 Haiying Zou
Haiying Zou Li Yang
Li Yang- Department of Endocrinology, Genetics and Metabolism, Jiangxi Provincial Children’s Hospital, Nanchang, Jiangxi, China
Introduction: Holocarboxylase synthetase deficiency (HLCSD) is a rare autosomal recessive genetic disorder caused by mutations in the holocarboxylase synthetase (HLCS) gene, which affects multiple systems. Common clinical manifestations include metabolic acidosis, rash, feeding difficulties, and growth retardation, with predominant involvement of the nervous system, skin, and hair. However, respiratory symptoms as the initial manifestation are relatively rare.
Case Presentation: We report the case of a 1 year and 4-month-old Chinese male patient who presented with a 2-day history of cough, followed by half a day of wheezing and shortness of breath. Despite supportive treatment with antibiotics upon admission, the infant continued to experience rapid and deep breathing accompanied by groaning, and obvious wheezing. Blood gas analysis revealed metabolic acidosis that was difficult to correct. Blood tandem mass spectrometry showed elevations in C50H, C3, C4OH, and urine organic acid analysis revealed elevations in lactate, 3-hydroxybutyric acid, 3-hydroxyisovaleric acid, acetoacetic acid, 3-methylcrotonylglycine, and methylcitric acid. Genetic testing revealed two variants in the HLCS gene in the infant: NM_001352514: exon6: c.1088T>A: p.V363D variant and exon11: c.2434C>T: p.R812* heterozygous variant, resulting in HLCSD. Ultimately, the diagnosis of HLCSD was established, and oral biotin treatment achieved good clinical efficacy.
Conclusion: This article summarizes the clinical data of a case of HLCSD in an infant, primarily presenting with respiratory symptoms. It provides a comprehensive summary of the etiology, diagnosis, and treatment, offering insights for the diagnosis of rare diseases by clinical physicians.
1 Introduction
Holocarboxylase synthetase (HLCS) deficiency (HLCSD), a rare autosomal recessive metabolic disorder, disrupts the binding of biotin to four specific apocarboxylases: pyruvate carboxylase, acetyl-CoA carboxylase, propionyl-CoA carboxylase, and methylcrotonyl-CoA carboxylase (L et al., 1985). HLCSD (OMIM#253270), also referred to as early-onset multiple carboxylase deficiency (MCD), typically becomes apparent shortly after birth. In contrast, biotinidase deficiency (BTD) is the form of MCD that presents later in life (Canda et al., 2020). Common manifestations of HLCSD include eczema, alopecia, lactic acidosis, hyperammonemia, seizures, and developmental delay (Tammachote et al., 2010). Treatment with biotin has demonstrated effective results (Wolf, 2022). Individuals who do not receive biotin therapy may experience psychomotor delay and severe metabolic acidosis, which can lead to unconsciousness or even fatality. It is important to note that HLCSD with respiratory tract involvement as the initial symptom is a rare occurrence.
HLCS deficiency may impacts respiratory health primarily through three interconnected mechanisms. Firstly, energy metabolism dysfunction occurs due to carboxylases, which are essential for energy generation, being impaired (Ferreira et al., 2021). This leads to an inadequate energy supply that can affect the normal function of the lungs and other organs (Chen et al., 2023). Secondly, HLCS deficiency can cause an accumulation of organic acids, potentially leading to metabolic acidosis (van der Knaap et al., 1998). This condition disrupts respiratory function as the body attempts to correct the blood pH imbalance by increasing the respiratory rate. Thirdly, biotin’s critical role in cell growth and DNA repair means its deficiency can compromise the immune system, increasing susceptibility to respiratory infections. Together, these effects highlight the complex ways in which HLCS deficiency can compromise respiratory health (Sullivan et al., 2023).
