 Lihua Yu
Lihua Yu Chaoxiang Yang
Chaoxiang Yang Ning Shang
Ning Shang Hongke Ding1
Hongke Ding1 Haowen Tan
Haowen Tan Yan Zhang
Yan Zhang- 1Medical Genetics Centre, Guangdong Women and Children Hospital, Guangzhou, China
- 2Department of Radiology, Guangdong Women and Children Hospital, Guangzhou, China
- 3Department of Ultrasound, Guangdong Women and Children Hospital, Guangzhou, China
- 4Aegicare (Shenzhen) Technology Co., Ltd., Shenzhen, China
A dilated lateral ventricle is a relatively common finding on prenatal ultrasound, and the causes are complex. We aimed to explore the etiology of a fetus with a dilated lateral ventricle. Trio whole-exome sequencing was performed to detect causative variants. A de novo variant of TAOK1 (NM_020791.2: c.227A>G) was detected in the proband and evaluated for potential functional impacts using a variety of prediction tools. Droplet digital polymerase chain reaction was used to exclude the parental mosaicism and to verify the phasing of the de novo variant. Based on peripheral blood analysis, the parents did not exhibit mosaicism at this site, and the de novo variant was paternally derived. Here, we describe a fetus with a de novo likely pathogenic variant of TAOK1 who had a dilated lateral ventricle and a series of particular phenotypes. This case expands the clinical spectrum of TAOK1-associated disorders. We propose a method for solving genetic disorders in which the responsible genes have not yet gone through ClinGen curation, particularly for prenatal cases.
Introduction
The TAO kinase family consists of three genes, TAOK1, TAOK2, and TAOK3, which encode TAOK1, TAOK2, and TAOK3, respectively (Dan et al., 2001; Miller et al., 2019). TAO kinases play multifunctional roles in many molecular and cellular events and can regulate neuronal survival and development in the nervous system (Fang et al., 2020; Hu et al., 2021). TAOK1 is highly expressed in the human brain and plays a role in the establishment of neuronal polarity, neuronal differentiation, and early brain development (Biernat et al., 2002; Timm et al., 2006; Draviam et al., 2007; Breuss and Keays, 2014; Poon et al., 2016). Many studies have provided evidence that TAOK1 dysfunction can result in neurodevelopmental disorders (NDDs) (Cooper et al., 2011; Xie et al., 2016; Deciphering Developmental Disorders Study, 2017; Dulovic-Mahlow et al., 2019; Satterstrom et al., 2020; van Woerden et al., 2021; Hunter et al., 2022). However, dysfunction of this kinase in prenatal cases has not been reported.
Here, we report structural brain abnormalities in a fetus with a de novo variant of TAOK1. To our knowledge, this is the first report of TAOK1 dysfunction as a prenatal diagnosis.
Case Presentation
A healthy 32-year-old gravida 3, para 2 (G3P2) woman underwent a prenatal examination at Guangdong Women and Children Hospital. She delivered two normal male infants, in 2013 and 2018, through uncomplicated vaginal deliveries. At 25 weeks of gestation for the current pregnancy, routine ultrasound scanning showed that the left lateral ventricle of the fetus was widened (10.1 mm compared to a reference of <10 mm) (Figure 1A). Common factors, such as infection and anemia, were ruled out, and COVID-19 nucleic acid tests were negative. Noninvasive prenatal testing (NIPT) indicated a low risk of fetal trisomy 13, 18, and 21. At 31 weeks of gestation, ultrasound scanning showed slight widening of the left lateral ventricle (11.0 mm compared to reference of <10 mm), with the umbilical cord surrounding the neck, of the fetus (Figure 1B). As shown in Figure 1C, magnetic resonance imaging (MRI) revealed poor bilateral and frontal operculum formation and shallow bilateral lateral fissures, which were more obvious on the right side. Bilateral polymicrogyria of the lateral fissure area could not be ruled out. The left ventricle was slightly wider. No abnormalities were observed in the corpus callosum, septum pellucidum, cerebellar vermis, or posterior fossa. Due to the abnormal cortical structure detected by MRI, interventional prenatal diagnosis was performed, along with chromosomal microarray analysis (CMA) and trio whole-exome sequencing (trio WES), simultaneously. The CMA result was negative, but trio WES detected a de novo missense variant of TAOK1 in the fetus. Whole-genome sequencing (WGS) and droplet digital polymerase chain reaction (ddPCR) was then performed to identify the source of the variation. Finally, the de novo variant of TAOK1 was found to originate from the paternal allele.
