 Aditya Nugroho1
Aditya Nugroho1 Evayusvita Rustam1
Evayusvita Rustam1 Nurin Widyani1
Nurin Widyani1 Fitri Indriani1*
Fitri Indriani1* Dede J. Sudrajat1
Dede J. Sudrajat1 Mohammad Agus Salim1
Mohammad Agus Salim1 Muhammad Majiidu2
Muhammad Majiidu2 Fifi Gus Dwiyanti2,3
Fifi Gus Dwiyanti2,3 Rahadian Pratama2,4
Rahadian Pratama2,4 Iskandar Z. Siregar2,3*
Iskandar Z. Siregar2,3*- 1Research Center for Applied Botany, National Research and Innovation Agency, Cibinong, West Java, Indonesia
- 2Molecular Science Research Group, Advanced Research Laboratory, IPB University, Bogor, West Java, Indonesia
- 3Department of Silviculture, Faculty of Forest and Environment, IPB University, Bogor, West Java, Indonesia
- 4Department of Biochemistry, Faculty of Mathematics and Natural Science, IPB University, Bogor, West Java, Indonesia
1 Introduction
Rubroshorea johorensis (Foxw.) P.S. Ashton and J. Heck, which is a synonym of Shorea johorensis Foxw, commonly referred to as red meranti, is a species belonging to the Dipterocarpaceae family (Ashton and Heckenhauer, 2022). This species is found in the tropical rainforests of the Indo-Malayan region (Aisha et al., 2014; Fathiya et al., 2018). R. johorensis is naturally distributed across Peninsular Malaysia, Sumatra, and Borneo, particularly in South and East Kalimantan, Brunei, and Central and Western Sarawak (Widiyatno et al., 2014). Typically, R. johorensis grows on hillsides at altitudes below 600 m above sea level, on undulating terrain, fertile clayey loam soils, and well-drained alluvial soils (Wahyudi et al., 2014). Shorea species, including R. johorensis, are essential timber trees known for their high economic value and significant presence in the international tropical timber market (Fathiya et al., 2018; Harahap et al., 2018). These species from the Dipterocarpaceae family are key products of tropical rainforests and serve as symbols of these ecosystems (Widiyatno et al., 2013; Yu et al., 2021). The wood of R. johorensis is widely used as a raw material for buildings, furniture, and various other applications (Purwaningsih, 2018; Suciyati et al., 2021).
The IUCN (International Union for Conservation of Nature) Red List of Threatened 2024 categorizes R. johorensis as vulnerable (VU) (IUCN, 2024). The population of species within the Dipterocarpaceae family, including the Shorea species, R. johorensis, has been declining due to logging, the conversion of forest land into plantations, and the establishment of industrial plantations (Widiyatno et al., 2013; Gaveau et al., 2016; Harahap et al., 2018). These activities are contributing to a continuous decrease in the global population of R. johorensis. Projections indicate that over three generations (1,860–2,100), the population of this species is expected to decline by 30%–50% (IUCN, 2024). This situation highlights the urgent need for conservation efforts to protect and sustain the existing populations of R. johorensis.
Data on the genomic resources of R. johorensis are still limited, which poses a significant challenge to advancing research in genetic conservation and forest landscape restoration. Unlocking genomic data for R. johorensis is vital for enhancing both genetic conservation strategies and tree breeding programs. Among these genomic resources, organellar genomes are particularly valuable, providing crucial insights into genetic variation among closely related species. In plants, organellar genomes, including mitogenome and plastome, are semi-autonomous structures encased in double membranes, housing independent genetic material. These organelles possess molecular machinery that regulates gene expression (Chevigny et al., 2020) and play a vital role in various physiological processes within plants (Mahapatra et al., 2021).