The mutation spectrum of the HLCS gene is associated with various clinical phenotypes, making molecular genetic analysis essential for a definitive diagnosis. These mutations can include missense mutations, deletions, as well as some total deletions and complex rearrangements (Yang et al., 2001). Recently, there have been reports of an increasing number of novel pathogenic variants (Wu et al., 2020). Interestingly, two mutations appear to be commonly observed in Chinese populations with HLCS deficiency (Zheng et al., 2020; Ling et al., 2023; Li et al., 2023). Furthermore, a study identified a novel heterozygous variant, c.996G > C (p.Gln332His), and a paracentric inversion on chromosome 21 (Quinonez et al., 2017). Additionally, there were five cases of HLCS deficiency with varying initial presentations and phenotypes (Donti et al., 2016). The sequencing of the HLCS gene helps confirm diagnoses and offer genetic counseling, while early treatment with pharmacological doses of oral biotin can prevent further decompensation in most cases before irreversible neurological damage occurs (Shao et al., 2022). In this report, we present a case of HCSD with respiratory tract symptoms as the initial manifestation, accompanied by metabolic acidosis and absence of a rash. The diagnosis of HCSD was established through molecular analysis.
2 Case description
A 17-month-old Chinese male infant presented with a worsening cough and shortness of breath over 2 days, alongside wheezing. Symptoms started with a wet cough, progressing to rapid breathing and deteriorating mental state. Treatments with traditional Chinese medicines “Qing Kai Ling” and “Fei Li Kou” showed no improvement. On admission, he had decreased appetite and was dehydrated but had no fever. Laboratory findings showed elevated white blood cells and neutrophils, hypoglycemia, and mild acidosis. A chest X-ray confirmed bronchitis. Further details are provided in Figure 1. Moreover, Urine analysis confirmed increased levels of lactate, 3-hydroxybutyric acid, 3-hydroxyisovaleric acid, acetoacetic acid, and 3-methylglutaconic acid, and decreased levels of methylmalonic acid. Table 1 presents the biochemical parameters measured at relevant time points. With the family’s consent, comprehensive whole-exome genetic testing was performed, identifying compound heterozygous variants in the HLCS gene: NM_001352514: exon6: c.1088T>A: p.V363D and exon11: c.2434C>T: p.R812*, which are considered pathogenic mutations. The diagnosis of HCSD was established, and treatment included the administration of biotin (10 mg, twice daily), and specialized formula feeding. Following treatment, the acidosis resolved, and the lactate level decreased to 2.4 mmol/L. The patient’s condition improved, and they were subsequently discharged.
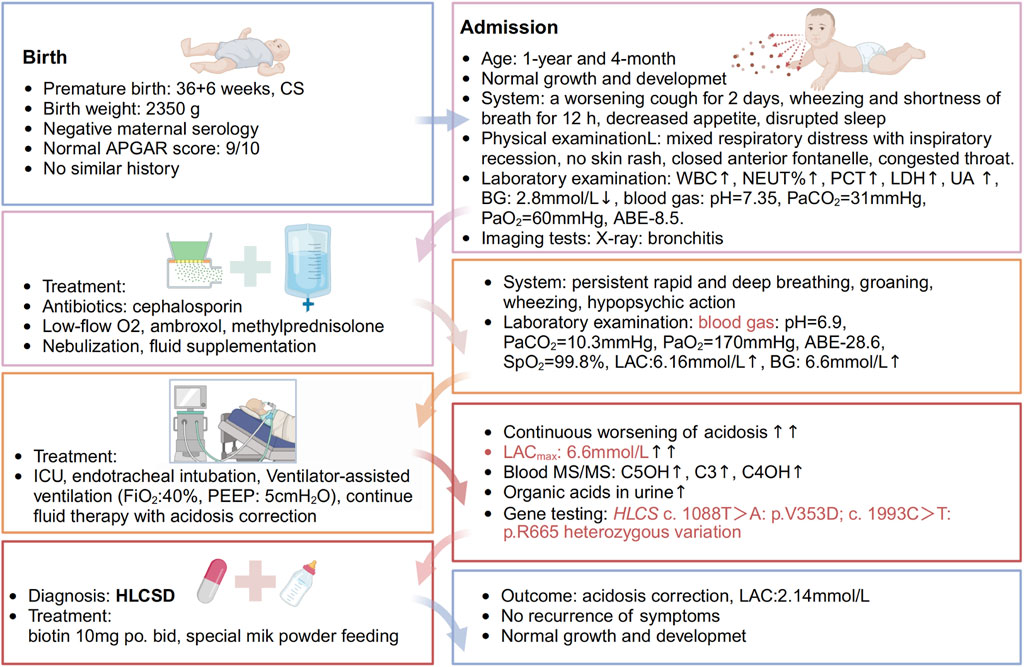
Figure 1. Timeline of events. A 1-year and 4-month-old Chinese male infant presented with a worsening cough for 2 days, accompanied by wheezing and shortness of breath for half a day. The patient initially experienced episodes of wet coughing, starting 2 days ago, with mild symptoms, and without fever, convulsions, or rash. For 6 h before admission, the patient also had a runny nose and one episode of non-projectile vomiting with moderate stomach contents. The patient underwent oral administration of Chinese patent medicines “Qing Kai Ling” and “Fei Li Kou” at a local hospital; however, no discernible improvement was observed. The