 Yuka Shiota1,2*
Yuka Shiota1,2* Tomoaki Nishiyama3
Tomoaki Nishiyama3 Shigeru Yokoyama2,4*
Shigeru Yokoyama2,4* Yuko Yoshimura4,5
Yuko Yoshimura4,5 Chiaki Hasegawa2,4
Chiaki Hasegawa2,4 Sanae Tanaka2,4
Sanae Tanaka2,4 Sumie Iwasaki1,5
Sumie Iwasaki1,5 Mitsuru Kikuchi4,6
Mitsuru Kikuchi4,6- 1Japan Society for the Promotion of Science, Tokyo, Japan
- 2Research Center for Child Mental Development, Kanazawa University, Kanazawa, Japan
- 3Research Center for Experimental Modeling of Human Disease, Kanazawa University, Kanazawa, Japan
- 4United Graduate School of Child Development, Osaka University, Kanazawa University, Hamamatsu University School of Medicine, Chiba University, and University of Fukui, Kanazawa, Japan
- 5Institute of Human and Social Sciences, Kanazawa University, Kanazawa, Japan
- 6Department of Psychiatry and Neurobiology, Graduate School of Medical Science, Kanazawa University, Kanazawa, Japan
Introduction: Autism spectrum disorders (ASD) represent a heterogeneous group of neurodevelopmental disorders with strong genetic predispositions. Although an increasing number of genetic variants have been implicated in the pathogenesis of ASD, little is known about the relationship between ASD-associated genetic variants and individual ASD traits. Therefore, we aimed to investigate these relationships.
Methods: Here, we report a case-control association study of 32 Japanese children with ASD (mainly with high-functioning autism [HFA]) and 36 with typical development (TD). We explored previously established ASD-associated genes using a next-generation sequencing panel and determined the association between Social Responsiveness Scale (SRS) T-scores and intelligence quotient (IQ) scores.
Results: In the genotype-phenotype analyses, 40 variants of five genes (SCN1A, SHANK3, DYRK1A, CADPS, and SCN2A) were associated with ASD/TD phenotypes. In particular, 10 SCN1A variants passed permutation filtering (false discovery rate <0.05). In the quantitative association analyses, 49 variants of 12 genes (CHD8, SCN1A, SLC6A1, KMT5B, CNTNAP2, KCNQ3, SCN2A, ARID1B, SHANK3, DYRK1A, FOXP1, and GRIN2B) and 50 variants of 10 genes (DYRK1A, SCN2A, SLC6A1, ARID1B, CNTNAP2, SHANK3, FOXP1, PTEN, SCN1A, and CHD8) were associated with SRS T- and IQ-scores, respectively.
Conclusion: Our data suggest that these identified variants are essential for the genetic architecture of HFA.
1 Introduction
Autism spectrum disorder (ASD) is a common neurodevelopmental disorder characterized by impaired social communication and restricted, repetitive behaviors or interests (Masi et al., 2017; Lord et al., 2018; Hirota and King, 2023). In the US, ASD affects approximately 2.3% of children aged 8 years (Hirota and King, 2023). One important aspect of ASD is its heterogeneous clinical features. For example, some individuals exhibit intellectual disability (ID) and limited language ability, while others display motor impairments (Hirota and King, 2023). This heterogeneity is attributed to multiple genetic variations and environmental factors (Baron-Cohen, 2017; Bai et al., 2019).
Genetic variations are associated with varying allelic effect sizes and population frequencies. In addition, genetic variations exist in a continuum, ranging from single nucleotide changes to genomic changes at the chromosomal level. Small-scale variations include single-nucleotide polymorphisms (SNPs) and insertions or deletions (indels) of a short DNA sequence, while typical large-scale variations include copy number variations (CNVs) and chromosomal rearrangement (Sebat et al., 2007). In general, rare variants tend to have a larger effect size than common variants (Grove et al., 2019). The presence of numerous variants with different effect sizes and allele frequencies, along with their interactions with environmental factors, generates a highly complex genetic architecture of ASD (Timpson et al., 2018). Although whole-genome and whole-exome next-generation sequencing (NGS) have been utilized for comprehensive analyses of such polygenic disorders, these approaches are relatively expensive (Mellone et al., 2022). Targeted panel sequencing allows the efficient and accurate detection of variants with high sensitivity (Mellone et al., 2022).
To date, several large cohort studies have identified an increasing number of ASD-associated genetic variants. More than one thousand ASD-associated rare variants have been identified using whole-genome sequencing and transmission and de novo association tests (Murtaza et al., 2022). By exome analyses in a Japanese population, Kimura et al. (2022) identified rare synaptic function-related variants. However, these large-scale cohort studies might have included individuals with various degrees of autistic traits and severity of ID. Moreover, relationships between genetic variants and individual ASD traits, such as speech development, social responsiveness, and intelligence remain largely unexplored.
In this study, we performed case-control NGS analyses combined with psychological assessments on Japanese children with ASD, mainly with high-functioning autism (HFA). As HFA is not considered to be a distinct and absolute diagnostic category, we assessed it as a form of ASD, which was highly heterogeneous in phenotype. Children with ASD with an intelligence quotient (IQ) of 70 or above were defined as having HFA (Alvares et al., 2020). Using short-read-based methods, we analyzed SNPs and short indels, regardless of whether they were rare or common in population frequency. We aimed to further our understanding of ASD by clarifying the relationship between ASD-associated variants and individual ASD traits.
2 Materials and methods
2.1 Participants
Participants, all of whom lived in the Hokuriku District of Japan, were called upon to participate in the Bambi Plan at Kanazawa University and its affiliated hospitals. The participants were children diagnosed with ASD and typically developing (TD) children who did not exhibit apparent developmental delay. Additionally, those with low birth weight were referred to participate in the Bambi Plan by the Department of Pediatrics of Kanazawa University Hospital. The children were 5–8 years old and were able to sit for 1 h, which is a requirement for the IQ assessment as it must be taken seated. Children with low birth weight and who exhibited developmental delay were assessed with the assessments, which were established according to the Diagnostic and Statistical Manual of Mental Disorders (fourth edition) (American Psychiatric Association, 2000) using the Diagnostic Interview for Social and Communication Disorders (Wing et al., 2002), Autism Diagnostic Observation Schedule-Generic (Lord et al., 2000), and Autism Diagnostic Observation Schedule 2 (Lord C et al., 2012), as well as children with ASD. Those who did not fully meet the criteria of these assessments were regarded as individuals with sub-threshold autistic traits (Shiota et al., 2021). In addition, children enrolled in TD were referred to as TD without the formal assessments. IQ was assessed using the mental processing composite scale of the Kaufman Assessment Battery for Children (K-ABC) (Kaufman AS, 1983). Overall, 118 children with ASD (92 males, 26 females), 104 with TD (59 males, 45 females), 3 with sub-threshold autistic traits (3 males), and 10 with low birth weight (3 males, 7 females) were enrolled (Supplementary Figure S1).
2.2 Target gene regions for enrichment
Based on the SFARI gene database (https://gene.sfari.org/) and previous reports (Satterstrom et al., 2020; Fóthi et al., 2022; Murtaza et al., 2022; Qiu et al., 2022), 16 highly confident ASD-associated genes, one promoter region, and 20 intergenic regions containing ASD-associated SNPs were selected for the biotinylated oligonucleotide probe design for enrichment (Table 1).
