 Daxing Xu†
Daxing Xu† Yongdun Xie†
Yongdun Xie† Huijun Guo
Huijun Guo Weiwei Zeng
Weiwei Zeng Hongchun Xiong
Hongchun Xiong Linshu Zhao
Linshu Zhao Jiayu GuShirong ZhaoYuping DingLuxiang Liu*
Jiayu GuShirong ZhaoYuping DingLuxiang Liu*- National Engineering Laboratory for Crop Molecular Breeding, Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, National Center of Space Mutagenesis for Crop Improvement, Beijing, China
The stems of cereal crops provide both mechanical support for lodging resistance and a nutrient supply for reproductive organs. Elongation, which is considered a critical phase for yield determination in winter wheat (Triticum aestivum L.), begins from the first node detectable to anthesis. Previously, we characterized a heavy ion beam triggered wheat mutant qd, which exhibited an altered stem elongation pattern without affecting mature plant height. In this study, we further analyzed mutant stem developmental characteristics by using transcriptome data. More than 40.87 Mb of clean reads including at least 36.61 Mb of unique mapped reads were obtained for each biological sample in this project. We utilized our transcriptome data to identify 124,971 genes. Among these genes, 4,340 differentially expressed genes (DEG) were identified between the qd and wild-type (WT) plants. Compared to their WT counterparts, qd plants expressed 2,462 DEGs with downregulated expression levels and 1878 DEGs with upregulated expression levels. Using DEXSeq, we identified 2,391 counting bins corresponding to 1,148 genes, and 289 of them were also found in the DEG analysis, demonstrating differences between qd and WT. The 5,199 differentially expressed genes between qd and WT were employed for GO and KEGG analyses. Biological processes, including protein-DNA complex subunit organization, protein-DNA complex assembly, nucleosome organization, nucleosome assembly, and chromatin assembly, were significantly enriched by GO analysis. However, only benzoxazinoid biosynthesis pathway-associated genes were enriched by KEGG analysis. Genes encoding the benzoxazinoid biosynthesis enzymes Bx1, Bx3, Bx4, Bx5, and Bx8_9 were confirmed to be differentially expressed between qd and WT. Our results suggest that benzoxazinoids could play critical roles in regulating the stem elongation phenotype of qd.
Introduction
Sessile plants produce a wide variety of chemical compounds to regulate their growth, stress resistance, and environmental response. Among all metabolic chemicals, benzoxazinoids represent a predominant family produced by numerous Poaceae family species including maize, rye, and wheat (Frey et al., 2009; Sue et al., 2011), and those compounds can account for 1% or more of the dry weight of crop seedlings (Zhou et al., 2018). Since benzoxazinoids are important compounds in crops, efforts to elucidate their biosynthetic pathway have been undertaken for more than 50 years (Makowska et al., 2015). Benzoxazinoid biosynthesis involves the chloroplast, endoplasmic reticulum, cytoplasm, and vacuole (Dutartre et al., 2012). It was also suggested that benzoxazinoids can be transported among different tissues (Ahmad et al., 2011). The biochemical process of benzoxazinoid synthesis in plants is catalyzed by enzymes encoded by the Bx genes Bx1-Bx9, while all the identified genes in this process have been mapped in maize, rye, and wheat (Makowska et al., 2015). Among the Bx genes, Bx1 is considered to be responsible for the first committed step of benzoxazinoid biosynthesis in chloroplasts (Tzin et al., 2017). Although the BX1 enzymes in maize and wheat exhibit little similarity in their amino acid sequences, they have comparable catalytic properties (Schullehner et al., 2008). The expression of the Bx1-Bx5 genes was considered to be co-regulated in hexaploid wheat (Nomura et al., 2005).
Benzoxazinoids in plants are traditionally characterized as crucial signaling molecules involved in the defense against herbivorous insects and pathogens (Niemeyer, 2009). Pathogen infection of plants can activate the benzoxazinoid biosynthesis pathway (Huffaker et al., 2011). In addition to pathogenic stress, the biosynthesis of benzoxazinoids can be regulated by a variety of abiotic factors, such as temperature, light conditions, photoperiod, drought, nitrogen status, plant hormones, and cultivation methods (Oikawa et al., 2002; Villagrasa et al., 2006; Niemeyer, 2009; Bakera et al., 2020). Benzoxazinoids are also constitutively produced in maize plants, even in the absence of a pathogen threat (Zhou et al., 2018), and they are observed to accumulate around the emerging roots of maize, possibly reducing pathogen infections (Park et al., 2004). Since there are multiple factors that could affect the biosynthesis of benzoxazinoids, these compounds may also be involved in the regulation of other plant developmental processes.
Wheat (Triticum aestivum L.) serves as an important food crop that provides nearly 55% of the carbohydrates and 20% of the calories consumed globally (Pfeifer et al., 2014). Increasing wheat yield potential remains one of the major breeding objectives for ensuring world food security (Foulkes et al., 2011). The stem elongation phase (SEP) is critical for final yield formation and has been proposed as an important site for improving yield potential in wheat (González et al., 2003; Borras-Gelonch et al., 2012). Based on floral development and the abortion process, the SEP can be dissected into seven stages, known as the terminal spikelet stage, white anther stage, green anther stage, yellow anther stage, tipping stage, heading stage, and anthesis stage (Guo et al., 2018a). During the SEP, the plant height increases quickly, and wheat floret primordia undergo a complicated developmental process. During these seven stages, 75 plant growth-associated traits were quantified (Guo et al., 2018b). The elongation of stems is regulated by various chemical components including plant hormones, such as GA and BR (Oliva et al., 2013; Claeys et al., 2014). Therefore, it was suggested that there may be multiple ways to regulate plant growth during the SEP to increase wheat yield potential (Guo et al., 2018b).
Along with the rapid development of sequencing technologies and the rapid decrease in unit price, transcriptomic analysis has become a powerful tool for elucidating the molecular mechanisms responsible for critical plant growth processes (Yu et al., 2016; Curci et al., 2017). Zhang et al. (2014) demonstrated distinct gene activation in wheat in response to pathogen infection. Using global transcriptome analysis, a gene co-expression regulation network and key genes involved in wheat grain development and heterosis were successfully characterized (Liu et al., 2018; Chi et al., 2019).
The wheat quick development mutant was characterized by a quicker stem elongation rate than that of wild-type plants (Zhang et al., 2016). In this study, through transcriptome analysis methods, we demonstrated that benzoxazinoids may play roles in regulating the stem elongation progress. Our results provide a new perspective for manipulating wheat stem growth to achieve better performance.
Materials and Methods
Plant Materials and Sample Preparation
The quick development mutant (qd) and its wild type Lunxuan987 were the same as previously described (Zhang et al., 2016). Briefly, we irradiated dry seeds of Lunxuan987 with 40 Gy of 7Li ion-beam, and screened the mutant exhibiting an altered stem elongation pattern from the M2 generation. Every generation of both wild-type and qd mutant plants were covered by paper bags during the anthesis period to ensure their self-pollination. The WT and qd seeds were sown in the experimental field of the Institute of Crop Sciences of Chinese Academy of Agricultural Sciences with normal cultivation management. Both qd and wild-type plants were planted in 20 rows 1.5 m in length. Thirty plants were uniformly planted in each row.
When the plant height difference between WT and qd reached its maximum value at the booting stage, the shoot tip meristem parts were collected as samples. We prepared three biological replicates for both wild-type plants (named WT-1, WT-2, and WT-3) and qd plants (named qd-1, qd-2, and qd-3) for RNA isolation, and each replicate contained 15 individual stems from five plants with uniform phenotypes.
RNA Isolation and Library Construction
Total RNA was extracted using the RNeasy® plant Mini Kit (QIAGEN, Germany) according to the manufacturer’s instructions. After the total RNA was extracted, the residual DNA was digested with DNase I (TaKaRa, Japan). Next, the total RNA was purified by an RNA cleanup kit (TIANGEN, China). The purity, concentration, and integrity of the RNA samples were analyzed by the Nanodrop, Qubit 2, and Agilent 2,100 methods, respectively, as described previously (Xiong et al., 2020).
