 Shunxian Zhou1
Shunxian Zhou1 Lei Wang
Lei Wang- 1College of Information Science and Engineering, Hunan Women’s University, Changsha, China
- 2The First Hospital of Hunan University of Chinese Medicine, Changsha, China
- 3Intelligent Equipment School, Changsha Rail Transit Institute, Changsha, China
- 4Changsha Technology Innovation Center of Artificial Intelligence Large Model Training, Changsha University, Changsha, China
Introduction: In recent years, lots of computational models have been proposed to infer potential lncRNA-disease associations.
Methods: In this manuscript, we introduced a novel end-to-end learning framework named CNMCLDA, in which, we first adopted two convolutional neural networks to extract hidden features of diseases and lncRNAs separately. And then, by combining these hidden features of diseases and lncRNAs with known lncRNA-disease associations, we designed five different loss functions. Next, based on errors obtained by these loss functions, we would perform back propagation to fit parameters in CNMCLDA, and complete those missing values in lncRNA-disease relational matrix according to these fitted parameters. In order to demonstrate the prediction performance of CNMCLDA, intensive experiments have been carried out and experimental results show that CNMCLDA can achieve better performances than state-of-the-art competitive predictive models in frameworks of five-fold cross validation, ten-fold cross validation and leave-one-disease-out cross validation respectively.
Results and Discussion: Moreover, in case studies of gastric cancer, glioma and breast cancer, there are 19, 17 and 16 out of top 20 candidate lncRNAs inferred by CNMCLDA having been confirmed by recent relevant literatures separately, which demonstrated the outstanding performance of CNMCLDA as well. Hence, it is obvious that CNMCLDA may be an effective tool for prediction of potential lncRNA-disease associations in the future.
1 Introduction
In the last few years, more and more researches have pointed out that lncRNAs play a significant role in some biological processes (Statello et al., 2021) and are associated with many human diseases including HIV (Zhou et al., 2023), cardiovascular diseases (Congrains et al., 2012), leukemia (Calin et al., 2007), various cancers (Yang et al., 2023; Gupta et al., 2010; Ci et al., 2024), etc. Hence, prediction of possible associations between lncRNAs and diseases can not only contribute to understand the pathogenesis of human diseases at the molecular level, but also provide a novel perspective for new drug development and personalized medication (Wu et al., 2021). Up to now, researchers have established a series of publicly available databases including lncRNAdb (Amaral et al., 2011; Cheng et al., 2015), NONCODE (Bu et al., 2012), LncRNADisease (Bao et al., 2019), and NRED (Dinger et al., 2009) etc., and based on these databases, lots of computational methods have been proposed successively, which can be roughly classified into three major categories according to their implementation strategies (Chen et al., 2017; Fan et al., 2022). The first type of approach is mainly based on different machine learning models, for instance, Zhou and Peng et al. established a prediction model by using a boosting-based ensemble learning model (Zhou et al., 2024). Yu and Wang et al. adopted the Naïve Bayes classifier to predict potential associations between lncRNAs and diseases (Jingwen et al., 2018; Yu et al., 2019). Xuan and Wang et al. utilized probability matrix decomposition to infer latent lncRNA-disease associations (Xuan et al., 2019). Wang et al. developed a novel model named gGATLDA for lncRNA-disease association prediction based on graph-level graph attention network (Wang and Zhong, 2022). Zhang et al. designed a lncRNA-disease association prediction tool development based on bridge heterogeneous information network via graph representation learning for family medicine and primary care (Zhang et al., 2023). The second type of approach is mainly based on the network topologies, for example, Sun et al. predicted potential lncRNA-disease association by applying random walk with restart on the lncRNA functional similarity network (Sun et al., 2014). Zhang et al. proposed a computational model by implementing flow propagation algorithm on multiple heterogeneous networks (Zhang et al., 2017). Chen et al. constructed an effective prediction model named KATZLDA by integrating the lncRNA functional similarity and the disease semantic similarity with known lncRNA-disease associations (Chen, 2015a). Different from the above two types of methods, which mainly rely on known lncrNa-disease associations verified by biological experiments to infer potential lncrNa-disease associations, the third type of approach mainly focuses on adopting indirect biological information to infer potential lncRNA-disease associations, which can achieve satisfactory predictive performance while lack of known lncRNA-disease associations. For example, Liu et al. established a novel computational model by combining disease genes and expression profiles of lncRNA (Liu et al., 2014). Through above descriptions, it is easy to know that those existing computational models exist the following limitations: (1) Lots of existing methods are strongly dependent on known lncRNA-disease associations. (2) Machine learning based methods randomly select unlabeled samples as negative samples, or directly take all unlabeled samples as negatives. (3) Most existing methods cannot predict potential associations between lncRNAs and diseases having no known associations with lncRNAs.
Therefore, in order to overcome above limitations of traditional forecasting models, in this paper, the prediction of potential diseases related lncRNAs will first be regarded as completion of missing values in a lncRNA-disease relational matrix, which has been demonstrated to be practical and effective in many bioinformatics fields. For example, in 2022, Yan et al. proposed a matrix completion model for drug repositioning (Yan et al., 2022), which can achieve satisfactory prediction performance. In 2024, Shi et al. designed a novel prediction model, which can effectively infer potential associations between microbes and diseases based on graph autoencoder and inductive matrix completion (Shi et al., 2024). Certainly, there are also some computational models designed to predict potential lncRNA-disease associations based on the idea of matrix completion. For instances, Lu et al. constructed a lncRNA-disease association prediction model based on the inductive matrix completion (Lu et al., 2018). Different from these existing matrix completion based models, in this paper, we developed a novel end-to-end learning framework called CNMCLDA to complete the lncRNA-disease relational matrix, in which, we combined known lncRNA-disease associations with known lncRNA-miRNA associations and known miRNA-disease associations, which ensured that CNMCLDA could achieve better performance than those existing prediction models based only on known lncRNA-disease associations. And at the same time, we further integrated five different loss functions to update parameters in CNMCLDA, and considered the balance between positive and passive samples in CNMCLDA, thus ensuring that CNMCLDA would be more powerful and effective. Finally, in order to demonstrate the effectiveness and superiority of CNMCLDA, we first compared it with nine state-of-the-art models under frameworks of 5-fold CV (cross-validation) and 10-fold CV respectively, and experimental results showed that CNMCLDA achieved reliable AUC values of 0.9235 and 0.9446 in 5-fold CV and 10-fold CV separately, which were higher than all those competitive models. Secondly, in view of limitation that some existing models cannot be applied to infer potential associations between lncRNAs and diseases without known associated lncRNAs, we further adopted a novel evaluation index named LODOCV (leave-one-disease-out cross validation) to assess the predictive performance between CNMCLDA and four of above nine state-of-the-art models that can be applied to infer potential associations between lncRNAs and diseases without known associated lncRNAs, and experimental results illustrated that CNMCLDA achieved better performance than all these competitive models simultaneously. Furthermore, in order to verify the adaptability of CNMCLDA, we downloaded and applied another different dataset to evaluate the prediction performance of CNMCLDA, and experimental results showed that CNMCLDA achieved satisfactory performance as well. Finally, in case studies of gastric cancer, glioma and breast cancer, experimental results illustrated that there were 19,17 and 16 of top 20 candidate lncRNAs predicted by CNMCLDA having been confirmed by recent literatures, which also demonstrated that CNMCLDA may become a vital tool to explore potential relationships between lncRNAs and diseases in the future.