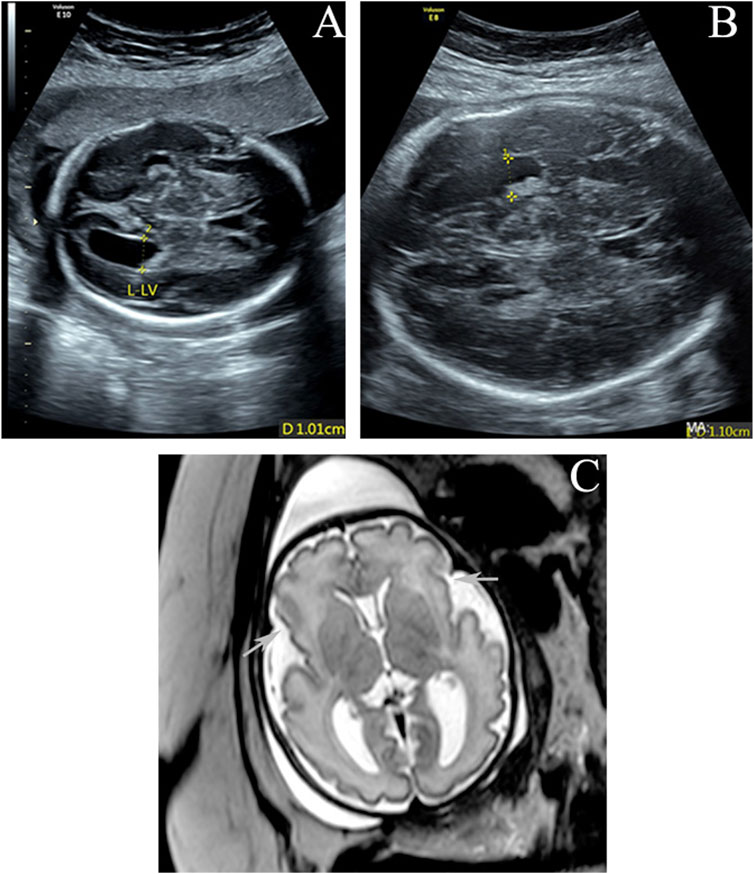
FIGURE 1. (A,B) Enlarged left lateral ventricle (10.1 and 11 mm) at 25 and 31 gestational weeks. (C) Axial T2-weighted imaging at 32 weeks of gestation shows poor bilateral and frontal operculum formation and shallow bilateral lateral fissures (arrows). The left ventricle is slightly wider.
Methods and Results
Trio Whole-Exome Sequencing
Genomic DNA was extracted from amniotic fluid and peripheral blood from the fetus and parents, respectively, using the QIAamp DNA Mini Kit (Qiagen), following the manufacturer’s instructions. Trio WES was performed to detect genetic variants (see Supplementary Methods), and a de novo variant of TAOK1 [GRCh37/hg19 chr17: 27802710, NM_020791.2: c.227A>G (p.Glu76Gly)] was found. No other variants were considered to contribute to the phenotype.
The de novo variant c.227A>G is absent in the general population according to public databases (gnomAD, 1000 Genomes Project, NHLBI Exome Sequencing Project 6500, and Exome Aggregation Consortium). This variation has not been previously reported in the ClinVar or PubMed databases (retrieved 15 January 2022). A variety of prediction tools (SIFT, DANN, and REVEL) were used to evaluate the possible functional impact of c.227A>G, and it is predicted to be a damaging variation by all three tools. Furthermore, various algorithms (GERP, phyloP, phastCons, and SiPhy) and multiple sequence alignments from the UCSC genome browser predicted that this position is conserved across multiple vertebrate species (from zebrafish to human). The variant p.Glu76Gly is located in the “Protein kinase” domain of TAOK1 (UniProt ID #Q7L7X3) in which benign variants are not found in ClinVar database. Although the protein structure of TAOK1 was not available in the PDB database, the structure of the kinase domain (amino acids 28–281) was predicted with the I-TASSER server, as illustrated in Figure 2 (Roy et al., 2010). The model with the highest confidence (C-score) and topological similarity (Tm-score) is used. The identified variant p.Glu76Gly is predicted to be located in an alpha-helix of the protein (Figure 2).
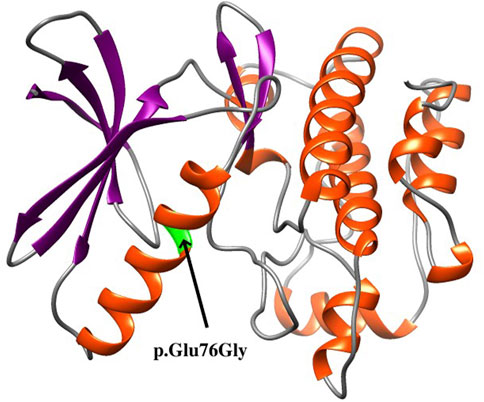
FIGURE 2. Structure of the kinase domain of the TAOK1 protein from amino acids 28 to 281, showing the localization of the identified variant p.Glu76Gly.
Whole-Genome Sequencing
To identify the phase of the de novo variant, WGS was performed for the proband (see Supplementary Methods). A heterozygous variant, c.306+468G>T (GRCh37/hg19 chr17: 27803257G>T), in TAOK1 was found, which is 547 bp downstream of c.227A>G and was used as the reference for ddPCR analysis.