The plastome is a valuable tool for genetic studies due to its slow evolutionary rate, maternal inheritance in most angiosperms, and its conserved structure and gene sequences (Zulfahmi et al., 2015; Fathiya et al., 2018; Song et al., 2019; Lim et al., 2020). In angiosperms, the plastome typically ranges from 107 kb to 218 kb (Daniell et al., 2016; Li J. et al., 2024) and contains 120 to 130 genes that are crucial for transcription, translation, and photosynthesis (Daniell et al., 2016; Lim et al., 2020). Due to its conserved nature, the plastome is a reliable resource for molecular identification, genetic diversity assessments, and phylogenetic studies (Chew et al., 2023; Kim et al., 2024). In contrast, plant mitogenome exhibit greater variability in size and structural complexity (Burger et al., 2003; Sloan et al., 2012). Their sizes can range from 200 kb to 2,900 kb, with larger genomes typically found in seed plants, which have an abundance of introns, intergenic regions, and repetitive sequences (Mower et al., 2012; Morley et al., 2019). The high rates of sequence repetition, recombination, and rearrangement contribute to the structural diversity of plant mitogenome (Johnston, 2019). Despite these challenges, mitogenome remain invaluable for understanding genome architecture and evolutionary dynamics (Drouin et al., 2008). Therefore, this study aims to sequence, assemble, and characterize the plastome and mitogenome of Rubroshorea johorensis. This research enhances understanding of its genetic diversity and contributes to conservation and breeding efforts. It represents a vital step toward preserving this species and enriching the knowledge of plant genetics.
2 Methods
2.1 Plant material, DNA extraction and sequencing
Fresh leaves of R. johorensis were collected from the Bogor Botanical Garden in West Java, Indonesia (6° 35′51.46″S, 106° 47′58.44″E), specifically from plot XXV, with collection number 237. The herbarium voucher for this sample has been deposited in the Herbarium Bogoriense (BO) with passport data BO 1997911. A total of 100 mg of fresh leaves was used for genomic DNA isolation, following the CTAB protocol (Doyle and Doyle, 1990). The initial quality of the extracted genomic DNA was assessed by visualizing it on a 1% agarose gel electrophoresis in TAE buffer, run for 30 min at 100 V. Subsequently, the purity and quantity of the extracted genomic DNA were determined using a NanoPhotometer® NP80 (IMPLEN) and a Qubit 1.0 Fluorometer (Thermo Fisher Scientific), utilizing the Qubit dsDNA BR assay kit. The long-read sequencing library was prepared according to the protocol provided by Oxford Nanopore Technologies (ONT) for Ligation Sequencing DNA V14 (SQK-LSK114), version ACDE_9163_v114_revQ_29 Jun 2022. Sequencing was conducted using a MinION Mk1C device equipped with an R10.4.1 flow cell (FLO-MIN114). All laboratory work was carried out at Forest Genetics and Molecular Forestry, Department of Silviculture, Faculty of Forestry and Environment, IPB University in Bogor, West Java.
2.2 Organelle genome assembly and annotation
Raw reads from MinION sequencing (POD5) were base-called into raw Fastq files using Dorado Basecaller v0.8.0. The resulting Fastq files were subsequently processed using Porechop v0.2.4 (Wick et al., 2017) and Chopper v0.5.0 (De Coster, 2018) to trim adaptors, remove low-quality bases (Phred score <9), and eliminate potential sequence contamination. The parameters used for Chopper included -l 500, -q 9, --head crop 10, and–tail crop 10. The cleaned Fastq data were then analyzed statistically using NanoPlot v1.31.0 (De Coster et al., 2018) to calculate and visualize the read distribution.
The de novo assembly of the R. johorensis organelle genome was performed using Flye v2.9.4 (Kolmogorov et al., 2019) to construct both the plastome and mitogenome. To enhance accuracy, the assembled contigs were polished using the Pilon v1.20.1 (Walker et al., 2014). These contigs were then mapped to the reference plastome of Shorea macrophylla, a synonym of Rubroshorea macrophylla (GenBank accession: ON321899), and to the mitogenome of Arabidopsis thaliana (GenBank accession: GCF_000001735.4).
The annotation was carried out using CPGAVAS2 (http://47.96.249.172:16019/analyzer/annotate) (Shi et al., 2019), with the plastome of S. macrophylla (accession number: ON321899) serving as the reference. The results of the annotation were manually verified using Unipro UGENE v45.1 (Okonechnikov et al., 2012) and NCBI Genomic Workbench v3.8.2 (Kuznetsov and Bollin, 2021). For the mitogenome, annotation was conducted using PMGA (http://47.96.249.172:16084/annotate.html) (Li L. F. et al., 2024). These results were also manually verified using Unipro UGENE v45.1 and NCBI Genomic Workbench v3.8.2. The circular genome was visualized using Organellar GenomeDRAW (OGDRAW), accessible through the MPI-MP Chlorobox platform (Greiner et al., 2019).
