 Youqiong Li1*
Youqiong Li1* Lihua Ye2Liang Liang1Lihong Zheng1Yongjun Xiao3Zhongchan Lao4Jinping Bai1Xi He1
Lihua Ye2Liang Liang1Lihong Zheng1Yongjun Xiao3Zhongchan Lao4Jinping Bai1Xi He1 Qixun Fang5Ting Qin1
Qixun Fang5Ting Qin1- 1Center for Medical Genetics and Prenatal Diagnosis, People’s Hospital of Guangxi Zhuang Autonomous Region, Nanning, China
- 2Department of Clinical Laboratory, Women and Children Care Hospital of Laibin, Laibin, China
- 3Department of Clinical Laboratory, The Second Nanning People’s Hospital, Nanning, China
- 4Department of Clinical Laboratory, Women and Children Care Hospital of Lingshan, Qinzhou, China
- 5Department of Research and Development, Yaneng Bioscience (Shenzhen) Co. Ltd., Shenzhen, China
Objectives: δ-thalassemia and δ-globin variants are rare hemoglobinopathies. However, co-inheritance of β-thalassemia and δ-globin gene mutations may affect the diagnosis of β-thalassemia carriers when based on the elevated Hb A2. This study aimed to identify and characterize δ-thalassemia and δ-globin variants in Southern China.
Methods: Ninety samples with suspected δ-globin gene mutations from 15,642 participants were selected for further molecular analysis based on their Hb A2 level (≦1.8%) and hematological parameters. Additionally, 37 samples with suspected δ-globin gene mutations were sent from other hospital to our laboratory for identification. GAP-PCR and PCR-reverse dot blot (PCR-RDB) were used to detect common α- and β-thalassemia in the Chinese population, and Sanger sequencing was used to identify δ-globin gene mutations.
Results: Among 15,642 samples examined, samples with δ-globin gene mutations were identified in 127 (0.81%) cases with as many as 28 different genotypes, including 81 (0.52%) cases of δ-thalassemia and 46 (0.29%) cases of δ-globin variants. The most prevalent δ-thalassemia and δ-globin variants of this study were HBD:c.−127T>C (75.3%, 61/81) and Hb A2-Melbourne (54.3%, 25/46). Most of the samples were heterozygous (87.4%, 111/127), and only two cases of homozygous were detected. There were three double heterozygotes and 11 cases of combined α/β-globin mutations. Notably, we also identified eight cases of novel mutations in the δ-globin gene. In both heterozygous and homozygous cases, δ-globin mutations maintained hematological parameters within normal ranges, while their co-occurrence with α- or β-thalassemia manifested as a thalassemia phenotype characterized by significantly reduced MCV and MCH values.
Conclusion: The study reveals that δ-globin gene mutations are prevalence in the South China and necessitates integration of δ-globin screening into existing thalassemia prevention protocols.
Introduction
Hemoglobinopathies are the most common hereditary diseases in China, with a higher prevalence in region south of the Yangtze River (Huang et al., 2019; Chen et al., 2022; Wang et al., 2022). These disorders can be classified into two types: thalassemia and structural hemoglobin (Hb) variant. Thalassemia is characterized by a reduction or absence in the production of normal globin chains, while structural Hb variants are caused by amino acid substitutions in the globin chains (Viprakasit and Ekwattanakit, 2018; Vijian et al., 2021). α- and β-globin variants, along with thalassemia, are the most common and severe types of hemoglobinopathies (Lou et al., 2023; Paiboonsukwong et al., 2022). In contrast, δ-globin variant and thalassemia are less frequently reported, as the carriers of these conditions are less common in the population (Kordafshari et al., 2016; Morgado et al., 2007). The clinical presentation of these hemoglobinopathies can range from being asymptomatic to causing severe transfusion-dependent anemia accompanied by other complications (Harteveld et al., 2022). Therefore, early detection and accurate diagnosis are essential for preventing the development of severe forms of hemoglobinopathies.
Hb A2 level is a crucial hematological marker for distinguishing between α- and β-thalassemia carriers (Srivorakun et al., 2020; Colaco and Nadkarni, 2021). In normal individuals, Hb A2 accounts for less than 3.5% of total Hb and is composed of α- and δ-globin chains (α2δ2). Elevated Hb A2 levels (Hb A2>3.5%) are usually considered β-thalassemia carrier in Chinese clinical laboratories. Genetic defects in the δ-globin gene (HBD gene) can lead to a reduced Hb A2 level (Hanart et al., 2023). Our laboratory experience revealed that HbA2 values for δ-globin mutant heterozygotes ranged from 1.1% to 1.8% using the CE assay. Clinical implications are not associated with either structural Hb A2 variants (δ-globin variant) or δ-thalassemia caused by mutations in the δ-globin gene. However, co-inheritance of a mutation in the δ-globin gene and β-thalassemia might cause the phenotype of elevated Hb A2 characteristic of β-thalassemia carriers to decrease to normal or borderline level, thus causing diagnostic results to be misinterpreted (Panyasai and Pornprasert, 2020; Chen et al., 2017). In previous studies, we reported that δ-globin variants combined with β-thalassemia affected the diagnosis of β-thalassemia (Lin et al., 2024; Li et al., 2020). To improve the identification of β-thalassemia phenotype and at-risk couples in regions with high thalassemia prevalence, it is important to establish a regional database of δ-globin gene mutations.
In this study, we aim to identify the δ-thalassemia and δ-globin variants in southern China, based on reduced HbA2 levels as quantified by capillary electrophoresis. We also report eight novel mutations for the first time.
