 Diana Marcela Vasquez-Forero1,2
Diana Marcela Vasquez-Forero1,2 Barbara Masotto3
Barbara Masotto3 Rosario Ferrer-Avargues3Christian Martin Moya3
Rosario Ferrer-Avargues3Christian Martin Moya3 Harry Pachajoa1,2,4*
Harry Pachajoa1,2,4*- 1Facultad de ciencia de la salud, Universidad Icesi, Cali, Colombia
- 2Departamento de Genética, Fundacion Valle del Lili, Cali, Colombia
- 3Medical Genetics Unit, Sistemas Genómicos, Paterna, Spain
- 4Centro de Investigaciones en Anomalías Congénitas y Enfermedades Raras Universidad Icesi, Cali, Colombia
Zhu–Tokita–Takenouchi–Kim syndrome is a multisystem disorder resulting from haploinsufficiency in the SON gene, which is characterized by developmental delay/intellectual disability, seizures, facial dysmorphism, short stature, and congenital malformations, primarily in the central nervous system, along with ophthalmic, dental, pulmonary, cardiologic, renal, gastrointestinal, and musculoskeletal anomalies. In this study, we describe the first Colombian patient with ZTT harboring a novel mutation that has not been previously reported and review the clinical and molecular features of previously reported patients in the literature.
Introduction
Zhu–Tokita–Takenouchi–Kim (ZTTK) syndrome is a rare autosomal dominant hereditary disease with multisystemic manifestations, caused by pathogenic variants in the SON gene, which is located on chromosome 21q22.11. The syndrome was first reported in 2015 by Zhu et al. (2015), who analyzed 119 trios based on Whole Exome Sequencing (WES) of patients with undiagnosed genetic diseases and identified a frameshift mutation in the SON gene in a 5-year-old girl with developmental delay, epilepsy, megalencephalic white matter dysplasia, mild dysmorphisms, intestinal atresia, and ventricular septal defect. Later in 2016, Takenouchi et al. (2016) reported a 13-year-old boy with the same frameshift mutation in SON, who had macrocephaly, facial dysmorphisms, severe intellectual disability, growth deficiency, and aortic valve regurgitation, but without brain abnormalities. That same year, Tokita et al. (2016) described seven individuals between 3 and 23 years of age with de novo truncating variants in SON, two of them with the previously reported frameshift mutation, presenting with developmental delay, short stature, hypotonia, and congenital malformations in the brain, heart, lungs, kidneys, and gastrointestinal tract.
In 2016, Kim et al. (2016) recruited 20 non-related individuals with de novo mutations in the SON gene and observed that all patients exhibited intellectual disability and facial dysmorphisms, with 17 of them also presenting with brain abnormalities, seizures or hypotonia, and other associated musculoskeletal, eye and vision, heart, urogenital, and gastrointestinal malformations. Thereby, ZTTK syndrome was named after the first authors who correlated the phenotypic findings with the pathogenic mutation in the SON gene (Tan et al., 2020).
Recently, several reports have contributed to establishing the phenotypic spectrum related to ZTTK syndrome. Dingemans in 2022 described 17 new patients who displayed the previously reported multisystem malformations, along with new abnormalities of the nails, skin, and hair, and also confirmed the presence of renal malformations and growth hormone deficiency in these patients (Dingemans et al., 2022). Additionally, in 2022, Kushary reported that hearing loss is an important feature observed in two out of 15 newly identified individuals with ZTTK syndrome, as well as in three previously reported patients (Kushary et al., 2021).
Here, we report the first case of ZTTK syndrome in Latin America, a 3-year-old boy with a de novo non-sense mutation in the SON gene, never reported in the literature.
Case description
The proband is a 3-year-old boy, who is the only offspring of healthy, non-consanguineous parents of advanced age (mother aged 39 years and father aged 46 years). The pregnancy was uneventful, with no abnormalities detected in ultrasound scans. However, due to fetal distress, the child was delivered via Cesarean section at 29 weeks of gestation. At birth, the child weighed 700 g and measured 33 cm in length. He required hospitalization for 3 months due to bronchopulmonary dysplasia, neonatal feeding difficulties, and severe hypotonia. Cardiac imaging revealed no abnormalities. Additionally, he exhibited developmental delay and experienced seizures. During the first year of life, he had three episodes of pneumonia and gastroesophageal reflux. At the age of 2, a brain MRI was conducted due to persistent seizures, which revealed agenesis of the corpus callosum; an EEG reported an inappropriate configuration of the background rhythm, and as a result, was evaluated by a pediatric neurologist who diagnosed focal structural epilepsy with clones on the left side of the face during sleep. Treatment was initiated with levetiracetam, which successfully controlled the seizures.