To construct libraries, we first used oligo(dT) magnetic beads to enrich the mRNA, and then the mRNA fragments were processed using a fragmentation buffer. Then, six-mer random primers were used to synthesize first-strand cDNA. Buffer, dNTPs, RNaseH, and DNA polymerase I were used to synthesize second-strand cDNA. AMPure XP breads were employed to purify cDNA. Next, the purified double-stranded cDNA was subjected to end repair, tailing, and ligation to a sequencing linker followed by the use of AMPure XP beads for fragment size selection and PCR enrichment to obtain a cDNA library. The purified double-stranded cDNA was subsequently subjected to terminal repair, dA tailing, and adaptor ligation. A total of six cDNA libraries were constructed.
Transcriptome Sequencing and Assembly
The libraries were sequenced as 150-bp paired-end reads using the Illumina high-throughput sequencing platform. After the sequencing linker was removed, the primer sequence was removed, and the low-quality reads were filtered out, a total of 53.73 Gb of clean data were obtained. The clean data of each sample were more than 7.49 Gb, and the percentage of Q30 bases was not less than 94.90%. The datasets presented in this study can be found in online repositories. The names of the repositories and accession number can be found at: https://www.ncbi.nlm.nih.gov/sra/; PRJNA682455.
Using the TopHat2 software, clean reads were compared to the wheat reference genome (IWGSC RefSeq v1.0, http://www.wheatgenome.org/). The results from TopHat 2 were spliced using the Cufflinks software, and the stitching results were compared with the original genome annotation information.
Identification and Annotation of Differentially Expressed Genes and Differential Exon Usage
FPKM (fragments per kilobase of transcript per million fragments mapped) was used as an indicator of transcript or gene expression levels. DEGs were identified using the DESeq2 package. The screening criteria that we used were as follows: the ratio of fold change (FC) was equal to or greater than 2 in the two groups (wild-type vs. qd), and the FDR was less than 0.01. To identify differential exon usage (DEUs), the DEXSeq package in R was used for analysis. We used FDR < 0.01 as a screening standard for counting bins to control false positives. After combining the genes from DEGs and DEUs, and setting FDR to less than 0.05, we used OmicShare tools1 to detect GO terms and KEGG pathways that were significantly enriched in the genome-wide context. A hypergeometric test was employed to define the significance of enrichment analysis for GO categories.
Verification of Gene Expression
To confirm the RNA-Seq results, we selected several upregulated and downregulated genes for qRT-PCR validation. After RNA extraction, genomic DNA was removed using DNase I (TaKaRa, Japan) and purified with an RNA clean Kit (TIANGEN, China). Reverse transcription was performed using an iScriptTM cDNA Synthesis Kit (Bio-Rad, America), and SsoFast™ EvaGreen® Supermix reagents (Bio-Rad, America) were used in a Bio-Rad CFX96 real-time fluorescence quantitative PCR instrument. qRT-PCR was performed with the following protocol: 95°C for 3 min, followed by 40 cycles of 95°C for 30 s, 59°C for 30 s, and 72°C for 10 s. Actin was used as a reference gene, and relative quantification was performed using the 2−ΔΔCt method. The primers for actin were as follows: forward, 5'-GTAGGAAATGGCTGACGGTG-3' and reverse, 5'-ATGCTAGGGAAAACAGCCCT-3'. The primers used to verify benzoxazinoid synthesis gene expression are listed in Supplementary Table 1.
Results
Clean Reads Data From Wild-Type and qd Plants
From the transcriptome sequencing data, we obtained at least 40.87 M of clean reads for each wild-type and qd sample. At least 36.61 M of unique mapped reads were detected in our samples, and the unique ratio ranged from 79.47 to 80.95, as listed in Table 1.
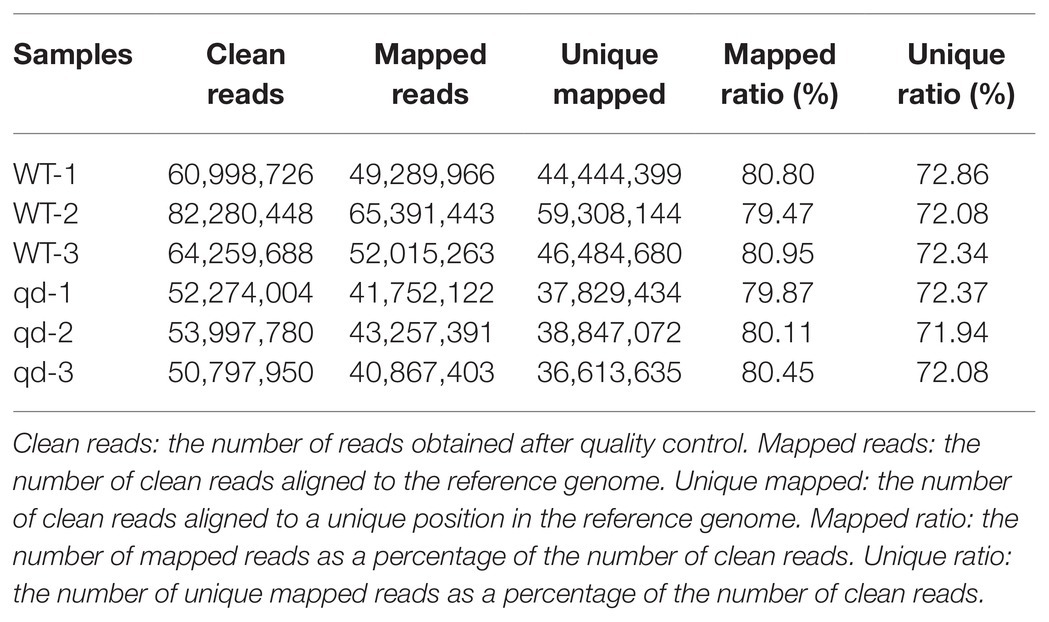
Table 1. Comparison of clean reads and the reference genome.
Identification of DEGs and DEUs
In total, we identified 4,340 DEGs (listed in Supplementary Table 2) between wild-type and qd plants. Among these genes, 2,462 DEGs showed downregulated expression levels in qd than in wild-type plants, and 1878 DEGs showed upregulated expression levels in qd than in wild-type plants, as shown in Figure 1A. By using DEXSeq, we identified 2,931 counting bins (listed in Supplementary Table 3), representing 1,148 genes. However, only 289 genes were detected in both DEGs and DEUs, as shown in Figures 1B, 2 and Supplementary Table 4. When we combined the results from DEGs and DEUs, 5,199 genes were identified that could be utilized in GO and KEGG analyses.