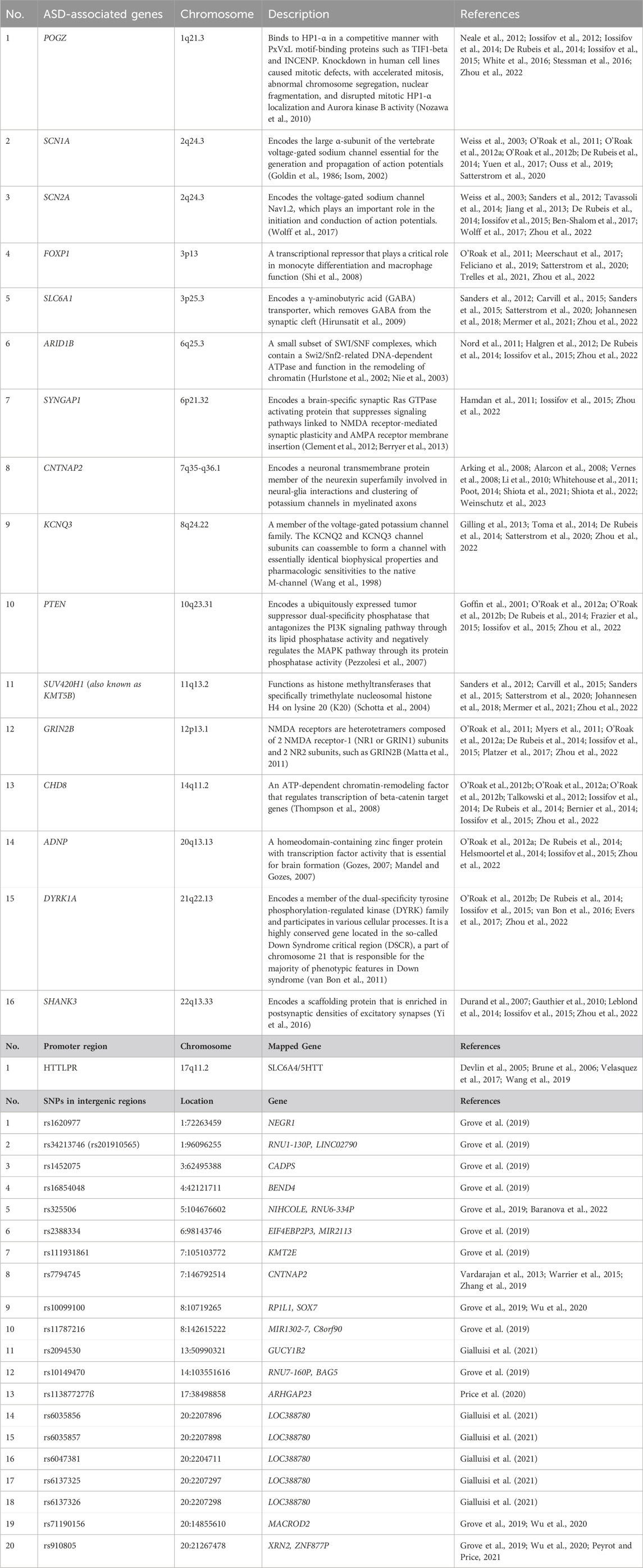
Table 1. Genes and SNPs targeted in this study.
2.3 Sequencing and data processing
Buccal mucosa was collected using a swab. Genomic DNA was extracted from the buccal mucosa using a Maxwell RSC Instrument (Promega, Madison, WI, United States) with the Maxwell RSC Blood DNA Kit (Promega), according to the manufacturer’s instructions. Subsequently, whole-genome amplification was performed on 147 genomic DNA samples (88 were excluded) using a PicoPLEX WGA kit (TaKaRa Bio, Mountain View, CA, United States), according to the manufacturer’s instructions. An Illumina paired-end sequencing library was constructed using the KAPA HyperPlus Kit (Kapa Biosystems, Wilmington, MA, United States), followed by enrichment of target sites with xGen Custom Hyb Panel (Integrated DNA Technologies) and sequencing using an Illumina iSeq 100 System. Adapter sequences were trimmed from the reads using Trimmomatic v0.39 and mapped to the human reference genome (hg38) using Burrows–Wheeler Aligner v0.7.17 (https://github.com/lh3/bwa). Libraries from the amplified DNA resulted in insufficient sequencing and mapping rates; thus, the data were not used for further analysis. Libraries constructed directly from the unamplified DNA yielded sufficient read-count mapping rates. Variant calls were carried out using HaplotypeCaller in GATK version 4.2.6.1 (McKenna et al., 2010) to obtain the gVCF file for each individual. Further, individual gVCF files were combined with CombineGVCFs (GATK version 4.4.0.0), and final genotype sets were called from the whole cohort of preselected target regions (Supplementary Table S1) using GenotypeGVCFs with 3-bp expansion (command: ‘--interval-padding’) to obtain the cohort genotype VCF file. Cohort genotype VCF files were read with PLINK ver. 1.9 (Purcell et al., 2007) to generate data files named plink.{bed,fam,bim}. Sex data were manually recorded on the. fam file. The phenotype data were encoded as 01 (TD), 02 (ASD), G1 (sub-threshold autistic traits), and d1 (low birth weight) (Supplementary Table S2). Using these files, case-control association tests were performed with PLINK. Genotype associations with IQ scores and Social Responsiveness Scale (SRS) (see Supplementary Material for details) scores, as quantitative phenotypes, were analyzed with PLINK. Statistically significant associations at the variant sites were further investigated using the Integrative Genomics Viewer (Robinson et al., 2011), and the validity was confirmed.
2.4 Statistical analyses
Statistical analyses were performed using R version 4.3.0 (R Core Team, 2021). SRS and IQ were treated as continuous variables for the quantitative association analyses. False discovery rates (FDRs) were calculated with the p. adjust function in R with the “BH” method, as previously described (Benjamini and Hochberg, 1995).
3 Results
3.1 Genotyping
Target-enrichment sequencing following whole-genome amplification resulted in low-quality reads. Subsequent direct library construction and enrichment yielded high-quality reads in 79 of the 88 samples. Consequently, genomic DNA from 79 children (32 with ASD, 36 with TD, 3 with sub-threshold autistic traits, and eight children with low birth weight; 53 males and 26 females aged 5–8 years; Supplementary Table S2) were processed for further analyses (Supplementary Figure S1).
3.2 Participants’ characteristics
The mean age of the children was 5.6 and 5.4 years for ASD and TD, respectively (Table 2). The sex ratio was biased toward males in participants with ASD (27:5) but comparable in those with TD (20:16). The IQ scores of three children were not obtained. SRS T-scores and IQ scores of these 79 children are plotted in Supplementary Figure S2 with ASD phenotype diagnosis (Supplementary Table S3), showing positive and negative correlation of SRS T-scores and IQ scores with ASD diagnosis, as demonstrated through Welch’s two-sample t-test (Table 2).
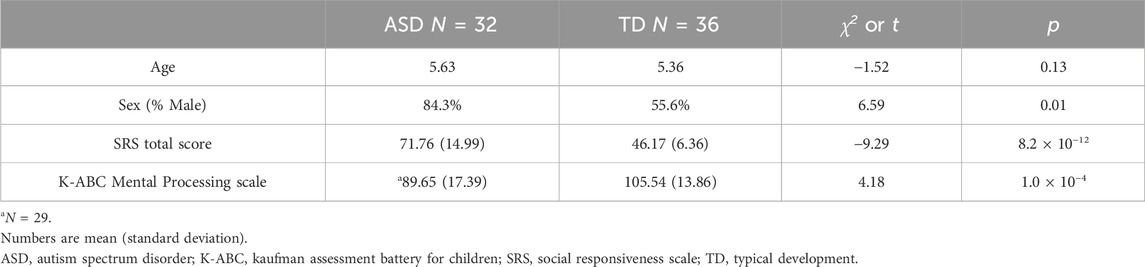
Table 2. Participants’ characteristics.
3.3 Genotype–phenotype association analyses
We detected 1,418 variant sites with an average call rate of 70% (Supplementary Data). In total, 748 variants were called at a genotype call rate of 95% (number of individuals = 79). Forty variants were associated with ASD/TD (p < 0.05, chi-square tests); low birth weight and sub-threshold autistic traits were not included in the analyses because the autistic phenotypes were not strictly defined. Thus, 68 children were included in the final analyses (Supplementary Figure S1). Among these, variants within the SCN1A gene, which encodes the pore-forming α-subunit of the Nav 1.1 voltage-gated sodium channel, exhibited the strongest association with ASD/TD (χ2 = 18.8–5.80, p =1.45 × 10-5-0.016) (Figure 1). In addition, ASD/TD was correlated with variants in SHANK3 (χ2 = 5.84–5.04, p = 0.016–0.025), DYRK1A (χ2 = 4.64, p = 0.03), CADPS (χ2 = 4.52, p = 0.03), and SCN2A (χ2 = 4.43–3.93, p = 0.04–0.05) genes. Compared with SCN2A (encodes the pore-forming α-subunit of the Nav 1.2 voltage-gated sodium channel) variants rs2304010, rs10930160, rs2304012, rs767942624, rs1252589686, and rs59934051 (Figure 1), the SCN1A variant rs79990586 exhibited lower p-values under dominant model (p = 0.01–0.03). Although 37.4 sites were expected to result in p < 0.05, if 748 sites are independent of ASD, then the associations of 10 variants (dbSNP: Chromosome: BP; rs28663047: 2: 166052559, rs11691603: 2: 166035836, rs10168027: 2: 166039309, rs10198801: 2: 166041507, rs67636132: 2: 166045389, rs11690962: 2: 166047552, rs1021999648: 2: 166049124, rs1187504368: 2: 166037655, rs1541783: 2: 166055076, and rs1019723: 2: 166039251) of SCN1A are calculated to have FDRs <0.05 (q = 2.8 × 10-3-0.02) and permutation test (p = 2.1 × 10-3-0.04), as summarized in Table 3 (also see Supplementary Table S4 for details). We found that 42 variants were associated with HFA (29 children with HFA) excluding individuals with ASD with IQ < 70/TD (p < 0.05, chi-square test), as summarized in Supplementary Table S5.