To validate the de novo variant c.227A>G and the reference variant c.306+468G>T, Sanger sequencing was performed for the family (see Supplementary Methods). The results showed that the proband carried c.227A>G but that neither parent did (Figure 3A); c.306+468G>T was found in the proband and mother but not in the father (Figure 3B).
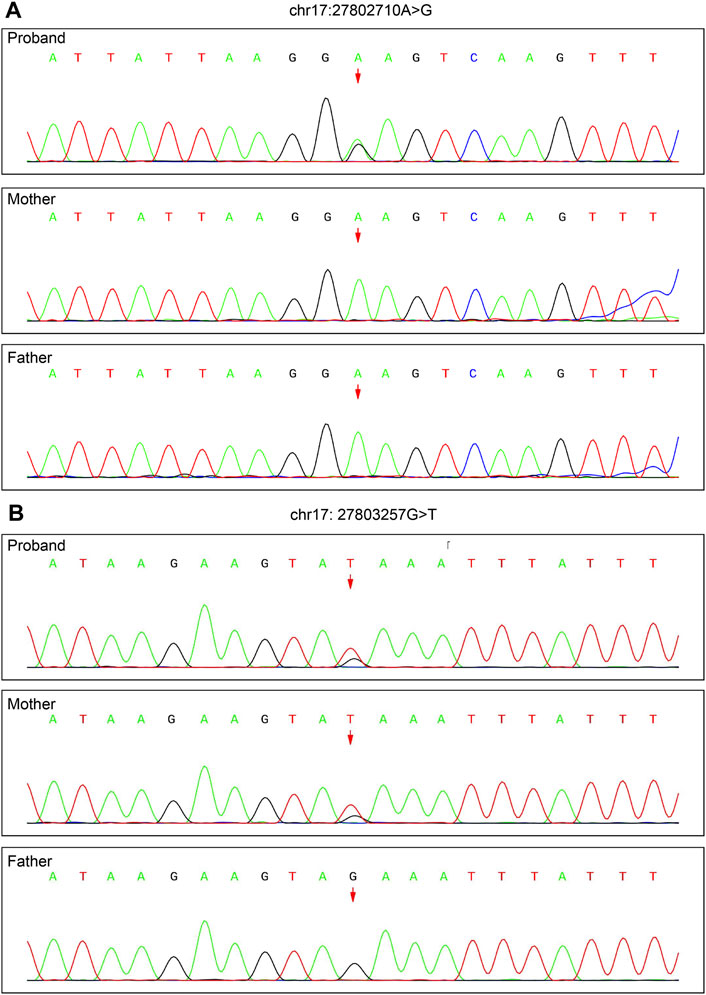
FIGURE 3. Validation of the de novo variant c.227A>G of TAOK1 (chr17: 27802710) identified by trio WES and the reference variant c.306+468G>T (chr17: 27803257) identified by WGS by Sanger sequencing. (A) c.227A>G was found in the proband, but not in either parent. (B) c.306+468G>T was detected in the proband and mother, but not in the father.
Droplet Digital PCR
ddPCR was used to assess parental mosaicism (see Supplementary Methods). As shown in Figure 4, the peripheral blood samples of the parents did not show mosaicism at the site of the de novo variant of TAOK1.
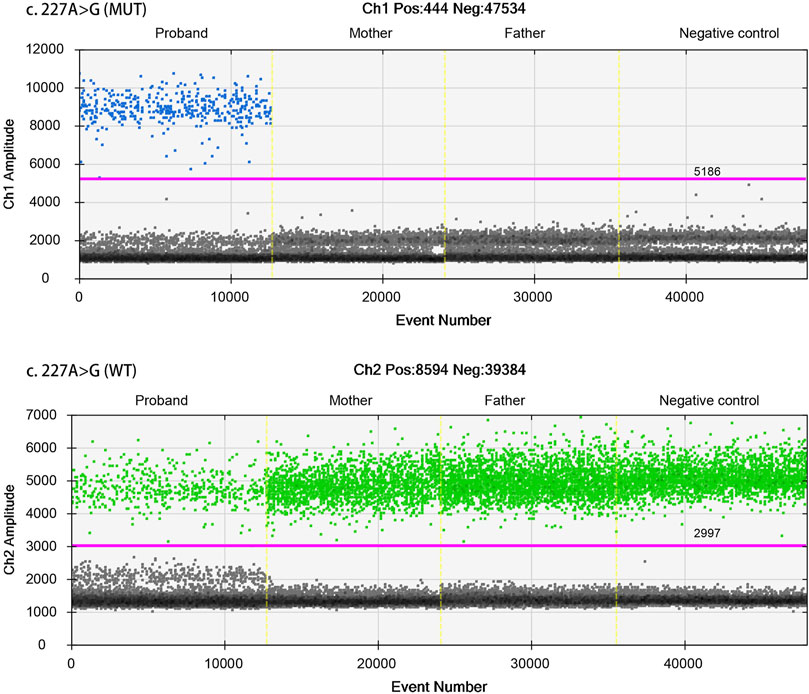
FIGURE 4. Droplet digital PCR (ddPCR) for mosaic variation detection. The four ddPCRs are divided by vertical dotted yellow lines for the proband, mother, father, and negative control. The pink line is the threshold, above which are positive droplets (blue and green), and below which are negative droplets (gray) without any target DNA. There is no target DNA for the mutant locus c.227A>G in the mother and father (top panel).