3 Result
Long-read sequencing of R. johorensis yielded a total of 1,657,747 reads, amounting to 5,759, 232, 230 base pairs (bp) of raw data. The mean read length was 3,474.1 bp, and the N50 value for read length was 5,104 bp. The mean read quality of the raw data was recorded as 12.7. After filtering, the number of reads that passed the quality assessment reached 1,463,146, totaling 5,198,148,860 bp. The mean read length increased to 3,552.7 bp, with the N50 value remaining stable at 5,105 bp. Additionally, the mean read quality improved to 15.1 after filtering. A total of 29,886 reads (30.94%) were successfully mapped to the plastome, resulting in a mean depth of coverage of 104.4×. For the mitogenome, mapping of sequencing reads to the A. thaliana reference genome resulted in a mean coverage depth of 44×, with 23.4% of the genome covered by at least one read. In contrast, the assembly of the R. johorensis mitogenome yielded an average coverage depth of 31×, indicating reliable sequencing depth across the majority of the mitochondrial genome.
The assembled plastome of R. johorensis has a total length of 149,968 bp, while its mitogenome spans 296,595 bp, with a GC content of 44.63% (Figure 1). The plastome structure consists of a Large Single Copy (LSC) region of 82,920 bp with a GC content of 35.39%, a Small Single Copy (SSC) region of 19,837 bp with a GC content of 31.55%, and two Inverted Repeat (IR) regions, each measuring 23,584 bp, with a GC content of 43.36%. Among these regions, the IR regions exhibited the highest GC content. Overall, the average GC content of the plastome was 37.39%.
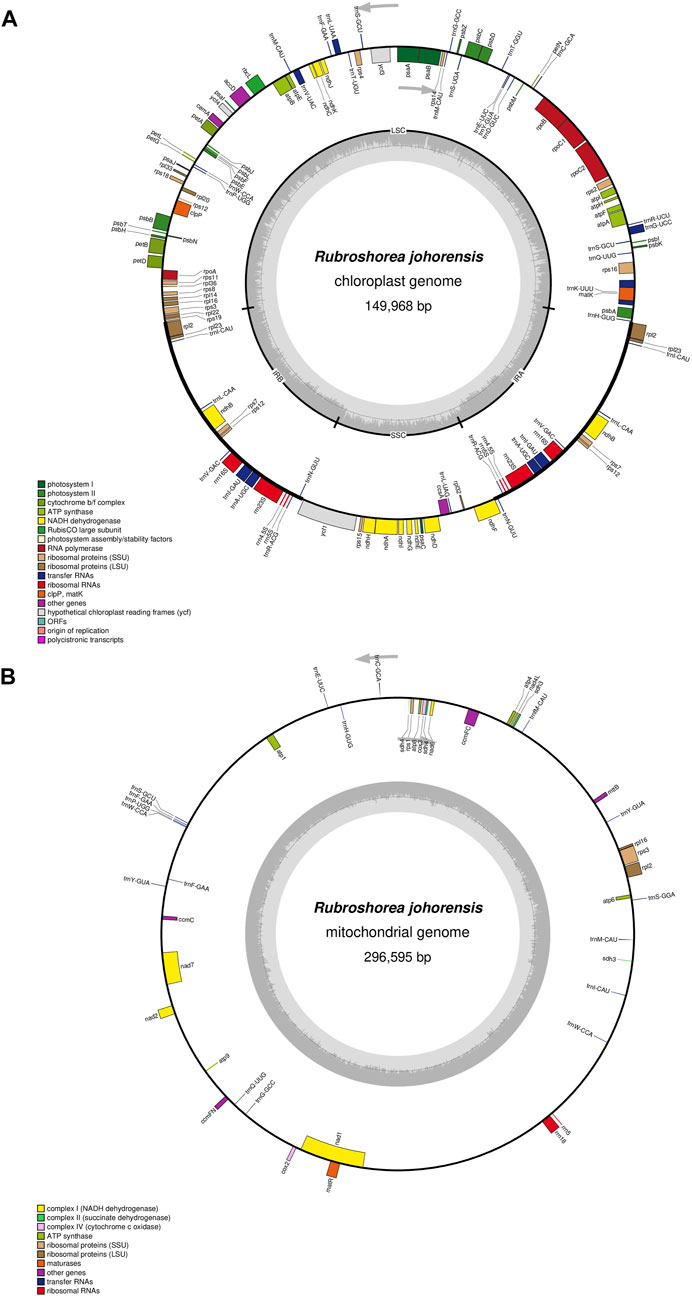
Figure 1. The organelle genome map of Rubroshorea johorensis. (A) The circular map of plastome genome in R. johorensis. (B) The circular map of the mitogenome in R. johorensis.