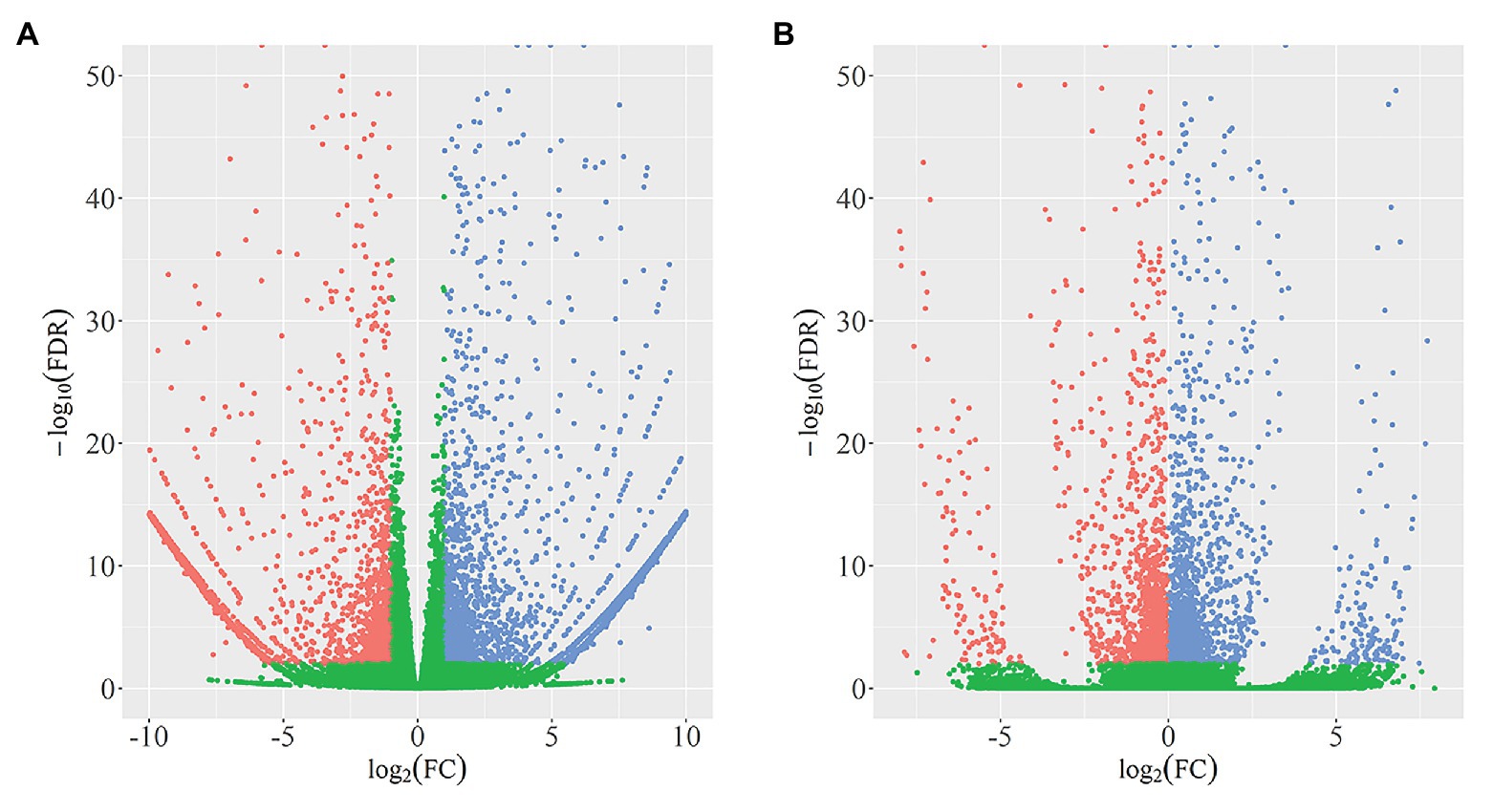
Figure 1. Identification and distribution of differentially expressed genes (DEGs; A) and differential exon usage (DEUs; B). Red dots represent DEGs or DEUs with upregulated expression levels in qd, blue dots represent DEGs or DEUs with downregulated expression levels in qd, green dots represent genes or exons without significant differences in expression levels between wild-type and qd.
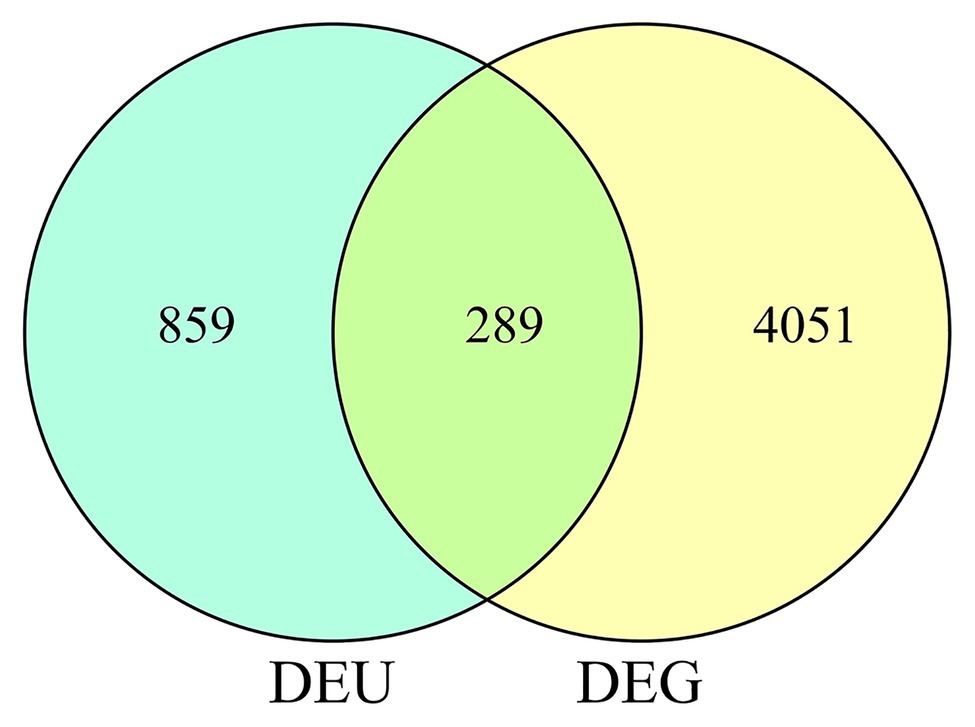
Figure 2. DEUs and DEGs corresponding to differential genes.
Go Term Analysis
The top GO biological process enrichment analysis showed that the differentially expressed genes were primarily concentrated in the formation of protein-DNA and other complex macromolecules in the nucleus, as shown in Figure 3 and listed in Supplementary Table 5. The terms included protein-DNA complex subunit organization, protein-DNA complex assembly, nucleosome organization, nucleosome assembly, chromatin assembly, and so on.
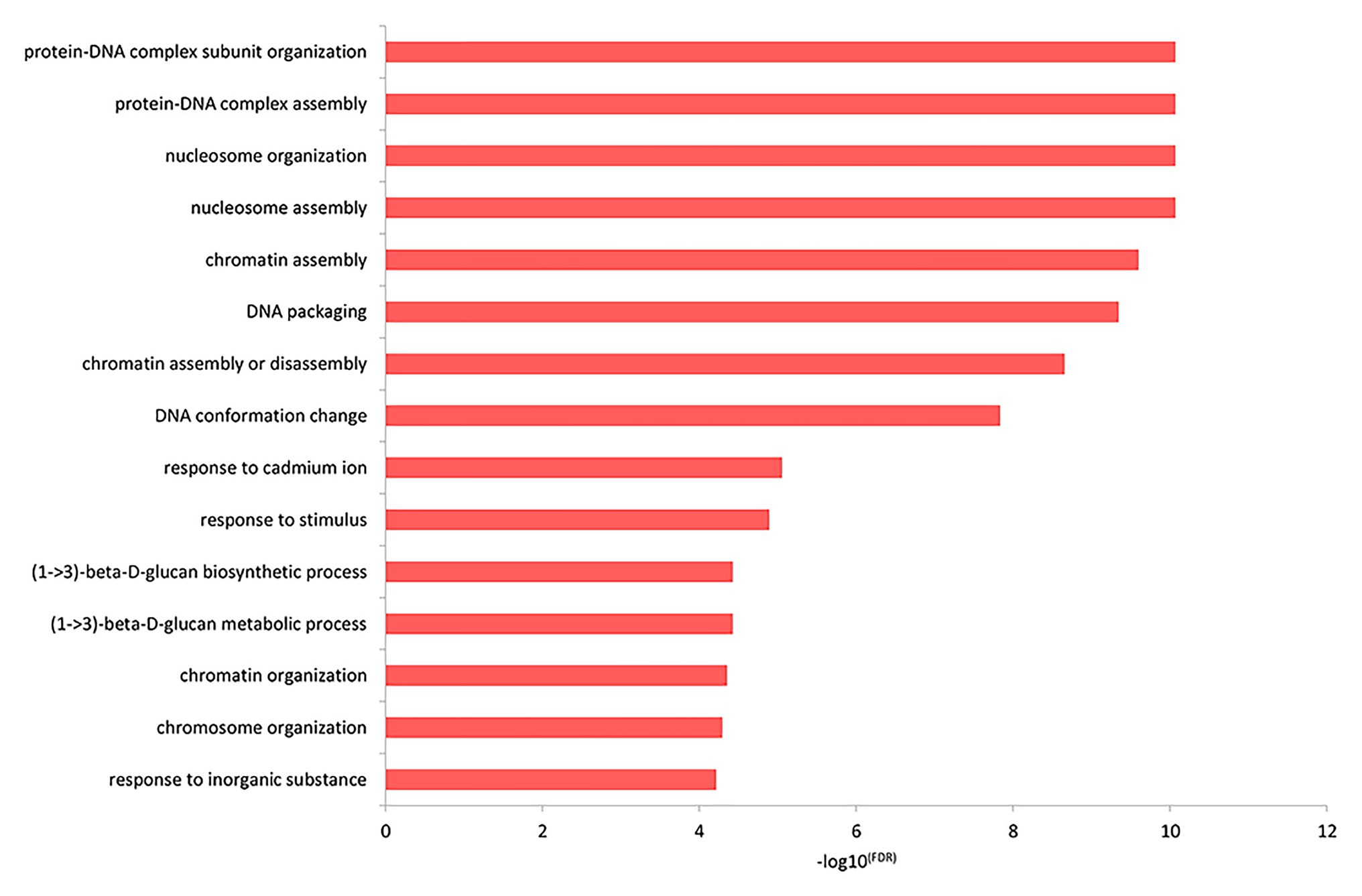
Figure 3. The functional enrichment of the differentially expressed genes in the top GO biological processes.