Materials and methods
Samples
The population of this study included 15,642 individuals who underwent routine screening for thalassemia in our hospital from January 2020 to December 2024. Out of a total of 15,642 samples processed, 90 samples were selected for further molecular analysis based on their Hb A2 level and hematological parameters. Based on our laboratory experience, an Hb A2 level of ≤1.8 can be used to as a screening criterion for δ-globin gene mutations after exclusion of other diseases (e.g., iron deficiency anemia). In addition, 37 samples with suspected δ-globin gene mutations were sent from outside hospitals to our laboratory for identification. This study was approved by the Ethics Committee of People’s hospital of Guangxi Zhuang Autonomous Region. Informed consents were collected from the participants.
Hematological parameters and Hb analysis
The automated blood cell counters (Sysmex, kobe, Japan) were used to assess the hematological parameters of red blood cell counts. Hb fractions separation and quantification were carried out using by capillary electrophoresis (CE) system (Sebia capillarys2 Flex Piercing; Sebia, Paris, France). The reference range for normal hematologic parameters are mean corpuscular volume (MCV) 82∼100 fL and mean corpuscular Hb (MCH) 27∼35 pg. After excluding iron deficiency anemia, subjects with low Hb A2 levels (<2.4%) were considered α-thalassemia carriers, and ≤1.8% are suspected to be carriers of the δ-globin gene mutations. The reference interval is 2.4% < Hb A2 < 3.5%.
Routine genetic test for thalassemia
Genomic DNA was extracted from peripheral blood according to the kit protocol (Yaneng Biotechnology Company, Shenzhen, China). The gap-polymerase chain reaction (Gap-PCR) was used to identify the four prevalent forms of deletional α-thalassemia in the Chinese population: --SEA, --THAI, -α3.7, and -α4.2 (Yaneng Biotechnology Company, Shenzhen, China). PCR and reverse dot blot (PCR-RDB) were used for determining the three common mutations of the α-globin gene: Hb Westmead (Hb WS), Hb Quong Sze (Hb QS), and Hb Constant Spring (Hb CS) (Yaneng Biotechnology Company, Shenzhen, China). The 17 known β-thalassemia mutations including −32 (C→A), −30 (T→C),−29 (A→G), −28 (A→G), CD14/15 (+G), CD17 (A→T), CD26 (G→A) (Hb E), CD27/28 (+C), CD31 (−C), CD41/42 (-TTCT), CD43 (G→T), CD71/72 (+A), IVS-Ⅰ-1 (G→T), IVS-Ⅰ-5 (G→C), IVS-Ⅱ-654 (C→T), 5′UTR+40–43 (−AAAC) (CAP), and Initiation codon (ATG>ACG) were analyzed by PCR-RDB (Yaneng Biotechnology Company, Shenzhen, China).
Sanger sequencing of the δ-globin gene
Sanger sequencing of the δ-globin gene was performed to ascertain the existence of mutations in the gene. The amplification primers, conditions and system were as described in our previous reports (Li et al., 2020). The PCR fragments were sequenced by an 3500XL automated genetic analyzer (ABI, Foster City, CA, United States).
Bioinformatic analysis
To assess the pathogenicity of novel δ-globin gene mutations, we employed three established computational prediction algorithms: PolyPhen-2 (probabilistic classification of missense variants), SIFT (Sorting Intolerant From Tolerant), and MutationTaster. PolyPhen-2, applicable exclusively to missense variants, generates normalized scores (0–1) reflecting deleterious potential, with classifications defined per db SNP/HGMD (The Human Gene Mutation Database) standards as benign (score ≤0.446), possibly damaging (0.447–0.908), or probably damaging (≥0.909). Parallel analysis using SIFT quantified amino acid substitution impacts through evolutionary conservation metrics, designating variants as deleterious (score ≤0.05) or tolerated (>0.05). MutationTaster provided complementary functional predictions through a Bayesian framework, categorizing variants into four clinically relevant classes: disease-causing automatic (A), disease-causing (D), polymorphism (N), or polymorphism automatic (P). This multi-algorithm approach aligns with ACMG/AMP guidelines for clinical variant interpretation and demonstrates critical utility in resolving ambiguous δ-globin variants lacking population frequency data.
Results
Prevalence of δ-globin gene
In this study, a total of 127 cases of δ-globin gene mutations were detected, including 37 samples sent to our laboratory from outside hospitals for characterization. In our hospital, 90 samples with δ-globin gene mutations were detected. Based on the number of screened cases, we can deduce that the carrier rate of the population in southern China is 0.81% (127/15,642), of which 0.52% (81/15,642) is in the case of δ-thalassemia, and the δ-globin gene variants is 0.29% (46/15,642). Of the 37 cases of δ-globin gene mutations sent from outside hospitals to our laboratory for detection, 17 were δ-thalassemia and 20 were δ-globin gene variants.
Genotype and phenotype features of δ-thalassemia
This study identified 11 distinct mutations across 81 δ-thalassemia individuals, with an overall carrier rate of 0.52% (81/15,642) in the screened poplulation (Table 1). The most prevalent δ-thalassemia mutation was HBD:c.−127T>C, representing 75.3% (61/81) of cases, followed by HBD:c.-80T>C (n = 5, 6.2%). In the heterozygous mutation state, the hematological parameters were observed as follows: Hb 144.0 ± 16.9 g/L, MCV 89.5 ± 3.8 fL, and MCH 30.1 ± 1.8 pg. A significant reduction in Hb A2 levels (range: 1.1%–1.8%; 1.3% ± 0.1%) was characteristic of δ-thalassemia in heterozygous carriers. Only one homozygous case of δ-thalassemia (HBD: c.−127T>C) was identified in this study, with CE analysis demonstrating the absence of Hb A2 peak (Figure 1A).
