 Oscar Fraile-Martinez1,2†
Oscar Fraile-Martinez1,2† Diego De Leon-Oliva1,2†Diego Liviu Boaru1,2
Diego De Leon-Oliva1,2†Diego Liviu Boaru1,2 Patricia De Castro-Martinez1,2
Patricia De Castro-Martinez1,2 Cielo Garcia-Montero1,2Silvestra Barrena-Blázquez1,2
Cielo Garcia-Montero1,2Silvestra Barrena-Blázquez1,2 Joaquin García-García3
Joaquin García-García3 Natalio García-Honduvilla1,2
Natalio García-Honduvilla1,2 Melchor Alvarez-Mon1,2,4,5Laura Lopez-Gonzalez2,3
Melchor Alvarez-Mon1,2,4,5Laura Lopez-Gonzalez2,3 Raul Diaz-Pedrero2,3,6*Luis G. Guijarro2,4,6,7
Raul Diaz-Pedrero2,3,6*Luis G. Guijarro2,4,6,7 Miguel A. Ortega1,2,4,8*
Miguel A. Ortega1,2,4,8*- 1Department of Medicine and Medical Specialities, Faculty of Medicine and Health Sciences, University of Alcalá, Alcala deHenares, Spain
- 2Ramón y Cajal Institute of Sanitary Research (IRYCIS), Madrid, Spain
- 3Department of Surgery, Medical and Social Sciences, Faculty of Medicine and Health Sciences, University of Alcalá, Alcala deHenares, Spain
- 4Network Biomedical Research Center for Liver and Digestive Diseases (CIBEREHD), Madrid, Spain
- 5Immune System Diseases-Rheumatology, Oncology Service an Internal Medicine (CIBEREHD), University Hospital Príncipe de Asturias, Alcala deHenares, Spain
- 6Department of General and Digestive Surgery, General and Digestive Surgery, Príncipe de Asturias Universitary Hospital, Alcala deHenares, Spain
- 7Unit of Biochemistry and Molecular Biology, Department of System Biology (CIBEREHD), University of Alcalá, Alcala deHenares, Spain
- 8Cancer Registry and Pathology Department, Principe de Asturias University Hospital, Alcala deHenares, Spain
Vascular diseases pose major health challenges, and understanding their underlying molecular mechanisms is essential to advance therapeutic interventions. Cellular senescence, a hallmark of aging, is a cellular state characterized by cell-cycle arrest, a senescence-associated secretory phenotype macromolecular damage, and metabolic dysregulation. Vascular senescence has been demonstrated to play a key role in different vascular diseases, such as atherosclerosis, peripheral arterial disease, hypertension, stroke, diabetes, chronic venous disease, and venous ulcers. Even though cellular senescence was first described in 1961, significant gaps persist in comprehending the epigenetic mechanisms driving vascular senescence and its subsequent inflammatory response. Through a comprehensive analysis, we aim to elucidate these knowledge gaps by exploring the network of epigenetic alterations that contribute to vascular senescence. In addition, we describe the consequent inflammatory cascades triggered by these epigenetic modifications. Finally, we explore translational applications involving biomarkers of vascular senescence and the emerging field of senotherapy targeting this biological process.
1 Introduction to cellular senescence
By definition, cellular senescence is the state of the cell induced by stress signals characterized by cell-cycle arrest, senescence-associated secretory phenotype (SASP), metabolic dysregulation, and macromolecular damage (Gorgoulis et al., 2019). Cellular senescence was first formally described in 1961 by Hayflick and Moorhead (Hayflick and Moorhead, 1961). They observed that cultured human diploid fibroblasts exhibited a finite number of divisions (40–60 population doublings), a phenomenon attributed to intrinsic factors, which were later identified as telomere shortening. Different external and internal stress and developmental signals trigger cellular senescence in response to cellular damage, including telomere shortening, DNA damage, oncogenic activation, radiation, oxidative and genotoxic stress, epigenetic changes, perturbed proteostasis, mitochondrial dysfunction, inflammation, nutrient deprivation and mechanical stress (Kuilman et al., 2010; Muñoz-Espín et al., 2013; Storer et al., 2013; Hernandez-Segura et al., 2018; Kumari and Jat, 2021; Huang et al., 2022; Zhou et al., 2023).
On the one hand, cellular senescence displays diverse physiological roles, mainly in embryonic development, wound healing, and prevention of tumor development (Muñoz-Espín et al., 2013; Rhinn et al., 2019; Wilkinson and Hardman, 2020; Yang et al., 2021). On the other hand, cellular senescence is a hallmark of aging due to the accumulation of senescent cells in old tissues compared to young tissues (Tuttle et al., 2020; López-Otín et al., 2023). Senescent cells signal to be removed by immune cells from the tissues, triggering both innate and adaptive responses (Kale et al., 2020). However, with age, the cell-intrinsic damage and the failure of immune clearance increase the number of senescent cells in the tissues and contribute to the development and progression of aging-related diseases such as cancer, neurodegenerative diseases, atherosclerosis, osteoarthritis or pulmonary fibrosis (Burton and Stolzing, 2018; Ovadya et al., 2018; Mylonas and O’Loghlen, 2022).
Senescence is characterized by a cell-cycle arrest that can occur in the G1 or G2 phase (Gire and Dulic, 2015). The cell-cycle withdrawal is generally irreversible, although under certain circumstances senescent cells can re-entry the cell-cycle, such as tumor cells, or be reprogrammed into induced pluripotent stem cells (Lapasset et al., 2011; Saleh et al., 2019). Senescence differs from quiescent and terminally differentiated cells, which also withdraw from the cell-cycle, at the level of signaling pathways, and SASP and neither shows macromolecular damage (Terzi et al., 2016; Fujimaki et al., 2019). The cell-cycle arrest is achieved through different tumor suppressor pathways, especially p53/p21WAF1/CIP1 and p16INK4A/pRB tumor suppressor pathways (Kumari and Jat, 2021). p53 acts as a transcription factor (TF), which is activated in response to DNA damage caused by factors like telomere attrition, oxidative stress, or oncogenic stress, and orchestrates a genetic response that leads to the induction of cellular senescence and inhibits other alternatives such as apoptosis (Childs et al., 2014; Sheekey and Narita, 2023). Of the downstream effectors, the key role is predominantly played by p21CIP1, which is an inhibitor of cyclin-dependent kinase (CDK)-cyclin complex activity leading to cell-cycle withdrawal (Herranz and Gil, 2018). On the other hand, activation of p16 through epigenetic regulation leads to the inhibition of the formation of cyclin D–CDK4/6 complexes, which prevents the phosphorylation of the retinoblastoma protein (pRb) (Rayess et al., 2012). Hypophosphorylated pRb maintains in the cytoplasm in complex with the TF E2F and cell-cycle genes are not transcribed. Moreover, the two pathways present an extensive crosstalk. The hypothesis is that while the p53/p21WAF1/CIP1 signaling pathway contributes to the onset of cellular senescence, the p16INK4A/pRB pathway is in charge of maintaining this state (Kohli et al., 2021).
Almost all somatic cell types and tissues can undergo cellular senescence. The evidence of cellular senescence in the vasculature is termed vascular senescence and plays a critical role in the development of vascular diseases, including atherosclerosis, peripheral arterial disease, hypertension, stroke, diabetes, chronic venous disease and venous ulcers (Yamazaki et al., 2016; Katsuumi et al., 2018; Shakeri et al., 2018; McCarthy et al., 2019; Ortega et al., 2021b; Lim et al., 2021).
Epigenetics regulates cellular senescence through chromatin remodeling, DNA methylation, and the involvement of non-coding RNAs (Sidler et al., 2017; Crouch et al., 2022). Senescent cells undergo chromatin restructuring, altering DNA packaging around histones, which impacts gene accessibility through modifications such as methylation and acetylation (Roger et al., 2021). DNA methylation contributes to establishing and maintaining the senescent state by silencing specific genes related to cell proliferation (Sakaki et al., 2017). In addition, non-coding RNAs, such as microRNAs and long non-coding RNAs, modulate fundamental pathways in senescence by targeting genes related to cellular aging, DNA repair, and the cell cycle (Puvvula, 2019). Together, these epigenetic modifications coordinate the transition to cellular senescence by regulating genes related to cell cycle arrest, DNA damage response, SASP, and inflammation, thus shaping the phenotype of senescent cells.
Inflammation is a protective mechanism, essential for the immune system to detect and eliminate harmful agents while initiating the healing process. Inflammatory responses can manifest as acute or chronic, each serving different purposes (Li et al., 2023). However, not all inflammatory processes benefit the body; in some cases, diseases trigger harmful inflammation, in which the immune system inadvertently attacks the body’s cells. In this sense, senescent cells often release several inflammatory substances, such as matrix metalloproteinases (MMPs), growth factors (GFs), and cytokines (CKs), which form the SASP (Giacconi et al., 2015; Lee et al., 2021). This paracrine signaling contributes to a variety of adverse outcomes, such as cancer development, persistent inflammation called “inflammaging” and tissue restructuring (Olivieri et al., 2018). Moreover, senescent cells are indeed a strong nexus between cellular aging and development of cancer (Campisi, 2013; Schmitt et al., 2022; López-Otín et al., 2023). Pharmacological removal of senescent cells expressing p16INK4A in aging mice delayed tumorigenesis and mitigated age-related decline in multiple organs, showing no apparent adverse effects (Baker et al., 2016).
In this comprehensive review, we aim to understand the interplay between epigenetic modifications and immune activation mechanisms underlying vascular senescence. First, we present an overview of the histology of the vascular wall and vascular senescence. Then, we explore the epigenetic alterations identified in the context of vascular senescence and elucidate the critical role played by immune activation. Subsequently, we sought to synthesize the connections between epigenetic modifications, the immune response, and the phenomenon of vascular senescence. Finally, we describe the translational implications arising from the fields of epigenetics, inflammation, and cellular senescence in the context of vascular disorders, including biomarkers and senotherapeutics.
2 Histology of vascular wall and vascular senescence
The vascular wall, a complex structure comprising three distinct layers, the intima, media, and adventitia, exhibits diverse histological, biochemical, and functional characteristics essential for maintaining vascular homeostasis and modulating the vascular response to stress or injury. From a histological point of view, the differences in thickness and composition of them distinguish the different types of blood vessels. In general, the tunica intima is formed by a single layer of endothelial cells (ECs) or endothelium, which surrounds the internal vascular lumen acting as a functional barrier (Taylor and Bordoni, 2023). The endothelium is supported by a basement membrane composed of collagen, proteoglycans, and glycoproteins (Halper, 2018). The subendothelial layer is composed of loose connective tissue, and sometimes harbors vascular smooth muscle cells (VSMCs), and an internal elastic lamina in arteries and some veins (Mazurek et al., 2017). The tunica media is stabilized by an elastic external lamina, in arteries, and an extracellular matrix composed mainly of elastin and collagen fibers (Munjal and Bordoni, 2023). The tunica media houses predominantly VSMCs arranged in circumferentially organized layers with elastin, reticular fibers, and proteoglycans between them (Frismantiene et al., 2018). The outermost layer, the adventitia, has myofibroblasts and fibroblasts as the main cellular constituents, responsible for generating the fibroelastic extracellular matrix that defines the appearance of this layer (Stenmark et al., 2013). In addition, in larger vessels, the adventitia harbors a network of small blood vessels and nerves, which provide irrigation and innervation to the vascular wall, respectively. Figure 1 illustrates the histological representation of these layers in both arteries and veins, as well as their main differences.

FIGURE 1. Representation of the histological structure of the vascular wall of arteries and veins. Arteries present a relatively thick tunica media and more VSMCs, whereas veins show a larger tunica adventitia. The internal elastic lamina appears in arteries and only in the median veins, where is thinner and discontinuous. The external elastic lamina of arteries is a layer composed of elastin that separates the tunica media and the adventitia. Finally, venous valves are folds of the tunica intima composed of dense connective tissue present in the middle veins, especially abundant in the lower extremities, to prevent the backflow of blood.
Cellular senescence in blood vessels plays a critical role in vascular aging, and the elimination of senescent cells is a promising approach to prevent or mitigate age-related vascular diseases (Figure 2) (Ungvari et al., 2020). Manifestations of vascular aging include arterial and capillary stiffness, endothelial dysfunction, increased oxidative stress, decreased capacity for angiogenesis, early signs of atherosclerosis, and low-grade chronic inflammation (Ghebre et al., 2016). Vascular aging constitutes a progressive decline in both the structural integrity and functional capacity of blood vessels, which contributes to damage to the heart, brain, kidneys, and other organs (Climie et al., 2023; Ya and Bayraktutan, 2023). It is important that, while there is extensive literature on arterial aging and related diseases, there is a significant knowledge gap on venous aging. However, research on the role of venous aging is of great importance in the understanding of the development of venous conditions prevalent among older adults, such as varicose veins, chronic venous insufficiency, and deep vein thrombosis (Ortega et al., 2021a; Ortega et al., 2021b; Molnár et al., 2021; Mühlberger et al., 2022).
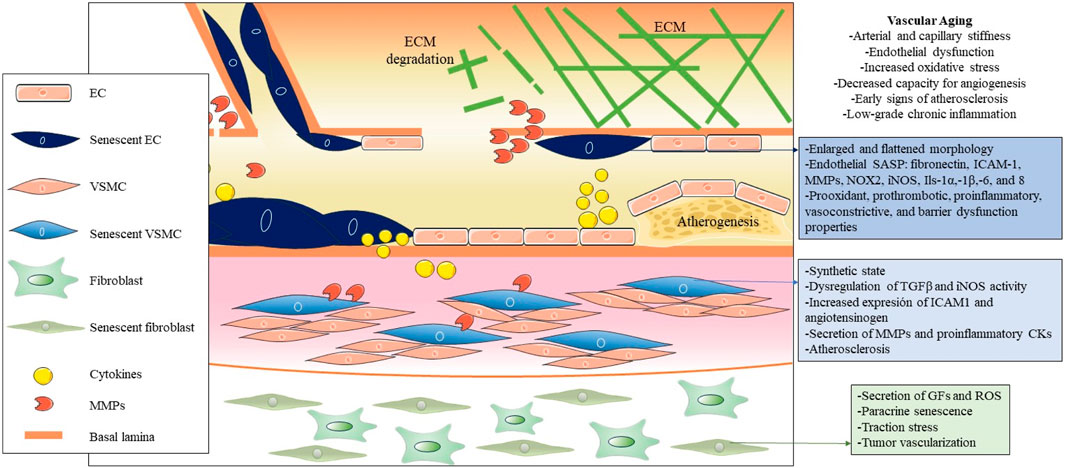
FIGURE 2. Pathogenic influence of cellular senescence in vascular aging. The figure illustrates the pathogenic impact of cellular senescence in vascular aging, depicting the main cellular and molecular entities associated with senescence in this context. Throughout the three main layers that compose the vascular wall, distinct senescent cell types, predominantly endothelial cells (ECs), vascular smooth muscle cells (VSMCs), and fibroblasts, are distinguished from the inside out. Due to their decreased functionality and secretion of SASP factors, these cells collectively contribute to vascular dysfunction, altered permeability, increased inflammation and oxidative stress, and the onset of atherogenesis in arteries.