To determine whether the mutant alleles of the variants, c.227A>G and c.306+468G>T, in the proband were located on the same chromosome, ddPCR was used to verify the phase (see Supplementary Methods). First, the T allele of c.306+468G>T was used as a reference, and phasing was performed. The results showed that these alleles were not located on the same chromosome (Figure 5A). Subsequently, the wild-type allele (G) of c.306+468G>T was used as a reference. The results showed that the G allele of c.306+468G>T and the mutant allele (G) of c.227A>G were located on the same chromosome (Figure 5B). The phasing analysis confirmed that the de novo variant c.227A>G derived from the paternal chromosome.
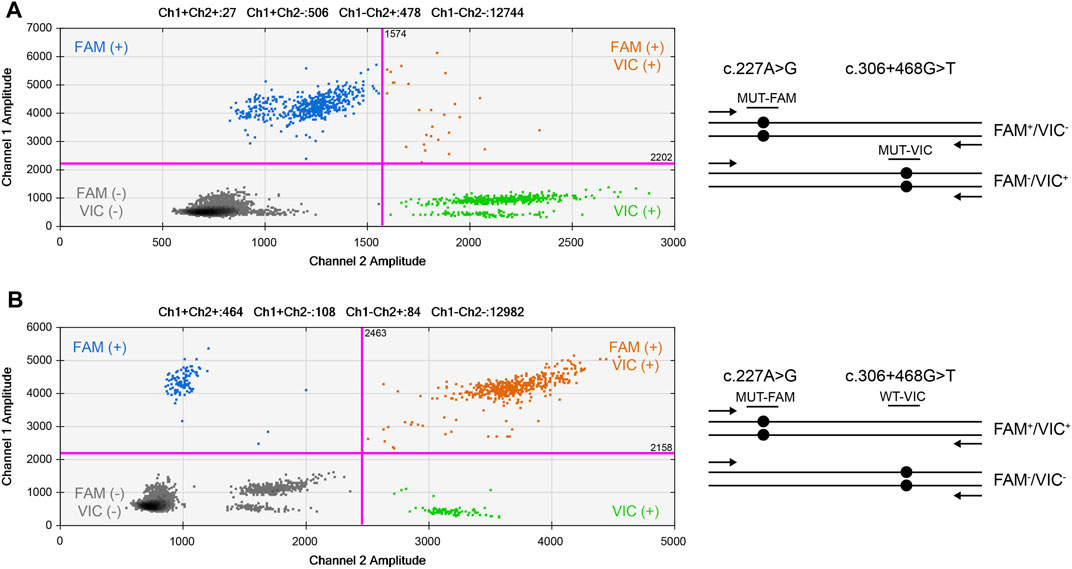
FIGURE 5. 2D cluster plot of droplet fluorescence for the de novo locus and reference locus. (A) The result of c.306+468G>T mutant and c.227A>G mutant probes. (B) The result of c.306+468G>T wild-type and c.227A>G mutant probes. FAM™ positive (Channel 1, mt−) droplets form the top-left blue cluster, HEX™ positive (Channel 2, mt+) droplets form the bottom-right green cluster, negative droplets for both targets form the bottom-left gray cluster, and positive droplets for both targets form the top-right orange cluster.
Discussion
To the best of our knowledge, only six studies have reported variants of TAOK1 in 40 patients with NDDs (Xie et al., 2016; Dulovic-Mahlow et al., 2019; Satterstrom et al., 2020; Basel-Salmon et al., 2021; van Woerden et al., 2021; Hunter et al., 2022). The phenotypes of the affected individuals are summarized in Table 1. All had NDDs, mainly involving global developmental delay, intellectual disability, hypotonia, and behavior problems, as well as brain MRI abnormalities and eye/visual problems. In this study, the fetus with a variant of TAOK1 had a dilated left lateral ventricle, and brain MRI imaging in six previously reported postnatal cases revealed dilated lateral ventricles. However, the published studies did not differentiate between unilateral and bilateral ventricular dilation. Therefore, it is uncertain whether variants of TAOK1 are associated with asymmetric ventricles.
