 George Hajishengallis1*
George Hajishengallis1* Tetsuhiro Kajikawa1
Tetsuhiro Kajikawa1 Evlambia Hajishengallis2
Evlambia Hajishengallis2 Tomoki Maekawa3
Tomoki Maekawa3 Edimara S. Reis4
Edimara S. Reis4 Dimitrios C. Mastellos5
Dimitrios C. Mastellos5 Despina Yancopoulou6
Despina Yancopoulou6 Hatice Hasturk7
Hatice Hasturk7 John D. Lambris4*
John D. Lambris4*- 1Department of Microbiology, Penn Dental Medicine, University of Pennsylvania, Philadelphia, PA, United States
- 2Division of Pediatric Dentistry, Department of Preventive and Restorative Sciences, Penn Dental Medicine, University of Pennsylvania, Philadelphia, PA, United States
- 3Research Center for Advanced Oral Science, Graduate School of Medical and Dental Sciences, Niigata University, Niigata, Japan
- 4Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States
- 5Division of Biodiagnostic Sciences and Technologies, National Center for Scientific Research “Demokritos”, Athens, Greece
- 6Amyndas Pharmaceuticals, Glyfada, Greece
- 7Center for Clinical and Translational Research, Forsyth Institute, Cambridge, MA, United States
Periodontitis is a prevalent inflammatory disease that leads to the destruction of the tooth-supporting tissues. Current therapies are not effective for all patients and this oral disease continues to be a significant public health and economic burden. Central to periodontal disease pathogenesis is a reciprocally reinforced interplay between microbial dysbiosis and destructive inflammation, suggesting the potential relevance of host-modulation therapies. This review summarizes and discusses clinical observations and pre-clinical intervention studies that collectively suggest that complement is hyperactivated in periodontitis and that its inhibition provides a therapeutic benefit. Specifically, interception of the complement cascade at its central component, C3, using a locally administered small peptidic compound (Cp40/AMY-101) protected non-human primates from induced or naturally occurring periodontitis. These studies indicate that C3-targeted intervention merits investigation as an adjunctive treatment of periodontal disease in humans.
Introduction
Complement represents an interactive network of soluble, cell surface-associated and intracellular molecules that activate, amplify, and regulate immunity and inflammation (1, 2). In addition to the classic serum proteins (C1-9), the network contains overall some 50 proteins, including pattern-recognition molecules, convertases and other proteases, receptors and regulators. Complement activation is initiated via distinct pathways, the classical, lectin, or alternative (Figure 1). The classical and lectin pathways are activated following the binding of complement-associated pattern-recognition molecules (e.g., C1q and mannose-binding lectin, respectively) to immune complexes (classical pathway) or to carbohydrate moieties exposed on microbial or damaged/necrotic host cells (lectin pathway). The alternative pathway is initiated by a tick-over mechanism and moreover amplifies the initial response induced by the other two complement pathways.
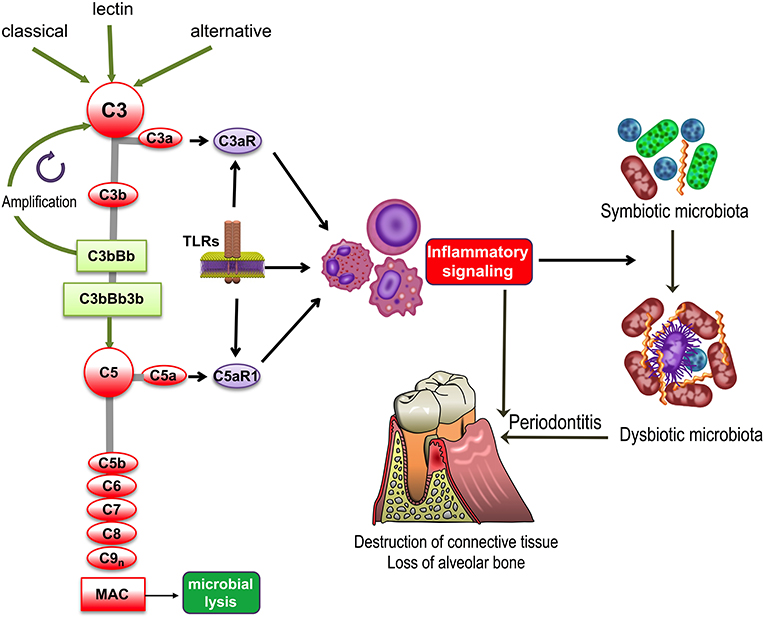
Figure 1. Complement and TLR involvement in dysbiosis and inflammatory bone loss in periodontitis. The classic, lectin, and alternative pathways converge at and activate the central complement component, C3, resulting in the generation of various effectors, such as, the C3b opsonin, the inflammatory anaphylatoxins C3a and C5a, and the C5b-9 membrane attack complex (MAC). The C3a and C5a activation fragments activate respectively C3aR and C5aR1, which cross-talk with Toll-like receptors (TLRs) and synergistically activate inflammatory leukocytes. TLRs also upregulate the expression of C3aR and C5aR1. Inflammation induced by complement-TLR crosstalk interactions not only causes gingival tissue destruction and bone loss in periodontitis but also contributes to the remodeling of a symbiotic microbiota into a dysbiotic one, thereby further potentiating destructive inflammation.
All three pathways converge at the central component of the complement system, C3, the activation of which leads to the generation of effectors that facilitate the ability of antibodies and phagocytes to clear microbial pathogens (via C3b opsonization), induce chemotaxis and inflammation (via the C3a and C5a anaphylatoxins), and lyse susceptible microbial targets (via the C5b-9 membrane attack complex [MAC]) (Figure 1). Furthermore, in cooperation with other immune and physiological systems, complement contributes to normal tissue and organ development, integrates and coordinates innate and adaptive immunity, mediates apoptotic cell clearance, and promotes tissue repair following injury (1, 3). In recent years, it has also become increasingly appreciated that complement is not exclusively produced in the liver and its actions are not restricted in the intravascular and extracellular compartments. Indeed, it has been shown that complement components can be produced locally by resident tissue cells as well as by recruited leukocytes. Moreover, an intracellular complement system was identified and shown to have novel homeostatic and immune functions, such as regulation of CD4+ T cell activation (2, 4).
Despite its importance in homeostatic immunity, complement may become dysregulated or excessively activated (e.g., due to host genetic or microbial virulence factors), thereby turning from a homeostatic to a pathological effector that can drive or exacerbate a number of disorders, such as cancer or inflammatory diseases, including periodontitis (Figure 1) as discussed in detail in the next section (5–8). Periodontitis is a common chronic inflammatory disease that causes destruction of the periodontium, i.e., the tissues that surround and support the teeth, namely, gingiva, periodontal ligament, and alveolar bone. If left untreated, periodontitis can lead to tooth loss and possibly impaired mastication (9). It is estimated that severe periodontitis afflicts ~10% of adults (10, 11). In its severe form, periodontal disease is associated with elevated risk for certain systemic conditions, such as atherosclerosis (12).
Recent evidence from clinical microbiome studies and mechanistic studies in animal models have shown that periodontitis is a dysbiotic disease rather than an infection attributed to a select few species (13–15). Connective tissue damage and loss of alveolar bone is mediated by a dysregulated and excessive inflammatory response, which includes components of both innate and adaptive immunity but fails to control the dysbiotic microbial challenge that induced it (16). In fact, the destructive periodontal inflammatory response is exploited by the dysbiotic microbial communities to procure nutrients from tissue breakdown products (17, 18). The fact that the disease is predominantly mediated by the host inflammatory response and that inflammation is necessary to support dysbiotic microbial communities justifies the rationale for developing host-modulation strategies to treat periodontitis. Such novel interventions may be used as adjuncts to improve current therapies (e.g., mechanical removal of the pathogenic biofilm), which are not always sufficient to treat periodontitis (19–22), thus accentuating its significant public health and economic burden (9, 11, 23, 24). Here, we discuss clinical and preclinical studies that have collectively linked complement overactivation to periodontitis and provided a mechanistic understanding of this relationship, paving the way to complement-targeted therapies to treat this oral inflammatory disease with strong associations to increased risk for other systemic conditions.
