 Francesco Paduano1,2,3*
Francesco Paduano1,2,3* Fernanda Fabiani1Emma Colao1
Fernanda Fabiani1Emma Colao1 Francesco Trapasso1,4Nicola Perrotti1,2Vito Barbieri5
Francesco Trapasso1,4Nicola Perrotti1,2Vito Barbieri5 Francesco Baudi4
Francesco Baudi4 Rodolfo Iuliano1,2*
Rodolfo Iuliano1,2*- 1Medical Genetics Unit, University “Magna Graecia”, Catanzaro, Italy
- 2Department of Health Sciences, University “Magna Graecia”, Catanzaro, Italy
- 3Tecnologica Research Institute and Marrelli Health, Biomedical Section, Stem Cells and Medical Genetics Units, Crotone, Italy
- 4Department of Experimental and Clinical Medicine, University Magna Graecia of Catanzaro, Catanzaro, Italy
- 5Medical Oncology Unit, Mater Domini Hospital, Catanzaro, Italy
Li–Fraumeni syndrome (LFS) is an inherited autosomal dominant disease characterized by a predisposition to many cancers. Germline pathogenic variants in TP53 are primarily responsible for LFS. By performing a targeted sequencing panel in a proband with liver carcinoma having a deceased son affected by osteosarcoma, we found the novel heterozygous frameshift variant c.645del (p.Ser215Argfs*32) in the TP53 gene. This variant co-segregated with typical LFS cancers in the family pedigree, consistent with the pathogenicity of this novel and previously undescribed TP53 variant.
Introduction
Li–Fraumeni syndrome (LFS) is a rare autosomal dominant disorder in which patients have an increased susceptibility to developing several childhood- and adult-onset tumors compared with other cancer syndromes (McBride et al., 2014; Schneider et al., 2019). LFS is usually associated with a family history of multiple malignancies, mainly the so-called core LFS cancers, including osteosarcomas, soft-tissue sarcomas, brain tumors, adrenocortical carcinomas, and early-onset female breast cancers (Egan et al., 2012; Zhou et al., 2017). Recent studies show that LFS is also associated with an increased risk of several additional cancers, including liver, lung, prostate, ovarian, and pancreatic gastrointestinal cancers; leukemia; and lymphoma as well as cancers of the kidney, larynx, lung, skin, testis, and thyroid (McBride et al., 2014; Schneider et al., 2019). Therefore, LFS is associated with the development of tumors at various sites compared with other hereditary syndromes that predispose subjects to cancer, commonly limited to specific tumor sites (McBride et al., 2014; Young et al., 2019).
LFS is mainly associated with germline variants in the TP53 gene, a tumor-suppressor gene whose protein product is a transcription factor involved in several cell functions such as DNA repair, cell cycle arrest, and senescence as well as autophagy, apoptosis, and necrosis (Bougeard et al., 2015; Miller et al., 2016).
Importantly, p53 protein works not only as a transcriptional regulator, but can also act in the cytosol and mitochondria to promote apoptosis through transcription-independent mechanisms (Barnoud et al., 2021).
Because wild-type p53 protein works as a tumor suppressor, variants in the tumor-suppressor gene TP53 that disrupt protein function or stability could be responsible for the accumulation of genomic alterations, culminating in the development of tumors, especially LFS-related cancers (Zhou et al., 2017; Di Agostino et al., 2019).
In addition to the loss of function that a mutation in TP53 may cause, certain mutant forms of p53 can exert dominant-negative effects over the coexpressed WT p53 allele, whereas others can exert additional oncogenic activity by a gain-of-function mechanism (Barnoud et al., 2021).
Heterozygous TP53 germline variants are reported in several families affected by LFS (Egan et al., 2012; Valdez et al., 2017; Kharaziha et al., 2019). Subjects who are carriers of a pathogenic TP53 variant possess a variable lifetime risk of developing cancer, and the phenotype can differ from cancer-free over a lifetime to fully penetrant, depending on the functional and structural effects of the causative TP53 variant (Olivier et al., 2010; de Andrade et al., 2017; Bittar et al., 2019). Here, we report a novel and rare heterozygous TP53 germline pathogenic variant (c.645del) found in the proband, his son, and three relatives from one non-consanguineous south Italian family having an aggregated history of typical LFS cancers.