The main cellular populations of the blood vessels that can undergo cellular senescence due to various stressors like oxidative stress, DNA damage, and signaling from nearby cells are ECs, VSMCs, adventitial fibroblasts, immune cells and endothelial progenitor cells (EPCs) (Katsuumi et al., 2018; Vellasamy et al., 2022; Suda et al., 2023).
2.1 Endothelial cell senescence
EC senescence is a key feature of the aging process in blood vessels. These cells, which are vital for vascular homeostasis, undergo senescence during aging or due to stimuli such as reactive oxygen species (ROS), chronic inflammation, altered blood flow patterns, metabolic influences such as glucose, insulin and specific lipid subfractions (Khan et al., 2017; Hwang et al., 2022), while molecules like polyphenols, amino acids, and omega-3 fatty acids display senescence-inhibiting effects (Sakai et al., 2017; Furuuchi et al., 2018; Tsuboi et al., 2018). This transition confers prooxidant, prothrombotic, proinflammatory, vasoconstrictive, and barrier dysfunction properties on ECs, altering their crucial role in maintaining healthy blood flow and vessel integrity (Ramírez et al., 2022; Han and Kim, 2023). Senescent ECs show the typical enlarged and flattened morphology of senescence. Senescence in ECs shows changes in molecular profiles and contributes to the distinctive endothelial SASP, including higher expression of fibronectin, intercellular adhesion molecule 1 (ICAM-1), matrix metalloproteinases (MMPs), NADPH oxidase 2 (NOX2), inducible nitric oxide synthase (iNOS), interleukins 1α 1β, 6, and 8, and decreased production of nitric oxide (NO) (Hayashi et al., 2006; Zhou et al., 2006; Yin and Pickering, 2016; Rojas et al., 2017). Interestingly, exercise counteracts EC senescence by modulating oxidative stress and inflammatory pathways, preserving vascular function during aging, and offering a promising avenue to counteract age-related vascular complications (Rossman et al., 2017; Meng et al., 2023). Also, circulating endothelial progenitor cells in patients with coronary artery disease exhibited reduced telomere length and telomerase activity via oxidative DNA damage, which may be related to EC senescence (Satoh et al., 2008).
2.2 Vascular smooth muscle cells senescence
Aged blood vessels show decreased compliance, elasticity, and distensibility, as well as increased stiffness (Kohn et al., 2015). These alterations contribute to higher systolic blood pressure and lower diastolic blood pressure. This is explained by the accumulation of collagen and decay of elastin in the layers of the arterial wall (Mitchell, 2021).
However, senescent VSMCs switch from a contractile to a synthetic state. This alteration involves dysregulated transforming growth factor-β (TGF-β) signaling, increased iNOS activity, and increased expression of ICAM-1 and angiotensinogen under stress conditions (Gorgoulis et al., 2005; Lakatta, 2015; You et al., 2019). Particularly, senescent VSMCs tend to accumulate in atherosclerotic plaques, contributing to inflammation, extension and vulnerability of the plaque by different mechanisms: reduced collagen production, secretion of MMPs, proinflammatory cytokines (IL-1α,-6 and,-8 and MCP1), chemotaxis of monocytes/macrophages, acquisition of an “osteoblast-like” phenotype (Nakano-Kurimoto et al., 2009; Gardner et al., 2015; Lacolley et al., 2017). Recent findings indicate that senescent VSMCs in atherosclerotic plaque have decreased levels of telomeric repeat-binding factor-2 (TRF2), a protein crucial for telomere protection (Wang et al., 2015; Uryga et al., 2021). In vivo studies with transgenic mice confirm that inhibition of VSMC senescence, by modulating TRF2, has the potential to prevent atherosclerotic disease progression.
2.3 Fibroblasts senescence
Senescent vascular fibroblasts, found in the adventitia of blood vessels, secrete proinflammatory molecules and contribute to vascular remodeling. Their secretion of bioactive substances, such as GFs and ROS, creates a pro-oxidant environment that may favor age-related vascular pathologies, such as pulmonary hypertension (Li et al., 2021). Furthermore, it has been demonstrated in fibroblasts that senescent cells can influence intact neighboring cells through a bystander effect, which induces a DNA damage response, propagating senescence in the vascular microenvironment (Nelson et al., 2012). Additionally, it was demonstrated in A 3D tissue model-on-a-chip, that senescent fibroblasts also exert excessive traction stress on the surrounding extracellular matrix (ECM) (Pauty et al., 2021). In particular, senescent fibroblasts also promote tumor vascularization by inducing increased expression of vascular endothelial growth factor (VEGF), showing a relevant influence on the tumor microenvironment (Coppe et al., 2006).
2.4 Immune cells senescence
Immunosenescence is a phenomenon in which the immune system gradually declines with age (Wang Y. et al., 2022). The senescence of immune cells plays a pivotal role in the development of atherosclerosis (Figure 3) (Vellasamy et al., 2022). The cellular senescence of monocytes and macrophages changes their functionality, contributing to chronic inflammation and pathological processes associated with atherosclerosis, which ultimately impact plaque development and stability and the risk of cardiovascular events. All three monocyte subsets, classical (CD14++CD16−), intermediate (CD14++C, D16+), and non-classical (CD14+CD16++), show characteristics of senescence, particularly the nonclassical subset, which increases with age (Ong et al., 2018). This subset had markedly elevated levels of TNF-α, CCL3, and CCL4, whereas IL-6, IL-8, IL-1β, and CCL5 were secreted at comparatively high levels in both the intermediate and non-classical subsets. This subset also expresses membrane-bound IL-1α and exhibits increased NF-kB signaling (Zawada et al., 2011). Senescent monocytes, especially the non-classical subset, express proatherogenic chemokine receptors (CCR2, CCR5, CCR7, and CX3CR1) and endothelial adhesion molecules (VCAM-1 and ICAM-1), which increases their adherence to vascular walls and contributes to the development of atherosclerotic plaque (Merino et al., 2011).
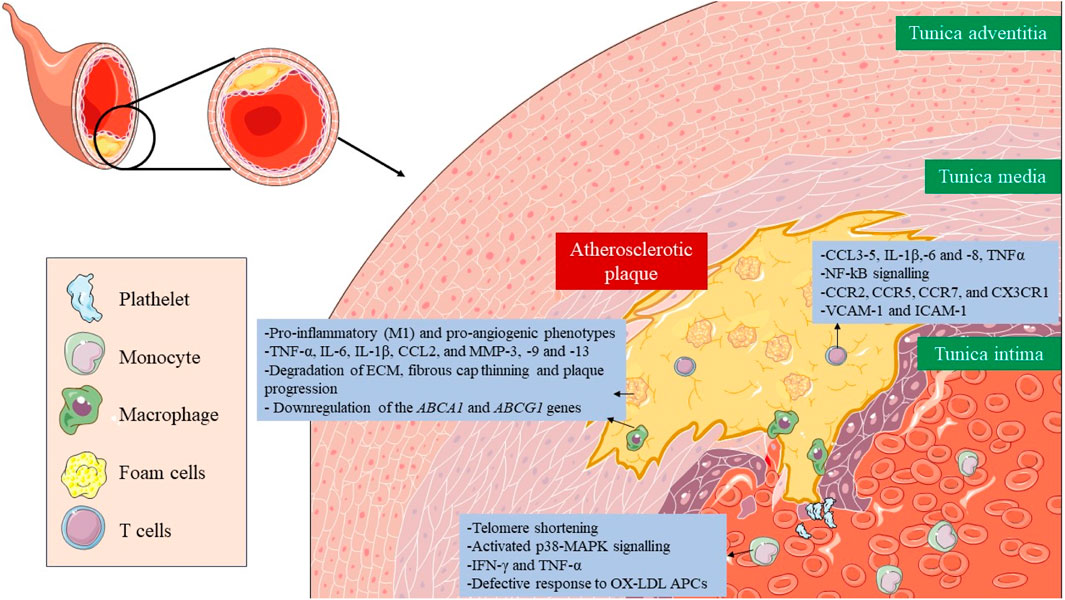
FIGURE 3. Role of senescent immune cells in atherosclerotic plaque. The senescence of immune cells, including monocytes, macrophages, foam cells, and T lymphocytes, plays a critical role in the formation, development and rupture of atherosclerotic plaque. Altered cellular functionality due to immunosenescence results in the secretion of proinflammatory cytokines, amplifying plaque inflammation and facilitating the recruitment of additional immune cells. Taken together, immunosenescence has a major impact on plaque stability, interactions, and potential therapeutic approaches targeting these senescent immune components.
In the case of senescent macrophages, they adopt pro-inflammatory (M1) and pro-angiogenic phenotypes characterized by the secretion of SASP factors, such as TNF-α, IL-6, IL-1β, CCL2, and MMP-3, -9 and -13, which also lead to degradation of ECM, fibrous cap thinning, plaque progression, and instability (Sun et al., 2022; Zhu et al., 2023). They also present a disruption in cholesterol efflux due to downregulation of the ABCA1 and ABCG1 genes (Sene et al., 2013; Lin et al., 2018). Lastly, senescent foam cells accumulate in the subendothelial space and work in a very similar way to macrophages contributing to atherosclerosis (Childs et al., 2016).
Lastly, senescence in T cells presents implications for vascular disorders. Senescence is induced after oxidative stress leads to a reduction in telomerase activity, and consequently, telomere shortening, and the acquisition of a proinflammatory phenotype, with the secretion of IFN-γ and TNF-α (Mittelbrunn and Kroemer, 2021; Shirakawa and Sano, 2021). They have activated the p38 mitogen-activated protein kinase (p38-MAPK) signaling pathway (Reustle and Torzewski, 2018). Different subsets of senescent T cells appear also within the atherosclerotic plaque, such as CD4+/CD8+ TEMRA, CD8+/CD4+CD28− or CD8+CD57+CD27−CD28null T cells, where they promote inflammation and present a defective response to oxidized low-density lipoproteins (oxLDL) antigen presenting cells (APCs) (Kaplan et al., 2011; Yu et al., 2016).
3 Epigenetic modifications and vascular senescence
Epigenetic mechanisms are fundamental drivers of cellular senescence (Nacarelli et al., 2017; Crouch et al., 2022). Expression of the “Yamanaka factors” demonstrates their potential to restore cellular pluripotency by effectively eliminating epigenetic memory in differentiated cells through loss of heterochromatin and reducing the levels of repressive histones H3K9me2, H3K9me3, and 5-methylcytosine (Takahashi and Yamanaka, 2006; Wang K. et al., 2022). Furthermore, epigenetic alterations are also a hallmark of aging (López-Otín et al., 2023). Therefore, it is of great interest to study their connection with cellular senescence, with a special focus on vascular senescence in this review. The epigenetic alterations include DNA methylation, histone modifications, non-coding RNA-based gene regulation, and remodeling of chromatin landscape, and are correlated with cardiovascular risk factors and vascular aging (Ding et al., 2018; Zhang et al., 2018; Lin et al., 2022). To deepen our understanding of the importance of epigenetic regulation, cellular senescence can be induced in vitro by epigenetic modifiers, which activate the corresponding molecular pathways, based on the upregulation of p16INK4A (Petrova et al., 2016). These include DNA methyltransferases inhibitors (5-aza-2- deoxycytidine), histone deacetylases inhibitors (sodium butyrate, trichostatin A), histone acetyltransferases inhibitors (curcumin, C646) or histone methyltransferases inhibitors (e.g., BRD4770).
DNA methylation is an important regulator of gene expression in vascular senescence (Ding et al., 2020; Xu et al., 2021). Environmental signals influence the activity of DNA methyltransferases (DNMT1) 1, 3a, and 3b (Denis et al., 2011). For instance, the deficiency of folic acid, a DNMT inhibitor, is correlated with an increased risk of cardiovascular disease (CVD), including coronary heart disease, atherosclerosis, and anemia (Glier et al., 2014; Voelter-Mahlknecht, 2016). The folic acid supplementation delays atherosclerotic lesion development by modulating MCP1 and VEGF DNA methylation levels (Cui et al., 2017). DNMT1 has been identified as an essential factor in the formation of senescence-associated heterochromatin foci (SAHF) through the upregulation of HMGA2 (Sati et al., 2020). SAHF are domains of facultative heterochromatin in senescent cells that repress the expression of genes related to proliferation and are another biomarker of cellular senescence (Aird and Zhang, 2013). Abnormal DNA methylation patterns, prevalent in aging cells, have been widely associated with various age-related vascular diseases, hence their importance in vascular senescence (Tabaei and Tabaee, 2019; Xu et al., 2021).
Histone acetylation stands as a critical epigenetic regulator in vascular senescence, modulating chromatin accessibility and gene expression. The regulation of histone acetylation involves histone deacetylases (HDACs) and histone acetyltransferases (HATs). Regarding EC senescence, HDAC3 activated by laminar flow and VEGF through the VEGF receptor 2/protein kinase B pathway, stimulates EPC proliferation and EC differentiation (Zeng et al., 2006). In contrast, suppression of HDAC3 disrupts VEGF-induced EPC function. Furthermore, shear stress increases HAT activity, driving differentiation of the mouse embryonic stem cells into EC lineage (Illi et al., 2005). On the other hand, the interaction of Sirt1 with the PAI-1 promoter inhibits the acetylation of lysine 16 of histone H4, exerting a protective effect against vascular endothelial cell senescence (Wan et al., 2014). Also, Ang II induced protein kinase B-mediated phosphorylation and lysine acetylation of PGC-1 through the general control histone acetyltransferase nonderepressible 5, resulting in reduced PGC-1 activity and catalase expression in vascular cells (Xiong et al., 2010). Different studies have highlighted specific histone methylation markers, such as H4K20me3, H3K9me3, and H3K27me3, associated with the aging process (Yi and Kim, 2020). Decreased expression of H3K27me3 has been observed in aged hematopoietic stem cells (HSCs), whereas decreased expression of H3K9me3 has been observed in mesenchymal stem cells (MSCs) from aged individuals (Cakouros and Gronthos, 2019; Wang K. et al., 2022).
Non-coding RNAs, microRNAs (miRNAs), and long non-coding RNAs (lncRNAs, play important regulatory roles in vascular senescence. The miRNAs, approximately 25 nucleotides in length, play a key role in regulating EC senescence and vascular aging (Lee et al., 2015; Lin et al., 2016; Nikolajevic et al., 2022). Several miRNAs are involved in processes such as vascular growth, angiogenesis, inflammation, and fibrosis (Table 1). In addition, miRNAs may play a role in senescence distinct from their traditional functions. For example, Argonaute 2 (AGO2) forms a complex with pRB in the nucleus by binding to let-7f, resulting in restrictive chromatin at the CD2 and CDCA8 promoters (Benhamed et al., 2012). lncRNAs also influence vascular diseases by regulating ECs and VSMCs. Loss of lncRNA H19 increases p21 and p16 expression, leading to EC senescence (Hofmann et al., 2019). LncRNA MEG3, elevated in the aged atrium and human umbilical vein endothelial cells (HUVECs), suppresses the proliferation of ECs through interaction with miR-9 and has a regulatory role on angiogenesis through the notch signaling pathway (Jayasuriya et al., 2022). Exosomal lncRNA GAS5 is shown to regulate the apoptosis of macrophages and ECs in atherosclerotic plaques (Chen et al., 2017).

TABLE 1. Role of different miRNAs with impact on vascular senescence.