The results of the top GO molecular functional enrichment analysis also showed that nucleic acid-protein binding terms were significantly enriched, as shown in Figure 4 and listed in Supplementary Table 5. The top GO cell component enrichment analysis further indicated that chromatin, nucleosomes, and other terms were significantly enriched, as shown in Figure 5 and listed in Supplementary Table 5.
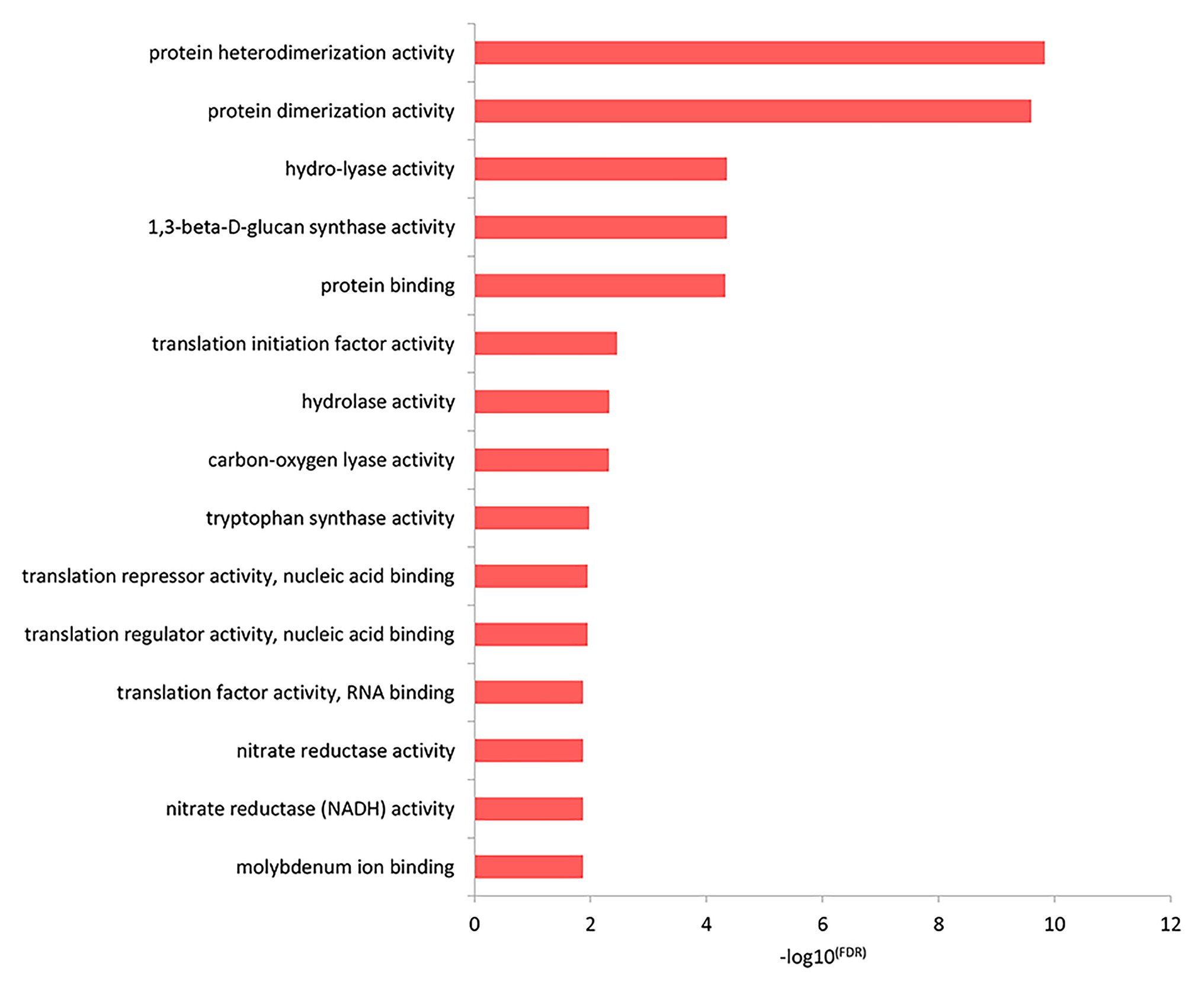
Figure 4. Molecular function enrichment analysis of differentially expressed genes in the top GO terms.
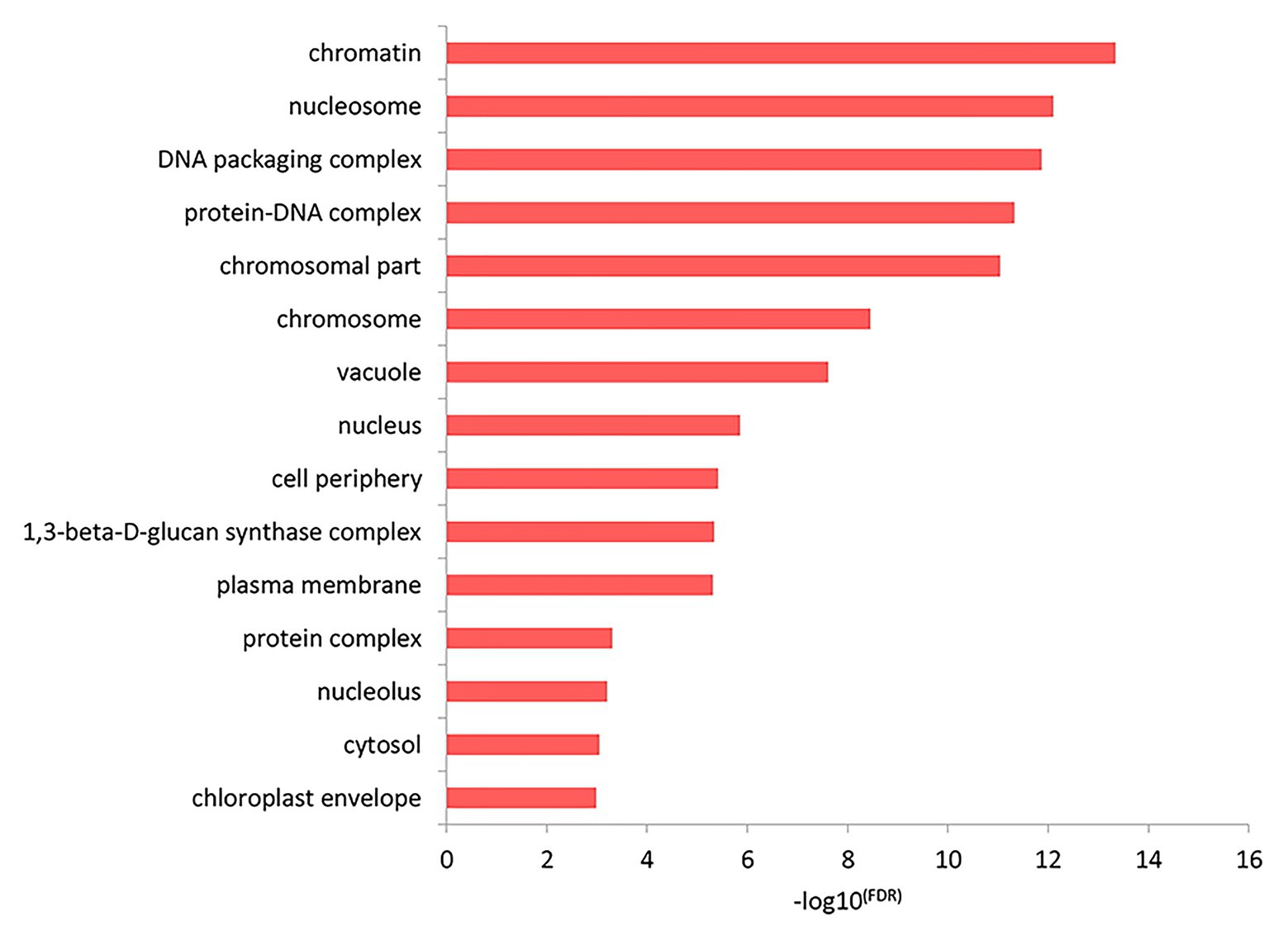
Figure 5. Cell component enrichment analysis of differentially expressed genes in the top GO terms.
