 Yang Lyu
Yang Lyu Zhongqi Liufu
Zhongqi Liufu Juan Xiao
Juan Xiao Tian Tang
Tian Tang- State Key Laboratory of Biocontrol and Guangdong Key Laboratory of Plant Resources, School of Life Sciences, Sun Yat-sen University, Guangzhou, China
New miRNAs are evolutionarily important but their functional evolution remains unclear. Here we report that the evolution of a microRNA cluster, mir-972C rewires its downstream regulatory networks in Drosophila. Genomic analysis reveals that mir-972C originated in the common ancestor of Drosophila where it comprises six old miRNAs. It has subsequently recruited six new members in the melanogaster subgroup after evolving for at least 50 million years. Both the young and the old mir-972C members evolved rapidly in seed and non-seed regions. Combining target prediction and cell transfection experiments, we found that the seed and non-seed changes in individual mir-972C members cause extensive target divergence among D. melanogaster, D. simulans, and D. virilis, consistent with the functional evolution of mir-972C reported recently. Intriguingly, the target pool of the cluster as a whole remains relatively conserved. Our results suggest that clustering of young and old miRNAs broadens the target repertoires by acquiring new targets without losing many old ones. This may facilitate the establishment of new miRNAs in existing regulatory networks.
Introduction
Newly evolved genes constitute at least 10–20% of the genome in every taxonomic group (Khalturin et al., 2009; Kondo et al., 2017), and they play a significant role in the innovations of biological traits (Kaessmann, 2010; Chen et al., 2013). Increasing evidence suggests that a large fraction of the new genes are functionally important (Kaessmann, 2010; McLysaght and Hurst, 2016; Kondo et al., 2017; Xia et al., 2021). For instance, they are primarily expressed in testes, and are often involved in reproductive functions including male fertility (Gubala et al., 2017; Kondo et al., 2017; Lange et al., 2021), sperm competition (Yeh et al., 2012), courtship (Dai et al., 2008), and pheromone metabolism (Zhang J. et al., 2004). The functional importance of these testes-biased genes is also supported by the prominent signatures of positive selection at these loci (Zhao et al., 2014). Despite the evolutionary significance of the new genes, we know few about the mechanisms through which these novel elements integrated into the regulatory networks. Transcriptomic and protein-protein interaction studies suggest that the targets of some new genes changed dramatically even among closely related species (Chen et al., 2012; Ross et al., 2013), yet the underlying mechanisms are unclear.
While the techniques involved in determining the targets of young protein-coding genes are challenging, it is feasible to predict the targets of newly evolved microRNAs (miRNAs). miRNAs are a class of ubiquitous post-transcriptional regulators that participate in diverse biological processes in eukaryotes (Bartel, 2004; DeVeale et al., 2021). In animals, mature miRNAs (about 22 nt long) prevent the protein accumulation of the targets by either repressing translation or inducing mRNA degradation, through binding to the 3′ untranslated region (3′ UTR) of the transcripts with their seed region (the 2nd–8th nucleotides of the mature sequence) (Bartel, 2009, 2018). Collectively, miRNAs have broad impacts on the transcriptome, as each of them potentially have hundreds of targets (Agarwal et al., 2015). Their effects on individual targets, however, are usually weak (Baek et al., 2008; Selbach et al., 2008). Even for the most highly-expressed miRNAs, the repression effects on individual targets are usually less than 50% (Guo et al., 2010; Stadler et al., 2012).
Using next-generation sequencing techniques, previous studies have identified a large cohort of new miRNAs across taxa (Berezikov, 2011). In Drosophila, we have reported that the birth and death of miRNAs is extremely rapid (Lyu et al., 2014). It has been shown that over 40% of the miRNAs are only observed in the specific lineages (Lyu et al., 2014). Among these evolutionarily young miRNAs, 95% of them likely arose from scratch, as their seeds and precursors are different from that of the existing miRNAs (Lyu et al., 2014). It appears that these newly-evolved miRNAs have introduced a wide array of novel miRNA-mRNA interactions. Similar to new protein-coding genes, young miRNAs are inclined to express in testes, and they exhibit strong signatures of positive selection (Lyu et al., 2014; Mohammed et al., 2014). Understanding the mechanisms through which new miRNAs and targets evolve will provide key insights into the evolutionary processes of new genes. For example, how novel components originated and integrated into biological networks.
Our previous study has identified a Drosophila-specific miRNA cluster that we refer as mir-972C (Lyu et al., 2014). In Drosophila melanogaster, it consists of at least 12 miRNA members including mir-972, making it the largest new miRNA cluster in this species by far (Marco et al., 2013; Lyu et al., 2014; Mohammed et al., 2018). We speculated that the mir-972C is evolutionarily important, as it is highly expressed in testes and its DNA sequences exhibit strong signal of positive selection (Lyu et al., 2014). To understand how newly-evolved miRNAs influence gene regulatory networks, we investigated the evolution of miR-972C sequences and the regulatory networks within Drosophila. We found that not only the members of mir-972C vary across species, their sequences also undergo rapid changes, which cause the evolution of the target repertoire. In the end, we discussed the selective forces that may drive the evolution of new miRNA clusters in the long-term.
Materials and Methods
Genomic Data
mir-972C sequences and coordinates were obtained from miRBase (mirbase.org, Release 22.1) (Kozomara et al., 2019). Genome sequences were retrieved from UCSC (genome.ucsc.edu). The genome versions used here are: D. melanogaster, dm6; D. simulans, droSim1; D. sechellia, droSec1; D. yakuba, droYak2; D. erecta, droEre2; D. ananassae, droAna3; D. pseudoobscura, dp4; D. virilis, droVir3; Anopheles gambiae, MOZ2; Apis mellifera, Amel_2.0. GTF annotation files, 3′UTR sequences and 3′UTR locations were downloaded from flyBase (flybase.org, file version: D. melanogaster, r6.17; D. simulans, r2.02; D. virilis, r.1.06). Small RNA and mRNA testes deep-sequence libraries from D. melanogaster, D. simulans, D. pseudoobscura, and D. virilis (Czech et al., 2008; Rozhkov et al., 2010; Brown et al., 2014; Lyu et al., 2014; Ahmed-Braimah et al., 2017; Zhao et al., 2018) were retrieved from the GEO database (GEO accession IDs are listed in Supplementary Table S1).
miRNA Homolog Search, Reads Validation, and Phylogenetic Inference
We searched for mir-972C sequences in the Drosophila genomes using BLAT (Kent, 2002) with default parameters and an E-value threshold of 0.001. Homologs of the mir-972C members in each species were identified using BLAST (Altschul et al., 1990) with queries of the known precursor sequences (miRBase Release 22.1) and an E-value threshold of 0.001. miRNA homologs from different species were aligned using MUSCLE (Edgar, 2004) with default parameters. To validate these miRNAs in D. melanogaster, D. simulans, D. pseudoobscura, and D. virilis, we used miRDeep2 (Friedländer et al., 2012) to map the small RNA sequencing reads (see Supplementary Table S1 for the information of the libraries) back to the genomic sequences of the entire cluster with default parameters. We used five standards that derived from a publication (Fromm et al., 2015) to validate miRNAs: 1) at least one miR* read; 2) at least 20 reads mapping to miR and miR* in total; 3) a hairpin structure with at least 13 paired nucleotides in miR:miR* duplex; 4) The top 3 iso-miR reads account for 85% of the miR arm reads and 5) the miR:miR* duplex to background reads ratio is >1. A maximum parsimony analysis was used to infer the origination of the mir-972C members by assuming that a miRNA emerged in the most recent common ancestor of all species bearing verified homologs.