Annotation of the R. johorensis plastome identified 126 genes, comprising 76 protein-coding genes, 29 tRNA genes, and four rRNA genes. Among them, 15 genes contain a single intron, while three genes (rps12, ycf3, and clpP) contain two introns. These genes were categorized into four functional groups: self-replicating genes, photosynthetic genes, genes with other functions, and genes of unknown function (Supplementary Table S1).
In the mitogenome of R. johorensis, a total of 44 genes were identified, which include two rRNA genes, 17 tRNA genes, and 25 protein-coding genes. Additionally, six of these genes contain introns, suggesting that RNA processing events may play a crucial role in gene expression and regulation, with further details provided in Supplementary Table S2. Similar to the mitogenome of other plants, R. johorensis likely exhibits high rates of recombination and structural rearrangements, features that are characteristic of plant mitogenome and contribute to genome plasticity. The number of protein-coding genes (22) in R. johorensis is comparable to that found in other land plants, supporting the idea that core mitochondrial functions are conserved across species. However, variations in gene content and the presence of introns may indicate species-specific adaptations that could be linked to environmental factors or evolutionary history. The results of the mitogenome sequencing revealed that the assembled genome is still limited. This finding is significant as this study represents the first comprehensive characterization of the mitogenome for a species within the Dipterocarpaceae family. It underscores the need for further research in this area. Conducting additional comparative genomic studies could provide deeper insights into the functional implications of these genomic features in R. johorensis.
Microsatellites (SSRs), or simple sequence repeats (1–6 bp), are widely distributed throughout genomes and hold significant importance in genomic analysis. This study identified various types of SSRs in the genome, with trinucleotide repeats being the most prevalent at 54%, followed by mononucleotide repeats at 31%. Dinucleotide, tetranucleotide, and pentanucleotide repeats were less common, comprising 8%, 4%, and 3%, respectively. Adenine-rich sequences were particularly abundant, with A (57), AAA (39), AAG (26), and AAT (25) emerging as the most frequent types. The analysis of longer repeats showed a dominance of forward repeats (51.82%), followed by palindromic repeats (32.12%). Reverse and complementary repeats were less prominent, representing for 10.22% and 5.84%, respectively (Figure 2).