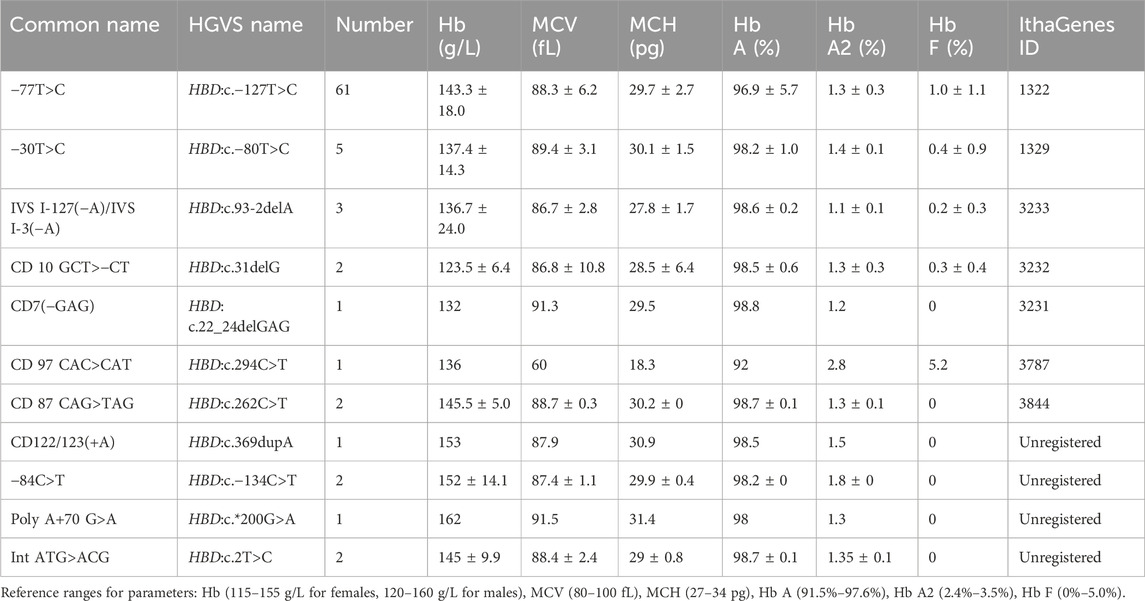
Table 1. Hematological characteristics and Hb analysis of δ-thalassemia in this study.
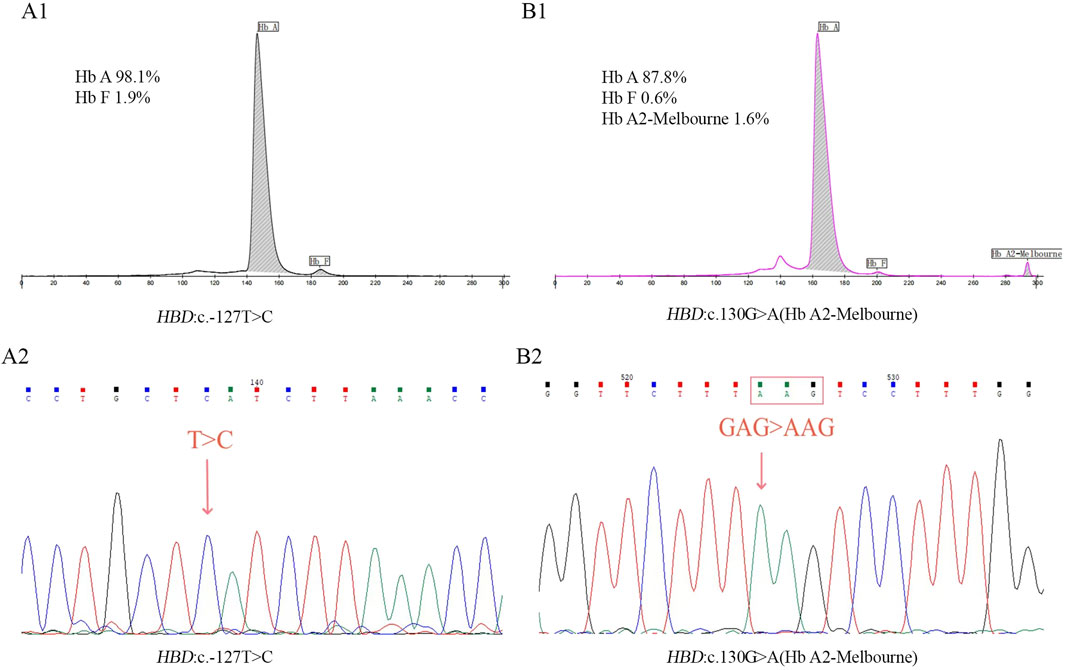
Figure 1. Results of CE (A1) and Sanger sequencing (A2) in homozygous mutation of HBD:c.-127T>C. Results of CE (B1) and Sanger sequencing (B2) in homozygous mutation of Hb A2-Melbourne.
Genotype and phenotype features of δ-globin variants
Seventeen different mutations were found in 46 δ-globin variants in this investigation, and the population that was screened had an overall carrier rate of 0.29% (46/15,642) (Table 2). Among δ-globin variants, Hb A2-Melbourne (HBD: c.130G>A) was the most frequent (n = 25, 54.3%), while other variants such as Hb A2-Coburg and Hb A2-Henan occurred in smaller cohorts (n = 2–3). In CE analyses, the majority of δ-globin variants are characterized by resolvable Hb A2 variants, while six variants remained undetectable. A homozygous case of Hb A2-Melbourne was identified, with CE revealed the lack of a visible Hb A2 peak (Figure 1B). Normal hematological parameters were preserved by δ-globin variants in both heterozygous and homozygous situations.