2 Materials
2.1 Data collection and preprocessing
In this section, we firstly collected known miRNA-disease associations, miRNA-lncRNA associations and lncRNA-disease associations from public databases of HMDD (Cui et al., 2023), starBasev2.0 (Li et al., 2014) and MNDRv2.0 (Cui et al., 2018) respectively. After removing duplicated associations, we finally obtained 246 different miRNAs, 1,089 different lncRNAs, 373 different diseases, a dataset SMD consisting of 4,704 known miRNA-disease associations between all these 246 miRNAs and 373 diseases, a dataset SML consisting of 9,086 known miRNA-lncRNA associations between all these 246 miRNAs and 1,089 lncRNAs, and a dataset SLD consisting of 407 known lncRNA-disease associations between 77 of all these 1,089 lncRNAs and 95 of all these 373 diseases. For convenience, let nm, nd, nl, nl_ld and nd_ld denote the numbers of all these 246 miRNAs, 373 diseases, 1,089 lncRNAs, 77 lncRNAs and 95 diseases separately, and NM, ND, NL, NL_LD and ND_LD represent the sets consisting of all these 246 miRNAs, 373 diseases, 1,089 lncRNAs, 77 lncRNAs and 95 diseases respectively, then a nm
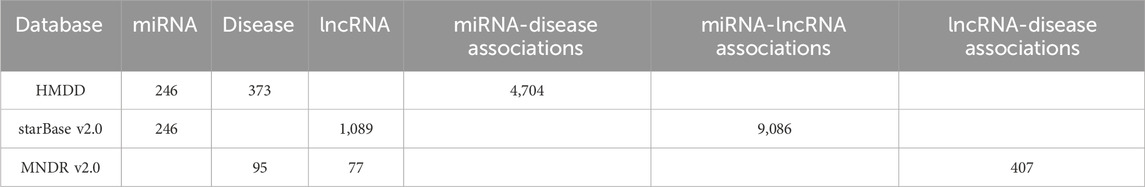
Table 1. Data downloaded from public databases HMDD, starBasev2.0 and MNDRv2.0.
2.2 Calculation of disease semantic similarity and lncRNA function similarity
In recent years, the semantic similarity of disease has been widely utilized in the field of bioinformatics, and especially in prediction of associations between diseases and lncRNAs (Wang et al., 2019; Xiao et al., 2020). In this section, we would adopt the semantic similarity of disease in CNMCLDA in the following way: Firstly, for each disease downloaded above, we would obtain its corresponding MESH (Medical Subject Headings) descriptors from the U.S. National Library of Medicine (http://www.nlm.nih.gov/), which was denoted as a Directed Acyclic Graph (DAG). And then, base on these DAGs, we would obtain the semantic similarity scores across all diseases, and a corresponding semantic similarity score matrix SD∈Rnd×nd. Next, by combining the matrix SD with the matrix LD obtained previously, we would further adopt the method proposed in reference (Xiao et al., 2020) to calculate the functional similarity of lncRNA, and obtain a corresponding functional similarity score matrix SL∈Rnl x nl as well.
3 Construction of the CNMCLDA
The goal of CNMCLDA is to fill those missing values in the original lncRNA-disease relational matrix LD. The traditional solution is to find two matrix W and H that satisfy the following Formula 1:
Different from above traditional method, CNMCLDA introduced a novel learning framework to fill in the matrix LD. As shown in Figure 1, CNMCLDA consists of three major parts including the disease sub-model part, the lncRNA sub-model part and the part of matrix completion. Among them, the disease sub-model part and the lncRNA sub-model part are utilized to extract hidden features of diseases and lncRNAs by adopting CNNs separately, while the part of matrix completion is designed to obtain the predicted scores of possible lncRNA-disease associations.
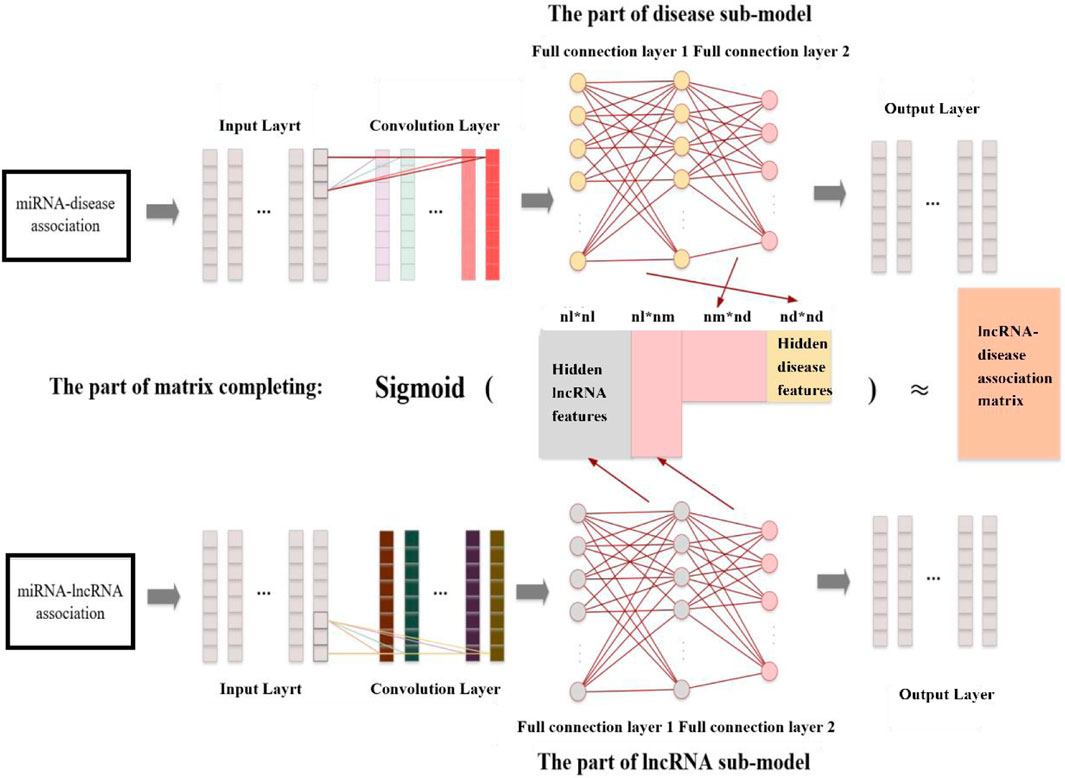
Figure 1. Flowchart of CNMCLDA.
