 Gizem Onder1,2,3
Gizem Onder1,2,3 Ozkan Ozdemir2,4
Ozkan Ozdemir2,4 Fulya Taylan3,5Cengiz Canpolat6
Fulya Taylan3,5Cengiz Canpolat6 Koray Yalcin7,8Fatih Erbey9,10Banu Oflaz Sozmen9,10Fikret Asarcikli9,10Turan Bayhan11,12Yunus Murat Akcabelen11Nese Yarali11,12Namik Yasar Ozbek11Ikbal Ok Bozkaya11Dilek Kacar11Berk Ergun13Alper Akkus2,14Davut Albayrak15Elif Ince16Ugur Demirsoy17Gul Nihal Ozdemir18
Koray Yalcin7,8Fatih Erbey9,10Banu Oflaz Sozmen9,10Fikret Asarcikli9,10Turan Bayhan11,12Yunus Murat Akcabelen11Nese Yarali11,12Namik Yasar Ozbek11Ikbal Ok Bozkaya11Dilek Kacar11Berk Ergun13Alper Akkus2,14Davut Albayrak15Elif Ince16Ugur Demirsoy17Gul Nihal Ozdemir18 Omer Dogru19Seda Aras20Eylul Aydin2,14Busra Unal21Ufuk Amanvermez2,22
Omer Dogru19Seda Aras20Eylul Aydin2,14Busra Unal21Ufuk Amanvermez2,22 Ozlem Akgun Dogan2,23Sezer Akyoney24
Ozlem Akgun Dogan2,23Sezer Akyoney24 Muge Sayitoglu25
Muge Sayitoglu25 Ann Nordgren3,5,26,27Nihat Bugra Agaoglu21,28*
Ann Nordgren3,5,26,27Nihat Bugra Agaoglu21,28* Ugur Ozbek2,29
Ugur Ozbek2,29 Ozden Hatirnaz Ng2,4*
Ozden Hatirnaz Ng2,4*- 1Department of Biochemistry and Molecular Biology, Health Sciences Institute, Acıbadem Mehmet Ali Aydınlar University, Istanbul, Türkiye
- 2Rare Diseases and Orphan Drugs Application and Research Center (ACURARE), Rare Diseases and Orphan Drugs Application and Research Center (ACURARE), Acıbadem University, Istanbul, Türkiye
- 3Department of Molecular Medicine and Surgery, Karolinska Institutet, Stockholm, Sweden
- 4Department of Medical Biology, School of Medicine, Acıbadem Mehmet Ali Aydınlar University, Istanbul, Türkiye
- 5Department of Clinical Genetics and Genomics, Karolinska University Hospital, Stockholm, Sweden
- 6Department of Pediatric Oncology, School of Medicine, Acıbadem Mehmet Ali Aydınlar University, Istanbul, Türkiye
- 7Department of Pediatric Hematology, Bahçeşehir University, Goztepe Medical Park Hospital, Istanbul, Türkiye
- 8Department of Medical Biotechnology, Health Sciences Institute, Acıbadem Mehmet Ali Aydınlar University, Istanbul, Türkiye
- 9Department of Pediatric Hematology and Oncology, Hospital of Koç University, Istanbul, Türkiye
- 10Department of Pediatric, School of Medicine, Koç University, Istanbul, Türkiye
- 11Department of Pediatric Hematology and Oncology, Ankara Bilkent City Hospital, Ankara, Türkiye
- 12Department of Pediatric Hematology and Oncology, Ankara Yıldırım Beyazıt University, Ankara, Türkiye
- 13GENIVA Information Health Services Company, Istanbul, Türkiye
- 14Department of Translational Medicine, Institute of Health Sciences, Acıbadem Mehmet Ali Aydınlar University, Istanbul, Türkiye
- 15Department of Pediatric Hematology, Samsun Medical Park Hospital, Samsun, Türkiye
- 16Department of Pediatric Hematology and Oncology, Faculty of Medicine, Ankara University, Ankara, Türkiye
- 17Department of Pediatric Oncology, Faculty of Medicine, Kocaeli University, Izmit, Kocaeli, Türkiye
- 18Department of Pediatric Hematology, Faculty of Medicine, Istinye University, Istanbul, Türkiye
- 19Department of Pediatric Hematology, Biruni University, Istanbul, Türkiye
- 20Department of Pediatric Hematology Oncology, Hatay Training and Research Hospital, Hatay, Türkiye
- 21Department of Cancer Genetics, Umraniye Traning and Research Hospital, Istanbul, Türkiye
- 22Department of Genome Studies, Institute of Health Sciences, Acıbadem Mehmet Ali Aydınlar University, Istanbul, Türkiye
- 23Department of Medical Genetics, School of Medicine, Acıbadem Mehmet Ali Aydınlar University, Istanbul, Türkiye
- 24Department of Bioinformatics and Biostatistic, Institute of Health Sciences, Acıbadem Mehmet Ali Aydınlar University, Istanbul, Türkiye
- 25Department of Genetics, Istanbul University, Institute of Aziz Sancar Experimental Medicine, Istanbul, Türkiye
- 26Department of Clinical Genetics and Genomics, Sahlgrenska University Hospital, Gothenburg, Sweden
- 27Department of Laboratory Medicine, Institute of Biomedicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden
- 28Department of Neurology, Krankenhaus Nordwest, Frankfurt, Germany
- 29International Biomedicine and Genome Institute (iBG), Izmir Dokuz Eylül University, Izmir, Türkiye
Background: Leukemia is the most common cancer in children, and 10%–15% of patients with leukemia/lymphoma carry pathogenic germline cancer-predisposing variants. Identifying these variants is critical for understanding the genetic predisposition and optimizing clinical management.
Methods: We performed germline short-read sequencing in 36 individuals from 20 families with suspected leukemia/lymphoma predisposition, including 20 index cases, 9 affected relatives, and 7 unaffected members.
Results: We identified 13 clinically relevant germline variants in known cancer predisposition genes including TP53, ETV6, MSH6, MLH1, and BRCA1. Notably, we uncovered novel candidate variants in ATR, TNFRSF9, ETAA1, and KSR1, which was supported by segregation analysis, consanguinity patterns, and secondary malignancy phenotypes. Several index cases exhibited striking familial cancer syndromes involving both hematologic and solid tumors, with progression from ALL to AML or glioma. Deep clinical–genomic correlation enabled reclassification of variants and refined diagnostic and therapeutic decision-making in multiple cases. The patients were referred to genetic counseling for surveillance of carriers and risk assessment for various family members.
Conclusion: These findings emphasize the clinical utility of germline testing in pediatric hematologic cancers by providing novel insights into the predisposition to leukemia/lymphoma and contributing to treatment regimens, donor selection, and diagnostic refinement, particularly in populations with high consanguinity.
1 Introduction
Leukemia is characterized by the arrest and clonal proliferation of hematopoietic stem cells in a specific stage of normal hematopoiesis and subsequent accumulation of neoplastic cells in the bone marrow, peripheral blood, and other tissues. Lymphoma is a malignancy caused by a similar process that starts in the lymphatic system. Of the childhood cancers, leukemia is the most common, followed by central nervous system tumors and lymphomas (Zhang et al., 2015). Although the development of leukemia and lymphoma has not yet been fully clarified, the role of germline variants in cancer predisposition genes is becoming clearer (Bloom et al., 2020). The diagnosis of a malignancy necessitates immediate intervention, and the underlying factors, such as a germline variant, occasionally go undetected by treatment centers. However, recent genomic studies have identified germline predispositions in 5%–18% of all childhood cancers. The diagnosis is influenced by differences in study cohorts, study design, and the definition of a positive germline finding (Zhang et al., 2015; Bloom et al., 2020; Tesi et al., 2024; Bakhuizen et al., 2024). Because of decreasing costs and continuous improvements in technology, next-generation sequencing (NGS) can be incorporated into the routine diagnostic work-up for these diseases (Brozou et al., 2022; Byrjalsen et al., 2020; Gröbner et al., 2018).
The incidence rate of childhood cancers in Türkiye is reported as 3.1% (http://iicc.iarc.fr). However, since the registry studies in Türkiye are limited and the NGS-based cancer predisposition evaluation is not covered by the general healthcare system, the expected incidence is much higher. In addition, the general rate of consanguineous marriages in Türkiye has been reported to be as high as 24% (Hacettepe University Institute of Population Studies, et al., 2019; Dündar and Karabulut, 2010). Such consanguinity increases the risk of constitutional mismatch repair deficiency (CMMRD) and CMMRD-like conditions, including Lynch syndrome, while also complicating the analysis of numerous rare homozygous variants. Evaluating these rare homozygous variants is challenging as the Turkish population is underrepresented in major public datasets such as gnomAD.
Multicenter studies initiated by the European Framework Programs in different countries have increased the awareness of germline predisposition factors in malignancies, including leukemias/lymphomas, and our study group was involved in one such study (https://www.cost.eu/actions/CA16223/). Additionally, we started a close collaboration with the Swedish Childhood Cancer Predisposition (ChiCaP) project (Tesi et al., 2024) for further analysis through whole-genome sequencing (WGS).
In this study, we aimed to identify both known and novel gene variants associated with childhood leukemia/lymphoma predisposition in Türkiye. Using short-read sequencing technologies, we studied children with leukemia/lymphoma along with their affected and unaffected family members. This study represents the first systematic analysis of families with high predisposition risk to childhood cancer in Türkiye and adds to the limited literature on germline predisposition to leukemia/lymphoma in the region.