The patient underwent a medical genetic consultation at the age of 3. The child weighed 8.5 kg (−5.85 SD), had a height of 90 cm (−3.09 SD), and had an OFC of 48.5 cm (−2.08 SD). Physical examination revealed several dysmorphic features, including plagiocephaly, hypertelorism, hyperesotropia of the left eye, epicanthal folds, smooth philtrum, depressed nasal bridge, bilateral clinodactyly of the fifth finger, and superposition of the second and fifth finger over the third and fourth finger on the left foot. Moreover, he presented with inguinal hernia, non-descended testicles, and phimosis (Figure 1). Additionally, the patient presented with severe psychomotor development delay, stereotyped movements, persistent bruxism, generalized hypotonia, no interaction with the interlocutors, lack of gaze fixation, and absence of language.
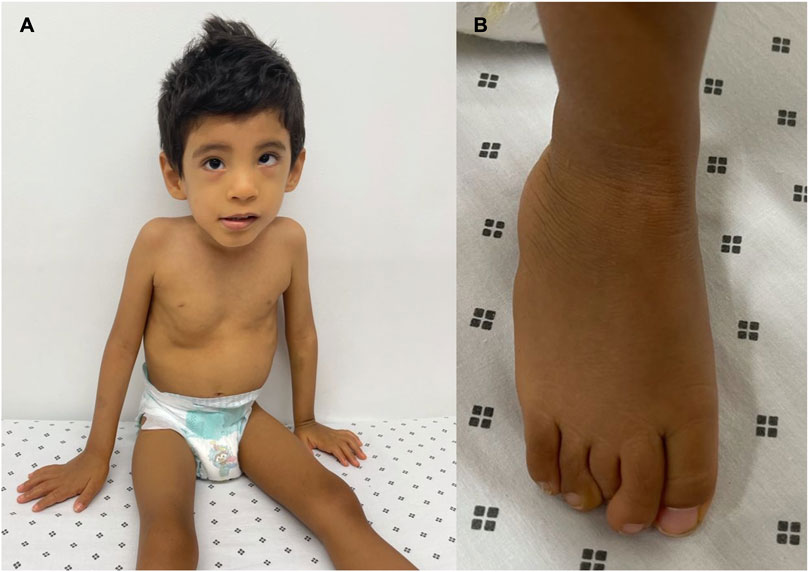
FIGURE 1. Phenotypic manifestations: (A) dysmorphic features such as hypertelorism, epicanthal folds, smooth philtrum, and depressed nasal bridge. (B) Superposition of the second and fifth finger over the third and fourth finger on the left foot.
The proband’s karyotype was normal for a male. The chromosome microarray analysis did not reveal any chromosomal numerical abnormalities or pathogenic CNVs. The underlying cause was unknown, so a trio whole-exome analysis was performed.
The WES analysis revealed an average read depth of 133.25x for this sample, and the coding regions of the genes included in this test had a coverage (≥20x) of 97.44%. A pathogenic, de novo, heterozygous variant SON: c.5743C>T; p.Arg1915Ter was identified in the patient and confirmed by Sanger sequencing (Figure 2). The parents tested negative.