Clinical Studies
The possible involvement of complement in human periodontitis was first recognized in the 1970s and 1980s by histological and clinical studies analyzing the gingival crevicular fluid (GCF) in periodontal health and disease. GCF represents the inflammatory exudate which bathes the gingival crevice or pocket, i.e., the space between the free gingiva and the tooth surfaces (25). GCF samples from periodontitis patients were shown to have complement-dependent hemolytic activity, suggesting that a functional complement system is present in this inflammatory exudate (26, 27). Activated complement fragments were shown to be highly abundant in the GCF from patients, but were undetectable or present in lower concentrations in GCF from healthy control individuals (28–32). Similarly, complement components and cleavage products were also readily detected in chronically inflamed gingiva but were undetected or at lower abundance in healthy tissue samples; the complement components detected in diseased gingiva (and also in GCF) were representative of the entire cascade (e.g., C1q, factor B, Bb, C3, C3a, C3b, C3c, C3d, C4, C5, C5a, C5b, C9) (26–37).
Importantly, periodontal therapy that resulted in decreased clinical indices of periodontal inflammation and tissue destruction also led to decreased C3 activation in the GCF (38). Conversely, and consistently, the progression of gingival inflammation during an experimental human gingivitis study was associated with elevated C3 cleavage in the GCF (32). Specifically, this study examined the cleavage of factor B, C3, and C4 in GCF collected during the experimental period and demonstrated, respectively, their conversion to Bb and C3c but not to C4c, thus implying selective activation of the alternative pathway (32). An immunohistochemical study showed that the complement regulator CD59 is expressed at lower levels in the gingiva of periodontitis patients as compared to healthy individuals, which might imply compromised protection of periodontitis-involved tissues against MAC-mediated autologous tissue damage (35).
More recent studies also support an association between complement and periodontitis. A case of aggressive periodontitis with severe angioedema localized to the gingiva was linked to dysregulated complement function, specifically deficiency of the C1-esterase inhibitor (C1INH) (39). A single nucleotide polymorphism affecting C5 (rs17611), which was previously linked to elevated C5 in serum and susceptibility to the complement-associated disease liver fibrosis (40), was shown to be more prevalent in patients with periodontitis than in healthy controls (41). In terms of its expression, C3 was shown to be among the top 5% genes that are most strongly downregulated following periodontal therapy (42). Another study has used integrative gene prioritization and databases from genome-wide association studies and microarray experiments, and identified C3 among the top 21 most promising candidate genes involved in periodontal disease (43). Interestingly, partial C4 gene deficiencies are significantly more frequent in periodontitis patients than in healthy individuals (44). This finding might suggest a protective function associated with C4, the activation of which occurs via the classical or the lectin pathways. However, it should be noted that C4a was recently shown to bind and activate protease-activated receptors (PAR) 1 and 4 (45) which are expressed by platelets and endothelial cells (46). Thus, C4-mediated effects may not necessarily involve downstream triggering of C3-dependent activities. Whether C4a might mediate complement crosstalk with the coagulation and/or the endothelial barrier system is currently uncertain as is the impact of such interactions on periodontitis.
The aforementioned clinical studies collectively indicate a role for complement activation in periodontal disease pathogenesis. However, the correlative nature of these human studies could not safely establish a causal relationship between complement and periodontitis and distinguish it from the alternative possibility that complement activation could simply be a marker of local periodontal inflammation. Causal evidence was derived from mechanistic animal model-based studies described below.
Mechanistic Studies in Animal Models
Animals models can be engaged to determine causative links between potential mechanisms and disease pathogenesis (47), thereby not only promoting knowledge on pathogenesis but also identifying therapeutic targets and paving the way to human clinical trials. As the triggering of the complement cascade is intertwined with TLR activation (3), the two systems are discussed together in the studies presented here.
In response to microbial infection or tissue damage, complement and TLRs are swiftly activated, frequently by the same agonists. In this regard, bacterial lipopolysaccharide (LPS; a TLR4 agonist), fungal zymosan (TLR2/6 agonist) and bacterial CpG DNA (TLR9 agonist) not only induce TLR signaling but also can activate complement (48). In fact, complement and TLRs are not only co-activated in response to microbial infection and other types of insult, such as tissue injury, but they also engage in signaling crosstalk interactions in several myeloid cell types (monocytes, macrophages, neutrophils, and dendritic cells) (49–54) (Figure 1). In a pioneering study, different TLR agonists systemically given to mice lacking a major membrane-associated complement regulator, the decay-accelerating factor, induced significantly higher plasma levels of pro-inflammatory cytokines than wild-type controls (55). Similarly, mice systemically co-injected with TLR agonists (specifically TLR2, TLR4, and TLR9 ligands) and a potent complement activator (cobra venom factor) display remarkably high plasma levels of pro-inflammatory cytokines (55). In the complement-TLR crosstalk, the activated signaling pathways converge at mitogen-activated protein kinases (extracellular signal-regulated kinase-1 and-2 and c-Jun N-terminal kinase), which in turn activate key transcription factors, namely activator protein-1 (AP-1) and nuclear factor-κB (NF-κB) (55). Although this synergy has the potential to invigorate innate immunity against infection, it may also contribute to destructive inflammatory responses.
In line with these findings, the concomitant activation of C5aR1 and TLR2 in the mouse gingiva by local co-administration of specific ligands (C5a and Pam3Cys, respectively) resulted in the induction of significantly higher levels of IL-1β, IL-6, IL-17, and TNF than stimulation of either receptor alone (56). These data suggested that a synergy between complement and TLRs may be a major contributor to the induction of periodontal inflammation. In support of this notion, mice lacking C5aR1 are quite resistant against inflammatory bone loss regardless of the presence of TLR2 (57) and, in an analogous manner, mice lacking TLR2 are protected from inflammatory bone loss regardless of the presence of C5aR1 (58). TLR9-deficient mice are also protected against experimental periodontitis (59), which could be attributed, at least in part, to complement-TLR9 synergy (55). These studies utilized a murine model of periodontitis in which the disease is initiated by dysbiosis following oral gavage with the keystone pathogen Porphyromonas gingivalis (60, 61). Consistent with the importance of complement involvement in periodontal disease pathogenesis, C3-deficient mice were protected against periodontitis in three distinct models, ligature-induced periodontitis, P. gingivalis-induced periodontitis, and aging-associated periodontitis (62). The ligature model involves the placement of silk ligatures around molar teeth leading to massive accumulation of indigenous bacteria and induction of inflammation and alveolar bone loss in specific-pathogen-free (but not germ-free) animals (60, 63–65). In the aging-associated periodontitis model, periodontal inflammation and bone loss develops naturally as a function of old age when homeostatic mechanisms break down (66–68). Interestingly, C3-deficient mice also exhibited reduced periodontal bacterial load in P. gingivalis-induced periodontitis as compared to wild-type littermate controls (62). These data suggest that lack of complement activation does not lead to defective control of the periodontal microbiota and, moreover, are consistent with the concept that destructive inflammation is required to sustain a quantitatively and compositionally altered dysbiotic microbiota (18).
Translational Preclinical Studies
The studies discussed earlier suggested that complement may be a promising target for the treatment of periodontitis. Indeed, in the oral gavage model of P. gingivalis-induced periodontitis, intra-gingival microinjection of wild-type mice with PMX-53, a C5aR1 antagonist (69), suppressed the induction of inflammatory cytokines (IL-1β, IL-6 and IL-17, and TNF) in the gingival tissue and inhibited alveolar bone loss (56). This protective effect occurred despite the presence of intact TLR2, in other words, inflammatory bone loss can be effectively inhibited by blocking only one of the two cross-talking receptors (56). PMX-53 was also tested in the ligature-induced periodontitis model where disease can be initiated independently of P. gingivalis (63). Although substantial inflammatory bone loss was induced after 5 days at the ligated areas of control-treated mice, mice locally microinjected (at the ligated sites) with PMX-53 exhibited significant protection against periodontal inflammation and bone loss (56). Rats given PMX-205 [another C5aR1 antagonist (70)] via the drinking water were also protected from ligature-induced bone loss (71), although with reduced efficacy perhaps due to the different route of drug administration and/or the use of a different animal species.
It is important to note that the same inflammatory mediators (e.g., TNF, IL-1β, prostaglandin E2) have been implicated in inflammatory periodontal bone loss across different species, such as mice, rats, dogs, non-human primates, and humans (72–77). Therefore, mice appear to be a useful model for human periodontitis especially for mechanistic studies, since mice currently represent the only available species with engineered knock-in or knock-out mutations for a whole panel of key immune response genes. However, promising results obtained in higher animals, such as non-human primates, increase the possibility that candidate drugs can be protective also in humans. In this regard, the periodontal tissue anatomy and immune system of non-human primates are similar to those of humans, and periodontitis in monkeys displays clinical, microbiological, and immuno-histological features that are highly similar to those of human periodontal disease (78–82). In fact, the use of non-human primates becomes necessary for testing drugs that lack specificity for the widely used rodent models and other small animals.