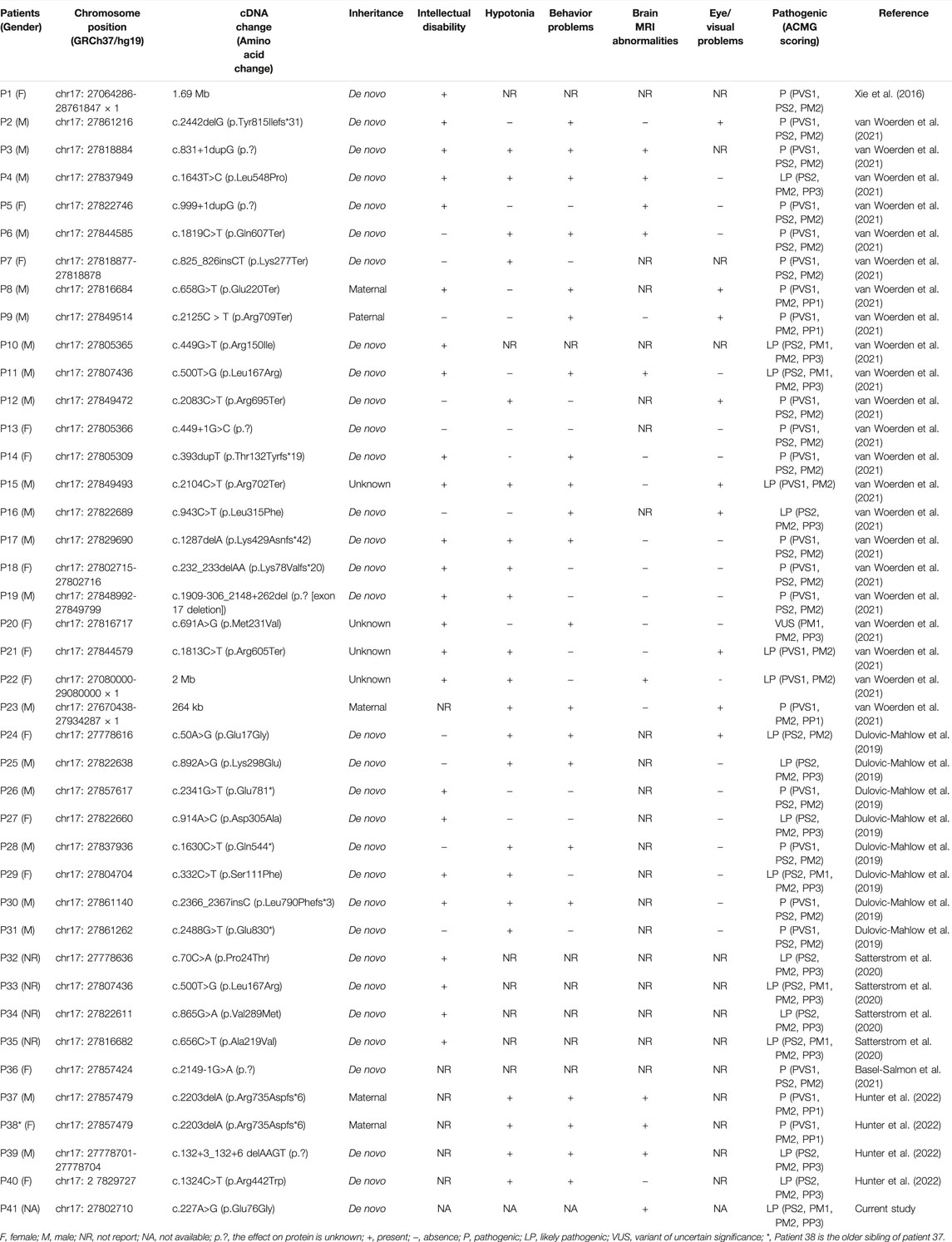
TABLE 1. Overview of the identified variants of TAOK1.
This is the first report of a variant of TAOK1 in the prenatal stage. We sought to determine whether prenatal de novo variants of TAOK1 can predict the risk of NDDs. To evaluate associations between the de novo variant of TAOK1 we found and phenotypes in the prenatal stage, we compared the pregnancy statuses of the patients with variants of TAOK1 (Table 2). As shown, few abnormal pregnancy statuses were found. MRI abnormalities in the fetus are relatively prevalent in patients with TAOK1-associated NDDs. We suggest that fetuses with brain MRI abnormalities accompanied by de novo variants of TAOK1 have a higher risk for NDDs, and should be carefully managed. Our study not only fills the gap between the variant of TAOK1 and the prenatal phenotypes but also provides valuable information for disease management, prognosis judgment and prenatal consultation.
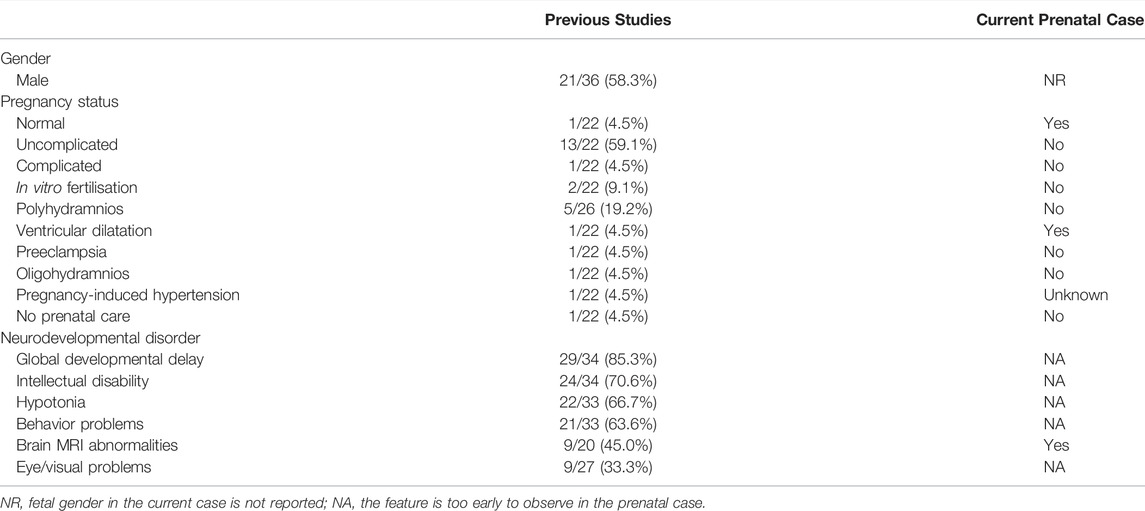
TABLE 2. Clinical features of patients with TAOK1 variants.
