 Thayne Woycinck Kowalski1,2,3,4,5,6†
Thayne Woycinck Kowalski1,2,3,4,5,6† Gabriela Barreto Caldas-Garcia1,7†
Gabriela Barreto Caldas-Garcia1,7† Julia do Amaral Gomes1,2,3,4
Julia do Amaral Gomes1,2,3,4 Lucas Rosa Fraga3,4,8,9,10
Lucas Rosa Fraga3,4,8,9,10 Lavínia Schuler-Faccini1,2,4,10
Lavínia Schuler-Faccini1,2,4,10 Mariana Recamonde-Mendoza5,11
Mariana Recamonde-Mendoza5,11 Vanessa Rodrigues Paixão-Côrtes7*
Vanessa Rodrigues Paixão-Côrtes7* Fernanda Sales Luiz Vianna1,2,3,4,9,10*
Fernanda Sales Luiz Vianna1,2,3,4,9,10*- 1Post-Graduation Program in Genetics and Molecular Biology, PPGBM, Universidade Federal do Rio Grande do Sul, UFRGS, Porto Alegre, Brazil
- 2Laboratory of Medical Genetics and Evolution, Genetics Department, Universidade Federal do Rio Grande do Sul, UFRGS, Porto Alegre, Brazil
- 3Laboratory of Genomic Medicine, Center of Experimental Research, Hospital de Clínicas de Porto Alegre, HCPA, Porto Alegre, Brazil
- 4National Institute of Medical Population Genetics, INAGEMP, Porto Alegre, Brazil
- 5Bioinformatics Core, Hospital de Clínicas de Porto Alegre, HCPA, Porto Alegre, Brazil
- 6Centro Universitário CESUCA, Cachoeirinha, Brazil
- 7Post-Graduation Program in Biodiversity and Evolution, PPGBioEvo Institute of Biology, Universidade Federal da Bahia, UFBA, Salvador, Brazil
- 8Department of Morphological Sciences, Institute of Health Sciences, Universidade Federal do Rio Grande do Sul (UFRGS), Porto Alegre, Brazil
- 9Post-Graduation Program in Medical Science, Universidade Federal do Rio Grande do Sul, UFRGS, Porto Alegre, Brazil
- 10Teratogen Information System, SIAT, Medical Genetics Service, Hospital de Clínicas de Porto Alegre, HCPA, Porto Alegre, Brazil
- 11Institute of Informatics, Universidade Federal do Rio Grande do Sul, UFRGS, Porto Alegre, Brazil
The identification of thalidomide–Cereblon-induced SALL4 degradation has brought new understanding for thalidomide embryopathy (TE) differences across species. Some questions, however, regarding species variability, still remain. The aim of this study was to detect sequence divergences between species, affected or not by TE, and to evaluate the regulated gene co-expression in a murine model. Here, we performed a comparative analysis of proteins experimentally established as affected by thalidomide exposure, evaluating 14 species. The comparative analysis, regarding synteny, neighborhood, and protein conservation, was performed in 42 selected genes. Differential co-expression analysis was performed, using a publicly available assay, GSE61306, which evaluated mouse embryonic stem cells (mESC) exposed to thalidomide. The comparative analyses evidenced 20 genes in the upstream neighborhood of NOS3, which are different between the species who develop, or not, the classic TE phenotype. Considering protein sequence alignments, RECQL4, SALL4, CDH5, KDR, and NOS2 proteins had the biggest number of variants reported in unaffected species. In co-expression analysis, Crbn was a gene identified as a driver of the co-expression of other genes implicated in genetic, non-teratogenic, limb reduction defects (LRD), such as Tbx5, Esco2, Recql4, and Sall4; Crbn and Sall4 were shown to have a moderate co-expression correlation, which is affected after thalidomide exposure. Hence, even though the classic TE phenotype is not identified in mice, a deregulatory Crbn-induced mechanism is suggested in this animal. Functional studies are necessary, especially evaluating the genes responsible for LRD syndromes and their interaction with thalidomide–Cereblon.
Introduction
Since the 1960s, when thalidomide teratogenicity was discovered, many attempts have been tried to identify its molecular mechanisms, aiming to use this knowledge to avoid new cases of the embryopathy. However, fully understanding how thalidomide causes malformations remains an urgent challenge (Asatsuma-Okumura et al., 2019).
One of the many aspects that intrigue scientists is the variability of thalidomide teratogenesis among species (Table 1). The most commonly used animal models, rat and mouse, do not develop the classic thalidomide embryopathy (TE) phenotype, identified in a range of organisms including zebrafish (Siamwala et al., 2012), chicken (Therapontos et al., 2009), frogs (Fort et al., 2000), rabbits (Fabro et al., 1967), sea urchins (Reichard-Brown et al., 2009), and opossum (Sorensen et al., 2017). Mice were tested even in a dose of 4,000 mg/kg and did not develop TE, whereas humans are sensitive to its teratogenesis in a dose of 0.5 mg/kg (Cahen, 1966; Stephens and Fillmore, 2000). Other insensitive species are bushbabies, pigs, and cats (Fink et al., 2018). However, the original studies with bushbabies and pigs have a small sample (n = 4) (Jonsson, 1972; Butler, 1977), and experiments with pregnant cats treated with thalidomide resulted in fetal loss and a variety of cardiovascular anomalies in the offspring (Khera, 1975).
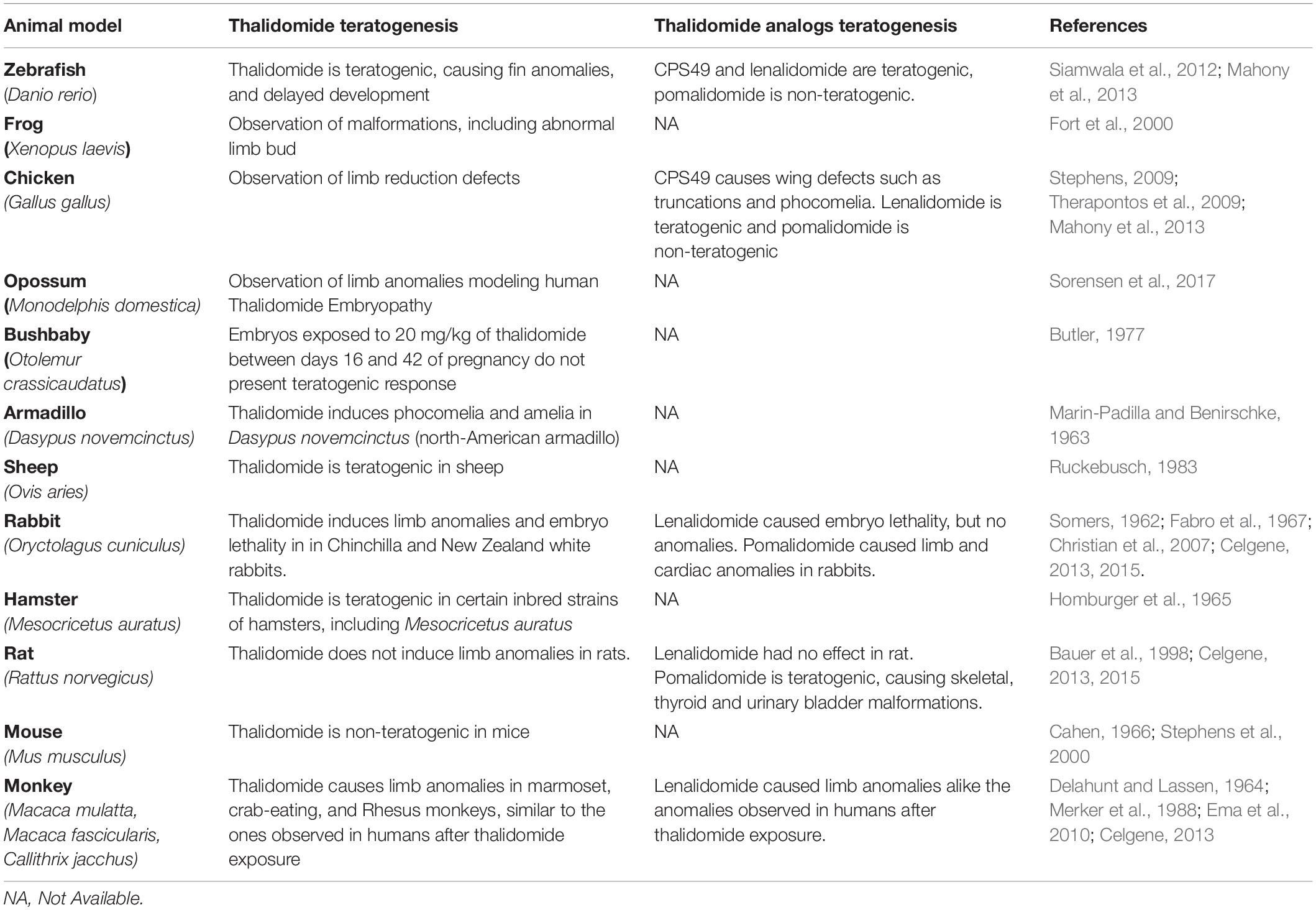
Table 1. Comparative table of the effects of thalidomide, and its analogs, exposure in different animal models.