In this regard, compstatin and new generation analogs are small peptidic inhibitors that have an exquisite specificity for human and non-human primate C3 (83–85). Given the absence of available C3 inhibitors in mice, the appropriateness of C3 as a therapeutic target in periodontitis could only be tested in primates. Specifically, the third-generation compstatin analog Cp40 was tested in cynomolgus monkeys (Macaca fascicularis) (62). Cp40 has a subnanomolar affinity for C3 (KD = 0.5 nM; 6,000-fold greater than that of the original compstatin) and a plasma human half-life (48 h) that exceeds expectations for most peptidic drugs. Mechanistically, the original compstatin and new generation analogs bind C3 and block its interaction with and cleavage by the C3 convertase into its active fragments, C3a and C3b (86) (Figure 2). In other words, the compstatin family of C3 inhibitors protect the C3 substrate rather than interfere with the C3 convertase. As a consequence, the compstatins prevent propagation and amplification of complement activation and generation of effector molecules regardless of the mechanism that initiated complement activation (83, 84).
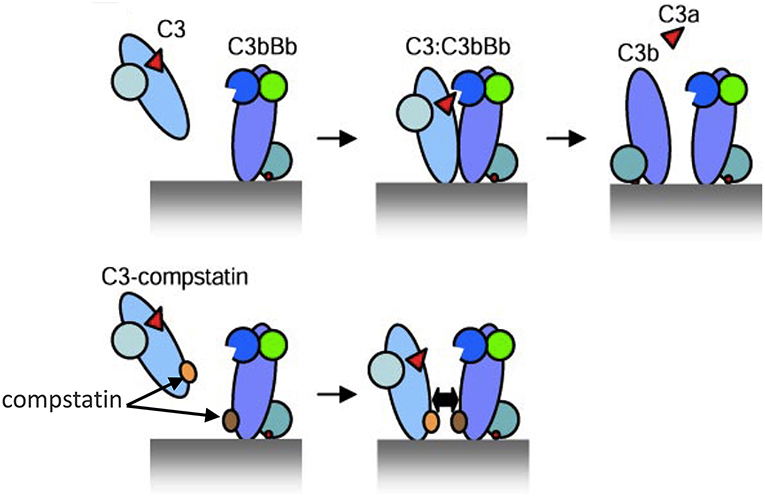
Figure 2. Model of C3 activation and its inhibition by compstatin. (Top) Depiction of key protein interactions resulting in the formation of C3 convertase on a target surface (e.g., a microbial cell surface). Native C3 binds to the convertase (C3bBb) and is cleaved into its active fragments, C3a and C3b. (Bottom) Compstatin acts by blocking protein-protein interaction. Specifically, compstatin binds both native C3 and C3b and sterically hinders the binding of native C3 by C3 convertases, hence preventing C3 cleavage into its active fragments. From ref. 87. Used by permission.
Periodontitis in adult cynomolgus monkeys was induced by placing silk ligatures around posterior teeth on both halves of the mandible (lower jaw) for a period of 6 weeks. Local treatment (through intra-gingival injection) with Cp40 started 3 days following ligature placement. A split-mouth experimental design was applied, where one side was treated with Cp40 and the other with an inactive control peptide, thus each animal served as its own control. The disease was monitored clinically by analyzing clinical indices that assess periodontal inflammation and tissue destruction (87). Cp40 treatment resulted in significant decrease of gingival index and clinical attachment loss, which correlated with lower levels of proinflammatory and osteoclastogenic cytokines (e.g., TNF, IL-1β, IL-17, and RANKL) in the GCF, as well as with decreased numbers of osteoclasts in bone biopsy specimens (62). In contrast to RANKL, the GCF content of osteoprotegerin (OPG), a natural inhibitor of RANKL, was maintained at increased levels in Cp40-treated as compared to control sites. Therefore, Cp40 appeared to cause a favorable reversal of the RANKL/OPG ratio, which is a potential indicator of periodontitis (88). Consistent with these data, radiographic analysis showed that Cp40-treated sites had significantly less bone loss as compared to control-treated sites (62).
To determine the potential usefulness of Cp40 in a therapeutic setting, the drug was administered to adult cynomolgus monkeys with pre-existing, naturally-occurring chronic periodontitis (89). The animals were locally injected in the gingiva with Cp40 either once a week (group of 5 animals) or three times per week (group of 10 animals) for a 6 weeks treatment period followed by a 6 weeks follow-up period in the absence of Cp40 treatment. Clinical examinations and collections of GCF samples were conducted at baseline and throughout the study. In both groups, treatment with Cp40 led to significant decrease in clinical indices that assess periodontal inflammation (gingival index and bleeding on probing), tissue destruction (probing pocket depth and clinical attachment loss) or tooth mobility which is often linked to bone loss. The improvement of clinical disease as reflected by reduced clinical indices correlated with decreased levels of proinflammatory and osteoclastogenic mediators (e.g., IL-17 and RANKL) in the GCF and decreased osteoclast numbers in bone biopsies. The protective effects mediated by Cp40 endured, although with reduced effectiveness, for at least 6 weeks after the drug was discontinued. Cp40 could therefore reverse pre-existing chronic periodontal inflammation without additional treatments, such as scaling and root planing (SRP) (89). Proteomic analysis of GCF samples collected from that study showed involvement of both the alternative and classical pathways of complement activation in naturally occurring non-human primate periodontitis; however, the alternative pathway was the most enriched of all biological pathways identified by gene ontology analysis (90). These proteomic findings are consistent with early clinical reports indicating that the complement alternative pathway is predominantly activated in GCF samples from human periodontitis patients, although the classical pathway is also activated (28, 29, 38). Based on this consideration (and the likelihood that carbohydrate or glycoprotein components of periodontal bacteria may activate the lectin pathway) the concomitant inhibition of all three pathways (as can be done by Cp40) is likely to provide increased protection against periodontitis as compared to inhibition of individual pathways of complement activation. Another main target revealed by the proteomic fingerprinting of GCF samples from Cp40-treated NHPs was leukocyte degranulation. Neutrophils account for considerable tissue damage in human periodontitis, in great part through degranulation of tissue-degrading proteases and cytotoxic molecules (76, 91–94). In this regard, the ability of Cp40 to suppress exocytosis likely represents another host protective mechanism.
In a follow-up study in cynomolgus monkeys with naturally-occurring chronic periodontitis, it was shown that an effective therapeutic dose of locally administered Cp40 [100 μg/site; used in the study by Maekawa et al. (89)] does not cause local irritation and has long-lasting protective effects even when given as infrequently as once per 3 weeks (95). Therefore, taken together, clinical observations in humans and pre-clinical intervention studies in non-human primates suggest that complement is overactivated in periodontitis and that C3 inhibition by Cp40 is a promising host-modulatory therapy that warrants investigation as a potential treatment of human periodontitis.
Given the potential for synergism between complement and TLRs, C3 inhibition in periodontitis can also inhibit inflammation that is activated by TLR signaling either in response to microbial TLR ligands (e.g., LPS, lipoproteins, and bacterial DNA) (96, 97) or in response to endogenous TLR ligands (e.g., biglycan, hyaluronan, or heparan sulfate fragments) that are released upon tissue injury and act as danger-associated molecular patterns (DAMPs) (98, 99). The latter suggests that complement inhibition may also suppress damaged tissue-induced inflammation, thereby blocking also the progression of periodontitis. Interestingly, several TLRs (TLR2, TLR3, TLR4, and TLR9), when activated by bacterial molecules or DAMPs released from stressed/damaged tissues, were shown to induce local expression of complement components (e.g., macrophage production of factor B and C3), thereby further promoting complement activity in an inflammatory environment (100–103). For instance, LPS induces production and release of factor B through a TLR4-TRIF pathway in macrophages (100). Moreover, TLR signaling suppresses the desensitization of G-protein-coupled receptors (GPCRs) by downregulating the expression of G-protein-coupled receptor kinases, required for inducing GPCR phosphorylation and internalization (104). This suggests a mechanism by which TLRs may potentially prolong the activation of GPCRs, such as C3aR and C5aR1. Furthermore, TLR-induced cytokines, such as IL-6, promote the expression of C3aR and C5aR (105). Therefore, TLRs regulate the expression of complement factors and both the expression and activation of complement receptors, which—as alluded to earlier—in turn can amplify TLR-dependent responses. This pro-inflammatory and potentially destructive feed-forward loop can be potentially disrupted by complement inhibition (Figure 3), a notion that may underlie the success of Cp40 treatment in the non-human primate preclinical model. Complement inhibition at the C3 level may also inhibit inflammasome-dependent inflammation since complement pathways (C3aR signaling and sublytic membrane attack complex) were shown to promote the activation of the NLRP3 inflammasome and IL-1β release (106, 107).