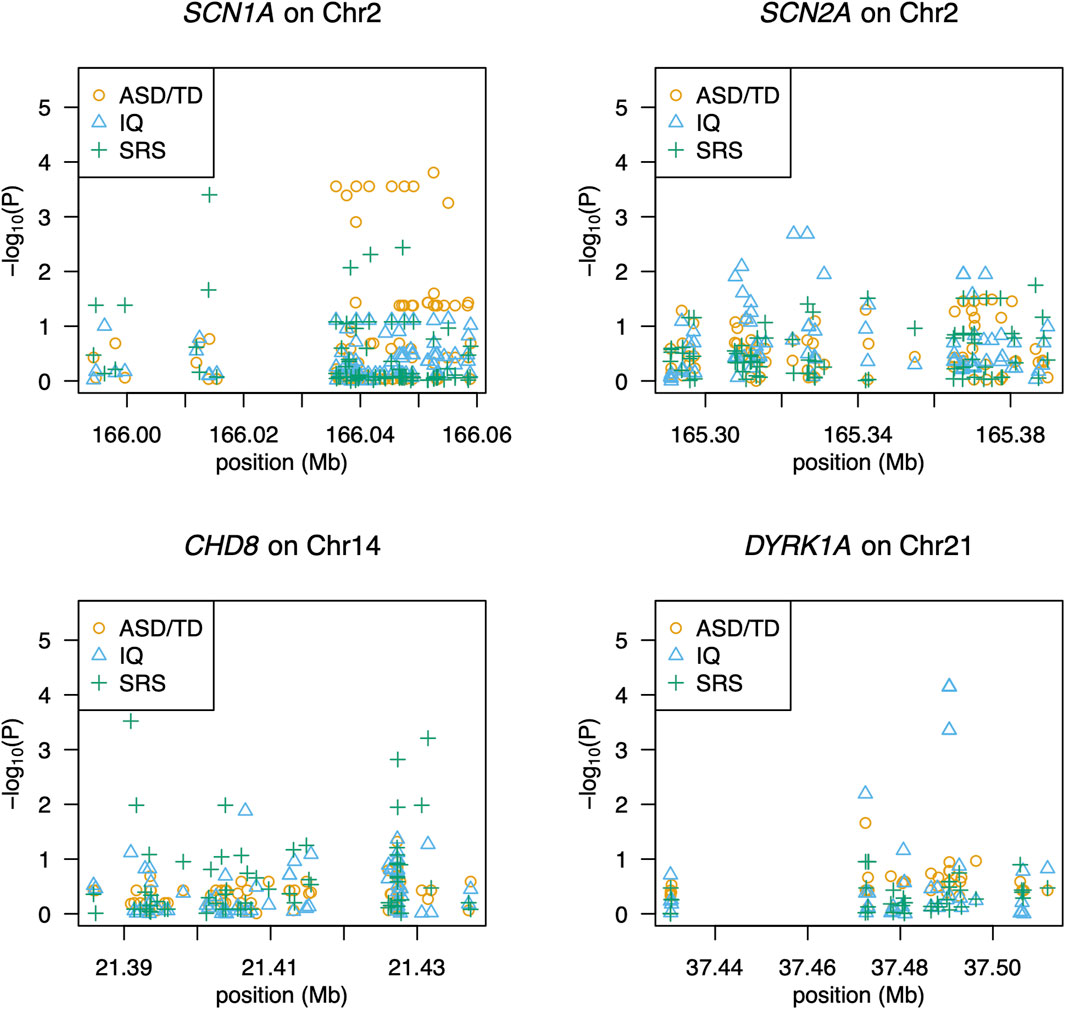
Figure 1. Association of SNPs with ASD, social responsiveness, or intelligence. The gene name and its chromosomal location are given above each panel. The x-axis shows the locations of SNPs, and the y-axis shows significance as -log10(P). Each symbol represents an individual. Circles (in orange), triangles (in blue), and pluses (in green) indicate association with ASD, IQ scores, and SRS-T scores, respectively.
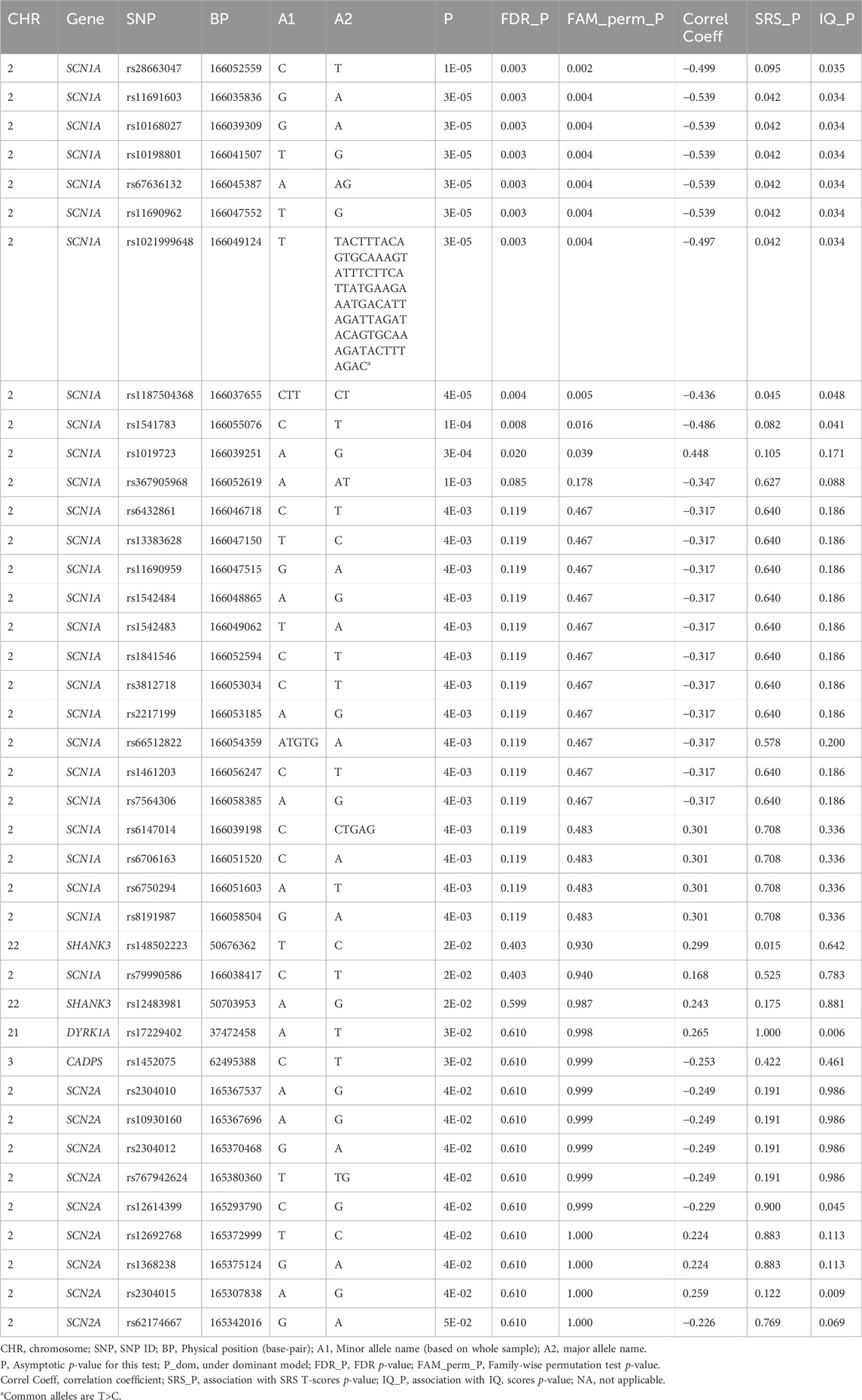
Table 3. Association analysis of genotype and ASD.
3.4 Association with SRS T-scores
Quantitative association analyses revealed 49 associated variants (p = 9.7 × 10-4-0.05; Supplementary Table S6). The strongest association (p = 9.7 × 10-4-0.02) was found in variants of the CHD8 gene, which encodes chromodomain helicase DNA-binding protein 8 (Figure 1). Additionally, rs148502223 (SHANK3; SH3 and multiple ankyrin repeat domains 3) was more closely associated with ASD than with TD (χ2 = 5.84, p = 0.02). Welch’s two-sample t-test revealed that individuals with the C/T genotype at rs148502223 had higher mean SRS T-scores than those with the C/C genotype (p = 0.01328). However, this association with rs148502223 and all detected variants was not significant after multiple-testing correction (FDRs >0.05).