2 Materials and methods
2.1 Study design and patient cohort
Between 2019 and 2024, children under the age of 18 years who developed leukemia/lymphoma and exhibited increased risk for cancer predisposition according to Jongmans’ criteria (Jongmans et al., 2016) were included in the study. All participants and/or their legal guardians provided written informed consent. Patients were recruited from eight pediatric hemato-oncology departments across hospitals in Türkiye. Additional clinical information and laboratory investigation results were systematically recorded in an in-house form. The samples and data generated in this study were archived in the Acibadem University Biobank Unit. An overview of the study design is presented in Figure 1. This study was approved by the Acibadem Healthcare Institutions Medical Research Ethics Committee (ATADEK; no. 2017-16/5 and no. 2024-11/496).
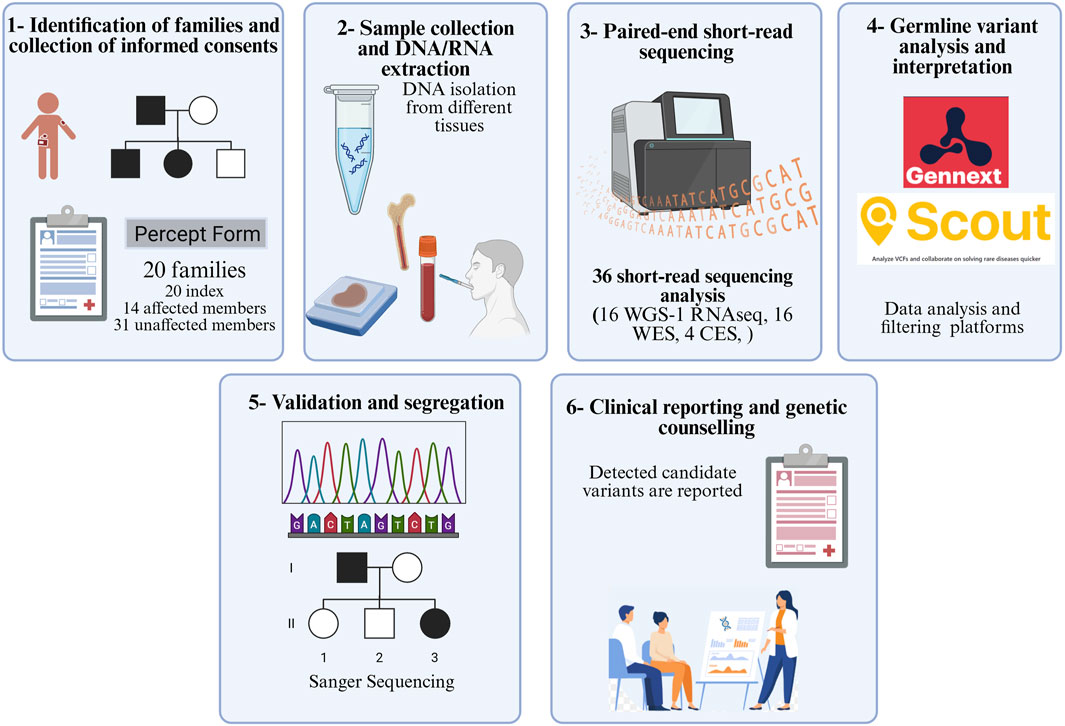
Figure 1. Workflow of the study (WGS: whole-genome sequencing; WES: whole-exome sequencing; CES: clinical exome sequencing).
2.2 Sample collection and DNA extraction
Peripheral blood or bone marrow samples were obtained during remission, and when available, buccal swab samples were collected from the index patients and their family members who were diagnosed with a hematological malignancy in childhood. For segregation analysis, blood samples were collected from the parents and other available family members. Germline DNA from remission blood or bone marrow samples was extracted using the QIAamp/QIAquick DNA extraction and purification kit (QIAGEN, Hilden, Germany). DNA from buccal swabs was extracted using the buccal swab DNA isolation kit (Hibrigen Biotechnology, Kocaeli, Türkiye). Genomic DNA from formalin-fixed paraffin-embedded samples was extracted using the QIAamp DNA FFPE Tissue Kit (QIAGEN, Hilden, Germany).
2.3 Paired-end short-read sequencing
Standard paired-end short-read sequencing was performed on germline DNA samples. Whole-exome sequencing (WES) was performed using the NovaSeq 6000 system (Illumina, Inc., San Diego, CA, United States) with the S1 reagent kit v1.5. Clinical exome sequencing (CES), which covers exons and flanking intronic regions of 4,490 clinically relevant genes, was performed using the SOPHiA Clinical Exome Solution v2 kit (SOPHiA Genetics, Lausanne, Switzerland) in Umraniye Training and Research Hospital, Istanbul, Türkiye. Additionally, short-read RNA sequencing (RNA-seq) was performed for a single case (case #13). WGS with 30x coverage was conducted at Science for Life Laboratory (SciLifeLab; Stockholm, Sweden), as previously described (Stranneheim et al., 2021).
2.4 Bioinformatics analysis
All sequencing data were subjected to bioinformatic processing, annotated, and filtered for candidate variants using the GenNext platform (https://app.gennext.bio/auth) (https://github.com/GenivaInformatics/gennext-workflows, Geniva, Istanbul, Türkiye). WGS data analysis was performed using the mutation identification pipeline (Stranneheim et al., 2021). Annotated and ranked variants in WGS data were further filtered and interpreted using the visualization tool Scout (https://github.com/Clinical-Genomics/scout). WGS was performed using the hg19 (GRCh37) reference genome, while WES was conducted using the hg38 (GRCh38) reference genome. The WGS data were analyzed using the ChiCaP clinical gene panel, which includes 189 well-established childhood cancer predisposition genes (Tesi et al., 2024). A detailed description of sequencing data analysis is provided in Supplementary File 1 and Supplementary Figures 1–2.
The Turkish Variome dataset was used to evaluate the variant allele frequencies of the candidate variants (Kars et al., 2021). Missense variants of unknown significance (VUS) were evaluated using variant analysis with multiple pathogenicity predictors (VAMPP), as described previously (Ozdemir et al., 2024). VAMPP scores higher than 0.35 indicated moderate evidence for pathogenicity (PP3) of variants (https://vamppscore.com/).
Moreover, we utilized AlphaFold (Jumper et al., 2021) to determine the predicted local distance difference test (pLDDT) scores and spliceAI (Jaganathan et al., 2019) for the splice site variants. All the candidate variants were validated, and segregation analyses were performed by Sanger sequencing for parental DNA samples using standard protocols. Additionally, ΔΔG values were calculated for missense variants with the DynaMut2 tool (https://biosig.lab.uq.edu.au/dynamut2/). A negative ΔΔG value indicates that the mutation is destabilizing, indicating a decrease in stability. In this context, the ΔΔG value was calculated for 15 missense variants.
The variant classifications were finalized with the combined analysis of these findings according to the American College of Medical Genetics (ACMG) (Richards et al., 2015).
3 Results
We identified 20 children with leukemia/lymphoma with suspected cancer predisposition and no known genetic diagnosis at the time of inclusion. The study also included 14 family members who were diagnosed with leukemia or any cancer, along with 31 unaffected family members. Five (25%) patients had congenital abnormalities, three children experienced treatment toxicity, six children developed an additional primary tumor alongside their hematological malignancies, and one child had more than two additional primary tumors (Table 1). We performed 16 WGS (four index cases, five members with previous cancer diagnosis, and seven healthy members), 16 WES (12 index cases and four members with previous cancer diagnosis), and four CES.