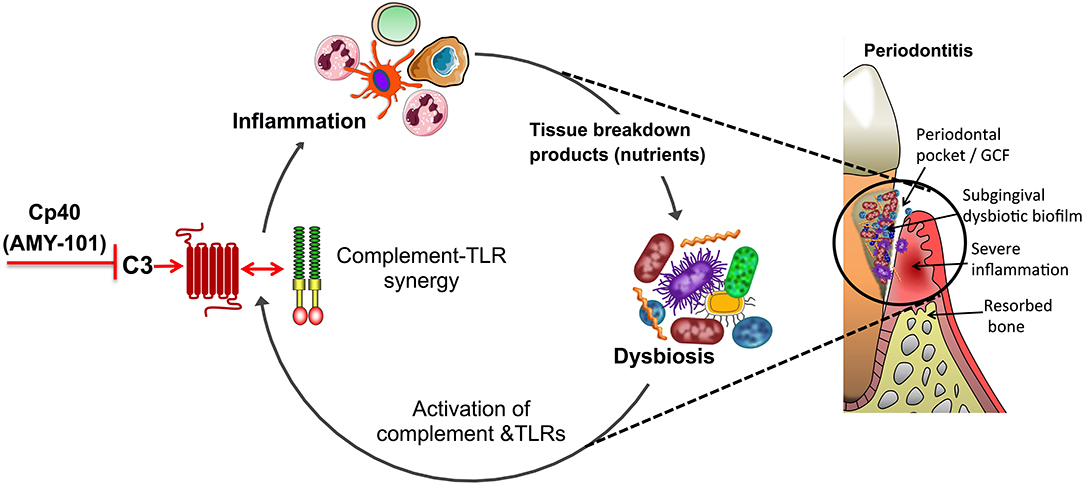
Figure 3. Complement-targeted inhibition blocks a vicious cycle linking destructive inflammation and dysbiosis in periodontitis. Periodontitis is driven by a reciprocally reinforced interplay between dysbiotic microbial communities and inflammation. Whereas, dysbiosis induces inflammation, the nutritionally favorable inflammatory environment is a major ecological factor that exacerbates dysbiosis. Studies in different preclinical models showed that complement inhibition abrogates complement-TLR crosstalk signaling and resulting inflammation, thereby breaking the disease-provoking vicious cycle regardless of the presence of intact TLR signaling. For instance, blocking the central complement component C3 through the use of the peptidic compound Cp40/AMY-101 has blocked experimental or naturally occurring periodontitis in non-human primates.
Safety and Other Considerations for Clinical Use
Given the participation of the dysbiotic periodontal microbiota in the pathogenesis of periodontitis, the targeting of complement may not appear as an intuitive therapeutic option for this oral disease. In general, a potential concern regarding the therapeutic use of complement inhibitors is whether complement blockade may undermine the competency of host antimicrobial defenses and thus increase the risk of infection. Although this possibility may not be an issue in acute conditions that require transient complement inhibition [e.g., in hemodialysis (108)], it has to be carefully considered in conditions that will require long-term use of complement inhibitors. In this regard, individuals with primary C3 deficiencies have increased risk of certain infections (e.g., Neisseria meningitidis and Streptococcus pneumoniae) although this enhanced susceptibility appears to subside in adulthood, presumably owing to the development of compensatory defense mechanisms (109–111). Current experience from FDA-approved anti-complement drugs, such as eculizumab that blocks C5 activation, shows that immunization against encapsulated bacteria (such as meningococci) can largely diminish infectious risks. Therefore, vaccinations as well as prophylactic use of antibiotics may be included to enable safe use of complement inhibitors in chronic settings. Importantly, in cases of complement inhibition with small-molecule inhibitors, such as compstatin, the compound can more readily phased out (than an antibody for instance) if necessary, thus enabling swift recovery of complement-dependent antimicrobial functions. Importantly, the monitoring of non-human primates under prolonged (up to 3 months) systemic treatment with Cp40 revealed no significant differences in biochemical, hematological, or immunological parameters in their blood or tissues as compared to those of vehicle alone-treated controls, despite complete inhibition of C3 in the plasma. Intriguingly, moreover, wounds inflicted in the skin of the Cp40-treated animals did not show any signs of infection but rather exhibited a trend toward faster wound healing as compared with the vehicle-treated controls (112). This finding is consistent with earlier observations in mice in which C3 deficiency resulted in faster skin wound healing as compared to C3-sufficient control mice (113).
Although a chronic condition, periodontitis is a local inflammatory disease and thus can be treated via local complement inhibition, a much safer approach than systemic administration of the same inhibitor. Systemic exposure with complement inhibitors following local injection into the periodontal tissues should be negligible and thus not impair complement activity in circulation or other tissues. This notion can be exemplified by experience with Cp40. As C3 is the most abundant protein of the complement system in blood (1.0 to 1.5 mg/ml), small amounts of locally injected Cp40 that could “escape” from the periodontal tissue should be readily bound by excess C3 in blood, hence not reaching other tissues at active (inhibitory) concentrations. In the treatment regimen used in the above-described NHP study by Maekawa et al. (89), a total of 1.5 mg Cp40 was injected (15 sites at 100 μg/site). Even if the full local dose were to be given systemically, this would only result to an amount of 0.2–0.3 mg/kg bodyweight in non-human primates (0.02–0.03 mg/kg bodyweight in humans), whereas a systemic Cp40 dose of 1–2 mg/kg bodyweight is necessary to reliably attain target-exceeding drug levels in non-human primates (114).
Even at the local level, complement inhibition is unlikely to lead to defective control of the periodontitis-associated microbiota. As discussed above, C3-deficient mice have decreased periodontal bacterial load compared to C3-sufficient controls during experimental periodontitis (62). These data are consistent with the notion that inflammation is an ecological driver of dysbiosis in periodontitis (Figure 3). Indeed, destructive periodontal inflammation causes the generation of tissue breakdown products (such as degraded collagen peptides or heme-containing compounds) that are used as a nutrient source by a subset of bacterial species associated with dysbiosis; these are mainly proteolytic/asaccharolytic organisms with iron-acquisition mechanisms and/or can thrive by utilizing other inflammatory byproducts, such as nitrate for anaerobic respiration, thereby outcompeting health-associated bacteria and exacerbating dysbiosis (18, 115, 116). Therefore, complement inhibition by Cp40 may not simply inhibit inflammation but may additionally interfere with the outgrowth of the dysbiotic microbiota (Figure 3). Experimental support of the notion that anti-inflammatory approaches can have indirect anti-microbial effects has been obtained in mouse and rabbit models of periodontitis, where the control of inflammation not only protected against disease but also decreased the bacterial load and reversed dysbiosis (76, 77, 117–119). Conversely, and in line with the previous statement, the bacterial biomass of biofilms associated with human periodontitis increases with increasing clinical inflammation (120).
Conclusions and Outlook
The studies discussed above suggest a clinical value of inhibiting all three main pathways of complement activation in periodontitis, which can be achieved by targeting the central component C3. C3 inhibition can directly inhibit inflammation and indirectly counteract dysbiosis. The safety and efficacy of Cp40 in non-human primate periodontitis (62, 89, 95) paves the way to clinical trials for the treatment of human periodontitis. To this end, aspects that need to be considered include questions regarding administration frequency, dosing, and selection of those patients who would most benefit from such a treatment. Even though Cp40 was successfully tested as a stand-alone treatment for both induced and naturally-occurring periodontitis in monkeys, the drug is more likely to be used as an adjunctive therapy to the management of human periodontal disease. Future clinical trials may investigate the combined potential of Cp40 and SRP to treat periodontal inflammation and suppress bone loss as compared to SRP alone. In very severe cases of periodontitis, combined Cp40 and SRP therapy could be compared to periodontal surgery, to determine whether the Cp40/SRP treatment can obviate the need for a surgical approach. It should be noted that a Cp40-based treatment (and host-modulation interventions in general) may not necessarily be applied in a therapeutic setting but also on a preventive basis (before the onset of periodontitis) to high-risk patients, such as cigarette smokers and diabetic patients (121–123).