3.5 Association with IQ scores
Using quantitative association analyses, we identified 50 variants (p = 3.0 × 10-5-0.05; Supplementary Table S7). The variants with the strongest association were DYRK1A (p = 3.0 × 10-5-6.3 × 10−3), which encodes dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A (Figure 1). rs17229402 of DYRK1A was associated with IQ scores (ASD vs. TD, p = 0.0313). Although the association of seven DYRK1A variants with IQ scores had FDR <0.05, (q = 4.4 × 10-3-0.02) (Supplementary Table S6), we did not detect reads through BAM file inspection, to support the presence of the variants.
3.6 Variants showing association with multiple phenotypes
Associations with both ASD/TD and SRS T-scores were observed in seven SCN1A variants (SNPs: rs11691603, rs10168027, rs10198801, rs11690962, and rs1187504368; indel: rs67636132 and rs1021999648) and one SHANK3 SNP (rs148502223) (Table 3). Nine SCN1A variants (SNPs: rs28663047, rs11691603, rs10168027, rs10198801, rs67636132, rs11690962, rs1187504368, and rs1541783; indel: rs1021999648), one DYRK1A SNP (rs17229402), and two SCN2A SNPs (rs12614399 and rs2304015) showed association with both ASD/TD and IQ scores (Table 3). One CHD8 SNP (rs10467770), six SCN1A SNPs (rs11691603, rs10168027, rs10198801, rs11690962, and rs1187504368; indel: rs1021999648) and one unconfirmed SCN2A (BP: 166045387 on Chromosome 2) showed association with both SRS T-scores and IQ scores (Table 3).
4 Discussion
In our cohort, mainly containing children with HFA, we identified 40 variants within five genes that are associated with ASD. In addition, we observed an association of 49 and 50 variants including CHD8 and DYRK1A with SRS T-scores and IQ scores, respectively. With the exception of seven unregistered DYRK1A variants and one unregistered SCN2A variant, all other identified variants were reported in dbSNP (Table 3; Supplementary Tables S5, S6). In particular, 10 variants of SCN1A passed filtering of the permutation test and the FDR was below 0.05, as determined through BH correction. These data provide some insights into the genetic architecture of HFA.
Of the variants found to be associated with the phenotypes, none were present in exons; all were found in introns and predicted to be benign variations that are not expected to cause medically important phenotypes. Given the nature of association studies, variants may constitute a linked haplotype group that results in improper expression levels in specific cell types. Confirmation of the expression levels is not possible in humans and is beyond the scope of the current report. Notably, HFA may be caused by a small effect size rather than a large effect size of the major ASD genes; that is, the genes are highly functional because gene function is only subtly impaired. The cumulative impact of hundreds of risk alleles from common variations (both SNPs and CNVs) has been observed to lower penetrance effects in HFA (Toma, 2020). A large genome-wide association study reported that the polygenic contribution of common SNPs may be more prominent in patients with HFA than in patients with both ASD and ID (Grove et al., 2019). Thus, the accumulation of genetic SNPs with small effect sizes may contribute to HFA. All ASD-related variations were already in dbVAR and are known polymorphisms with little impact on fitness. Given that the subtle effects of intronic variation may be linked to the phenotype, it may be worth designing probes on introns and flanking regions in addition to those in the exons of the candidate target genes.
However, in our cohort, variants in SCN2A were associated with ASD, and those on SCN1A were most strongly associated with the ASD phenotype. Among them, SNP rs3812718 is a known risk factor for epilepsy (Tang et al., 2014; Wang et al., 2018; Zhi et al., 2018). SCN1A and SCN2A, both of which are located on chromosome 2, encode voltage-gated sodium channel α-subunits Nav1.1 and Nav1.2, respectively. Both proteins play important roles in the initiation and propagation of action potentials, thereby modulating the neural network activity (Barbieri et al., 2023). Missense mutations in these genes have been identified in inherited seizure disorders, including generalized epilepsy with febrile seizures plus (GEFS+) (Meisler et al., 2001), as well as in ASD (O'Roak et al., 2011; Reynolds et al., 2020). Loss-of-function mutations in SCN1A have been observed in most cases of Dravet syndrome (DS), manifesting as epileptic seizures, hyperactivity, autistic traits, and cognitive decline. Loss of function of the Nav1.1 channel from familial febrile seizures to GEFS+ and finally DS results in a progressive and severe DS phenotype (Catterall, 2014; Ishii et al., 2017). In our study, no individuals had a clinical history of epilepsy (unpublished data), and all variants were detected in introns. A possible explanation is that these intronic variants, which do not destroy the channel structure, may contribute only to autistic traits in HFA but not to ASD with epileptic seizures.
One key aspect of the heterogeneity of ASD may result from diverse IQ (Wolff et al., 2022). In clinical reports of ASD cases, more individuals with ASD had above-average IQ and fewer had below-average IQ, with the latter including -ID (Mayes and Calhoun, 2003; Rommelse et al., 2015). Autistic traits can be found and examined in any type of psychiatric or neurodevelopmental disorder, as well as in individuals without a disorder (Mottron and Bzdok, 2020). This could account for the increase in reports of individuals with ASD with an average or above-average IQ (Wolff et al., 2022).
The cumulative effect of diverse common risk alleles from common variants is thought to cause HFA (Toma, 2020). For example, 22 rare SNPs were documented in delayed speech development in Spanish children with HFA (Alvarez-Mora et al., 2016). Nakata et al. (2019) found that the downregulation of miR-6126 in HFA was correlated with the severity of social deficits. More recently, Wang et al. (2023) noted that ASD-associated genes were responsible for impairments in social communication but not in cognitive functions. Individuals with HFA carried fewer disruptive de novo variants than those with both ASD and ID (Wang et al., 2023). These observations imply that HFA-related genes are distinct from low-functioning autism-related genes. In addition, nine new ASD-associated genes were revealed, including SLC35G1, in which they identified recurrent mutations in HFA probands (Wang et al., 2023). In the future, an HFA group should be compared carefully with an ASD + ID group to identify HFA-specific factors.
We also found that CHD8 variants were strongly associated with SRS T-scores. The protein product of CHD8, one of the major ASD-associated genes, functions as a chromatin-remodeling factor that regulates the expression of many genes including those for β-catenin and several components of the p53 pathway (Krumm et al., 2014; Katayama et al., 2016). A large cohort of a two-stage analysis of rare de novo and inherited coding SNPs identified CHD8 as a gene with exome-wide significance (p < 2.5 × 10−6) (Zhou et al., 2022). Furthermore, CHD8 regulates the expression of ASD-associated genes related to synaptic function and neurodevelopment during neurogenesis (Sugathan et al., 2014; Cotney et al., 2015; Paulsen et al., 2022). DYRK1A variants were strongly associated with IQ scores. DYRK1A encodes a dual-specificity tyrosine phosphorylation-regulated kinase 1A that contributes to neural development by phosphorylating various substrates including transcription, splicing, and synaptic proteins (Park et al., 2009). DYRK1A has also been implicated in the development of intelligence (Earl et al., 2017; Neumann et al., 2018; Trost et al., 2022); however, the site variations discovered in the present analysis have not been confirmed through alignment.
Both SRS T-score and IQ are important criteria for the classification of ASD. However, it remains unclear whether the same genes are associated with both of them. Previous studies have reported a relationship between IQ and autistic traits. Bölte et al. (2008) reported a low correlation between SRS and IQ. In contrast, Marinopoulou et al. (2023) demonstrated that intellectual functioning was negatively associated with SRS in children with ASD. Presumably, these SNPs detected in this study may independently affect both social responsiveness and intelligence through complex interactions.
This study has a few limitations. First, the sample size was small. To detect the small-effect size genes by high-multiplicity comparison, a sample size of a few thousand would be necessary. Second, variants were determined as substitutions or small indels, and such SNPs were not phased; thus, the haplotype of the entire gene structure was not determined. Haplotypes can be reconstructed using long-read sequencing or family genetic analysis. Finally, a targeted panel was constructed by the combination of highly-confident ASD-associated genes known at the beginning of this study. Optimizing the NGS panel by incorporating new variants and/or by removing inappropriate ones would enable the precise elucidation of genetic architectures of different subtypes of ASD.
In summary, our results indicate the presence of core HFA-associated genes in the Japanese population. Future studies, combined with a trio analysis of children and their parents at a larger scale, would define in detail the genetic architecture of HFA.
Data availability statement
The data presented in the study are deposited in the NBDC repository, accession number JGAS000731.
Ethics statement
This study was approved by the Kanazawa University Medical Ethics Committee , and were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the minors’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
YS: Writing–original draft, Methodology, Funding acquisition, Formal Analysis, Data curation, Conceptualization. TN: Writing–review and editing, Formal Analysis, Data curation. SY: Writing–review and editing, Supervision, Methodology. YY: Writing–review and editing, Resources, Investigation. CH: Writing–review and editing, Resources, Investigation. ST: Writing–review and editing, Resources, Investigation. SI: Writing–review and editing, Resources, Investigation. MK: Writing–review and editing, Supervision, Funding acquisition.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by Grant-in-Aid for JSPS Fellows (grant Number: 22J14602) and Kawano Masanori Memorial Public Interest Incorporated Foundation for Promotion of Pediatrics. Furthermore, the work was partially supported by the Moonshot Research and Development Program (grant number: JPMJMS2297) from Japan Science and Technology Agency, JST, and the Collaborative Research Program of the Collaborative Research Network for Asian Children with Developmental Disorders: MEXT Policy Initiative FY 2021, under joint research conducted through the initiative.