Molecular mechanisms previously identified in thalidomide are different across species, such as the induction of oxidative stress (Hansen and Harris, 2004), driven by several genes including NFKB1, NFKB2, DKK1, GSTP1, and GSS. Other mechanisms for thalidomide teratogenesis have been proposed, such as anti-angiogenesis, which led to several studies evaluating the role of the vascular endothelium mainly expressing genes (KDR, VEGFA, and CDH5) and nitric oxide synthase genes NOS2 and NOS3 (Therapontos et al., 2009; Siamwala et al., 2012). All the studies have aimed to understand how the molecular disruptions directed by thalidomide could impact in limb development (Hansen and Harris, 2004; Therapontos et al., 2009; Ito et al., 2010; Siamwala et al., 2012). However, only recently, Donovan et al. (2018) and Matyskiela et al. (2018) elucidated an important mechanism that has helped in the understanding of this mystery: Cereblon, a protein which is a thalidomide primary target (Ito et al., 2010), mediates SALL4 degradation (Donovan et al., 2018; Matyskiela et al., 2018).
SALL4 is a transcription factor (TF) involved in limb development; in humans, loss-of-function mutations in SALL4 cause Duane-radial ray syndrome (Kohlhase et al., 2002), an autosomal recessive condition characterized by limb anomalies, similar to the ones observed in TE (Kohlhase et al., 2003). Because of the pattern of anomalies, TE is considered a phenocopy of other genetic syndromes, mainly characterized by limb reduction defects (Cassina et al., 2017), caused by mutations in ESCO2, TBX5, RBM8A, and RECQL4 genes (Li et al., 1997; Vega et al., 2005; Van Maldergem et al., 2006).
Recently, it was proposed that, in mice, variants in Crbn and Sall4 genes interrupt the degradation of Sall4 protein mediated by Cereblon–thalidomide (Donovan et al., 2018; Matyskiela et al., 2018). In this scenario, genetic alterations in both SALL4 and CRBN would be pivotal for thalidomide teratogenic activity (Gao et al., 2020). However, only a few species were evaluated in the mentioned research. Besides SALL4, other neosubstrates have recently been proposed, including limb development genes such as MEIS2 (Fischer et al., 2014) and TP63 (Asatsuma-Okumura et al., 2019).
Before it was identified as a thalidomide primary target (Ito et al., 2010), a Cereblon stop-codon mutation was implicated in a mild mental retardation autosomal recessive disorder (Higgins et al., 2004). Notwithstanding, its canonical function is related to a E3–ubiquitin–ligase complex, which comprises DDB1, ROC1, and CUL4A proteins as well; Cereblon acts as a substrate receptor, ubiquitinating proteins to be degraded (Angers et al., 2006). In mouse brains, Cereblon mRNA was detected especially in serotoninergic and adrenergic neurons, as well as in hippocampal and cerebellum regions (Aizawa et al., 2011). Worthy of note is that Crbn-null mice are viable and do not present major malformations (Rajadhyaksha et al., 2012; Lee et al., 2013); however, memory and learning deficits are apparent in these animals (Bavley et al., 2018). Because of Cereblon binding and the outstanding immunomodulatory capacity of thalidomide, the drug has been implicated in the treatment of neurodegenerative and neuropsychiatric disorders; however, thalidomide neuroimmunomodulatory mechanisms do not appear to be Cereblon-dependent and probably occur through TNFa mRNA degradation (Jung et al., 2021).
Notwithstanding, TE in humans is considered heterogeneous, because the phenotypic presentation is variable, an effect that is not explained only by SALL4 or CRBN variants (Gomes et al., 2019; Kowalski et al., 2020). Considering TE intra- and interspecific variability, the aim of this research was to perform a comparative analysis in the main proteins already associated with thalidomide mechanisms of action, across different species, affected or not by classic TE. We have evaluated 42 genes, including SALL4, regarding synteny, neighborhood, and protein conservation, which might be relevant in the understanding of TE divergences. Moreover, we also performed differential gene expression and co-expression analyses, focused in the mice to better comprehend the thalidomide action mechanisms in this species; we combined these bioinformatics analyses with systems biology strategies, to evaluate the main targets of thalidomide exposure in mice and rats.
Materials and Methods
A full description of the performed analyses is available in Supplementary File 1. Figure 1A presents a step-by-step scheme of the analysis.
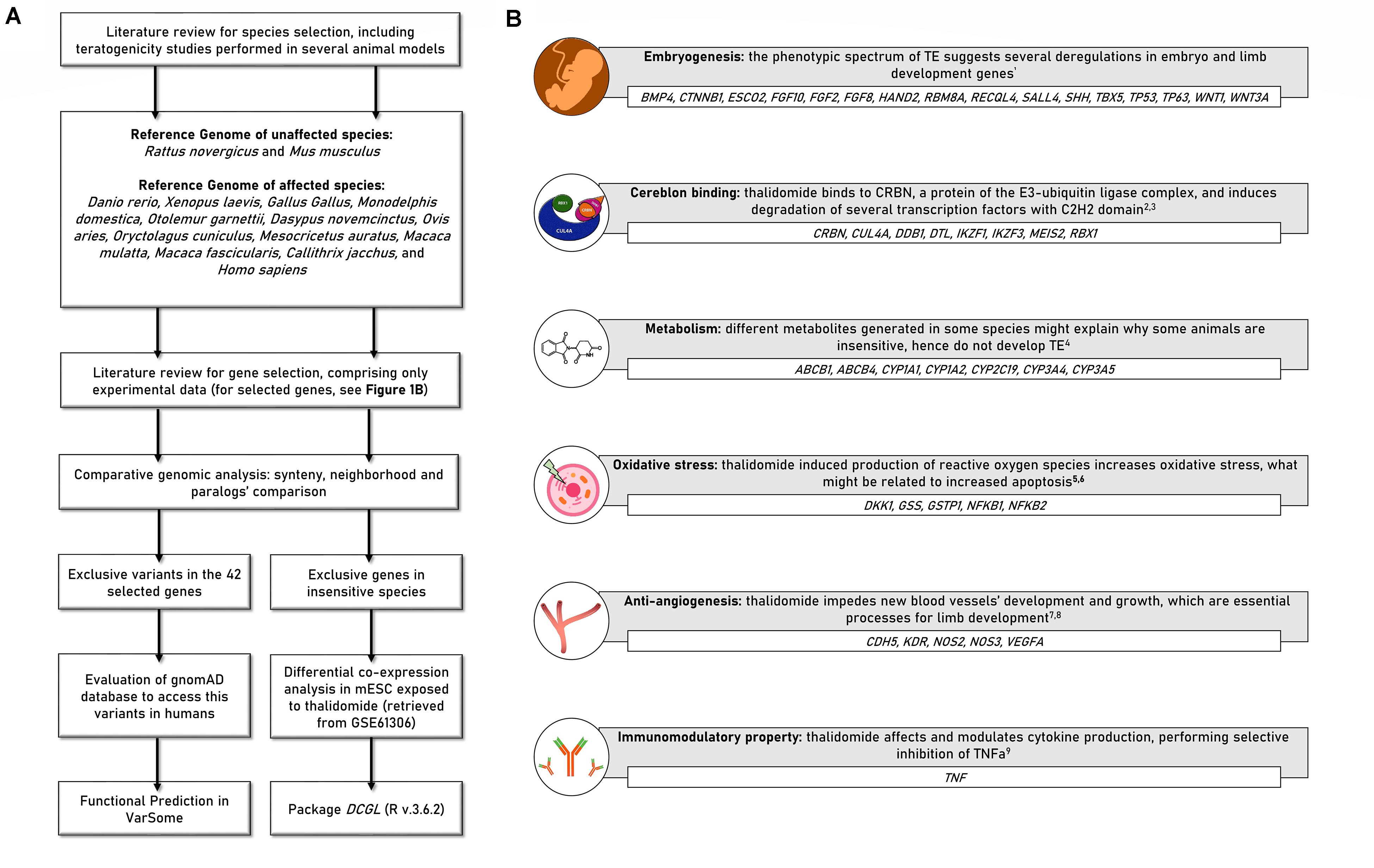
Figure 1. (A) Scheme presenting the study delineation and analyses. (B) Forty-two candidate genes selected for the comparative analyses. BMP4, Bone morphogenetic protein 4; CTNNB1, Catenin beta-1; ESCO2, Establishment of Sister Chromatid Cohesion N-Acetyltransferase 2; FGF10, Fibroblast growth factor 10; FGF2, Fibroblast Growth Factor 2; FGF8, Fibroblast Growth Factor 8; HAND2, Heart And Neural Crest Derivatives Expressed 2; RBM8A, RNA Binding Motif Protein 8A; RECQL4, RecQ Like Helicase 4; SALL4, Sal-like protein 4; SHH, Sonic Hedgehog Signaling Molecule; TBX5, T-Box Transcription Factor 5; TP53, Tumor Protein P53; TP63, Tumor Protein P63; WNT1, Wnt Family Member 1; WNT3A, Wnt Family Member 3A; CRBN, Cereblon; CUL4A, Cullin 4A; DDB1, Damage Specific DNA Binding Protein 1; DTL, Denticleless E3 Ubiquitin Protein Ligase Homolog; IKZF1, IKAROS Family Zinc Finger 1; IKZF3, IKAROS Family Zinc Finger 3; MEIS2, Meis Homeobox 2; RBX1, Ring-Box 1; ABCB1, ATP Binding Cassette Subfamily B Member 1; ABCB4, ATP Binding Cassette Subfamily B Member 4; CYP1A1, Cytochrome P450 1A1; CYP1A2, Cytochrome P450 1A2; CYP2C19, Cytochrome P450 2C19; CYP3A4, Cytochrome P450 3A4; CYP3A5, Cytochrome P450 3A5; CDH5, Cadherin 5; KDR, Kinase Insert Domain Receptor; NOS2, Nitric oxide synthase 2; NOS3, Nitric Oxide Synthase 3; VEGFA, Vascular endothelial growth factor A; DKK1, Dickkopf WNT Signaling Pathway Inhibitor 1; GSS, Glutathione synthetase; GSTP1, Glutathione S-transferase P; NFKB1, Nuclear factor NF-kappa-B p105 subunit; NFKB2, Nuclear factor NF-kappa-B p100 subunit; TNF, Tumor necrosis factor; TE, thalidomide embryopathy. References—1Knobloch et al., 2011; 2Ito et al., 2010; 3Donovan et al., 2018; 4Stephens et al., 2000; 5Hansen and Harris, 2004; 6Knobloch et al., 2007; 7D’Amato et al., 1994; 8Vargesson, 2009; 9Sampaio et al., 1991.