Target Evolution and Functional Analyses
We predicted the target sites using TargetScan (Lewis et al., 2005). “8mer” and “7mer-8A” targets were used for the following analyses. To select testes-expressed genes, we used the published testes RNA-seq data (Supplementary Table S1) and mapped the reads to the genome using STAR (parameters: -runThreadN 4 -runMode genome Generate) (Dobin et al., 2013). Read counts at gene level were calculated by counting all the reads that overlapped any exon for each gene using featureCounts (Liao et al., 2014) followed by TPM (Transcripts Per Kilobase Million) normalization. Genes with an average TPM (across multiple replicates)
In vitro Validation of miR-975 Targets
To construct pUAST-mir-975 plasmids from each species, we amplified mir-975 genes from the genomic sequences of D. melanogaster (ISO-1), D. simulans (simNC48S), and D. virilis (V46) and cloned the fragments into a pUAST vector (see Supplementary Table S3 for primers and restriction sites). PCR reactions were carried out using the EX-Taq DNA Polymerase (TAKARA). Cells were transfected in a 48-well plate with 100 ng of ub-GAL4 and 200 ng of conspecific pUAST-mir-975 or the control vector (pUAST only) using Lipofectamine 2000 (Thermo Fisher Scientific, catalog no.12566014). Samples were collected 48 h after transfection.
Total RNAs were extracted from the samples using TRIzol (Thermo Fisher Scientific, catalog no. 15596026) for qRT-PCR and RNA-seq analyses. To quantify miRNA expression, total RNAs were reverse-transcribed into cDNAs using stem-loop reverse transcription and analyzed using the TaqMan qRT-PCR method following the miRNA UPL (Roche Diagnostics) probe assay protocol (He et al., 2016). The 2S RNA was used as the endogenous control (see Supplementary Table S4 for the qRT-PCR primers). cDNA libraries for each RNA sample were sequenced using the Illumina HiSeq 2000 at the Beijing Genomics Institute (Shenzhen). Reads were mapped to the genomes using TopHat (v.1.3.1) with a parameter -r 20 (Trapnell et al., 2009). Gene expression was estimated by FPKM (Fragments Per Kilobase per Million) using Cufflinks (v.2.1.1) with default parameters (Trapnell et al., 2010). Differentially expressed genes were determined using Cuffdiff (v.2.1.1) with default parameters (Trapnell et al., 2010). Non-expressed genes (FPKM = 0) were removed from the further analyses.
To confirm the trans effects of miRNA evolution on target repression, we expressed dme-mir-975 and dsi-mir-975 in S2 cells, respectively, and measured the expression of nine predicted targets of dme-mir-975. gDNA was removed from total RNAs by using TURBO DNA-free kit (Thermo Fisher Scientific, catalog no. AM 1907). Total RNAs were transcribed into cDNAs with the PrimeScript first strand cDNA synthesis kit (TAKARA Bio, catalog no. 6110A) and followed by a qPCR analysis using the SYBR Premix Ex Taq II kit (TAKARA Bio, catalog no. RR82WR). rp49 was used as an internal control. Primers used are listed in Supplementary Table S5.
Results
Gain and Loss of Members in a Fast-Evolving miRNA Cluster
The mir-972C cluster of D. melanogaster comprises 12 miRNAs spanning a 10.8-kb region located in the 18C-D band of the X chromosome. Based on the genomic proximity among members, the cluster was further divided into three sub-clusters (mir-972/9369/973/974, mir-4966-1/4966-2/975/976/977, mir-978/979), spanning less than 1 kb of each, along with an orphan miRNA mir-2499 (Figure 1). The mir-972C members most likely originated de novo, as no sequence similarity was found either between the cluster members except for the mir-4966 duplicates, or between them and other miRNAs that have been characterized in D. melanogaster (BLAST search, E < 0.001).
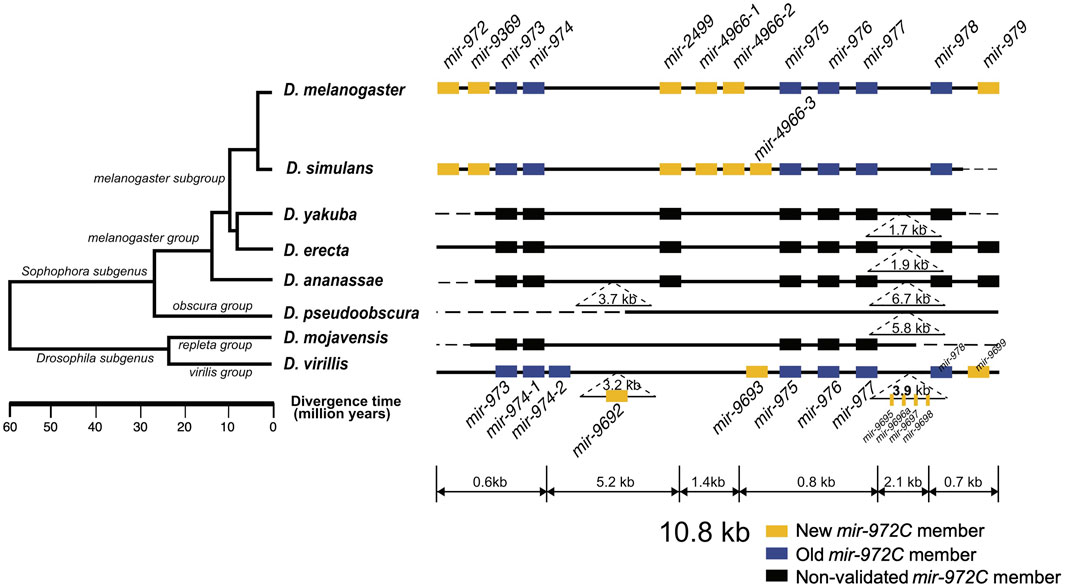
FIGURE 1. Evolutionary history of the mir-972 cluster (mir-972C) in Drosophila. This cluster includes twelves miRNAs with distinct seeds. New (yellow boxes) and old (blue boxes) mir-972C members validated through deep sequencing (see Methods) are shown. miRNA homologs are colored in black. Deletions and insertions observed in sequence alignments are represented by dashed lines and inverted triangles. The genomic region is not drawn to scale. The phylogenetic tree was previously reconstructed based on whole-genome sequences (Clark et al., 2007).
To investigate the origin and evolution of mir-972C, we searched for the orthologs of these miRNA genes in other Drosophila species, and in Aedes (mosquito) and Apis (honey bee) which have diverged from Drosophila 250 and 300 million years ago, respectively (Honeybee Genome Sequencing Consortium 2006; Yeates and Wiegmann, 1999). We found homologous sequences in all the seven Drosophila genomes surveyed (D. simulans, D. yakuba, D. erecta, D. ananassae, D. pseudoobscura, D. mojavensis, and D. virilis), but failed to detect any homologs in the mosquito or the honey bee genomes. This result indicates that mir-972C most likely emerged in the common ancestor of Drosophila between 60 and 250 million years ago. After origination, individual members of mir-972C have undergone rapid birth and death. In the D. pseudoobscura genome, the homologous sequences of mir-2499 and mir-979 were identified. Using the conspecific testes library, we were unable to detect the expression of these sequences, suggesting the loss of the entire cluster in this species (Figure 1). The distribution of individual miRNAs also varies across the remaining species. For example, mir-973/974/975/976/977/978 sequences are represented in all the species except D. pseudoobscura, while other miRNAs have been lost in various lineages (Figure 1).
To date the origin time of each miRNA, we validated the expression of individual mir-972C members in the Drosophila species where small RNA sequencing data from testes were available, including: D. melanogaster, D. simulans, D. pseudoobscura, and D. virilis (see Supplementary Table S1 for data information). Using the recently proposed criteria for miRNA annotation (Fromm et al., 2015), we found that mir-973/974/975/976/977/978 are expressed in D. melanogaster, D. simulans, and D. virilis; mir-972/9369/2499/4966 are expressed in D. melanogaster and D. simulans, mir-979 is only expressed in D. melanogaster, and mir-9692/9693/9695/9696a/9697/9698/9699 only exist in D. virilis (see Supplementary Data for arm occupancy). miRNA expression in D. pseudoobscura was not detected.