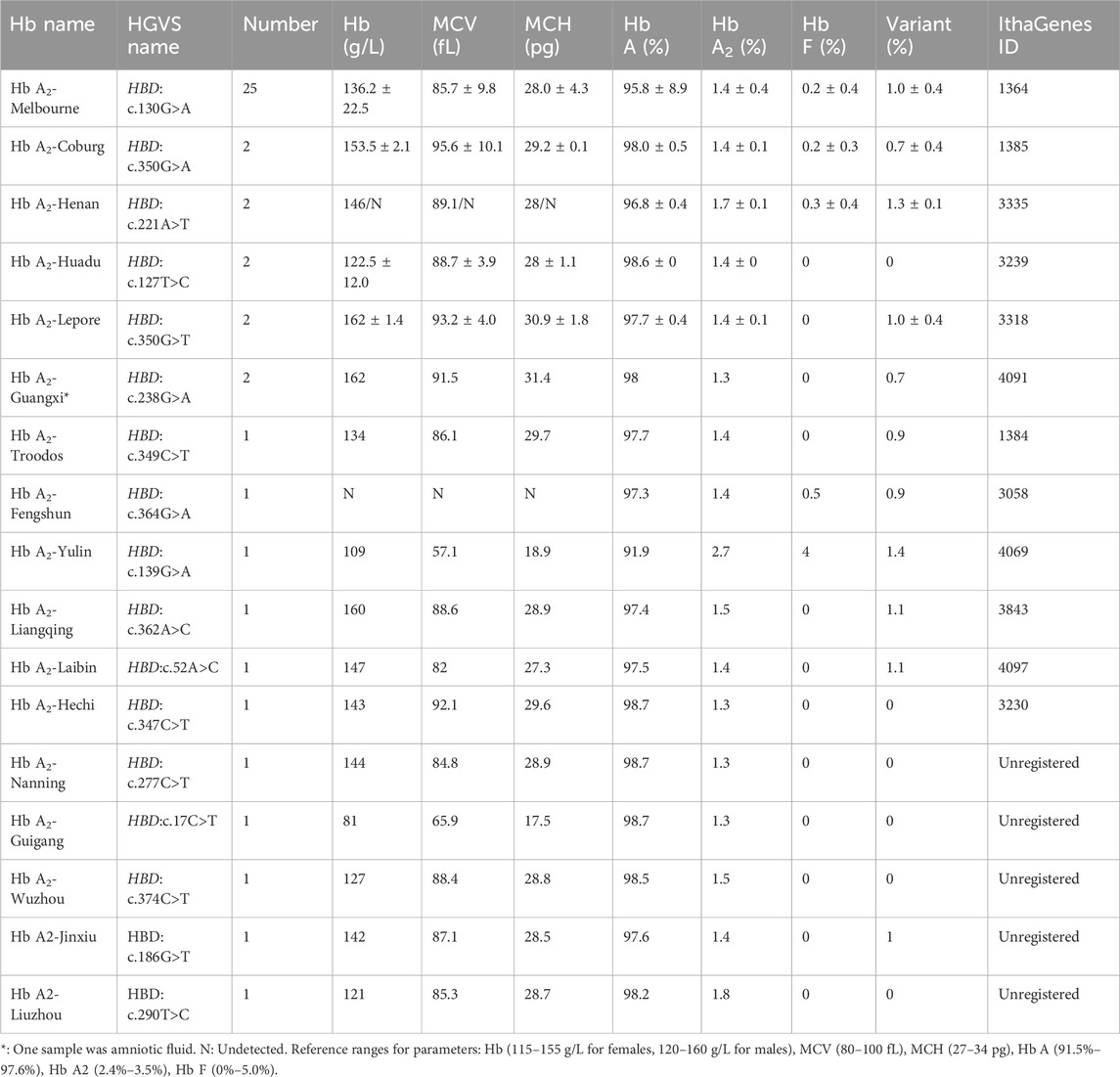
Table 2. Hematological characteristics and Hb analysis of δ-globin variants in this study.
Co-inherited δ-globin gene andα/β-globin gene mutations
Eleven cases harbored compound heterozygosity involving δ-globin and α/β-globin mutations, including 5 cases with α-globin mutations and 6 cases with β-globin mutations (Table 3). Co-inherited δ-globin gene and α/β-thalassemia resulted in a hematological phenotype that was comparable to the thalassemia it was associated with.
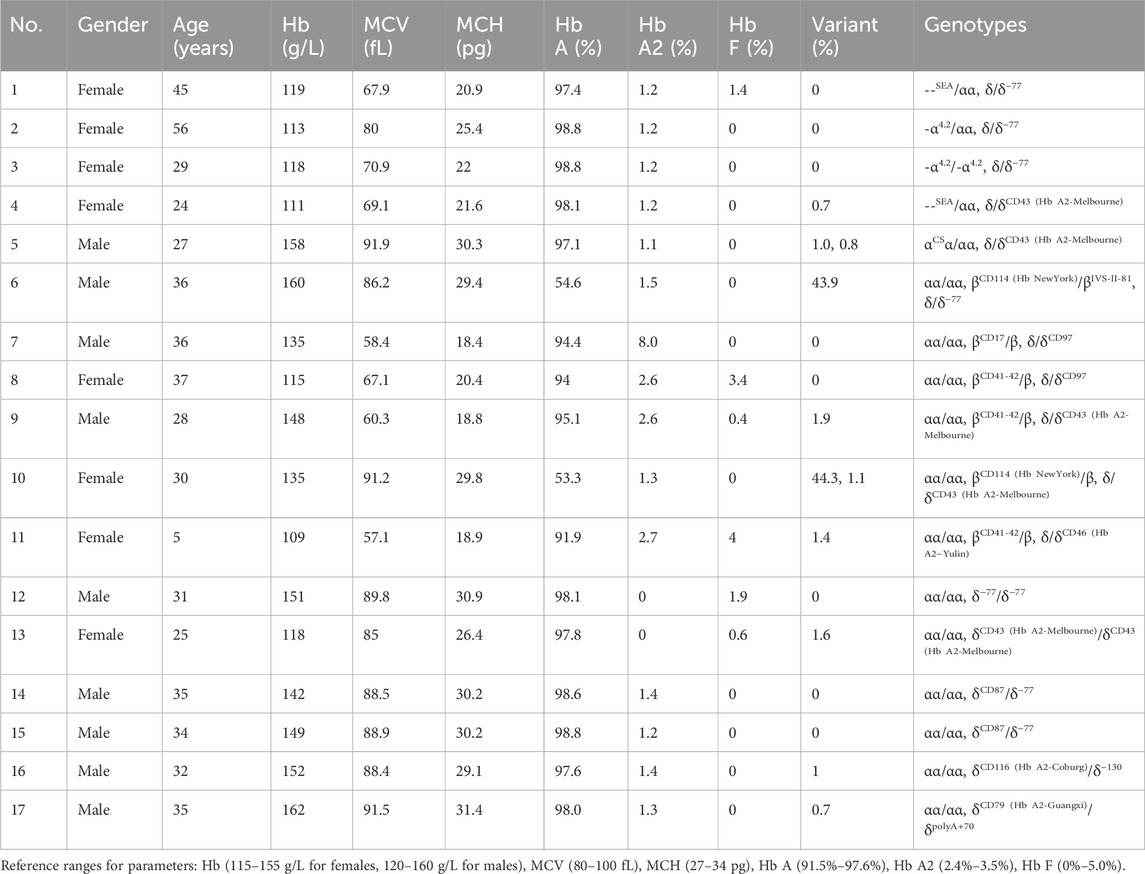
Table 3. Hematological characteristics and Hb analysis of homozygous mutation/double heterozygous mutations in δ-globin gene or coinherited δ-globin gene and α/β-globin genes.