Species and Gene Selection
Fourteen species were included according to previous reports regarding thalidomide exposure during embryo development (Table 1). Forty-two candidate genes were selected from previous studies in animal models; hence, only genes previously accessed by in vitro or in vivo experiments were included (D’Amato et al., 1994; Hansen and Harris, 2004; Therapontos et al., 2009; Ito et al., 2010; Siamwala et al., 2012; Donovan et al., 2018; Matyskiela et al., 2018). These genes are related to (1) thalidomide metabolism; (2) embryonic and/or limb development; and (3) thalidomide molecular mechanisms: anti-angiogenesis, oxidative stress, binding to Cereblon protein, and immunomodulatory property (Figure 1B).
Comparative Genomics Analysis
Synteny, neighborhood reports, and comparison of paralog amount were done for all 42 genes based on genomic databases. The main comparative method used was searching for differences present in the genetic maps and sequences of 12 affected vs. two non-affected species (Table 1 and Figure 2). Thus, species-specific variations were not described. We retrieved transcript sequences from Ensembl release 100 and/or BLAST on NCBI. We performed alignments using MUSCLE (Edgar, 2004) in MEGA 7 (Kumar et al., 2016). gnomAD was assessed to verify variants in humans. Functional prediction was performed in VarSome. PubTator, Ensembl, and PharmGKB databases were used to evaluate clinical and/or pharmacogenetic associations.
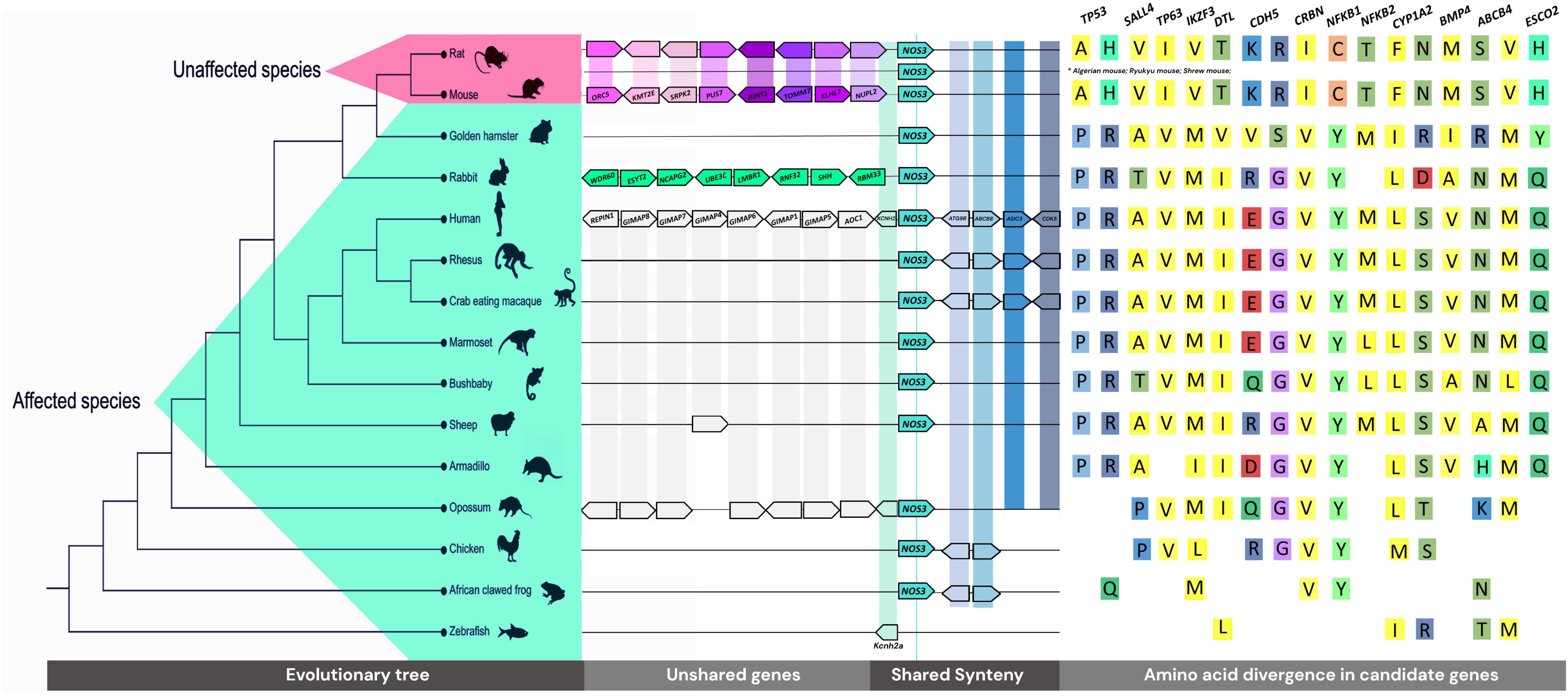
Figure 2. Genomic tree of the evaluated species, synteny and neighborhood of NOS3 gene, and the main variants encountered in mice and rats, which are reported in gnomAD database.
Differential Co-expression Analysis
Differential co-expression analysis was performed in the GSE61306 study (Gao et al., 2015), obtained from the GEO database. RNA from mESC was collected 24, 48, and 72 h after thalidomide or saline exposure. Confirmatory analyses were performed in valproic acid, retinoic acid, or warfarin-exposed mESC, available in the ArrayExpress database (accession numbers: E-TABM-903, E-TABM-1205, and E-MTAB-300). DCGL R package was used for differential co-expression analysis (Yang et al., 2013). Gene–gene co-expression was evaluated using Pearson correlation, including only Pearson’s r of at least |± 0.5| in exposed or control samples (Mukaka, 2012). P-values of < 0.05 were included, after false discovery rate (FDR) adjustment. For the TF analysis, we incorporated the TRRUST database (Han et al., 2015) in the DCGL package, to obtain Mus musculus interactions. Co-expression networks were assembled Cytoscape v.3.7.2. Heatmaps were generated using diffcoexp and ggplot2 R packages.
Results
Synteny, Neighborhood, and Gene Copy Number Differences Across Species
Twelve species who develop TE and two non-affected (mouse and rat) were compared as two groups, affected vs. unaffected. Except for gene copy number differences, the results are related to differences of these groups. Hence, our synteny and neighborhood analysis either was conserved for all species or did not show a pattern of conservation between the groups. To illustrate, the vicinity of SHH in mouse and rat only has five downstream shared genes, while 20 upstream genes are divergent between these rodents. Consequently, we could not find any reasonable relation to thalidomide teratogenesis in such cases.