Benzoxazinoid Biosynthesis Pathway Was Enriched by KEGG Analysis
The 5,199 differentially expressed genes were further analyzed by KEGG enrichment analysis. As shown in Figure 6, nine DEGs and one DEU were significantly enriched in the benzoxazinoid biosynthesis pathway with FDR < 0.01. These nine DEGs and one DEU were distributed among five enzymes of this pathway, namely, Bx1, Bx3, Bx4, Bx5, and Bx8_9. The Bx1 gene contained four of these nine DEGs, while Bx3, Bx4, and Bx5 contained one of the nine DEGs. The Bx8_9 gene contained two DEGs and one DEU in this analysis. As shown in Figure 7, the Bx1 gene contained both upregulated and downregulated DEGs expression, while the DEGs in the other four genes exhibited downregulated expression levels in qd plants than in the wild-type. The expression patterns of six of these nine DEGs were successfully verified by qPCR analysis, as shown in Figure 8.
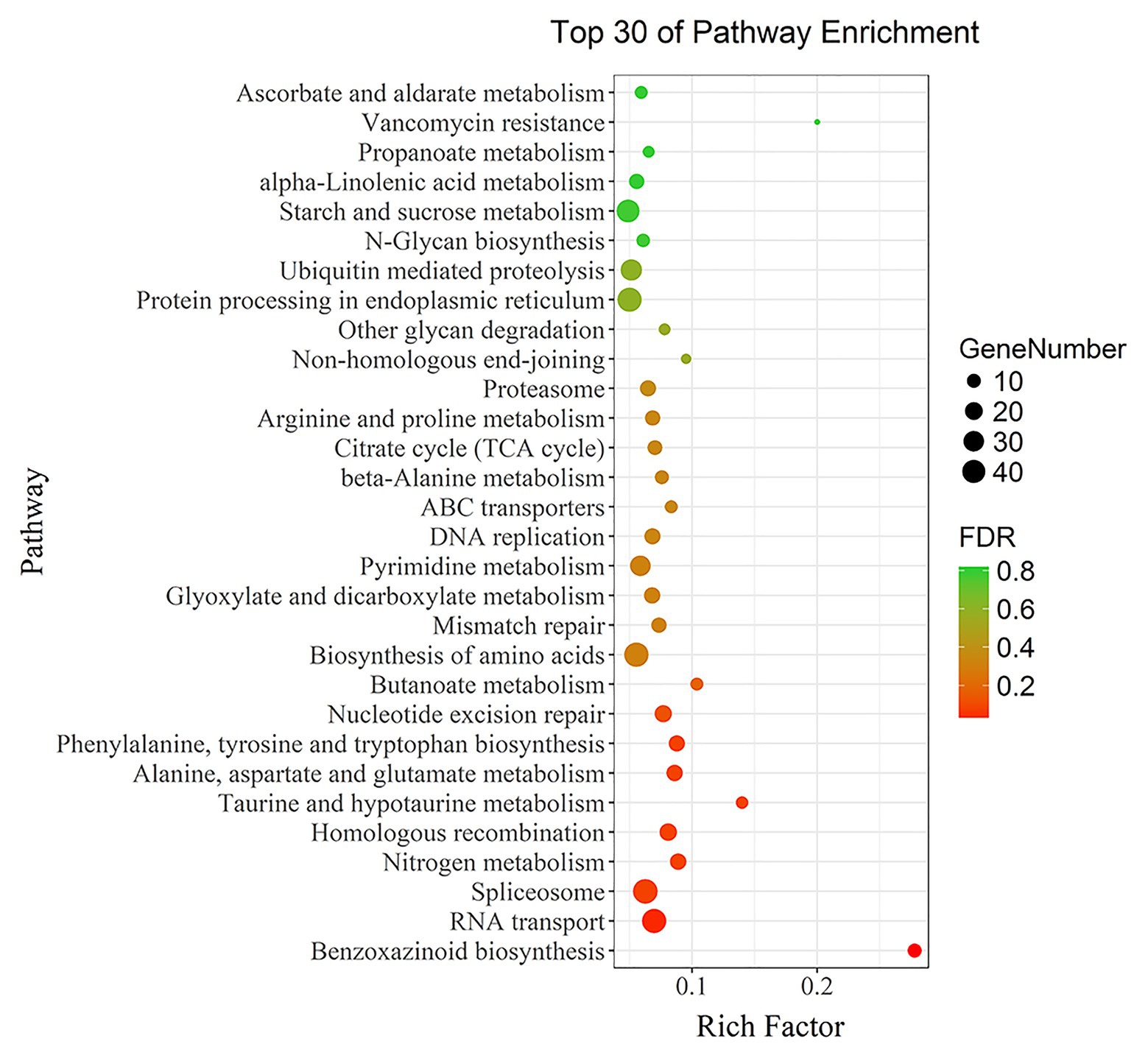
Figure 6. KEGG functional enrichment analysis of differentially expressed genes. Each point in the graph represents a pathway. The size of the spot represents the number of differentially expressed genes enriched in the pathway. The color of the point represents the significance of enrichment, where the red color represents a significantly enriched pathway.
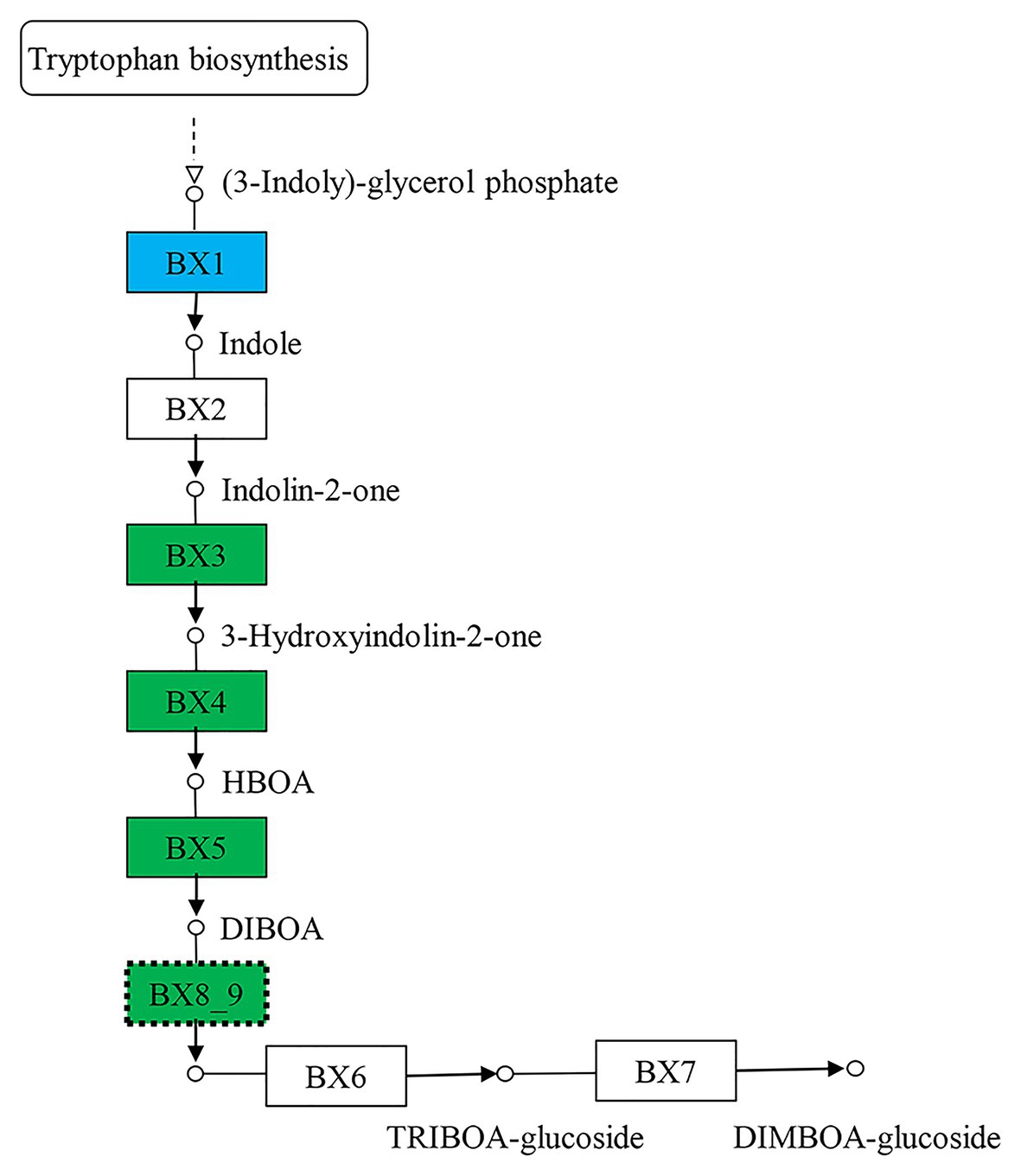
Figure 7. The biosynthesis pathway of benzoxazinoids. The boxes filled with green represent the enzymes containing significantly downregulated DEGs in qd. The box filled with blue represents the enzymes that contained both significantly upregulated and downregulated DEGs in qd. The dotted boxes represent enzymes containing DEUs.