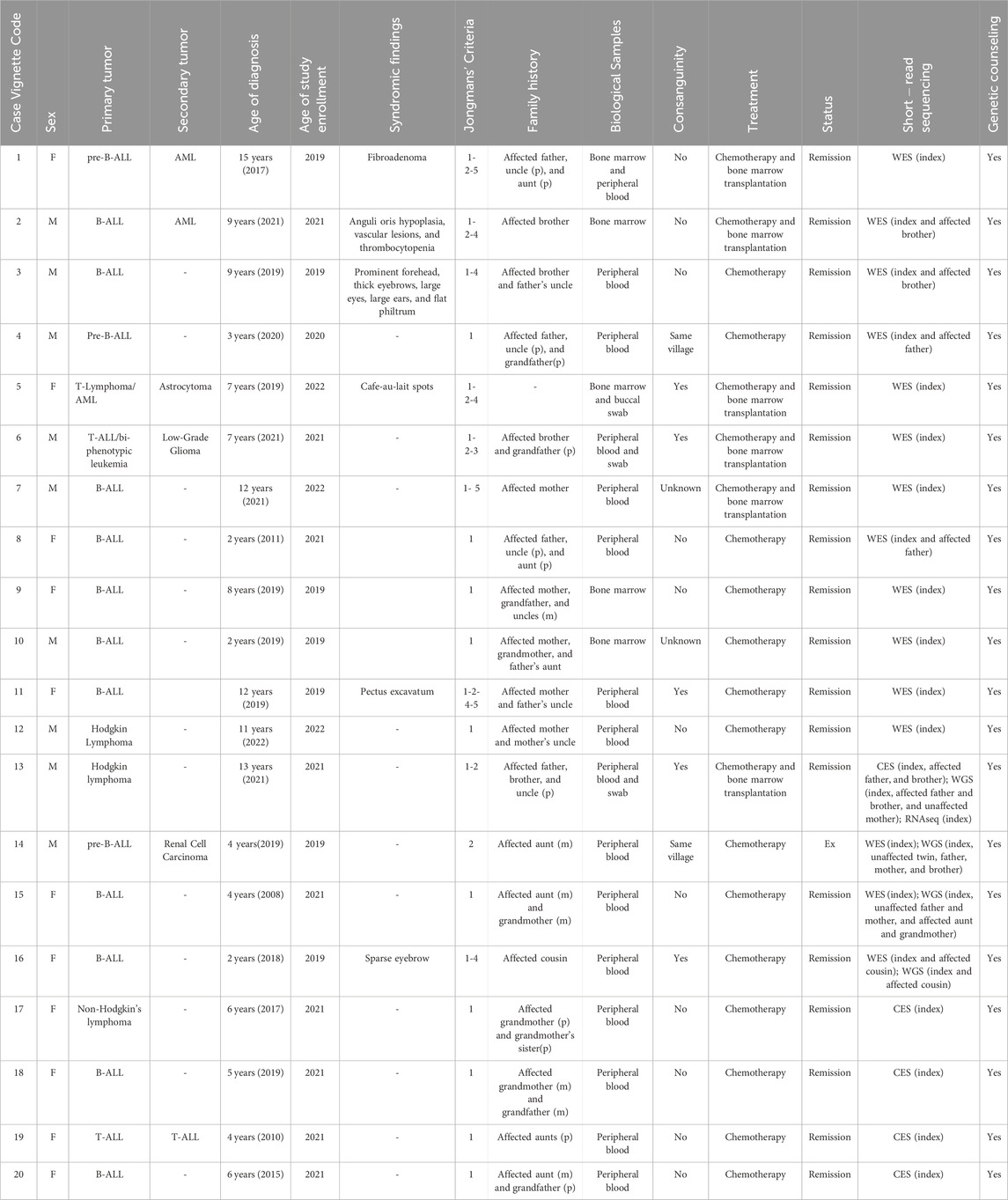
Table 1. Clinical features of the cases. The clinical features of the index cases and the indications of enrollment according to Jongmans’ criteria are summarized. The biological sample that was studied and the short-read sequencing methodology are also described. (WES; whole-exome sequencing, WGS; whole-genome sequencing, CES; clinical exome sequencing, RNA Seq; RNA sequencing; p: paternal; m: maternal).
3.1 Baseline characteristics
We analyzed 36 individuals, including 20 index cases (diagnosis age median: 8 years, female/male: 11/9) and nine affected and seven unaffected family members. Seven out of 20 families (35%) reported consanguinity. Of the index cases, 14 were diagnosed with B-ALL (acute lymphoblastic leukemia), two with T-ALL, one with T-lymphoblastic lymphoma, two were Hodgkin’s lymphoma cases, and one was a non-Hodgkin’s lymphoma case (Table 1; Supplementary File 2).
3.2 Frequency and spectrum of germline variants
Twenty index patients presented 30 candidate variants (Figure 2; Table 2). The candidate genes observed in the B-ALL cases were ABCC6, BCNP1, BMP6, BRAT1, BRCA1, DNHD, ETAA1, ETV6, JAK2, JAK3, KSR1, MAP2K2, MLLT10, MUTYH, MYH11, MVP, PAPSS2, RAD52, RECQL, TGFBRS1, TP53, and WRN. The candidate genes observed in the T-ALL cases were ATR, BRCA2, BIRC6, and NOTCH1, and those observed in lymphoma cases were MLH1, MSH6, RAD52, and TNFRSF9 (Figure 3).
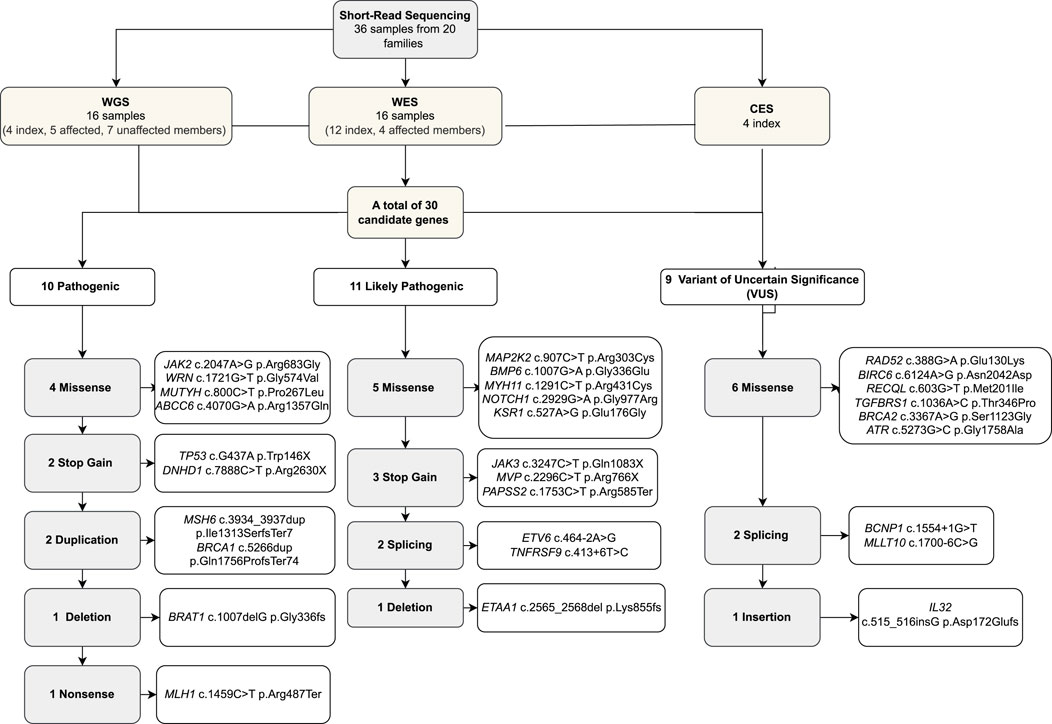
Figure 2. Cohort of the study.

Table 2. Candidate gene variants. A total of 30 candidate variants were determined in 20 index cases. (Chr; chromosome, Het; heterozygous, Hom; homozygous).
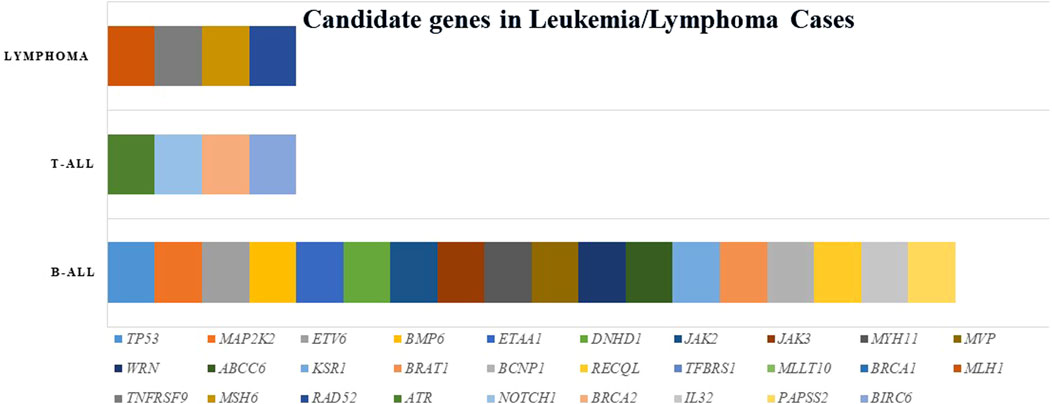
Figure 3. Candidate genes in leukemia and lymphoma.
AlphaFold provided pLDDT scores for 28 variants (DNHD1 and BIRC6 were not available), and 13 of these (TP53, BMP6, JAK2/3, MYH11, MVP, RAD52, WRN, MUTYH, TGFBRS1, RECQL, ABCC6, and PAPSS2) had very high scores. To assess the impact on protein dynamics and stability, we analyzed the stabilization status using DynaMut2 (https://biosig.lab.uq.edu.au/dynamut2/). Eleven missense variants were found to be destabilizing (BMP6, JAK2, MYH11, ATR, NOTCH, RAD52, WRN, MUTYH, RECQL, BRCA2, and ABCC6) (Supplementary File 3).
Additionally, 15 missense variants were also evaluated by the AlphaMissense tool, and eight of them had a score of >0.5 (Supplementary File 1). Two splice-site variants (ETV6 and BCNP1) were at the essential splice site (Supplementary file 2). The spliceAI algorithm yielded higher than 0.5 scores of TNFRSF9 c.413 + 6T>C variant in case #13, and the RNA sequencing of the index revealed an alternative splice site between exons 5 and 6 (Supplementary File 2.15–2.16).
Seven candidate variants (MAP2K2, BMP6, DNHD1, RAD52, MUTYH, RECQL, and ABCC6) presented allele frequencies <0.001 in Turkish Variome (Table 2).
Altogether, based on all the provided evidence, 10 variants were classified as pathogenic (P), 11 were likely pathogenic (LP), and nine were VUS. Eleven out of the 16 missense variants were suitable for VAMPP score evaluation, and four of them (MYH11, MUTYH, TGFBRS1, and ABCC6) showed scores >0.35 but did not change the variant classification of VUS (Table 2; Figure 2).