cough worsened, accompanied by rapid breathing and wheezing, and the patient’s mental status and appetite gradually deteriorated. As a result, the decision was made to seek treatment at our hospital. The patient has exhibited decreased appetite, disrupted sleep, daily yellow soft stools, pale yellow urine, and decreased urine output. There is no known history of recurrent respiratory distress in the patient, who is the second infant in the family. The patient was delivered by Cesarean section at 36 weeks and 6 days of gestation, with a birth weight of 2.35 kg. The patient’s growth and development have been observed to be similar to other infant of the same age. Both parents of the infant are healthy and non-consanguineous, and there is no record of any similar illness within the family. The patient was afebrile, with no diarrhea. He demonstrates normal development and clear consciousness, but presents with a weak mental state and rapid breathing. There is no presence of skin rash. The anterior fontanelle is closed. The patient has red lips, dry oral mucosa, congested throat, and no enlarged tonsils or purulent discharge. Mixed respiratory distress with inspiratory recession is observed. Coarse respiratory sounds and audible wheezing are noted in both lungs. The heart rate is recorded at 145 beats per minute with a regular rhythm and strong heart sounds, and no murmurs are present. The abdomen is soft with no masses, tenderness, or rebound tenderness, and the subcutaneous fat thickness is measured at 8 mm. No other abnormalities are found during the physical examination. The patient’s admission laboratory examination revealed several abnormalities. There was an increase in white blood cells and neutrophil percentage, as well as elevated levels of procalcitonin, lactate dehydrogenase, and uric acid. Additionally, the patient had hypoglycemia with a blood glucose level of 2.8 mmol/L. The blood gas analysis showed a pH of 7.35, PaCO2 = 31 mmHg, PaO2 = 60 mmHg, and a base deficit of −8.5. Mild decreases were observed in the levels of IgG and complement C3. Coagulation, erythrocyte sedimentation rate, routine urine and stool tests, and pathogen testing yielded normal results. Liver function, electrolytes, and blood lipids were also within the normal range. Allergen testing came back negative. The chest X-ray showed evidence of bronchitis, while the abdominal color Doppler ultrasound did not reveal any abnormalities in the liver, gallbladder, or spleen. Echocardiography showed no apparent abnormalities in the heart. Brain magnetic resonance imaging demonstrated patchy T2 flair hyperintensities in the subcortical white matter of the bilateral lateral ventricular posterior corner and frontal lobe, suggesting possible delayed myelination. Furthermore, there was local enlargement in the left temporal horn and a smaller volume of the left hippocampus compared to the right side. Initially, the patient received cephalosporin to treat the infection, along with low-flow oxygen, ambroxol to aid expectoration, and methylprednisolone to reduce airway hyperreactivity. Treatment also included budesonide, salbutamol, and ipratropium bromide nebulization, as well as fluid supplementation. Despite these interventions, the infant exhibited persistent rapid and deep breathing with groaning, obvious wheezing, and severely impaired mental response. As a result, the infant was transferred to the intensive care unit for further evaluation.