Nuclear lamins are structural proteins of the nuclear envelope and are classified as V-type intermediate filaments. These lamins are divided into two main types based on their isoelectric points: A-type (which includes lamins A and C) and B-type (B1 and B2). Among these, lamin B1 is of particular importance in ensuring organogenesis and survival of the organism (Kim et al., 2011). Research indicates that cellular senescence, observed in both human and mouse cells, leads to a depletion of lamin B1 (Freund et al., 2012). Interestingly, this depletion occurs due to direct stimulation of the p53 or pRB pathway, independent of several typical cellular markers of senescence, such as p38, NF-κB, DNA damage response, or ROS. In contrast to apoptosis, in which caspases cleave lamins, senescent cells do not show lamin B1 cleavage products. Furthermore, inhibition of caspases does not affect the loss of lamin B1 during senescence (Shah et al., 2013). Instead, decreased stability of lamin B1 mRNA contributes to reduced levels of lamin B1 mRNA during the senescence process. Thus, lamin B1 depletion emerges as a robust indicator of cellular senescence, applicable both in vitro and in vivo.
4 Connecting inflammation with vascular senescence
A significant aspect of the changes in gene expression associated with aging is the increased activation of genes related to inflammation and immune responses. Indeed, chronic inflammation is another hallmark of aging and increases with age, known as inflammaging (Li et al., 2023). This phenomenon is related to the interaction between inflammation and cellular senescence, which favors the pathogenesis of numerous age-related vascular diseases (Del Pinto and Ferri, 2018; Liberale et al., 2022). This interaction is bidirectional, as inflammation spreads cellular senescence and senescent cells promote inflammation by secreting SASP factors (Stojanovic et al., 2020; Zhu et al., 2021). In addition, the function of the immune system decreases with age, termed immunosenescence, and thus the elimination of senescent cells, which promotes senescence-induced inflammation. The relationship between inflammation and vascular senescence can be explained by the involvement of specific biomarkers and signaling pathways.
IL-6 is emerging as a member of the interleukin family showing promise as a biomarker for aging and a reliable indicator of low-grade inflammation (Puzianowska-Kuźnicka et al., 2016). It is correlated with markers of aging such as carotid intima-media thickness (cIMT) and plaque progression (Huang et al., 2016). Moreover, IL-6 has been implicated in hypertension as its levels decrease with angiotensin II receptor blockade therapy suggesting a link to blood pressure in hypertensive patients (Stevenson et al., 2018). C-reactive protein (CRP), an acute phase reactant regulated by IL-6 and IL-1, does not act as a general marker of inflammation but also poses a significant risk factor for age-related conditions. Elevated CRP levels have been associated with declines in physical abilities. They are correlated with conditions such as CVD, hypertension, diabetes mellitus, and kidney disease (Tang et al., 2017). GDF 15, a member of the superfamily transforming growth factor β (TGF β), stands out particularly due to its controversial association with age and EC senescence. On the one hand, GDF15 can promote EC senescence through a p16 ROS-mediated pathway and contribute to atherosclerosis through pro-senescent activity (Park et al., 2016). On the other hand, the paracrine effects of GDF15 in non-senescent ECs showed that GDF15 increased proliferation, migration, and NO production and activated several signaling pathways such as AKT, ERK1/,2, and SMAD2 without triggering any oxidative stress, and therefore, preventing the endothelial dysfunction (Ha et al., 2019).
The SASP comprises a set of various substances released by senescent cells, which play a key role in maintaining tissue balance or precipitating dysfunction. This phenomenon is one of the main ones responsible for the adverse effects associated with senescent cells. Within the SASP, these cells release an array of substances, such as growth factors, metalloproteinases, cytokines, chemokines, and extracellular vesicles (EVs), which profoundly influence immune signaling and cell-cell communication (Ohtani, 2022; Sun et al., 2022). We have reviewed, i.e., section 2 the main SASP factors secreted by senescent cells affecting the vascular wall, which contribute to inflammation, and oxidative stress and lead to vascular dysfunction and disease. Mechanisms such as the NF-κB, C/EBPbd GATA4, mTOR, p38 MAPK, and cGAS-STING pathways and cytoplasmic chromatin fragments contribute to SASP activation (Salminen et al., 2012; Kang et al., 2015; Cuollo et al., 2020; Miller et al., 2021). Interactions of senescent cells with the microenvironment occur through cytoplasmic bridges, exosomes, NOTCH/JAG1 signaling, and ROS release, reflecting the complexity and importance of their impact on tissue function and health outcomes (Coppé et al., 2010; Biran et al., 2014; Takasugi et al., 2017).
5 Translational applications derived from epigenetics, inflammation, and cellular senescence in vascular disorders
5.1 Biomarkers of cellular senescence. Relevance in vascular aging
The study of cellular senescence has expanded considerably in recent years due to the discovery of numerous novel roles that this phenomenon plays in physiology and disease. The cell-cycle withdrawal is just one of the hallmarks of cellular senescence, which can be triggered by several stressors, such as telomere shortening, oxidative stress, DNA damage, and, oncogene activation (Muñoz-Espín and Serrano, 2014). Accordingly, cellular senescence has been reinterpreted as the process by which a dividing cell, in response to a stressful or damaging stimulus, enters a stable cell cycle arrest and usually secretes a complex mixture of substances that affect the surrounding tissue, while maintaining metabolic activity and resisting the signals of mitosis and apoptosis. Among other things, senescent cells have an enlarged and flattened shape, an expanded lysosomal compartment, and certain chromatin and epigenetic alterations (Beck et al., 2020). To accurately delineate cellular senescence in tissues and cell cultures, an optimal multifactorial approach requires meticulous organization and compilation of previously published methodologies and diverse approaches (González-Gualda et al., 2021). Senescence invites investigation of its involvement in diverse cellular pathophysiological processes, thus elucidating its four distinguishing features. Recently, single-cell time-lapse imaging revealed us that cell cycle withdrawal occurs gradually and not in a clear binary step (Ashraf et al., 2023). In addition, the intensity levels of senescence biomarkers appear to integrate the duration of earlier cell cycle withdrawal. The biomarkers related to epigenetic mechanisms and inflammation/SASP that regulate vascular senescence have been discussed in the previous sections. However, now we will take a look at the more general senescence biomarkers (Table 2).
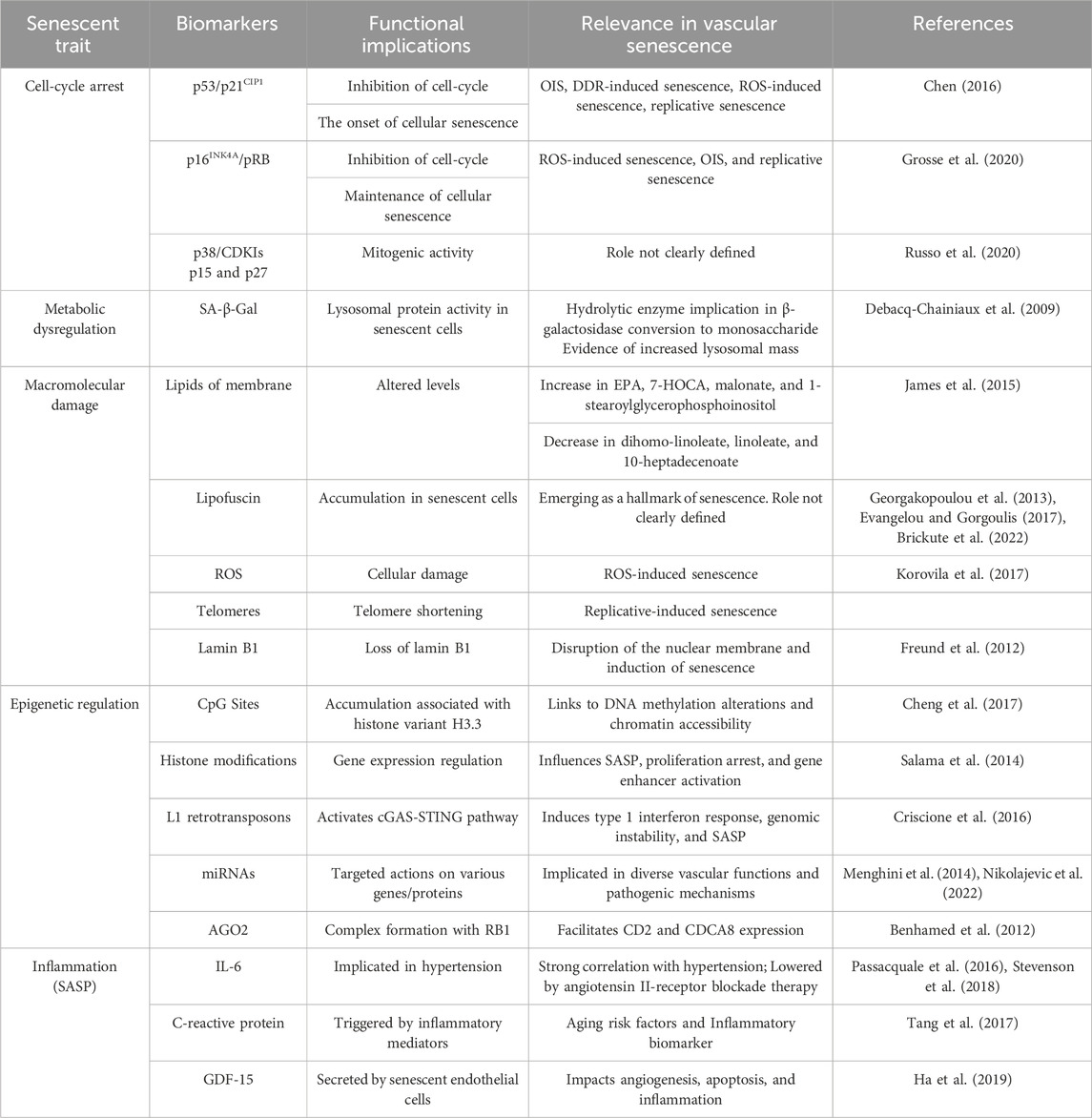
TABLE 2. Biomarkers of cellular senescence and their respective functions.
The cell-cycle arrest is one of the most distinctive features of cellular senescence and is indicated by a decrease in phosphorylated pRB and the protein markers p16, p21, and p53 (González-Gualda et al., 2021). As we have reviewed in the Introduction, the two main pathways that control this are the p53/p21CIP1 and p16INK4A/pRB axes. The former is observed in different pathways, such as oncogene-induced senescence (OIS), DNA damage response (DDR)-induced senescence, reactive oxygen species (ROS)-induced senescence, and replicative senescence. Following a DNA-damaging stimulus, p53 is activated through phosphorylation (p-p53), which then increases the temporary expression of the CDKI p21CIP1. After that, p21CIP1 suppresses CDK2-cyclin E, enabling pRB dephosphorylation and E2F sequestration to stop the cell cycle (Chen, 2016). On the other hand, the p16INK4A/pRB pathway is assumed to be more important in maintaining the quiescent state because it is normally engaged during ROS-induced senescence, OIS, and replicative senescence, meanwhile, it is not expressed during DDR-induced senescence. In this case, the protein p16lnk4a directly inhibits the CDK4-Cyclin D complex upon activation of the INK4a/ARF genetic locus. This allows dephosphorylation and stability of the pRB-E2F complex, and consequently the inhibition of the cell cycle (Grosse et al., 2020). In both networks, pRB is the ultimate downstream target because, in its hypophosphorylated state, it binds E2F, a transcription factor that facilitates cell-cycle progression and entry into S-phase. Furthermore, the MAPK p38 and the CDKIs p15INK4b and p27KIP1 can also be used as markers, however, their role in the senescent program is not as clear or widespread (Zhao et al., 2016; Russo et al., 2020).
To confirm the senescence phenotype or type of senescence, several markers associated with cell cycle arrest and SASP are usually analyzed along with other biomarkers. The presence of decreased expression of cyclins CCNA2, CCNE2, and LMNB1, a,s well as increased expression of a selection of SASP genes and the cyclin-dependent kinase inhibitors CDKN1A (p21WAF1/Cip1), CDKN2A (p16INK4A) and CDK2B (p15INK4B) need to be identified (Casella et al., 2019). These gene signatures are likely to be modified in the coming years due to the current lack of transcriptome datasets and the availability of further single-cell research to assess intrapopulation heterogeneity.
Secondly, there is a metabolic dysregulation. About this hallmark, the pathway that is most affected is the β-galactosidase (SA-β-Gal) (Cai et al., 2020; de Mera-Rodríguez et al., 2021). It is a hydrolase enzyme that catalyzes the conversion of β-galactosidase into monosaccharide in the lysosomes. The most frequent substrates for SA-β-Gal activity are galactose and 5-bromo-4-chloro-3-hydroxyindole-1. This enzyme assay, which uses X-Gal as a chromogenic substrate, tracks the elevated expression and activity of this lysosomal protein in senescent cells and provides evidence of an increase in lysosomal mass (Debacq-Chainiaux et al., 2009; Valieva et al., 2022). The senescent cells differ in several ways from normal in terms of mitochondrial dynamics, function, and appearance. Senescent cells have fewer functioning mitochondria, which exhibit reduced membrane potential higher proton leakage, decreased rates of fusion and fission, increased bulk, and a greater quantity of metabolites related to the tricarboxylic acid cycle (TCA cycle) (Kaplon et al., 2013). Also, it can be brought on by damaging elements of mitochondrial biology, like the electron transport chain (ETC.), complex I assembly, and/or mitochondrial sirtuins (Correia-Melo et al., 2016). On the other hand, senescent cells frequently create higher levels of ROS, which can lead to telomere shorting DDR activation in addition to the protein and lipid degradation covered in earlier sections. SASP regulation is also associated with mitochondrial dysfunction during senescence (Martini and Passos, 2023). Senescent cells appear to reduce the SASP by mitophagy, or mitochondrial clearing. Even when cells do not express important proinflammatory SASP factors like IL-6 and IL-8, senescence can still be induced by genetic or pharmacological suppression of the ECT (Wiley et al., 2016; Wiley and Campisi, 2021).
The macromolecular damage in cellular senescence comprises lipids, proteins, and nucleic acids affecting the cellular structures. Regarding the plasma membrane, both its integrity and signal transduction depend on the lipid content. Changes in lipid profiles result from altered lipid metabolism, a characteristic of several age-related illnesses (Ademowo et al., 2017). Senescent fibroblasts exhibit an increase in fatty acids, their precursors, and phospholipid catabolites, such as eicosapentaenoic (EPA), 1-stear-oylglycerophosphoinositol, malonate, and 7-alpha-hydroxy-3-oxo-4-cholestenoate (7-HOCA), whereas dihomo-linoleate, linoleate, and 10-heptadecenoate decrease (James et al., 2015). ROS is a well-known cause of protein damage because it oxidizes cysteine and methionine residues, changing the way proteins fold and function (Korovila et al., 2017). Threonine, proline, lysine, and arginine residues are carbonylated. Protein carbonylation makes hydrophobic surfaces visible, which causes unfolding and aggregation. Furthermore, carbonyl residues can contribute to protein aggregation by reacting with amino groups to create Schiff bases. After further cross-linking with lipids and carbohydrates, lipofuscin insoluble aggregate is created (Evangelou et al., 2017). It accumulates as a byproduct of senescence in senescent cells and ought to be regarded as a new “hallmark” of senescence (Georgakopoulou et al., 2013). It is commonly recognized that the Sudan-Black-B (SBB) histochemical stain will only react with lipofuscin, which is a mixture of oxidized proteins, lipids, and metals (Evangelou and Gorgoulis, 2017). It has also been observed to build up in senescent cells. It is demonstrated that the use of SBB staining yields very specific results when it comes to senescent cell imaging (Brickute et al., 2022). This offers special benefits for understanding the physiological mechanism and the pathophysiology of different age-related diseases as well as for predicting how well treatment approaches will work.