3.3 Variants causing cancer predisposition
In the 13 index cases, we hypothesized that the candidate variants caused cancer predisposition. The detailed information, pedigrees, and genotypes of the cases and variants are provided as case vignettes in Supplementary File 2.
Index case #1 (Supplementary File 2.1) was diagnosed with B-ALL, and we determined a germline pathogenic stop-gain variant in TP53 c.437G>A p.Trp146Ter. The patient successfully completed treatment for B-ALL in 2019; however, she was diagnosed with acute myeloid leukemia (AML) in 2021. In index case #2 (Supplementary File 2.2), who had B-ALL, we found a maternally inherited LP ETV6 c.464-2A>G variant. It was also detected in the patient’s affected brother, who died of AML after a T-ALL diagnosis.
The LP MAP2K2 c.907C>T p.R303C and LP BMP6 c.1007G>A p.Gly336Glu variants were detected in B-ALL index case #3 (Supplementary File 2.4) and his brother with Hodgkin’s lymphoma. In addition, our case presented elevated iron levels. Their parents were first-degree cousins and were heterozygous for both variants.
In index case #4 (Supplementary File 2.5), the diagnosis was of B-ALL. The patient’s father and one paternal uncle had been treated for Hodgkin’s lymphoma; they suffered relapse after 10 and 20 years, respectively, after the first diagnosis, and additional thyroid papillary carcinoma and synovial sarcoma were diagnosed in the father. The patient’s parents were consanguineous, and all the affected family members were positive for the likely pathogenic variant ETAA1 c.2565_2568del p.K855fs and the pathogenic variant DNHD1 c.7888C>T p.R2630X, whereas the mother and the siblings, who were unaffected, were negative.
Index case #5 (Supplementary File 2.6) was found to be homozygous for the pathogenic variant MSH6 c.3934_3937dup p.Ile1313SerfsTer7. T-ALL was diagnosed when the patient was 12 years old, and 3 years later, primary AML and astrocytoma were diagnosed. Their parents were first-degree cousins, and both were heterozygous for the candidate variant.
We hypothesized that the VUS ATR c.5273G>C p.Gly1758Ala was the causative factor in case #6 (Supplementary File 2.7), who had T-ALL. Both the patient and his brother, who also had T-ALL and died due to treatment toxicity, were homozygous for this variant. The buccal swab analysis confirmed the homozygous status of the variant in the index case. A year later, the index case developed a low-grade glioma. The parents were first-degree cousins, and both were heterozygous for this variant. The patient was also heterozygous for the LP NOTCH1 c.2929G>A p.Gly977Arg and homozygous VUS BIRC6 c.6124A>G p.Asn2042Asp variants. NOTCH1 was also heterozygous in the affected brother and the father, but the mother was wild-type (WT), whereas BIRC6 was heterozygous in the mother and homozygous in the index. The affected brother’s DNA material was insufficient to check the BIRC6 variant.
In index case #7 (B-ALL, Supplementary File 2.8) and in the mother (Hodgkin’s lymphoma), the pathogenic variant present was WRN c.1721G>T p. Gly574Val was detected. His parents were second-degree cousins. B-ALL was diagnosed in index case #8; AML had been diagnosed in her father. They were positive for the LP MYH11 c.1291C>T p.Arg431Cys variant. The family reported members on the paternal side who had died of lymphoma and osteosarcoma.
We found heterozygosity for the LP MVP c.2296C>T p.Arg766X and IL32 c.515_516insG p.Asp172Glufs* variants in both the index case #11 (Supplementary File 2.12) and her mother, in whom colon cancer had been diagnosed at an early age. The parents were consanguineous, and bladder cancer had been diagnosed in the father’s paternal uncle.
Hodgkin’s lymphoma was diagnosed not only in index case #13 (Supplementary File 2.14) but also in his brother, his father, and two of his paternal uncles. WGS revealed homozygosity for the likely pathogenic variant TNFRSF9 c.413 + 6T>C in all the affected cases, and the variant was comprehensively evaluated by RNA sequencing for possible alternative transcripts. The variant was confirmed in the buccal swab of the index patient. The parents were consanguineous, and the mother was heterozygous for the variant.
WES revealed heterozygosity for the pathogenic variant BRAT1 c.1007delGG336fs in index case #15 (Supplementary File 2.19), who had B-ALL. The mother was also heterozygous for the same variant, whereas the maternal aunt, who had had ovarian cancer, and the maternal grandmother, who had breast cancer, were WT. Subsequent WGS analysis revealed the LP KSR1 c.527A>G p.Glu176Gly in all affected family members, and this variant was, therefore, considered causative. Another clinically relevant variant was pathogenic MLH1 c.1459C>T p.Arg487Ter. We determined that index case #17(Supplementary File 2.21), who had non-Hodgkin’s lymphoma, was heterozygous for this variant, as was her unaffected father. The father’s mother, maternal aunt, and maternal grandmother had died of breast cancer.
We also determined the heterozygous pathogenic BRCA1 c.5266dup p.Gln1756ProfsTer74 variant in both the index case #18 (Supplementary File 2.22) and her mother by CES analysis. The maternal grandmother had died of breast cancer at the age of 45; the mother’s father was WT, which confirms that the variant was maternally inherited by the patient’s mother.
For case #9, case #10, case #12, and case #19 (Supplementary File 2.10, 2.11, 2.13, and 2.23, respectively), the analysis revealed pathogenic JAK2, likely pathogenic JAK3, VUS RAD52, and VUS BRCA2 variants, respectively, but only the index case’s samples were available, and further analysis could not be performed within the families. The evidence was insufficient to support the potential causative effect of these variants, and they remained unsolved (Table 2). Case #14 was diagnosed with B-ALL, and soon after the initial diagnosis, brain metastasis occurred, and the patient died following the development of renal cell carcinoma. The WGS analysis of the peripheral blood sample revealed candidate variants in BCNP1, RECQL, TGFBRS1, PAPSS2, and MLLT10, but none showed strong enough evidence to be considered causative. Likewise, in case #16, an incidental pathogenic heterozygous MUTYH variant was identified. However, pathogenicity in MUTYH is typically associated with bi-allelic variants, particularly in the context of MUTYH-associated polyposis. This variant was heterozygous and not present in the affected cousin. Furthermore, the family had no history of gastrointestinal symptoms suggestive of polyposis. Therefore, we did not consider this variant to be related to leukemia predisposition. Although we detected candidate variants, we found no strong evidence for variants detected in cases #14, #16 (Supplementary File 2.20), and #20 (Supplementary File 2.24), and they remained unsolved.
3.4 Clinical utility of germline findings
For the clinical utility of these results, we evaluated the genetic findings with the patient’s primary hematologist/oncologist. For instance, in case #1, the TP53 variant changed the diagnosis to Li–Fraumeni syndrome, and the family was referred to a clinical geneticist for counseling and surveillance plan. In case #5, the homozygous MSH6 variant changed the diagnosis from neurofibromatosis to constitutional mismatch repair deficiency (CMMRD), and the family was referred to a clinical geneticist for counseling and surveillance plan for the risk of Lynch syndrome. Our findings also led to a change in the treatment or donor candidate. In cases #2, #3, #5, #6, and #7, the unaffected donor siblings were also found to be carriers of the candidate genes, and the transplant planning was changed to be performed either from the WT siblings or unrelated donors.
Genetic counseling was provided by clinical geneticists at the referral centers, in accordance with the NCCN Guidelines for genetic/familial high-risk assessment (https://www.nccn.org/guidelines/category_1). For index cases with established cancer predisposition syndromes—such as Li–Fraumeni syndrome (TP53) or constitutional mismatch repair deficiency (CMMRD, MSH6)—counseling included a structured discussion of the associated cancer risks, current clinical guidelines, and established surveillance protocols (e.g., whole-body MRI for Li–Fraumeni and colonoscopy for CMMRD). Families were offered cascade testing and referred to relevant specialty clinics, including adult genetics services, to ensure long-term coordination of care.
For variants of uncertain significance or novel candidate variants without established disease associations or management protocols, clinical geneticists focused on transparent communication regarding the current level of evidence, limitations in interpretation, and potential—but not definitive—implications for cancer risk. These sessions emphasized the importance of regular phenotypic monitoring and re-evaluation as new data become available. Surveillance recommendations in these cases were tailored based on the family history, clinical phenotype, and expert consensus. To support clinical integration of the genetic results, all cases with potential diagnostic or therapeutic relevance were presented at multidisciplinary tumor boards, where available. These boards typically included pediatric oncologists, hematologists, pathologists, and clinical geneticists. Discussions focused on treatment implications, variant reinterpretation, and potential surveillance strategies for both the index cases and at-risk relatives. This collaborative setting enabled shared decision-making, particularly in complex or uncertain scenarios.
In centers lacking access to a clinical geneticist, the patient’s primary physician assumed the responsibility of conveying results and implementing follow-up care, supported by structured reports and consultation with the central study team. Families with asymptomatic carriers were counseled on possible future risks and the need for longitudinal follow-up, while psychosocial concerns were addressed through individualized discussions.
Overall, the genetic counseling process—supported by multidisciplinary input—was critical in translating germline findings into personalized clinical action, guiding diagnosis, treatment, and long-term risk management for families affected by childhood hematologic malignancies.