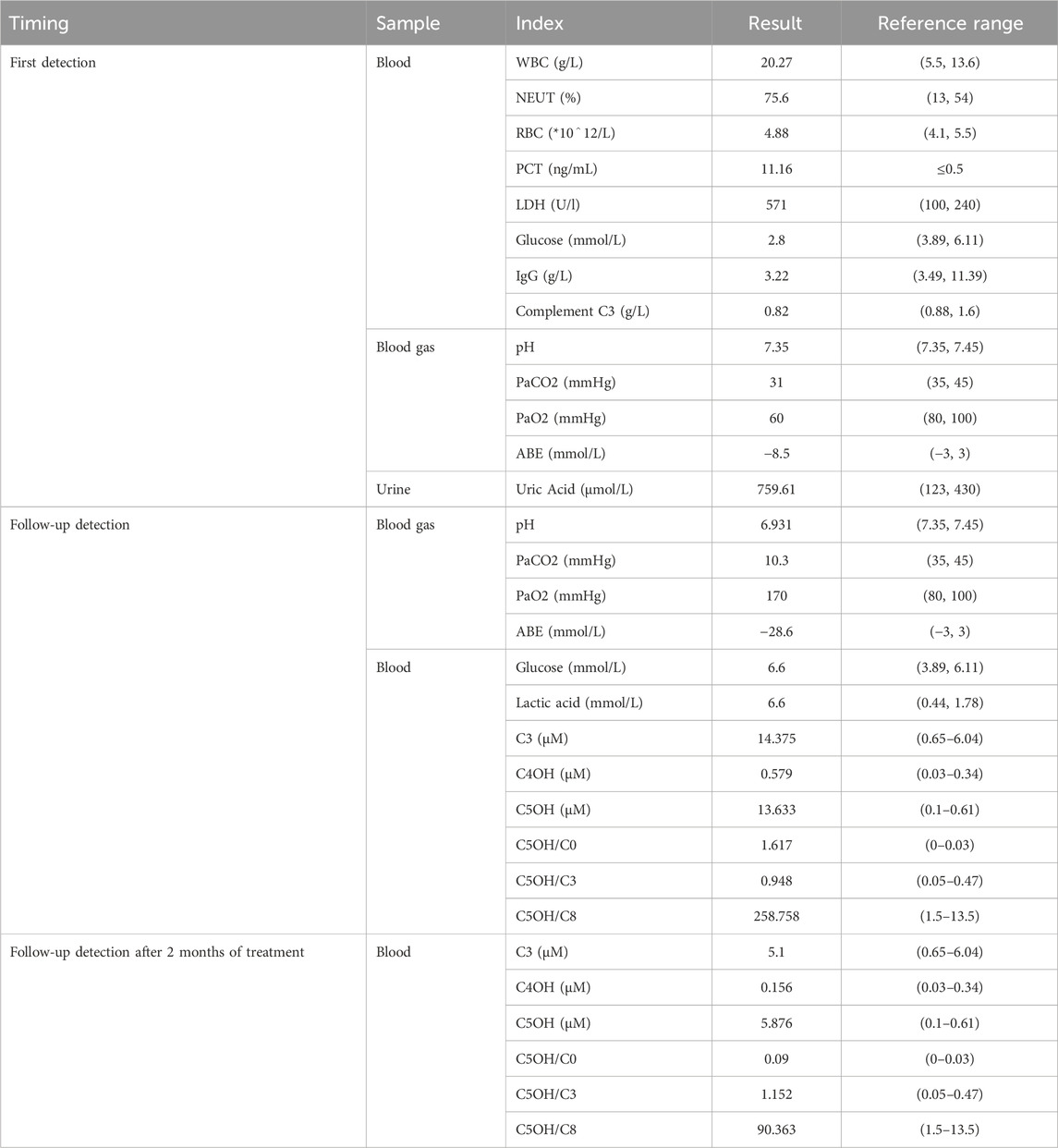
Table 1. The biochemical parameters measured at relevant time points.