Taken together, these results indicate that mir-972C initially originated in the common ancestor of Drosophila and subsequently diverged among different clades. Although the cluster originated more than 60 million years ago, the youngest member, mir-979, emerged in the recent 4 million years. Based on the phylogeny, we classified the miRNAs into the new members that originated after the Sophophora/Drosophila split (mir-972/9369/2499/4966/979) and the old members that arose before that event (mir-973/974/975/976/977/978) (Figure 1).
Evolution of miR-972C Targets Mediated by Seed Innovation
After showing the evolution of the mir-972C members, we continued to investigate the sequence changes in individual miRNAs among D. melanogaster, D. simulans, and D. virilis. We are particularly interested in the alterations in the seed sequences as they are responsible for target recognition. The precursor alignments (Supplementary Figure S1) reveal two types of seed changes: 1) seed shifting, in which the dominant mature miRNA is shifted due to the changes in Drosha and Dicer processing (e.g., mir-976), and 2) arm switching, in which the mature miRNA switches to the other arms of the precursor (e.g., mir-975). We further inferred the time that the these two types of events occurred on the phylogenetic tree (Figure 2A). As shown in Figure 2A, six of the nine events occurred after the split of D. melanogaster and D. simulans in the recent 4 million years. Both the new and the old mir-972C members are involved in this seed innovation. The arm switching of mir-975 occurred after the split of D. virilis and D. melanogaster/D. simulans but it is unclear on which branch (Figure 2A). mir-978 is the only member that experienced both seed shifting and arm switching, and its seed is different among all three species (Supplementary Figure S1).
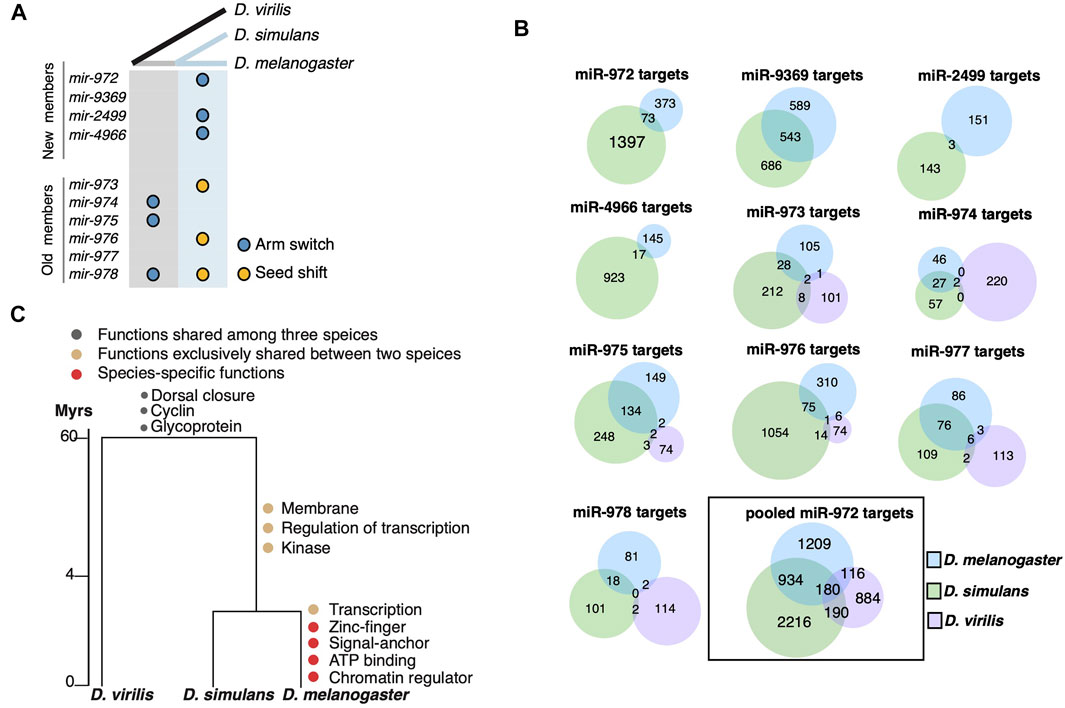
FIGURE 2. Evolution of miR-972C targets. (A) miR-972C seed innovations. Arm switching (blue circles) and seed shifts (yellow circles) were inferred and denoted along ancestral (grey) and recent (light blue) branches. (B) Venn diagrams depict the number of shared targets of individual miR-972C member targets or pooled cluster targets. (C) Functional evolution of targets. GO categories of shared and lineage-specific targets are indicated on the corresponding evolutionary branches.
Observing the significant changes in the seed region, we studied the evolution of miR-972C targets. Since mir-975C appears exclusively expressed in testes, we first excluded genes that are not expressed in this tissue from the target analysis. To this end, RNA-seq reads from the testes of D. melanogaster, D. simulans, and D. virilis (Brown et al., 2014; Ahmed-Braimah et al., 2017) were mapped to the conspecific genome and the number of reads within each gene was normalized to TPM (Transcript Per Million). After removing the genes whose expression was not supported by enough reads (TPM <1), we retained 11,149 genes in D. melanogaster, 11,832 in D. simulans, and 10,453 in D. virilis for further analyses. Overall, 37.9% of the transcripts expressed in testes were overlapped among the three species (see Supplementary Figure S2A for the numbers of overlapping and species-specific transcripts. 37.9% is as the proportion of the overlap targets relative to the total targets in the species). It has been reported that testes-expressed genes exhibit lineage-specific bursts of rapid evolution and positive selection (Haerty et al., 2007; Parsch and Ellegren, 2013), which likely to contributes to the limited overlap among the Drosophila species. Next, we searched for the miR-972C target sites on the 3′-UTR of these testes-specific transcripts using TargetScan (Agarwal et al., 2018). As shown in Figure 2B, the number of the overlapping targets between the D. melanogaster/D. simulans branch and D. virilis is extremely small (<10 for each miRNA). Since TargetScan searches for miRNA target sites on the 3′UTR that pair with the seed sequences, target divergence among species is either due to the changes in the seed sequences themselves, or due to the alterations of miRNA binding sites on the 3′-UTRs. As for mir-974 and mir-977, their seeds are fully conserved among the Drosophila species. Even so, their targets are not shared much among the species, suggesting significant changes in the miRNA binding sites on the 3′UTRs. Considering the sequences of testes-expressed genes have evolved much faster (Haerty et al., 2007; Parsch and Ellegren, 2013), the limited number of sharing targets is not completely surprising. In addition, 3′UTRs of testes-expressed genes are significantly shorter (Sanfilippo et al., 2017), which makes them less likely to be targeted by miRNAs. The 3′UTR shortening might contribute to the low number of sharing targets as well. These results suggest that 3′UTR divergence also plays an important role in the target evolution of miR-972C.
We took a closer look at the target divergence between D. melanogaster and D. simulans. While the mir-9369/974/975/977 seeds are identical between these species (Supplementary Figure S1), the proportion of overlapping targets ranges from 22.0 to 29.9% (Figure 2B). Both seed shifting and arm switching significantly reduced the overlap: only 1.0–4.0% of targets are shared between the two species after arm switching (mir-972/2499/4966, Figure 2B), and 5.2–8.8% are shared after seed shifting (mir-973/976/978) (Figure 2B). Although the number of overlapping targets between D. melanogaster and D. simulans was small for each miRNA after seed changes, the overlap in targets for the entire cluster (22.8%) was largely comparable with that of the miRNAs with identical seeds (Figure 2B). This is likely because a 3′UTR targeted by a mir-972C member in one species can be targeted by a different member in another. These observations support the idea that although the targets of each miRNA evolve rapidly, the entire miRNA cluster keeps a relatively conserved target pool.