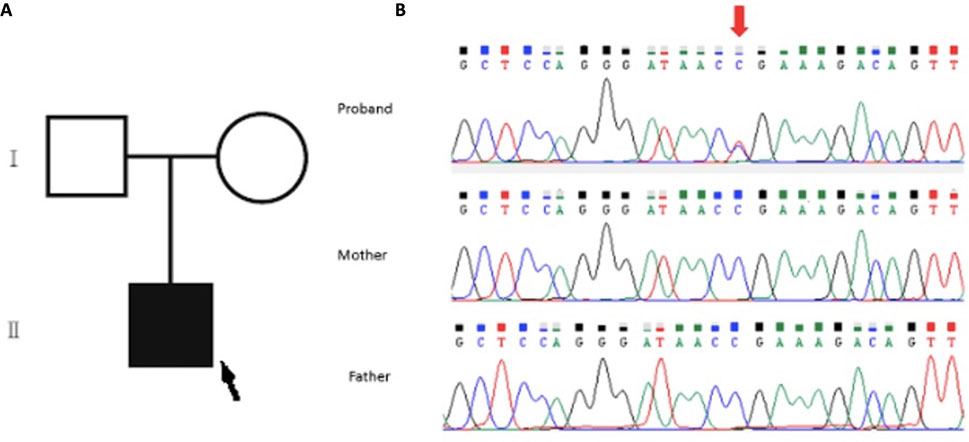
FIGURE 2. (A) Family pedigree of the patient: the proband is indicated with a black arrow. (B) Sanger sequencing analysis of the family. Upper panel (proband): segments of genomic DNA sequences showing the diagnostic variant SON:c.5743C>T indicated by the red arrow. Note the absence of such a variant in the middle and lower panel for the father and mother, respectively.
Material and methods
Chromosome karyotyping
The karyotype was determined in the child. A blood sample of 0.3 mL was incubated in a culture medium for complete separation of lymphocytes (20% fetal calf serum in Roswell Park Memorial Institute 1640 medium with 2.5% phytohemagglutinin and 2% L-glutamine and 1% penicillin–streptomycin in an incubator at 37°C for 72 h). Metaphases were harvested by adding colcemid for 60 min, followed by the addition of hypotonic potassium chloride (0.075 M) (treatment for 10 min and fixation, using standard 3:1 methanol–acetic fixative—Merck). Chromosome preparations were stained with G-banding. Chromosomal analysis revealed 46, XY.
DNA extraction
Peripheral blood samples were obtained from the proband and his parents. The genomic DNA was extracted using a Qiagen QIAamp DNA Mini Kit according to the manufacturer’s protocol.
Chromosomal microarray analysis
The Affymetrix CytoScan 750 K gene chip (Applied Biosystems) was used for DNA digestion, amplification, purification, fragmentation, labeling, hybridization, rinsing, staining, and scanning. ChAS software was used to analyze the scanned data. The established size threshold for reporting copy number variants (CNVs) was ≥100 Kb for gains and losses and ≥5 Mb for reduction of heterozygosity (ROH).
Whole exome sequencing (WES)
Genomic DNA of the proband and his parents was sequenced using next-generation sequencing (NGS). A trio whole exome sequencing was performed. Target regions were captured using the SureSelectXT Human All Exon V6 (Agilent Technologies), and sequencing was performed on an Illumina NovaSeq, with a read length of 2 × 150 bp.
The sequencing reads were aligned to the human reference genome (GRCh38) using the Burrows–Wheeler Aligner. The Genome Analysis Toolkit (GATK) was used to perform variant calling (VCF_files). Variants were filtered using GeneSystems® analysis software according to their predicted effects, allele frequencies, with a read count≥ 10x, and a variant reads/total reads ratio ≥ 0.2. The analyzed regions include the coding exons and adjacent intronic regions (±8bp) of the captured genes. The Integrative Genomics Viewer was used for visual exploration of the clinically relevant variants.
The American College of Medical Genetics and Genomics (ACMG) criteria was used for variant classification (Richards et al., 2015).
Sanger sequencing
Clinically relevant variants were confirmed by Sanger sequencing in the proband and his parents. The 3730xl DNA Analyzer (Applied Biosystems) was used, and the sequence of the primers was as follows: forward 5′ TTCACGGTCACGTTCAAGAC and reverse 5′AAAGACAGAAACATTTATCCTAAACCA.