5.2 Senotherapy
Given the great importance that scientific evidence has given to senescence in the development of age-related pathological processes, the field of senotherapy has experienced exponential growth throughout the last decade. At first, two big groups of senotherapeutic compounds were identified: senolytics and senomorphics. Senolytics selectively induce cell death of senescent cells, mostly by targeting elements of anti-apoptotic and pro-survival pathways of the cell (tyrosine kinases, Bcl-2 protein family, MDM2, FOXO4, HDACs) (Table 3), while senomorphics modulate the SASP, responsible of many of the deleterious effects associated to senescence (by targeting telomerase, sirtuins, mTOR, NF-kB, ATM JAK/STAT, p38/MAPK) (Table 4) (Ya and Bayraktutan, 2023).
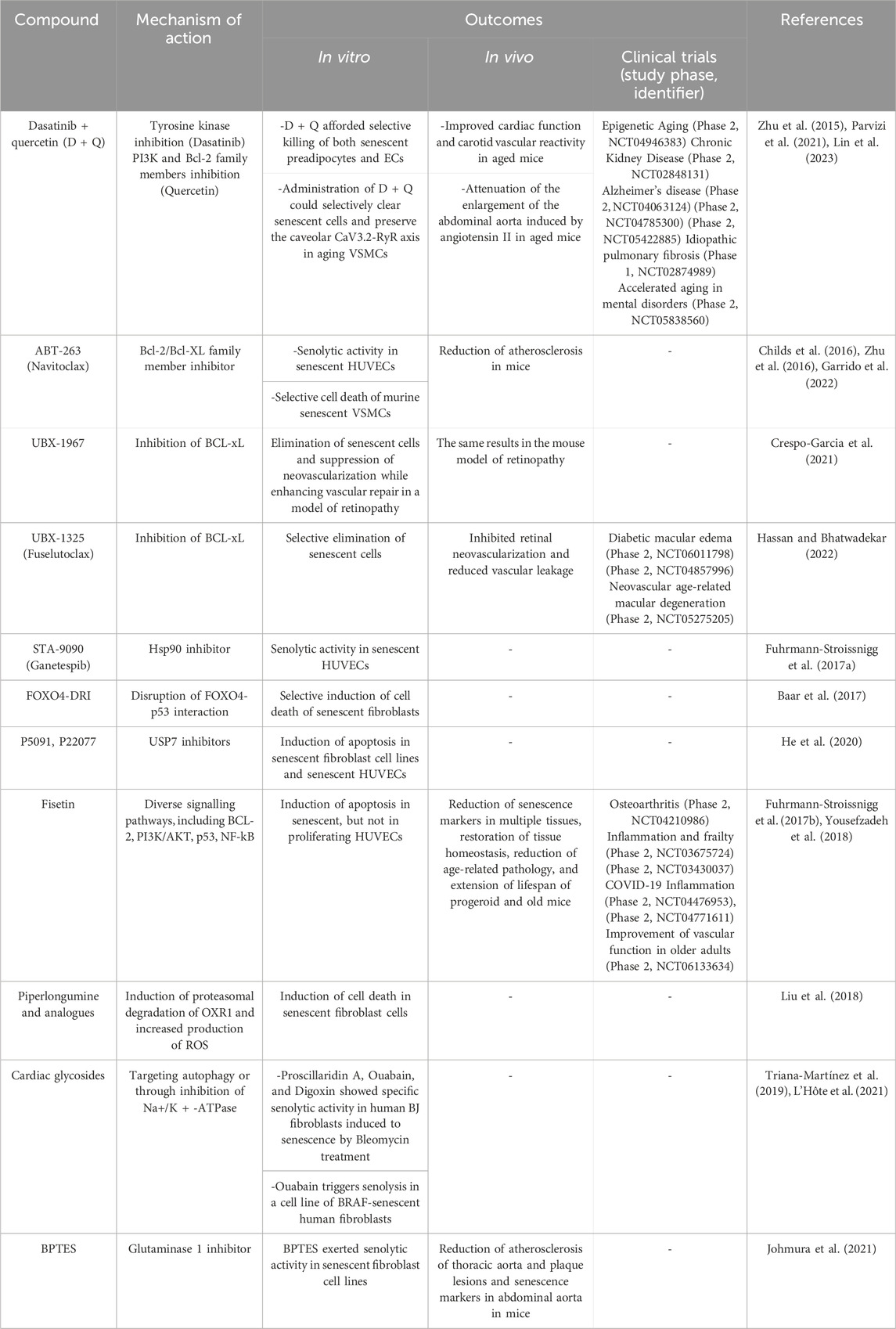
TABLE 3. Senolytics: effects in vascular cells and clinical trials for vascular diseases. BPTES [bis-2-(5-phenyl- acetamido-1,3,4-thiadiazol-2-yl) ethyl sulfide].
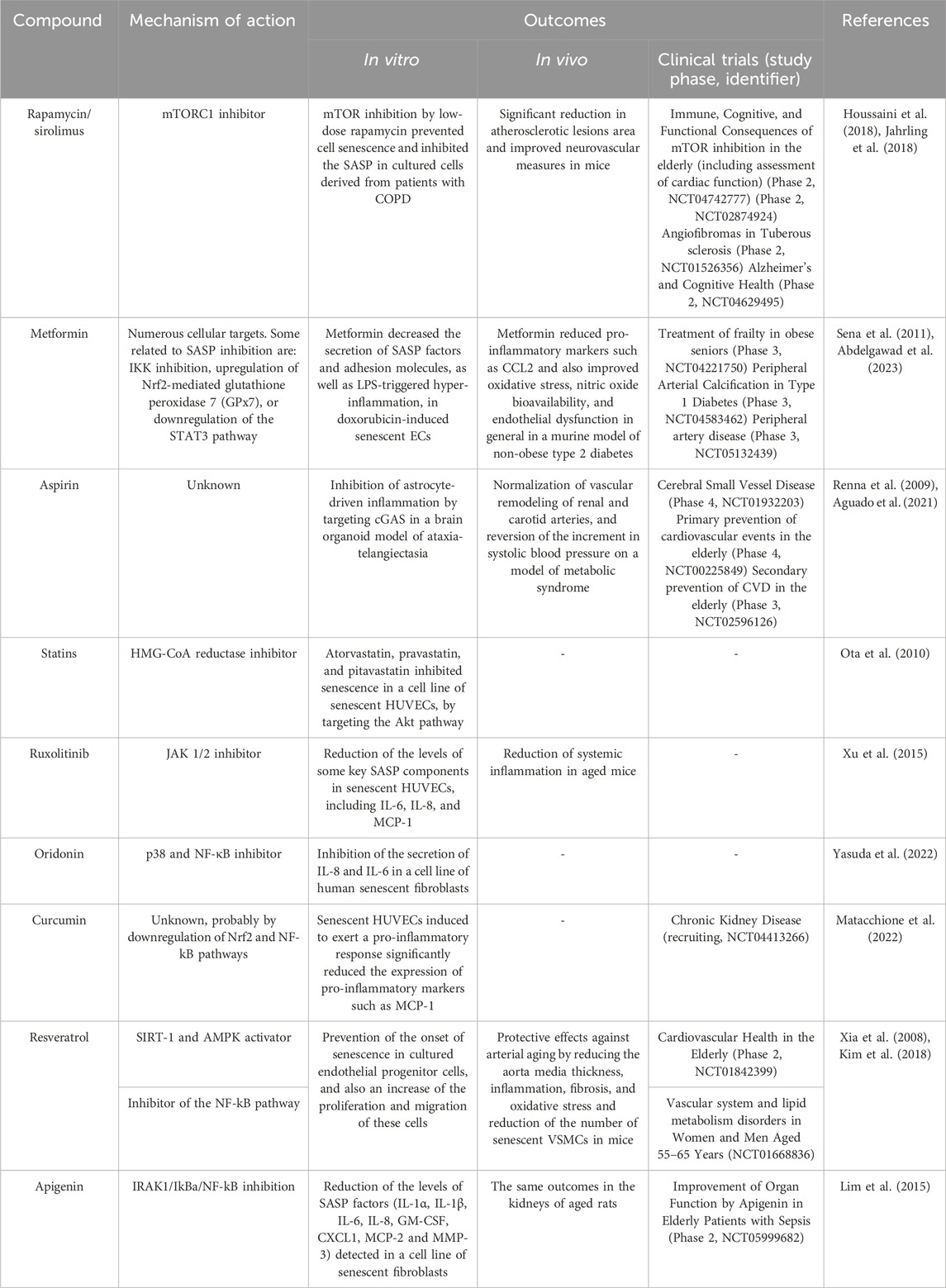
TABLE 4. Senomorphics: effects in vascular cells and clinical trials for vascular diseases. COPD: chronic obstructive pulmonary disease.
Over time, new senotherapeutic approaches emerged, addressing some of the limitations that senolytics or senomorphics compounds presented. For example, the development of galactose-modified senolytic prodrugs allowed guiding the action of these molecules more specifically toward senescent cells (Zhang et al., 2022). It consists of conjugating the senolytic compound with a galactose moiety, which would be cleaved by the senescent cells, given the increased β-galactosidase activity displayed by these cells; this way, the active compound would be released, selectively killing the senescent cell. This strategy has been already tested with some senolytics such as Navitoclax (González-Gualda et al., 2020). Other approaches involve the use of small peptides targeting surface molecules of these cells, like CD47, implicated in the inhibition of phagocytosis of these cells by the immune system, and therefore restoring the clearance of senescent cells (Jatal et al., 2022). Also worth mentioning is the development of senescence immunotherapy. One example of this is the use of functionalized nanoparticles with antibodies to CD9, a surface cell protein upregulated in endothelial cell senescence, to deliver rosuvastatin, a senomorphic agent, to these cells (Pham et al., 2021). Lastly, it is important to highlight the potential of therapeutic strategies that attempt to induce senescence, particularly interesting to enhance the effectiveness of other treatments, such as anti-cancer therapy. Certain studies provide evidence that inducing senescence of endothelial cells in a preclinical model leads to the production of pro-inflammatory and angiogenic factors by these cells, stimulating the immune response and sensitizing unresponsive tumors to immunotherapy (Ruscetti et al., 2020).
However, it is important to mention that senotherapy has still numerous challenges to confront. Firstly, senescent cells are heterogeneous between species, and cell types, even within the same cell type, and rely on different senescence-mediating pathways from 1 cell to another (Zhang et al., 2023). Secondly, not all senescence-associated metabolic alterations are cell-specific, which may lead to undesired off-target events of senotherapeutics, like rapamycin, which apart from its senomorphic properties, has been shown to accelerate the progression of cultured endothelial progenitor cells to a senescent state or cause impaired vasorelaxation by dysregulation of superoxide and NO production (Imanishi et al., 2006; Jabs et al., 2008). Added to this is the important role that senescent cells play in processes such as tissue renewal, wound healing, tumor suppression, and others, which is why senotherapeutics must allow beneficial senescent cell populations to persist (Zhang et al., 2023). Indiscriminate elimination of senescent cells in some situations and conditions may have harmful effects, as described in this study where elimination of senescent pulmonary endothelial cells by FOXO4-DRI worsened pulmonary hemodynamics in a murine model of pulmonary hypertension (Born et al., 2023).
Nevertheless, a multitude of pathologies can potentially be alleviated by the use of senotherapeutics, as shown by the favorable results of many current clinical trials. Many of these are vascular diseases such as fibrotic conditions, atherosclerotic disease, pulmonary arterial hypertension, or peripheral artery disease, among others (Suda et al., 2023). Table 3 and Table 4 list some of the most studied senolytic and senomorphic compounds in pathologies related to vascular alterations, as well as their mechanisms of action, main outcomes of the studies performed, and clinical trials carried out.
6 Conclusion
Cellular senescence is a state in which the cell leaves the cell cycle in response to damage signals or developmental cues to prevent tumor proliferation and promote cell survival. The senescent cell remains viable despite macromolecular damage and dysregulated metabolism and acquires a SASP. Cellular senescence is a hallmark of aging because its accumulation in tissues is detrimental to tissue and organismal homeostasis. Some studies that selectively eliminate these cells in animal models manage to prolong their lifespan. In this regard, we have reviewed the impact of cellular senescence on the vascular wall, termed vascular senescence, which appears to be one of the most important drivers of vascular aging. The best characterized and studied cell types that become senescent and contribute to vascular dysfunction are endothelial cells, vascular smooth muscle cells, adventitial fibroblasts, and immune cells. In general, they produce oxidative stress and secretion of proinflammatory factors that damage the vascular wall. This has been found in age-related vascular diseases such as atherosclerosis, peripheral artery disease, hypertension, chronic venous disease, or venous ulcers.
Multiple epigenetic mechanisms are activated during the senescence state, including the expression of microRNAs, chromatin remodeling, DNA methylation, histone modification, and loss of nuclear integrity. This implies upregulation in the expression of inflammation-related genes, mainly those that promote SASP, such as proinflammatory cytokines, chemokines, chemokine receptors, and endothelial adhesion molecules. Of interest is the investigation of specific biomarkers of vascular senescence in different blood vessel types and cell types. Identifying unique signatures in different cell types and blood vessels can improve disease detection and response to treatments of vascular diseases. Moreover, exploring the inflammatory response activated during vascular senescence may reveal new therapeutic targets to treat age-related vascular diseases. In addition, understanding this complex process allows us the application of a promising therapeutic approach, senotherapy. Further research is needed to better characterize the different senotherapeutic options, senolytics and senomorphics, and to translate the results of research in preclinical models and clinical trials into clinical practice, improving diagnostic accuracy, prognostic ability, and therapeutic interventions for vascular senescence-related conditions.