Results
Case Report and Genetic Analysis
The proband (III.2) is a 64-year-old deceased male affected by liver cancer, having two healthy daughters and one 20-year-old deceased son affected by osteosarcoma (Figure 1). Because of this latter finding as well as an aggregated family cancer history suggestive of p53 dysfunction, we analyzed the proband with a next-generation sequencing (NGS) tumor panel containing the TP53, BRCA2, BRCA1, ATM, CHEK2, and PALB2 genes (Supplementary Methods). NGS analysis showed a heterozygous frameshift TP53 variant ([GRCh37/hg19] chr17: g.7578204delA; NM_000546.6: c.645del (p.Ser215Argfs*32) in the exon 6 of the TP53 (III.2, Figure 2A). Sanger sequencing confirmed the presence in the proband of the heterozygous frameshift c.645del variant in the exon 6 of the TP53 gene (Figure 2B). No genetic alterations were detected within the other genes included in the panel.
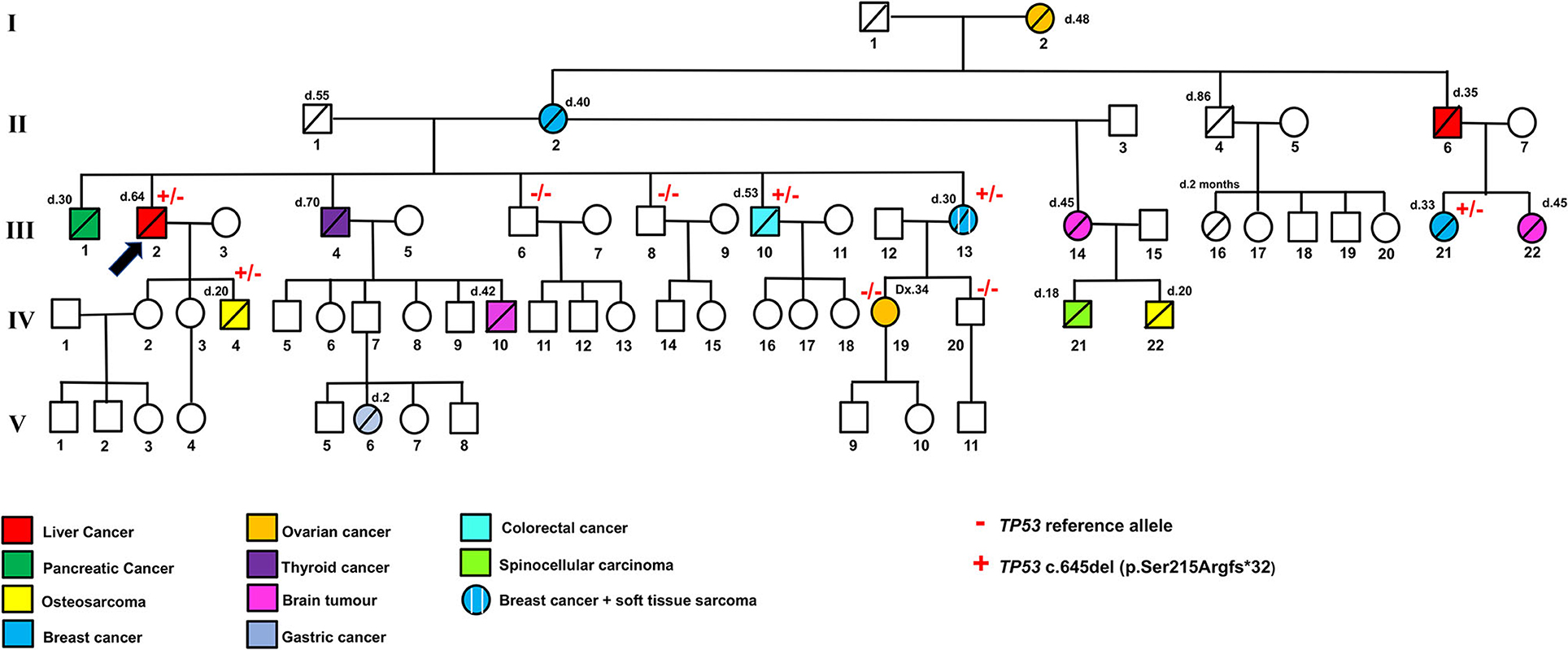
Figure 1. Family pedigree indicating the genotype and phenotype of each individual. + and – indicate the TP53 pathogenic variant c.645del (p.Ser215Argfs*32) and TP53 reference allele, respectively. Different colors indicate liver cancers, pancreatic cancer, osteosarcoma, breast cancer, ovarian cancer, thyroid cancer, brain tumor, gastric cancer, colorectal cancer, spinocellular carcinoma, and soft tissue sarcoma. d, age of death; Dx, age of diagnosis.
