remove false-positive primers.
Conclusions
With the released Arachis genome sequence, more gene families can be identified and characterized. This study offers advice on gene family identification and characterization in Arachis. The HMM-based method can be used to identify members of a given gene family. Full-length sequences were used for evolutionary analysis. Homologs can be identified by a BLAST-based method. Non-specific amplification can be avoided in qRT-PCR.
Author Contributions
HS and YZ conceived the study. HS wrote the paper. HS and DY approved the final version.
Funding
This work was supported by the First Class Grassland Science Discipline Program of Shandong Province, China, and the Natural Science Foundation of Shandong Province, China (ZR2019QC017).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00525/full#supplementary-material
Table S1. Identification of 12 gene families in Arachis.
Table S2. Comparison of four Arachis gene families using HMM-based and BLAST-based methods. aThe E-value was set to 10 in both the HMM-based and BLAST-based methods.
Table S3. Gene names in the four Arachis gene families. aThe E-value was set to 10 in both the HMM-based and BLAST-based methods.
Table S4. False positive rates in HMM-based and BLAST-based methods.
References
Bertioli, D. J., Cannon, S. B., Froenicke, L., Huang, G., Farmer, A. D., Cannon, E. K. S., et al. (2016). The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. 48, 438–446. doi: 10.1038/ng.3517
Bertioli, D. J., Jenkins, J., Clevenger, J., Dudchenko, O., Gao, D., Seijo, G., et al. (2019). The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat. Genet. 51, 877–884. doi: 10.1038/s41588-019-0405-z
Chen, X., Li, H., Pandey, M. K., Yang, Q., Wang, X., Garg, V., et al. (2016). Draft genome of the peanut A-genome progenitor (Arachis duranensis) provides insights into geocarpy, oil biosynthesis, and allergens. Proc. Natl. Acad. Sci. U.S.A. 113, 6785–6790. doi: 10.1073/pnas.1600899113
Chen, X., Lu, Q., Liu, H., Zhang, J., Hong, Y., Lan, H., et al. (2019). Sequencing of cultivated peanut, Arachis hypogaea, yields insights into genome evolution and oil improvement. Mol. Plant. 12, 920–934. doi: 10.1016/j.molp.2019.03.005
Clevenger, J., Chu, Y., Scheffler, B., and Ozias-Akins, P. (2016). A developmental transcriptome map for allotetraploid Arachis hypogaea. Front. Plant. Sci. 7:1446. doi: 10.3389/fpls.2016.01446
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Gao, C., Sun, J., Wang, C., Dong, Y., Xiao, S., Wang, X., and Jiao, Z. (2017). Genome-wide analysis of basic/helix-loop-helix gene family in peanut and assessment of its roles in pod development. PLoS ONE 12:e0181843. doi: 10.1371/journal.pone.0181843
Guimaraes, L. A., Mota, A. P. Z., Araujo, A. C. G., de Alencar Figueiredo, L. F., Pereira, B. M., de Passos Saraiva, M. A., et al. (2017). Genome-wide analysis of expansin superfamily in wild Arachis discloses a stress-responsive expansin-like B gene. Plant Mol. Biol. 94, 79–96. doi: 10.1007/s11103-017-0594-8
Jin, H., Tang, X., Xing, M., Zhu, H., Sui, J., Cai, C., and Li, S. (2019). Molecular and transcriptional characterization of phosphatidyl ethanolamine-binding proteins in wild peanuts Arachis duranensis and Arachis ipaensis. BMC Plant Biol. 19:484. doi: 10.1186/s12870-019-2113-3
Kende, H., Bradford, K. J., Brummell, D. A., Cho, H. T., Cosgrove, D. J., Fleming, A. J., et al. (2004). Nomenclature for members of the expansin superfamily of genes and proteins. Plant Mol. Biol. 55, 311–314. doi: 10.1007/s11103-004-0158-6
Lu, Q., Li, H., Hong, Y., Zhang, G., Wen, S., Li, X., et al. (2018). Genome sequencing and analysis of the peanut B-genome progenitor (Arachis ipaensis). Front. Plant. Sci. 9:604. doi: 10.3389/fpls.2018.00604