The protective effects of Cp40 in non-human primate periodontitis are maintained for many weeks following drug withdrawal from treated monkeys (89, 95). This is an encouraging finding although the optimal frequency of Cp40 administration for long-term treatment of human periodontitis may need to be decided empirically. The unique pharmacokinetic properties of Cp40 described earlier are consistent with a “target-driven” model, where an initial rapid clearance of excess free peptide (i.e., not bound by C3) is followed by slow clearance of C3-bound peptide. The tight binding of Cp40 to C3 thus appears to delay its clearance and, indeed, the determined half-life values of different compstatin analogs correlate with their C3-binding affinities (85). Similarly, strong binding of Cp40 to abundant C3 in the inflamed periodontal tissue could contribute to delayed clearance of the drug from the tissue, thus contributing to sustained protection from periodontal inflammation. Moreover, the ability of Cp40 to arrest inflammation and presumably to reverse dysbiosis may reset the balance toward tissue homeostasis, which on its own (despite the absence of the drug) could resiliently inhibit or delay the recurrence of pathological processes.
Amyndas Pharmaceuticals is developing Cp40, a third-generation non-PEGylated compstatin analog, as the clinical candidate drug AMY-101. AMY-101 is developed for therapeutic interventions in C3 glomerulopathy (C3G), complications of ABO-incompatible kidney transplantation, paroxysmal nocturnal hemoglobinuria (PNH), and periodontal disease (124, 125). The PEGylated version of an earlier compstatin analog, POT-4/4(1MeW) (APL-2, Apellis Pharmaceuticals) is clinically developed for use in complement-mediated disorders including age-related macular degeneration and PNH. In a Phase II trial, prolonged APL-2 treatment was shown to be safe and reduced the growth rate of geographic atrophy associated with age-related macular degeneration. AMY-101 has obtained orphan drug designation for C3G and PNH from the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) in 2016 and more recently, in 2017, successfully completed a phase I safety trial (125, 126). Targeted modifications of the N- and C-terminus of Cp40/AMY-101 have led to a series of fourth-generation compstatins with higher solubility, improved PK profiles thus broadening the spectrum of administration routes and likely reducing the dosing frequency of these peptidic drugs in chronic regimens (127). Overall, more than 20 candidate complement-targeted drugs that inhibit distinct points of the cascade are currently being tested in clinical trials for a variety of inflammatory and degenerative diseases (125). The documented safety of Cp40/AMY-101 and its protective effects in highly relevant preclinical models of periodontitis merits investigation in future clinical trials for the treatment of human periodontitis.
Author Contributions
GH and JL: conceptualization; GH: original draft. All authors listed made a substantial intellectual contribution to the manuscript and edited and approved it for publication.
Conflict of Interest Statement
JL is the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors (including third-generation compstatin analogs, such as AMY-101). JL and GH are inventors of patents or patent applications that describe the use of complement inhibitors for therapeutic purposes, some of which are developed by Amyndas Pharmaceuticals. JL is also the inventor of the compstatin technology licensed to Apellis Pharmaceuticals [i.e., 4(1MeW)7W/POT-4/APL-1 and PEGylated derivatives].
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are supported by grants from the U.S. National Institutes of Health (AI068730 and AI030040 to JL; DE015254, DE021685, and DE024716 to GH) and the European Commission (FP7-DIREKT 602699 to JL).
References
1. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
2. Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD. Novel mechanisms and functions of complement. Nat Immunol. (2017) 18:1288–98. doi: 10.1038/ni.3858
3. Hajishengallis G, Lambris JD. More than complementing tolls: complement-toll-like receptor synergy and crosstalk in innate immunity and inflammation. Immunol Rev. (2016) 274:233–44. doi: 10.1111/imr.12467
4. Arbore G, Kemper C, Kolev M. Intracellular complement–the complosome–in immune cell regulation. Mol Immunol. (2017) 89:2–9. doi: 10.1016/j.molimm.2017.05.012
5. Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol. (2016) 12:383–401. doi: 10.1038/nrneph.2016.70
6. Hovingh ES, van den Broek B, Jongerius I. Hijacking complement regulatory proteins for bacterial immune evasion. Front Microbiol. (2016) 7:2004. doi: 10.3389/fmicb.2016.02004
7. Reis ES, Mastellos DC, Ricklin D, Mantovani A, Lambris JD. Complement in cancer: untangling an intricate relationship. Nat Rev Immunol. (2018) 18:5–18. doi: 10.1038/nri.2017.97
8. Hajishengallis G, Abe T, Maekawa T, Hajishengallis E, Lambris JD. Role of complement in host-microbe homeostasis of the periodontium. Semin Immunol. (2013) 25:65–72. doi: 10.1016/j.smim.2013.04.004
10. Eke PI, Dye BA, Wei L, Slade GD, Thornton-Evans GO, Borgnakke WS, et al. Update on prevalence of periodontitis in adults in the United States: NHANES 2009 to 2012. J Periodontol. (2015) 86:611–22. doi: 10.1902/jop.2015.140520
11. Kassebaum NJ, Bernabe E, Dahiya M, Bhandari B, Murray CJ, Marcenes W. Global burden of severe periodontitis in 1990–2010: a systematic review and meta-regression. J Dent Res. (2014) 93:1045–53. doi: 10.1177/0022034514552491
12. Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol. (2015) 15:30–44. doi: 10.1038/nri3785
13. Diaz PI, Hoare A, Hong BY. Subgingival microbiome shifts and community dynamics in periodontal diseases. J Calif Dent Assoc. (2016) 44:421–35.
14. Lamont RJ, Hajishengallis G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol Med. (2015) 21:172–83. doi: 10.1016/j.molmed.2014.11.004
15. Lamont RJ, Koo H, Hajishengallis G. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol. (2018) 16:745–59. doi: 10.1038/s41579-018-0089-x
16. Hajishengallis G, Korostoff JM. Revisiting the page & schroeder model: the good, the bad and the unknowns in the periodontal host response 40 years later. Periodontol 2000. (2017) 75:116–51. doi: 10.1111/prd.12181
17. Rosier BT, Marsh PD, Mira A. Resilience of the oral microbiota in health: mechanisms that prevent dysbiosis. J Dent Res. (2017) 97:371–80. doi: 10.1177/0022034517742139
18. Hajishengallis G. The inflammophilic character of the periodontitis-associated microbiota. Mol Oral Microbiol. (2014) 29:248–57. doi: 10.1111/omi.12065
19. Colombo AP, Bennet S, Cotton SL, Goodson JM, Kent R, Haffajee AD, et al. Impact of periodontal therapy on the subgingival microbiota of severe periodontitis: comparison between good responders and individuals with refractory periodontitis using the human oral microbe identification microarray. J Periodontol. (2012) 83:1279–87. doi: 10.1902/jop.2012.110566
20. Armitage GC. Classifying periodontal diseases: a long-standing dilemma. Periodontol 2000. (2002) 30:9–23. doi: 10.1034/j.1600-0757.2002.03002.x
21. Rams TE, Degener JE, van Winkelhoff AJ. Antibiotic resistance in human chronic periodontitis microbiota. J Periodontol. (2014) 85:160–9. doi: 10.1902/jop.2013.130142
22. Tonetti MS, Chapple IL Working group 3 of seventh European workshop on P. Biological approaches to the development of novel periodontal therapies–consensus of the seventh European workshop on periodontology. J Clin Periodontol. (2011) 38Suppl 11:114–8. doi: 10.1111/j.1600-051X.2010.01675.x
23. Beikler T, Flemmig TF. Oral biofilm-associated diseases: trends and implications for quality of life, systemic health and expenditures. Periodontol 2000. (2011) 55:87–103. doi: 10.1111/j.1600-0757.2010.00360.x
24. Petersen PE, Ogawa H. The global burden of periodontal disease: towards integration with chronic disease prevention and control. Periodontol 2000. (2012) 60:15–39. doi: 10.1111/j.1600-0757.2011.00425.x
25. Lamster IB, Ahlo JK. Analysis of gingival crevicular fluid as applied to the diagnosis of oral and systemic diseases. Ann N Y Acad Sci. (2007) 1098:216–29. doi: 10.1196/annals.1384.027
26. Courts FJ, Boackle RJ, Fudenberg HH, Silverman MS. Detection of functional complement components in gingival crevicular fluid from humans with periodontal diseases. J Dent Res. (1977) 56:327–31. doi: 10.1177/00220345770560032001
27. Boackle RJ. The interaction of salivary secretions with the human complement system–a model for the study of host defense systems on inflamed mucosal surfaces. Crit Rev Oral Biol Med. (1991) 2:355–67. doi: 10.1177/10454411910020030401
28. Attstrom R, Laurel AB, Lahsson U, Sjoholm A. Complement factors in gingival crevice material from healthy and inflamed gingiva in humans. J Periodont Res. (1975) 10:19–27. doi: 10.1111/j.1600-0765.1975.tb00003.x
29. Schenkein HA, Genco RJ. Gingival fluid and serum in periodontal diseases. II. evidence for cleavage of complement components C3, C3 proactivator (factor B) and C4 in gingival fluid. J Periodontol. (1977) 48:778–84. doi: 10.1902/jop.1977.48.12.778
30. Schenkein HA, Genco RJ. Gingival fluid and serum in periodontal diseases. i. quantitative study of immunoglobulins, complement components, and other plasma proteins. J Periodontol. (1977) 48:772–7. doi: 10.1902/jop.1977.48.12.772
31. Challacombe SJ, Shirlaw PJ. Immunology of diseases of the oral cavity. In: Mestecky J, Lamm ME, Strober W, Bienenstock J, McGhee JR, Mayer L, editors. Mucosal Immunology. Elsevier Academic Press (2005). p. 1517–46.