To understand the biological consequences of miR-972C target evolution, we examined the functional enrichment of the predicted targets on each evolutionary branch using DAVID (Huang et al., 2009). Mutual targets that are shared among the three species are enriched in “dorsal closure” (p = 1.0E-05), “Cyclin” (p = 0.046) and “Glycoprotein” (p = 0.039), indicating a possible role of this cluster in the common ancestor (Figure 2C, Supplementary Table S2). After the split of D. virilis and the D. melanogaster/D. simulans branch, we observed a burst of new GO categories on the D. melanogaster/D. simulans branch and also in D. melanogaster (i.e., regulation of transcription, p = 0.027), consistent with the increase in the target number of this lineage (Figure 2B). Interestingly, D. melanogaster continued to gain new targets that are involved in “transcription” (p = 1.1E-03), suggesting a reinforcement of the ancestral functions. These results indicate that the evolution of miR-972C targets may contribute to functional innovation between species.
Both Seed and Non-seed Mutations Contributed to the Target Evolution of miR-975
Among the members of miR-972C, miR-975 is especially intriguing as it has distinct effects on male reproduction between D. melanogaster and D. simulans (Zhao et al., 2021), and thus offers an opportunity to study the functional evolution of miRNAs. mir-975 has undergone substitutions in both seed and non-seed regions (Figure 2A, Supplementary Figure S1). The mir-975 seed in D. virilis is completely different from those in D. melanogaster and D. simulans as a result of arm switching (Figure 2A, Supplementary Figure S1). Furthermore, there is a single nucleotide substitution right next the seed region in the mature miR-975 sequences between D. melanogaster and D. simulans (Supplementary Figure S1).
To understand the impact of mir-975 sequence evolution on its target repertoire, we overexpressed the conspecific mir-975 sequences in cells derived from D. melanogaster (S2), D. simulans (ML-82-19a), and D. virilis (WR-Dv-1) and monitored the expression changes of both miR-975 itself and the transcriptome as a whole. Quantitative PCR confirmed that miR-975 was only expressed in cells transfected with the pUAST-mir-975 vector but not in cells transfected with the control pUAST vector (Figure 3A). Venn diagram shows that the vast majority of the transcripts expressed in the three cell lines were overlapped (Supplementary Figure S2B). When miR-975 was overexpressed, predicted targets were significantly down-regulated compared to the transcriptomes of the D. melanogaster and the D. simulans cells (Figures 3B,C, Kolmogorov-Smirnov test, both p < 0.05). The repression magnitude is small, consistent with the weak repression from miRNAs (Zhao et al., 2017; Chen et al., 2019). Target repression is not significant in D. virilis cells (Kolmogorov-Smirnov test, p = 0.41, Figure 3D), probably because there are only a few predicted targets in this species (n = 65).
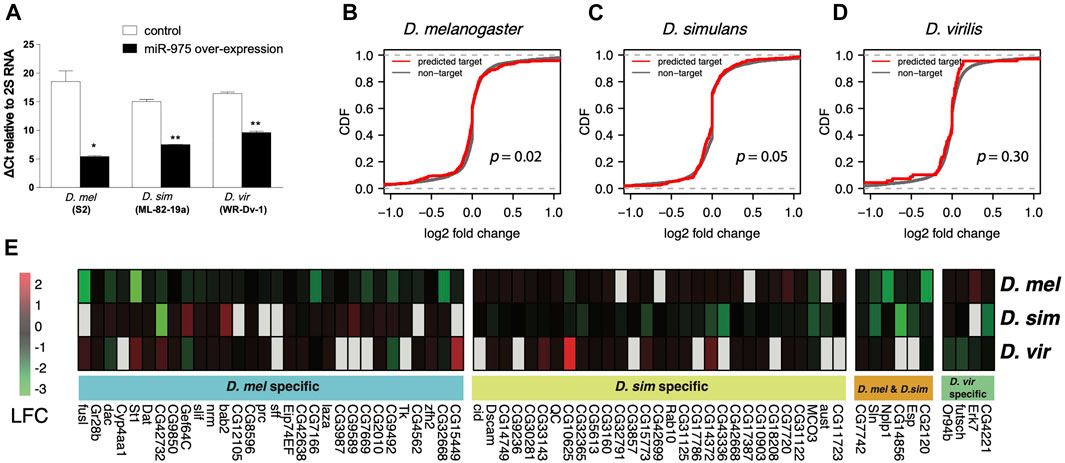
FIGURE 3. In vitro validation of miR-975 target divergence. (A) Relative miR-975 expression levels in D. melanogaster (S2), D. simulans (ML-82-19a), and D. virilis (WR-Dv-1) cell lines after transfection with the pUAST-only vector (control) or conspecific pUAST-mir-975. The bar plot shows ΔCt values (CtmiR-975—Ct2S RNA) from the qRT-PCR assays. (B–D) Effect of miR-975 on predicted targets and non-targets in cell lines derived from D. melanogaster (B), D. simulans (C), and D. virilis (D). (CDF: cumulative distribution function. p-values from the Kolmogorov-Smirnov test are shown). (E) Heatmap shows the log2 fold change (LFC) of validated miR-975 targets in the three species.
Using 1.2-fold repression as a cut-off (Wen et al., 2011), we found 36 targets that were down-regulated in D. melanogaster, 35 in D. simulans, and four in D. virilis (Figure 3E). As expected, none of these targets were shared between D. virilis and the other two species. We also confirmed arm switching of dvi-miR-975 in WR-Dv-1 cells using qPCR (Supplementary Figure S3). These results indicated that the target pool was completely changed by arm switching. Between D. melanogaster and D. simulans, although the seed of miR-975 is identical, only six (9.2%) of the down-regulated targets are identical (Figure 3E). Taken together, our in vitro experiments demonstrate that both seed and non-seed changes in mir-975 contribute to the evolution of target regulatory networks among the Drosophila species.
D. simulans miR-975 Has a Weaker Effect on the D. melanogaster Transcriptome Than the Conspecific miR-975
Since the evolution of miRNA sequences and 3′UTRs could both contribute to the changes in miRNA targeting (Hausser and Zavolan, 2014), we next investigated the trans effects of miR-975 on the D. melanogaster transcriptome. As shown in Supplementary Figure S1, the ninth base of the mir-975 mature sequences underwent a transversion (G ->U) after the split of D. melanogaster and D. simulans (Figure 4A). As a result, target sites complementary to the ninth base of the mir-975 mature sequence were enriched for adenine (A) in D. melanogaster but enriched for cytosine (C) in D. simulans (p < 0.05, Fisher’s exact test, Figures 4B,C). To determine the effects of this mutation, we conducted reporter assays by transferring either the dme-mir-975 or the dsi-mir-975 fragment to the D. melanogaster S2 cells, along with a ub-Gal4 driver (see Materials and Methods). qPCR assay shows that both dme-miR-975 and dsi-miR-975 were highly expressed in the S2 cells after transfection (Supplementary Figure S4), and miR-975 was not detected in the S2 cells transfected with the ub-Gal4 control. We examined the expression of nine D. melanogaster specific targets that have an “A” site complementary to the ninth base of the mature dme-miR-975. Six out of the nine targets were significantly down-regulated by overexpressing dme-miR-975 (p < 0.05, student’s t-test), whereas none was repressed by overexpressing dsi-miR-975 (Figure 4D), despite the expression level of dsi-miR-975 was much higher than dme-miR-975 (Supplementary Figure S4). These results suggest that even the innovation of non-seed region mediates significant changes in target repertoire. Importantly, since 70.6–86.0% of the transcripts that expressed (TPM ≥ 1) in the S2 cells were also detected in the testes (Supplementary Figure S2C), our in vitro analysis has strong implications on the evolution of miRNA target repertoire in vivo.