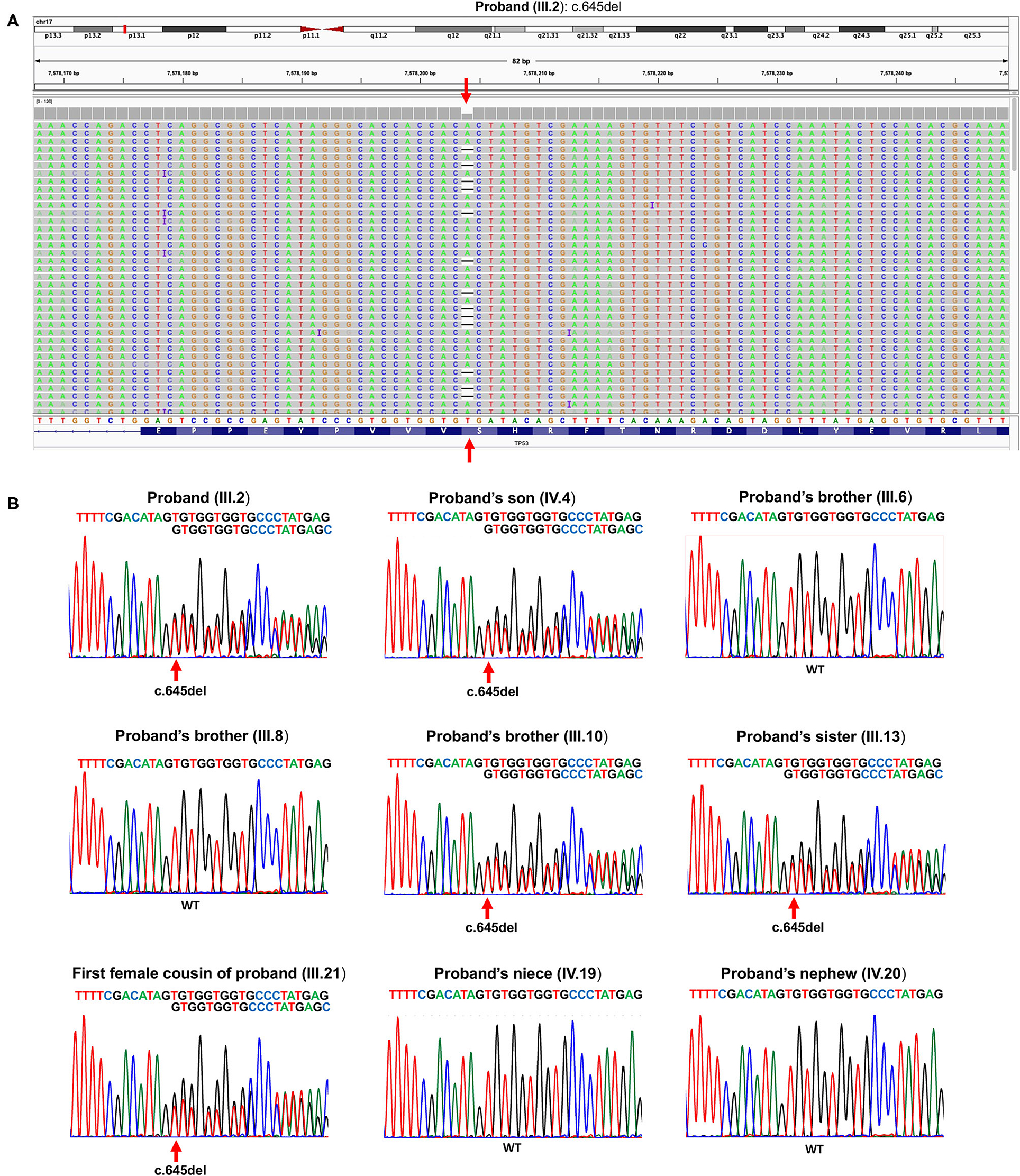
Figure 2. (A) Snapshot of Integrative Genomics Viewer, showing the TP53 germline variant c.645del (p.Ser215Argfs*32) in the proband (III.2). (B) Sanger sequencing showing the c.645del pathogenic heterozygous TP53 variant in the proband (III.2), his son (IV.4), his brother (III.10), his sister (III.13), and his first female cousin (III.21). WT TP53 allele is shown in proband's brother (III.6), proband's brother (III.8), proband's niece (IV.19), and proband's nephew (IV.20). The red arrow indicates the heterozygous deletion.