Discussion
Zhu–Tokita–Takenouchi–Kim syndrome is an autosomal dominant hereditary disease characterized by facial dysmorphisms, short stature, intellectual disability/development delay, and hypotonia, often accompanied by congenital malformations affecting the brain, heart, kidneys, gastrointestinal tract, and musculoskeletal system. To date, 70 patients, including our patient, have been reported with a pathogenic frameshift mutation in SON, and they exhibit similar phenotypes, including dysmorphic features, developmental delay/intellectual disability (91.5%), hypotonia (65.7%), and brain malformation (77%) such as ventricular enlargement, leukodystrophy, cerebellar dysplasia, and corpus callosum abnormality, with or without the presence of seizures and hypotonia, EEG abnormalities, regression, and an autism spectrum disorder (Tokita et al., 2016). Eye abnormalities (52.8%) described are strabismus, myopia, and hypermetropia (Dingemans et al., 2022) (Slezak et al., 2020), while hearing loss (11.5%) is the main otorhinolaryngological feature, typically moderate to severe, and sometimes congenital (Kushary et al., 2021). Heart malformations (21.4%) include septal defects, aberrant subclavian artery, valvopathies, and patent ductus arteriosus (Tokita et al., 2016). Concomitant gastrointestinal malformations (18.5%) include intestinal atresia and duodenal malformation (Dingemans et al., 2022), while urogenital malformations (24.2%) such as single kidney, horseshoe kidney, and kidney dysplasia have been reported (Dingemans et al., 2022) (Slezak et al., 2020). Musculoskeletal abnormalities (68.5%) include short stature, craniosynostosis, joint laxity, cubitus valgus, scoliosis, hemivertebrae, cervical cord compression, lumbar hyperlordosis, contractures, small hands and feet, flat feet, aplasia/hypoplasia of the thumb, genu valgum, and abnormal rib defects (Slezak et al., 2020). Skin, hair, and nail alterations (15.7%) and teeth alterations (4.3%) such as retronychia, onychodystrophy, hyperkeratosis, dental enamel hypoplasia, and microdontia have also been reported (Table 1) (Quintana Castanedo et al., 2020).

TABLE 1. Phenotype summary of clinical features of ZTTK syndrome.
Furthermore, certain authors have reported on psychiatric conditions that were not previously included in the spectrum of the disease, but are present in most patients, such as autism spectrum disorder, destructive behavior, severe maladjusted behavior, hyperactivity, and neural regression (Yang and Yang, 2020). A small number of cases have also been noted to exhibit feeding difficulties after birth (52.8%): dysphagia, poor suckling, gastroesophageal reflux, and immunoglobulin deficiency (8.5%); mainly IgA deficiency (Tokita et al., 2016); as well as growth hormone deficiency (5.7%) (Slezak et al., 2020).
The principal clinical features of our patient overlap with those previously reported. However, it is noteworthy that our patient presented with bronchodysplasia, and pulmonary manifestations have only been reported in one other patient with agenesis of one lung (Tokita et al., 2016). While a specific facial dysmorphism for ZTTK syndrome has not been established yet, we observed some recurrent characteristics such as microcephaly and craniosynostosis, which have been reported in 16% and 8.5% of previously reported patients, respectively; other features that were observed include hypertelorism, epicanthal folds, strabismus, smooth philtrum, and thin lips.
The SON gene is located on the chromosome 21q22.11 and consists of 34,478 base pairs (bp) distributed across 12 exons. Exon 3 constitutes 82% of the gene’s coding region (Yang et al., 2019). The gene comprises three significant domains: an arginine/serine-rich domain, a G-patch domain, and a double-stranded RNA-binding motif (Tokita et al., 2016). SON is a nuclear speckle-localized protein that functions as a splicing co-factor and participates in the regulation of cell cycle progression. It plays a crucial role in neural development (Yang and Yang, 2020). Studies conducted in murine animal models have shown that SON knockdown causes alterations in neuronal migration and decreases the dendritic spine density in the cerebral cortex (Ueda et al., 2020). Additionally, in zebrafish models, haploid deficiency of the SON gene causes alteration in the tail length and body axis, associated with eye malformation and microcephaly (Kim et al., 2016). The SON gene is also recognized as a key neurodevelopment regulator, controlling the expression of other neurodevelopment, metabolism, and neuronal cell transition genes (Yang et al., 2019).
Until now, the 70 cases described in the literature reported 53 different mutations in SON. Of these mutations, 33 are frameshift mutations, with the most common mutations being c.5753_5756del and p (Val1918GlufsTer87), located in the exon 3 on the arginine/serine rich domain, which may have a genotype–phenotype correlation with cardiac defects (Kushary et al., 2021); Additionally, there were four missense, two in-frame deletions, eight non-sense variants, a 0.19 Mb deletion, and a whole-gene deletion.