As shown in Table 1, de novo variants of TAOK1 have been found in thirty-one of 40 previously reported patients (77.5%). An additional de novo variant of TAOK1 was detected in the fetus in this study. Variants in five (12.5%) affected individuals, P8, P9, P23, P37, and P38, were considered to be inherited from an affected mother or father (Table 1), which are classified as pathogenic herein. The very mild cognitive phenotypes of some affected parents might be explained by incomplete penetrance and variable expressivity (Hunter et al., 2022). Furthermore, no recurrent variants were reported in the region of 17q11.2 (chr17: 27064286-28761847), indicating that TAOK1 is not prone to hotspot variant, which was also mentioned by van Woerden et al. (2021). The variant identified in this study is located at chr17: 27802710, within the range of previous findings. Based on the protein structure modeling result (Figure 2), p.Glu76Gly is predicted to be located in an alpha-helix structure, and glycine is generally considered to destabilize an alpha-helix. Accordingly, we predicted that this novel missense variant affects protein kinase function, though more functional experiments are needed to validate this assumption.
In this study, the de novo variant was confirmed to originate from the paternal chromosome by a ddPCR phasing strategy, consistent with the findings that de novo variants arise more frequently in paternal germ cells than in maternal germ cells (Kong et al., 2012; Goldmann et al., 2016). The primarily de novo variants on the paternal chromosome could be explained by fundamental differences in germ cell biology in the female and male lineages. Spermatogenesis requires regular mitotic cell divisions of spermatogonial stem cells throughout male reproductive life (Goriely and Wilkie, 2012). However, the influence of maternal chromosomes on de novo variants in offspring cannot be ignored (Gao et al., 2019; Goldmann et al., 2019). In recent years, de novo variants have been found to be a prominent cause of NDDs, including intellectual disability (ID), autism, and schizophrenia (SCZ) (Veltman and Brunner, 2012; Acuna-Hidalgo et al., 2016). The relationship between paternal-age-related de novo variants and the risk for psychiatric and developmental disorders has been assessed, including for autism spectrum disorder (ASD), congenital heart disease (CHD), NDDs with epilepsy (EPI), ID, and SCZ (Taylor et al., 2019). Recurrent risk of a de novo variant should be considered if a germline mosaic variant is detected in parental samples, and the sibling recurrent risk can be as low as 0.5% if absent from samples of both parents by highly sensitive screening technology (Wright et al., 2019).
Variants of a gene of uncertain significance should always be classified as having uncertain significance of pathogenicity (Richards et al., 2015). When we obtained the trio WES results in November 2020, the TAOK1 gene has not been associated with any Mendelian disorder in the OMIM (Online Mendelian Inheritance in Man) database. We further explored research articles and found that Dulovic-Mahlow et al. (2019) first reported eight patients, all with de novo variants considered pathogenic due to loss of function of the TAO kinase family. We then evaluated the gene-disease association following the ClinGen Gene-Disease Validity Standard Operating Procedures (Strande et al., 2017), and curated the TAOK1 gene to “moderate” grade. Finally, the de novo variant c.227A>G of TAOK1 in our case was classified as likely pathogenic (PS2+PM1+PM2+PP3) based on ACMG guidelines (Zhang et al., 2020). A clear understanding of the clinical validity of the gene-disease relationship is critical for accurate interpretation of variants and successful medical decision-making based on genetic testing results. Because of limitations of the prenatal phenotype, accurate genetic variant classification in prenatal diagnosis is especially important. It would benefit from cross-laboratory data sharing and evaluating the strength of a gene-disease relationship based on the ClinGen Gene-Disease Validity Standard Operating Procedures. During the revision of the manuscript, the definitive classification of the gene-disease relationship between TAOK1 and syndromic intellectual disability was curated by the ClinGen Intellectual Disability and Autism Gene Curation Expert Panel on 4 August 2021. In addition, the TAOK1 gene was associated with OMIM disease (developmental delay with or without intellectual impairment or behavioral abnormalities, MIM #619575) starting from 19 October 2021. All of these are essential for future work.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: Genome Sequence Archive (Genomics, Proteomics, and Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2021), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA001877) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human.