Acknowledgments
We are grateful to all study participants and our colleagues at the Bambi Plan for their support. We thank Mari Okuno, Yoko Morita, and Nao Oura for their secretarial assistance. Illumina library construction, enrichment, and sequencing through initial genotype calling were performed at Kubix (kubix.co.jp). We thank Editage (www.editage.com) for English language editing.
Conflict of interest
The authors declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1352480/full#supplementary-material
References
Alarcón, M., Abrahams, B. S., Stone, J. L., Duvall, J. A., Perederiy, J. V., Bomar, J. M., et al. (2008). Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 82 (1), 150–159. doi:10.1016/j.ajhg.2007.09.005
Alvares, G. A., Bebbington, K., Cleary, D., Evans, K., Glasson, E. J., Maybery, M. T., et al. (2020). The misnomer of 'high functioning autism': intelligence is an imprecise predictor of functional abilities at diagnosis. Autism 24, 221–232. doi:10.1177/1362361319852831
Alvarez-Mora, M. I., Calvo Escalona, R., Puig Navarro, O., Madrigal, I., Quintela, I., Amigo, J., et al. (2016). Comprehensive molecular testing in patients with high functioning autism spectrum disorder. Mutat. Res. 784-785, 46–52. doi:10.1016/j.mrfmmm.2015.12.006
American Psychiatric Association (2000). Diagnostic and statistical manual of mental disorders: dsm-IV-tr. Washington, DC: American Psychiatric Association.
Arking, D. E., Cutler, D. J., Brune, C. W., Teslovich, T. M., West, K., Ikeda, M., et al. (2008). A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet. 82 (1), 160–164. doi:10.1016/j.ajhg.2007.09.015
Bai, D., Yip, B. H. K., Windham, G. C., Sourander, A., Francis, R., Yoffe, R., et al. (2019). Association of genetic and environmental factors with autism in a 5-country cohort. JAMA Psychiatry 76, 1035–1043. doi:10.1001/jamapsychiatry.2019.1411
Baranova, A., Wang, J., Cao, H., Chen, J. H., Chen, J., Chen, M., et al. (2022). Shared genetics between autism spectrum disorder and attention-deficit/hyperactivity disorder and their association with extraversion. Psychiatry Res. 314, 114679. doi:10.1016/j.psychres.2022.114679
Barbieri, R., Nizzari, M., Zanardi, I., Pusch, M., and Gavazzo, P. (2023). Voltage-gated sodium channel dysfunctions in neurological disorders. Life (Basel) 13, 1191. doi:10.3390/life13051191
Baron-Cohen, S. (2017). Editorial perspective: neurodiversity - a revolutionary concept for autism and psychiatry. J. Child. Psychol. Psychiatry 58, 744–747. doi:10.1111/jcpp.12703
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300. doi:10.1111/j.2517-6161.1995.tb02031.x
Ben-Shalom, R., Keeshen, C. M., Berrios, K. N., An, J. Y., Sanders, S. J., and Bender, K. J. (2017). Opposing effects on Na(V)1.2 function underlie differences between SCN2A variants observed in individuals with autism spectrum disorder or infantile seizures. Biol. Psychiatry 82 (3), 224–232. doi:10.1016/j.biopsych.2017.01.009
Bernier, R., Golzio, C., Xiong, B., Stessman, H. A., Coe, B. P., Penn, O., et al. (2014). Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158 (2), 263–276. doi:10.1016/j.cell.2014.06.017
Berryer, M. H., Hamdan, F. F., Klitten, L. L., Møller, R. S., Carmant, L., Schwartzentruber, J., et al. (2013). Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum. Mutat. 34 (2), 385–394. doi:10.1002/humu.22248
Bölte, S., Poustka, F., and Constantino, J. N. (2008). Assessing autistic traits: cross-cultural validation of the social responsiveness scale (SRS). Autism Res. 1, 354–363. doi:10.1002/aur.49
Brune, C. W., Kim, S. J., Salt, J., Leventhal, B. L., Lord, C., and Cook, E. H. (2006). 5-HTTLPR genotype-specific phenotype in children and adolescents with autism. Am. J. Psychiatry 163 (12), 2148–2156. doi:10.1176/ajp.2006.163.12.2148
Carvill, G. L., McMahon, J. M., Schneider, A., Zemel, M., Myers, C. T., Saykally, J., et al. (2015). Mutations in the GABA transporter SLC6A1 cause epilepsy with myoclonic-atonic seizures. Am. J. Hum. Genet. 96 (5), 808–815. doi:10.1016/j.ajhg.2015.02.016
Catterall, W. A. (2014). Sodium channels, inherited epilepsy, and antiepileptic drugs. Annu. Rev. Pharmacol. Toxicol. 54, 317–338. doi:10.1146/annurev-pharmtox-011112-140232
Clement, J. P., Aceti, M., Creson, T. K., Ozkan, E. D., Shi, Y., Reish, N. J., et al. (2012). Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151 (4), 709–723. doi:10.1016/j.cell.2012.08.045
Cotney, J., Muhle, R. A., Sanders, S. J., Liu, L., Willsey, A. J., Niu, W., et al. (2015). The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat. Commun. 6, 6404. doi:10.1038/ncomms7404
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515 (7526), 209–215. doi:10.1038/nature13772
Devlin, B., Cook, E. H., Coon, H., Dawson, G., Grigorenko, E. L., McMahon, W., et al. (2005). Autism and the serotonin transporter: the long and short of it. Mol. Psychiatry 10 (12), 1110–1116. doi:10.1038/sj.mp.4001724
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39 (1), 25–27. doi:10.1038/ng1933
Earl, R. K., Turner, T. N., Mefford, H. C., Hudac, C. M., Gerdts, J., Eichler, E. E., et al. (2017). Clinical phenotype of ASD-associated DYRK1A haploinsufficiency. Mol. Autism 8, 54. doi:10.1186/s13229-017-0173-5
Evers, J. M., Laskowski, R. A., Bertolli, M., Clayton-Smith, J., Deshpande, C., Eason, J., et al. (2017). Structural analysis of pathogenic mutations in the DYRK1A gene in patients with developmental disorders. Hum. Mol. Genet. 26 (3), 519–526. doi:10.1093/hmg/ddw409
Feliciano, P., Zhou, X., Astrovskaya, I., Turner, T. N., Wang, T., Brueggeman, L., et al. (2019). Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genom Med. 4, 19. doi:10.1038/s41525-019-0093-8
Fóthi, Á., Pintér, C., Pollner, P., and Lőrincz, A. (2022). Peripheral gene interactions define interpretable clusters of core ASD genes in a network-based investigation of the omnigenic theory. NPJ Syst. Biol. Appl. 8, 28. doi:10.1038/s41540-022-00240-x
Frazier, T. W., Embacher, R., Tilot, A. K., Koenig, K., Mester, J., and Eng, C. (2015). Molecular and phenotypic abnormalities in individuals with germline heterozygous PTEN mutations and autism. Mol. Psychiatry 20 (9), 1132–1138. doi:10.1038/mp.2014.125
Gauthier, J., Champagne, N., Lafrenière, R. G., Xiong, L., Spiegelman, D., Brustein, E., et al. (2010). De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. U. S. A. 107 (17), 7863–7868. doi:10.1073/pnas.0906232107
Gialluisi, A., Andlauer, T. F. M., Mirza-Schreiber, N., Moll, K., Becker, J., Hoffmann, P., et al. (2021). Genome-wide association study reveals new insights into the heritability and genetic correlates of developmental dyslexia. Mol. Psychiatry 26 (7), 3004–3017. doi:10.1038/s41380-020-00898-x
Gilling, M., Rasmussen, H. B., Calloe, K., Sequeira, A. F., Baretto, M., Oliveira, G., et al. (2013). Dysfunction of the heteromeric KV7.3/KV7.5 potassium channel is associated with autism spectrum disorders. Front. Genet. 4, 54. doi:10.3389/fgene.2013.00054
Goffin, A., Hoefsloot, L. H., Bosgoed, E., Swillen, A., and Fryns, J. P. (2001). PTEN mutation in a family with Cowden syndrome and autism. Am. J. Med. Genet. 105 (6), 521–524. doi:10.1002/ajmg.1477
Goldin, A. L., Snutch, T., Lübbert, H., Dowsett, A., Marshall, J., Auld, V., et al. (1986). Messenger RNA coding for only the alpha subunit of the rat brain Na channel is sufficient for expression of functional channels in Xenopus oocytes. Proc. Natl. Acad. Sci. U. S. A. 83 (19), 7503–7507. doi:10.1073/pnas.83.19.7503