In contrast, an outstanding difference between the upstream neighborhood of the NOS3 gene in sensitive vs. insensitive species was identified: at least 20 genes composing that region differ from other species, in the same region (Figure 2). The rodents, except the golden hamster, share a subset of genes (Supplementary Table 1). The rabbit, exclusively, has the following genes related to the TE phenotype at that region: WDR60, ESYT2, NCAPG2, UBE3C, LMBR1, RNF32, SHH, RBM33, and NOM1. Other sensitive species share GIMAPs (GTPase, IMAP Family Members), which may be associated with Behcet’s syndrome, a condition treated with thalidomide (Shek and Lim, 2002), as well as cancer-related genes, such as RARRES2 and TMEM176A (Supplementary Table 2).
We found a great difference between the number of copies of some genes related to metabolism among the animals, being affected or not by TE (Supplementary Table 3). CYP2C19, the main gene responsible for thalidomide metabolism (Teo et al., 2000), has 10 tandem copies (7 paralogs > 70% id) in the mouse, while only five (>76% id) in humans. CYP3A4 and CYP3A5 also vary in number of copies; mice have 9 (>68% id), while humans have 5 (>75% id) (Supplementary Table 4). There is a core set of 103 and 77 CYPs (Cytochrome P450’s-seed PF00067-pFam) in mouse and human genomes (Supplementary Table 5), respectively, enzymes involved directly in the oxidative metabolism.
Gene Alignments Revealed Exclusive Unaffected Species’ Variants
Considering that thalidomide is not a selective pressure factor, positive selection sites were not searched. Thus, even a neutral substitution for intrinsic processes of the organisms could add some effect to molecular interactions of this medication into the cell. As a result, we found 299 amino acid substitutions exclusive in insensitive species in 34/42 analyzed genes. Proteins with a higher number of variants in the insensitive group were RECQL4 (65), SALL4 (35), CDH5 (22), KDR (18), and NOS2 (17). A detailed description of all substitutions and protein regions is available in Supplementary Table 6.
Human Genetic Variability Shares Exclusive Unaffected Species’ Variants
For all the gene alignments, the reference genome sequence of each species was used. However, humans have great genetic variability; hence, missense alterations are known to exist in the human species, without being contemplated in the reference genome sequence. Based on this reasoning, all the variants exclusive for insensitive species, according to the comparative sequence alignment, were evaluated in the gnomAD database, aiming to identify similar alterations in humans.
Twenty-four variants, which led to the same missense alterations observed in mice and rats, were encountered in gnomAD registers, distributed across 13 genes (Supplementary Table 7). One missense substitution, p.P72A in Trp53 of mice and rats, has been registered to occur after two possible genomic alterations in humans: (I) rs587782769, changing the first nucleotide of the codon; and (II) rs 1042522, modifying the second nucleotide of the codon. In mice and rats, the variant occurs by the exchange of the first nucleotide and the last nucleotide of the codon, being CCC in humans and GCA in mice and rats.
Functional prediction was performed in the VarSome database according to the criteria established by the American College of Medical Genetics (Richards et al., 2015). Fourteen variants were predicted as “likely benign” and 10 as “variants of unknown significance.” The variant rs587782769 of TP53 was the only one predicted as “likely pathogenic,” because of previous associations with Li-Fraumeni syndrome, a predisposing syndrome of hereditary cancer. The other TP53 variant that leads to p.P72A, rs 1042522, is of unknown significance in Li-Fraumeni syndrome. However, rs1042522 was the only variant identified with high frequency in human populations, having a global minor allele frequency of 0.6629. This was also the only alteration to be registered in the PharmGKB database, with a pharmacogenetic association for different drugs, all antineoplastic agents. The CRBN gene presented one variant of unknown significance, and SALL4 presented two likely benign variants.
Overall, some variants encountered in insensitive species are reported as exclusive for these animals; however, when evaluating the immense variability of the human genome, it was possible to identify the same alterations in humans.
Crbn Identified as a Deregulator of Gene-Pair Co-expression
The original results for the differential gene expression analysis are available in Gao et al. (2015). Here, we evaluated the co-expression of gene pairs through Pearson correlation, aiming to identify whether this correlation is disrupted in thalidomide exposure. Forty-two genes previously selected, and the genes identified to be exclusive of the insensitive species in the evolutionary analysis, were included, totalizing 71 genes. A heatmap comprising the Pearson correlation for all the gene pairs obtained is available in Figure 3A (for controls) and Figure 3B (for thalidomide exposure). Evaluating the red and blue areas, it is possible to visualize stronger expression correlations in controls, when compared to the white areas of the thalidomide-exposed sample heatmap. Nevertheless, aiming for a more robust statistical evaluation, a differential co-expression analysis was performed. This strategy aims to identify patterns of deregulation between genes, which could point to a dysfunctional pathway or mechanism and prioritize the main candidates in a list of hypothesized genes (de la Fuente, 2010; Savino et al., 2020).
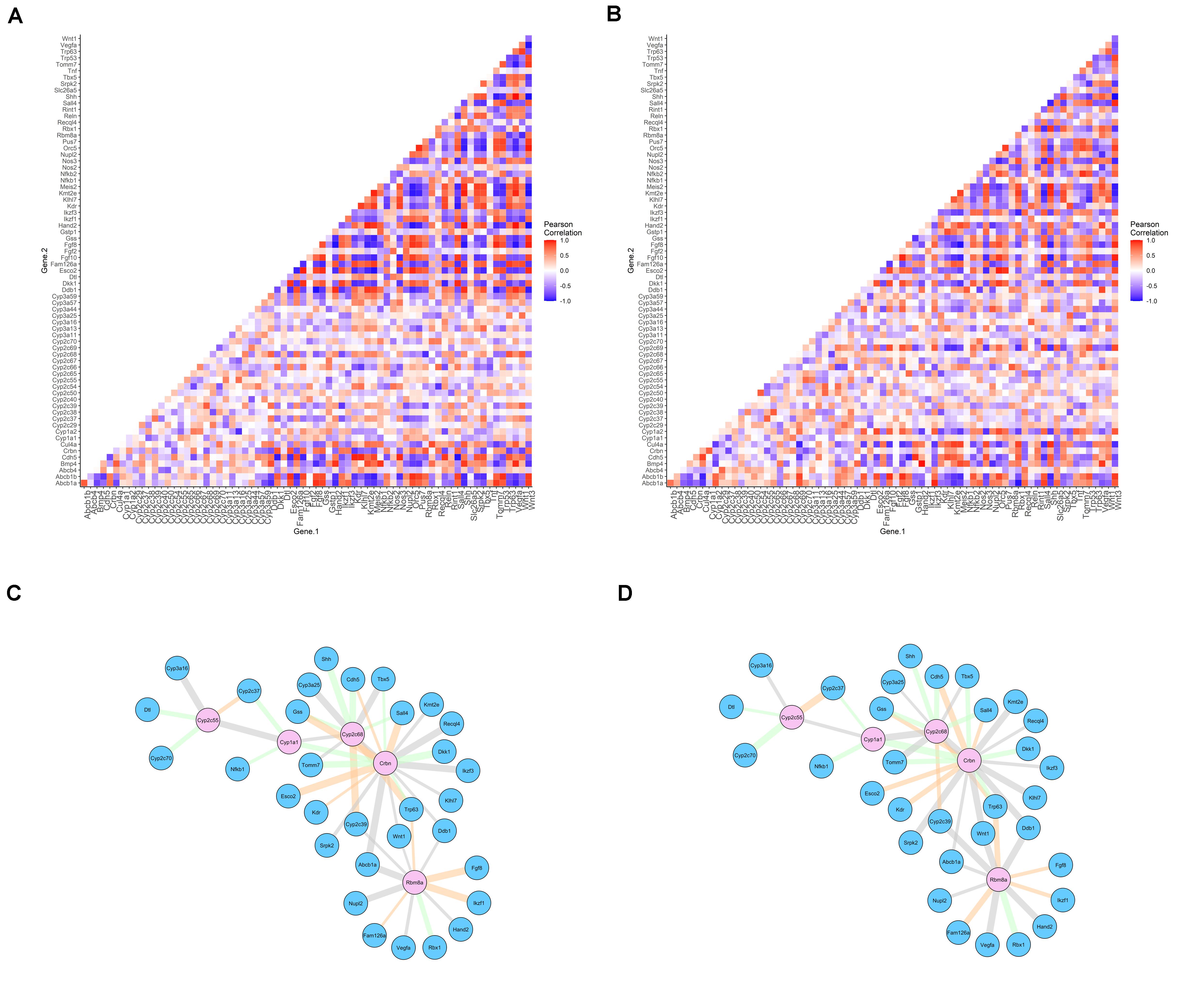
Figure 3. Pearson correlation for differentially co-expressed gene pairs in controls (A) and thalidomide (B) exposed cells. Networks of differentially co-expressed gene pairs for controls (C) and thalidomide (D) exposed cells. Legend: (A,B) positive correlations represented in red; negative correlations in blue; absence of correlation in white. (C,D) genes that drive co-expression are represented in pink nodes; the other gene that composes the pair is represented in blue nodes. Edges’ thickness represents Pearson’s r correlation coefficient; gray edges = switched opposites (r change > 1); orange edges = differentially signed (r change < 1, but altering correlation from positive to negative, or contrariwise); green edges = same signed (correlation was changed, but maintained as positive or negative).