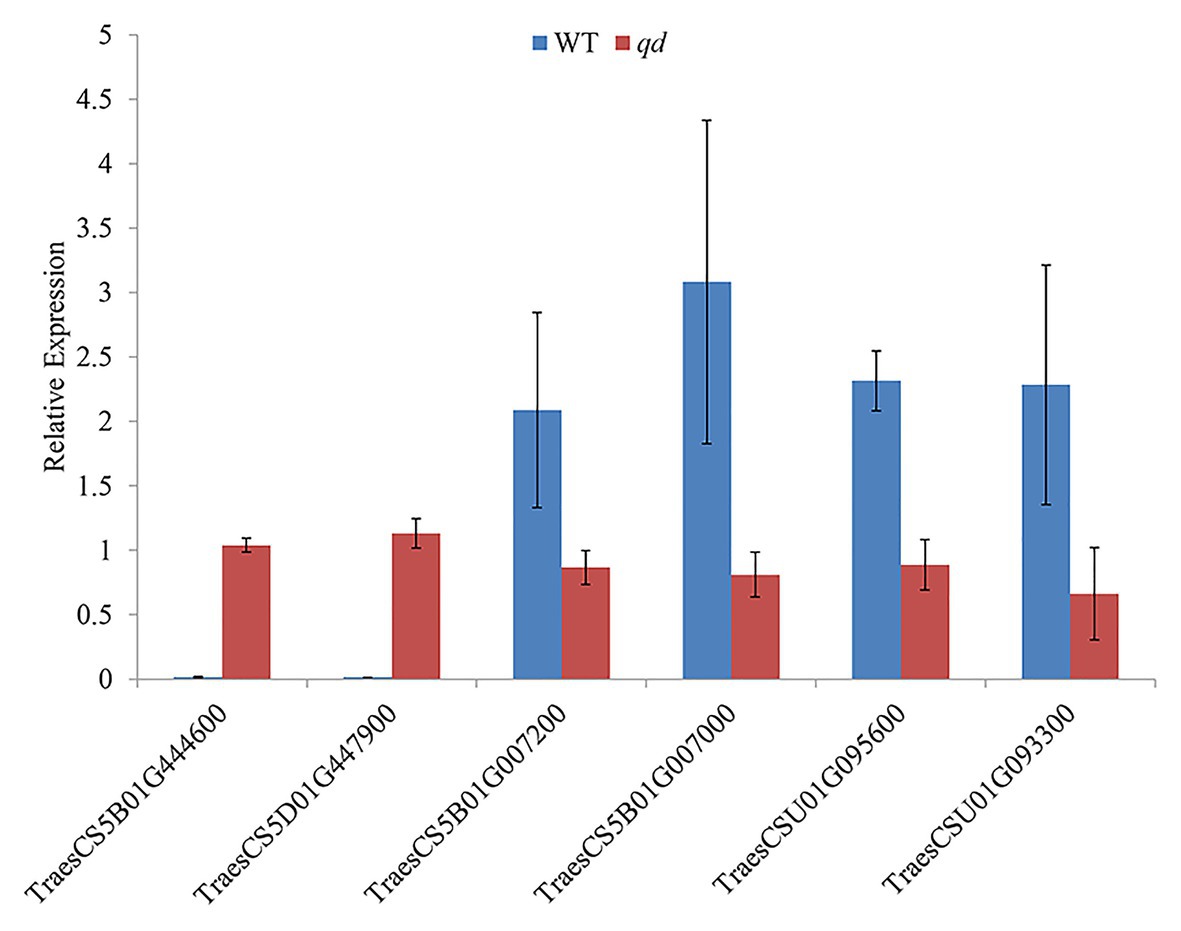
Figure 8. Verification of the DEGs in the benzoxazinoid biosynthesis pathway by qPCR. Bx1 contained two DEGs, namely, TraesCS5B01G444600 and TraesCS5D01G447900. Bx3 contained one DEG, namely, TraesCS5B01G007200. Bx5 contained one DEG, namely, TraesCS5B01G007000. Bx8_9 contained two DEGs, namely, TraesCSU01G095600 and TraesCSU01G093300.
Discussion
The stems of cereal crops provide mechanical support for lodging resistance and nutrient supply for reproductive organs. The development of crop stems is closely associated with final yield formation (Hirano et al., 2017). Along with stem elongation, inflorescence formation occurs (Gomez-Ariza et al., 2019), which has exhibited great dynamic changes in plant and floret growth-related traits in wheat during SEP (Guo et al., 2018a), and the duration of the SEP influences the number of fertile florets in wheat (Miralles et al., 2000). The wheat mutant qd studied in this research was obtained from ion beam mutagenesis, and compared with wild-type plants, the mutant showed an increased stem elongation rate (Zhang et al., 2016). Considerable efforts have been made to obtain a better understanding of the genetic and physical basis of stem elongation regulation in cereal crops. The overexpression of a lectin gene, OsJAC1, clearly suppressed rice stem elongation (Jiang et al., 2007). Stem elongation is also regulated by some transcription factors (Xiang et al., 2017; Gomez-Ariza et al., 2019). In recent years, several studies have helped to elucidate the genetic background that affects stem elongation traits, which provides useful information for manipulating stem growth (Borras-Gelonch et al., 2012; Kaur et al., 2017; Guo et al., 2018b). Although we have obtained considerable information on stem elongation regulation, the molecular mechanism governing stem elongation has not been thoroughly elucidated to date. The wheat mutant qd may be a useful material to characterize the regulation of stem elongation.
In this study, the benzoxazinoid biosynthesis pathway was identified by transcriptome sequencing analysis, which means that benzoxazinoids may play a role in regulating wheat stem elongation. Benzoxazinoids are traditional crucial elements for crop disease resistance (Ahmad et al., 2011; Tzin et al., 2017). Although benzoxazinoids are considered protective secondary metabolites, secondary metabolites are multifunctional and can function as potent regulators of plant growth (Erb and Kliebenstein, 2020). Benzoxazinoids have been indicated to play a role in improving plant tolerance to soil salinity (Makleit, 2005). In maize, the benzoxazinoid synthesis gene Bx12 was observed to affect both male and female flowering time (Romero Navarro et al., 2017).
One way in which benzoxazinoids regulate plant growth and development is through their interactions with plant hormones. Jasmonic acid was proven to regulate benzoxazinoid biosynthesis in plants (Oikawa et al., 2002). Benzoxazinoids and their breakdown products affect auxin-induced plant growth by regulating auxin signaling (Zhou et al., 2018). It has also been demonstrated that benzoxazinoids can inhibit gibberellin-induced α-amylase activity (Kato-Noguchi, 2008). We observed that gibberellins played an important role in regulating the phenotype of the qd mutant (Zhang et al., 2016). In this study, we also identified DEGs representing the CPS and GA20ox enzymes, as shown in Figure 9. For the qd mutant, the molecular mechanism by which benzoxazinoids are involved in regulating stem elongation in association with the gibberellin signaling pathway remains to be elucidated.