Specifically, the main processes of CNMCLDA can be described as follows:
Step 1 : Designing a CNN for the disease sub-model part to extract hidden features of diseases by inputting MD.
Step 2 : Designing a CNN for the lncRNA sub-model part to extract hidden features of lncRNAs by inputting ML.
Step 3 : Calculating the predicted score matrix of lncRNA-disease associations based on the newly obtained hidden features of diseases and lncRNAs.
Step 3.1: Balancing positive and negative samples in LD.
Step 3.2: Constructing loss functions for CNNs.
Step 3.3: Calculating error values based on loss functions and updating parameters in CNMCLDA by back propagation.
Step 4 : Repeating steps 1 through 3 until CNMCLDA reaches a steady state.
3.1 Design of CNN for the disease sub-model to extract hidden features of diseases
CNN is a common deep learning architecture that excels in image recognition, natural language processing, etc (Gu et al., 2018). In this section, we would design a CNN consisting of a convolutional layer and three fully-connected layers for the disease-sub model part to extract hidden features of diseases firstly. For convenience, we would set the number of convolutional kernels to nd, and let
where
Thereafter, we can integrate all these feature maps as outputs of the convolutional layer.
Additionally, in these three fully-connected layers, the inputs of each layer can be derived by combining outputs of the previous layer with the weight matrix
3.2 Design of CNN for the lncRNA sub-model to extract hidden features of lncRNAs
In this section, we would further design a CNN consisting of a convolutional layer and three fully-connected layers for the lncRNA sub-model part to extract hidden features of lncRNAs. In a similar way, For convenience, we would the number of convolutional kernels to nl, and let
Thereafter, by combining all these feature maps, we can obtain the output of the convolutional layer as well. And moreover, the dimension of the weight matrix
3.3 Calculating the predicted score matrix of lncRNA-disease associations
Firstly, as the number of known lncrNa-disease associations is very limited, the number of elements equal to 0 in the original lncrNa-disease association matrix LD is far greater than the number equal to 1. For convenience, we call these elements equal to 0 or 1 as negative samples and positive samples, respectively, it is obvious that the proportion of positive samples and negative samples in the original lncrNa-disease relationship matrix LD is quite unbalanced, which makes it unreasonable to directly implement CNMCLDA on the original lncrNa-disease relationship matrix LD. Therefore, before implementing CNMCLDA, we will implement a division on the positive and negative samples of LD to ensure the approximate balance of positive and negative samples. Inspired by the method of KATZ (Sun et al., 2014), we will first construct a matrix FLD as the Formula 7:
And then, we will randomly select negative samples with amount equaling to the number of positive samples from the part of the matrix FLD with element of 0. Obviously, in this way, the positive and negative samples will be approximately balanced.
Next, considering that our main objective is to fill in the missing values in LD, therefore, based on features extracted from two CNNs, we will define the main loss function as the Formulas 8, 9:
where
Obviously, the above Formula 8 can only be used to calculate the error values of heterogeneous nodes (i.e., the positive and negative samples in LD) in the sample set.
However, in CNNs of the disease-sub model and lncRNA sub-model, since we hope that the output of each CNN will be equivalent to the associations between the current node and all miRNA nodes, hence, we will define another loss function for this purpose as Formula 10:
where
Moreover, by combining the semantic similarity of disease with the functional similarity of lncRNA, we can define a novel loss function as Formula 11:
Additionally, based on the framework of general matrix completion model, we can define a novel loss function as Formula 12:
Finally, based on the weights and bias in the CNN, we can further define a novel loss function as Formula 13:
Thereafter, by integrating above five loss functions, we can obtain a total loss function as Formula 14:
Finally, based on the total loss function, we can further adopt the Adam optimization method (Shi et al., 2024) to iteratively optimize the hyper-parameters in CNMCLDA. And In the actual deployment CNMCLDA, considering the time cost and precision requirements, the iteration process will stop when the value of losstotal is less than 10−3. Hence, we can finally obtain the predicted scores of possible lncRNA-disease associations as Formula 15:
4 Performance evaluation
In this section, we compared CNMCLDA with seven state-of-the-art models including NBCLDA (Jingwen et al., 2018), CFNBC (Yu et al., 2019), PMFILDA (Xuan et al., 2019), gGATLDA (Wang and Zhong, 2022), LDAGRL (Zhang et al., 2023), IIRWR (Wang et al., 2019), FVTLDA (Xiao et al., 2020), BIWALK (Hu et al., 2019), and LRWHLDA (Li et al., 2021). Among these competitive models, IIRWR, BIWALK and LRWHLDA adopt network propagation-based methods to infer potential lncRNA-disease associations, while NBCLDA, CFNBC, PMFILDA, gGATLDA, LDAGRL and FVTLDA adopt machine learning-based methods to predict potential associations between lncRNAs and diseases.
During experiments, frameworks of K-fold CV including 5-fold CV and 10-fold CV would be employed first to compare the prediction performances between CNMCLDA and all these competing models. While implementing K-fold CV, known lncRNA-disease associations would be divided into K equal subsets randomly, and each subset was left out as the test sample, whereas the remaining K-1 subsets were retained as training samples (Zhou et al., 2018). Moreover, all test samples and unknown lncRNA-disease associations would be considered as candidate samples. Hence, after ranking these candidate samples according to their predicted scores obtained by experiments, for a given threshold, we could obtain the TPR (True Positive Rate) and FPR (False Positive Rate) of each method according to the following Formulas 16, 17 separately:
where TP (True Positive) and FP (False Positive) represent the numbers of known and unknown lncRNA-disease associations with scores above the given threshold respectively, while FN (False Negative) and TN (True Negative) denote the numbers of known and unknown lncRNA-disease associations with scores below the given threshold respectively.