32. Patters MR, Niekrash CE, Lang NP. Assessment of complement cleavage in gingival fluid during experimental gingivitis in man. J Clin Periodontol. (1989) 16:33–7. doi: 10.1111/j.1600-051X.1989.tb01609.x
33. Toto PD, Lin L, Gargiulo A. Identification of C3a, IgG, IgM in inflamed human gingiva. J Dent Res. (1978) 57:696. doi: 10.1177/00220345780570050501
34. Nikolopoulou-Papaconstantinou AA, Johannessen AC, Kristoffersen T. Deposits of immunoglobulins, complement, and immune complexes in inflamed human gingiva. Acta Odontol Scand. (1987) 45:187–93. doi: 10.3109/00016358709098858
35. Rautemaa R, Meri S. Protection of gingival epithelium against complement-mediated damage by strong expression of the membrane attack complex inhibitor protectin (CD59). J Dent Res. (1996) 75:568–74. doi: 10.1177/00220345960750010901
36. Schenkein HA, Genco RJ. Complement cleavage products in inflammatory exudates from patients with periodontal diseases. J Immunol. (1978) 120:1796.
37. Lally ET, McArthur WP, Baehni PC. Biosynthesis of complement components in chronically inflamed gingiva. J Periodontal Res. (1982) 17:257–62. doi: 10.1111/j.1600-0765.1982.tb01152.x
38. Niekrash CE, Patters MR. Simultaneous assessment of complement components C3, C4, and B and their cleavage products in human gingival fluid. II. longitudinal changes during periodontal therapy. J Periodontal Res. (1985) 20:268–75. doi: 10.1111/j.1600-0765.1985.tb00434.x
39. Roberts A, Shah M, Chapple IL. C-1 esterase inhibitor dysfunction localised to the periodontal tissues: clues to the role of stress in the pathogenesis of chronic periodontitis? J Clin Periodontol. (2003) 30:271–7. doi: 10.1034/j.1600-051X.2003.01266.x
40. Hillebrandt S, Wasmuth HE, Weiskirchen R, Hellerbrand C, Keppeler H, Werth A, et al. Complement factor 5 is a quantitative trait gene that modifies liver fibrogenesis in mice and humans. Nat Genet. (2005) 37:835–43. doi: 10.1038/ng1599
41. Chai L, Song Y-Q, Zee K-Y, Leung WK. Single nucleotide polymorphisms of complement component 5 and periodontitis. J Periodont Res. (2010) 45:301–8. doi: 10.1111/j.1600-0765.2009.01234.x
42. Beikler T, Peters U, Prior K, Eisenacher M, Flemmig TF. Gene expression in periodontal tissues following treatment. BMC Med Genomics. (2008) 1:30. doi: 10.1186/1755-8794-1-30
43. Zhan Y, Zhang R, Lv H, Song X, Xu X, Chai L, et al. Prioritization of candidate genes for periodontitis using multiple computational tools. J Periodontol. (2014) 85:1059–69. doi: 10.1902/jop.2014.130523
44. Seppanen M, Lokki ML, Notkola IL, Mattila K, Valtonen V, Nieminen A, et al. Complement and c4 null alleles in severe chronic adult periodontitis. Scand J Immunol. (2007) 65:176–81. doi: 10.1111/j.1365-3083.2006.01886.x
45. Wang H, Ricklin D, Lambris JD. Complement-activation fragment C4a mediates effector functions by binding as untethered agonist to protease-activated receptors 1 and 4. Proc Natl Acad Sci USA. (2017) 114:10948–53. doi: 10.1073/pnas.1707364114
46. Roumenina LT, Rayes J, Frimat M, Fremeaux-Bacchi V. Endothelial cells: source, barrier, and target of defensive mediators. Immunol Rev. (2016) 274:307–29. doi: 10.1111/imr.12479
47. Hajishengallis G, Lamont RJ, Graves DT. The enduring importance of animal models in understanding periodontal disease. Virulence. (2015) 6:229–35. doi: 10.4161/21505594.2014.990806
48. Hajishengallis G, Lambris JD. Crosstalk pathways between Toll-like receptors and the complement system. Trends Immunol. (2010) 31:154–63. doi: 10.1016/j.it.2010.01.002
49. Seow V, Lim J, Iyer A, Suen JY, Ariffin JK, Hohenhaus DM, et al. Inflammatory responses induced by lipopolysaccharide are amplified in primary human monocytes but suppressed in macrophages by complement protein C5a. J Immunol. (2013) 191:4308–16. doi: 10.4049/jimmunol.1301355
50. Fang C, Zhang X, Miwa T, Song WC. Complement promotes the development of inflammatory T-helper 17 cells through synergistic interaction with Toll-like receptor signaling and interleukin-6 production. Blood. (2009) 114:1005–15. doi: 10.1182/blood-2009-01-198283
51. Maekawa T, Krauss JL, Abe T, Jotwani R, Triantafilou M, Triantafilou K, et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe. (2014) 15:768–78. doi: 10.1016/j.chom.2014.05.012
52. Zaal A, Lissenberg-Thunnissen SN, van Schijndel G, Wouters D, van Ham SM, ten Brinke A. Crosstalk between toll like receptors and C5a receptor in human monocyte derived DCs suppress inflammatory cytokine production. Immunobiology. (2013) 218:175–80. doi: 10.1016/j.imbio.2012.02.014
53. Bosmann M, Sarma JV, Atefi G, Zetoune FS, Ward PA. Evidence for anti-inflammatory effects of C5a on the innate IL-17A/IL-23 axis. FASEB J. (2011) 26:1640–51. doi: 10.1096/fj.11-199216
54. Yamada M, Oritani K, Kaisho T, Ishikawa J, Yoshida H, Takahashi I, et al. Complement C1q regulates LPS-induced cytokine production in bone marrow-derived dendritic cells. Eur J Immunol. (2004) 34:221–30. doi: 10.1002/eji.200324026
55. Zhang X, Kimura Y, Fang C, Zhou L, Sfyroera G, Lambris JD, et al. Regulation of toll-like receptor-mediated inflammatory response by complement in vivo. Blood. (2007) 110:228–36 doi: 10.1182/blood-2006-12-063636
56. Abe T, Hosur KB, Hajishengallis E, Reis ES, Ricklin D, Lambris JD, et al. Local complement-targeted intervention in periodontitis: proof-of-concept using a C5a receptor (CD88) antagonist. J Immunol. (2012) 189:5442–8. doi: 10.4049/jimmunol.1202339
57. Liang S, Krauss JL, Domon H, McIntosh ML, Hosur KB, Qu H, et al. The C5a receptor impairs IL-12-dependent clearance of Porphyromonas gingivalis and is required for induction of periodontal bone loss. J Immunol. (2011) 186:869–77. doi: 10.4049/jimmunol.1003252
58. Burns E, Bachrach G, Shapira L, Nussbaum G. Cutting Edge: TLR2 is required for the innate response to Porphyromonas gingivalis: activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol. (2006) 177:8296–300. doi: 10.4049/jimmunol.177.12.8296
59. Kim PD, Xia-Juan X, Crump KE, Abe T, Hajishengallis G, Sahingur SE. Toll-like receptor 9-mediated inflammation triggers alveolar bone loss in experimental murine periodontitis. Infect Immun. (2015) 83:2992–3002. doi: 10.1128/IAI.00424-15
60. Graves DT, Fine D, Teng YT, Van Dyke TE, Hajishengallis G. The use of rodent models to investigate host-bacteria interactions related to periodontal diseases. J Clin Periodontol. (2008) 35:89–105. doi: 10.1111/j.1600-051X.2007.01172.x
61. Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. (2011) 10:497–506. doi: 10.1016/j.chom.2011.10.006
62. Maekawa T, Abe T, Hajishengallis E, Hosur KB, DeAngelis RA, Ricklin D, et al. Genetic and intervention studies implicating complement C3 as a major target for the treatment of periodontitis. J Immunol. (2014) 192 6020–7. doi: 10.4049/jimmunol.1400569
63. Abe T, Hajishengallis G. Optimization of the ligature-induced periodontitis model in mice. J Immunol Methods. (2013) 394:49–54. doi: 10.1016/j.jim.2013.05.002
64. Jiao Y, Darzi Y, Tawaratsumida K, Marchesan JT, Hasegawa M, Moon H, et al. Induction of bone loss by pathobiont-mediated nod1 signaling in the oral cavity. Cell Host Microbe. (2013) 13:595–601. doi: 10.1016/j.chom.2013.04.005
65. Rovin S, Costich ER, Gordon HA. The influence of bacteria and irritation in the initiation of periodontal disease in germfree and conventional rats. J Periodontal Res. (1966) 1:193–204. doi: 10.1111/j.1600-0765.1966.tb01860.x
66. Liang S, Hosur KB, Domon H, Hajishengallis G. Periodontal inflammation and bone loss in aged mice. J Periodontal Res. (2010) 45:574–8. doi: 10.1111/j.1600-0765.2009.01245.x
67. Eskan MA, Hajishengallis G, Kinane DF. Differential activation of human gingival epithelial cells and monocytes by Porphyromonas gingivalis fimbriae. Infect Immun. (2007) 75:892–8. doi: 10.1128/IAI.01604-06
68. Hajishengallis G. Aging and its impact on innate immunity and inflammation: Implications for periodontitis. J Oral Biosci. (2014) 56:30–7. doi: 10.1016/j.job.2013.09.001