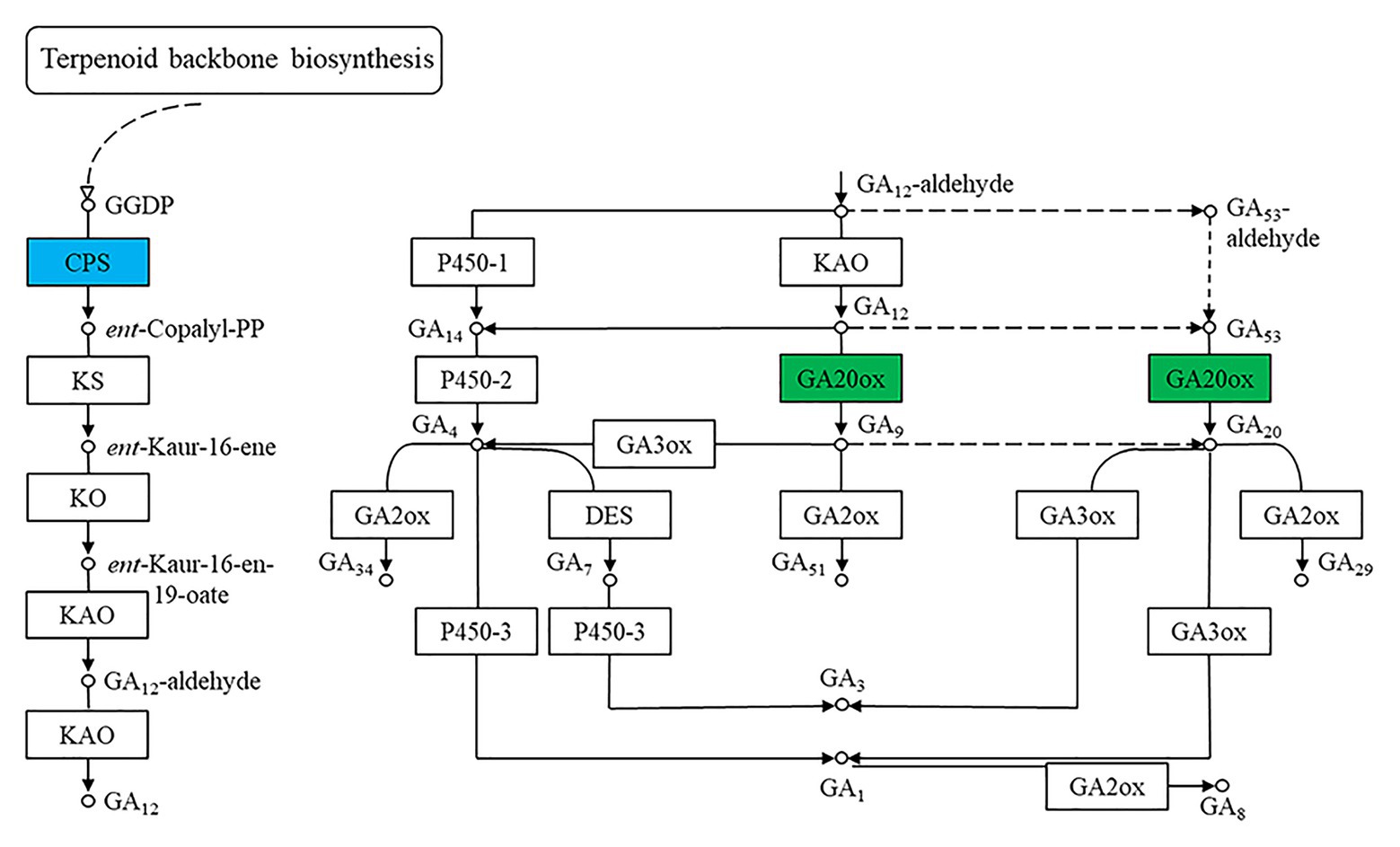
Figure 9. Differentially expressed genes of the gibberellin synthesis pathway. The boxes filled with green represent enzymes containing significantly downregulated DEGs in qd. The box filled with blue represents the enzymes that contained significantly upregulated and downregulated DEGs in qd.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/sra/; PRJNA682455.
Author Contributions
DX did most of the experiments of this study. YX screened the mutant and wrote the manuscript. HG and WZ did some experiments. HX, LZ, JG, SZ, and YD helped in experiments and writing. LL designed this project. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Project No. 11775304) and the National Key Research and Development Project (Project No. 2016YFD0102101).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.623861/full#supplementary-material
Footnotes
References
Ahmad, S., Veyrat, N., Gordon-Weeks, R., Zhang, Y., Martin, J., Smart, L., et al. (2011). Benzoxazinoid metabolites regulate innate immunity against aphids and fungi in maize. Plant Physiol. 157, 317–327. doi: 10.1104/pp.111.180224
Bakera, B., Święcicka, M., Stochmal, A., Kowalczyk, M., Bolibok, L., and Rakoczy-Trojanowska, M. (2020). Benzoxazinoids biosynthesis in rye (Secale cereale L.) is affected by low temperature. Agronomy 10:1260. doi: 10.3390/agronomy10091260
Borras-Gelonch, G., Rebetzke, G. J., Richards, R. A., and Romagosa, I. (2012). Genetic control of duration of pre-anthesis phases in wheat (Triticum aestivum L.) and relationships to leaf appearance, tillering, and dry matter accumulation. J. Exp. Bot. 63, 69–89. doi: 10.1093/jxb/err230
Chi, Q., Guo, L., Ma, M., Zhang, L., Mao, H., Wu, B., et al. (2019). Global transcriptome analysis uncovers the gene co-expression regulation network and key genes involved in grain development of wheat (Triticum aestivum L.). Funct. Integr. Genomics 19, 853–866. doi: 10.1007/s10142-019-00678-z
Claeys, H., De Bodt, S., and Inze, D. (2014). Gibberellins and DELLAs: central nodes in growth regulatory networks. Trends Plant Sci. 19, 231–239. doi: 10.1016/j.tplants.2013.10.001
Curci, P. L., Aiese Cigliano, R., Zuluaga, D. L., Janni, M., Sanseverino, W., and Sonnante, G. (2017). Transcriptomic response of durum wheat to nitrogen starvation. Sci. Rep. 7:1176. doi: 10.1038/s41598-017-01377-0
Dutartre, L., Hilliou, F., and Feyereisen, R. (2012). Phylogenomics of the benzoxazinoid biosynthetic pathway of Poaceae: gene duplications and origin of the Bx cluster. BMC Evol. Biol. 12:64. doi: 10.1186/1471-2148-12-64
Erb, M., and Kliebenstein, D. J. (2020). Plant secondary metabolites as defenses, regulators, and primary metabolites: the blurred functional trichotomy. Plant Physiol. 184, 39–52. doi: 10.1104/pp.20.00433
Foulkes, M. J., Slafer, G. A., Davies, W. J., Berry, P. M., Sylvester-Bradley, R., Martre, P., et al. (2011). Raising yield potential of wheat. III. Optimizing partitioning to grain while maintaining lodging resistance. J. Exp. Bot. 62, 469–486. doi: 10.1093/jxb/erq300
Frey, M., Schullehner, K., Dick, R., Fiesselmann, A., and Gierl, A. (2009). Benzoxazinoid biosynthesis, a model for evolution of secondary metabolic pathways in plants. Phytochemistry 70, 1645–1651. doi: 10.1016/j.phytochem.2009.05.012
Gomez-Ariza, J., Brambilla, V., Vicentini, G., Landini, M., Cerise, M., Carrera, E., et al. (2019). A transcription factor coordinating internode elongation and photoperiodic signals in rice. Nat. Plants 5, 358–362. doi: 10.1038/s41477-019-0401-4
González, F. G., Slafer, G. A., and Miralles, D. J. (2003). Floret development and spike growth as affected by photoperiod during stem elongation in wheat. Field Crop Res. 81, 29–38. doi: 10.1016/S0378-4290(02)00196-X
Guo, Z., Chen, D., and Schnurbusch, T. (2018a). Plant and floret growth at distinct developmental stages during the stem elongation phase in wheat. Front. Plant Sci. 9:330. doi: 10.3389/fpls.2018.00330
Guo, Z., Liu, G., Roder, M. S., Reif, J. C., Ganal, M. W., and Schnurbusch, T. (2018b). Genome-wide association analyses of plant growth traits during the stem elongation phase in wheat. Plant Biotechnol. J. 16, 2042–2052. doi: 10.1111/pbi.12937
Hirano, K., Ordonio, R. L., and Matsuoka, M. (2017). Engineering the lodging resistance mechanism of post-green revolution rice to meet future demands. Proc. Jpn. Acad. Ser. B: Phys. Biol. Sci. 93, 220–233. doi: 10.2183/pjab.93.014
Huffaker, A., Dafoe, N. J., and Schmelz, E. A. (2011). ZmPep1, an ortholog of Arabidopsis elicitor peptide 1, regulates maize innate immunity and enhances disease resistance. Plant Physiol. 155, 1325–1338. doi: 10.1104/pp.110.166710
Jiang, J.-F., Xu, Y.-Y., and Chong, K. (2007). Overexpression of OsJAC1, a lectin gene, suppresses the coleoptile and stem elongation in rice. J. Integr. Plant Biol. 49, 230–237. doi: 10.1111/j.1744-7909.2007.00428.x
Kato-Noguchi, H. (2008). Effects of four benzoxazinoids on gibberellin-induced α-amylase activity in barley seeds. J. Plant Physiol. 165, 1889–1894. doi: 10.1016/j.jplph.2008.04.006
Kaur, S., Zhang, X., Mohan, A., Dong, H., Vikram, P., Singh, S., et al. (2017). Genome-wide association study reveals novel genes associated with culm cellulose content in bread wheat (Triticum aestivum, L.). Front. Plant Sci. 8:1913. doi: 10.3389/fpls.2017.01913
Liu, Y. -J., Gao, S. -Q., Tang, Y. -M., Gong, J., Zhang, X., Wang, Y. -B., et al. (2018). Transcriptome analysis of wheat seedling and spike tissues in the hybrid Jingmai 8 uncovered genes involved in heterosis. Planta 247, 1307–1321. doi: 10.1007/s00425-018-2848-3
Makleit, P. (2005). Changes in cyclic hydroxamic acid content of various rye varieties for the effect of abiotic stress. Acta Biol. Szeged. 49, 103–104.