Control vs. thalidomide-exposed mESC were evaluated, to determine the gene pairs with differential co-expression, and the gene of the pair that is probably driving this differential co-expression. As a result, 49 gene pairs differentially co-expressed were significant. The network of differential co-expression correlations can be visualized in Figure 3C (controls) and Figure 3D (thalidomide-exposed). Only Crbn, Cyp2c68, and Rbm8a genes were considered as significant drivers of the co-expression after FDR P-value adjustment (Supplementary Table 8). In 19 gene pairs, Crbn acted as the driver of this co-expression correlation; the other genes in the pairs are associated with limb reduction defects’ syndromes.
In regard to Crbn-Sall4 Pearson correlation, it is set in r = 0.75 in control samples and r = –0.48, after thalidomide exposure. Hence, a positive correlation is altered to negative, with a correlation difference of 1.23. The biggest correlation difference encountered when Crbn was a co-expression gene driver was 1.37, for the Crbn-Recql4 pair.
To better comprehend the co-expression correlations obtained, the same thalidomide-affected genes were analyzed in mESC cells exposed to three different teratogens, separately: valproic acid, retinoic acid, or warfarin. No statistically significant association was identified for evaluated genes (Supplementary Table 9). Another differential co-expression analysis was performed in the thalidomide-exposed mESC array (GSE61306), using a different set of genes, from the NF-kB pathway, known to be affected by thalidomide (Hansen and Harris, 2004). Five genes were identified as co-expression drivers, providing 70 gene pairs differentially co-expressed in control vs. thalidomide-exposure samples (Supplementary Table 10). These results indicate that the gene selection performed was careful and relevant to evaluate thalidomide-induced mechanisms; it also suggests that the positive association between Crbn and the limb reduction defects’ genes is not spurious.
In sequence, we evaluated which TFs could be regulators of the differentially co-expressed gene pairs. The results of this strategy are available in Supplementary Figure 1. Ikzf1, a C2H2 transcription factor degraded by thalidomide, is proposed as a driver of the co-expression, forming gene pairs with Dkk1, Vegfa, and Kdr. These genes have three other TFs in common: Bmi1, Ets1, and Runx2 (Supplementary Table 11), hence being suggested as potential regulators that may also alter the candidate gene expression.
In summary, differential co-expression analysis provided insights regarding thalidomide effects in mESC. First, Crbn drives the co-expression of other genes implicated in genetic, non-teratogenic, limb reduction defects, such as Tbx5, Esco2, Recql4, and Sall4; a fifth gene implicated in limb reduction defects, Rbm8a, was also considered a driver of the co-expression correlation. Second, Crbn and Sall4 have a moderate co-expression correlation, which is affected in thalidomide exposure, even though the typical TE phenotype is not identified in mice. Finally, Ikzf1 might be a regulator of two essential genes for angiogenesis: Vegfa and its receptor Kdr.
Discussion
A comparative genomics analysis was associated with an evaluation of gene–gene co-expression to identify candidate genes for thalidomide teratogenesis variability across species. Twenty genes upstream NOS3 were different when comparing sensitive and insensitive species neighborhoods. Co-expression analysis suggests that Crbn drives the co-expression of limb reduction defects’ genes, including Sall4, in mESC exposed to thalidomide. This might indicate a deregulation between Crbn-Sall4 even in a species known to be insensitive to thalidomide exposure.
During more than 50 years of research, properties of thalidomide and its analogs have been elucidated through animal model studies. Some mechanisms such as anti-angiogenesis (D’Amato et al., 1994; Therapontos et al., 2009) and oxidative stress (Hansen and Harris, 2004), evaluated in these studies, have an extremely relevant role in the whole understanding of the TE specificity. Mammals have an additional layer of complexity: the maternal—fetal interface, the mother metabolism genes affecting the drug availability and placental transfers. Recently, a new light was brought to the understanding of the TE scenario, due to the discovery of SALL4 degradation mediated by Cereblon–thalidomide (Donovan et al., 2018; Matyskiela et al., 2018). Mouse resistance to the TE development was justified by the presence of missense variants both in CRBN and in SALL4 (Donovan et al., 2018; Matyskiela et al., 2018). In the present study, we identified differences among sensitive and insensitive animals in all mechanisms studied associated with thalidomide teratogenic effects, which could explain the heterogeneous outcome in the embryos, as we reported in humans (Kowalski et al., 2020).
Anti-angiogenesis is one of the most studied properties in TE. Our analysis of differential regulation suggests a co-expression correlation between an angiogenesis gene, Vegfa, and its receptor, Kdr, driven by the Ikzf1 gene. Ikaros, the protein encoded by Ikzf1, degradation is demonstrated to be an important event in thalidomide therapeutics in multiple myeloma (Krönke et al., 2014). Furthermore, thalidomide anti-angiogenic property is suggested as independent of Cereblon binding (Beedie et al., 2020; Peach et al., 2020). Hence, the differential co-expression correlations and variants encountered in insensitive species might help to understand thalidomide anti-angiogenic effects in these animals.
Differences in anti-angiogenesis genes also have a relevant role in the whole understanding of TE specificity. In previous studies, evaluating individuals with TE, our group identified variants of susceptibility in NOS3 (Vianna et al., 2013; Kowalski et al., 2016). Our synteny and neighborhood analyses demonstrated nearby genes to NOS3 differ in unaffected rodent species, rabbit species, and other TE species. Eight genes close to NOS3 in the rabbit are involved in conditions similar to TE, including acheiropody and polydactyly. A hypothesis is that alterations in the pattern of expression of these genes during development could favor the establishment of TE characteristics, considering TE conserved regulatory mechanisms. Interestingly, van den Boogaard et al. (2019) identified, in mice, a transcribed distal enhancer involved in the KCNH2 gene regulation (the first gene upstream to NOS3, present in all species in Figure 2). In vitro knockdown of the ncRNA enhancer transcript results in reduced expression of Kcnh2b and two neighboring mRNAs, Nos3 and Abcb8. Also, multiple partially redundantly active enhancers interfere in the complex regulatory landscape of Kcnh2. However, this evidence of a connection between the regulation and expression of NOS3 immediately nearby genes refers to the syntenic region described in our results. Whether TE-tested species’ variable NOS3 upstream regions share similar regulatory relationships according to their genes content remains to be explored.
Historical reports estimated that 20–50% of the human embryos exposed to thalidomide actually developed TE (Newman, 1986). Our studies have reported variants in the TE individuals when compared to Brazilians without congenital anomalies (Vianna et al., 2016; Gomes et al., 2019; Kowalski et al., 2020). The rare variants we identified in the canonical sequences of insensitive species were absent in that TE sample. The polymorphism rs1042522 of TP53 was genotyped in TE and unaffected individuals; however, its frequency was similar in both groups (Gomes et al., 2017). Despite that, rs1042522 is a functionally well-characterized polymorphism (Dumont et al., 2003) that could also help to explain interspecific variability to thalidomide-teratogenesis. p53 is implicated as a regulator of the embryo’s susceptibility to teratogens (Torchinsky and Toder, 2010). Here, we demonstrate three variants in CRBN and 35 alterations in SALL4 which are exclusive in mice and rats. The implication of these variants in Cereblon–thalidomide-induced degradation must be further evaluated.
The gene-specific approach used here to evaluate potential regulators in the co-expression analysis is the most fine-grained method in this type of analysis, usable for identification of genes that more strongly change their connectivity with other genes (Savino et al., 2020). In cancers, this approach has been used to prioritize therapeutic targets (Savino et al., 2020). Global analysis could be useful for the selection of new gene candidates; however, the prioritization of candidate regulators by biological relevance improves the chance of identifying true associated factors (Yang et al., 2013). Here, this prioritization was provided by accessing only genes that had been previously, experimentally validated in thalidomide exposure. Hence, differential co-expression analysis proposed Crbn as a gene that drives co-expression when evaluating different gene pairs.