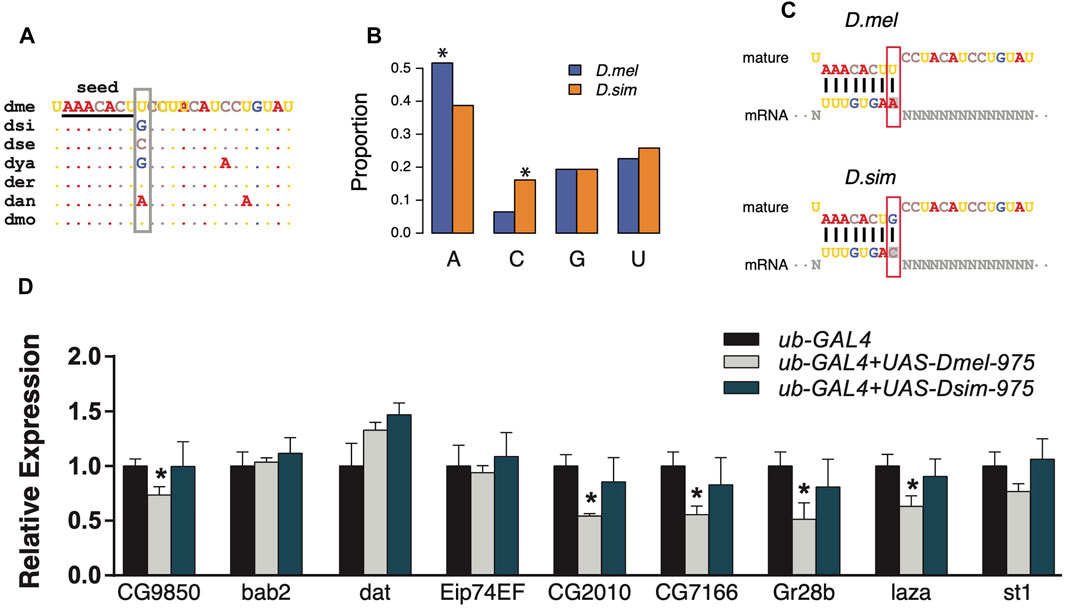
FIGURE 4. Divergent regulation effects of dme-miR-975 and dsi-miR-975 in S2 cells. (A) miR-975 mature sequences from seven Drosophila species. They share the same seed (underlined), but their 9th nucleotide (grey box) varies among species. Species abbreviations: dme, D. melanogaster; dsi, D. simulans; dse, D. sechellia; dya, D. yakuba; der, D. erecta; dan, D. ananassae; dmo, D. mojavensis. (B, C) Bar plot demonstrates that the 3′UTR sites bound to the 9th base of mature sequences are enriched for A in D. melanogaster but are enriched for C in D. simulans (see C for the sequence match). (D) qPCR results validated that evolution of the 9th nucleotide led to the differential regulation of dme-miR-975 and dsi-miR-975 in S2 cells.
Discussion
New genes continuously contribute to genetic novelty and offer a unique opportunity to understand the phenotypic divergence between species and the evolution of genetic regulatory networks (Tang et al., 2010; Chen et al., 2012; Ross et al., 2013; Zhao et al., 2021). As key players in gene regulation, miRNAs repress their targets weakly but broadly in animals (Zhao et al., 2017; Chen et al., 2019). However, it remains unclear how their functions have evolved, which might ultimately determine their evolutionary fate (Wu et al., 2009; Lyu et al., 2014; Penso-Dolfin et al., 2018). Some debates even have been focused on whether new miRNAs have biological functions at all (Nozawa et al., 2010). Here we show that the adaptive evolution of the mir-972C is accompanied by dramatic evolution of the target repertoires between distantly and closely related Drosophila species. Importantly, we found that changes in both seed and non-seed regions contribute to the evolution of the target pool. While the sequence evolution of each member has recruited new targets that represent novel functions in specific lineages, the vast majority of old targets are conserved when we consider the cluster as a whole. These results shed light on the survival and evolution of new genes in general.
Our results suggest that in a clustering form, new miRNAs may have a higher chance to survive and integrate into the regulatory networks (Zhang et al., 2007; Wang et al., 2016). Individual miRNAs, especially the evolutionarily young ones, have little effects on their targets (Sood et al., 2006; Chen and Rajewsky, 2007; Chen et al., 2019). However in a cluster, new miRNAs are co-expressed and function as a unit (Kabekkodu et al., 2018). As a result, the target pool is predicted to expand greatly. Indeed, while only 0.7–10.1% of the testes-specific transcriptome is potentially targeted by individual mir-972C members in D. melanogaster (Figure 2B), 21.1% can be influenced by the whole cluster together. Recent studies have shown that large miRNA target pools are evolutionarily beneficial in maintaining stability of gene expression through broad and weak regulation (Zhao et al., 2017; Chen et al., 2019). Consistent with this notion, a significant proportion of the miR-972C target pool remains unchanged (e.g., 22.8% of the targets are conserved between D. melanogaster and D. simulans, Figure 2B), despite arm switching and seed shifting occurred frequently between species.
Functional analysis of the target repertoire shows a reinforcement of the ancestral functional categories of miR-972C targets (Figure 2C). It also suggests that this miRNA cluster continues to recruit additional targets either through the evolution of existing miRNAs or the birth of new hairpins. Such processes may also bring novel functions. It is thus not unexpected that members of mir-972C does not follow the reported pattern for insect miRNAs that conserved miRNAs tend to express at a higher level and possess more targets than lineage-specific miRNAs (Chen and Rajewsky, 2007; Ylla et al., 2016). We found no significant difference in expression levels between young and old members of mir-972C in any of the three species on survey (all p > 0.05, Mann-Whitney U test, Supplementary Figure S5A). Old members have more targets than the young ones in D. simulans (p = 0.044, Mann-Whitney U test, Supplementary Figure S5B) but not in D. melanogaster or D. virilis (p > 0.05, Mann-Whitney U test, Supplementary Figure S5B). It is likely that new miRNAs in this cluster have quickly increased their expression levels and recruited a large number of targets.
Fast-evolving targeting implies that these miRNAs have never been deeply integrated into the existing gene regulatory networks. The long-term survival of these novel miRNAs remains unclear. Previously we have shown that miR-975 exerts different influences on male fertility between D. melanogaster and D. simulans, and its loss-of-function might be adaptive in some lineages (Lu et al., 2018a, 2018b; Zhao et al., 2021). Another good example in Drosophila is the mir-310/311/312/313 cluster (mir-310C), which is another adaptive miRNA cluster with a same age as mir-972C (Lu et al., 2008; Lyu et al., 2014). mir-310C is known to affect egg morphology, hatchability, and male fertility (Pancratov et al., 2013; Liufu et al., 2017). Redundant and incoherent regulation of multiple phenotypes by mir-310C suggests that these miRNAs play a role in stability control (Liufu et al., 2017). It is thus not surprising that the miRNA-target interactions could be readily changed. Unlike mir-310C that was duplicated from mir-92a/b, mir-972C seems to have evolved from non-functional sequences and transcribed specifically in testes (Marco, 2014; Mohammed et al., 2014). The cost of gene loss is more acceptable when the expression of the gene is restricted in fewer tissues (Fraïsse et al., 2019). For this reason, the loss of the entire mir-972 cluster in the D. pseudoobscura lineage is not surprising (Figure 1).
It should be noted that as a testes-biased miRNA cluster, the fast evolution of mir-972C may be associated with the rapid turnover of cellular environments in this tissue. It is well established that testis is the most rapidly evolving tissue due to the selective forces associated with sperm competition, reproductive isolation, and sexual conflict (Kaessmann, 2010). Previous investigations in many taxa have demonstrated that male-biased genes evolve relatively quickly at both sequence and expression level (Meiklejohn et al., 2003; Zhang Z. et al., 2004; Ellegren and Parsch, 2007; Yang et al., 2016). Changes of chromatin states during spermatogenesis allow aberrant transcription which makes testis a hotspot for new gene origination (Kaessmann, 2010). This cellular environment may boost the evolutionary rate of genes with which it has co-evolved, including miRNAs (Tang et al., 2010; Wang et al., 2016). Interestingly, mir-972C targets do not show GO enrichment in male functions (Figure 2C), despite the testes-specific expression of this cluster (Mohammed et al., 2014; Lu et al., 2018b). Why would mir-972C be beneficial to the male reproductive system? One plausible explanation is that the high complexity of the testes transcriptome (Vibranovski et al., 2009) that requires substantial regulations to stabilize the system (Wu et al., 2009). mir-972C would be an excellent candidate to do so as it is highly abundant and broadly tied to the testes transcriptome.