3.5 Use of NetworkAnalyst for leukemia/lymphoma predisposition
The STRING interaction network within NetworkAnalyst revealed that 26 genes were interconnected either directly or indirectly. Separate network analyses were conducted for all candidate and causative genes (Supplementary Figure 3a, b), as well as for genes associated specifically with leukemia and lymphoma (Supplementary Figure 3c,d). TP53 was both the central hub and the most connected gene in the network. In the leukemia-specific network, TP53, NOTCH1, JAK2, and BRCA1/2 were found to be key hub genes; in the lymphoma-specific network, MLH1 was identified as a key hub gene. Of the 18 causative genes, nine (TP53, ATR, ETAA1, BRCA1, BRCA2, BRAT1, WRN, MLH1, and MSH6) were identified as being responsible for DNA repair and damage response (Figures 4a,b).
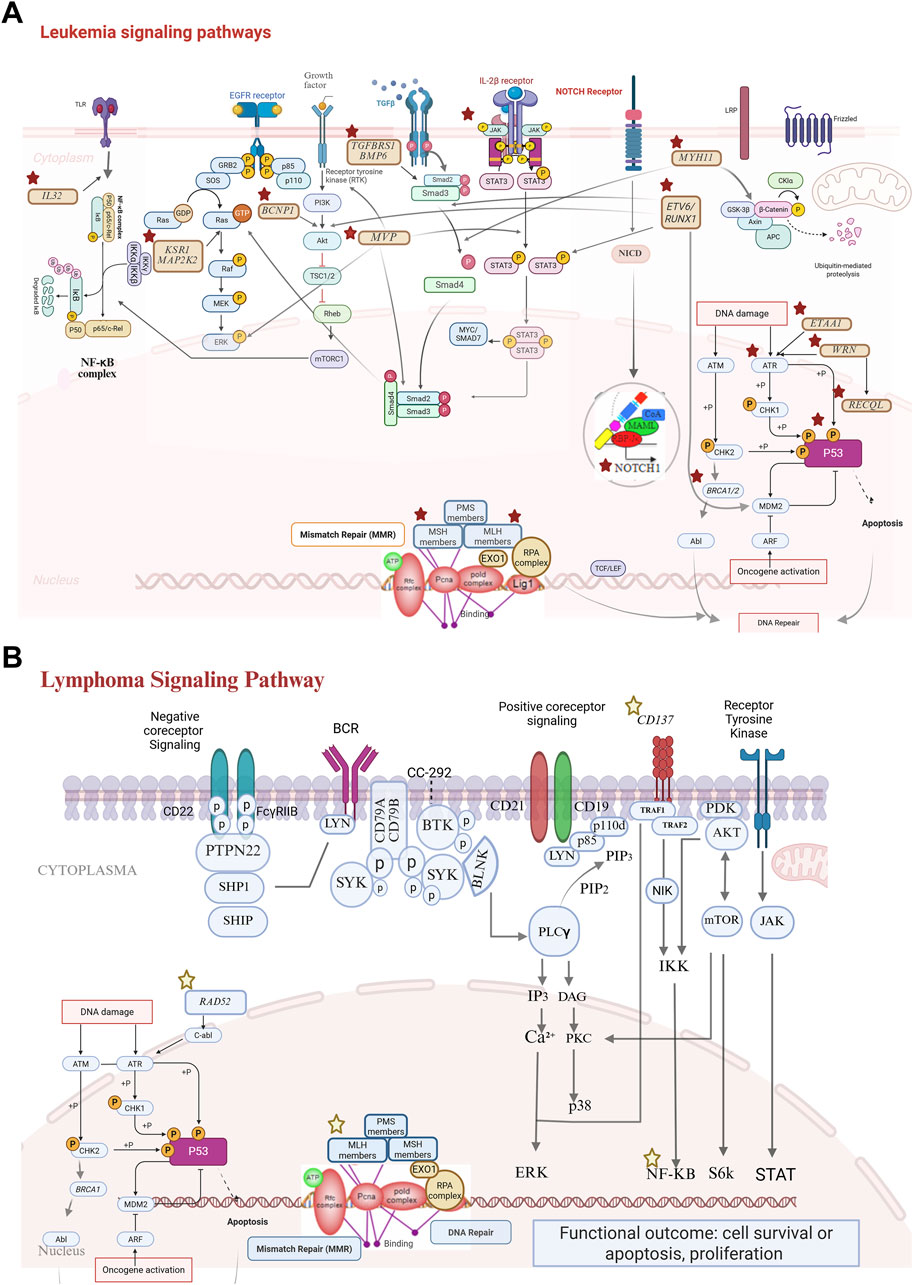
Figure 4. Signaling pathways of candidate genes (stars: genes identified as potential candidates in leukemia/lymphoma cases). (A) Leukemia signaling pathways: (cytoplasmic pathways; NF-κB pathway: activation through IL32 signaling leads to transcriptional regulation via the NF-κB complex. MAPK pathway: initiated through receptor tyrosine kinases, including EGFR, and mediated by KSR1 and MAP2K2, driving cell proliferation. PI3K/AKT/mTORC1 pathway: a critical survival and growth axis influenced by BCNP1 and TGFβ signaling. NOTCH and IL-2β signaling; NOTCH1: NICD (notch intracellular domain) signaling contributes to transcriptional regulation associated with leukemia progression. STAT3 activation: IL-2β receptor engagement results in STAT3 phosphorylation, which impacts the transcriptional activity. DNA repair mechanisms; mismatch repair (MMR): key components such as PMS, MLH, MSH, and EXO1 ensure genomic stability. DNA damage response (DDR): genes such as WRN, RECQL, and BRCA1/2 coordinate repair and apoptotic signaling via the ATM and ATR pathways. β-catenin pathway: dysregulation of WNT signaling (via GSK-3β inhibition) contributes to leukemic transformation. Tumor suppressor pathways; P53: central to apoptosis and cell cycle arrest and frequently inactivated in leukemias. ETV6: common in childhood leukemia and drives oncogenic processes.). (B) Lymphoma signaling pathways: NF-κB signaling via TNFRSF9: TNFRSF9 activation recruits adapter proteins (e.g., TRAF molecules), leading to the phosphorylation and degradation of IκB. This allows the NF-κB complex to translocate to the nucleus, where it regulates genes involved in inflammation, survival, and proliferation. Dysregulation of this pathway is a hallmark of many lymphomas. DNA repair pathways; RAD52: critical for homologous recombination repair and for addressing DNA double-strand breaks that arise during replication or due to genotoxic stress. MSH6 and MLH1: ensure replication fidelity by correcting mismatched base pairs. Loss of MMR function increases mutation rates and contributes to lymphomagenesis.
4 Discussion
Leukemia and lymphoma are the most common childhood hematological malignancies. Approximately 5% of childhood hematological malignancies arise from germline cancer-predisposing gene variants (Zhang et al., 2015; Bakhuizen et al., 2024; Brady et al., 2022; Salmoiraghi et al., 2016) Many cancer-predisposing genes and their association with syndromes or isolated tumors have been described in the literature (Rahman, 2014; Li et al., 2021). Despite these advancements, awareness of genetic predisposition to cancer remains low.
Various guidelines (Baliakas et al., 2019; Goudie et al., 2021) and tools are utilized to assess the childhood predisposition to cancer (Maese et al., 2024; Schlegelberger et al., 2021; Lazic et al., 2023). For the patient selection, we utilized the Jongmans’ criteria (Jongmans et al., 2016) due to its clarity and applicability in clinical settings, particularly in centers where cancer or medical geneticists are not available. We enrolled cases presenting at least one of these following signs: cancer affecting multiple family members across three generations, patients developing multiple primary tumors, bilateral cancer in paired organs, cancer appearing earlier than expected, or patients showing severe toxicity to cancer treatment.
We found strong evidence of the clinical correlation between the variants and cancer predisposition in 13 out of 20 cases. A germline stop-gain TP53 variant was identified in a B-ALL patient, a mutation known to cause Li–Fraumeni syndrome and recently associated with leukemia (Qian et al., 2018). In our case, we could not obtain samples from the affected family members form the paternal side, but the mother was WT, and we assumed paternal inheritance. Similarly, ETV6 is one of the most well-known leukemia-predisposing genes. Germline variants are found in the ETV6 gene and comprehensively described the clinical features in children with ALL who carry these risk variants (Moriyama et al., 2015). We determined the variant at the essential splice site of the ETV6 gene in our case #2 and his brother, who died during the study. In addition to B-ALL, the patient exhibited syndromic features and thrombocytopenia. The germline ETV6 variant has been described previously in leukemia/lymphoma cases in a family with thrombocytopenia (Rampersaud et al., 2019). We showed that the variant was maternally inherited. ETV6 germline variants show reduced penetrance, which may explain why the carrier mother was unaffected (Rampersaud et al., 2019).
Case #3, who had B-ALL, had variants of both the MAP2K2 and BMP6 genes, which were also detected in his brother, who had Hodgkin’s lymphoma. MAP2K2 plays an important role in the RAS signaling pathway, and germline variants in this pathway lead to RASopathies (Tidyman and Rauen, 2016). The activation of the MAPK pathway promotes cell proliferation and survival, and MAP2K2 is also considered a cancer-predisposing gene, as in Noonan syndrome (Alba-Pavón et al., 2023; Cavé et al., 2016). The additional findings of mild facial dysmorphism may correspond to the effect of the MAP2K2 homozygous variant. The MAP2K2 variants were detected in primary central nervous system lymphoma (Fukumura et al., 2016). Among our study cohort, two siblings, one with syndromic leukemia (case #3) and one with Hodgkin’s lymphoma, were homozygous for the MAP2K2 variant. We also identified a second variant at the pro-peptide domain of BMP6, which is necessary for proper folding of the protein and maturation. The variations in this domain have been reported to show a dominant negative effect and are related to iron overload (Daher et al., 2016). Case #3 also exhibited elevated iron levels; however, this might also have been a result of transfusions. NetworkAnalyst revealed MAP2K2 as a central hub, and it was connected to TP53 via other MAPK pathway members, which strengthens MAP2K2 as a predisposing factor.