Author contributions
OF-M: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Validation, Writing–original draft, Writing–review and editing. DDL-O: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Validation, Writing–original draft, Writing–review and editing. DL: Data curation, Investigation, Methodology, Writing–original draft, Writing–review and editing. PDC-M: Investigation, Writing–original draft, Writing–review and editing. CG: Data curation, Formal Analysis, Investigation, Methodology, Software, Writing–original draft, Writing–review and editing. SB-B: Formal Analysis, Investigation, Methodology, Writing–original draft, Writing–review and editing. JG-G: Data curation, Formal Analysis, Investigation, Methodology, Writing–original draft, Writing–review and editing. NG-H: Conceptualization, Data curation, Investigation, Methodology, Validation, Writing–original draft, Writing–review and editing. MA-N: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Project administration, Resources, Writing–original draft, Writing–review and editing. LL-G: Data curation, Investigation, Methodology, Writing–original draft, Writing–review and editing. RD-P: Conceptualization, Data curation, Formal Analysis, Investigation, Software, Validation, Visualization, Writing–original draft, Writing–review and editing. LG: Conceptualization, Investigation, Validation, Writing–original draft, Writing–review and editing. MO: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Validation, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The study (FIS-PI21/01244) was supported by the Instituto de Salud Carlos III (grant no. Estatal de I + D + I 2020–2027) and co-financed by the European Development Regional Fund “A way to achieve Europe”, as well as P2022/BMD-7321 (Comunidad de Madrid) and ProACapital, Halekulani S. L. and MJR. The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdelgawad, I. Y., Agostinucci, K., Sadaf, B., Grant, M. K. O., and Zordoky, B. N. (2023). Metformin mitigates SASP secretion and LPS-triggered hyper-inflammation in Doxorubicin-induced senescent endothelial cells. Front. Aging 4, 1170434–1170514. doi:10.3389/fragi.2023.1170434
Ademowo, O. S., Dias, H. K. I., Burton, D. G. A., and Griffiths, H. R. (2017). Lipid (per) oxidation in mitochondria: an emerging target in the ageing process? Biogerontology 18, 859–879. doi:10.1007/s10522-017-9710-z
Aguado, J., Chaggar, H. K., Gómez-Inclán, C., Shaker, M. R., Leeson, H. C., Mackay-Sim, A., et al. (2021). Inhibition of the cGAS-STING pathway ameliorates the premature senescence hallmarks of Ataxia-Telangiectasia brain organoids. Aging Cell 20, 134688–e13516. doi:10.1111/acel.13468
Aird, K. M., and Zhang, R. (2013). “Detection of senescence-associated heterochromatin foci (SAHF),” in Cell senescence. Methods in molecular biology. Editors L. Galluzzi, I. Vitale, O. Kepp, and G. Kroemer (Totowa: Humana Press), Vol. 965, 185–196. doi:10.1007/978-1-62703-239-1_12
Ashraf, H. M., Fernandez, B., and Spencer, S. L. (2023). The intensities of canonical senescence biomarkers integrate the duration of cell-cycle withdrawal. Nat. Commun. 14, 4527–4613. doi:10.1038/s41467-023-40132-0
Baar, M. P., Brandt, R. M. C., Putavet, D. A., Klein, J. D. D., Derks, K. W. J., Bourgeois, B. R. M., et al. (2017). Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169, 132–147. doi:10.1016/j.cell.2017.02.031
Baker, D. J., Childs, B. G., Durik, M., Wijers, M. E., Sieben, C. J., Zhong, J., et al. (2016). Naturally occurring p16 Ink4a-positive cells shorten healthy lifespan. Nature 530, 184–189. doi:10.1038/nature16932
Beck, J., Horikawa, I., and Harris, C. (2020). Cellular senescence: mechanisms, morphology, and mouse models. Vet. Pathol. 57, 747–757. doi:10.1177/0300985820943841
Benhamed, M., Herbig, U., Ye, T., Dejean, A., and Bischof, O. (2012). Senescence is an endogenous trigger for microRNA-directed transcriptional gene silencing in human cells. Nat. Cell Biol. 14, 266–275. doi:10.1038/ncb2443
Biran, A., Perelmutter, M., Gal, H., Burton, D. G. A., Ovadya, Y., Vadai, E., et al. (2014). Senescent cells communicate via intercellular protein transfer. Genes Dev. 29, 791–802. doi:10.1101/gad.259341.115
Borgdorff, V., Lleonart, M. E., Bishop, C. L., Fessart, D., Bergin, A. H., Overhoff, M. G., et al. (2010). Multiple microRNAs rescue from Ras-induced senescence by inhibiting p21 Waf1/Cip1. Oncogene 29, 2262–2271. doi:10.1038/onc.2009.497
Born, E., Lipskaia, L., Breau, M., Houssaini, A., Beaulieu, D., Marcos, E., et al. (2023). Eliminating senescent cells can promote pulmonary hypertension development and progression. Circulation 147, 650–666. doi:10.1161/CIRCULATIONAHA.122.058794
Brickute, D., Chen, C., Braga, M., Barnes, C., Wang, N., Allott, L., et al. (2022). Design, synthesis, and evaluation of a novel PET imaging agent targeting lipofuscin in senescent cells. RSC Adv. 12, 26372–26381. doi:10.1039/d2ra04535d
Burns, D. M., D’Ambrogio, A., Nottrott, S., and Richter, J. D. (2011). CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature 473, 105–108. doi:10.1038/nature09908
Burton, D. G. A., and Stolzing, A. (2018). Cellular senescence: immunosurveillance and future immunotherapy. Ageing Res. Rev. 43, 17–25. doi:10.1016/j.arr.2018.02.001
Cai, Y., Zhou, H., Zhu, Y., Sun, Q., Ji, Y., Xue, A., et al. (2020). Elimination of senescent cells by β-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Res. 30, 574–589. doi:10.1038/s41422-020-0314-9
Cakouros, D. D., and Gronthos, S. (2019). Epigenetic regulation of bone marrow stem cell aging: revealing epigenetic signatures associated with hematopoietic and mesenchymal stem cell aging. Aging Dis. 10, 174–189. doi:10.14336/AD.2017.1213
Campisi, J. (2013). Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 75, 685–705. doi:10.1146/annurev-physiol-030212-183653
Casella, G., Munk, R., Kim, K. M., Piao, Y., De, S., Abdelmohsen, K., et al. (2019). Transcriptome signature of cellular senescence. Nucleic Acids Res. 47, 7294–7305. doi:10.1093/nar/gkz555
Chen, J. (2016). The cell-cycle arrest and apoptotic and progression. Cold Spring Harb. Perspect. Med. 6, 1–16. doi:10.1101/cshperspect.a026104
Chen, L., Yang, W., Guo, Y., Chen, W., Zheng, P., Zeng, J., et al. (2017). Exosomal lncRNA GAS5 regulates the apoptosis of macrophages and vascular endothelial cells in atherosclerosis. PLoS One 12, e0185406. doi:10.1371/journal.pone.0185406
Cheng, L. Q., Zhang, Z. Q., Chen, H. Z., and Liu, D. P. (2017). Epigenetic regulation in cell senescence. J. Mol. Med. 95, 1257–1268. doi:10.1007/s00109-017-1581-x
Childs, B. G., Baker, D. J., Kirkland, J. L., Campisi, J., and van Deursen, J. M. (2014). Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 15, 1139–1153. doi:10.15252/embr.201439245
Childs, B. G., Baker, D. J., Wijshake, T., Conover, C. A., Campisi, J., and van Deursen, J. M. (2016). Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354, 472–477. doi:10.1126/science.aaf6659
Climie, R. E., Alastruey, J., Mayer, C. C., Schwarz, A., Laucyte-Cibulskiene, A., Voicehovska, J., et al. (2023). Vascular ageing: moving from bench towards bedside. Eur. J. Prev. Cardiol. 30, 1101–1117. doi:10.1093/eurjpc/zwad028
Coppé, J. P., Desprez, P. Y., Krtolica, A., and Campisi, J. (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis. 5, 99–118. doi:10.1146/annurev-pathol-121808-102144
Coppe, J. P., Kauser, K., Campisi, J., and Beauséjour, C. M. (2006). Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 281, 29568–29574. doi:10.1074/jbc.M603307200
Correia-Melo, C., Marques, F. D., Anderson, R., Hewitt, G., Hewitt, R., Cole, J., et al. (2016). Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 35, 724–742. doi:10.15252/embj.201592862
Crespo-Garcia, S., Tsuruda, P. R., Dejda, A., Ryan, R. D., Fournier, F., Chaney, S. Y., et al. (2021). Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL inhibition. Cell Metab. 33, 818–832.e7. doi:10.1016/j.cmet.2021.01.011
Criscione, S. W., Teo, Y. V., and Neretti, N. (2016). The chromatin landscape of cellular senescence. Trends Genet. 32, 751–761. doi:10.1016/j.tig.2016.09.005
Crouch, J., Shvedova, M., Thanapaul, R. J. R. S., Botchkarev, V., and Roh, D. (2022). Epigenetic regulation of cellular senescence. Cells 11, 672–716. doi:10.3390/cells11040672
Cui, S., Li, W., Lv, X., Wang, P., Gao, Y., and Huang, G. (2017). Folic acid supplementation delays atherosclerotic lesion development by modulating MCP1 and VEGF DNA methylation levels in vivo and in vitro. Int. J. Mol. Sci. 18, 990–1018. doi:10.3390/ijms18050990
Cuollo, L., Antonangeli, F., Santoni, A., and Soriani, A. (2020). The senescence-associated secretory phenotype (Sasp) in the challenging future of cancer therapy and age-related diseases. Biol. (Basel) 9, 485–516. doi:10.3390/biology9120485
Debacq-Chainiaux, F., Erusalimsky, J. D., Campisi, J., and Toussaint, O. (2009). Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 4, 1798–1806. doi:10.1038/nprot.2009.191
Del Pinto, R., and Ferri, C. (2018). Inflammation-accelerated senescence and the cardiovascular system: mechanisms and perspectives. Int. J. Mol. Sci. 19, 3701. doi:10.3390/ijms19123701
de Mera-Rodríguez, J. A., Álvarez-Hernán, G., Gañán, Y., Martín-Partido, G., Rodríguez-León, J., and Francisco-Morcillo, J. (2021). Is senescence-associated β-galactosidase a reliable in vivo marker of cellular senescence during embryonic development? Front. Cell Dev. Biol. 9, 1–12. doi:10.3389/fcell.2021.623175
Denis, H., Ndlovu, M. N., and Fuks, F. (2011). Regulation of mammalian DNA methyltransferases: a route to new mechanisms. EMBO Rep. 12, 647–656. doi:10.1038/embor.2011.110
Ding, Q., Shao, C., Rose, P., and Zhu, Y. Z. (2020). Epigenetics and vascular senescence–potential new therapeutic targets? Front. Pharmacol. 11, 535395–535411. doi:10.3389/fphar.2020.535395
Ding, Y., Tang, X., Chen, H., and Liu, D. (2018). “Epigenetic regulation of vascular aging and age-related vascular diseases,” in Aging and aging-related diseases. Advances in experimental medicine and biology. Editor Z. Wang (Singapore: Springer), Vol. 1086.
Du, W. W., Li, X., Li, T., Li, H., Khorshidi, A., Liu, F., et al. (2015). The microRNA miR-17-3p inhibits mouse cardiac fibroblast senescence by targeting Par4. J. Cell Sci. 128, 293–304. doi:10.1242/jcs.158360
Evangelou, K., and Gorgoulis, V. G. (2017). Sudan black B, the specifi c histochemical stain for lipofuscin: a novel method to detect senescent cells. Oncogene-Induced Senescence, 175–184. doi:10.1007/978-1-4939-6670-7
Evangelou, K., Lougiakis, N., Rizou, S. V., Kotsinas, A., Kletsas, D., Muñoz-Espín, D., et al. (2017). Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 16, 192–197. doi:10.1111/acel.12545
Freund, A., Laberge, R. M., Demaria, M., and Campisi, J. (2012). Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 23, 2066–2075. doi:10.1091/mbc.E11-10-0884
Frismantiene, A., Philippova, M., Erne, P., and Resink, T. J. (2018). Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell. Signal. 52, 48–64. doi:10.1016/j.cellsig.2018.08.019
Fuhrmann-Stroissnigg, H., Ling, Y. Y., Zhao, J., McGowan, S. J., Zhu, Y., Brooks, R. W., et al. (2017a). Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 8, 422. doi:10.1038/s41467-017-00314-z
Fuhrmann-Stroissnigg, H., Niedernhofer, L. J., Robbins, P. D., Tchkonia, T., Kirkland, J. L., et al. (2017b). New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany. NY) 9, 955–963. doi:10.18632/aging.101202
Fujimaki, K., Li, R., Chen, H., Croce, K. D., Zhang, H. H., Xing, J., et al. (2019). Graded regulation of cellular quiescence depth between proliferation and senescence by a lysosomal dimmer switch. Proc. Natl. Acad. Sci. U. S. A. 116, 22624–22634. doi:10.1073/pnas.1915905116
Furuuchi, R., Shimizu, I., Yoshida, Y., Hayashi, Y., Ikegami, R., Suda, M., et al. (2018). Boysenberry polyphenol inhibits endothelial dysfunction and improves vascular health. PLoS One 13, e0202051. doi:10.1371/journal.pone.0202051
Gardner, S. E., Humphry, M., Bennett, M. R., and Clarke, M. C. H. (2015). Senescent vascular smooth muscle cells drive inflammation through an interleukin-1α-dependent senescence-associated secretory phenotype. Arterioscler. Thromb. Vasc. Biol. 35, 1963–1974. doi:10.1161/ATVBAHA.115.305896
Garrido, A. M., Kaistha, A., Uryga, A. K., Oc, S., Foote, K., Shah, A., et al. (2022). Efficacy and limitations of senolysis in atherosclerosis. Cardiovasc. Res. 118, 1713–1727. doi:10.1093/cvr/cvab208
Georgakopoulou, E. A., Tsimaratou, K., Evangelou, K., Fernandez, M. P. J., Zoumpourlis, V., Trougakos, I. P., et al. (2013). Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging (Albany. NY) 5, 37–50. doi:10.18632/aging.100527
Ghebre, Y. T., Yakubov, E., Wong, W. T., Krishnamurthy, P., Sayed, N., Sikora, A. G., et al. (2016). Vascular aging: implications for cardiovascular disease and therapy. Transl. Med. 06, 183. doi:10.4172/2161-1025.1000183
Giacconi, R., Malavolta, M., Costarelli, L., and Provinciali, M. (2015). Cellular senescence and inflammatory burden as determinants of mortality in elderly people until the extreme old age. EBioMedicine 2, 1316–1317. doi:10.1016/j.ebiom.2015.09.015
Gire, V., and Dulic, V. (2015). Senescence from G2 arrest, revisited. Cell Cycle 14, 297–304. doi:10.1080/15384101.2014.1000134
Glier, M. B., Green, T. J., and Devlin, A. M. (2014). Methyl nutrients, DNA methylation, and cardiovascular disease. Mol. Nutr. Food Res. 58, 172–182. doi:10.1002/mnfr.201200636
González-Gualda, E., Baker, A. G., Fruk, L., and Muñoz-Espín, D. (2021). A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 288, 56–80. doi:10.1111/febs.15570
González-Gualda, E., Pàez-Ribes, M., Lozano-Torres, B., Macias, D., Wilson, J. R., González-López, C., et al. (2020). Galacto-conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 19, 131422–e13219. doi:10.1111/acel.13142
Gorgoulis, V., Adams, P. D., Alimonti, A., Bennett, D. C., Bischof, O., Bishop, C., et al. (2019). Cellular senescence: defining a path forward. Cell 179, 813–827. doi:10.1016/j.cell.2019.10.005
Gorgoulis, V. G., Pratsinis, H., Zacharatos, P., Demoliou, C., Sigala, F., Asimacopoulos, P. J., et al. (2005). p53-Dependent ICAM-1 overexpression in senescent human cells identified in atherosclerotic lesions. Lab. Investig. 85, 502–511. doi:10.1038/labinvest.3700241
Grosse, L., Wagner, N., Emelyanov, A., Molina, C., Lacas-Gervais, S., Wagner, K. D., et al. (2020). Defined p16High senescent cell types are indispensable for mouse healthspan. Cell Metab. 32, 87–99. doi:10.1016/j.cmet.2020.05.002