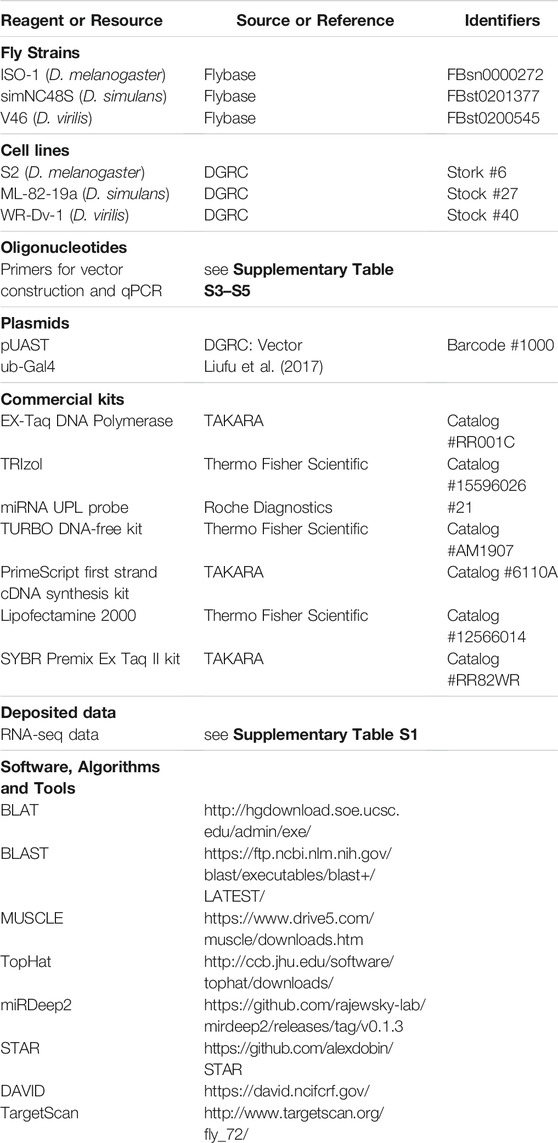
Key Resources Table
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE107390.
Author Contributions
YL and TT conceived the study, YL, ZL and JX conducted the research, YL and ZL analyzed the data, YL, ZL and TT wrote the paper.
Funding
This study was funded by the National Science Foundation of China (31801081, 31770246), Guangdong Natural Science Foundation (2020A1515010467).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.760530/full#supplementary-material
References
Agarwal, V., Bell, G. W., Nam, J.-W., and Bartel, D. P. (2015). Predicting Effective microRNA Target Sites in Mammalian mRNAs. eLife 4, e05005. doi:10.7554/elife.05005
Agarwal, V., Subtelny, A. O., Thiru, P., Ulitsky, I., and Bartel, D. P. (2018). Predicting microRNA Targeting Efficacy in Drosophila. Genome Biol. 19, 152. doi:10.1186/s13059-018-1504-3
Ahmed-Braimah, Y. H., Unckless, R. L., and Clark, A. G. (2017). Evolutionary Dynamics of Male Reproductive Genes in the Drosophila virilis Subgroup. G3 (Bethesda) 7, 3145–3155. doi:10.1534/g3.117.1136
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic Local Alignment Search Tool. J. Mol. Biol. 215, 403–410. doi:10.1016/S0022-2836(05)80360-2
Baek, D., Villén, J., Shin, C., Camargo, F. D., Gygi, S. P., and Bartel, D. P. (2008). The Impact of microRNAs on Protein Output. Nature 455, 64–71. doi:10.1038/nature07242
Bartel, D. P. (2009). MicroRNAs: Target Recognition and Regulatory Functions. Cell 136, 215–233. doi:10.1016/j.cell.2009.01.002
Berezikov, E. (2011). Evolution of microRNA Diversity and Regulation in Animals. Nat. Rev. Genet. 12, 846–860. doi:10.1038/nrg3079
Brown, J. B., Boley, N., Eisman, R., May, G. E., Stoiber, M. H., Duff, M. O., et al. (2014). Diversity and Dynamics of the Drosophila Transcriptome. Nature 512, 393–399. doi:10.1038/nature12962
Chen, K., and Rajewsky, N. (2007). The Evolution of Gene Regulation by Transcription Factors and microRNAs. Nat. Rev. Genet. 8, 93–103. doi:10.1038/nrg1990
Chen, S., Krinsky, B. H., and Long, M. (2013). New Genes as Drivers of Phenotypic Evolution. Nat. Rev. Genet. 14, 645–660. doi:10.1038/nrg3521
Chen, S., Ni, X., Krinsky, B. H., Zhang, Y. E., Vibranovski, M. D., White, K. P., et al. (2012). Reshaping of Global Gene Expression Networks and Sex-Biased Gene Expression by Integration of a Young Gene. EMBO J. 31, 2798–2809. doi:10.1038/emboj.2012.108
Chen, Y., Shen, Y., Lin, P., Tong, D., Zhao, Y., Allesina, S., et al. (2019). Gene Regulatory Network Stabilized by Pervasive Weak Repressions: microRNA Functions Revealed by the May-Wigner Theory. Natl. Sci. Rev. 6, 1176–1188. doi:10.1093/nsr/nwz076
Clark, A. G., Eisen, M. B., Smith, D. R., Bergman, C. M., Oliver, B., Markow, T. A., et al. (2007). Evolution of Genes and Genomes on the Drosophila Phylogeny. Nature 450, 203–218. doi:10.1038/nature06341
Czech, B., Malone, C. D., Zhou, R., Stark, A., Schlingeheyde, C., Dus, M., et al. (2008). An Endogenous Small Interfering RNA Pathway in Drosophila. Nature 453, 798–802. doi:10.1038/nature07007
Dai, H., Chen, Y., Chen, S., Mao, Q., Kennedy, D., Landback, P., et al. (2008). The Evolution of Courtship Behaviors through the Origination of a New Gene in Drosophila. Proc. Natl. Acad. Sci. 105, 7478–7483. doi:10.1073/pnas.0800693105
DeVeale, B., Swindlehurst-Chan, J., and Blelloch, R. (2021). The Roles of microRNAs in Mouse Development. Nat. Rev. Genet. 22, 307–323. doi:10.1038/s41576-020-00309-5
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 29, 15–21. doi:10.1093/bioinformatics/bts635
Edgar, R. C. (2004). MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 32, 1792–1797. doi:10.1093/nar/gkh340
Ellegren, H., and Parsch, J. (2007). The Evolution of Sex-Biased Genes and Sex-Biased Gene Expression. Nat. Rev. Genet. 8, 689–698. doi:10.1038/nrg2167
Fraïsse, C., Puixeu Sala, G., and Vicoso, B. (2019). Pleiotropy Modulates the Efficacy of Selection inDrosophila Melanogaster. Mol. Biol. Evol. 36, 500–515. doi:10.1093/molbev/msy246
Friedländer, M. R., Mackowiak, S. D., Li, N., Chen, W., and Rajewsky, N. (2012). miRDeep2 Accurately Identifies Known and Hundreds of Novel microRNA Genes in Seven Animal Clades. Nucleic Acids Res. 40, 37–52. doi:10.1093/nar/gkr688
Fromm, B., Billipp, T., Peck, L. E., Johansen, M., Tarver, J. E., King, B. L., et al. (2015). A Uniform System for the Annotation of Vertebrate microRNA Genes and the Evolution of the Human microRNAome. Annu. Rev. Genet. 49, 213–242. doi:10.1146/annurev-genet-120213-092023
Gubala, A. M., Schmitz, J. F., Kearns, M. J., Vinh, T. T., Bornberg-Bauer, E., Wolfner, M. F., et al. (2017). The Goddard and Saturn Genes Are Essential for Drosophila Male Fertility and May Have Arisen De Novo. Mol. Biol. Evol. 34, msx057–1082. doi:10.1093/molbev/msx057
Guo, H., Ingolia, N. T., Weissman, J. S., and Bartel, D. P. (2010). Mammalian microRNAs Predominantly Act to Decrease Target mRNA Levels. Nature 466, 835–840. doi:10.1038/nature09267
Haerty, W., Jagadeeshan, S., Kulathinal, R. J., Wong, A., Ravi Ram, K., Sirot, L. K., et al. (2007). Evolution in the Fast Lane: Rapidly Evolving Sex-Related Genes in Drosophila. Genetics 177, 1321–1335. doi:10.1534/genetics.107.078865
Hausser, J., and Zavolan, M. (2014). Identification and Consequences of miRNA-Target Interactions - beyond Repression of Gene Expression. Nat. Rev. Genet. 15, 599–612. doi:10.1038/nrg3765
He, L., Xie, M., Huang, J., Zhang, T., Shi, S., and Tang, T. (2016). Efficient and Specific Inhibition of Plant microRNA Function by Anti-microRNA Oligonucleotides (AMOs) In Vitro and In Vivo. Plant Cel Rep 35, 933–945. doi:10.1007/s00299-016-1933-y