Eight novel mutations of δ-globin gene
In this study, eitght novel δ-globin gene mutations (three δ-thalassemia and five δ-globin variants) were identified through systematic molecular and Hb analyses (Figures 2, 3). Hematological profiling presented nomal erythrocyte indices in most carriers. Quantification of Hb fractions by CE demostrated uniformly reduced Hb A2 levels across all variants (1.3%–1.8%). All variants were classified as novel mutations based on absence in population databases. Bioinformatic analysis showed that seven of the eight novel mutations were deleterious (Table 4).
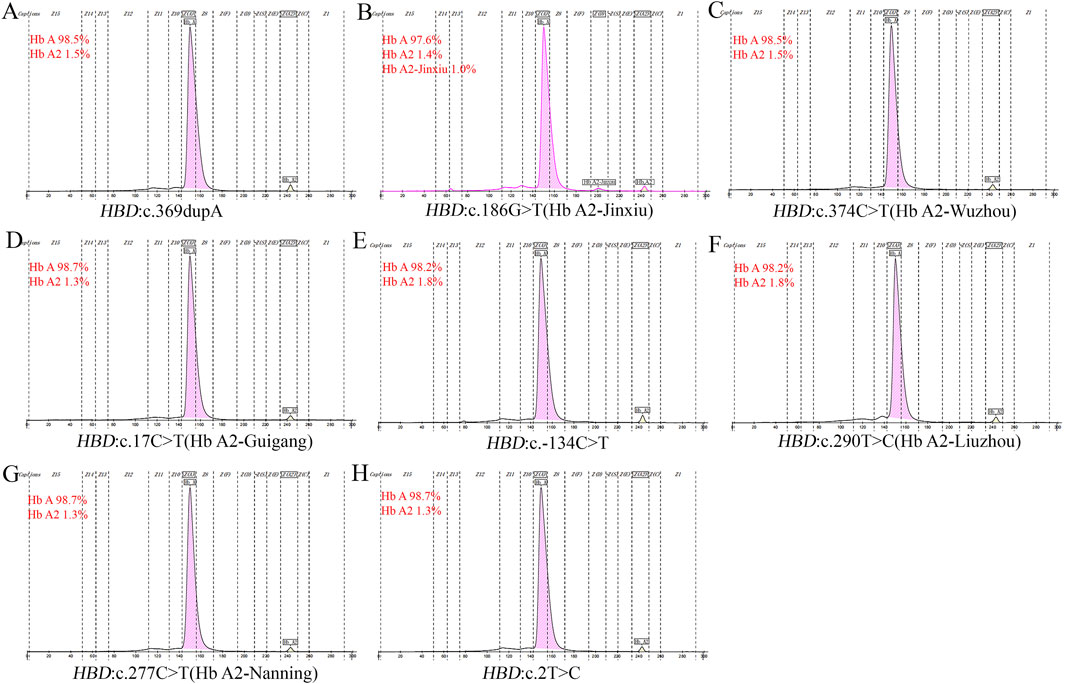
Figure 2. Electropherograms of eight novel mutations. (A) HBD:c.369dupA; (B) HBD:c.186G>T (Hb A2-Jinxiu); (C) HBD:c.374C>T (Hb A2-Wuzhou); (D) HBD:c.17 C>T (Hb A2-Guigang); (E) HBD:c.−134 C>T; (F) HBD:c.290T>C (Hb A2-Liuzhou); (G) HBD:c.277C>T (Hb A2-Nanning); (H) HBD:c.2T>C.