In humans, ESCO2, TBX5, and RECQL4 cause Roberts, Holt–Oram, and Baller–Gerold syndromes, respectively (Li et al., 1997; Vega et al., 2005; Van Maldergem et al., 2006). Besides, mutations in Rbm8a lead to thrombocytopenia-absent radius syndrome (Albers et al., 2012). All these genetic conditions are phenotypically very similar to TE (Cassina et al., 2017) and require differential diagnosis (Mansour et al., 2019). Despite being an exploratory result, this is the first suggestion of a thalidomide-driven effect in these genes, which might help to explain the phenotype similarities. It is known that Sall4 expression is not reduced in thalidomide exposure, because the degrading mechanism occurs posttranslationally (Donovan et al., 2018). Here we demonstrate Crbn-Sall4 gene co-expression being affected after thalidomide exposure in mESC. This result does not invalidate the idea of a genetic resistance driven by both Crbn and Sall4 variants; however, the altered co-expression in mice raises the hypothesis whether this differential co-expression is also present in TE-affected species. A positive result in the affected species would point to an associated pre-transcriptional mechanism that may help to understand the TE susceptibility, as we have reported in humans (Kowalski et al., 2016).
Concluding, we found several differences between sensitive and insensitive animals regarding known mechanisms of thalidomide teratogenicity: we identified gene copy number divergences among species; non-syntenic genes at the upstream vicinity of NOS3 in rodent, rabbit, and other TE species; and 299 variants in mice and rats within genes of interest. Some variants are in domains, and, until now, they are original data toward the thalidomide teratogenesis molecular context. Consequently, we could not predict if they would be neutral or functional for TE differential outcomes. However, 24 substitutions are rare missense alterations in humans. These results can inspire in vitro and in vivo additional investigations. We also performed a differential co-expression analysis in mESC exposed to thalidomide and identified Crbn as a gene that drives the expression correlation of a series of genes reported previously as affected by thalidomide, as well as genes related to limb reduction defects (of which TE is a phenocopy). These correlated gene pairs include Crbn-Sall4. Finally, differential regulation analysis suggests Ikzf1 as a driver gene for co-expression as well, a result that deserves to be explored in depth in other studies, especially in vivo.
The research here conducted complements the recent results regarding thalidomide comparative teratogenesis. Our study is limited by the lack of experimental data of thalidomide exposure in other affected and unaffected species. However, this is an exploratory analysis that provides new insights for this challenge. The outburst of babies born with TE led to many changes in legislation concerning animal model research and drug commercialization around the world (Daemmrich, 2002). In order to guarantee safe commercial liberation of a medicine, all drugs must be tested at least in two animal species, one non-rodent (Cook and Fairweather, 1968), and results in these models must not be completely extrapolated to human species, once teratogenesis is not a species-specific process (De Santis et al., 2004). Hence, studies of comparative teratogenesis like this are important to trace strategies for avoiding a new thalidomide tragedy.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s. Data studied was obtained from the public genomic databases Ensembl and GenBank (NCBI). Gene expression data is available in Gene Expression Omnibus (GEO) database, series number GSE61306. Confirmatory analyses were performed in valproic acid, retinoic acid or warfarin-exposed mESC, available in the ArrayExpress database (accession numbers: E-TABM-903, E-TABM-1205, and E-MTAB-300).
Ethics Statement
Ethical review and approval was not required for the animal study because the present study uses only secondary data, available in GEO database (GSE61306) and ENA database (E-TABM-903, E-TABM-1205, and E-MTAB-300), as reported in the Data Availability Statement.
Author Contributions
TK and GC-G contributed in devising the concept, designing and conducting the analyses, and writing the manuscript. JG contributed in devising the concept, designing the analysis, and performing the analyses. LS-F contributed in devising and supervising the analyses. LF, MR-M, VP-C, and FV contributed in devising the concept, designing the analyses, supervising the analyses, and correcting the manuscript. All authors discussed the results and contributed scientifically to the manuscript.
Funding
This research was funded by the Instituto Nacional de Genética Médica Populacional (INAGEMP), grant nos. CNPq 573993/2008-4 and FAPERGS 17/2551.0000521-0; Hospital de Clínicas de Porto Alegre (HCPA)—Fundo de Incentivo a Pesquisa e Eventos (FIPE), grant nos. 2010-0244, 2017-0248, and 2019-0792; and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) grant no. 423249/2016-9. FV was a recipient of a CNPq scholarship grant (grant no. CNPq 312993/2017-0). TK was firstly recipient of a Coordination of Improvement of Higher Education Personnel (CAPES) scholarship (grant no. 88881.132344/2016-01), and then of a CNPq scholarship (grant no. 156158/2018-3).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We deeply thank L. Neto, A. Dupont, M. Furtado, and B. Rengel for the technical assistance, and all the members of the Laboratory of Genomic Medicine (HCPA), Laboratory of Medical Genetics and Evolution (UFRGS), and Bioinformatics Core (HCPA) for all the helpful discussions and scientific support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.680217/full#supplementary-material
References
Aizawa, M., Abe, Y., Ito, T., Handa, H., and Nawa, H. (2011). mRNA distribution of the thalidomide binding protein cereblon in adult mouse brain. Neurosci. Res. 69, 343–347. doi: 10.1016/j.neures.2010.12.019
Albers, C. A., Paul, D. S., Schulze, H., Freson, K., Stephens, J. C., Smethurst, P. A., et al. (2012). Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat. Genet. 44, s431–s432. doi: 10.1038/ng.1083
Angers, S., Li, T., Yi, X., MacCoss, M. J., Moon, R. T., and Zheng, N. (2006). Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443, 590–593. doi: 10.1038/nature05175
Asatsuma-Okumura, T., Ando, H., De Simone, M., Yamamoto, J., Sato, T., Shimizu, N., et al. (2019). p63 is a cereblon substrate involved in thalidomide teratogenicity. Nat. Chem. Biol. 15, 1077–1084. doi: 10.1038/s41589-019-0366-7
Bauer, K. S., Dixon, S. C., and Figg, W. D. (1998). Inhibition of angiogenesis by thalidomide requires metabolic activation, which is species-dependent. Biochem. Pharmacol. 55, 1827–1834. doi: 10.1016/s0006-2952(98)00046-x
Bavley, C. C., Rice, R. C., Fischer, D. K., Fakira, A. K., Byrne, M., Kosovsky, M., et al. (2018). Rescue of learning and memory deficits in the human nonsyndromic intellectual disability cereblon knock-out mouse model by targeting the AMP-activated protein kinase-mTORC1 translational pathway. J. Neurosci. 38, 2780–2795. doi: 10.1523/JNEUROSCI.0599-17.2018
Beedie, S. L., Huang, P. A., Harris, E. M., Strope, J. D., Mahony, C., Chau, C. H., et al. (2020). Role of cereblon in angiogenesis and in mediating the antiangiogenic activity of immunomodulatory drugs. FASEB J. 34, 11395–11404. doi: 10.1096/fj.201903060RR
Butler, H. (1977). The effect of thalidomide on a prosimian: the greater galago (Galago crassicaudatus). J. Med. Primatol. 6, 319–324. doi: 10.1159/000459764
Cahen, R. L. (1966). Experimental and clinical chemoteratogenesis. Adv. Pharmacol. 4, 263–349. doi: 10.1016/s1054-3589(08)60101-5
Cassina, M., Cagnoli, G. A., Zuccarello, D., Di Gianantonio, E., and Clementi, M. (2017). Human teratogens and genetic phenocopies. Understanding pathogenesis through human genes mutation. Eur. J. Med. Genet. 60, 22–31. doi: 10.1016/j.ejmg.2016.09.011
Celgene (2013). “Revlimid (lenalidomide) prescribing information,” in Food and Drug Administration Access Data. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/021880s028lbl.pdf
Celgene (2015). “POMALYST (pomalidomide) prescribing information,” in Food and Drug Administration Access Data. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/204026s019lbl.pdf