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009). Bioinformatics Enrichment Tools: Paths toward the Comprehensive Functional Analysis of Large Gene Lists. Nucleic Acids Res. 37, 1–13. doi:10.1093/nar/gkn923
Hulsen, T., de Vlieg, J., and Alkema, W. (2008). BioVenn - a Web Application for the Comparison and Visualization of Biological Lists Using Area-Proportional Venn Diagrams. BMC Genomics 9, 488. doi:10.1186/1471-2164-9-488
Kabekkodu, S. P., Shukla, V., Varghese, V. K., D' Souza, J., Chakrabarty, S., and Satyamoorthy, K. (2018). Clustered miRNAs and Their Role in Biological Functions and Diseases. Biol. Rev. 93, 1955–1986. doi:10.1111/brv.12428
Kaessmann, H. (2010). Origins, Evolution, and Phenotypic Impact of New Genes. Genome Res. 20, 1313–1326. doi:10.1101/gr.101386.109
Kent, W. J. (2002). BLAT-The BLAST-like Alignment Tool. Genome Res. 12, 656–664. doi:10.1101/gr.229202
Khalturin, K., Hemmrich, G., Fraune, S., Augustin, R., and Bosch, T. C. G. (2009). More Than Just Orphans: Are Taxonomically-Restricted Genes Important in Evolution. Trends Genet. 25, 404–413. doi:10.1016/j.tig.2009.07.006
Kondo, S., Vedanayagam, J., Mohammed, J., Eizadshenass, S., Kan, L., Pang, N., et al. (2017). New Genes Often Acquire Male-specific Functions but Rarely Become Essential in Drosophila. Genes Dev. 31, 1841–1846. doi:10.1101/gad.303131.117
Kozomara, A., Birgaoanu, M., and Griffiths-Jones, S. (2019). miRBase: from microRNA Sequences to Function. Nucleic Acids Res. 47, D155–D162. doi:10.1093/nar/gky1141
Lange, A., Patel, P. H., Heames, B., Damry, A. M., Saenger, T., Jackson, C. J., et al. (2021). Structural and Functional Characterization of a Putative De Novo Gene in Drosophila. Nat. Commun. 12, 1667. doi:10.1038/s41467-021-21667-6
Lewis, B. P., Burge, C. B., and Bartel, D. P. (2005). Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes Are microRNA Targets. Cell 120, 15–20. doi:10.1016/j.cell.2004.12.035
Liao, Y., Smyth, G. K., and Shi, W. (2014). featureCounts: an Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 30, 923–930. doi:10.1093/bioinformatics/btt656
Liufu, Z., Zhao, Y., Guo, L., Miao, G., Xiao, J., Lyu, Y., et al. (2017). Redundant and Incoherent Regulations of Multiple Phenotypes Suggest microRNAs' Role in Stability Control. Genome Res. 27, 1665–1673. doi:10.1101/gr.222505.117
Lu, G.-A., Zhao, Y., Liufu, Z., and Wu, C.-I. (2018a). On the Possibility of Death of New Genes - Evidence from the Deletion of De Novo microRNAs. BMC genomics 19, 388. doi:10.1186/s12864-018-4755-1
Lu, G.-A., Zhao, Y., Yang, H., Lan, A., Shi, S., Liufu, Z., et al. (2018b). Death of New microRNA Genes inDrosophilavia Gradual Loss of Fitness Advantages. Genome Res. 28, 1309–1318. doi:10.1101/gr.233809.117
Lu, J., Fu, Y., Kumar, S., Shen, Y., Zeng, K., Xu, A., et al. (2008). Adaptive Evolution of Newly Emerged Micro-RNA Genes in Drosophila. Mol. Biol. Evol. 25, 929–938. doi:10.1093/molbev/msn040
Lyu, Y., Shen, Y., Li, H., Chen, Y., Guo, L., Zhao, Y., et al. (2014). New MicroRNAs in Drosophila-Birth, Death and Cycles of Adaptive Evolution. Plos Genet. 10, e1004096. doi:10.1371/journal.pgen.1004096
Marco, A., Ninova, M., Ronshaugen, M., and Griffiths-Jones, S. (2013). Clusters of microRNAs Emerge by New Hairpins in Existing Transcripts. Nucleic Acids Res. 41, 7745–7752. doi:10.1093/nar/gkt534
Marco, A. (2014). Sex-biased Expression of microRNAs in Drosophila melanogaster. Open Biol. 4, 140024. doi:10.1098/rsob.140024
McLysaght, A., and Hurst, L. D. (2016). Open Questions in the Study of De Novo Genes: what, How and Why. Nat. Rev. Genet. 17, 567–578. doi:10.1038/nrg.2016.78
Meiklejohn, C. D., Parsch, J., Ranz, J. M., and Hartl, D. L. (2003). Rapid Evolution of Male-Biased Gene Expression in Drosophila. Proc. Natl. Acad. Sci. 100, 9894–9899. doi:10.1073/pnas.1630690100
Mohammed, J., Bortolamiol-Becet, D., Flynt, A. S., Gronau, I., Siepel, A., and Lai, E. C. (2014). Adaptive Evolution of Testis-specific, Recently Evolved, Clustered miRNAs inDrosophila. RNA 20, 1195–1209. doi:10.1261/rna.044644.114
Mohammed, J., Flynt, A. S., Panzarino, A. M., Mondal, M. M. H., DeCruz, M., Siepel, A., et al. (2018). Deep Experimental Profiling of microRNA Diversity, Deployment, and Evolution across the Drosophila Genus. Genome Res. 28, 52–65. doi:10.1101/gr.226068.117
Nozawa, M., Miura, S., and Nei, M. (2010). Origins and Evolution of microRNA Genes in Drosophila Species. Genome Biol. Evol. 2, 180–189. doi:10.1093/gbe/evq009
Pancratov, R., Peng, F., Smibert, P., Yang, J.-S., Olson, E. R., Guha-Gilford, C., et al. (2013). The miR-310/13 Cluster Antagonizes β-catenin Function in the Regulation of Germ and Somatic Cell Differentiation in the Drosophila Testis. Development 140, 2904–2916. doi:10.1242/dev.092817
Parsch, J., and Ellegren, H. (2013). The Evolutionary Causes and Consequences of Sex-Biased Gene Expression. Nat. Rev. Genet. 14, 83–87. doi:10.1038/nrg3376
Penso-Dolfin, L., Moxon, S., Haerty, W., and Di Palma, F. (2018). The Evolutionary Dynamics of microRNAs in Domestic Mammals. Sci. Rep. 8, 1–13. doi:10.1038/s41598-018-34243-8
Ross, B. D., Rosin, L., Thomae, A. W., Hiatt, M. A., Vermaak, D., de la Cruz, A. F. A., et al. (2013). Stepwise Evolution of Essential Centromere Function in a Drosophila Neogene. Science 340, 1211–1214. doi:10.1126/science.1234393
Rozhkov, N. V., Aravin, A. A., Zelentsova, E. S., Schostak, N. G., Sachidanandam, R., McCombie, W. R., et al. (2010). Small RNA-Based Silencing Strategies for Transposons in the Process of Invading Drosophila Species. RNA 16, 1634–1645. doi:10.1261/rna.2217810