In case #4 (B-ALL), along with his father and paternal uncle who had Hodgkin’s lymphoma, we detected variants of both ETAA1 and DNHD1 genes. To date, ETAA1 has been reported to cause predisposition to pancreatic cancer and nonpolyposis colorectal cancer. ETAA1 is a key activator of the ATR-dependent DNA damage response. It directly binds to RPA-coated single-stranded DNA at the stalled replication forks and activates ATR-CHK1 signaling. Germline or somatic mutations in ETAA1 may impair genome integrity and replication stress response, increasing susceptibility to tumorigenesis, particularly in rapidly proliferating tissues such as hematopoietic cells (Yu et al., 2018; Childs et al., 2015). However, the COSMIC database includes variants of ETAA1 in lymphoma cell lines. The ETAA1 gene may be a novel predisposition gene that emerges in hematologic cancers in both children and adults. Somatic variants of both candidate genes have been associated with soft-tissue sarcomas and lymphomas in the St. Jude database (https://pecan.stjude.cloud/). Thyroid papillary carcinoma and synovial sarcoma developed in the father, both of which could be related to the germline DNHD1 variant. DNHD1 encodes a protein that belongs to the dynein family, which is involved in intracellular transport and, potentially, centrosome stability. Although not previously linked to hematologic malignancies directly, somatic variants of DNHD1 have been reported in soft-tissue sarcomas and lymphomas in the St Jude database (https://pecan.stjude.cloud/variants/proteinpaint?gene=DNHD1). Aberrant intracellular transport and spindle checkpoint dysregulation may contribute to chromosomal instability, thus indirectly promoting malignant transformation in hematopoietic cells.
Case #5, with cafe-au-lait–like spots on the skin, received a diagnosis of T-ALL at another center and was found negative for the NF1 gene. Subsequent WES at our institution revealed a homozygous MSH6 variant. MSH6 is a member of the DNA mismatch repair system, and its somatic variations have been related to various tumors (Ercan et al., 2024). Similarly, the germline MSH6 variants are related to Lynch syndrome and CMMRD. T-ALL was diagnosed in case #5, followed by the development of AML and astrocytoma. Consanguinity increases the incidence of CMMRD, and hematologic malignancies are detected frequently in such families (Ercan et al., 2024; Ripperger and Schlegelberger, 2016). The parents were related, and all siblings, except one, were heterozygous for the same mutation; one sister was homozygous. The family was informed about the risk of Lynch syndrome, and surveillance was suggested. In addition, the homozygous sister, who was 4 years old at the time of the study, was referred to related clinics for close monitoring for CMMRD.
We determined a homozygous VUS ATR variant, together with the paternally inherited NOTCH1 and maternally inherited BIRC6 variants, in case #6, who had T-ALL. This patient’s older brother had had T-ALL and had died of treatment-related toxicity at the age of 5 years. Homozygous ATR gene variants are also observed in patients with Seckel syndrome. AML development was reported in a patient with Seckel syndrome who died of severe treatment-related toxicity (Hayani et al., 1994). In our patient, deep phenotyping revealed no dysmorphic features consistent with Seckel syndrome. The amino acid change appeared to have a mild effect; this may explain the non-syndromic but cancer-predisposing effect of the variant. In addition, our patient exhibited loss of chromosome 5q, as reported previously (Hayani et al., 1994). ATR plays a role in the DNA damage sensor; therefore, chromosomal instability is expected in the affected patients. ATR is a central kinase in the DNA damage response pathway, and it is especially activated by replication stress. It phosphorylates multiple targets, including CHK1, to halt the cell cycle and repair damaged DNA. Miao et al. showed that NOTCH1 restores the cell cycle deficiency in BRCA1-mutated triple-negative breast cancer through ATR/CHK1 signaling (Miao et al., 2020). In our case, both genes were affected, and further studies are needed to investigate the combined effects of NOTCH1 and ATR variants. Because the father was WT for BIRC6, the patient’s homozygous status must have resulted from a novel second mutation and led to astrocytoma development (Chen et al., 1999).
We performed WGS for four families whose WES/CES panels were negative, and we were able to define a possible predisposing factor in two cases. Samples from case #13, the affected brother, and the affected father were studied with CES, and no candidate variant could be determined. Later, WGS revealed a novel TNFRSF9 variant. The TNFRSF9 gene encodes the cell surface receptor CD137, which contributes to the proliferation, survival, and development of T-cells (Shen et al., 2023). We detected homozygosity for the variant of this gene in this Hodgkin’s lymphoma family; all affected family members were homozygous for the mutation. The constitutive expression of the CD137 ligand has been shown in the serum of patients with B-cell lymphomas (Souza-Fonseca-Guimaraes et al., 2016), which may explain the contribution of this variant to cancer predisposition. Moreover, homozygosity for variants of this gene was shown to lead to immunodeficiency with lymphoproliferation, but Shen et al. described wide clinical heterogeneity, including differences in age at onset and abnormalities ranging from severe to mild (Shen et al., 2023). Our patients did not present any immunodeficiency yet, which may develop later in life.
In case #15, WES revealed a heterozygous BRAT1 variant both in the patient and in the unaffected mother. However, the maternal grandmother with breast cancer and the maternal aunt with ovarian cancer were WT for BRAT1. Subsequent WGS revealed a novel KSR1 variant in all the affected cases. It falls within the N-terminal regulatory domain of the KSR1 protein, which plays a role in scaffold-mediated regulation of the MAPK/ERK signaling pathway. This domain is essential for mediating protein–protein interactions with RAF and MEK, which is crucial for transmitting RAS signaling. Variants in this region may impair MAPK cascade regulation, contributing to oncogenic processes including leukemogenesis. KSR1 consists of 22 exons, and translation begins at exon 4 (Liu et al., 2023), where our variant is also located. Disruption of this region has been reported to inhibit protein expression, which may also be occurring in our patients and should be checked by further studies.
Both leukemia and lymphoma are disorders of hematopoietic cells, and they share common molecular pathways. However, their organs of origin and the affected cells differ. Members of the DNA repair or JAK/STAT pathways are deregulated in both conditions (Fernández et al., 2023; Liang et al., 2024), whereas NFKB is prominently involved in lymphoma development (Grondona et al., 2018), and TP53 or MAPK/RAS is prominently involved in leukemia development (Yu et al., 2020; Pikman and Stieglitz, 2021). Our study findings were consistent with these characteristics. In both case #5 and case #17, we detected variants in genes (MLH1 and MSH6) that play a role in DNA repair mechanisms. However, the TNFRSF9 variant, an NFKB pathway member, was found in case #13, who belonged to family with Hodgkin’s lymphoma.
Some of the affected family members did not provide samples for segregation analysis. This major limitation emphasizes the critical need for biobanking of samples from affected individuals. However, after the variant was detected, some families declined to provide additional samples due to anxiety. Beyond the general concerns about knowing one’s cancer susceptibility, families also worry about the cancer risk in unaffected but variant-positive siblings (Bon et al., 2022). While the patients in this study were enrolled from the main hematology/oncology centers, the demographic distribution was diverse. However, patients living in rural areas were often unable to travel, and the clinical features could not be assessed, limiting detailed phenotyping. Furthermore, the underrepresentation of Turkish genomes in public databases complicates variant analysis and interpretation, posing significant challenges for precision medicine and genetic diagnostics in Türkiye. Thus, the genetic background of approximately 35% of the cohort remained unclear.
In conclusion, we identified known and novel variants that may contribute to genetic predisposition to childhood leukemia/lymphoma. Our findings can guide physicians in developing treatment strategies, surveillance plans, and genetic counseling approaches for the family members at risk. Our study also raised awareness of childhood cancer predisposition among the participating hemato-oncology clinics across multiple hospitals in Türkiye. This study represents the largest cohort in Türkiye for investigating genetic predisposition to childhood leukemia/lymphoma.
Data availability statement
The raw data supporting the findings of this article will be made available by the authors upon reasonable request.