Following this TP53 mutation assessment, genetic counseling was also extended to proband family members, and several first- and second-degree relatives affected by tumors specifically associated with LFS were reported. Importantly, the proband's son (IV.4, Figure 2B), deceased at the age of 20 due to osteosarcoma, was a heterozygous carrier of the c.645del TP53 variant. The proband's brother (III.1), who died at the age of 30 due to a pancreatic tumor, as well as the proband brother (III.4), deceased at the age of 70 due to thyroid carcinoma, did not receive any genetic testing. Two more proband's brothers (III.6) and (III.8) were referred to find health status and were found to have wild-type TP53 alleles. The proband's brother (III.10), deceased at the age of 53 due to colorectal cancer, was himself a heterozygous carrier of the c.645del TP53 variant. Also, a 30-year-old deceased proband's sister (III.13) affected by both breast cancer and soft tissue sarcoma and his first female cousin (III.21) deceased at the age of 33 due to breast cancer were both heterozygous carriers of the c.645del TP53 variant. Finally, the proband's niece (IV.19), who has a diagnosis of breast cancer at the age of 34, and the proband's nephew (IV.20), who was healthy, were found to have wild-type TP53 alleles (Figure 2B).
Interestingly, other typical LFS tumors were described in the family pedigree. The proband's mother (II.2), who died at the age of 40, and his grandmother (I.2), who died at the age of 48, had breast and ovarian cancer, respectively. His half-sister (III.14), who had a 20-year-old deceased son due to osteosarcoma (IV.22) and an 18-year-old dead daughter due to spinocellular carcinoma (IV.21), was affected by a brain tumor and died at the age of 45. In addition, a brain tumor was found in the proband's nephew (IV.10), and the proband's first female cousin (III.22); they were deceased at the age of 42 and 45, respectively. All the proband's relatives affected by typical LFS cancers (I.2, II.2, III.14, III.22, IV.10, IV.21, IV.22, Figure 1) were deceased and did not receive any genetic testing.
Discussion
Here, we describe for the first time a family with a novel and rare germline variant in the TP53 gene responsible for LFS syndrome. Because the proband had a deceased affected son with osteosarcoma and a family having an aggregated history of typical LFS cancers, a mutational screening of the TP53 gene was offered to him and his family members. Genetic testing revealed a heterozygous pathogenic germline TP53 variant c.645del in the proband, his son, and three probands' relatives. At the protein level, the revealed c.645del variant causes a frameshift with the introduction of a premature stop codon within the p53 DNA binding domain (DBD) (p.Ser215Argfs*32). The DBD domain, located in the central part of the p53 protein, contains most of the LFS-associated missense variants (Bouaoun et al., 2016; AlHarbi et al., 2018). However, also frameshift pathogenic variants in the p53 DBD, such as c.685dup (p.Cys229Leufs*11), have been observed in a family with LFS (Ji et al., 2018).
The TP53 frameshift variant identified here, not previously described in any database including HGMD, LOVD, and ClinVar, was classified as pathogenic based on the American College of Medical Genetics (ACMG) criteria (PVS1+PM2+PP1_Moderate) (Richards et al., 2015).
Notably, a duplication of two bases at the same region in the TP53 gene (c.643_644dup), creating a frameshift starting at codon 215 (p.Ser215fs), was classified as pathogenic in the Clinvar database (accession ID: VCV000492747.1).