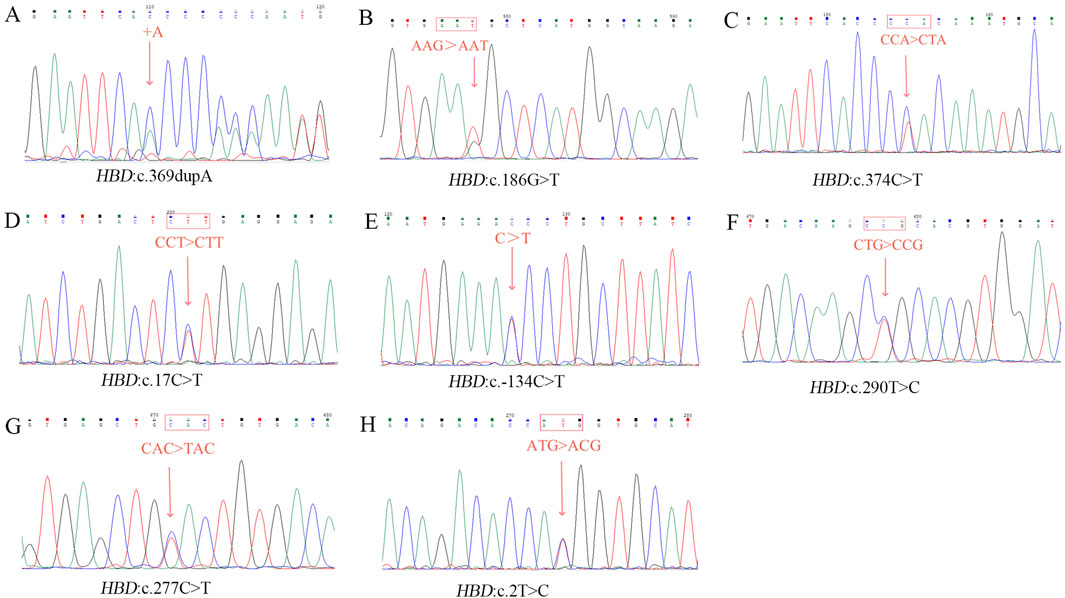
Figure 3. Sanger sequencing result of eight novel mutations. (A) HBD:c.369dupA; (B) HBD:c.186G>T; (C) HBD:c.374C>T; (D) HBD:c.17 C>T; (E) HBD:c.−134 C>T; (F) HBD:c.290T>C; (G) HBD:c.277C>T; (H) HBD:c.2T>C.
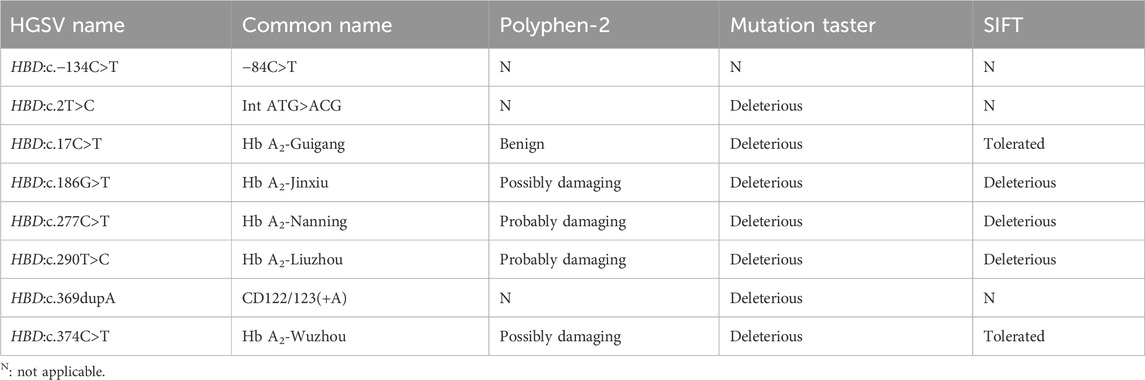
Table 4. Bioinformatics analysis using three different predictive tools.
Discussion
In this study, our analysis identified 28 distinct δ-globin variants and δ-thalassemia mutations among 127 carriers, corresponding to a population-level carrier frequency of 0.81% (127/15,642). The prevalence is higher than in Thailand and Tunisia, as well as that reported by other Chinese investigators (Hanart et al., 2023; Kasmi et al., 2021; Xu et al., 2023; Liu et al., 2013). The most common discovery was δ-thalassemia (81 cases, 0.52% carrier rate), which was followed by δ-globin variant (46 cases, 0.29% carrier rate). Notably, eight novel mutations were detected; comprising five δ-globin variants and three δ-thalassemia mutations, none previously cataloged in the HbVar or IthaGenes databases. The finding of eight novel mutations (6.3% of the total number of δ-globin gene mutations) further highlights the limitations of existing variant databases. This study indicates that a need for integration of δ-globin mutations screening into existing thalassemia prevetion protocols in the region.
The molecular epidemiology of δ-thalassemia in this cohort revealed a predominance of the HBD:c.−127T>C variant, accounting for 67.8% (61/90) of cases. This prevalence consisted with previous reports from China but exceeds Chinese regional frequencies documented in earlier studies (63.2% and 51.6%) (Xu et al., 2023; Liu et al., 2013). Surprisingly, the second most frequent mutations, Hb A2-Melbourne (27.8%, 25/90), demonstrated a disproportionately higher incidence compared to historical Chinese cohort data. This finding represents a notable deviation from established mutation profiles in China and may be attributed to the genetic heterogeneity of regional populations. Genotypic analysis indicated that the majority of δ-globin mutations occurred in heterozygous states, with a few co-inheritances of α/β-globin mutations. Only two isolated homozygous cases, one δ-thalassemia (HBD:c.−127T>C) and one δ-globin variant (Hb A2-Melbourne), were identified, both exhibiting undetectable Hb A2 peak by CE. δ-thalassemia and δ-globin variant heterozygotes, all of them showed reduced A2 values in their CE results (1.1%–1.8%), and some of the δ-globin variants additionally detected low levels of Hb A2 variant peaks. In this study, six δ-globin variants failed to produce resolvable Hb A2 variant peaks (Table 2). Hematological parameters of heterozygous and homozygous cases remained within normal range, reinforcing the clinically silent phenotype associated with δ-globin mutations.
Eleven cases with co-inherited δ-globin and α/β-globin mutations displayed clinically diverse symptoms ranging from asymptomatic to mild anemia. δ-globin combined with thalassemia showed a thalassemia phenotype with decreased values of hematological parameters MCV and MCH, whereas in the case of the combined α/β-globin variant, hematological parameters were normal, with separation of abnormal peaks only during electrophoresis. Elevated Hb A2 levels were considered as a diagnostic criterion for typical β-thalassemia trait. However, when combined with the δ-globin mutation, the Hb A2 level may be normal, thus masking the β-thalassemia trait and resulting in underdiagnosis. Our results emphasize the importance of integrated α/β/δ-globin genotyping in regions with high thalassemia prevalence for accurate diagnosis and counseling.