Ha, G., De Torres, F., Arouche, N., Benzoubir, N., Ferratge, S., Hatem, E., et al. (2019). GDF15 secreted by senescent endothelial cells improves vascular progenitor cell functions. PLoS One 14, e0216602. doi:10.1371/journal.pone.0216602
Halper, J. (2018). Basic components of vascular connective tissue and extracellular matrix. 1st ed. Netherlands: Elsevier Inc. doi:10.1016/bs.apha.2017.08.012
Han, Y., and Kim, S. Y. (2023). Endothelial senescence in vascular diseases: current understanding and future opportunities in senotherapeutics. Exp. Mol. Med. 55, 1–12. doi:10.1038/s12276-022-00906-w
Hassan, J. W., and Bhatwadekar, A. D. (2022). Senolytics in the treatment of diabetic retinopathy. Front. Pharmacol. 13, 896907–896909. doi:10.3389/fphar.2022.896907
Hayashi, T., Matsui-Hirai, H., Miyazaki-Akita, A., Fukatsu, A., Funami, J., Ding, Q. F., et al. (2006). Endothelial cellular senescence is inhibited by nitric oxide: implications in atherosclerosis associated with menopause and diabetes. Proc. Natl. Acad. Sci. U. S. A. 103, 17018–17023. doi:10.1073/pnas.0607873103
Hayflick, L., and Moorhead, P. S. (1961). The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621. doi:10.1016/0014-4827(61)90192-6
He, Y., Li, W., Lv, D., Zhang, X., Zhang, X., Ortiz, Y. T., et al. (2020). Inhibition of USP7 activity selectively eliminates senescent cells in part via restoration of p53 activity. Aging Cell 19, 131177–e13211. doi:10.1111/acel.13117
Hernandez-Segura, A., Brandenburg, S., and Demaria, M. (2018). Induction and validation of cellular senescence in primary human cells. J. Vis. Exp. 136, 57782–57810. doi:10.3791/57782
Herranz, N., and Gil, J. (2018). Mechanisms and functions of cellular senescence. J. Clin. Invest. 128, 1238–1246. doi:10.1172/JCI95148
Hofmann, P., Sommer, J., Theodorou, K., Kirchhof, L., Fischer, A., Li, Y., et al. (2019). Long non-coding RNA H19 regulates endothelial cell aging via inhibition of STAT3 signalling. Cardiovasc. Res. 115, 230–242. doi:10.1093/cvr/cvy206
Houssaini, A., Breau, M., Kebe, K., Abid, S., Marcos, E., Lipskaia, L., et al. (2018). mTOR pathway activation drives lung cell senescence and emphysema. JCI insight 3, e93203–e93220. doi:10.1172/jci.insight.93203
Huang, L. C., Lin, R. T., Chen, C. F., Chen, C. H., Hank Juo, S. H., and Lin, H. F. (2016). Predictors of carotid intima-media thickness and plaque progression in a Chinese population. J. Atheroscler. Thromb. 23, 940–949. doi:10.5551/jat.32177
Huang, W., Hickson, L. T. J., Eirin, A., Kirkland, J. L., and Lerman, L. O. (2022). Cellular senescence: the good, the bad and the unknown. Nat. Rev. Nephrol. 18, 611–627. doi:10.1038/s41581-022-00601-z
Hutcheson, R., Chaplin, J., Hutcheson, B., Borthwick, F., Proctor, S., Gebb, S., et al. (2014). MiR-21 normalizes vascular smooth muscle proliferation and improves coronary collateral growth in metabolic syndrome. FASEB J. 28, 4088–4099. doi:10.1096/fj.14-251223
Hwang, H. J., Kim, N., Herman, A. B., Gorospe, M., and Lee, J. S. (2022). Factors and pathways modulating endothelial cell senescence in vascular aging. Int. J. Mol. Sci. 23, 10135. doi:10.3390/ijms231710135
Illi, B., Scopece, A., Nanni, S., Farsetti, A., Morgante, L., Biglioli, P., et al. (2005). Epigenetic histone modification and cardiovascular lineage programming in mouse embryonic stem cells exposed to laminar shear stress. Circ. Res. 96, 501–508. doi:10.1161/01.RES.0000159181.06379.63
Imanishi, T., Kobayashi, K., Kuki, S., Takahashi, C., and Akasaka, T. (2006). Sirolimus accelerates senescence of endothelial progenitor cells through telomerase inactivation. Atherosclerosis 189, 288–296. doi:10.1016/j.atherosclerosis.2005.12.031
Jabs, A., Göbel, S., Wenzel, P., Kleschyov, A. L., Hortmann, M., Oelze, M., et al. (2008). Sirolimus-induced vascular dysfunction. Increased mitochondrial and nicotinamide adenosine dinucleotide phosphate oxidase-dependent superoxide production and decreased vascular nitric oxide formation. J. Am. Coll. Cardiol. 51, 2130–2138. doi:10.1016/j.jacc.2008.01.058
Jahrling, J. B., Lin, A. L., DeRosa, N., Hussong, S. A., Van Skike, C. E., Girotti, M., et al. (2018). mTOR drives cerebral blood flow and memory deficits in LDLR−/− mice modeling atherosclerosis and vascular cognitive impairment. J. Cereb. Blood Flow. Metab. 38, 58–74. doi:10.1177/0271678X17705973
James, E. L., Michalek, R. D., Pitiyage, G. N., De Castro, A. M., Vignola, K. S., Jones, J., et al. (2015). Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J. Proteome Res. 14, 1854–1871. doi:10.1021/pr501221g
Jatal, R., Mendes Saraiva, S., Vázquez-Vázquez, C., Lelievre, E., Coqueret, O., López-López, R., et al. (2022). Sphingomyelin nanosystems decorated with TSP-1 derived peptide targeting senescent cells. Int. J. Pharm. 617, 121618. doi:10.1016/j.ijpharm.2022.121618
Jayasuriya, R., Ganesan, K., Xu, B., and Ramkumar, K. M. (2022). Emerging role of long non-coding RNAs in endothelial dysfunction and their molecular mechanisms. Biomed. Pharmacother. 145, 112421. doi:10.1016/j.biopha.2021.112421
Johmura, Y., Yamanaka, T., Omori, S., Wang, T. W., Sugiura, Y., Matsumoto, M., et al. (2021). Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 371, 265–270. doi:10.1126/science.abb5916
Kale, A., Sharma, A., Stolzing, A., Stolzing, A., Desprez, P. Y., Desprez, P. Y., et al. (2020). Role of immune cells in the removal of deleterious senescent cells. Immun. Ageing 17, 16–19. doi:10.1186/s12979-020-00187-9
Kang, C., Xu, Q., Martin, T. D., Li, M. Z., Demaria, M., Aron, L., et al. (2015). The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349, aaa5612. doi:10.1126/science.aaa5612
Kaplan, R. C., Sinclair, E., Landay, A. L., Lurain, N., Sharrett, A. R., Gange, S. J., et al. (2011). T cell activation and senescence predict subclinical carotid artery disease in HIV-infected women. J. Infect. Dis. 203, 452–463. doi:10.1093/infdis/jiq071
Kaplon, J., Zheng, L., Meissl, K., Chaneton, B., Selivanov, V. A., MacKay, G., et al. (2013). A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498, 109–112. doi:10.1038/nature12154
Katsuumi, G., Shimizu, I., Yoshida, Y., and Minamino, T. (2018). Vascular senescence in cardiovascular and metabolic diseases. Front. Cardiovasc. Med. 5, 18–13. doi:10.3389/fcvm.2018.00018
Khan, S. Y., Awad, E. M., Oszwald, A., Mayr, M., Yin, X., Waltenberger, B., et al. (2017). Premature senescence of endothelial cells upon chronic exposure to TNFα can be prevented by N-acetyl cysteine and plumericin. Sci. Rep. 7, 39501–39513. doi:10.1038/srep39501
Kim, E. N., Kim, M. Y., Lim, J. H., Kim, Y., Shin, S. J., Park, C. W., et al. (2018). The protective effect of resveratrol on vascular aging by modulation of the renin–angiotensin system. Atherosclerosis 270, 123–131. doi:10.1016/j.atherosclerosis.2018.01.043
Kim, Y., Sharov, A. A., McDole, K., Cheng, M., Hao, H., Fan, C.-M., et al. (2011). Mouse B-type lamins are required for proper organogenesis but not by embryonic stem cells. Science 334, 1706–1710. doi:10.1126/science.1211222
Kohli, J., Wang, B., Brandenburg, S. M., Basisty, N., Evangelou, K., Varela-Eirin, M., et al. (2021). Algorithmic assessment of cellular senescence in experimental and clinical specimens. Berlin, Germany: Springer US. doi:10.1038/s41596-021-00505-5
Kohn, J. C., Lampi, M. C., and Reinhart-King, C. A. (2015). Age-related vascular stiffening: causes and consequences. Front. Genet. 6, 112–117. doi:10.3389/fgene.2015.00112
Korovila, I., Hugo, M., Castro, J. P., Weber, D., Höhn, A., Grune, T., et al. (2017). Proteostasis, oxidative stress and aging. Redox Biol. 13, 550–567. doi:10.1016/j.redox.2017.07.008
Kuilman, T., Michaloglou, C., Mooi, W. J., and Peeper, D. S. (2010). The essence of senescence. Genes Dev. 24, 2463–2479. doi:10.1101/gad.1971610
Kumari, R., and Jat, P. (2021). Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front. Cell Dev. Biol. 9, 645593–645624. doi:10.3389/fcell.2021.645593
Lacolley, P., Regnault, V., Segers, P., and Laurent, S. (2017). Vascular smooth muscle cells and arterial stiffening: relevance in development, aging, and disease. Physiol. Rev. 97, 1555–1617. doi:10.1152/physrev.00003.2017
Lai, Z., Lin, P., Weng, X., Su, J., Chen, Y., He, Y., et al. (2018). MicroRNA-574-5p promotes cell growth of vascular smooth muscle cells in the progression of coronary artery disease. Biomed. Pharmacother. 97, 162–167. doi:10.1016/j.biopha.2017.10.062
Lakatta, E. G. (2015). So! What’s aging? Is cardiovascular aging a disease? J. Mol. Cell. Cardiol. 83, 1–13. doi:10.1016/j.yjmcc.2015.04.005
Lapasset, L., Milhavet, O., Prieur, A., Besnard, E., Babled, A., Ät-Hamou, N., et al. (2011). Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 25, 2248–2253. doi:10.1101/gad.173922.111
Lee, H. J., Lee, W. J., Hwang, S. C., Choe, Y., Kim, S., Bok, E., et al. (2021). Chronic inflammation-induced senescence impairs immunomodulatory properties of synovial fluid mesenchymal stem cells in rheumatoid arthritis. Stem Cell Res. Ther. 12, 502–516. doi:10.1186/s13287-021-02453-z
Lee, S., Choi, E., Cha, M. J., Park, A. J., Yoon, C., and Hwang, K. C. (2015). Impact of miRNAs on cardiovascular aging. J. Geriatr. Cardiol. 12, 569–574. doi:10.11909/j.issn.1671-5411.2015.05.011
L’Hôte, V., Courbeyrette, R., Pinna, G., Cintrat, J. C., Le Pavec, G., Delaunay-Moisan, A., et al. (2021). Ouabain and chloroquine trigger senolysis of BRAF-V600E-induced senescent cells by targeting autophagy. Aging Cell 20, 134477–e13514. doi:10.1111/acel.13447
Li, K., Li, Y., Yu, Y., Ding, J., Huang, H., Chu, C., et al. (2021). Bmi-1 alleviates adventitial fibroblast senescence by eliminating ROS in pulmonary hypertension. BMC Pulm. Med. 21, 80–10. doi:10.1186/s12890-021-01439-0
Li, X., Li, C., Zhang, W., Wang, Y., Qian, P., and Huang, H. (2023). Inflammation and aging: signaling pathways and intervention therapies. Signal Transduct. Target. Ther. 8, 239. doi:10.1038/s41392-023-01502-8
Liberale, L., Badimon, L., Montecucco, F., Lüscher, T. F., Libby, P., and Camici, G. G. (2022). Inflammation, aging, and cardiovascular disease: JACC review topic of the week. J. Am. Coll. Cardiol. 79, 837–847. doi:10.1016/j.jacc.2021.12.017
Lim, D. X. E., Richards, T., Kanapathy, M., Sudhaharan, T., Wright, G. D., Phillips, A. R. J., et al. (2021). Extracellular matrix and cellular senescence in venous leg ulcers. Sci. Rep. 11, 20168–20212. doi:10.1038/s41598-021-99643-9
Lim, H., Park, H., and Kim, H. P. (2015). Effects of flavonoids on senescence-associated secretory phenotype formation from bleomycin-induced senescence in BJ fibroblasts. Biochem. Pharmacol. 96, 337–348. doi:10.1016/j.bcp.2015.06.013
Lin, J., Guo, W., Luo, Q., Zhang, Q., Wan, T., Jiang, C., et al. (2023). Senolytics prevent caveolar CaV 3.2-RyR axis malfunction in old vascular smooth muscle. Aging Cell 22, e14002. doi:10.1111/acel.14002
Lin, J. B., Sene, A., Santeford, A., Fujiwara, H., Sidhu, R., Ligon, M. M., et al. (2018). Oxysterol signatures distinguish age-related macular degeneration from physiologic aging. EBioMedicine 32, 9–20. doi:10.1016/j.ebiom.2018.05.035
Lin, X., Zhan, J. K., Wang, Y. J., Tan, P., Chen, Y. Y., Deng, H. Q., et al. (2016). Function, role, and clinical application of MicroRNAs in vascular aging. Biomed. Res. Int. 2016, 6021394. doi:10.1155/2016/6021394
Lin, Z., Ding, Q., Li, X., Feng, Y., He, H., Huang, C., et al. (2022). Targeting epigenetic mechanisms in vascular aging. Front. Cardiovasc. Med. 8, 806988–807016. doi:10.3389/fcvm.2021.806988
Liu, X., Wang, Y., Zhang, X., Gao, Z., Zhang, S., Shi, P., et al. (2018). Senolytic activity of piperlongumine analogues: synthesis and biological evaluation. Bioorg. Med. Chem. 26, 3925–3938. doi:10.1016/j.bmc.2018.06.013
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2023). Hallmarks of aging: an expanding universe. Cell 186, 243–278. doi:10.1016/j.cell.2022.11.001
Martini, H., and Passos, J. F. (2023). Cellular senescence: all roads lead to mitochondria. FEBS J. 290, 1186–1202. doi:10.1111/febs.16361
Matacchione, G., Valli, D., Silvestrini, A., Giuliani, A., Sabbatinelli, J., Giordani, C., et al. (2022). Curcumin, polydatin and quercetin synergistic activity protects from high-glucose-induced inflammation and oxidative stress. Antioxidants 11, 1037. doi:10.3390/antiox11061037
Mazurek, R., Dave, J. M., Chandran, R. R., Misra, A., Sheikh, A. Q., and Greif, D. M. (2017). Vascular cells in blood vessel wall development and disease. 1st ed. Netherlands: Elsevier Inc. doi:10.1016/bs.apha.2016.08.001
McCarthy, C. G., Wenceslau, C. F., Webb, R. C., and Joe, B. (2019). Novel contributors and mechanisms of cellular senescence in hypertension-associated premature vascular aging. Am. J. Hypertens. 32, 709–719. doi:10.1093/ajh/hpz052
Meng, J., Geng, Q., Jin, S., Teng, X., Xiao, L., Wu, Y., et al. (2023). Exercise protects vascular function by countering senescent cells in older adults. Front. Physiol. 14, 1138162–1138210. doi:10.3389/fphys.2023.1138162
Menghini, R., Stöhr, R., and Federici, M. (2014). MicroRNAs in vascular aging and atherosclerosis. Ageing Res. Rev. 17, 68–78. doi:10.1016/j.arr.2014.03.005
Merino, A., Buendia, P., Martin-Malo, A., Aljama, P., Ramirez, R., and Carracedo, J. (2011). Senescent CD14+CD16+ monocytes exhibit proinflammatory and proatherosclerotic activity. J. Immunol. 186, 1809–1815. doi:10.4049/jimmunol.1001866
Miller, K. N., Dasgupta, N., Liu, T., Adams, P. D., and Vizioli, M. G. (2021). Cytoplasmic chromatin fragments—from mechanisms to therapeutic potential. Elife 10, 637288–e63810. doi:10.7554/eLife.63728
Mitchell, G. F. (2021). Arterial stiffness in aging: does it have a place in clinical practice? recent advances in hypertension. Hypertension 77, 768–780. doi:10.1161/HYPERTENSIONAHA.120.14515
Mittelbrunn, M., and Kroemer, G. (2021). Hallmarks of T cell aging. Nat. Immunol. 22, 687–698. doi:10.1038/s41590-021-00927-z
Molnár, A. Á., Nádasy, G. L., Dörnyei, G., Patai, B. B., Delfavero, J., Fülöp, G. Á., et al. (2021). The aging venous system: from varicosities to vascular cognitive impairment. GeroScience 43, 2761–2784. doi:10.1007/s11357-021-00475-2
Mühlberger, D., Zumholz, A. K., Brenner, E., Mumme, A., Stücker, M., Falkenstein, T., et al. (2022). Cellular senescence at the saphenofemoral junction in patients with healthy, primary varicose and recurrent varicose veins – a pilot study. Vascular 30, 559–567. doi:10.1177/17085381211012882
Munjal, A., and Bordoni, B. (2023). “Histology, vascular,” in StatPearls - NCBI bookshelf (Treasure Island (FL): StatPearls Publishing).