Christian, M. S., Laskin, O. L., Sharper, V., Hoberman, A., Stirling, D. I., and Latriano, L. (2007). Evaluation of the developmental toxicity of lenalidomide in rabbits. Birth Defects Res. B Dev. Reprod. Toxicol. 80, 188–207. doi: 10.1002/bdrb.20115
Cook, M. J., and Fairweather, F. A. (1968). Methods used in teratogenic testing. Lab. Anim. 2, 219–228. doi: 10.1258/002367768781082834
Daemmrich, A. (2002). A tale of two experts: thalidomide and political engagement in the United States and West Germany. Soc. Hist. Med. 15, 137–158. doi: 10.1093/shm/15.1.137
D’Amato, R. J., Loughnan, M. S., Flynn, E., and Folkman, J. (1994). Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. U. S. A. 91, 4082–4085. doi: 10.1073/pnas.91.9.4082
de la Fuente, A. (2010). From ‘differential expression’ to ‘differential networking’ – identification of dysfunctional regulatory networks in diseases. Trends Genet. 26, 326–333. doi: 10.1016/j.tig.2010.05.001
Delahunt, C. S., and Lassen, L. J. (1964). Thalidomide syndrome in monkeys. Science 146, 1300–1305. doi: 10.1126/science.146.3649.1300
De Santis, M., Straface, G., Carducci, B., Cavaliere, A. F., De Santis, L., Lucchese, A., et al. (2004). Risk of drug-induced congenital defects. Eur. J. Obstet. Gynecol. Reprod. Biol. 117, 10–19. doi: 10.1016/j.ejogrb.2004.04.022
Donovan, K. A., An, J., Nowak, R. P., Yuan, J. C., Fink, E. C., Berry, B. C., et al. (2018). Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. Elife 7:e38430. doi: 10.7554/eLife.38430
Dumont, P., Leu, J. I., Della Pietra, A. C. III, George, D. L., and Murphy, M. (2003). The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat. Genet. 33, 357–365. doi: 10.1038/ng1093
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Ema, M., Ise, R., Kato, H., Oneda, S., Hirose, A., Hirata-Koizumi, M., et al. (2010). Fetal malformations and early embryonic gene expression response in cynomolgus monkeys maternally exposed to thalidomide. Reprod. Toxicol. 29, 49–56. doi: 10.1016/j.reprotox.2009.09.003
Fabro, S., Smith, R. L., and Williams, R. T. (1967). The fate of the hydrolysis products of thalidomide in the pregnant rabbit. Biochem. J. 104, 570–574. doi: 10.1042/bj1040570
Fink, E. C., McConkey, M., Adams, D. N., Haldar, S. D., Kennedy, J. A., Guirguis, A. A., et al. (2018). Crbn (I391V) is sufficient to confer in vivo sensitivity to thalidomide and its derivatives in mice. Blood 132, 1535–1544. doi: 10.1182/blood-2018-05-852798
Fischer, E. S., Böhm, K., Lydeard, J. R., Yang, H., Stadler, M. B., Cavadini, S., et al. (2014). Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53. doi: 10.1038/nature13527
Fort, D. J., Stover, E. L., Bantle, J. A., and Finch, R. A. (2000). Evaluation of the developmental toxicity of thalidomide using frog embryo teratogenesis assay-xenopus (FETAX): biotransformation and detoxification. Teratog. Carcinog. Mutagen. 20, 35–47. doi: 10.1002/(sici)1520-6866200020:1<35::aid-tcm4<3.0.co;2-i
Gao, S., Wang, S., Fan, R., and Hu, J. (2020). Recent advances in the molecular mechanism of thalidomide teratogenicity. Biomed. Pharmacother. 127:110114. doi: 10.1016/j.biopha.2020.110114
Gao, X., Sprando, R. L., and Yourick, J. J. (2015). Transcriptomic changes in mouse embryonic stem cells exposed to thalidomide during spontaneous differentiation. Data Brief 4, 199–202. doi: 10.1016/j.dib.2015.05.014
Gomes, J. D. A., Kowalski, T. W., Fraga, L. R., Macedo, G. S., Sanseverino, M. T. V., Schuler-Faccini, L., et al. (2019). The role of ESCO2, SALL4 and TBX5 genes in the susceptibility to thalidomide teratogenesis. Sci. Rep. 9:11413. doi: 10.1038/s41598-019-47739-8
Gomes, J. D. A., Kowalski, T. W., Fraga, L. R., Tovo-Rodrigues, L., Sanseverino, M. T. V., Schuler-Faccini, L., et al. (2017). Genetic susceptibility to thalidomide embryopathy in humans: study of candidate development genes. Birth Defects Res. 110, 456–461. doi: 10.1002/bdr2.1163
Han, H., Shim, H., Shin, D., Shim, J. E., Ko, Y., Shin, J., et al. (2015). TRRUST: a reference database of human transcriptional regulatory interactions. Sci. Rep. 5:11432. doi: 10.1038/srep11432
Hansen, J. M., and Harris, C. (2004). A novel hypothesis for thalidomide-induced limb teratogenesis: redox misregulation of the NF-kappaB pathway. Antioxid. Redox Signal. 6, 1–14. doi: 10.1089/152308604771978291
Higgins, J. J., Pucilowska, J., Lombardi, R. Q., and Rooney, J. P. (2004). A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology 63, 1927–1931. doi: 10.1212/01.wnl.0000146196.01316.a2
Homburger, F., Chaube, S., Eppenberger, M., Bogdonoff, P. D., and Nixon, C. W. (1965). Susceptibility of certain inbred strains of hamsters to teratogenic effects of thalidomide. Toxicol. Appl. Pharmacol. 7, 686–693. doi: 10.1016/0041-008x(65)90126-2
Ito, T., Ando, H., Suzuki, T., Ogura, T., Hotta, K., Imamura, Y., et al. (2010). Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350. doi: 10.1126/science.1177319
Jonsson, B. G. (1972). Thalidomide teratology in swine: a preparatory study. Acta Pharmacol. Toxicol. (Copenh.) 31, 24–26. doi: 10.1111/j.1600-0773.1972.tb00692.x
Jung, Y. J., Tweedie, D., Scerba, M. T., Kim, D. S., Palmas, M. F., Pisanu, A., et al. (2021). Repurposing immunomodulatory imide drugs (IMiDs) in neuropsychiatric and neurodegenerative disorders. Front. Neurosci. 15:656921. doi: 10.3389/fnins.2021.656921
Khera, K. S. (1975). Fetal cardiovascular and other defects induced by thalidomide in cats. Teratology 11, 65–69. doi: 10.1002/tera.1420110108
Knobloch, J., Jungck, D., and Koch, A. (2011). Apoptosis induction by thalidomide: critical for limb teratogenicity but therapeutic potential in idiopathic pulmonary fibrosis? Curr. Mol. Pharmacol. 4, 26–61. doi: 10.2174/1874467211104010026
Knobloch, J., Shaughnessy, J. D. Jr., and Rüther, U. (2007). Thalidomide induces limb deformities by perturbing the Bmp/Dkk1/Wnt signaling pathway. FASEB J. 21, 1410–1421. doi: 10.1096/fj.06-7603com
Kohlhase, J., Heinrich, M., Schubert, L., Liebers, M., Kispert, A., Laccone, F., et al. (2002). Okihiro syndrome is caused by SALL4 mutations. Hum. Mol. Genet. 11, 2979–2987. doi: 10.1093/hmg/11.23.2979
Kohlhase, J., Schubert, L., Liebers, M., Rauch, A., Becker, K., Mohammed, S. N., et al. (2003). Mutations at the SALL4 locus on chromosome 20 result in a range of clinically overlapping phenotypes, including Okihiro syndrome, Holt-Oram syndrome, acro-renal-ocular syndrome, and patients previously reported to represent thalidomide embryopathy. J. Med. Genet. 40, 473–478. doi: 10.1136/jmg.40.7.473
Kowalski, T. W., Fraga, L. R., Tovo-Rodrigues, L., Sanseverino, M. T., Hutz, M. H., Schuler-Faccini, L., et al. (2016). New findings in eNOS gene and Thalidomide Embryopathy suggest pre-transcriptional effect variants as susceptibility factors. Sci. Rep. 6:23404. doi: 10.1038/srep23404
Kowalski, T. W., Gomes, J. D. A., Garcia, G. B. C., Fraga, L. R., Paixao-Cortes, V. R., Recamonde-Mendoza, M., et al. (2020). CRL4-cereblon complex in Thalidomide Embryopathy: a translational investigation. Sci. Rep. 10, 851–851. doi: 10.1038/s41598-020-57512-x
Krönke, J., Udeshi, N. D., Narla, A., Grauman, P., Hurst, S. N., McConkey, M., et al. (2014). Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305. doi: 10.1126/science.1244851
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lee, K. M., Yang, S. J., Kim, Y. D., Choi, Y. D., Nam, J. H., Choi, C. S., et al. (2013). Disruption of the cereblon gene enhances hepatic AMPK activity and prevents high-fat diet-induced obesity and insulin resistance in mice. Diabetes 62, 1855–1864. doi: 10.2337/db12-1030
Li, Q. Y., Newbury-Ecob, R. A., Terrett, J. A., Wilson, D. I., Curtis, A. R., Yi, C. H., et al. (1997). Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat. Genet. 15, 21–29. doi: 10.1038/ng0197-21
Mahony, C., Erskine, L., Niven, J., Greig, N. H., Figg, W. D., and Vargesson, N. (2013). Pomalidomide is nonteratogenic in chicken and zebrafish embryos and nonneurotoxic in vitro. Proc. Nat. Acad. Sci. U.S.A. 110, 12703–12708. doi: 10.1073/pnas.1307684110
Mansour, S., Baple, E., and Hall, C. M. (2019). A clinical review and introduction of the diagnostic algorithm for thalidomide embryopathy (DATE). J. Hand. Surg. Eur. Vol. 44, 96–108. doi: 10.1177/1753193418800631
Marin-Padilla, M., and Benirschke, K. (1963). Thalidomide induced alterations in the blastocyst and placenta of the armadillo, dasypus novemcinctus mexicanus, including a choriocarcinoma. Am. J. Pathol. 43, 999–1016.