Molecular characterization confirmed all novel mutations adhere to Human Genome Variation Society (HGVS) nomenclature standards, including promoter mutations (e.g., HBD:c.−134C>T), initiation codon alterations (e.g., HBD:c .2T>C), and missense substitutions (e.g., HBD:c.347C>T). Bioinformatic analysis of eight novel mutations revealed diverse pathogenic potentials. HBD: c.277C>T and HBD: c.290T>C were unanimously predicted as deleterious by three tools, while the prediction of the two software programs for HBD: c.186G>T and HBD: c.374C>T were inconsistent. Surprisingly, for the prediction of HBD: c.17C>T, they had opposite conclusions, with Mutation Taster considered deleterious, while PolyPhen-2 and SIFT were recognized as benign and tolerated. These findings underscored the limitations of prediction tools and the need for functional studies to validate predictions, particularly for variants with atypical phenotypes. Based on the residence of the probands, we named the δ-globin variants HBD:c.17C>T, HBD:c.186G>T, HBD:c.277C>T, HBD:c.290T>C, and HBD:c.374C>T as Hb A2-Guigang, Hb A2-Jinxiu, Hb A2-Nanning, Hb A2 -Liuzhou, Hb A2-Wuzhou, respectively. In addition, HBD: c.347C>T has been registered in the Ithagene database, but did not name the Hb name; we try to name it as Hb A2-Hechi to facilitate the study and communication.
This study expanded the molecular spectrum of δ-globin gene defects and highlighted critical discrepancies in mutation frequencies across populations, underscoring the necessity for ethnically tailored genetic databases to optimize hemoglobinopathy diagnostics. While our findings did not represent a comprehensive epidemiological survey of δ-globin molecular defects in China, they revealed a substantial carrier burden (0.81% aggregate frequency) within the studied cohort, challenging historical perceptions of δ-globin variants as clinically negligible in Chinese populations. To address diagnostic challenges, we implemented a standardized δ-globin screening protocol (Figure 4) integrating CE, multiplex gap-PCR, andSanger sequencing, which successfully resolved β-thalassemia cases masked by conventional screening methods, thereby mitigating risks of underdiagnosis. The workflow’s efficacy in detecting co-inherited α/β-globin defects (8.7% of resolved cases) demonstrated its utility for refining carrier risk stratification and informing precision prenatal counseling.
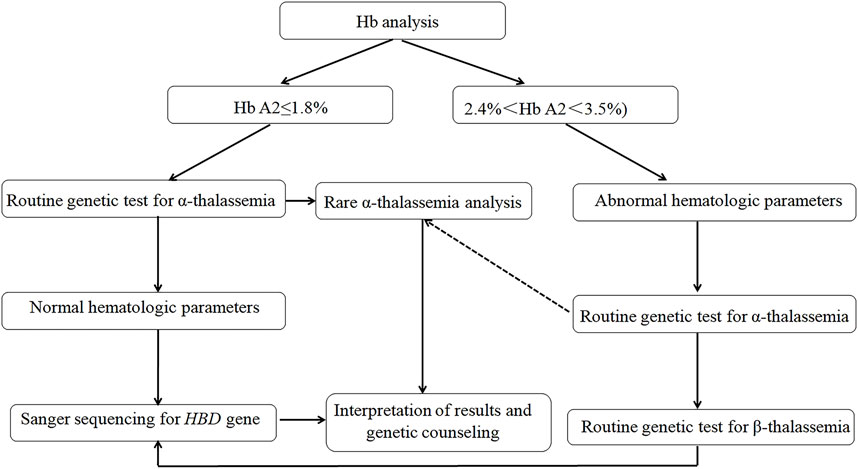
Figure 4. Recommended workflow for the δ-globin gene mutations.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics statement
The studies involving humans were approved by Ethics Committee of People’s hospital of Guangxi Zhuang Autonomous Region. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from a by- product of routine care or industry. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YL: Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Writing – original draft, Writing – review and editing. LY: Data curation, Funding acquisition, Methodology, Resources, Writing – review and editing. LL: Data curation, Methodology, Resources, Writing – review and editing. LZ: Data curation, Methodology, Writing – review and editing. YX: Methodology, Resources, Writing – review and editing. ZL: Methodology, Resources, Writing – review and editing. JB: Methodology, Resources, Writing – review and editing. XH: Methodology, Writing – review and editing. QF: Methodology, Writing – review and editing. TQ: Methodology, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Natural Science Foundation of Guangxi (2023GXNSFAA026102), the Laibin scientific research and technology development project (Laikezhuan 241,534), and the Health Department Research Fund of Guangxi (Z20200076).