Obviously, through setting different thresholds, a unique ROC (Receiver operating characteristic) curve could be obtained by plotting TPRs against FPRs for each method. Thereafter, the AUC (Area Under the ROC Curve) could be used to evaluate the prediction performance of the given method (Hand and Till, 2001). As shown in Figures 2, 3, while implementing the 5-fold CV, CNMCLDA achieved reliable AUC value of 0.92350, which was significantly higher than all competitive models such as IIRWR (0.8653), CFNBC (0.85608), PMFILDA (0.90849), NBCLDA (0.85236), BIWALK (0.91453), LRWHLDA (0.82103), gGATLDA (0.92216), LDAGRL (0.92158), and FVTLDA (0.89050). While implementing the 10-fold CV, CNMCLDA achieved reliable AUC value of 0.94464, which was much better than all competitive models such as IIRWR (0.87302), CFNBC (0.85697), PMFILDA (0.92369), NBCLDA (0.85219), BIWALK (0.91778), LRWHLDA (0.82959), gGATLDA (0.94420), LDAGRL (0.93162), and FVTLDA (0.89360) as well.
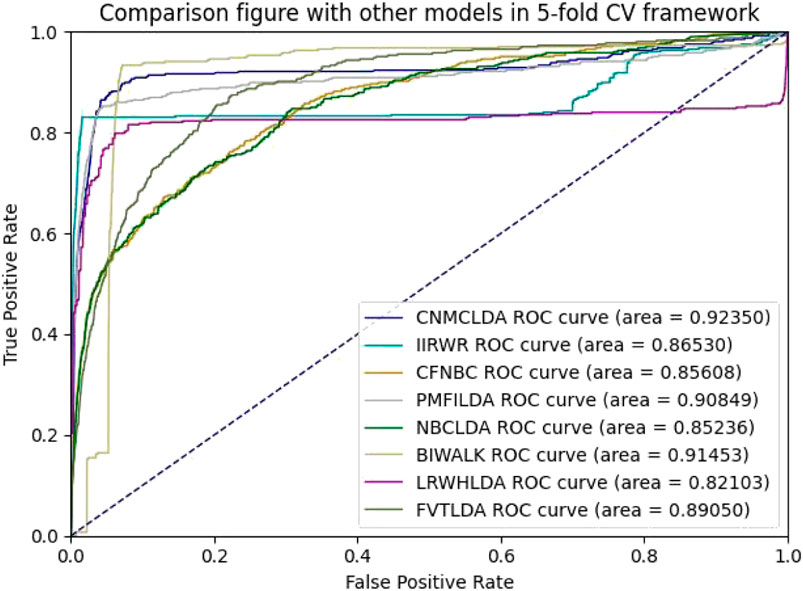
Figure 2. The AUCs achieved by CNMCLAD, IIRWR, PMFILDA, NBCLDA, CFNBC, BIWALK, LRWHLDA and FVTLDA in framework of five-fold CV.
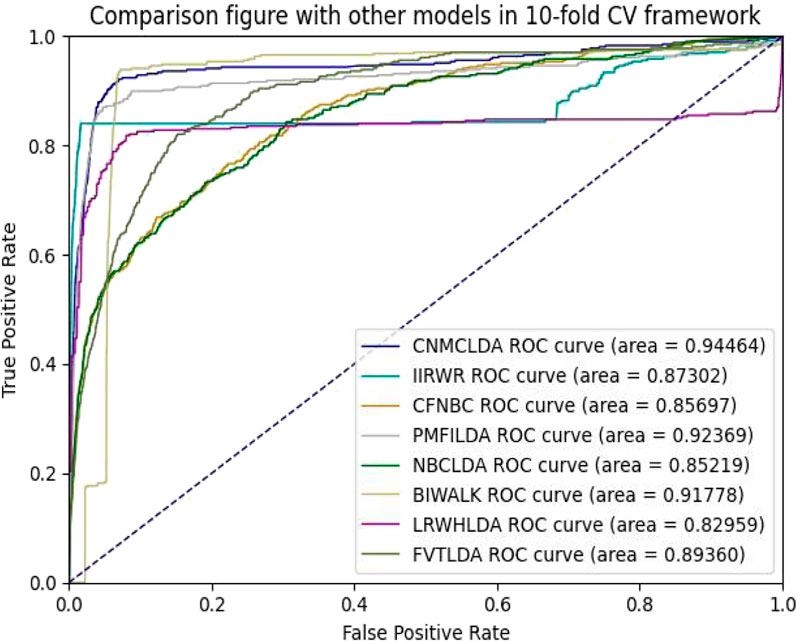
Figure 3. The AUCs achieved by CNMCLAD, IIRWR, PMFILDA, NBCLDA, CFNBC, BIWALK, LRWHLDA and FVTLDA in framework of ten-fold CV.
Moreover, in order to further evaluate the predictive ability of CNMCLDA, we introduced a novel evaluation metric called LODOCV, which could be implemented as follows: for a given disease d, all lncRNAs having known associations with d would be left out as test samples, while the remaining lncRNAs were utilized for prediction. Especially, considering that IIRWR, BIWALK and LRWHLDA are RW (Random Walk)-based methods, which cannot be used to predict lncRNAs that have no known associations with any disease, we compared CNMCLDA only with the remaining four predictive models such as CFNBC, FVTLDA, NBCLDA and PMFILDA. As shown in Figure 4, CNMCLDA achieved much better predictive performance than all these four competitive models. And meanwhile, in order to show the prediction performance of CNMCLDA more intuitively, we illustrated the means and variances of AUCs of CNMCLDA and all these four competitive models in Table 2, and the statistical significance of the predictive performance difference between CNMCLDA and all these four competitive models in Table 3, respectively.