Gozes, I. (2007). Activity-dependent neuroprotective protein: from gene to drug candidate. Pharmacol. Ther. 114 (2), 146–154. doi:10.1016/j.pharmthera.2007.01.004
Grove, J., Ripke, S., Als, T. D., Mattheisen, M., Walters, R. K., Won, H., et al. (2019). Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444. doi:10.1038/s41588-019-0344-8
Halgren, C., Kjaergaard, S., Bak, M., Hansen, C., El-Schich, Z., Anderson, C. M., et al. (2012). Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin. Genet. 82 (3), 248–255. doi:10.1111/j.1399-0004.2011.01755.x
Hamdan, F. F., Daoud, H., Piton, A., Gauthier, J., Dobrzeniecka, S., Krebs, M. O., et al. (2011). De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol. Psychiatry 69 (9), 898–901. doi:10.1016/j.biopsych.2010.11.015
Helsmoortel, C., Vulto-van Silfhout, A. T., Coe, B. P., Vandeweyer, G., Rooms, L., van den Ende, J., et al. (2014). A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet. 46 (4), 380–384. doi:10.1038/ng.2899
Hirota, T., and King, B. H. (2023). Autism spectrum disorder: a review. JAMA 329, 157–168. doi:10.1001/jama.2022.23661
Hirunsatit, R., George, E. D., Lipska, B. K., Elwafi, H. M., Sander, L., Yrigollen, C. M., et al. (2009). Twenty-one-base-pair insertion polymorphism creates an enhancer element and potentiates SLC6A1 GABA transporter promoter activity. Pharmacogenet Genomics 19 (1), 53–65. doi:10.1097/FPC.0b013e328318b21a
Hurlstone, A. F., Olave, I. A., Barker, N., van Noort, M., and Clevers, H. (2002). Cloning and characterization of hELD/OSA1, a novel BRG1 interacting protein. Biochem. J. 364 (1), 255–264. doi:10.1042/bj3640255
Iossifov, I., Levy, D., Allen, J., Ye, K., Ronemus, M., Lee, Y. H., et al. (2015). Low load for disruptive mutations in autism genes and their biased transmission. Proc. Natl. Acad. Sci. U. S. A. 112 (41), E5600–E5607. doi:10.1073/pnas.1516376112
Iossifov, I., O'Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515 (7526), 216–221. doi:10.1038/nature13908
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74 (2), 285–299. doi:10.1016/j.neuron.2012.04.009
Ishii, A., Watkins, J. C., Chen, D., Hirose, S., and Hammer, M. F. (2017). Clinical implications of SCN1A missense and truncation variants in a large Japanese cohort with Dravet syndrome. Epilepsia 58, 282–290. doi:10.1111/epi.13639
Isom, L. L. (2002). The role of sodium channels in cell adhesion. Front. Biosci. 7, 12–23. doi:10.2741/isom
Jiang, Y. H., Yuen, R. K., Jin, X., Wang, M., Chen, N., Wu, X., et al. (2013). Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet. 93 (2), 249–263. doi:10.1016/j.ajhg.2013.06.012
Johannesen, K. M., Gardella, E., Linnankivi, T., Courage, C., de Saint Martin, A., Lehesjoki, A. E., et al. (2018). Defining the phenotypic spectrum of SLC6A1 mutations. Epilepsia 59 (2), 389–402. doi:10.1111/epi.13986
Katayama, Y., Nishiyama, M., Shoji, H., Ohkawa, Y., Kawamura, A., Sato, T., et al. (2016). CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 537, 675–679. doi:10.1038/nature19357
Kaufman, A. S. (1983). Kaufman assessment Battery for children (K- ABC) administration and scoring manual. Circle Pines: American Guidance Service.
Kimura, H., Nakatochi, M., Aleksic, B., Guevara, J., Toyama, M., Hayashi, Y., et al. (2022). Exome sequencing analysis of Japanese autism spectrum disorder case-control sample supports an increased burden of synaptic function-related genes. Transl. Psychiatry 12, 265. doi:10.1038/s41398-022-02033-6
Krumm, N., O'Roak, B. J., Shendure, J., and Eichler, E. E. (2014). A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 37, 95–105. doi:10.1016/j.tins.2013.11.005
Leblond, C. S., Nava, C., Polge, A., Gauthier, J., Huguet, G., Lumbroso, S., et al. (2014). Meta-analysis of SHANK mutations in autism spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genet. 10 (9), e1004580. doi:10.1371/journal.pgen.1004580
Li, X., Hu, Z., He, Y., Xiong, Z., Long, Z., Peng, Y., et al. (2010). Association analysis of CNTNAP2 polymorphisms with autism in the Chinese Han population. Psychiatr. Genet. 20 (3), 113–117. doi:10.1097/YPG.0b013e32833a216f
Lord, C., Elsabbagh, M., Baird, G., and Veenstra-Vanderweele, J. (2018). Autism spectrum disorder. Lancet 392, 508–520. doi:10.1016/s0140-6736(18)31129-2
Lord, C., Risi, S., Lambrecht, L., Cook, E. H., Leventhal, B. L., DiLavore, P. C., et al. (2000). The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J. Autism Dev. Disord. 30, 205–223. doi:10.1023/A:1005592401947
Lord C, R. M., DiLavore, P. C., Risi, S., Gotham, K., and Bishop, S. (2012). Autism diagnostic observation Schedule. Torrence: Western Psychological Services.
Mandel, S., and Gozes, I. (2007). Activity-dependent neuroprotective protein constitutes a novel element in the SWI/SNF chromatin remodeling complex. J. Biol. Chem. 282 (47), 34448–34456. doi:10.1074/jbc.M704756200
Marinopoulou, M., Billstedt, E., Wessman, C., Bornehag, C. G., and Hallerbäck, M. U. (2023). Association between intellectual functioning and autistic traits in the general population of children. Child. Psychiatry Hum. Dev. 1-12. doi:10.1007/s10578-023-01562-5
Masi, A., DeMayo, M. M., Glozier, N., and Guastella, A. J. (2017). An overview of autism spectrum disorder, heterogeneity and treatment options. Neurosci. Bull. 33, 183–193. doi:10.1007/s12264-017-0100-y
Matta, J. A., Ashby, M. C., Sanz-Clemente, A., Roche, K. W., and Isaac, J. T. (2011). mGluR5 and NMDA receptors drive the experience- and activity-dependent NMDA receptor NR2B to NR2A subunit switch. Neuron 70 (2), 339–351. doi:10.1016/j.neuron.2011.02.045
Mayes, S. D., and Calhoun, S. L. (2003). Analysis of WISC-III, Stanford-Binet:IV, and academic achievement test scores in children with autism. J. Autism. Dev. Disord. 33, 329–341. doi:10.1023/a:1024462719081
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi:10.1101/gr.107524.110
Meerschaut, I., Rochefort, D., Revençu, N., Pètre, J., Corsello, C., Rouleau, G. A., et al. (2017). FOXP1-related intellectual disability syndrome: a recognisable entity. J. Med. Genet. 54 (9), 613–623. doi:10.1136/jmedgenet-2017-104579
Meisler, M. H., Kearney, J., Ottman, R., and Escayg, A. (2001). Identification of epilepsy genes in human and mouse. Annu. Rev. Genet. 35, 567–588. doi:10.1146/annurev.genet.35.102401.091142
Mellone, S., Puricelli, C., Vurchio, D., Ronzani, S., Favini, S., Maruzzi, A., et al. (2022). The usefulness of a targeted next generation sequencing gene panel in providing molecular diagnosis to patients with a broad spectrum of neurodevelopmental disorders. Front. Genet. 13, 875182. doi:10.3389/fgene.2022.875182
Mermer, F., Poliquin, S., Rigsby, K., Rastogi, A., Shen, W., Romero-Morales, A., et al. (2021). Common molecular mechanisms of SLC6A1 variant-mediated neurodevelopmental disorders in astrocytes and neurons. Brain 144 (8), 2499–2512. doi:10.1093/brain/awab207
Mottron, L., and Bzdok, D. (2020). Autism spectrum heterogeneity: fact or artifact? Mol. Psychiatry 25, 3178–3185. doi:10.1038/s41380-020-0748-y
Murtaza, N., Cheng, A. A., Brown, C. O., Meka, D. P., Hong, S., Uy, J. A., et al. (2022). Neuron-specific protein network mapping of autism risk genes identifies shared biological mechanisms and disease-relevant pathologies. Cell Rep. 41, 111678. doi:10.1016/j.celrep.2022.111678
Myers, R. A., Casals, F., Gauthier, J., Hamdan, F. F., Keebler, J., Boyko, A. R., et al. (2011). A population genetic approach to mapping neurological disorder genes using deep resequencing. PLoS Genet. 7 (2), e1001318. doi:10.1371/journal.pgen.1001318