The characteristic phenotype was observed in the individuals with a variant that causes loss of function in SON, leading to haploinsufficiency (Dingemans et al., 2022) (Table 2). Although patients with a missense variant present with a similar phenotype, at the cellular level, it does not cause neuronal migration defects, indicating that the pathophysiological mechanisms are different and are yet to be described (Dingemans et al., 2022).
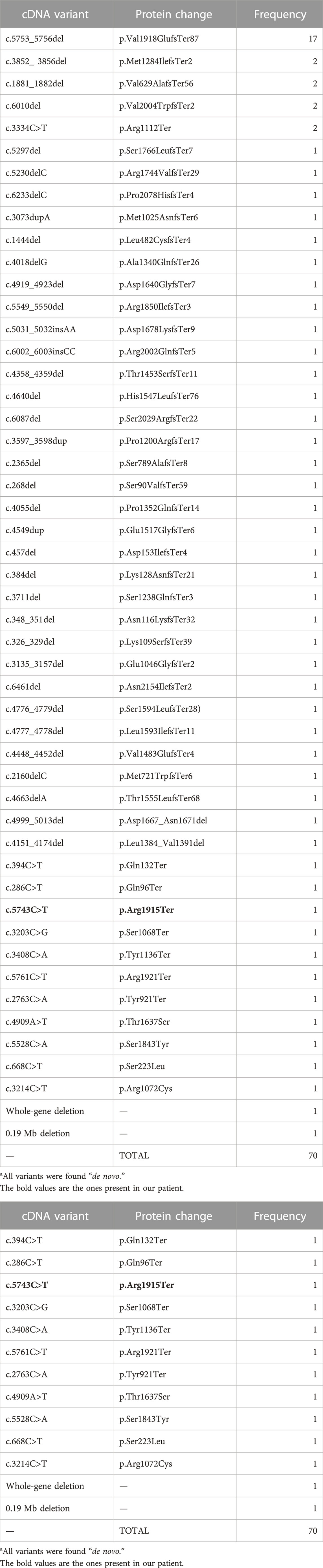
TABLE 2. Genotype summary of mutation types identified in SON (NM_138927.4) in association with ZTTK syndrome reported in the literature to date.
Our patient presents with a non-sense variant: c.5743C>T (p.Arg1915Ter), causing a truncated protein; according to the ACMG guidelines (Richards et al., 2015), the mutation is classified as a pathogenic mutation, which had not been previously reported in the literature, the first described case being the ZTTK case in Colombia.
After the diagnosis, paraclinical tests were conducted to investigate the phenotypic findings described in previous reports. However, these tests did not reveal any additional abnormalities in the gastrointestinal tract, renal system, or hearing ability. Since the seizures were well-managed with levetiracetam, no adjustments were made to the medication.