It is well-known that carriers of TP53 pathogenic variants have an increased risk of early onset cancers, often recognized as LFS syndrome (Kharaziha et al., 2019; Yamamoto et al., 2020). Although LFS is a highly penetrant cancer syndrome, its penetrance in subjects with a TP53 germline variant varies depending on the variant type (Varley et al., 2001; Bougeard et al., 2015). For example, the most severe variants of TP53 associated with earlier tumor onset are the dominant-negative missense variants because of their ability to produce malfunctioning or non-functioning p53 tetramers (Frebourg et al., 2020). Instead, null variants, including frameshift variants, such as that described here, are mainly observed in families with typically adult cancers with a lower disease penetrance (Bougeard et al., 2015; Frebourg et al., 2020).
The novel inherited frameshift TP53 variant described here results in a predicted truncated protein with a premature stop codon. There are two hypotheses explaining the consequence of this TP53 frameshift variant on p53 protein function. One suggests that the modified mRNA could be unstable and, thus, degraded by non-sense-mediated mRNA decay. The other is that the modified mRNA could be transcribed into a truncated protein having a dominant-negative effect that disrupts the p53 tumor-suppressor function.
Here, we describe a family carrying a novel pathogenic germline variant of the TP53 gene. This inherited frameshift variant is extremely rare because more than 70% of the pathogenic TP53 variants in LFS families were observed to be missense variants (AlHarbi et al., 2018). This family case report highlights the importance of offering genetic counseling and genetic testing to all family members at high risk to be carriers of TP53 inherited pathogenic variants.
Data Availability Statement
The datasets presented in this article are not readily available due to ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
The study was conceived by RI, FP, and NP. VB, FF, FP, and EC performed the experiments. FP, EC, FF, FB, VB, and RI analyzed and interpreted the data. FP, RI, FT, FF, and NP wrote the manuscript. VB, NP, and FT supervised the project. All authors contributed to the article and approved the submitted version.
Conflict of Interest
FP is employed by Tecnologica Research Institute and Marrelli Health.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Debora Caprella for her technical contribution.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.734809/full#supplementary-material
References
AlHarbi, M., Mubarak, N., AlMubarak, L., Aljelaify, R., AlSaeed, M., Almutairi, A., et al. (2018). Rare TP53 variant associated with Li-Fraumeni syndrome exhibits variable penetrance in a Saudi family. NPJ Genomic Med. 3, 1–6. doi: 10.1038/s41525-018-0074-3
Barnoud, T., Indeglia, A., and Murphy, M. E. (2021). Shifting the paradigms for tumor suppression: lessons from the p53 field. Oncogene 40, 4281–4290. doi: 10.1038/s41388-021-01852-z
Bittar, C. M., Vieira, I. A., Sabato, C. S., Andreis, T. F., Alemar, B., Artigalás, O., et al. (2019). TP53 variants of uncertain significance: increasing challenges in variant interpretation and genetic counseling. Familial Cancer 18, 451–456. doi: 10.1007/s10689-019-00140-w
Bouaoun, L., Sonkin, D., Ardin, M., Hollstein, M., Byrnes, G., Zavadil, J., et al. (2016). TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum. Mutat. 37, 865–876. doi: 10.1002/humu.23035
Bougeard, G., Renaux-Petel, M., Flaman, J.-M., Charbonnier, C., Fermey, P., Belotti, M., et al. (2015). Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J. Clin. Oncol. 33, 2345–2352. doi: 10.1200/JCO.2014.59.5728
de Andrade, K. C., Mirabello, L., Stewart, D. R., Karlins, E., Koster, R., Wang, M., et al. (2017). Higher-than-expected population prevalence of potentially pathogenic germline TP53 variants in individuals unselected for cancer history. Hum. Mutat. 38, 1723–1730. doi: 10.1002/humu.23320
Di Agostino, S., Fontemaggi, G., Strano, S., Blandino, G., and D'Orazi, G. (2019). Targeting mutant p53 in cancer: the latest insights. J. Exp. Clin. Cancer Res. 38, 1–3. doi: 10.1186/s13046-019-1302-0
Egan, K. M., Nabors, L. B., Olson, J. J., Monteiro, A. N., Browning, J. E., Madden, M. H., et al. (2012). Rare TP53 genetic variant associated with glioma risk and outcome. J. Med. Genet. 49, 420–421. doi: 10.1136/jmedgenet-2012-100941
Frebourg, T., Lagercrantz, S. B., Oliveira, C., Magenheim, R., and Evans, D. G. (2020). Guidelines for the Li-Fraumeni and heritable TP53-related cancer syndromes. Eur. J. Hum. Genet. 28, 1379–1386. doi: 10.1038/s41431-020-0638-4
Ji, M., Wang, L., Shao, Y., Cao, W., Xu, T., Chen, S., et al. (2018). A novel dysfunctional germline P53 mutation identified in a family with Li-Fraumeni syndrome. Am. J. Cancer Res. 8:165–169.