Sanfilippo, P., Wen, J., and Lai, E. C. (2017). Landscape and Evolution of Tissue-specific Alternative Polyadenylation across Drosophila Species. Genome Biol. 18, 229. doi:10.1186/s13059-017-1358-0
Selbach, M., Schwanhäusser, B., Thierfelder, N., Fang, Z., Khanin, R., and Rajewsky, N. (2008). Widespread Changes in Protein Synthesis Induced by microRNAs. Nature 455, 58–63. doi:10.1038/nature07228
Sood, P., Krek, A., Zavolan, M., Macino, G., and Rajewsky, N. (2006). Cell-type-specific Signatures of microRNAs on Target mRNA Expression. Proc. Natl. Acad. Sci. 103, 2746–2751. doi:10.1073/pnas.0511045103
Stadler, M., Artiles, K., Pak, J., and Fire, A. (2012). Contributions of mRNA Abundance, Ribosome Loading, and post- or Peri-Translational Effects to Temporal Repression of C. elegans Heterochronic miRNA Targets. Genome Res. 22, 2418–2426. doi:10.1101/gr.136515.111
Tang, T., Kumar, S., Shen, Y., Lu, J., Wu, M.-L., Shi, S., et al. (2010). Adverse Interactions between Micro-RNAs and Target Genes from Different Species. Proc. Natl. Acad. Sci. 107, 12935–12940. doi:10.1073/pnas.1007591107
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: Discovering Splice Junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi:10.1093/bioinformatics/btp120
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript Assembly and Quantification by RNA-Seq Reveals Unannotated Transcripts and Isoform Switching during Cell Differentiation. Nat. Biotechnol. 28, 511–515. doi:10.1038/nbt.1621
Vibranovski, M. D., Lopes, H. F., Karr, T. L., and Long, M. (2009). Stage-specific Expression Profiling of Drosophila Spermatogenesis Suggests that Meiotic Sex Chromosome Inactivation Drives Genomic Relocation of Testis-Expressed Genes. Plos Genet. 5, e1000731. doi:10.1371/journal.pgen.1000731
Wang, Y., Luo, J., Zhang, H., and Lu, J. (2016). microRNAs in the Same Clusters Evolve to Coordinately Regulate Functionally Related Genes. Mol. Biol. Evol. 33, 2232–2247. doi:10.1093/molbev/msw089
Wen, J., Parker, B. J., Jacobsen, A., and Krogh, A. (2011). MicroRNA Transfection and AGO-Bound CLIP-Seq Data Sets Reveal Distinct Determinants of miRNA Action. RNA 17, 820–834. doi:10.1261/rna.2387911
Wu, C.-I., Shen, Y., and Tang, T. (2009). Evolution under Canalization and the Dual Roles of microRNAs-A Hypothesis. Genome Res. 19, 734–743. doi:10.1101/gr.084640.108
Xia, S., VanKuren, N. W., Chen, C., Zhang, L., Kemkemer, C., Shao, Y., et al. (2021). Genomic Analyses of New Genes and Their Phenotypic Effects Reveal Rapid Evolution of Essential Functions in Drosophila Development. Plos Genet. 17, e1009654. doi:10.1371/journal.pgen.1009654
Yang, L., Zhang, Z., and He, S. (2016). Both Male-Biased and Female-Biased Genes Evolve Faster in Fish Genomes. Genome Biol. Evol. 8, 3433–3445. doi:10.1093/gbe/evw239
Yeates, D. K., and Wiegmann, B. M. (1999). Congruence and Controversy: Toward a Higher-Level Phylogeny of Diptera. Annu. Rev. Entomol. 44, 397–428. doi:10.1146/annurev.ento.44.1.397
Yeh, S.-D., Do, T., Chan, C., Cordova, A., Carranza, F., Yamamoto, E. A., et al. (2012). Functional Evidence that a Recently Evolved Drosophila Sperm-specific Gene Boosts Sperm Competition. Proc. Natl. Acad. Sci. 109, 2043–2048. doi:10.1073/pnas.1121327109
Ylla, G., Fromm, B., Piulachs, M.-D., and Belles, X. (2016). The microRNA Toolkit of Insects. Sci. Rep. 6, 37736. doi:10.1038/srep37736
Zhang, J., Dean, A. M., Brunet, F., and Long, M. (2004a). Evolving Protein Functional Diversity in New Genes of Drosophila. Proc. Natl. Acad. Sci. 101, 16246–16250. doi:10.1073/pnas.0407066101
Zhang, R., Peng, Y., Wang, W., and Su, B. (2007). Rapid Evolution of an X-Linked microRNA Cluster in Primates. Genome Res. 17, 612–617. doi:10.1101/gr.6146507
Zhang, Z., Hambuch, T. M., and Parsch, J. (2004b). Molecular Evolution of Sex-Biased Genes in Drosophila. Mol. Biol. Evol. 21, 2130–2139. doi:10.1093/molbev/msh223
Zhao, L., Saelao, P., Jones, C. D., and Begun, D. J. (2014). Origin and Spread of De Novo Genes in Drosophila melanogaster Populations. Science 343, 769–772. doi:10.1126/science.1248286
Zhao, Y., Lin, P., Liufu, Z., Yang, H., Lyu, Y., Shen, X., et al. (2018). Regulation of Large Number of Weak Targets-New Insights from Twin-microRNAs. Genome Biol. Evol. 10, 1255–1264. doi:10.1093/gbe/evy079
Zhao, Y., Lu, G.-A., Yang, H., Lin, P., Liufu, Z., Tang, T., et al. (2021). Run or Die in the Evolution of New MicroRNAs-Testing the Red Queen Hypothesis on De Novo New Genes. Mol. Biol. Evol. 38, 1544–1553. doi:10.1093/molbev/msaa317
Keywords: new miRNA, miRNA cluster, miRNA evolution, regulatory network, testis
Citation: Lyu Y, Liufu Z, Xiao J and Tang T (2021) A Rapid Evolving microRNA Cluster Rewires Its Target Regulatory Networks in Drosophila. Front. Genet. 12:760530. doi: 10.3389/fgene.2021.760530
Received: 18 August 2021; Accepted: 11 October 2021;
Published: 28 October 2021.
Edited by:
Haipeng Li, Shanghai Institute of Nutrition and Health (CAS), ChinaReviewed by:
Jing Luo, Yunnan University, ChinaNikolaos Vakirlis, Alexander Fleming Biomedical Sciences Research Center, Greece
Copyright © 2021 Lyu, Liufu, Xiao and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tian Tang, bHNzdHRAbWFpbC5zeXN1LmVkdS5jbg==
†Present address: Yang Lyu, Department of Molecular and Integrative Physiology and Geriatrics Center, Biomedical Sciences and Research Building, University of Michigan, Ann Arbor, United States
‡These authors have contributed equally to this work and share first authorship