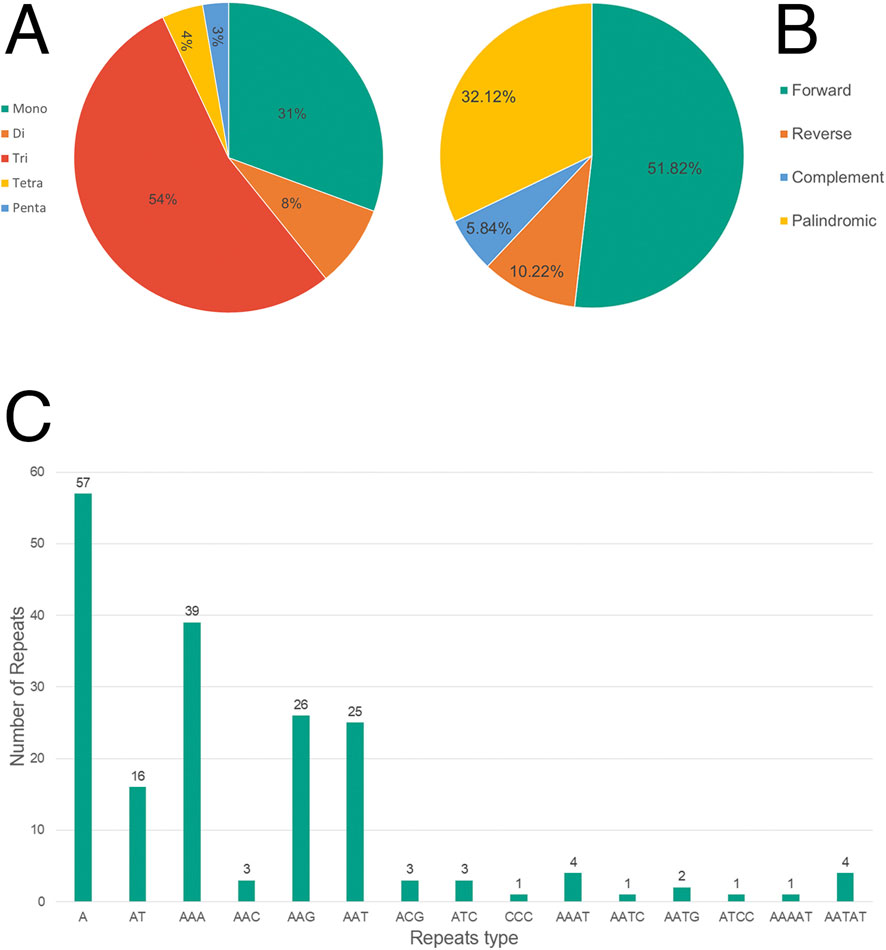
Figure 2. Frequency of simple sequence repeat (SSR) and repetitive regions in the Rubroshorea johorensis plastome. (A) Number of different SSRs types. (B) Distribution of repetitive regions. (C) Number of different SSRs motifs.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can, be found below: https://www.ebi.ac.uk/ena/browser/view/PRJEB73584, https://www.ebi.ac.uk/ena/browser/view/SAMEA115502444, https://www.ebi.ac.uk/ena/browser/view/ERR13245837, https://www.ncbi.nlm.nih.gov/genbank/, PV078022, https://www.ncbi.nlm.nih.gov/genbank/, PV540234.
Author contributions
AN: Formal Analysis, Writing – original draft. ER: Methodology, Writing – review and editing. NW: Methodology, Writing – review and editing. FI: Formal Analysis, Writing – original draft. DJS: Writing – review and editing. MAS: Writing – original draft. MM: Writing – review and editing. FGD: Data curation, Writing – review and editing. RP: Writing – review and editing. IZS: Conceptualization, Funding acquisition, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by the National Competitive Basic Research Scheme for 2022-2023 (Skema Penelitian Dasar Kompetitif Nasional 2022-2023) under Grant No: 001/E5/PG.02.00PT/2022 and 001/E5/PG.02.00.PL/2023 (between DIKBUDRISTEK and IPB University), as well as Grant No: 15850/IT3.D10/PT.01.02/P/T/2023 (between Directorate of Research and Innovation IPB University and the Principal Investigator, IZS), entitled “Pilot Sequencing of 100 Native Forest Tree Genomes to Support Ecosystem Restoration (Rintisan Sekuensing 100 Genom Pohon Hutan Asli untuk Mendukung Restorasi Ekosistem)”, which was awarded by the Ministry of Education, Culture, Research, and Technology of the Republic of Indonesia (DIKBUDRISTEK). Furthermore, the study was funded by the Early Career Researcher Internship Program, which was awarded by the National Research and Innovation Agency (BRIN). The ORG.one program by Oxford Nanopore Technologies (ONT) also provides support for flow cells used in genome sequencing.