Concluding is important to recognize ZTTK syndrome as a severe multisystem developmental disorder suspected in patients with developmental delay/intellectual disability, short stature, facial dysmorphism, and congenital malformations: neuronal, ophthalmological, otorhinolaryngological, dental, pulmonary, cardiological, renal, gastrointestinal, and musculoskeletal. The diagnosis is established in a proband with suggestive findings and loss-of-function variants in the SON gene.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Fundación Valle del Lili, Colombia (human study protocol #617) and performed in accordance with the Declaration of Helsinki. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Acknowledgments
The authors would like to thank the patient’s family for agreeing to the publication of this report. They also thank the people who have contributed to the development and execution of this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1183362/full#supplementary-material
References
Dingemans, A. J. M., Truijen, K. M. G., Kim, J. H., Alaçam, Z., Faivre, L., Collins, K. M., et al. (2022). Establishing the phenotypic spectrum of ZTTK syndrome by analysis of 52 individuals with variants in SON. Eur. J. Hum. Genet. 30 (3), 271–281. doi:10.1038/s41431-021-00960-4
Kim, J. H., Shinde, D. N., Reijnders, M. R. F., Hauser, N. S., Belmonte, R. L., Wilson, G. R., et al. (2016). De novo mutations in SON disrupt RNA splicing of genes essential for brain development and metabolism, causing an intellectual-disability syndrome. Am. J. Hum. Genet. 99 (3), 711–719. doi:10.1016/j.ajhg.2016.06.029
Kushary, S. T., Revah-Politi, A., Barua, S., Ganapathi, M., Accogli, A., Aggarwal, V., et al. (2021). ZTTK syndrome: Clinical and molecular findings of 15 cases and a review of the literature. Am. J. Med. Genet. 185, 3740–3753. doi:10.1002/ajmg.a.62445
Quintana Castanedo, L., Sánchez Orta, A., Maseda Pedrero, R., Santos Simarro, F., Palomares Bralo, M., Feito Rodríguez, M., et al. (2020). Skin and nails abnormalities in a patient with ZTTK syndrome and a de novo mutation in SON. Pediatr. Dermatol. 37 (3), 517–519. doi:10.1111/pde.14113
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Slezak, R., Smigiel, R., Rydzanicz, M., Pollak, A., Kosinska, J., Stawinski, P., et al. (2020). Phenotypic expansion in Zhu-Tokita-Takenouchi-Kim syndrome caused by de novo variants in the SON gene. Mol. Genet. Genomic Med. 8 (10), e1432–e1437. doi:10.1002/mgg3.1432
Takenouchi, T., Miura, K., Uehara, T., Mizuno, S., and Kosaki, K. (2016). Establishing SON in 21q22.11 as a cause a new syndromic form of intellectual disability: Possible contribution to Braddock–Carey syndrome phenotype. Am. J. Med. Genet. Part A 170 (10), 2587–2590. doi:10.1002/ajmg.a.37761
Tan, Y., Duan, L., Yang, K., Liu, Q., Wang, J., Dong, Z., et al. (2020). A novel frameshift variant in SON causes Zhu-Tokita-Takenouchi-Kim Syndrome. J. Clin. Laboratory Analysis 34 (8), 233266–e23335. doi:10.1002/jcla.23326
Tokita, M. J. J., Braxton, A. A., Shao, Y., Lewis, A. M., Vincent, M., Küry, S., et al. (2016). De novo truncating variants in SON cause intellectual disability, congenital malformations, and failure to thrive. Am. J. Hum. Genet. 99 (3), 720–727. doi:10.1016/j.ajhg.2016.06.035
Ueda, M., Matsuki, T., Fukada, M., Eda, S., Toya, A., Iio, A., et al. (2020). Knockdown of Son, a mouse homologue of the ZTTK syndrome gene, causes neuronal migration defects and dendritic spine abnormalities. Mol. Brain 13 (1), 80–89. doi:10.1186/s13041-020-00622-4
Yang, L., and Yang, F. (2020). A de novo heterozygous variant in the SON gene is associated with Zhu-Tokita-Takenouchi-Kim syndrome. Mol. Genet. Genomic Med. 8 (11), e1496. doi:10.1002/mgg3.1496
Yang, Y., Xu, L., Yu, Z., Huang, H., and Yang, L. (2019). Clinical and genetic analysis of ZTTK syndrome caused by SON heterozygous mutation c.394C>T. Mol. Genet. Genomic Med. 7 (11), e953–e958. doi:10.1002/mgg3.953
Keywords: SON, Zhu–Tokita–Takenouchi–Kim syndrome, ZTTK syndrome, rare disease, Colombia
Citation: Vasquez-Forero DM, Masotto B, Ferrer-Avargues R, Moya CM and Pachajoa H (2023) Case report: A novel SON mutation in a Colombian patient with ZTTK syndrome. Front. Genet. 14:1183362. doi: 10.3389/fgene.2023.1183362
Received: 10 March 2023; Accepted: 12 April 2023;
Published: 05 July 2023.
Edited by:
Piero Pavone, University of Catania, ItalyReviewed by:
Volkan Okur, New York Genome Center, United StatesFadi F Hamdan, Montreal University, Canada
Copyright © 2023 Vasquez-Forero, Masotto, Ferrer-Avargues, Moya and Pachajoa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harry Pachajoa, aG1wYWNoYWpvYUBpY2VzaS5lZHUuY28=