Ethics Statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
All authors contributed to the study conception and design. Data collection was performed by LY, CY, NS, HD, and JZ. Bioinformatic analysis was performed by YZ and HT. The entire investigation was supervised by YZ. The first draft of the manuscript was written by LY, YZ, HT, and YZ, and all authors revised the manuscript. All authors read and approved the final version.
Funding
This work was supported by Guangzhou Municipal Science and Technology Project (202102080358).
Conflict of Interest
YZ and HT are employed by Aegicare (Shenzhen) Technology Co., Ltd., Shenzhen, China.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the pregnant woman and her family for sharing information.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.836853/full#supplementary-material
References
Acuna-Hidalgo, R., Veltman, J. A., and Hoischen, A. (2016). New Insights into the Generation and Role of De Novo Mutations in Health and Disease. Genome Biol. 17, 241. doi:10.1186/s13059-016-1110-1
Basel-Salmon, L., Ruhrman-Shahar, N., Orenstein, N., Goldberg, Y., Gonzaga-Jauregui, C., Shuldiner, A. R., et al. (2021). When Phenotype Does Not Match Genotype: Importance of “Real-time” Refining of Phenotypic Information for Exome Data Interpretation. Genet. Med. 23, 215–221. doi:10.1038/s41436-020-00938-5
Biernat, J., Wu, Y.-Z., Timm, T., Zheng-Fischhöfer, Q., Mandelkow, E., Meijer, L., et al. (2002). Protein Kinase MARK/PAR-1 Is Required for Neurite Outgrowth and Establishment of Neuronal Polarity. Mol. Biol. Cell. 13, 4013–4028. doi:10.1091/mbc.02-03-0046
Breuss, M., and Keays, D. A. (2014). Microtubules and Neurodevelopmental Disease: the Movers and the Makers. Adv. Exp. Med. Biol., 75–96. doi:10.1007/978-94-007-7687-6_5
Cooper, G. M., Coe, B. P., Girirajan, S., Rosenfeld, J. A., Vu, T. H., Baker, C., et al. (2011). A Copy Number Variation Morbidity Map of Developmental Delay. Nat. Genet. 43, 838–846. doi:10.1038/ng.909
Dan, I., Watanabe, N. M., and Kusumi, A. (2001). The Ste20 Group Kinases as Regulators of MAP Kinase Cascades. Trends Cell. Biol. 11, 220–230. doi:10.1016/s0962-8924(01)01980-8
Deciphering Developmental Disorders Study (2017). Prevalence and Architecture of De Novo Mutations in Developmental Disorders. Nature 542, 433–438. doi:10.1038/nature21062
Draviam, V. M., Stegmeier, F., Nalepa, G., Sowa, M. E., Chen, J., Liang, A., et al. (2007). A Functional Genomic Screen Identifies a Role for TAO1 Kinase in Spindle-Checkpoint Signalling. Nat. Cell. Biol. 9, 556–564. doi:10.1038/ncb1569
Dulovic-Mahlow, M., Trinh, J., Kandaswamy, K. K., Braathen, G. J., Di Donato, N., Rahikkala, E., et al. (2019). De Novo Variants in TAOK1 Cause Neurodevelopmental Disorders. Am. J. Hum. Genet. 105, 213–220. doi:10.1016/j.ajhg.2019.05.005
Fang, C.-Y., Lai, T.-C., Hsiao, M., and Chang, Y.-C. (2020). The Diverse Roles of TAO Kinases in Health and Diseases. Int. J. Mol. Sci. 21, E7463. doi:10.3390/ijms21207463
Gao, Z., Moorjani, P., Sasani, T. A., Pedersen, B. S., Quinlan, A. R., Jorde, L. B., et al. (2019). Overlooked Roles of DNA Damage and Maternal Age in Generating Human Germline Mutations. Proc. Natl. Acad. Sci. U. S. A. 116, 9491–9500. doi:10.1073/pnas.1901259116
Goldmann, J. M., Veltman, J. A., and Gilissen, C. (2019). De Novo Mutations Reflect Development and Aging of the Human Germline. Trends Genet. 35, 828–839. doi:10.1016/j.tig.2019.08.005
Goldmann, J. M., Wong, W. S. W., Pinelli, M., Farrah, T., Bodian, D., Stittrich, A. B., et al. (2016). Parent-of-origin-specific Signatures of De Novo Mutations. Nat. Genet. 48, 935–939. doi:10.1038/ng.3597
Goriely, A., and Wilkie, A. O. M. (2012). Paternal Age Effect Mutations and Selfish Spermatogonial Selection: Causes and Consequences for Human Disease. Am. J. Hum. Genet. 90, 175–200. doi:10.1016/j.ajhg.2011.12.017
Hu, C., Feng, P., Yang, Q., and Xiao, L. (2021). Clinical and Neurobiological Aspects of TAO Kinase Family in Neurodevelopmental Disorders. Front. Mol. Neurosci. 14, 655037. doi:10.3389/fnmol.2021.655037