Acknowledgments
The authors acknowledge the significant contributions of Bogor Botanical Gardens in granting access to research materials. The authors also express gratitude to the Forest Genetics and Molecular Forestry Laboratory at IPB University for providing the necessary research consumables and ORG.one for supplying research materials needed for the sequencing activities. We sincerely thank Siska Nur Fajri for her valuable assistance with DNA extraction.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1574266/full#supplementary-material
References
Aisha, F., Din, L. B., and Yaacob, W. A. (2014). Resveratrol tetramer of hopeaphenol isolated from Shorea johorensis (Dipterocarpaceae). AIP Conf. Proc. 1614, 302–308. doi:10.1063/1.4895212
Ashton, P. S., and Heckenhauer, J. (2022). Tribe shoreae Dipterocarpaceae subfamily dipterocarpoideae finally dissected. Kew Bull. 77, 885–903. doi:10.1007/s12225-022-10057-w
Burger, G., Gray, M. W., and Lang, B. F. (2003). Mitochondrial genomes: anything goes. Trends Genets 19, 709–716. doi:10.1016/j.tig.2003.10.012
Chevigny, N., Schatz-Daas, D., Lotfi, F., and Gualberto, J. M. (2020). DNA repair and the stability of the plant mitochondrial genome. Int. J. Mol. Sci. 21, 328. doi:10.3390/ijms21010328
Chew, I. Y. Y., Chung, H. H., Lim, L. W. K., Lau, M. M. L. L., Gan, H. M., Wee, B. S., et al. (2023). Complete chloroplast genome data of Shorea macrophylla (Engkabang): structural features, comparative and phylogenetic analysis. Data Brief. 47, 109029. doi:10.1016/j.dib.2023.109029
Daniell, H., Li, C.-S., Yu, M., and Chang, W.-J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 134. doi:10.1186/s13059-016-1004-2
De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., and Van Broeckhoven, C. (2018). NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34 (15), 2666–2669. doi:10.1093/bioinformatics/bty149
Drouin, G., Daoud, H., and Xia, J. (2008). Relative rates of synonymous substitutions in the mitochondrial, chloroplast, and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 49, 827–831. doi:10.1016/j.ympev.2008.09.009
Fathiya, N., Harnelly, E., Thomy, Z., and Iqbar, I. (2018). Molecular identification of Shorea johorensis in ketambe research station, gunung leuser national park. J. Nat. 18 (2), 57–64. doi:10.24815/jn.v18i2.10123
Gaveau, D. L., Sheil, D., Husnayaen, , Salim, M. A., Arjasakusuma, S., Ancrenaz, M., et al. (2016). Rapid conversions and avoided deforestation: examining four decades of industrial plantation expansion in Borneo. Sci. Rep. 6, 32017. doi:10.1038/srep32017
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47 (W1), W59–W64. doi:10.1093/nar/gkz238
Harahap, N. N., Siregar, I. Z., and Dwiyanti, F. G. (2018). Root architecture and its relation with the growth characteristics of three planted Shorea species (Dipterocarpaceae). IOP Conf. Ser. Earth Environ. Sci. 203, 012016. doi:10.1088/1755-1315/203/1/012016
IUCN (2024). The IUCN red list of threatened species version 2024-2. Available online at: https://www.iucnredlist.org/species/33119/68073900 (Accessed December 27, 2024).
Johnston, I. G. (2019). Tension and resolution: dynamic, evolving populations of organelle genomes within plant cells. Mol. Plant. 12, 764–783. doi:10.1016/j.molp.2018.11.002
Kim, J. E., Kim, K. M., Kim, Y. S., Chung, G. Y., Che, S. H., and Na, C. S. (2024). Chloroplast genomes of Vitis flexuosa and Vitis amurensis: molecular structure, phylogenetic, and comparative analyses for wild plant conservation. Genes 15, 761. doi:10.3390/genes15060761
Kolmogorov, M., Tuan, J., Lin, T., and Pevzner, P. (2019). Assembly of long error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi:10.1038/s41587-019-0072-8
Kuznetsov, A., and Bollin, C. J. (2021). NCBI genome workbench: desktop software for comparative genomics, visualization, and GenBank data submission. Methods Mol. Biol. 2231, 261–295. doi:10.1007/978-1-0716-1036-7_16
Li, J., Ni, Y., Lu, Q., Chen, H., and Liu, C. (2024). PMGA: a plant mitochondrial genome annotator. Plant Commun. 9, 101191. doi:10.1016/j.xplc.2024.101191