Panchy, N., Lehti-Shiu, M., and Shiu, S. H. (2016). Evolution of gene duplication in plants. Plant Physiol. 171, 2294–2316. doi: 10.1104/pp.16.00523
Rispail, N., and Rubiales, D. (2016). Genome-wide identification and comparison of legume MLO gene family. Sci. Rep. 6:32673. doi: 10.1038/srep32673
Shivaraj, S. M., Deshmukh, R., Sonah, H., and Bélanger, R. R. (2019). Identification and characterization of aquaporin genes in Arachis duranensis and Arachis ipaensis genomes, the diploid progenitors of peanut. BMC Genomics 20:222. doi: 10.1186/s12864-019-5606-4
Song, H., Guo, Z., Hu, X., Qian, L., Miao, F., Zhang, X., and Chen, J. (2019). Evolutionary balance between LRR domain loss and young NBS-LRR genes production governs disease resistance in Arachis hypogaea cv. Tifrunner. BMC Genom. 20:844. doi: 10.1186/s12864-019-6212-1
Song, H., Sun, W., Yang, G., and Sun, J. (2018). WRKY transcription factors in legumes. BMC Plant Biol. 18:243. doi: 10.1186/s12870-018-1467-2
Song, H., Wang, P., Li, C., Han, S., Lopez-Baltazar, J., Zhang, X., et al. (2016a). Identification of lipoxygenase (LOX) genes from legumes and their responses in wild type and cultivated peanut upon Aspergillus flavus infection. Sci. Rep. 6:35245. doi: 10.1038/srep35245
Song, H., Wang, P., Li, C., Han, S., Zhao, C., Xia, H., et al. (2017). Comparative analysis of NBS-LRR genes and their response to Aspergillus flavus in Arachis. PLoS ONE 12:e0171181. doi: 10.1371/journal.pone.0171181
Song, H., Wang, P., Lin, J. Y., Zhao, C., Bi, Y., and Wang, X. (2016b). Genome-wide identification and characterization of WRKY gene family in peanut. Front Plant Sci. 7:534. doi: 10.3389/fpls.2016.00534
Van de Peer, Y., Mizrachi, E., and Marchal, K. (2017). The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424. doi: 10.1038/nrg.2017.26
Wang, P., Song, H., Li, C., Li, P., Li, A., Guan, H., et al. (2017). Genome-wide dissection of the heat shock transcription factor family genes in Arachis. Front Plant Sci. 8:106. doi: 10.3389/fpls.2017.00106
Wang, Z., Yan, L., Wan, L., Huai, D., Kang, Y., Shi, L., et al. (2019). Genome-wide systematic characterization of bZIP transcription factors and their expression profiles during seed development and in response to salt stress in peanut. BMC Genom. 20:51. doi: 10.1186/s12864-019-5434-6
Yin, D., Ji, C., Ma, X., Li, H., Zhang, W., Li, S., et al. (2018). Genome of an allotetraploid wild peanut Arachis monticola: a de novo assembly. GigaScience 7:giy066. doi: 10.1093/gigascience/giy066
Yin, D., Ji, C., Song, Q., Zhang, W., Zhang, X., Zhao, K., et al. (2019). Comparison of Arachis monticola with diploid and cultivated tetraploid genomes reveals asymmetric subgenome evolution and improvement of peanut. Adv. Sci. 28:1901672. doi: 10.1002/advs.201901672
Zhao, K., Li, K., Ning, L., He, J., Ma, X., Li, Z., et al. (2019). Genome-wide analysis of the growth-regulating factor family in peanut (Arachis hypogaea L.). Int. J. Mol. Sci. 20:4120. doi: 10.3390/ijms20174120
Keywords: Arachis, gene family, evolution, expression, homology
Citation: Zhang Y, Yin D and Song H (2020) Genome-Wide Identification and Characterization of Gene Families in Arachis: Methods and Strategies. Front. Genet. 11:525. doi: 10.3389/fgene.2020.00525
Received: 01 January 2020; Accepted: 30 April 2020;
Published: 27 May 2020.
Edited by:
Ray Ming, University of Illinois at Urbana-Champaign, United StatesReviewed by:
Cheng Sun, Chinese Academy of Agricultural Sciences, ChinaWeilong Hao, Wayne State University, United States
Copyright © 2020 Zhang, Yin and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Song, Ymlvc29uZ2h1aUBvdXRsb29rLmNvbQ==