2.1 Genetic analysis
The patient and his non-consanguineous parents underwent comprehensive physical examinations and genetic testing. Whole-exome sequencing (WES) was conducted using the Verita Trekker® variant detection system and the Enliven® variant annotation and interpretation system developed by Berry Gene. PCR and capillary electrophoresis were employed for a comprehensive analysis of the WES results pertaining to dynamic mutations. Table 2 presents the single nucleotide variants (SNVs) and Insertions/deletions (InDel) findings. The patient’s sample revealed two variants of the HLCS gene: a pathogenic variant, HLCS: NM_001352514: exon6: c.1088T>A: p.V363D, and another pathogenic variant, HLCS: NM_001352514: exon11: c.2434C>T: p.R812*, which is associated with HLCSD (OMIM:253270). Following guidelines from the American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015), recommendations from the ClinGen Sequence Variant Interpretation (SVI) Expert Panel (Abou Tayoun et al., 2018; Biesecker and Harrison, 2018; Ghosh et al., 2018), and screening for phenotype-related gene variants in databases like HPO, OMIM, and GHR, the c.1088T>A mutation of the HLCS gene is categorized as pathogenic, and the c.1993C>T mutation is also deemed to be pathogenic. Figure 2 displays the SNV and InDel results for the patient and parents, illustrating that the patient inherited a heterozygous c.1088T>A mutation in the HLCS gene from the father and a heterozygous c.2434C>T mutation from the mother.
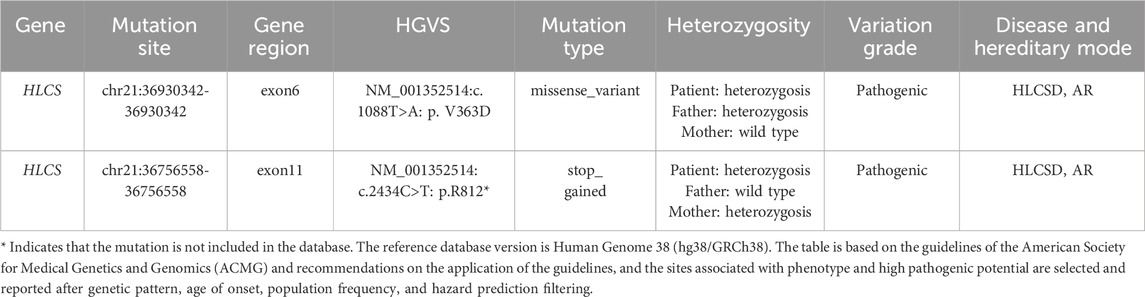
Table 2. SNV and InDel test results.

Figure 2. SNV and InDel site maps. (A) HLCS: NM_001352514: exon6: c.1088T>A: p.V363D. (B) HLCS: NM_001352514: exon11: c.2434C>T: p.R812*. * Indicates that the mutation is not included in the database.
3 Discussion
HCLS facilitates the covalent binding of biotin to an inactive apocarboxylase through its carboxyl group, thereby forming the active holoenzyme (León-Del-Río et al., 2017). HCLS deficiency is a rare autosomal recessive metabolic disorder that impairs gluconeogenesis, fatty acid metabolism, and amino acid catabolism, resulting in dermatological, metabolic, respiratory, and neurological abnormalities. The age at which HCLS deficiency presents varies, with over half of the cases occurring in newborns (Wiltink et al., 2016). In this study, patients initially presented with respiratory symptoms as the primary clinical manifestation, accompanied by metabolic acidosis, dysregulation of blood sugar, and other symptoms. Specific symptoms were not initially evident, leading to delayed diagnosis and treatment. However, once the diagnosis was confirmed through genetic testing, prompt biotin therapy was administered, resulting in the correction of lactic acidosis, alleviation of symptoms, and absence of subsequent complications.