Matyskiela, M. E., Couto, S., Zheng, X., Lu, G., Hui, J., Stamp, K., et al. (2018). SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Biol. 14, 981–987. doi: 10.1038/s41589-018-0129-x
Merker, H. J., Heger, W., Sames, K., Stürje, H., and Neubert, D. (1988). Embryotoxic effects of thalidomide-derivatives in the non-human primate Callithrix jacchus. I. Effects of 3-(1,3-dihydro-1-oxo-2H-isoindol-2-yl)-2,6-dioxopiperidine (EM12) on skeletal development. Arch. Toxicol. 61, 165–179. doi: 10.1007/BF00316631
Mukaka, M. M. (2012). Statistics corner: a guide to appropriate use of correlation coefficient in medical research. Malawi Med. J. 24, 69–71.
Newman, C. G. (1986). The thalidomide syndrome: risks of exposure and spectrum of malformations. Clin. Perinatol. 13, 555–573. doi: 10.1016/s0095-5108(18)30810-8
Peach, M. L., Beedie, S. L., Chau, C. H., Collins, M. K., Markolovic, S., Luo, W., et al. (2020). Antiangiogenic activity and in silico cereblon binding analysis of novel thalidomide analogs. Molecules 25:5683. doi: 10.3390/molecules25235683
Rajadhyaksha, A. M., Ra, S., Kishinevsky, S., Lee, A. S., Romanienko, P., DuBoff, M., et al. (2012). Behavioral characterization of cereblon forebrain-specific conditional null mice: a model for human non-syndromic intellectual disability. Behav. Brain Res. 226, 428–434. doi: 10.1016/j.bbr.2011.09.039
Reichard-Brown, J. L., Spinner, H., and McBride, K. (2009). Sea urchin embryos exposed to thalidomide during early cleavage exhibit abnormal morphogenesis later in development. Birth Defects Res. B Dev. Reprod. Toxicol. 86, 496–505. doi: 10.1002/bdrb.20215
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Ruckebusch, Y. (1983). Pharmacology of reticulo-ruminal motor function. J. Vet. Pharmacol. Ther. 6, 245–272. doi: 10.1111/j.1365-2885.1983.tb00001.x
Sampaio, E. P., Sarno, E. N., Galilly, R., Cohn, Z. A., and Kaplan, G. (1991). Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J. Exp. Med. 173, 699–703. doi: 10.1084/jem.173.3.699
Savino, A., Provero, P., and Poli, V. (2020). Differential Co-expression analyses allow the identification of critical signalling pathways altered during tumour transformation and progression. Int. J. Mol. Sci. 21:9461. doi: 10.3390/ijms21249461
Shek, L. P., and Lim, D. L. (2002). Thalidomide in Behçet’s disease. Biomed. Pharmacother. 56, 31–35. doi: 10.1016/s0753-3322(01)00154-8
Siamwala, J. H., Veeriah, V., Priya, M. K., Rajendran, S., Saran, U., Sinha, S., et al. (2012). Nitric oxide rescues thalidomide mediated teratogenicity. Sci. Rep. 2:679. doi: 10.1038/srep00679
Somers, G. S. (1962). Thalidomide and congenital abnormalities. Lancet 1, 912–913. doi: 10.1016/s0140-6736(62)91943-8
Sorensen, D., Sackett, A., Urban, D. J., Maier, J., Vargesson, N., and Sears, K. E. (2017). A new mammalian model system for thalidomide teratogenesis: monodelphis domestica. Reprod. Toxicol. 70, 126–132. doi: 10.1016/j.reprotox.2017.01.010
Stephens, T. D. (2009). The effect of thalidomide in chicken embryos. Birth Defects Res. B Dev. Reprod. Toxicol. 85, 725–731. doi: 10.1002/bdra.20597
Stephens, T. D., Bunde, C. J., and Fillmore, B. J. (2000). Mechanism of action in thalidomide teratogenesis. Biochem. Pharmacol. 59, 1489–1499. doi: 10.1016/s0006-2952(99)00388-3
Stephens, T. D., and Fillmore, B. J. (2000). Hypothesis: thalidomide embryopathy-proposed mechanism of action. Teratology 61, 189–195. doi: 10.1002/(SICI)1096-9926(200003)61:3<189::AID-TERA6<3.0.CO;2-W
Teo, S. K., Sabourin, P. J., O’Brien, K., Kook, K. A., and Thomas, S. D. (2000). Metabolism of thalidomide in human microsomes, cloned human cytochrome P-450 isozymes, and Hansen’s disease patients. J. Biochem. Mol. Toxicol. 14, 140–147. doi: 10.1002/(sici)1099-0461200014:3<140::aid-jbt3<3.0.co;2-p
Therapontos, C., Erskine, L., Gardner, E. R., Figg, W. D., and Vargesson, N. (2009). Thalidomide induces limb defects by preventing angiogenic outgrowth during early limb formation. Proc. Natl. Acad. Sci. U. S. A. 106, 8573–8578. doi: 10.1073/pnas.0901505106
Torchinsky, A., and Toder, V. (2010). Mechanisms of the embryo’s response to embryopathic stressors: a focus on p53. J. Reprod. Immunol. 85, 76–80. doi: 10.1016/j.jri.2010.01.003
van den Boogaard, M., van Weerd, J. H., Bawazeer, A. C., Hooijkaas, I. B., van de Werken, H., Tessadori, F., et al. (2019). Identification and characterization of a transcribed distal enhancer involved in cardiac Kcnh2 regulation. Cell Rep. 28, 2704–2714.e5. doi: 10.1016/j.celrep.2019.08.007
Van Maldergem, L., Siitonen, H. A., Jalkh, N., Chouery, E., De Roy, M., Delague, V., et al. (2006). Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J. Med. Genet. 43, 148–152. doi: 10.1136/jmg.2005.031781
Vargesson, N. (2009). Thalidomide-induced limb defects: resolving a 50-year-old puzzle. Bioessays 31, 1327–1336. doi: 10.1002/bies.200900103
Vega, H., Waisfisz, Q., Gordillo, M., Sakai, N., Yanagihara, I., Yamada, M., et al. (2005). Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat. Genet. 37, 468–470. doi: 10.1038/ng1548
Vianna, F. S., Fraga, L. R., Tovo-Rodrigues, L., Tagliani-Ribeiro, A., Biondi, F., Maximino, C. M., et al. (2013). Polymorphisms in the endothelial nitric oxide synthase gene in thalidomide embryopathy. Nitric Oxide 35, 89–92. doi: 10.1016/j.niox.2013.09.002
Vianna, F. S., Kowalski, T. W., Tovo-Rodrigues, L., Tagliani-Ribeiro, A., Godoy, B. A., Fraga, L. R., et al. (2016). Genomic and in silico analyses of CRBN gene and thalidomide embryopathy in humans. Reprod. Toxicol. 66, 99–106. doi: 10.1016/j.reprotox.2016.10.003
Keywords: IMiDs, teratogenesis, teratogens, comparative genomics, co-expression, C2H2, NOS3, synteny
Citation: Kowalski TW, Caldas-Garcia GB, Gomes JA, Fraga LR, Schuler-Faccini L, Recamonde-Mendoza M, Paixão-Côrtes VR and Vianna FSL (2021) Comparative Genomics Identifies Putative Interspecies Mechanisms Underlying Crbn-Sall4-Linked Thalidomide Embryopathy. Front. Genet. 12:680217. doi: 10.3389/fgene.2021.680217
Received: 13 March 2021; Accepted: 18 May 2021;
Published: 23 June 2021.
Edited by:
Md Saidur Rahman, Chung-Ang University, South KoreaReviewed by:
Jamila H. Siamwala, Brown University, United StatesKosuke Kataoka, Waseda University, Japan
Copyright © 2021 Kowalski, Caldas-Garcia, Gomes, Fraga, Schuler-Faccini, Recamonde-Mendoza, Paixão-Côrtes and Vianna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vanessa Rodrigues Paixão-Côrtes, dmFuZXNzYWNlekBnbWFpbC5jb20=; Fernanda Sales Luiz Vianna, ZnNsdmlhbm5hQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work