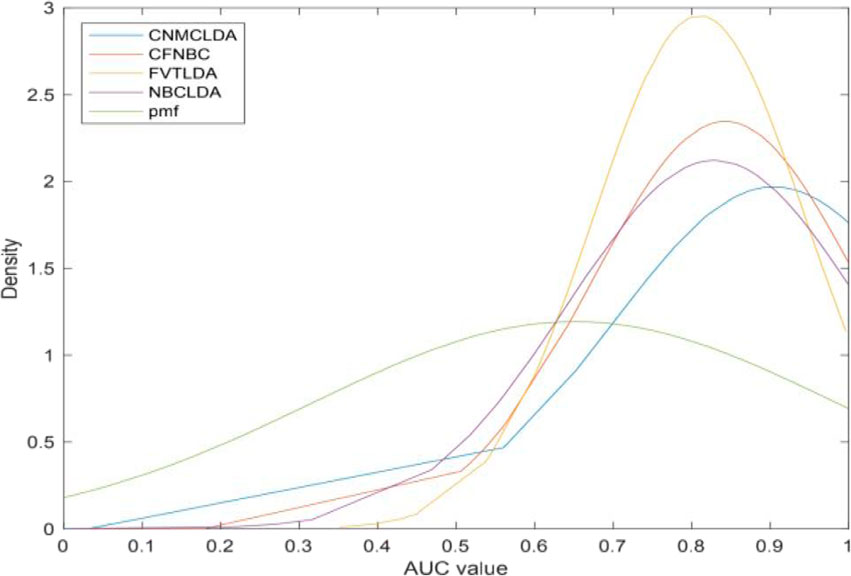
Figure 4. Performance comparison of CNMCLDA with the other four computational models in framework of LODOCV.
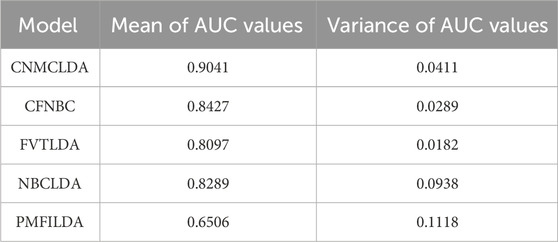
Table 2. Comparison of the means and variances of AUCs between CNMCLDA and four competitive models in the framework of LODOCV.

Table 3. Comparison of the statistical significance of performance differences between CNMCLDA and four competitive models in the framework of LODOCV.
Finally, under the framework of LOOCV (Leave-One-Out Cross Validation), we further compared the AUCs achieved by CNMCLDA and HGLDA (Chen, 2015b) based on the dataset proposed by HGLDA, which consists of 183 known lncRNA-disease associations that have been confirmed by experiments. While implementing LOOCV, each known lncRNA-disease association are selected out in turn as the test sample, and the rest associations are regarded as the training samples (Bo et al., 2006). As illustrated in Table 4, CNMCLDA can achieve an AUC of 0.8546, which is much higher that the AUC of 0.7621 achieved by HGLDA.
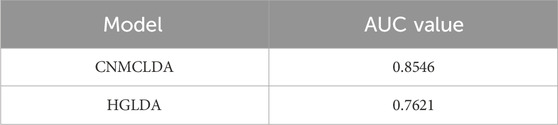
Table 4. Comparison of prediction performances between CNMCLDA and HGLDA based on the dataset proposed by HGLDA.
Therefore, it can be seen from above descriptions that CNMCLDA can achieve better prediction performance than existing state-of-the-art models.
5 Parameter analysis
As described in the method section, there are four hyper-parameters in CNMCLDA. In this section, we would evaluate the impacts of these parameters on the predictive performance of CNMCLDA under the framework of 5-fold CV. During experiments, we estimated the performance of remaining 3 parameters by fixing one parameter, and the range of each parameter would be set to {0.001,0.01,0.1,1,10} respectively. Finally, we found that CNMCLDA could achieve the best predictive results (see Supplementary Material S1) while these parameters were set as follows:
6 Case study
In this section, in order to demonstrate the effectiveness and practicability of CNMCLDA, we implemented case studies of gastric cancer, glioma, and breast cancer on known dataset having confirmed by experiments. During experiments of case studies, for a given disease d, we first regarded all lncRNAs having no known associations with d as candidates. Thereafter, all candidate lncRNAs would be ranked according to their prediction scores calculated by CNMCLDA. Finally, we would validate the relationships between top 20 candidate lncRNAs and d by the recently published papers in NCBI database (https://www.ncbi.nlm.nih.gov/).
Gastric cancer is the second most frequently leading cause of death in cancer, and it is also the fourth most common cancer in the word (Hartgrink et al., 2009; Guo et al., 2014). Recently, a larger number of literatures have confirmed the relationship between lncRNAs and gastric cancer, and lncRNAs may be therapeutic targets in patients with gastric cancer. For example, Chen et al. found that the upregulation of lncRNA XIST was related to aggressive tumor phenotypes and survive of gastric cancer (Chen et al., 2016). Yang et al. pointed out that the level of H19 in gastric cancer cells and tissues was significantly higher than that in normal control (Yang et al., 2012). As shown in Table 5(a), we listed top 20 candidate lncRNAs predicted by CNMCLDA, only one of these top 20 candidate lncRNAs has not been confirmed by recent relevant literatures. Moreover, all remaining 19 lncRNAs having been verified to be related to gastric cancer were attached with corresponding PMID (PubMed unique identifiers) in Table 5(a) as well.
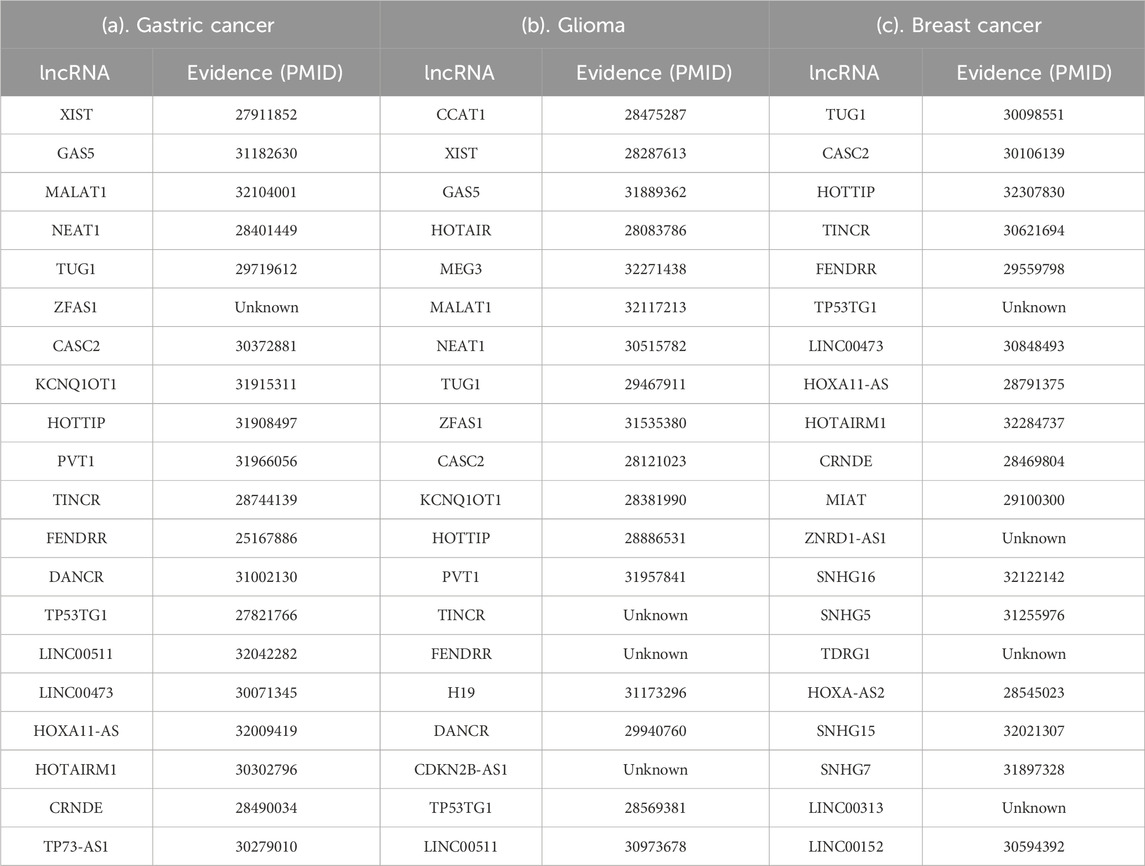
Table 5. Top 20 potential {gastric cancer, glioma, breast cancer}-related lncRNAs predicted by CNMCLDA and their PubMed unique identifiers.