Functional studies suggest that the HCLS gene variant exon6: c.1088T>A: p.V363D is associated with impaired gene function (Dupuis et al., 1999). This variant has been observed in compound heterozygosity with the c.2434C>T variant in multiple patients. Homozygous variants at this site have also been detected in some patients (Wu et al., 2020). Regarding the HLCS: NM_001352514: exon11: c.2434C>T: p.R812* variant, it has been found to be compound heterozygous with the c.1088T>A variant. Literature reports indicate the presence of an unknown phase pathogenic variant in one patient (Tangeraas et al., 2020). This variant is located in the last exon of the gene and is predicted to undergo Nonsense-Mediated mRNA Decay (NMD) in less than 10% of the protein region. This variant is classified as “Pathogenic” in the ClinVar database and as “DM” in the HGMD (Suzuki et al., 2005; Tabor et al., 2014; Xiong et al., 2015).
Mutations in the HLCS gene (OMIM:609018) lead to HLCSD (OMIM:253270). The main manifestations of HLCSD include metabolic acidosis, global developmental delay, increased muscle tone, hyperventilation, coma, hair loss, organic aciduria, vomiting, seizures, decreased muscle tone, generalized hypotonia, irritability, elevated levels of 3-methylcrotonylglycine in urine, 3-hydroxyisovaleric aciduria, difficulty with infant feeding, hyperammonemia, rash, tachypnea, thrombocytopenia, neonatal onset, and lactic acidosis. This case report emphasizes the respiratory symptoms as the initial manifestation, which is rare and lacks specificity, making it susceptible to misdiagnosis. The genetic testing results presented in this report will undoubtedly aid in the diagnosis and treatment of HLCS deficiency in China.
The limitations of this case report include the focus on detecting SNVs within the entire exome region and a 5 bp range of splice junctions, as well as InDels within 50 bp of the exonic region. This detection method does not consider poly structures, tandem repeat sequences, GC-rich regions, and homologous similar sequences (pseudogenes). Additionally, InDels exceeding 50 bp have certain limitations. The current whole exome sequencing, due to technical constraints of whole exome capture, cannot ensure complete coverage of the exonic region. Furthermore, there is a possibility of maternal contamination and low-level mosaicism that cannot be ruled out. In addition to the known pathogenic variant c.1088T>A: p.V363D, the variant c.2434C>T: p.R812* is likely to be a contributing factor in disease development. However, further studies are required to validate these findings.
4 Conclusion
HLCSD is a rare autosomal recessive genetic disease resulting from mutations in the HLCS gene. The lack of specific symptoms makes it susceptible to misdiagnosis and underdiagnosis by clinicians. We present a case of HLCSD in a patient who primarily presented with respiratory symptoms. Therefore, obtaining a comprehensive medical history and performing appropriate examinations is essential in infants and young infant with unexplained respiratory disorders characterized by rapid, deep breathing. Early diagnosis and prompt treatment can mitigate the disability and mortality associated with HLCSD.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by The Ethical Committee, Jiangxi Provincial Children’s Hospital (NO.330038). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
HZ: Data curation, Formal Analysis, Supervision, Writing–original draft, Conceptualization, Investigation, Methodology, Validation, Visualization. LY: Writing–original draft, Data curation, Investigation, Methodology, Software, Supervision, Validation, Visualization, Writing–review and editing. RZ: Data curation, Investigation, Methodology, Software, Writing–review and editing. YQ: Conceptualization, Formal Analysis, Investigation, Methodology, Supervision, Validation, Visualization, Writing–original draft.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article. N/A.