Ethics statement
These studies involving humans were approved by Acimath Mehmet Ali Aydimanlar University (ATADEK; no. 2017-16/5) and by the Acimath Healthcare Institutions Medical Research Ethics Committee (ATADEK; no. 2024-11/496). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
GO: Methodology, Writing – original draft, Investigation, Writing – review and editing. OO: Writing – review and editing, Investigation, Methodology. FT: Writing – review and editing, Investigation, Methodology. CC: Writing – review and editing. KY: Investigation, Methodology, Writing – original draft, Writing – review and editing. FE: Writing – review and editing. BS: Writing – review and editing. FA: Writing – review and editing. TB: Writing – review and editing. YA: Writing – review and editing. NY: Writing – review and editing. NO: Writing – review and editing. IB: Writing – review and editing. DK: Writing – review and editing. BE: Methodology, Writing – review and editing. AA: Methodology, Writing – review and editing. DA: Writing – review and editing. EI: Writing – review and editing. UD: Writing – review and editing. GO: Writing – review and editing. OD: Writing – review and editing. SA: Writing – review and editing. EA: Writing – review and editing. BU: Writing – review and editing. UA: Writing – review and editing. OD: Writing – review and editing. SA: Writing – review and editing. MS: Writing – review and editing. AN: Writing – review and editing. NB: Methodology, Writing – original draft, Investigation, Writing – review and editing. UO: Writing – review and editing. ON: Writing – review and editing, Writing – original draft, Methodology, Investigation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This project received support from the Acıbadem University Scientific Research Projects Commission (ABAPKO) unit (Project No: 2019/03/05), the Turkish Society of Hematology (Project no: 2021/04), and the İSTisNA (İstanbul Undiagnosed and Rare Diseases Solution Platform) Project (Project No: TR10/19/FZD/0003) (https://istisna.org/en/about-us/). The whole-genome sequencing component was funded by grants from the Swedish Childhood Cancer Fund (PR2022-0027), the Swedish Research Council (2021-02860), the Swedish Cancer Society (22 2057 PJ), the Cancer Society of Stockholm (211293), Hållsten Research Foundation, Berth von Kantzow Foundation, and Region Stockholm (grant number: 51024). The funders had no role in the study design, data collection, data analysis, data interpretation, or writing of the report.
Acknowledgments
This study was conducted as part of the COST-LEGEND (European Cooperation in Science and Technology-LEukemia GENe Discovery) and in collaboration with the Swedish ChiCaP (Childhood Cancer Predisposition) projects. Gizem Onder was supported by The Scientific and Technological research council of Türkiye (TUBITAK) 2214-A International Research Fellowship Program for PhD Students (2022/2) and EJP RD (European Joint Program on Rare Diseases)—The ERN (European Reference Networks) Research Mobility Fellowships. In addition, NB was supported by the scholarship number 1059B192100905 within the framework of the 2219—International Postdoctoral Research Scholarship Program provided by TUBITAK BIDEB. Eylul Aydin was supported by Acibadem University Kerem Aydınlar Foundation PhD Scholarship program.
The authors thank their colleagues at the Department of Molecular Medicine and Surgery, Karolinska Institutet, for hosting Gizem Onder as a short-term visiting researcher and Nurse Yesim Oren from Acıbadem Altunizade Hospital Department of Pediatric Oncology for supporting the sample and data collection and providing a professional but sincere bridge with the patients and families. They are also grateful to Furkan Baysal Atalay and Huma Gunay for their assistance and dedication in preparing the samples for downstream analysis in the laboratory.
Conflict of interest
Author BE was employed by GENIVA Information Health Services Company.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1624306/full#supplementary-material
References
Alba-Pavón, P., Alaña, L., Gutierrez-Jimeno, M., García-Obregón, S., Imízcoz, T., Panizo, E., et al. (2023). Identification of germline cancer predisposition variants in pediatric sarcoma patients from somatic tumor testing. Sci. Rep. 13 (1), 2959. doi:10.1038/s41598-023-29982-2
Bakhuizen, J. J., Bourdeaut, F., Wadt, K. A. W., Kratz, C. P., Jongmans, C. J. M., and Waespe, N. (2024). Genetic testing for childhood cancer predisposition syndromes: controversies and recommendations from the SIOPE host genome working group meeting 2022. EJC Paediatr. Oncol. 4, 100176. doi:10.1016/j.ejcped.2024.100176
Baliakas, P., Tesi, B., Wartiovaara-Kautto, U., Stray-Pedersen, A., Friis, L. S., Dybedal, I., et al. (2019). Nordic guidelines for germline predisposition to myeloid neoplasms in adults: recommendations for genetic diagnosis, clinical management and Follow-up. Hemasphere 3 (6), e321. doi:10.1097/HS9.0000000000000321
Bloom, M., Maciaszek, J. L., Clark, M. E., Pui, C. H., and Nichols, K. E. (2020). Recent advances in genetic predisposition to pediatric acute lymphoblastic leukemia. Expert Rev. Hematol. 13 (1), 55–70. doi:10.1080/17474086.2020.1685866
Bon, S. B. B., Wouters, R. H. P., Hol, J. A., Jongmans, M. C. J., van den Heuvel-Eibrink, M. M., and Grootenhuis, M. A. (2022). Parents' experiences with large-scale sequencing for genetic predisposition in pediatric renal cancer: a qualitative study. Psychooncology 31 (10), 1692–1699. doi:10.1002/pon.6016
Brady, S. W., Roberts, K. G., Gu, Z., Shi, L., Pounds, S., Pei, D., et al. (2022). The genomic landscape of pediatric acute lymphoblastic leukemia. Nat. Genet. 54 (9), 1376–1389. doi:10.1038/s41588-022-01159-z
Brozou, T., Yasin, L., Brandes, D., Picard, D., Walter, C., Varghese, J., et al. (2022). Resolving inherited and de novo germline predisposing sequence variants by means of whole exome trio analyses in childhood hematological malignancies. Front. Pediatr. 10, 1080347. doi:10.3389/fped.2022.1080347
Byrjalsen, A., Hansen, T. V. O., Stoltze, U. K., Mehrjouy, M. M., Barnkob, N. M., Hjalgrim, L. L., et al. (2020). Nationwide germline whole genome sequencing of 198 consecutive pediatric cancer patients reveals a high incidence of cancer prone syndromes. PLoS Genet. 16 (12), e1009231. doi:10.1371/journal.pgen.1009231
Cavé, H., Caye, A., Strullu, M., Aladjidi, N., Vignal, C., Ferster, A., et al. (2016). Acute lymphoblastic leukemia in the context of RASopathies. Eur. J. Med. Genet. 59 (3), 173–178. doi:10.1016/j.ejmg.2016.01.003
Chen, Z., Naito, M., Hori, S., Mashima, T., Yamori, T., and Tsuruo, T. (1999). A human IAP-family gene, apollon, expressed in human brain cancer cells. Biochem. Biophys. Res. Commun. 264 (3), 847–854. doi:10.1006/bbrc.1999.1585
Childs, E. J., Mocci, E., Campa, D., Bracci, P. M., Gallinger, S., Goggins, M., et al. (2015). Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat. Genet. 47 (8), 911–916. doi:10.1038/ng.3341
Daher, R., Kannengiesser, C., Houamel, D., Lefebvre, T., Bardou-Jacquet, E., Ducrot, N., et al. (2016). Heterozygous mutations in BMP6 pro-peptide lead to inappropriate hepcidin synthesis and moderate iron overload in humans. Gastroenterology 150 (3), 672–683. doi:10.1053/j.gastro.2015.10.049
Dündar, M., and Karabulut, S. Y. (2010). Rare disease and orphan drugs in Turkey; medical and social problem. Erciyes Med J. 32, 195–200.
Ercan, A. B., Aronson, M., Fernandez, N. R., Chang, Y., Levine, A., Liu, Z. A., et al. (2024). Clinical and biological landscape of constitutional mismatch-repair deficiency syndrome: an international replication repair deficiency consortium cohort study. Lancet Oncol. 25 (5), 668–682. doi:10.1016/S1470-2045(24)00026-3
Fernández, S., Solórzano, J. L., Díaz, E., Menéndez, V., Maestre, L., Palacios, S., et al. (2023). JAK/STAT blockade reverses the malignant phenotype of hodgkin and reed-sternberg cells. Blood Adv. 7 (15), 4135–4147. doi:10.1182/bloodadvances.2021006336
Fukumura, K., Kawazu, M., Kojima, S., Ueno, T., Sai, E., Soda, M., et al. (2016). Genomic characterization of primary central nervous system lymphoma. Acta Neuropathol. 131 (6), 865–875. doi:10.1007/s00401-016-1536-2
Goudie, C., Witkowski, L., Cullinan, N., Reichman, L., Schiller, I., Tachdjian, M., et al. (2021). Performance of the McGill interactive pediatric OncoGenetic guidelines for identifying cancer predisposition syndromes. JAMA Oncol. 7 (12), 1806–1814. doi:10.1001/jamaoncol.2021.4536
Gröbner, S. N., Worst, B. C., Weischenfeldt, J., Buchhalter, I., Kleinheinz, K., Rudneva, V. A., et al. (2018). The landscape of genomic alterations across childhood cancers. Nature 555 (7696), 321–327. doi:10.1038/nature25480
Grondona, P., Bucher, P., Schulze-Osthoff, K., Hailfinger, S., and Schmitt, A. (2018). NF-κB activation in lymphoid malignancies: Genetics, signaling, and targeted therapy. Biomedicines 6 (2), 38. doi:10.3390/biomedicines6020038
Hacettepe University Institute of Population Studies (2019). The scientific and technological research council of Turkey (TUBITAK), T.R. presidency of Turkey directorate of strategy and budget, and ICF. 2019. Turkey demographic and health survey 2018. Ankara, Turkey: Hacettepe university institute of population studies, presidency of Turkey directorate of strategy and budget. Available online at: https://www.dhsprogram.com/pubs/pdf/FR372/FR372.pdf.