As for glioma, it is a major type of adult intracranial tumors. High grade gliomas tend to infiltrate into brain extracellular matrix, which makes surgery and radiotherapy difficult (Gwak et al., 2012). A lot of evidences show that lncRNAs plays an important role in glioma. For instance, Wang et al. demonstrated that lncRNA CASC2 can play an anti-tumor role in glioma through negative regulation of MicroRNA-21 (Wang et al., 2015). Wang et al. pointed out that lncRNA HOXA11-AS is a biomarker to identify glioma and can be used as a therapeutic target for glioma patients (Wang et al., 2016). As illustrated in Table 5(b), we found 17 lncRNAs out of top 20 candidate lncRNAs predicted by CNMCLDA having been confirmed to be related to gastric cancer in recent relevant literatures.
Moreover, breast cancer is the most common cancer, and it is also the main cause of cancer death in women all over the world (DeSantis et al., 2015). Up to now, there are many relevant literatures demonstrating the relationship between lncRNA and breast cancer, such as lncRNA H19 (Sun et al., 2015), lncRNA UCA1 (Xiao et al., 2016), lncRNA HOTAIR (Xue et al., 2015) and so on. As illustrated in Table 5(c), there are 16 out of top 20 candidate lncRNAs predicted by CNMCLDA having been reported in recent literatures. Hence, based on experimental results of above case studies, we can conclude that CNMCLDA has excellent prediction ability.
7 Discussion
In this study, different from existing methods, we regarded the prediction of potential diseases-related lncRNAs as completion of missing values of the lncRNA-disease relational matrix, and defined a novel end-to-end learning framework CNMCLDA to infer potential lncRNA-disease associations. The main contribution of CNMCLDA includes: (1) lots of existing methods are strongly dependent on known lncRNA-disease associations, however, CNMCLDA combines a variety of biological information to ensure that it does not rely only on known lncRNA-disease associations, which makes it suitable for inferring potential associations between lncRNAs and isolated diseases. (2) traditional machine learning based methods randomly select unlabeled samples as negative samples, or directly take all unlabeled samples as negatives, while CNMCLDA takes into account the balance of positive and negative samples, enabling it to achieve better predictive performance. (3) five different loss functions are designed to optimize the parameters of CNMCLDA synchronously, which makes it more effective. Certainly, CNMCLDA still has rooms for improvement. For instance, the neural network can be designed more complicated by combining the symptoms and pathological stages of diseases, and multi-view learning can be carried out as well. Meanwhile, known lncRNA-disease associations can be divided into upregulation and downregulation parts for multi-label learning. Finally, how to balance the four trade-off parameters in loss functions to achieve global optimal solutions is still a challenging task. Moreover, One of the major characteristics of lncRNA is that it has good tissue specificity and cell type specificity (Grassi et al., 2021), so it is very suitable for the study of specific related mechanisms. In terms of tumors, inflammation, immune diseases, neurological diseases, lncRNA provides a good tool for the study of heterogeneity, and is particularly suitable for future studies in marker mining or target discovery.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
SZ: Conceptualization, Writing – original draft, Writing – review and editing, Methodology. SC: Conceptualization, Data curation, Methodology, Software, Validation, Writing – review and editing. JL: Data curation, Formal Analysis, Resources, Software, Validation, Writing – review and editing. YT: Formal Analysis, Software, Writing – review and editing. LW: Conceptualization, Project administration, Supervision, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was partly sponsored by the National Natural Science Foundation of China (No.62272064), the Natural Science Foundation of Hunan Province (No.2023JJ60185) and the Key project of Changsha Science and technology Plan (No. KQ2203001).