Acknowledgments
We thank the patient and her parents for their cooperation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abou Tayoun, A. N., Pesaran, T., DiStefano, M. T., Oza, A., Rehm, H. L., Biesecker, LG, and Harrison, SMClinGen Sequence Variant Interpretation Working Group (ClinGen SVI) Csviwg, C. G. (2018). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 39, 1517–1524. doi:10.1002/humu.23626
Biesecker, L. G., and Harrison, S. M.ClinGen Sequence Variant Interpretation Working Group (2018). The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet. Med. official J. Am. Coll. Med. Genet. 20, 1687–1688. doi:10.1038/gim.2018.42
Canda, E., Kalkan Uçar, S., and Çoker, M. (2020). Biotinidase deficiency: prevalence, impact and management strategies. Pediatr. Health Med. Ther. 11, 127–133. doi:10.2147/PHMT.S198656
Chen, S., Li, M., Zhang, R., Ye, L., Jiang, Y., Jiang, X., et al. (2023). Type 1 diabetes and diet-induced obesity predispose C57BL/6J mice to PM2.5-induced lung injury: a comparative study. Part Fibre Toxicol. 20, 10. doi:10.1186/s12989-023-00526-w
Donti, T. R., Blackburn, P. R., and Atwal, P. S. (2016). Holocarboxylase synthetase deficiency pre and post newborn screening. Mol. Genet. metabolism Rep., 7. doi:10.1016/j.ymgmr.2016.03.007
Dupuis, L., Campeau, E., Leclerc, D., and Gravel, R. A. (1999). Mechanism of biotin responsiveness in biotin-responsive multiple carboxylase deficiency. Mol. Genet. metabolism, 66. doi:10.1006/mgme.1998.2785
Ferreira, C. R., Martinelli, D., and Blau, N. (2021). Clinical and biochemical footprints of inherited metabolic diseases. VI. Metabolic dermatoses. Mol. Genet. Metab. 134, 87–95. doi:10.1016/j.ymgme.2021.07.005
Ghosh, R., Harrison, S. M., Rehm, H. L., Plon, S. E., and Biesecker, L. G. (2018). Updated recommendation for the benign stand-alone ACMG/AMP criterion. Hum. Mutat., 39. doi:10.1002/humu.23642
León-Del-Río, A., Valadez-Graham, V., and Gravel, R. A. (2017). Holocarboxylase synthetase: a moonlighting transcriptional coregulator of gene expression and a cytosolic regulator of biotin utilization. Annu. Rev. Nutr., 37. doi:10.1146/annurev-nutr-042617-104653
Li, K.-Y., Tang, J.-P., Jiang, Y.-L., Yue, S.-Z., Zhou, B., Wen, R., et al. (2023). Holocarboxylase synthetase deficiency induced by HLCS gene mutations: a rare disease study. Zhongguo Dang Dai Er Ke Za Zhi 25, 401–407. doi:10.7499/j.issn.1008-8830.2211062
Ling, S., Qiu, W., Zhang, H., Liang, L., Lu, D., Chen, T., et al. (2023). Clinical, biochemical, and genetic analysis of 28 Chinese patients with holocarboxylase synthetase deficiency. Orphanet J. Rare Dis. 18, 48. doi:10.1186/s13023-023-02656-y
L, S., Bj, B., and Wl, N. (1985). Biotin holocarboxylase synthetase deficiency. Ann. N. Y. Acad. Sci., 447. doi:10.1111/j.1749-6632.1985.tb18446.x
Quinonez, S. C., Seeley, A. H., Lam, C., Glover, T. W., Barshop, B. A., and Keegan, C. E. (2017). Paracentric inversion of chromosome 21 leading to disruption of the HLCS gene in a family with holocarboxylase synthetase deficiency. JIMD Rep. 34, 55–61. doi:10.1007/8904_2016_9
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Shao, J., Division of Genetics and Metabolism, Child Diseases and Health Care Branch, Chinese Association for Maternal and Child Health, , and Division of Genetics and Metabolism, Rare Diseases Committee of Beijing Medical Association, (2022). Expert consensus on screening, diagnosis and treatment of multiple carboxylase deficiency. J. Zhejiang Univ. Med. Sci. 51, 129–135. doi:10.3724/zdxbyxb-2022-0164