69. Kohl J. Drug evaluation: the C5a receptor antagonist PMX-53. Curr Opin Mol Ther. (2006) 8:529–38.
70. Delisle Milton RC, Milton SC, Chamberlin AR. Improving the fmoc solid phase synthesis of the cyclic hexapeptide complement C5a antagonist, PMX205. Int J Pept Res Ther. (2011) 17:337–42. doi: 10.1007/s10989-011-9273-9
71. Breivik T, Gundersen Y, Gjermo P, Taylor SM, Woodruff TM, Opstad PK. Oral treatment with complement factor C5a receptor (CD88) antagonists inhibits experimental periodontitis in rats. J Periodontal Res. (2011) 46:643–7. doi: 10.1111/j.1600-0765.2011.01383.x
72. Assuma R, Oates T, Cochran D, Amar S, Graves DT. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. J Immunol. (1998) 160:403–9.
73. Graves D. Cytokines that promote periodontal tissue destruction. J Periodontol. (2008) 79(8 Suppl):1585–91. doi: 10.1902/jop.2008.080183
74. Noguchi K, Ishikawa I. The roles of cyclooxygenase-2 and prostaglandin E2 in periodontal disease. Periodontol 2000. (2007) 43:85–101. doi: 10.1111/j.1600-0757.2006.00170.x
75. Glowacki AJ, Yoshizawa S, Jhunjhunwala S, Vieira AE, Garlet GP, Sfeir C, et al. Prevention of inflammation-mediated bone loss in murine and canine periodontal disease via recruitment of regulatory lymphocytes. Proc Natl Acad Sci USA. (2013) 110:18525–30. doi: 10.1073/pnas.1302829110
76. Eskan MA, Jotwani R, Abe T, Chmelar J, Lim JH, Liang S, et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat Immunol. (2012) 13:465–73. doi: 10.1038/ni.2260
77. Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17–driven inflammatory bone loss. Sci Transl Med. (2014) 6:229ra40. doi: 10.1126/scitranslmed.3007696
78. Brecx MC, Nalbandian J, Ooya K, Kornman KS, Robertson PB. Morphological studies on periodontal disease in the cynomolgus monkey. II. Light microscopic observations on ligature-induced periodontitis. J Periodontal Res. (1985) 20:165–75. doi: 10.1111/j.1600-0765.1985.tb00423.x
79. Page RC, Schroeder HE. Periodontitis in Man and Other Animals- A Comparative Review. Basel: Karger (1982).
80. Kornman KS, Holt SC, Robertson PB. The microbiology of ligature-induced periodontitis in the cynomolgus monkey. J Periodontal Res. (1981) 16:363–71. doi: 10.1111/j.1600-0765.1981.tb00987.x
81. Ebersole JL, Kirakodu S, Novak MJ, Stromberg AJ, Shen S, Orraca L, et al. Cytokine gene expression profiles during initiation, progression and resolution of periodontitis. J Clin Periodontol. (2014) 41:853–61. doi: 10.1111/jcpe.12286
82. Ebersole JL, Cappelli D, Mathys EC, Steffen MJ, Singer RE, Montgomery M, et al. Periodontitis in humans and non-human primates: oral-systemic linkage inducing acute phase proteins. Ann Periodontol. (2002) 7:102–11. doi: 10.1902/annals.2002.7.1.102
83. Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol. (2013) 190:3831–8. doi: 10.4049/jimmunol.1203487
84. Mastellos DC, Yancopoulou D, Kokkinos P, Huber-Lang M, Hajishengallis G, Biglarnia AR, et al. Compstatin: a C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur J Clin Invest. (2015) 45:423–40. doi: 10.1111/eci.12419
85. Qu H, Ricklin D, Bai H, Chen H, Reis ES, Maciejewski M, et al. New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology. (2013) 218:496–505. doi: 10.1016/j.imbio.2012.06.003
86. Janssen BJ, Halff EF, Lambris JD, Gros P. Structure of compstatin in complex with complement component C3c reveals a new mechanism of complement inhibition. J Biol Chem. (2007) 282:29241–7. doi: 10.1074/jbc.M704587200
87. Armitage GC. Periodontal diagnoses and classification of periodontal diseases. Periodontol 2000. (2004) 34:9–21. doi: 10.1046/j.0906-6713.2002.003421.x
88. Belibasakis GN, Bostanci N. The RANKL-OPG system in clinical periodontology. J Clin Periodontol. (2012) 39:239–48. doi: 10.1111/j.1600-051X.2011.01810.x
89. Maekawa T, Briones RA, Resuello RR, Tuplano JV, Hajishengallis E, Kajikawa T, et al. Inhibition of pre-existing natural periodontitis in non-human primates by a locally administered peptide inhibitor of complement C3. J Clin Periodontol. (2016) 43:238–49. doi: 10.1111/jcpe.12507
90. Bostanci N, Bao K, Li X, Maekawa T, Grossmann J, Panse C, et al. Gingival exudatome dynamics implicate inhibition of the alternative complement pathway in the protective action of the C3 inhibitor Cp40 in non-human primate periodontitis. J Prot Res. (2018) 17:3153–75. doi: 10.1021/acs.jproteome.8b00263
91. Hajishengallis G, Moutsopoulos NM, Hajishengallis E, Chavakis T. Immune and regulatory functions of neutrophils in inflammatory bone loss. Semin Immunol. (2016) 28:146–58. doi: 10.1016/j.smim.2016.02.002
92. Chapple IL, Matthews JB. The role of reactive oxygen and antioxidant species in periodontal tissue destruction. Periodontol 2000. (2007) 43:160–232. doi: 10.1111/j.1600-0757.2006.00178.x
93. Sima C, Glogauer M. Neutrophil dysfunction and host susceptibility to periodontal inflammation: current state of knowledge. Curr Oral Health Rep. (2014) 1:95–103. doi: 10.1007/s40496-014-0015-x
94. Dutzan N, Kajikawa T, Abusleme L, Greewell-Wild T, Zuazo CE, Ikeuchi T, et al. Dysbiotic microbiome triggers Th17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med. (2018) 10:eaat0797. doi: 10.1126/scitranslmed.aat0797
95. Kajikawa T, Briones RA, Resuello RRG, Tuplano JV, Reis ES, Hajishengallis E, et al. Safety and efficacy of the complement inhibitor AMY-101 in a natural model of periodontitis in non-human primates. Mol Ther Methods Clin Dev. (2017) 6:207–15. doi: 10.1016/j.omtm.2017.08.001
96. Hajishengallis G. Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends Immunol. (2014) 35:3–11. doi: 10.1016/j.it.2013.09.001
97. Hajishengallis G, Sahingur SE. Novel inflammatory pathways in periodontitis. Adv Dent Res. (2014) 26:23–9. doi: 10.1177/0022034514526240
98. Miyake K. Innate immune sensing of pathogens and danger signals by cell surface toll-like receptors. Semin Immunol. (2007) 19:3–10. doi: 10.1016/j.smim.2006.12.002
99. Schaefer L. Extracellular matrix molecules: endogenous danger signals as new drug targets in kidney diseases. Curr Opin Pharmacol. (2010) 10:185–90. doi: 10.1016/j.coph.2009.11.007
100. Kaczorowski DJ, Afrazi A, Scott MJ, Kwak JH, Gill R, Edmonds RD, et al. Pivotal advance: the pattern recognition receptor ligands lipopolysaccharide and polyinosine-polycytidylic acid stimulate factor B synthesis by the macrophage through distinct but overlapping mechanisms. J Leukoc Biol. (2010) 88:609–18. doi: 10.1189/jlb.0809588