Kharaziha, P., Ceder, S., Axell, O., Krall, M., Fotouhi, O., Böhm, S., et al. (2019). Functional characterization of novel germline TP53 variants in Swedish families. Clin. Genet. 96, 216–225. doi: 10.1111/cge.13564
McBride, K. A., Ballinger, M. L., Killick, E., Kirk, J., Tattersall, M. H., Eeles, R. A., et al. (2014). Li-Fraumeni syndrome: cancer risk assessment and clinical management. Nat. Rev. Clin. Oncol. 11:260. doi: 10.1038/nrclinonc.2014.41
Miller, M., Shirole, N., Tian, R., Pal, D., and Sordella, R. (2016). The evolution of TP53 mutations: from loss-of-function to separation-of-function mutants. J. Cancer Biol. Res. 4:1091.
Olivier, M., Hollstein, M., and Hainaut, P. (2010). TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor Perspect. Biol. 2:a001008. doi: 10.1101/cshperspect.a001008
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423. doi: 10.1038/gim.2015.30
Schneider, K., Zelley, K., Nichols, K. E., and Garber, J. (2019). Li-fraumeni syndrome. Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1311/
Valdez, J. M., Nichols, K. E., and Kesserwan, C. (2017). Li-Fraumeni syndrome: a paradigm for the understanding of hereditary cancer predisposition. Brit. J. Haematol. 176, 539–552. doi: 10.1111/bjh.14461
Varley, J., Attwooll, C., White, G., McGown, G., Thorncroft, M., Kelsey, A. M., et al. (2001). Characterization of germline TP53 splicing mutations and their genetic and functional analysis. Oncogene 20, 2647–2654. doi: 10.1038/sj.onc.1204369
Yamamoto, Y., Kanai, M., Kou, T., Sugiyama, A., Nakamura, E., Miyake, H., et al. (2020). Clinical significance of TP53 variants as possible secondary findings in tumor-only next-generation sequencing. J. Hum. Genet. 65, 125–132. doi: 10.1038/s10038-019-0681-6
Young, J. L., Pantaleao, A., Zaspel, L., Bayer, J., Peters, J. A., Khincha, P. P., et al. (2019). Couples coping with screening burden and diagnostic uncertainty in Li-Fraumeni syndrome: connection versus independence. J. Psychosoc. Oncol. 37, 178–193. doi: 10.1080/07347332.2018.1543376
Keywords: Li-Fraumeni, TP53, targeted sequencing, LFS cancers, germline TP53 variant
Citation: Paduano F, Fabiani F, Colao E, Trapasso F, Perrotti N, Barbieri V, Baudi F and Iuliano R (2021) Case Report: Identification of a Novel Pathogenic Germline TP53 Variant in a Family With Li–Fraumeni Syndrome. Front. Genet. 12:734809. doi: 10.3389/fgene.2021.734809
Received: 01 July 2021; Accepted: 02 August 2021;
Published: 01 September 2021.
Edited by:
Prashant Kumar Verma, All India Institute of Medical Sciences, IndiaReviewed by:
Saverio Alberti, University of Messina, ItalyRami I. Aqeilan, Hadassah Medical Center, Israel
Copyright © 2021 Paduano, Fabiani, Colao, Trapasso, Perrotti, Barbieri, Baudi and Iuliano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rodolfo Iuliano, aXVsaWFub0B1bmljei5pdA==; Francesco Paduano, ZnJhbmNlc2NvLnBhZHVhbm9AdW5pY3ouaXQ=