Makowska, B., Bakera, B., and Rakoczy-Trojanowska, M. (2015). The genetic background of benzoxazinoid biosynthesis in cereals. Acta Physiol. Plant. 37:176.
Miralles, D. J., Richards, R. A., and Slafer, G. A. (2000). Duration of the stem elongation period influences the number of fertile florets in wheat and barley. Funct. Plant Biol. 27, 931–940.
Niemeyer, H. M. (2009). Hydroxamic acids derived from 2-hydroxy-2H-1,4-benzoxazin-3(4H)-one: key defense chemicals of cereals. J. Agric. Food Chem. 57, 1677–1696. doi: 10.1021/jf8034034
Nomura, T., Ishihara, A., Yanagita, R. C., Endo, T. R., and Iwamura, H. (2005). Three genomes differentially contribute to the biosynthesis of benzoxazinones in hexaploid wheat. Proc. Natl. Acad. Sci. U. S. A. 102, 16490–16495. doi: 10.1073/pnas.0505156102
Oikawa, A., Ishihara, A., and Iwamura, H. (2002). Induction of HDMBOA-Glc accumulation and DIMBOA-Glc 4-O-methyltransferase by jasmonic acid in poaceous plants. Phytochemistry 61, 331–337. doi: 10.1016/s0031-9422(02)00225-x
Oliva, M., Farcot, E., and Vernoux, T. (2013). Plant hormone signaling during development: insights from computational models. Curr. Opin. Plant Biol. 16, 19–24. doi: 10.1016/j.pbi.2012.11.006
Park, W. J., Hochholdinger, F., and Gierl, A. (2004). Release of the benzoxazinoids defense molecules during lateral- and crown root emergence in Zea mays. J. Plant Physiol. 161, 981–985. doi: 10.1016/j.jplph.2004.01.005
Pfeifer, M., Kugler, K. G., Sandve, S. R., Zhan, B., Rudi, H., Hvidsten, T. R., et al. (2014). Genome interplay in the grain transcriptome of hexaploid bread wheat. Science 345:1250091. doi: 10.1126/science.1250091
Romero Navarro, J. A., Willcox, M., Burgueno, J., Romay, C., Swarts, K., Trachsel, S., et al. (2017). A study of allelic diversity underlying flowering-time adaptation in maize landraces. Nat. Genet. 49, 476–480. doi: 10.1038/ng.3784
Schullehner, K., Dick, R., Vitzthum, F., Schwab, W., Brandt, W., Frey, M., et al. (2008). Benzoxazinoid biosynthesis in dicot plants. Phytochemistry 69, 2668–2677. doi: 10.1016/j.phytochem.2008.08.023
Sue, M., Nakamura, C., and Nomura, T. (2011). Dispersed benzoxazinone gene cluster: molecular characterization and chromosomal localization of glucosyltransferase and glucosidase genes in wheat and rye. Plant Physiol. 157, 985–997. doi: 10.1104/pp.111.182378
Tzin, V., Hojo, Y., Strickler, S. R., Bartsch, L. J., Archer, C. M., Ahern, K. R., et al. (2017). Rapid defense responses in maize leaves induced by Spodoptera exigua caterpillar feeding. J. Exp. Bot. 68, 4709–4723. doi: 10.1093/jxb/erx274
Villagrasa, M., Guillamón, M., Labandeira, A., Taberner, A., Eljarrat, E., and Barceló, D. (2006). Benzoxazinoid allelochemicals in wheat: distribution among foliage, roots, and seeds. J. Agric. Food Chem. 54, 1009–1015. doi: 10.1021/jf050898h
Xiang, J., Tang, S., Zhi, H., Jia, G., Wang, H., and Diao, X. (2017). Loose Panicle1 encoding a novel WRKY transcription factor, regulates panicle development, stem elongation, and seed size in foxtail millet [Setaria italica (L.) P. Beauv.]. PLoS One 12:e0178730. doi: 10.1371/journal.pone.0178730
Xiong, H., Guo, H., Xie, Y., Gu, J., Zhao, L., Zhao, S., et al. (2020). Comparative transcriptome analysis of two common wheat varieties induced by 7Li-ion beam irradiation reveals mutation hotspot regions and associated pathways. Radiat. Phys. Chem. 170:108650. doi: 10.1016/j.radphyschem.2019.108650
Yu, K., Wang, X., Chen, F., Chen, S., Peng, Q., Li, H., et al. (2016). Genome-wide transcriptomic analysis uncovers the molecular basis underlying early flowering and apetalous characteristic in Brassica napus L. Sci. Rep. 6:30576. doi: 10.1038/srep30576
Zhang, N., Xie, Y. -D., Guo, H. -J., Zhao, L. -S., Xiong, H. -C., Gu, J. -Y., et al. (2016). Gibberellins regulate the stem elongation rate without affecting the mature plant height of a quick development mutant of winter wheat (Triticum aestivum L.). Plant Physiol. Biochem. 107, 228–236. doi: 10.1016/j.plaphy.2016.06.008
Zhang, H., Yang, Y., Wang, C., Liu, M., Li, H., Fu, Y., et al. (2014). Large-scale transcriptome comparison reveals distinct gene activations in wheat responding to stripe rust and powdery mildew. BMC Genomics 15:898. doi: 10.1186/1471-2164-15-898
Keywords: transcriptome, benzoxazinoids, stem elongation, mutant, wheat
Citation: Xu D, Xie Y, Guo H, Zeng W, Xiong H, Zhao L, Gu J, Zhao S, Ding Y and Liu L (2021) Transcriptome Analysis Reveals a Potential Role of Benzoxazinoid in Regulating Stem Elongation in the Wheat Mutant qd. Front. Genet. 12:623861. doi: 10.3389/fgene.2021.623861
Edited by:
Shuyu Liu, Texas A&M University System, United StatesReviewed by:
Waltram Second Ravelombola, University of Arkansas, United StatesLiu Dengcai, Sichuan Agricultural University, China
Copyright © 2021 Xu, Xie, Guo, Zeng, Xiong, Zhao, Gu, Zhao, Ding and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luxiang Liu, bGl1bHV4aWFuZ0BjYWFzLmNu
†These authors have contributed equally to this work