Muñoz-Espín, D., Cañamero, M., Maraver, A., Gómez-López, G., Contreras, J., Murillo-Cuesta, S., et al. (2013). Programmed cell senescence during mammalian embryonic development. Cell 155, 1104–1118. doi:10.1016/j.cell.2013.10.019
Muñoz-Espín, D., and Serrano, M. (2014). Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 15, 482–496. doi:10.1038/nrm3823
Mylonas, A., and O’Loghlen, A. (2022). Cellular senescence and ageing: mechanisms and interventions. Front. Aging 3, 866718–866810. doi:10.3389/fragi.2022.866718
Nacarelli, T., Liu, P., and Zhang, R. (2017). Epigenetic basis of cellular senescence and its implications in aging. Genes (Basel) 8, 343. doi:10.3390/genes8120343
Nakano-Kurimoto, R., Ikeda, K., Uraoka, M., Nakagawa, Y., Yutaka, K., Koide, M., et al. (2009). Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am. J. Physiol. - Hear. Circ. Physiol. 297, H1673–H1684. doi:10.1152/ajpheart.00455.2009
Nelson, G., Wordsworth, J., Wang, C., Jurk, D., Lawless, C., Martin-Ruiz, C., et al. (2012). A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11, 345–349. doi:10.1111/j.1474-9726.2012.00795.x
Nikolajevic, J., Ariaee, N., Liew, A., Abbasnia, S., Fazeli, B., and Sabovic, M. (2022). The role of MicroRNAs in endothelial cell senescence. Cells 11, 1185–1218. doi:10.3390/cells11071185
Ohtani, N. (2022). The roles and mechanisms of senescence-associated secretory phenotype (SASP): can it be controlled by senolysis? Inflamm. Regen. 42, 11. doi:10.1186/s41232-022-00197-8
Olivieri, F., Prattichizzo, F., Grillari, J., and Balistreri, C. R. (2018). Cellular senescence and inflammaging in age-Related diseases. Mediat. Inflamm. 2018, 9076485. doi:10.1155/2018/9076485
Ong, S. M., Hadadi, E., Dang, T. M., Yeap, W. H., Tan, C. T. Y., Ng, T. P., et al. (2018). The pro-inflammatory phenotype of the human non-classical monocyte subset is attributed to senescence article. Cell Death Dis. 9, 1–12. doi:10.1038/s41419-018-0327-1
Ono, K. (2016). Functions of microRNA-33a/b and microRNA therapeutics. J. Cardiol. 67, 28–33. doi:10.1016/j.jjcc.2015.10.017
Ortega, M. A., Fraile-Martínez, O., García-Montero, C., Álvarez-Mon, M. A., Chaowen, C., Ruiz-Grande, F., et al. (2021a). Understanding chronic venous disease: a critical overview of its pathophysiology and medical management. J. Clin. Med. 10, 3239–3242. doi:10.3390/jcm10153239
Ortega, M. A., Fraile-Martínez, O., Pekarek, L., Alvarez-Mon, M. A., Asúnsolo, Á., Sanchez-Trujillo, L., et al. (2021b). Defective expression of the peroxisome regulators PPARα receptors and lysogenesis with increased cellular senescence in the venous wall of chronic venous disorder. Histol. Histopathol. 36, 547–558. doi:10.14670/HH-18-322
Ota, H., Eto, M., Kano, M. R., Kahyo, T., Setou, M., Ogawa, S., et al. (2010). Induction of endothelial nitric oxide synthase, SIRT1, and catalase by statins inhibits endothelial senescence through the Akt pathway. Arterioscler. Thromb. Vasc. Biol. 30, 2205–2211. doi:10.1161/ATVBAHA.110.210500
Ovadya, Y., Landsberger, T., Leins, H., Vadai, E., Gal, H., Biran, A., et al. (2018). Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 9, 5435. doi:10.1038/s41467-018-07825-3
Pande, R., Parikh, A., Shenoda, B., Ramanathan, S., Alexander, G. M., Schwartzman, R. J., et al. (2021). Hsa-miR-605 regulates the proinflammatory chemokine CXCL5 in complex regional pain syndrome. Biomed. Pharmacother. 140, 111788. doi:10.1016/j.biopha.2021.111788
Park, H., Kim, C. H., Jeong, J. H., Park, M., and Kim, K. S. (2016). GDF15 contributes to radiation-induced senescence through the ros-mediated p16 pathway in human endothelial cells. Oncotarget 7, 9634–9644. doi:10.18632/oncotarget.7457
Parvizi, M., Franchi, F., Arendt, B. K., Ebtehaj, S., Rodriguez-Porcel, M., and Lanza, I. R. (2021). Senolytic agents lessen the severity of abdominal aortic aneurysm in aged mice. Exp. Gerontol. 151, 111416. doi:10.1016/j.exger.2021.111416
Passacquale, G., Di Giosia, P., and Ferro, A. (2016). The role of inflammatory biomarkers in developing targeted cardiovascular therapies: lessons from the cardiovascular inflammation reduction trials. Cardiovasc. Res. 109, 9–23. doi:10.1093/cvr/cvv227
Pauty, J., Nakano, S., Usuba, R., Nakajima, T., Johmura, Y., Omori, S., et al. (2021). A 3D tissue model-on-a-chip for studying the effects of human senescent fibroblasts on blood vessels. Biomater. Sci. 9, 199–211. doi:10.1039/d0bm01297a
Petrova, N. V., Velichko, A. K., Razin, S. V., and Kantidze, O. L. (2016). Small molecule compounds that induce cellular senescence. Aging Cell 15, 999–1017. doi:10.1111/acel.12518
Pham, L. M., Kim, E. C., Ou, W., Phung, C. D., Nguyen, T. T., Pham, T. T., et al. (2021). Targeting and clearance of senescent foamy macrophages and senescent endothelial cells by antibody-functionalized mesoporous silica nanoparticles for alleviating aorta atherosclerosis. Biomaterials 269, 120677. doi:10.1016/j.biomaterials.2021.120677
Puvvula, P. K. (2019). LncRNAs regulatory networks in cellular senescence. Int. J. Mol. Sci. 20, 2615. doi:10.3390/ijms20112615
Puzianowska-Kuźnicka, M., Owczarz, M., Wieczorowska-Tobis, K., Nadrowski, P., Chudek, J., Slusarczyk, P., et al. (2016). Interleukin-6 and C-reactive protein, successful aging, and mortality: the PolSenior study. Immun. Ageing 13, 21–12. doi:10.1186/s12979-016-0076-x
Ramírez, R., Ceprian, N., Figuer, A., Valera, G., Bodega, G., Alique, M., et al. (2022). Endothelial senescence and the chronic vascular diseases: challenges and therapeutic opportunities in atherosclerosis. J. Pers. Med. 12, 215. doi:10.3390/jpm12020215
Rayess, H., Wang, M. B., and Srivatsan, E. S. (2012). Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 130, 1715–1725. doi:10.1002/ijc.27316
Reddy, M. A., Das, S., Zhuo, C., Jin, W., Wang, M., Lanting, L., et al. (2016). Regulation of vascular smooth muscle cell dysfunction under diabetic conditions by MIR-504. Arterioscler. Thromb. Vasc. Biol. 36, 864–873. doi:10.1161/ATVBAHA.115.306770
Renna, N. F., Vazquez, M. A., Lama, M. C., González, E. S., and Miatello, R. M. (2009). Effect of chronic aspirin administration on an experimental model of metabolic syndrome. Clin. Exp. Pharmacol. Physiol. 36, 162–168. doi:10.1111/j.1440-1681.2008.05042.x
Reustle, A., and Torzewski, M. (2018). Role of p38 MAPK in atherosclerosis and aortic valve sclerosis. Int. J. Mol. Sci. 19, 3761–3820. doi:10.3390/ijms19123761
Rhinn, M., Ritschka, B., and Keyes, W. M. (2019). Cellular senescence in development, regeneration and disease. Dev 146, dev151837. doi:10.1242/dev.151837
Roger, L., Tomas, F., and Gire, V. (2021). Mechanisms and regulation of cellular senescence. Int. J. Mol. Sci. 22, 13173–13242. doi:10.3390/ijms222313173
Rojas, M., Lemtalsi, T., Toque, H., Xu, Z., Fulton, D., Caldwell, R., et al. (2017). NOX2-Induced activation of arginase and diabetes-induced retinal endothelial cell senescence. Antioxidants 6, 43. doi:10.3390/antiox6020043
Rossman, M. J., Kaplon, R. E., Hill, S. D., McNamara, M. N., Santos-Parker, J. R., Pierce, G. L., et al. (2017). Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am. J. Physiol. - Hear. Circ. Physiol. 313, H890–H895. doi:10.1152/ajpheart.00416.2017
Ruscetti, M., Morris, J. P., Mezzadra, R., Russell, J., Leibold, J., Romesser, P. B., et al. (2020). Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell 181, 424–441. doi:10.1016/j.cell.2020.03.008
Russo, G. L., Stampone, E., Cervellera, C., and Borriello, A. (2020). Regulation of p27kip1 and p57kip2 functions by natural polyphenols. Biomolecules 10, 1316–1331. doi:10.3390/biom10091316
Sakai, C., Ishida, M., Ohba, H., Yamashita, H., Uchida, H., Yoshizumi, M., et al. (2017). Fish oil omega-3 polyunsaturated fatty acids attenuate oxidative stress-induced DNA damage in vascular endothelial cells. PLoS One 12, e0187934. doi:10.1371/journal.pone.0187934
Sakaki, M., Ebihara, Y., Okamura, K., Nakabayashi, K., Igarashi, A., Matsumoto, K., et al. (2017). Potential roles of DNA methylation in the initiation and establishment of replicative senescence revealed by array-based methylome and transcriptome analyses. PLoS One 12, e0171431. doi:10.1371/journal.pone.0171431
Salama, R., Sadaie, M., Hoare, M., and Narita, M. (2014). Cellular senescence and its effector programs. Genes Dev. 28, 99–114. doi:10.1101/gad.235184.113
Saleh, T., Tyutyunyk-Massey, L., and Gewirtz, D. A. (2019). Tumor cell escape from therapy-induced senescence as a model of disease recurrence after dormancy. Cancer Res. 79, 1044–1046. doi:10.1158/0008-5472.CAN-18-3437
Salminen, A., Kauppinen, A., and Kaarniranta, K. (2012). Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 24, 835–845. doi:10.1016/j.cellsig.2011.12.006
Sati, S., Bonev, B., Szabo, Q., Jost, D., Bensadoun, P., Serra, F., et al. (2020). 4D genome rewiring during oncogene-induced and replicative senescence. Mol. Cell 78, 522–538. doi:10.1016/j.molcel.2020.03.007
Satoh, M., Ishikawa, Y., Takahashi, Y., Itoh, T., Minami, Y., and Nakamura, M. (2008). Association between oxidative DNA damage and telomere shortening in circulating endothelial progenitor cells obtained from metabolic syndrome patients with coronary artery disease. Atherosclerosis 198, 347–353. doi:10.1016/j.atherosclerosis.2007.09.040
Schmitt, C. A., Wang, B., and Demaria, M. (2022). Senescence and cancer — role and therapeutic opportunities. Nat. Rev. Clin. Oncol. 19, 619–636. doi:10.1038/s41571-022-00668-4
Schober, A., Nazari-Jahantigh, M., Wei, Y., Bidzhekov, K., Gremse, F., Grommes, J., et al. (2014). MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat. Med. 20, 368–376. doi:10.1038/nm.3487
Sena, C. M., Matafome, P., Louro, T., Nunes, E., Fernandes, R., and Seiça, R. M. (2011). Metformin restores endothelial function in aorta of diabetic rats. Br. J. Pharmacol. 163, 424–437. doi:10.1111/j.1476-5381.2011.01230.x
Sene, A., Khan, A. A., Cox, D., Nakamura, R. E. I., Santeford, A., Kim, B. M., et al. (2013). Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 17, 549–561. doi:10.1016/j.cmet.2013.03.009
Shah, P. P., Donahue, G., Otte, G. L., Capell, B. C., Nelson, D. M., Cao, K., et al. (2013). Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 27, 1787–1799. doi:10.1101/gad.223834.113
Shakeri, H., Lemmens, K., Gevaert, A. B., De Meyer, G. R. Y., and Segers, V. F. M. (2018). Cellular senescence links aging and diabetes in cardiovascular disease. Am. J. Physiol. - Hear. Circ. Physiol. 315, H448–H462. doi:10.1152/ajpheart.00287.2018
Sheekey, E., and Narita, M. (2023). P53 in senescence – it’S a marathon, not a sprint. FEBS J. 290, 1212–1220. doi:10.1111/febs.16325
Shirakawa, K., and Sano, M. (2021). T cell immunosenescence in aging, obesity, and cardiovascular disease. Cells 10, 2435–2516. doi:10.3390/cells10092435
Sidler, C., Kovalchuk, O., and Kovalchuk, I. (2017). Epigenetic regulation of cellular senescence and aging. Front. Genet. 8, 138. doi:10.3389/fgene.2017.00138
Stenmark, K. R., Yeager, M. E., El Kasmi, K. C., Nozik-Grayck, E., Gerasimovskaya, E. V., Li, M., et al. (2013). The adventitia: essential regulator of vascular wall structure and function. Annu. Rev. Physiol. 75, 23–47. doi:10.1146/annurev-physiol-030212-183802
Stevenson, A. J., McCartney, D. L., Harris, S. E., Taylor, A. M., Redmond, P., Starr, J. M., et al. (2018). Trajectories of inflammatory biomarkers over the eighth decade and their associations with immune cell profiles and epigenetic ageing. Clin. Epigenetics 10, 159–210. doi:10.1186/s13148-018-0585-x
Stojanovic, S. D., Fiedler, J., Bauersachs, J., Thum, T., and Sedding, D. G. (2020). Senescence-induced inflammation: an important player and key therapeutic target in atherosclerosis. Eur. Heart J. 41, 2983–2996. doi:10.1093/eurheartj/ehz919
Storer, M., Mas, A., Robert-Moreno, A., Pecoraro, M., Ortells, M. C., Di Giacomo, V., et al. (2013). Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155, 1119–1130. doi:10.1016/j.cell.2013.10.041
Suda, M., Paul, K. H., Minamino, T., Miller, J. D., Lerman, A., Ellison-Hughes, G. M., et al. (2023). Senescent cells: a therapeutic target in cardiovascular diseases. Cells 12, 1296–1337. doi:10.3390/cells12091296
Sun, Y., Wang, X., Liu, T., Zhu, X., and Pan, X. (2022). The multifaceted role of the SASP in atherosclerosis: from mechanisms to therapeutic opportunities. Cell Biosci. 12, 74–20. doi:10.1186/s13578-022-00815-5
Tabaei, S., and Tabaee, S. S. (2019). DNA methylation abnormalities in atherosclerosis. Artif. Cells, Nanomedicine Biotechnol. 47, 2031–2041. doi:10.1080/21691401.2019.1617724
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi:10.1016/j.cell.2006.07.024
Takasugi, M., Okada, R., Takahashi, A., Virya Chen, D., Watanabe, S., and Hara, E. (2017). Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 8, 15729–15811. doi:10.1038/ncomms15728
Tang, Y., Fung, E., Xu, A., and Lan, H. Y. (2017). C-reactive protein and ageing. Clin. Exp. Pharmacol. Physiol. 44, 9–14. doi:10.1111/1440-1681.12758
Taylor, A. M., and Bordoni, B. (2023). “Histology, blood vascular system,” in StatPearls - NCBI bookshelf (Treasure Island (FL): StatPearls Publishing).