Sullivan, M. R., McGowen, K., Liu, Q., Akusobi, C., Young, D. C., Mayfield, J. A., et al. (2023). Biotin-dependent cell envelope remodelling is required for Mycobacterium abscessus survival in lung infection. Nat. Microbiol. 8, 481–497. doi:10.1038/s41564-022-01307-5
Suzuki, Y., Yang, X., Aoki, Y., Kure, S., and Matsubara, Y. (2005). Mutations in the holocarboxylase synthetase gene HLCS. Hum. Mutat., 26. doi:10.1002/humu.20204
Tammachote, R., Janklat, S., Tongkobpetch, S., Suphapeetiporn, K., and Shotelersuk, V. (2010). Holocarboxylase synthetase deficiency: novel clinical and molecular findings. Clin. Genet., 78. doi:10.1111/j.1399-0004.2009.01357.x
Tabor, H. K., Auer, P. L., Jamal, S. M., Chong, J. X., Yu, J.-H., Gordon, A. S., et al. (2014). Pathogenic variants for Mendelian and complex traits in exomes of 6,517 European and African Americans: implications for the return of incidental results. Am. J. Hum. Genet. 95, 183–193. doi:10.1016/j.ajhg.2014.07.006
Tangeraas, T., Sæves, I., Klingenberg, C., Jørgensen, J., Kristensen, E., Gunnarsdottir, G., et al. (2020). Performance of expanded newborn screening in Norway supported by post-analytical bioinformatics tools and rapid second-tier DNA analyses. Int. J. neonatal Screen. 6, 6. doi:10.3390/ijns6030051
van der Knaap, M. S., Bakker, H. D., and Valk, J. (1998). MR imaging and proton spectroscopy in 3-hydroxy-3-methylglutaryl coenzyme A lyase deficiency. AJNR Am. J. Neuroradiol. 19, 378–382. doi:10.1016/s1076-6332(98)80137-2
Wiltink, R. C., Kruijshaar, M. E., van Minkelen, R., Onkenhout, W., Verheijen, F. W., Kemper, E. A., et al. (2016). Neonatal screening for profound biotinidase deficiency in The Netherlands: consequences and considerations. Eur. J. Hum. Genet. 24, 1424–1429. doi:10.1038/ejhg.2016.65
Wolf, B. (2022). Revisiting the administration of biotin to children with biotin-responsive disorders. Mol. Genet. Metab. 137, 225–227. doi:10.1016/j.ymgme.2022.07.004
Wu, H.-R., Chen, K.-J., Hsiao, H.-P., and Chao, M.-C. (2020). Impaired glucose homeostasis and a novel HLCS pathogenic variant in holocarboxylase synthetase deficiency: a report of two cases and brief review. J. Pediatr. Endocrinol. Metab. 33, 1481–1486. doi:10.1515/jpem-2020-0106
Xiong, H. Y., Alipanahi, B., Lee, L. J., Bretschneider, H., Merico, D., Yuen, R. K. C., et al. (2015). RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 347, 1254806. doi:10.1126/science.1254806
Yang, X., Aoki, Y., Li, X., Sakamoto, O., Hiratsuka, M., Kure, S., et al. (2001). Structure of human holocarboxylase synthetase gene and mutation spectrum of holocarboxylase synthetase deficiency. Hum. Genet., 109. doi:10.1007/s004390100603
Keywords: holocarboxylase synthetase deficiency (HLCSD), biotinidase deficiency, HLCS gene, mutation, biotin
Citation: Zou H, Yang L, Zhang R and Qin Y (2024) Case report: A case of holocarboxylase synthetase deficiency with respiratory tract as the initial symptom. Front. Genet. 15:1439343. doi: 10.3389/fgene.2024.1439343
Received: 27 May 2024; Accepted: 06 November 2024;
Published: 20 November 2024.
Edited by:
Sunita Bijarnia-Mahay, Sir Ganga Ram Hospital, IndiaReviewed by:
Patryk Lipiński, Maria Sklodowska-Curie Medical Academy, PolandHarvey L. Levy, Harvard Medical School, United States
Copyright © 2024 Zou, Yang, Zhang and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Yang, eWFuZ2xpMDUyMzJAMTYzLmNvbQ==