Li, L. F., Yang, M., Qi, Y., Gao, P.-H., Yang, S.-W., Zhao, Y.-T., et al. (2024). Chloroplast genome of four Amorphophallus species: genomic features, comparative analysis, and phylogenetic relationships among Amorphophallus species. BMC Genom 25, 1122. doi:10.1186/s12864-024-11053-z
Lim, L. W. K., Chung, H. H., and Hussain, H. (2020). Complete chloroplast genome sequencing of sago palm (Metroxylon sagu Rottb.): molecular structures, comparative analysis and evolutionary significance. Gene Rep. 19, 100662. doi:10.1016/j.genrep.2020.100662
Mahapatra, K., Banerjee, S., De, S., Mitra, M., Roy, P., and Roy, S. (2021). An insight into the mechanism of plant organelle genome maintenance and implications of organelle genome in crop improvement: an update. Front. Cell Dev. Biol. 9, 671698. doi:10.3389/fcell.2021.671698
Morley, S., Ahmad, N., and Nielsen, B. (2019). Plant organelle genome replication. Plants 8, 358. doi:10.3390/plants8100358
Mower, J. P., Sloan, D. B., and Alverson, A. J. (2012). “Plant mitochondrial genome diversity: the genomics revolution,” in Plant genome diversity volume 1. Editors J. Wendel, J. Greilhuber, J. Dolezel, and I. Leitch (Vienna: Springer). doi:10.1007/978-3-7091-1130-7_9
Okonechnikov, K., Golosova, O., and Fursov, M.Ugene Team (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28 (8), 1166–1167. doi:10.1093/bioinformatics/bts091
Purwaningsih, , and Kintamani, E. (2018). The diversity of Shorea spp. (meranti) at some habitats in Indonesia. IOP Conf. Ser. Earth Environ. Sci. 197, 012034. doi:10.1088/1755-1315/197/1/012034
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47 (W1), W65–W73. doi:10.1093/nar/gkz345
Sloan, D. B., Alverson, A. J., Chuckalovcak, J. P., Wu, M., McCauley, D. E., Palmer, J. D., et al. (2012). Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 10 (1), e1001241. doi:10.1371/journal.pbio.1001241
Song, Y., Chen, Y., Lv, J., Xu, J., Zhu, S., and Li, M. (2019). Comparative chloroplast genomes of sorghum species: sequence divergence and phylogenetic relationships. Biomed. Res. Int. 2019, 5046958–5047011. doi:10.1155/2019/5046958
Suciyati, A., Suryadarma, I. G. P., Paidi, , and Abrori, F. M. (2021). Ethnobotanical study based on the five dimensions of basic life needs in Tidung Tribe of North Kalimantan, Indonesia. Biodiversitas 22 (6), 3199–3208. doi:10.13057/biodiv/d220623
Wahyudi, A., Sari, N., Saridan, S., Cahyono, D. D. N., Rayan, N. M., Fernades, A. A., et al. (2014). Shorea leprosula miq dan Shorea johorensis Foxw: ekologi, silvikultur, budidaya dan pengembangan. Samarinda: Balai Besar Penelitian Dipterokarpa.
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one 9 (11), e112963. doi:10.1371/journal.pone.0112963
Widiyatno, P. S., Soekotjo, , Naiem, M., Hardiwinoto, S., and Kasmujiono, (2013). The growth of selected Shorea spp in secondary tropical rain forest: the effect of silviculture treatment to improve growth quality of Shorea spp. Procedia Environ. Sci. 17, 160–166. doi:10.1016/j.proenv.2013.02.024
Widiyatno, S., Naiem, M., Purnomo, S., and Setiayanto, P. E. (2014). Early performance of 23 dipterocarp species planted in logged-over rainforest. J. Trop. For. Sci. 26 (2), 259–266.
Yu, Y., Han, Y., Peng, Y., Tian, Z., Zeng, P., Zongm, H., et al. (2021). Comparative and phylogenetic analyses of eleven complete chloroplast genomes of Dipterocarpoideae. Chin. Med. 16, 125. doi:10.1186/s13020-021-00538-8
Keywords: circular genome, conservation, dipterocarpaceae, MinION, mitogenome, plastome
Citation: Nugroho A, Rustam E, Widyani N, Indriani F, Sudrajat DJ, Salim MA, Majiidu M, Dwiyanti FG, Pratama R and Siregar IZ (2025) Complete assembly of the organellar genome of Rubroshorea johorensis utilizing advanced long-read sequencing technologies. Front. Genet. 16:1574266. doi: 10.3389/fgene.2025.1574266
Received: 10 February 2025; Accepted: 25 April 2025;
Published: 14 May 2025.
Edited by:
Dawei Xue, Hangzhou Normal University, ChinaReviewed by:
Edi Sudianto, University of Liège, BelgiumJihan Li, Chinese Academy of Sciences (CAS), China
Copyright © 2025 Nugroho, Rustam, Widyani, Indriani, Sudrajat, Salim, Majiidu, Dwiyanti, Pratama and Siregar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fitri Indriani, Zml0cmkuaW5kcmlhbmlAYnJpbi5nby5pZA==; Iskandar Z. Siregar, c2lyZWdhckBhcHBzLmlwYi5hYy5pZA==