Acknowledgments
The authors thank the referees for suggestions that helped improve the paper substantially.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1580512/full#supplementary-material
References
Amaral, P. P., Clark, M. B., Gascoigne, D. K., Dinger, M. E., and Mattick, J. S. (2011). lncRNAdb: a reference database for long noncoding RNAs. Nucleic Acids Res. 39, D146–D151. doi:10.1093/nar/gkq1138
Bao, Z., Yang, Z., Huang, Z., Zhou, Y., Cui, Q., and Dong, D. (2019). LncRNADisease 2.0: an updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 47, D1034–d1037. doi:10.1093/nar/gky905
Bo, L., Wang, L., and Jiao, L. (2006). Feature scaling for kernel fisher discriminant analysis using leave-one-out cross validation. Neural Comput. 18, 961–978. doi:10.1162/089976606775774642
Bu, D., Yu, K., Sun, S., Xie, C., Skogerbø, G., Miao, R., et al. (2012). NONCODE v3.0: integrative annotation of long noncoding RNAs. Nucleic Acids Res. 40, D210–D215. doi:10.1093/nar/gkr1175
Calin, G. A., Liu, C., Ferracin, M., Hyslop, T., Spizzo, R., Sevignani, C., et al. (2007). Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell 12 (3), 215–229. doi:10.1016/j.ccr.2007.07.027
Chen, Dl., Ju, Hq., Lu, Yx., Chen, L. Z., Zeng, Z. L., Zhang, D. S., et al. (2016). Long non-coding RNA XIST regulates gastric cancer progression by acting as a molecular sponge of miR-101 to modulate EZH2 expression. J. Exp. Clin. Cancer Res. 35, 142. doi:10.1186/s13046-016-0420-1
Chen, X. (2015a). KATZLDA: KATZ measure for the lncRNA-disease association prediction. Sci. Rep. 5, 16840. doi:10.1038/srep16840
Chen, X. (2015b). Predicting lncRNA-disease associations and constructing lncRNA functional similarity network based on the information of miRNA. Sci. Rep. 5, 13186. doi:10.1038/srep13186
Chen, X., Yan, C. C., Zhang, X., and You, Z. H. (2017). Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief. Bioinforma. 18, 558–576. doi:10.1093/bib/bbw060
Cheng, Q. X., Thomson, D. W., Maag Jesper, L. V., Bartonicek, N., Signal, B., Clark, M. B., et al. (2015). lncRNAdb v2.0: expanding the reference database for functional long noncoding RNAs. Nuclc Acids Res. 43, 168–173. doi:10.1093/nar/gku988
Ci, Y., Zhang, Y., and Zhang, X. (2024). Correction: methylated lncRNAs suppress apoptosis of gastric cancer stem cells via the lncRNA–miRNA/protein axis. Cell. and Mol. Biol. Lett. 29, 102. doi:10.1186/s11658-024-00621-6
Congrains, A., Kamide, K., Oguro, R., Yasuda, O., Miyata, K., Yamamoto, E., et al. (2012). Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis 220, 449–455. doi:10.1016/j.atherosclerosis.2011.11.017
Cui, C., Zhong, B., Fan, R., and Cui, Q. (2023). HMDD v4.0: a database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 52, D1327–D1332. doi:10.1093/nar/gkad717
Cui, T., Zhang, L., Huang, Y., Yi, Y., Tan, P., Zhao, Y., et al. (2018). MNDR v2.0: an updated resource of ncRNA-disease associations in mammals. Nucleic Acids Res. 46, D371–D374. doi:10.1093/nar/gkx1025
DeSantis, C. E., Bray, F., Ferlay, J., Lortet-Tieulent, J., Anderson, B. O., and Jemal, A. (2015). International variation in female breast cancer incidence and mortality rates. Cancer Epidemiol. Biomarkers Prev. 24 (10), 1495–1506. doi:10.1158/1055-9965.EPI-15-0535
Dinger, M. E., Pang, K. C., Mercer, T. R., Crowe, M. L., Grimmond, S. M., and Mattick, J. S. (2009). NRED: a database of long noncoding RNA expression. Nucleic Acids Res. 37, D122–D126. doi:10.1093/nar/gkn617
Fan, Y., Chen, M., and Pan, X. (2022). Gcrflda: scoring lncRNA-disease associations using graph convolution matrix completion with conditional random field. Brief. Bioinforma. 23, bbab361. doi:10.1093/bib/bbab361
Grassi, L., Izuogu, O. G., Jorge, N. A. N., Seyres, D., Bustamante, M., Burden, F., et al. (2021). Cell type-specific novel long non-coding RNA and circular RNA in the BLUEPRINT hematopoietic transcriptomes atlas. Haematologica 106 (10), 2613–2623. doi:10.3324/haematol.2019.238147
Gu, J., Wang, Z., Kuen, J., Ma, L., Shahroudy, A., Shuai, B., et al. (2018). Recent advances in convolutional neural networks. Pattern Recognit. 77, 354–377. doi:10.1016/j.patcog.2017.10.013
Guo, X., Xia, J., and Deng, K. (2014). Long non-coding RNAs: emerging players in gastric cancer. Tumor Biol. 35 (11), 10591–10600. doi:10.1007/s13277-014-2548-y
Gupta, R. A., Shah, N., Wang, K. C., Kim, J., Horlings, H. M., Wong, D. J., et al. (2010). Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076. doi:10.1038/nature08975
Gwak, H. S., Kim, T. H., Jo, G. H., Kim, Y. J., Kwak, H. J., Kim, J. H., et al. (2012). Silencing of microRNA-21 confers radio-sensitivity through inhibition of the PI3K/AKT pathway and enhancing autophagy in malignant glioma cell lines. PLoS One 7, e47449. doi:10.1371/journal.pone.0047449
Hand, D. J., and Till, R. J. (2001). A simple generalisation of the area under the ROC curve for multiple class classification problems. Mach. Learn. 45, 171–186. doi:10.1023/a:1010920819831
Hartgrink, H. H., Jansen, E. P. M., Grieken, N. C. T. V., and van de Velde, C. J. H. (2009). Gastric cancer. Lancet 374 (9688), 477–490. doi:10.1016/S0140-6736(09)60617-6
Hu, J., Gao, Y., Li, J., Zheng, Y., Wang, J., and Shang, X. (2019). A novel algorithm based on bi-random walks to identify disease-related lncRNAs. BMC Bioinforma. 20, 569. doi:10.1186/s12859-019-3128-3
Jingwen, Y., Pengyao, P., Lei, W., Kuang, L., Li, X., and Wu, Z. (2018). A novel probability model for LncRNA–disease association prediction based on the Naïve bayesian classifier. Genes 9 (7), 345. doi:10.3390/genes9070345
Li, J., Zhao, H., Xuan, Z., Yu, J., Xiang, F., Liao, B., et al. (2021). A novel approach for potential human LncRNA-disease association prediction based on local random walk. IEEE/ACM Trans. Comput. Biol. Bioinforma. 18 (3), 1049–1059. doi:10.1109/TCBB.2019.2934958