Terzi, M. Y., Izmirli, M., and Gogebakan, B. (2016). The cell fate: senescence or quiescence. Mol. Biol. Rep. 43, 1213–1220. doi:10.1007/s11033-016-4065-0
Triana-Martínez, F., Picallos-Rabina, P., Da Silva-Álvarez, S., Pietrocola, F., Llanos, S., Rodilla, V., et al. (2019). Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 10, 1–12. doi:10.1038/s41467-019-12888-x
Tsuboi, T., Maeda, M., and Hayashi, T. (2018). Administration of L-arginine plus L-citrulline or L-citrulline alone successfully retarded endothelial senescence. PLoS One 13, e0192252. doi:10.1371/journal.pone.0192252
Tuttle, C. S. L., Waaijer, M. E. C., Slee-Valentijn, M. S., Stijnen, T., Westendorp, R., and Maier, A. B. (2020). Cellular senescence and chronological age in various human tissues: a systematic review and meta-analysis. Aging Cell 19, 130833–e13111. doi:10.1111/acel.13083
Ungvari, Z., Tarantini, S., Sorond, F., Merkely, B., and Csiszar, A. (2020). Mechanisms of vascular aging, A geroscience perspective: JACC focus seminar. J. Am. Coll. Cardiol. 75, 931–941. doi:10.1016/j.jacc.2019.11.061
Uryga, A. K., Grootaert, M. O. J., Garrido, A. M., Oc, S., Foote, K., Chappell, J., et al. (2021). Telomere damage promotes vascular smooth muscle cell senescence and immune cell recruitment after vessel injury. Commun. Biol. 4, 611. doi:10.1038/s42003-021-02123-z
Valieva, Y., Ivanova, E., Fayzullin, A., Kurkov, A., and Igrunkova, A. (2022). Senescence-associated β-galactosidase detection in pathology. Diagnostics 12, 2309. doi:10.3390/diagnostics12102309
Vellasamy, D. M., Lee, S. J., Goh, K. W., Goh, B. H., Tang, Y. Q., Ming, L. C., et al. (2022). Targeting immune senescence in atherosclerosis. Int. J. Mol. Sci. 23, 13059–13124. doi:10.3390/ijms232113059
Voelter-Mahlknecht, S. (2016). Epigenetic associations in relation to cardiovascular prevention and therapeutics. Clin. Epigenetics 8, 4–17. doi:10.1186/s13148-016-0170-0
Wan, Y. Z., Gao, P., Zhou, S., Zhang, Z. Q., Hao, D. L., Lian, L. S., et al. (2014). SIRT1-mediated epigenetic downregulation of plasminogen activator inhibitor-1 prevents vascular endothelial replicative senescence. Aging Cell 13, 890–899. doi:10.1111/acel.12247
Wang, H. W., Lo, H. H., Chiu, Y. L., Chang, S. J., Huang, P. H., Liao, K. H., et al. (2014). Dysregulated miR-361-5p/VEGF axis in the plasma and endothelial progenitor cells of patients with coronary artery disease. PLoS One 9, 980700–e98111. doi:10.1371/journal.pone.0098070
Wang, J., Uryga, A. K., Reinhold, J., Figg, N., Baker, L., Finigan, A., et al. (2015). Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation 132, 1909–1919. doi:10.1161/CIRCULATIONAHA.115.016457
Wang, K., Liu, H., Hu, Q., Wang, L., Liu, J., Zheng, Z., et al. (2022a). Epigenetic regulation of aging: implications for interventions of aging and diseases. Signal Transduct. Target. Ther. 7, 374. doi:10.1038/s41392-022-01211-8
Wang, M., Ji, Y., Cai, S., and Ding, W. (2016a). MiR-206 suppresses the progression of coronary artery disease by modulating vascular endothelial growth factor (VEGF) expression. Med. Sci. Monit. 22, 5011–5020. doi:10.12659/MSM.898883
Wang, S., He, W., and Wang, C. (2016b). MiR-23a regulates the vasculogenesis of coronary artery disease by targeting epidermal growth factor receptor. Cardiovasc. Ther. 34, 199–208. doi:10.1111/1755-5922.12187
Wang, Y., Dong, C., Han, Y., Gu, Z., and Sun, C. (2022b). Immunosenescence, aging and successful aging. Front. Immunol. 13, 942796–942818. doi:10.3389/fimmu.2022.942796
Wiley, C. D., and Campisi, J. (2021). The metabolic roots of senescence: mechanisms and opportunities for intervention. Nat. Metab. 3, 1290–1301. doi:10.1038/s42255-021-00483-8
Wiley, C. D., Velarde, M. C., Lecot, P., Liu, S., Sarnoski, E. A., Freund, A., et al. (2016). Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 23, 303–314. doi:10.1016/j.cmet.2015.11.011
Wilkinson, H. N., and Hardman, M. J. (2020). Senescence in wound repair: emerging strategies to target chronic healing wounds. Front. Cell Dev. Biol. 8, 773–813. doi:10.3389/fcell.2020.00773
Xia, L., Wang, X. X., Hu, X. S., Guo, X. G., Shang, Y. P., Chen, H. J., et al. (2008). Resveratrol reduces endothelial progenitor cells senescence through augmentation of telomerase activity by Akt-dependent mechanisms. Br. J. Pharmacol. 155, 387–394. doi:10.1038/bjp.2008.272
Xiong, S., Salazar, G., San Martin, A., Ahmad, M., Patrushev, N., Hilenski, L., et al. (2010). PGC-1 alpha serine 570 phosphorylation and GCN5-mediated acetylation by angiotensin II drive catalase down-regulation and vascular hypertrophy. J. Biol. Chem. 285, 2474–2487. doi:10.1074/jbc.M109.065235
Xu, H., Li, S., and Liu, Y. S. (2021). Roles and mechanisms of DNA methylation in vascular aging and related diseases. Front. Cell Dev. Biol. 9, 699374–699418. doi:10.3389/fcell.2021.699374
Xu, M., Tchkonia, T., Ding, H., Ogrodnik, M., Lubbers, E. R., Pirtskhalava, T., et al. (2015). JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. U. S. A. 112, E6301–E6310. doi:10.1073/pnas.1515386112
Ya, J., and Bayraktutan, U. (2023). Vascular ageing: mechanisms, risk factors, and treatment strategies. Int. J. Mol. Sci. 24, 11538. doi:10.3390/ijms241411538
Yamazaki, Y., Baker, D. J., Tachibana, M., Liu, C. C., Van Deursen, J. M., Brott, T. G., et al. (2016). Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke 47, 1068–1077. doi:10.1161/STROKEAHA.115.010835
Yang, J., Liu, M., Hong, D., Zeng, M., and Zhang, X. (2021). The paradoxical role of cellular senescence in cancer. Front. Cell Dev. Biol. 9, 722205. doi:10.3389/fcell.2021.722205
Yasuda, S., Horinaka, M., Iizumi, Y., Goi, W., Sukeno, M., and Sakai, T. (2022). Oridonin inhibits SASP by blocking p38 and NF-κB pathways in senescent cells. Biochem. Biophys. Res. Commun. 590, 55–62. doi:10.1016/j.bbrc.2021.12.098
Yi, S. J., and Kim, K. (2020). New insights into the role of histone changes in aging. Int. J. Mol. Sci. 21, 8241–8320. doi:10.3390/ijms21218241
Yin, H., and Pickering, J. G. (2016). Cellular senescence and vascular disease: novel routes to better understanding and therapy. Can. J. Cardiol. 32, 612–623. doi:10.1016/j.cjca.2016.02.051
You, W., Hong, Y., He, H., Huang, X., Tao, W., Liang, X., et al. (2019). TGF-β mediates aortic smooth muscle cell senescence in Marfan syndrome. Aging (Albany. NY) 11, 3574–3584. doi:10.18632/aging.101998
Yousefzadeh, M. J., Zhu, Y., McGowan, S. J., Angelini, L., Fuhrmann-Stroissnigg, H., Xu, M., et al. (2018). Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28. doi:10.1016/j.ebiom.2018.09.015
Yu, H. T., Park, S., Shin, E. C., and Lee, W. W. (2016). T cell senescence and cardiovascular diseases. Clin. Exp. Med. 16, 257–263. doi:10.1007/s10238-015-0376-z
Zawada, A. M., Rogacev, K. S., Rotter, B., Winter, P., Marell, R. R., Fliser, D., et al. (2011). SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood 118, 50–61. doi:10.1182/blood-2011-01-326827
Zeng, L., Xiao, Q., Margariti, A., Zhang, Z., Zampetaki, A., Patel, S., et al. (2006). HDAC3 is crucial in shear- and VEGF-induced stem cell differentiation toward endothelial cells. J. Cell Biol. 174, 1059–1069. doi:10.1083/jcb.200605113
Zhang, L., Pitcher, L. E., Prahalad, V., Niedernhofer, L. J., and Robbins, P. D. (2023). Targeting cellular senescence with senotherapeutics: senolytics and senomorphics. FEBS J. 290, 1362–1383. doi:10.1111/febs.16350
Zhang, Q., Li, S., Chen, F., Zeng, R., and Tong, R. (2022). Targeted delivery strategy: a beneficial partner for emerging senotherapy. Biomed. Pharmacother. 155, 113737. doi:10.1016/j.biopha.2022.113737
Zhang, W., Song, M., Qu, J., and Liu, G. H. (2018). Epigenetic modifications in cardiovascular aging and diseases. Circ. Res. 123, 773–786. doi:10.1161/CIRCRESAHA.118.312497
Zhao, G., Wang, H., Xu, C., Wang, P., Chen, J., Wang, P., et al. (2016). SIRT6 delays cellular senescence by promoting p27Kip1 ubiquitin-proteasome degradation. Aging (Albany. NY) 8, 2308–2323. doi:10.18632/aging.101038
Zhou, X., Perez, F., Han, K., and Jurivich, D. A. (2006). Clonal senescence alters endothelial ICAM-1 function. Mech. Ageing Dev. 127, 779–785. doi:10.1016/j.mad.2006.07.003
Zhou, Y., Nishiura, A., Morikuni, H., Deng, W., Tsujibayashi, T., Momota, Y., et al. (2023). RANKL+ senescent cells under mechanical stress: a therapeutic target for orthodontic root resorption using senolytics. Int. J. Oral Sci. 15, 20–25. doi:10.1038/s41368-023-00228-1
Zhu, H., Shen, F., Liao, T., Qian, H., and Liu, Y. (2023). Immunosenescence and macrophages: from basics to therapeutics. Int. J. Biochem. Cell Biol. 165, 106479–106512. doi:10.1016/j.biocel.2023.106479
Zhu, X., Chen, Z., Shen, W., Huang, G., Sedivy, J. M., Wang, H., et al. (2021). Inflammation, epigenetics, and metabolism converge to cell senescence and ageing: the regulation and intervention. Signal Transduct. Target. Ther. 6, 245–329. doi:10.1038/s41392-021-00646-9
Zhu, Y., Tchkonia, T., Fuhrmann-Stroissnigg, H., Dai, H. M., Ling, Y. Y., Stout, M. B., et al. (2016). Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15, 428–435. doi:10.1111/acel.12445
Keywords: cellular senescence, epigenetic modifications, immune activation, endothelial cells (ECs), vascular smooth muscle cells (VSMCs)
Citation: Fraile-Martinez O, De Leon-Oliva D, Boaru DL, De Castro-Martinez P, Garcia-Montero C, Barrena-Blázquez S, García-García J, García-Honduvilla N, Alvarez-Mon M, Lopez-Gonzalez L, Diaz-Pedrero R, Guijarro LG and Ortega MA (2024) Connecting epigenetics and inflammation in vascular senescence: state of the art, biomarkers and senotherapeutics. Front. Genet. 15:1345459. doi: 10.3389/fgene.2024.1345459
Received: 27 November 2023; Accepted: 15 February 2024;
Published: 26 February 2024.
Edited by:
Taicheng Zhou, The Sixth Affiliated Hospital of Sun Yat-sen University, ChinaReviewed by:
Zuo Wang, University of South China, ChinaYuji Ikeno, The University of Texas Health Science Center at San Antonio, United States
Copyright © 2024 Fraile-Martinez, De Leon-Oliva, Boaru, De Castro-Martinez, Garcia-Montero, Barrena-Blázquez, García-García, García-Honduvilla, Alvarez-Mon, Lopez-Gonzalez, Diaz-Pedrero, Guijarro and Ortega. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel A. Ortega, bWlndWVsLmFuZ2VsLm9ydGVnYTkyQGdtYWlsLmNvbQ==; Raul Diaz-Pedrero, cmF1bC5kaWF6cEB1YWguZXM=
†These authors have contributed equally to this work