Hayani, A., Suarez, C. R., Molnar, Z., LeBeau, M., and Godwin, J. (1994). Acute myeloid leukaemia in a patient with Seckel syndrome. J. Med. Genet. 31 (2), 148–149. doi:10.1136/jmg.31.2.148
Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J. F., Darbandi, S. F., Knowles, D., Li, Y. I., et al. (2019). Predicting splicing from primary sequence with deep learning. Cell 176 (3), 535–548. doi:10.1016/j.cell.2018.12.015
Jongmans, M. C., Loeffen, J. L., Waanders, E., Hoogerbrugge, P. M., Ligtenberg, M. J., Kuiper, R. P., et al. (2016). Recognition of genetic predisposition in pediatric cancer patients: an easy-to-use selection tool. Eur. J. Med. Genet. 59 (3), 116–125. doi:10.1016/j.ejmg.2016.01.008
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Kars, M. E., Basak, A. N., Onat, O. E., Bilguvar, K., Choi, J., Itan, Y., et al. (2021). The genetic structure of the Turkish population reveals high levels of variation and admixture. Proc. Natl. Acad. Sci. U. S. A. 118 (36), e2026076118. doi:10.1073/pnas.2026076118
Lazic, J., Haas, O. A., Özbek, U., Ripperger, T., Byrjalsen, A., Te, K. G., et al. (2023). Perception and management of cancer predisposition in pediatric cancer centers: a European-wide questionnaire-based survey. Pediatr. Blood Cancer 70 (5), e30229. doi:10.1002/pbc.30229
Li, H., Sisoudiya, S. D., Martin-Giacalone, B. A., Khayat, M. M., Dugan-Perez, S., Marquez-Do, D. A., et al. (2021). Germline cancer predisposition variants in pediatric rhabdomyosarcoma: a report from the children's oncology group. J. Natl. Cancer Inst. 113 (7), 875–883. doi:10.1093/jnci/djaa204
Liang, D., Wang, Q., Zhang, W., Tang, H., Song, C., Yan, Z., et al. (2024). JAK/STAT in leukemia: a clinical update. Mol. Cancer 23 (1), 25. doi:10.1186/s12943-023-01929-1
Liu, Z., Krstic, A., Neve, A., Casalou, C., Rauch, N., Wynne, K., et al. (2023). Kinase suppressor of RAS 1 (KSR1) maintains the transformed phenotype of BRAFV600E mutant human melanoma cells. Int. J. Mol. Sci. 24 (14), 11821. doi:10.3390/ijms241411821
Maese, L. D., Wlodarski, M. W., Kim, S. Y., Bertuch, A. A., Bougeard, G., Chang, V. Y., et al. (2024). Update on recommendations for surveillance for children with predisposition to hematopoietic malignancy. Clin. Cancer Res. 30 (19), 4286–4295. doi:10.1158/1078-0432.CCR-24-0685
Miao, K., Lei, J. H., Valecha, M. V., Zhang, A., Xu, J., Wang, L., et al. (2020). NOTCH1 activation compensates BRCA1 deficiency and promotes triple-negative breast cancer formation. Nat. Commun. 11 (1), 3256. doi:10.1038/s41467-020-16936-9
Moriyama, T., Metzger, M. L., Wu, G., Nishii, R., Qian, M., Devidas, M., et al. (2015). Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol. 16 (16), 1659–1666. doi:10.1016/S1470-2045(15)00369-1
Ozdemir, O., Bychkovsky, B. L., Unal, B., Onder, G., Amanvermez, U., Aydin, E., et al. (2024). Molecular and in silico analysis of the CHEK2 gene in individuals with high risk of cancer predisposition from Türkiye. Cancers (Basel) 16 (22), 3876. doi:10.3390/cancers16223876
Pikman, Y., and Stieglitz, E. (2021). Targeting the ras pathway in pediatric hematologic malignancies. Curr. Opin. Pediatr. 33 (1), 49–58. doi:10.1097/MOP.0000000000000981
Qian, M., Cao, X., Devidas, M., Yang, W., Cheng, C., Dai, Y., et al. (2018). TP53 germline variations influence the predisposition and prognosis of B-Cell acute lymphoblastic leukemia in children. J. Clin. Oncol. 36 (6), 591–599. doi:10.1200/JCO.2017.75.5215
Rahman, N. (2014). Realizing the promise of cancer predisposition genes. Nature 505 (7483), 302–308. doi:10.1038/nature12981
Rampersaud, E., Ziegler, D. S., Iacobucci, I., Payne-Turner, D., Churchman, M. L., Schrader, K. A., et al. (2019). Germline deletion of ETV6 in familial acute lymphoblastic leukemia. Blood Adv. 3 (7), 1039–1046. doi:10.1182/bloodadvances.2018030635
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Ripperger, T., and Schlegelberger, B. (2016). Acute lymphoblastic leukemia and lymphoma in the context of constitutional mismatch repair deficiency syndrome. Eur. J. Med. Genet. 59 (3), 133–142. doi:10.1016/j.ejmg.2015.12.014
Salmoiraghi, S., Montalvo, M. L., Ubiali, G., Tosi, M., Peruta, B., Zanghi, P., et al. (2016). Mutations of TP53 gene in adult acute lymphoblastic leukemia at diagnosis do not affect the achievement of hematologic response but correlate with early relapse and very poor survival. Haematologica 101 (6), e245–e248. doi:10.3324/haematol.2015.137059
Schlegelberger, B., Mecucci, C., and Wlodarski, M. (2021). Review of guidelines for the identification and clinical care of patients with genetic predisposition for hematological malignancies. Fam. Cancer 20 (4), 295–303. doi:10.1007/s10689-021-00263-z
Shen, K., Wang, J., Zhou, K., Mu, W., Zhang, M., Deng, X., et al. (2023). CD137 deficiency because of two novel biallelic TNFRSF9 mutations in a patient presenting with severe EBV-associated lymphoproliferative disease. Clin. Transl. Immunol. 12 (5), e1448. doi:10.1002/cti2.1448
Souza-Fonseca-Guimaraes, F., Blake, S. J., Makkouk, A., Chester, C., Kohrt, H. E., and Smyth, M. J. (2016). Anti-CD137 enhances anti-CD20 therapy of systemic B-cell lymphoma with altered immune homeostasis but negligible toxicity. Oncoimmunology 5 (7), e1192740. doi:10.1080/2162402X.2016.1192740
Stranneheim, H., Lagerstedt-Robinson, K., Magnusson, M., Kvarnung, M., Nilsson, D., Lesko, N., et al. (2021). Integration of whole genome sequencing into a healthcare setting: high diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. 13, 40. doi:10.1186/s13073-021-00855-5
Tesi, B., Robinson, K. L., Abel, F., Díaz de Ståhl, T., Orrsjö, S., Poluha, A., et al. (2024). Diagnostic yield and clinical impact of germline sequencing in children with CNS and extracranial solid tumors-a nationwide, prospective Swedish study. Lancet Reg. Health Eur. 39, 100881. doi:10.1016/j.lanepe.2024.100881
Tidyman, W. E., and Rauen, K. A. (2016). Pathogenetics of the RASopathies. Hum. Mol. Genet. 25 (R2), R123–R132. doi:10.1093/hmg/ddw191
Yu, L., Yin, B., Qu, K., Li, J., Jin, Q., Liu, L., et al. (2018). Screening for susceptibility genes in hereditary non-polyposis colorectal cancer. Oncol. Lett. 15 (6), 9413–9419. doi:10.3892/ol.2018.8504
Yu, C. H., Chang, W. T., Jou, S. T., Lin, T. K., Chang, Y. H., Lin, C. Y., et al. (2020). TP53 alterations in relapsed childhood acute lymphoblastic leukemia. Cancer Sci. 111 (1), 229–238. doi:10.1111/cas.14238
Keywords: germline variants, short-read sequencing, cancer predisposition, childhood leukemia, childhood lymphoma
Citation: Onder G, Ozdemir O, Taylan F, Canpolat C, Yalcin K, Erbey F, Sozmen BO, Asarcikli F, Bayhan T, Akcabelen YM, Yarali N, Ozbek NY, Bozkaya IO, Kacar D, Ergun B, Akkus A, Albayrak D, Ince E, Demirsoy U, Ozdemir GN, Dogru O, Aras S, Aydin E, Unal B, Amanvermez U, Dogan OA, Akyoney S, Sayitoglu M, Nordgren A, Bugra Agaoglu N, Ozbek U and Ng OH (2025) Genetic heterogeneity in childhood leukemia/lymphoma: a Turkish cohort with strong predisposition. Front. Genet. 16:1624306. doi: 10.3389/fgene.2025.1624306
Received: 07 May 2025; Accepted: 30 July 2025;
Published: 09 September 2025.
Edited by:
Jorge Melendez-Zajgla, National Institute of Genomic Medicine (INMEGEN), MexicoReviewed by:
Poonam Gera, Research and Education in Cancer (ACTREC), IndiaEric Nickels, Children’s Hospital of Los Angeles, United States
Copyright © 2025 Onder, Ozdemir, Taylan, Canpolat, Yalcin, Erbey, Sozmen, Asarcikli, Bayhan, Akcabelen, Yarali, Ozbek, Bozkaya, Kacar, Ergun, Akkus, Albayrak, Ince, Demirsoy, Ozdemir, Dogru, Aras, Aydin, Unal, Amanvermez, Dogan, Akyoney, Sayitoglu, Nordgren, Bugra Agaoglu, Ozbek and Ng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nihat Bugra Agaoglu, QWdhb2dsdS5OaWhhdEBraG53LmRl; Ozden Hatirnaz Ng, b3pkZW4uaGF0aXJuYXpAYWNpYmFkZW0uZWR1LnRy