101. Ostvik AE, Granlund A, Gustafsson BI, Torp SH, Espevik T, Mollnes TE, et al. Mucosal toll-like receptor 3-dependent synthesis of complement factor B and systemic complement activation in inflammatory bowel disease. Inflamm Bowel Dis. (2014) 20:995–1003. doi: 10.1097/MIB.0000000000000035
102. Zou L, Feng Y, Li Y, Zhang M, Chen C, Cai J, et al. Complement factor B is the downstream effector of TLRs and plays an important role in a mouse model of severe sepsis. J Immunol. (2013) 191:5625–35. doi: 10.4049/jimmunol.1301903
103. Pope MR, Hoffman SM, Tomlinson S, Fleming SD. Complement regulates TLR4-mediated inflammatory responses during intestinal ischemia reperfusion. Mol Immunol. (2010) 48:356–64. doi: 10.1016/j.molimm.2010.07.004
104. Fan J, Malik AB. Toll-like receptor-4 (TLR4) signaling augments chemokine-induced neutrophil migration by modulating cell surface expression of chemokine receptors. Nat Med. (2003) 9:315–21. doi: 10.1038/nm832
105. Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. (2008) 8:776–87. doi: 10.1038/nri2402
106. Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Kohl J, et al. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. (2013) 122:3473–81. doi: 10.1182/blood-2013-05-502229
107. Triantafilou K, Hughes TR, Triantafilou M, Morgan BP. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci. (2013) 126(Pt 13):2903–13. doi: 10.1242/jcs.124388
108. Reis ES, DeAngelis RA, Chen H, Resuello RR, Ricklin D, Lambris JD. Therapeutic C3 inhibitor Cp40 abrogates complement activation induced by modern hemodialysis filters. Immunobiology. (2015) 220:476–82. doi: 10.1016/j.imbio.2014.10.026
109. S Reis E, Falcao DA, Isaac L. Clinical aspects and molecular basis of primary deficiencies of complement component C3 and its regulatory proteins factor I and factor H. Scand J Immunol. (2006) 63:155–68. doi: 10.1111/j.1365-3083.2006.01729.x
110. Audemard-Verger A, Descloux E, Ponard D, Deroux A, Fantin B, Fieschi C, et al. Infections revealing complement deficiency in adults: a french nationwide study enrolling 41 patients. Medicine (Baltimore). (2016) 95:e3548. doi: 10.1097/MD.0000000000003548
111. Lachmann PJ, Smith RA. Taking complement to the clinic–has the time finally come? Scand J Immunol. (2009) 69:471–8. doi: 10.1111/j.1365-3083.2009.02258.x
112. Reis ES. Safety profile of non-human primates under prolonged C3 inhibition. Clin Immunol. (2018) 197:96–106. doi: 10.1016/j.clim.2018.09.004
113. Rafail S, Kourtzelis I, Foukas PG, Markiewski MM, DeAngelis RA, Guariento M, et al. Complement deficiency promotes cutaneous wound healing in mice. J Immunol. (2015) 194:1285–91. doi: 10.4049/jimmunol.1402354
114. Risitano AM, Ricklin D, Huang Y, Reis ES, Chen H, Ricci P, et al. Peptide inhibitors of C3 activation as a novel strategy of complement inhibition for the treatment of paroxysmal nocturnal hemoglobinuria. Blood. (2014) 123:2094–101. doi: 10.1182/blood-2013-11-536573
115. Marsh PD. Are dental diseases examples of ecological catastrophes? Microbiology. (2003) 149(Pt 2):279–94. doi: 10.1099/mic.0.26082-0
116. Nassar M, Tabib Y, Capucha T, Mizraji G, Nir T, Pevsner-Fischer M, et al. GAS6 is a key homeostatic immunological regulator of host-commensal interactions in the oral mucosa. Proc Natl Acad Sci USA. (2017) 114:E337–46. doi: 10.1073/pnas.1614926114
117. Hasturk H, Kantarci A, Goguet-Surmenian E, Blackwood A, Andry C, Serhan CN, et al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol. (2007) 179:7021–9. doi: 10.4049/jimmunol.179.10.7021
118. Abe T, Shin J, Hosur K, Udey MC, Chavakis T, Hajishengallis G. Regulation of osteoclast homeostasis and inflammatory bone loss by MFG-E8. J Immunol. (2014) 193:1383–91. doi: 10.4049/jimmunol.1400970
119. Lee C-T, Teles R, Kantarci A, Chen T, McCafferty J, Starr JR, et al. Resolvin E1 reverses experimental periodontitis and dysbiosis. J Immunol. (2016) 197:2796–806. doi: 10.4049/jimmunol.1600859
120. Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, et al. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. (2013) 7:1016–25. doi: 10.1038/ismej.2012.174
121. Heitz-Mayfield LJ. Disease progression: identification of high-risk groups and individuals for periodontitis. J Clin Periodontol. (2005) 6(32 Suppl):196–209. doi: 10.1111/j.1600-051X.2005.00803.x
122. Genco RJ, Genco FD. Common risk factors in the management of periodontal and associated systemic diseases: the dental setting and interprofessional collaboration. J Evid Based Dent Pract. (2014) (14 Suppl):4–16. doi: 10.1016/j.jebdp.2014.03.003
123. Joshi V, Matthews C, Aspiras M, de Jager M, Ward M, Kumar P. Smoking decreases structural and functional resilience in the subgingival ecosystem. J Clin Periodontol. (2014) 41:1037–47. doi: 10.1111/jcpe.12300
124. Mastellos DC, Ricklin D, Hajishengallis E, Hajishengallis G, Lambris JD. Complement therapeutics in inflammatory diseases: promising drug candidates for C3-targeted intervention. Mol Oral Microbiol. (2016) 31:3–17. doi: 10.1111/omi.12129
125. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol. (2018) 14:26–47. doi: 10.1038/nrneph.2017.156
126. Medicine UNLo. ClinicalTrials.gov. (2017). Available online at: https://clinicaltrials.gov/ct2/show/NCT03316521
Keywords: complement, C3, therapeutics, compstatin Cp40, AMY-101, primate models, inflammation, periodontitis
Citation: Hajishengallis G, Kajikawa T, Hajishengallis E, Maekawa T, Reis ES, Mastellos DC, Yancopoulou D, Hasturk H and Lambris JD (2019) Complement-Dependent Mechanisms and Interventions in Periodontal Disease. Front. Immunol. 10:406. doi: 10.3389/fimmu.2019.00406
Received: 02 December 2018; Accepted: 15 February 2019;
Published: 12 March 2019.
Edited by:
Liwu Li, Virginia Tech, United StatesReviewed by:
Roberta Bulla, University of Trieste, ItalyMaciej Cedzynski, Institute for Medical Biology (PAN), Poland
Copyright © 2019 Hajishengallis, Kajikawa, Hajishengallis, Maekawa, Reis, Mastellos, Yancopoulou, Hasturk and Lambris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George Hajishengallis, geoh@upenn.edu
John D. Lambris, lambris@pennmedicine.upenn.edu