Nakata, M., Kimura, R., Funabiki, Y., Awaya, T., Murai, T., and Hagiwara, M. (2019). MicroRNA profiling in adults with high-functioning autism spectrum disorder. Mol. Brain 12, 82. doi:10.1186/s13041-019-0508-6
Neale, B. M., Kou, Y., Liu, L., Ma'ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485 (7397), 242–245. doi:10.1038/nature11011
Neumann, F., Gourdain, S., Albac, C., Dekker, A. D., Bui, L. C., Dairou, J., et al. (2018). DYRK1A inhibition and cognitive rescue in a Down syndrome mouse model are induced by new fluoro-DANDY derivatives. Sci. Rep. 8, 2859. doi:10.1038/s41598-018-20984-z
Nie, Z., Yan, Z., Chen, E. H., Sechi, S., Ling, C., Zhou, S., et al. (2003). Novel SWI/SNF chromatin-remodeling complexes contain a mixed-lineage leukemia chromosomal translocation partner. Mol. Cell Biol. 23 (8), 2942–2952. doi:10.1128/mcb.23.8.2942-2952.2003
Nord, A. S., Roeb, W., Dickel, D. E., Walsh, T., Kusenda, M., O'Connor, K. L., et al. (2011). Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur. J. Hum. Genet. 19 (6), 727–731. doi:10.1038/ejhg.2011.24
Nozawa, R. S., Nagao, K., Masuda, H. T., Iwasaki, O., Hirota, T., Nozaki, N., et al. (2010). Human POGZ modulates dissociation of HP1alpha from mitotic chromosome arms through Aurora B activation. Nat. Cell Biol. 12 (7), 719–727. doi:10.1038/ncb2075
O'Roak, B. J., Deriziotis, P., Lee, C., Vives, L., Schwartz, J. J., Girirajan, S., et al. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589. doi:10.1038/ng.835
O'Roak, B. J., Vives, L., Fu, W., Egertson, J. D., Stanaway, I. B., Phelps, I. G., et al. (2012a). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338 (6114), 1619–1622. doi:10.1126/science.1227764
O'Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012b). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485 (7397), 246–250. doi:10.1038/nature10989
Ouss, L., Leunen, D., Laschet, J., Chemaly, N., Barcia, G., Losito, E. M., et al. (2019). Autism spectrum disorder and cognitive profile in children with Dravet syndrome: delineation of a specific phenotype. Epilepsia Open 4 (1), 40–53. doi:10.1002/epi4.12281
Park, J., Song, W. J., and Chung, K. C. (2009). Function and regulation of Dyrk1A: towards understanding Down syndrome. Cell Mol. Life Sci. 66, 3235–3240. doi:10.1007/s00018-009-0123-2
Paulsen, B., Velasco, S., Kedaigle, A. J., Pigoni, M., Quadrato, G., Deo, A. J., et al. (2022). Autism genes converge on asynchronous development of shared neuron classes. Nature 602, 268–273. doi:10.1038/s41586-021-04358-6
Peyrot, W. J., and Price, A. L. (2021). Identifying loci with different allele frequencies among cases of eight psychiatric disorders using CC-GWAS. Nat. Genet. 53 (4), 445–454. doi:10.1038/s41588-021-00787-1
Pezzolesi, M. G., Zbuk, K. M., Waite, K. A., and Eng, C. (2007). Comparative genomic and functional analyses reveal a novel cis-acting PTEN regulatory element as a highly conserved functional E-box motif deleted in Cowden syndrome. Hum. Mol. Genet. 16 (9), 1058–1071. doi:10.1093/hmg/ddm053
Platzer, K., Yuan, H., Schütz, H., Winschel, A., Chen, W., Hu, C., et al. (2017). GRIN2B encephalopathy: novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J. Med. Genet. 54 (7), 460–470. doi:10.1136/jmedgenet-2016-104509
Poot, M. (2014). A candidate gene association study further corroborates involvement of contactin genes in autism. Mol. Syndromol. 5 (5), 229–235. doi:10.1159/000362891
Price, K. M., Wigg, K. G., Feng, Y., Blokland, K., Wilkinson, M., He, G., et al. (2020). Genome-wide association study of word reading: overlap with risk genes for neurodevelopmental disorders. Genes Brain Behav. 19 (6), e12648. doi:10.1111/gbb.12648
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi:10.1086/519795
Qiu, S., Qiu, Y., Li, Y., and Cong, X. (2022). Genetics of autism spectrum disorder: an umbrella review of systematic reviews and meta-analyses. Transl. Psychiatry 12, 249. doi:10.1038/s41398-022-02009-6
R Core Team (2021). R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. Available: https://www.R-project.org/.
Reynolds, C., King, M. D., and Gorman, K. M. (2020). The phenotypic spectrum of SCN2A-related epilepsy. Eur. J. Paediatr. Neurol. 24, 117–122. doi:10.1016/j.ejpn.2019.12.016
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi:10.1038/nbt.1754
Rommelse, N., Langerak, I., van der Meer, J., de Bruijn, Y., Staal, W., Oerlemans, A., et al. (2015). Intelligence may moderate the cognitive profile of patients with ASD. PLoS One 10, e0138698. doi:10.1371/journal.pone.0138698
Sanders, S. J., He, X., Willsey, A. J., Ercan-Sencicek, A. G., Samocha, K. E., Cicek, A. E., et al. (2015). Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87 (6), 1215–1233. doi:10.1016/j.neuron.2015.09.016
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J., et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485 (7397), 237–241. doi:10.1038/nature10945
Satterstrom, F. K., Kosmicki, J. A., Wang, J., Breen, M. S., De Rubeis, S., An, J. Y., et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23. doi:10.1016/j.cell.2019.12.036
Schotta, G., Lachner, M., Sarma, K., Ebert, A., Sengupta, R., Reuter, G., et al. (2004). A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 18 (11), 1251–1262. doi:10.1101/gad.300704
Sebat, J., Lakshmi, B., Malhotra, D., Troge, J., Lese-Martin, C., Walsh, T., et al. (2007). Strong association of de novo copy number mutations with autism. Science 316, 445–449. doi:10.1126/science.1138659
Shi, C., Sakuma, M., Mooroka, T., Liscoe, A., Gao, H., Croce, K. J., et al. (2008). Down-regulation of the forkhead transcription factor Foxp1 is required for monocyte differentiation and macrophage function. Blood 112 (12), 4699–4711. doi:10.1182/blood-2008-01-137018
Shiota, Y., Hirosawa, T., Yoshimura, Y., Tanaka, S., Hasegawa, C., Iwasaki, S., et al. (2021). A common variant of CNTNAP2 is associated with sub-threshold autistic traits and intellectual disability. PLoS One 16, e0260548. doi:10.1371/journal.pone.0260548
Shiota, Y., Hirosawa, T., Yoshimura, Y., Tanaka, S., Hasegawa, C., Iwasaki, S., et al. (2022). Effect of CNTNAP2 polymorphism on receptive language in children with autism spectrum disorder without language developmental delay. Neuropsychopharmacol. Rep. 42 (3), 352–355. doi:10.1002/npr2.12267
Stessman, H. A. F., Willemsen, M. H., Fenckova, M., Penn, O., Hoischen, A., Xiong, B., et al. (2016). Disruption of POGZ is associated with intellectual disability and autism spectrum disorders. Am. J. Hum. Genet. 98 (3), 541–552. doi:10.1016/j.ajhg.2016.02.004
Sugathan, A., Biagioli, M., Golzio, C., Erdin, S., Blumenthal, I., Manavalan, P., et al. (2014). CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. U. S. A. 111, E4468–E4477. doi:10.1073/pnas.1405266111
Talkowski, M. E., Rosenfeld, J. A., Blumenthal, I., Pillalamarri, V., Chiang, C., Heilbut, A., et al. (2012). Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149 (3), 525–537. doi:10.1016/j.cell.2012.03.028
Tang, L., Lu, X., Tao, Y., Zheng, J., Zhao, P., Li, K., et al. (2014). SCN1A rs3812718 polymorphism and susceptibility to epilepsy with febrile seizures: a meta-analysis. Gene 533, 26–31. doi:10.1016/j.gene.2013.09.071
Tavassoli, T., Kolevzon, A., Wang, A. T., Curchack-Lichtin, J., Halpern, D., Schwartz, L., et al. (2014). De novo SCN2A splice site mutation in a boy with Autism spectrum disorder. BMC Med. Genet. 15, 35. doi:10.1186/1471-2350-15-35