Hunter, J. M., Massingham, L. J., Manickam, K., Bartholomew, D., Williamson, R. K., Schwab, J. L., et al. (2022). Inherited and De Novo Variants Extend the Etiology of TAOK1-Associated Neurodevelopmental Disorder. Cold Spring Harb. Mol. Case Stud. 8, a006180. doi:10.1101/mcs.a006180
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). Rate of De Novo Mutations and the Importance of Father’s Age to Disease Risk. Nature 488, 471–475. doi:10.1038/nature11396
Miller, C. J., Lou, H. J., Simpson, C., van de Kooij, B., Ha, B. H., Fisher, O. S., et al. (2019). Comprehensive Profiling of the STE20 Kinase Family Defines Features Essential for Selective Substrate Targeting and Signaling Output. PLoS Biol. 17, e2006540. doi:10.1371/journal.pbio.2006540
Poon, C. L. C., Mitchell, K. A., Kondo, S., Cheng, L. Y., and Harvey, K. F. (2016). The Hippo Pathway Regulates Neuroblasts and Brain Size in Drosophila melanogaster. Curr. Biol. 26, 1034–1042. doi:10.1016/j.cub.2016.02.009
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Roy, A., Kucukural, A., and Zhang, Y. (2010). I-TASSER: a Unified Platform for Automated Protein Structure and Function Prediction. Nat. Protoc. 5, 725–738. doi:10.1038/nprot.2010.5
Satterstrom, F. K., Kosmicki, J. A., Wang, J., Breen, M. S., De Rubeis, S., An, J.-Y., et al. (2020). Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell. 180, 568–584. e23. doi:10.1016/j.cell.2019.12.036
Strande, N. T., Riggs, E. R., Buchanan, A. H., Ceyhan-Birsoy, O., DiStefano, M., Dwight, S. S., et al. (2017). Evaluating the Clinical Validity of Gene-Disease Associations: An Evidence-Based Framework Developed by the Clinical Genome Resource. Am. J. Hum. Genet. 100, 895–906. doi:10.1016/j.ajhg.2017.04.015
Taylor, J. L., Debost, J.-C. P. G., Morton, S. U., Wigdor, E. M., Heyne, H. O., Lal, D., et al. (2019). Paternal-age-related De Novo Mutations and Risk for Five Disorders. Nat. Commun. 10, 3043. doi:10.1038/s41467-019-11039-6
Timm, T., Matenia, D., Li, X.-Y., Griesshaber, B., and Mandelkow, E.-M. (2006). Signaling from MARK to Tau: Regulation, Cytoskeletal Crosstalk, and Pathological Phosphorylation. Neurodegener. Dis. 3, 207–217. doi:10.1159/000095258
van Woerden, G. M., Bos, M., de Konink, C., Distel, B., Avagliano Trezza, R., Shur, N. E., et al. (2021). TAOK1 Is Associated with Neurodevelopmental Disorder and Essential for Neuronal Maturation and Cortical Development. Hum. Mutat. 42, 445–459. doi:10.1002/humu.24176
Veltman, J. A., and Brunner, H. G. (2012). De Novo mutations in Human Genetic Disease. Nat. Rev. Genet. 13, 565–575. doi:10.1038/nrg3241
Wright, C. F., Prigmore, E., Rajan, D., Handsaker, J., McRae, J., Kaplanis, J., et al. (2019). Clinically-relevant Postzygotic Mosaicism in Parents and Children with Developmental Disorders in Trio Exome Sequencing Data. Nat. Commun. 10, 2985. doi:10.1038/s41467-019-11059-2
Xie, B., Fan, X., Lei, Y., Chen, R., Wang, J., Fu, C., et al. (2016). A Novel De Novo Microdeletion at 17q11.2 Adjacent to NF1 Gene Associated with Developmental Delay, Short Stature, Microcephaly and Dysmorphic Features. Mol. Cytogenet. 9, 41. doi:10.1186/s13039-016-0251-y
Keywords: dilated lateral ventricle, TAOK1, trio-whole exome sequencing, rare disease, neurodevelopment disorder
Citation: Yu L, Yang C, Shang N, Ding H, Zhu J, Zhu Y, Tan H and Zhang Y (2022) Paternal De Novo Variant of TAOK1 in a Fetus With Structural Brain Abnormalities. Front. Genet. 13:836853. doi: 10.3389/fgene.2022.836853
Received: 16 December 2021; Accepted: 30 May 2022;
Published: 19 July 2022.
Edited by:
Emiliano González Vioque, University Clinical Hospital of Santiago, SpainReviewed by:
Yiran Guo, Children’s Hospital of Philadelphia, United StatesVictor Wei Zhang, Jiajian Medical Co., Ltd., China
Copyright © 2022 Yu, Yang, Shang, Ding, Zhu, Zhu, Tan and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Zhang, emhhbmd5YW4xOTgxXzIwMDNAYWxpeXVuLmNvbQ==
†ORCID: Lihua Yu orcid.org/0000-0002-8456-9493