Li, J.-H., Liu, S., Zhou, H., Qu, L.-H., and Yang, J.-H. (2014). starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42, D92–D97. doi:10.1093/nar/gkt1248
Liu, M. X., Chen, X., Chen, G., Cui, Q. H., and Yan, G. Y. (2014). A computational framework to infer human disease-associated long noncoding RNAs. PloS one 9 (1), e84408. doi:10.1371/journal.pone.0084408
Lu, C., Yang, M., Luo, F., Wu, F. X., Li, M., Pan, Y., et al. (2018). Prediction of lncRNA-disease associations based on inductive matrix completion. Bioinformatics 34 (19), 3357–3364. doi:10.1093/bioinformatics/bty327
Shi, K., Huang, K., Li, L., Liu, Q., Zhang, Y., and Zheng, H. (2024). Predicting microbe-disease association based on graph autoencoder and inductive matrix completion with multi-similarities fusion. Front. Microbiol. 15, 1438942. doi:10.3389/fmicb.2024.1438942
Statello, L., Guo, C. J., Chen, L. L., and Huarte, M. (2021). Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 22, 96–118. doi:10.1038/s41580-021-00330-4
Sun, H., Wang, G., Peng, Y., Zeng, Y., Zhu, Q. N., Li, T. L., et al. (2015). H19 lncRNA mediates 17β-estradiol-induced cell proliferation in MCF-7 breast cancer cells. Oncol. Rep. 33 (6), 3045–3052. doi:10.3892/or.2015.3899
Sun, J., Shi, H., Wang, Z., Zhang, C., Liu, L., Wang, L., et al. (2014). Inferring novel lncRNA–disease associations based on a random walk model of a lncRNA functional similarity network. Mol. Biosyst. 10 (8), 2074–2081. doi:10.1039/c3mb70608g
Wang, L., Xiao, Y., Li, J., Feng, X., Li, Q., and Yang, J. (2019). IIRWR: internal inclined random walk with restart for LncRNA-disease association prediction. IEEE Access 7, 54034–54041. doi:10.1109/access.2019.2912945
Wang, L., and Zhong, C. (2022). gGATLDA: lncRNA-disease association prediction based on graph-level graph attention network. BMC Bioinforma. 23, 11–24. doi:10.1186/s12859-021-04548-z
Wang, P., Liu, Y. H., Yao, Y. L., Li, Z., Li, Z. Q., Ma, J., et al. (2015). Long non-coding RNA CASC2 suppresses malignancy in human gliomas by miR-21. Cell. Signal. 27, 275–282. doi:10.1016/j.cellsig.2014.11.011
Wang, Q., Zhang, J., and Liu, Y. (2016). A novel cell cycle-associated lncRNA, HOXA11-AS, is transcribed from the 5-prime end of the HOXA transcript and is a biomarker of progression in glioma. Cancer Lett. 1, 251–259. doi:10.1016/j.canlet.2016.01.039
Wu, Q.-W., Xia, J.-F., Ni, J.-C., and Zheng, C.-H. (2021). GAERF: predicting lncRNA-disease associations by graph auto-encoder and random forest. Briefings Bioinforma. 22, bbaa391. doi:10.1093/bib/bbaa391
Xiao, C., Wu, C. H., and Hu, H. Z. (2016). LncRNA UCA1 promotes epithelial-mesenchymal transition (EMT) of breast cancer cells via enhancing Wnt/beta-catenin signaling pathway. Eur. Rev. Med. and Pharmacol. Sci. 20 (13), 2819–2824.
Xiao, Y., Xiao, Z., Feng, X., Chen, Z., Kuang, L., and Wang, L. (2020). A novel computational model for predicting potential LncRNA-disease associations based on both direct and indirect features of LncRNA-disease pairs. BMC Bioinforma. 21, 555. doi:10.1186/s12859-020-03906-7
Xuan, Z., Li, J., Yu, J., Feng, X., Zhao, B., and Wang, L. (2019). A probabilistic matrix factorization method for identifying lncRNA-disease associations. Genes 10 (2), 126. doi:10.3390/genes10020126
Xue, X., Yang, Y. A., Zhang, A., Fong, K. W., Kim, J., Song, B., et al. (2015). LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene 35 (21), 2746–2755. doi:10.1038/onc.2015.340
Yan, Y., Yang, M., Zhao, H., Duan, G., Peng, X., and Wang, J. (2022). Drug repositioning based on multi-view learning with matrix completion. Brief. Bioinform 23 (3), bbac054. doi:10.1093/bib/bbac054
Yang, C., Liang, Y., Shu, J., Wang, S., Hong, Y., Chen, K., et al. (2023). Long non-coding RNAs in multiple myeloma. Int. J. Oncol. 62 (6), 1–12. doi:10.3892/ijo.2023.5517
Yang, F., Bi, J., Xue, X., Zheng, L., Zhi, K., Hua, J., et al. (2012). Up-regulated long non-coding RNA H19 contributes to proliferation of gastric cancer cells. FEBS J. 279 (17), 3159–3165. doi:10.1111/j.1742-4658.2012.08694.x
Yu, J., Xuan, Z., Feng, X., Zou, Q., and Wang, L. (2019). A novel collaborative filtering model for LncRNA-disease association prediction based on the Naïve Bayesian classifier. BMC Bioinforma. 20 (1), 396. doi:10.1186/s12859-019-2985-0
Zhang, J., Zhang, Z., and Chen, Z. (2017). Integrating multiple heterogeneous networks for novel LncRNA-disease association inference. IEEE/ACM Trans. Comput. Biol. and Bioinforma. 99, 1–10. doi:10.1109/TCBB.2017.2701379
Zhang, P., Zhang, W., Sun, W., Li, L., Xu, J., Wang, L., et al. (2023). A lncRNA-disease association prediction tool development based on bridge heterogeneous information network via graph representation learning for family medicine and primary care. Front. Genet. 14, 1084482. doi:10.3389/fgene.2023.1084482
Zhou, L., Peng, X., Zeng, L., and Peng, L. (2024). Finding potential lncRNA-disease associations using a boosting-based ensemble learning model. Front. Genet. 10, 1356205. doi:10.3389/fgene.2024.1356205
Zhou, S., Xuan, Z., Wang, L., Ping, P., and Pei, T. (2018). A novel model for predicting associations between diseases and lncrna-mirna pairs based on A newly constructed bipartite network. Comput. Math. Methods Med. 4, 6789089–6789113. doi:10.1155/2018/6789089
Keywords: lncRNA-disease associations, computational model, prediction model, convolutional neural network, negative samples
Citation: Zhou S, Chen S, Le J, Xu Y and Wang L (2025) A novel end-to-end learning framework for inferring lncRNA-disease associations based on convolution neural network. Front. Genet. 16:1580512. doi: 10.3389/fgene.2025.1580512
Received: 20 February 2025; Accepted: 31 March 2025;
Published: 09 April 2025.
Edited by:
Yonggang Lu, Lanzhou University, ChinaCopyright © 2025 Zhou, Chen, Le, Xu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sisi Chen, NDM3MDc5NjUzQHFxLmNvbQ==; Jinhai Le, bGVqaW5oYWlAc29odS5jb20=; Lei Wang, d2FuZ2xlaUB4dHUuZWR1LmNu