Thompson, B. A., Tremblay, V., Lin, G., and Bochar, D. A. (2008). CHD8 is an ATP-dependent chromatin remodeling factor that regulates beta-catenin target genes. Mol. Cell Biol. 28 (12), 3894–3904. doi:10.1128/mcb.00322-08
Timpson, N. J., Greenwood, C. M. T., Soranzo, N., Lawson, D. J., and Richards, J. B. (2018). Genetic architecture: the shape of the genetic contribution to human traits and disease. Nat. Rev. Genet. 19, 110–124. doi:10.1038/nrg.2017.101
Toma, C. (2020). Genetic variation across phenotypic severity of autism. Trends Genet. 36, 228–231. doi:10.1016/j.tig.2020.01.005
Toma, C., Torrico, B., Hervás, A., Valdés-Mas, R., Tristán-Noguero, A., Padillo, V., et al. (2014). Exome sequencing in multiplex autism families suggests a major role for heterozygous truncating mutations. Mol. Psychiatry 19 (7), 784–790. doi:10.1038/mp.2013.106
Trelles, M. P., Levy, T., Lerman, B., Siper, P., Lozano, R., Halpern, D., et al. (2021). Individuals with FOXP1 syndrome present with a complex neurobehavioral profile with high rates of ADHD, anxiety, repetitive behaviors, and sensory symptoms. Mol. Autism 12 (1), 61. doi:10.1186/s13229-021-00469-z
Trost, B., Thiruvahindrapuram, B., Chan, A. J. S., Engchuan, W., Higginbotham, E. J., Howe, J. L., et al. (2022). Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell 185, 4409–4427.e18. doi:10.1016/j.cell.2022.10.009
van Bon, B. W., Coe, B. P., Bernier, R., Green, C., Gerdts, J., Witherspoon, K., et al. (2016). Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol. Psychiatry 21 (1), 126–132. doi:10.1038/mp.2015.5
van Bon, B. W., Hoischen, A., Hehir-Kwa, J., de Brouwer, A. P., Ruivenkamp, C., Gijsbers, A. C., et al. (2011). Intragenic deletion in DYRK1A leads to mental retardation and primary microcephaly. Clin. Genet. 79 (3), 296–299. doi:10.1111/j.1399-0004.2010.01544.x
Vardarajan, B. N., Eran, A., Jung, J. Y., Kunkel, L. M., and Wall, D. P. (2013). Haplotype structure enables prioritization of common markers and candidate genes in autism spectrum disorder. Transl. Psychiatry 3 (5), e262. doi:10.1038/tp.2013.38
Velasquez, F., Wiggins, J. L., Mattson, W. I., Martin, D. M., Lord, C., and Monk, C. S. (2017). The influence of 5-HTTLPR transporter genotype on amygdala-subgenual anterior cingulate cortex connectivity in autism spectrum disorder. Dev. Cogn. Neurosci. 24, 12–20. doi:10.1016/j.dcn.2016.12.002
Vernes, S. C., Newbury, D. F., Abrahams, B. S., Winchester, L., Nicod, J., Groszer, M., et al. (2008). A functional genetic link between distinct developmental language disorders. N. Engl. J. Med. 359 (22), 2337–2345. doi:10.1056/NEJMoa0802828
Wang, H., Yin, F., Gao, J., and Fan, X. (2019). Association between 5-HTTLPR polymorphism and the risk of autism: a meta-analysis based on case-control studies. Front. Psychiatry 10, 51. doi:10.3389/fpsyt.2019.00051
Wang, H. S., Pan, Z., Shi, W., Brown, B. S., Wymore, R. S., Cohen, I. S., et al. (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282 (5395), 1890–1893. doi:10.1126/science.282.5395.1890
Wang, J., Yu, J., Wang, M., Zhang, L., Yang, K., Du, X., et al. (2023). Discovery and validation of novel genes in a large Chinese autism spectrum disorder cohort. Biol. Psychiatry 94, 792–803. doi:10.1016/j.biopsych.2023.06.025
Wang, Z. J., Chen, J., Chen, H. L., Zhang, L. Y., Xu, D., and Jiang, W. T. (2018). Association between SCN1A polymorphism rs3812718 and valproic acid resistance in epilepsy children: a case-control study and meta-analysis. Biosci. Rep. 38. doi:10.1042/bsr20181654
Warrier, V., Chee, V., Smith, P., Chakrabarti, B., and Baron-Cohen, S. (2015). A comprehensive meta-analysis of common genetic variants in autism spectrum conditions. Mol. Autism 6, 49. doi:10.1186/s13229-015-0041-0
Weinschutz Mendes, H., Neelakantan, U., Liu, Y., Fitzpatrick, S. E., Chen, T., Wu, W., et al. (2023). High-throughput functional analysis of autism genes in zebrafish identifies convergence in dopaminergic and neuroimmune pathways. Cell Rep. 42 (3), 112243. doi:10.1016/j.celrep.2023.112243
Weiss, L. A., Escayg, A., Kearney, J. A., Trudeau, M., MacDonald, B. T., Mori, M., et al. (2003). Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol. Psychiatry 8 (2), 186–194. doi:10.1038/sj.mp.4001241
White, J., Beck, C. R., Harel, T., Posey, J. E., Jhangiani, S. N., Tang, S., et al. (2016). POGZ truncating alleles cause syndromic intellectual disability. Genome Med. 8 (1), 3. doi:10.1186/s13073-015-0253-0
Whitehouse, A. J., Bishop, D. V., Ang, Q. W., Pennell, C. E., and Fisher, S. E. (2011). CNTNAP2 variants affect early language development in the general population. Genes Brain Behav. 10 (4), 451–456. doi:10.1111/j.1601-183X.2011.00684.x
Wing, L., Leekam, S. R., Libby, S. J., Gould, J., and Larcombe, M. (2002). The diagnostic interview for social and communication disorders: background, inter-rater reliability and clinical use. J. Child. Psychol. Psychiatry 43, 307–325. doi:10.1111/1469-7610.00023
Wolff, M., Johannesen, K. M., Hedrich, U. B. S., Masnada, S., Rubboli, G., Gardella, E., et al. (2017). Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 140 (5), 1316–1336. doi:10.1093/brain/awx054
Wolff, N., Stroth, S., Kamp-Becker, I., Roepke, S., and Roessner, V. (2022). Autism spectrum disorder and IQ - a complex interplay. Front. Psychiatry 13, 856084. doi:10.3389/fpsyt.2022.856084
Wu, Y., Cao, H., Baranova, A., Huang, H., Li, S., Cai, L., et al. (2020). Multi-trait analysis for genome-wide association study of five psychiatric disorders. Transl. Psychiatry 10 (1), 209. doi:10.1038/s41398-020-00902-6
Yi, F., Danko, T., Botelho, S. C., Patzke, C., Pak, C., Wernig, M., et al. (2016). Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352 (6286), aaf2669. doi:10.1126/science.aaf2669
Yuen, C. Y., Merico, D., Bookman, M., Howe, J. L., Thiruvahindrapuram, B., Patel, R. V., et al. (2017). Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 20 (4), 602–611. doi:10.1038/nn.4524
Zhang, T., Zhang, J., Wang, Z., Jia, M., Lu, T., Wang, H., et al. (2019). Association between CNTNAP2 polymorphisms and autism: a family-based study in the Chinese han population and a meta-analysis combined with GWAS data of psychiatric genomics consortium. Autism Res. 12 (4), 553–561. doi:10.1002/aur.2078
Zhi, H., Wu, C., and Yang, Z. (2018). SCN1A rs3812718 polymorphism is associated with epilepsy: an updated meta-analysis. Epilepsy Res. 142, 81–87. doi:10.1016/j.eplepsyres.2018.03.016
Keywords: autism spectrum disorder, genetic architecture, high-functioning autism, next-generation sequencing, single-nucleotide polymorphism, common variant, social responsiveness scale
Citation: Shiota Y, Nishiyama T, Yokoyama S, Yoshimura Y, Hasegawa C, Tanaka S, Iwasaki S and Kikuchi M (2024) Association of genetic variants with autism spectrum disorder in Japanese children revealed by targeted sequencing. Front. Genet. 15:1352480. doi: 10.3389/fgene.2024.1352480
Received: 08 December 2023; Accepted: 04 March 2024;
Published: 30 August 2024.
Edited by:
Jacob Peedicayil, Christian Medical College and Hospital, IndiaReviewed by:
Kanako Ishizuka, Nagoya Institute of Technology, JapanMónica Alejandra Rosales-Reynoso, Centro de Investigación Biomédica de Occidente (CIBO), Mexico
Martina Arenella, King’s College London, United Kingdom
Copyright © 2024 Shiota, Nishiyama, Yokoyama, Yoshimura, Hasegawa, Tanaka, Iwasaki and Kikuchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuka Shiota, eXVrYXNAbWVkLmthbmF6YXdhLXUuYWMuanA=; Shigeru Yokoyama, c2hpZ2VydXlAbWVkLmthbmF6YXdhLXUuYWMuanA=