Conflict of interest
Author QF was employed by Yaneng Bioscience (Shenzhen) Co. Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Chen, G. L., Huang, L. Y., Zhou, J. Y., and Li, D. Z. (2017). Hb A2-tianhe (HBD: c.323G>A): first report in a Chinese family with normal Hb A2-β-thalassemia trait. Hemoglobin 41 (4-6), 291–292. doi:10.1080/03630269.2017.1398170
Chen, P., Lin, W. X., and Li, S. Q. (2022). THALASSEMIA in ASIA 2021: thalassemia in Guangxi province, people's Republic of China. Hemoglobin 46 (1), 33–35. doi:10.1080/03630269.2021.2008960
Colaco, S., and Nadkarni, A. (2021). Borderline HbA2 levels: dilemma in diagnosis of beta-thalassemia carriers. Mutat. Res. Rev. Mutat. Res. 788, 108387. doi:10.1016/j.mrrev.2021.108387
Hanart, C., Singha, K., Changtrakul, Y., Fucharoen, S., and Srivorakun, H. (2023). Prospective screening for δ-hemoglobinopathies associated with decreased hemoglobin A2 levels or hemoglobin A2 variants: a single center experience. Clin. Chim. Acta. 547, 117417. doi:10.1016/j.cca.2023.117417
Harteveld, C. L., Achour, A., Arkesteijn, S. J. G., Ter Huurne, J., Verschuren, M., Bhagwandien-Bisoen, S., et al. (2022). The hemoglobinopathies, molecular disease mechanisms and diagnostics. Int. J. Lab. Hematol. 44 (Suppl. 1), 28–36. doi:10.1111/ijlh.13885
Huang, H., Xu, L., Chen, M., Lin, N., Xue, H., Chen, L., et al. (2019). Molecular characterization of thalassemia and hemoglobinopathy in Southeastern China. Sci. Rep. 9 (1), 3493. doi:10.1038/s41598-019-40089-5
Kasmi, C., Amri, Y., Hadj-Fredj, S., Oueslati, S., Dabboussi, M., Mahjoub, R., et al. (2021). Analysis of δ-globin gene alleles in Tunisians: description of three new delta-thalassemia mutations. Mol. Biol. Rep. 48 (8), 5923–5933. doi:10.1007/s11033-021-06592-7
Kordafshari, A., Amirian, A., Zeinali, S., Valaei, A., Maryami, F., and Karimipoor, M. (2016). Molecular characterization of δ-thalassemia in Iran. Hemoglobin 40 (1), 44–47. doi:10.3109/03630269.2015.1092982
Li, Y., Huang, T., Mao, T., Zhang, X., Liang, L., and Meng, M. (2020). Detection of a Hb A2-Melbourne (HBD: c.130G>A) combined with β-thalassemia in a Chinese individual. J. Clin. Lab. Anal. 34 (9), e23401. doi:10.1002/jcla.23401
Lin, H. M., Liang, L., Cai, Y. J., Zheng, L. H., Qin, Q. P., and Li, Y. Q. (2024). A new δ-globin gene variant: Hb A2-yulin [δ46(CD5)Gly→Arg,HBD: C.139G > A]. Hemoglobin 48 (2), 121–124. doi:10.1080/03630269.2024.2325443
Liu, N., Xie, X. M., Zhou, J. Y., Li, R., Liao, C., and Li, D. Z. (2013). Analysis of δ-globin gene mutations in the Chinese population. Hemoglobin 37 (1), 85–93. doi:10.3109/03630269.2012.747965
Lou, J., Sun, M., Mao, A., Liu, Y., Zhao, Y., Fu, Y., et al. (2023). Molecular spectrum and prevalence of thalassemia investigated by third-generation sequencing in the Dongguan region of Guangdong Province, Southern China. Clin. Chim. Acta 551, 117622. doi:10.1016/j.cca.2023.117622
Morgado, A., Picanço, I., Gomes, S., Miranda, A., Coucelo, M., Seuanes, F., et al. (2007). Mutational spectrum of delta-globin gene in the Portuguese population. Eur. J. Haematol. 79 (5), 422–428. doi:10.1111/j.1600-0609.2007.00949.x
Paiboonsukwong, K., Jopang, Y., Winichagoon, P., and Fucharoen, S. (2022). Thalassemia in Thailand. Hemoglobin 46 (1), 53–57. doi:10.1080/03630269.2022.2025824
Panyasai, S., and Pornprasert, S. (2020). Association of Hb A2 variants with several forms of α- and β-thalassemia in Thailand. Hemoglobin 44 (3), 179–183. doi:10.1080/03630269.2020.1770099
Srivorakun, H., Thawinan, W., Fucharoen, G., Sanchaisuriya, K., and Fucharoen, S. (2020). Thalassemia and erythroid transcription factor KLF1 mutations associated with borderline hemoglobin A2 in the Thai population. Arch. Med. Sci. 18 (1), 112–120. doi:10.5114/aoms.2020.93392
Vijian, D., Wan, A., Rahman, W. S., Ponnuraj, K. T., Zulkafli, Z., and Mohd Noor, N. H. (2021). Molecular detection of alpha thalassemia: a review of prevalent techniques. Medeni. Med. J. 36 (3), 257–269. doi:10.5222/MMJ.2021.14603
Viprakasit, V., and Ekwattanakit, S. (2018). Clinical classification, screening and diagnosis for thalassemia. Hematol. Oncol. Clin. North Am. 32 (2), 193–211. doi:10.1016/j.hoc.2017.11.006
Wang, M., Zhang, X., Zhang, Y., and Xiao, M. (2022). Prevalence and genetic analysis of thalassemia and hemoglobinopathy in different ethnic groups and regions in hainan island, southeast China. Front. Genet. 13, 874624. doi:10.3389/fgene.2022.874624
Keywords: δ-thalassemia, δ-globin variants, HBD gene, Hb A2, thalassemia
Citation: Li Y, Ye L, Liang L, Zheng L, Xiao Y, Lao Z, Bai J, He X, Fang Q and Qin T (2025) Unveiling the molecular landscape of δ-thalassemia and δ-globin variants in southern China: novel mutations, gene spectrum, and implications for thalassemia diagnosis. Front. Genet. 16:1584310. doi: 10.3389/fgene.2025.1584310
Received: 27 February 2025; Accepted: 22 April 2025;
Published: 09 May 2025.
Edited by:
Catherine Lynn T. Silao, University of the Philippines Manila, PhilippinesReviewed by:
Guoxing Zhong, Huizhou First Maternal and Child Healthcare Hospital, ChinaRazan Hayati Zulkeflee, Universiti Sains Malaysia Health Campus, Malaysia
Copyright © 2025 Li, Ye, Liang, Zheng, Xiao, Lao, Bai, He, Fang and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youqiong Li, bGl5b3VxaW9uZzMyN0AxNjMuY29t