 Kevin O. Saunders
Kevin O. Saunders- Laboratory of Protein Expression, Departments of Surgery, Molecular Genetics and Microbiology, and Immunology, Duke University Medical Center, Duke Human Vaccine Institute, Durham, NC, United States
Antibodies and Fc-fusion antibody-like proteins have become successful biologics developed for cancer treatment, passive immunity against infection, addiction, and autoimmune diseases. In general these biopharmaceuticals can be used for blocking protein:protein interactions, crosslinking host receptors to induce signaling, recruiting effector cells to targets, and fixing complement. With the vast capability of antibodies to affect infectious and genetic diseases much effort has been placed on improving and tailoring antibodies for specific functions. While antibody:antigen engagement is critical for an efficacious antibody biologic, equally as important are the hinge and constant domains of the heavy chain. It is the hinge and constant domains of the antibody that engage host receptors or complement protein to mediate a myriad of effector functions and regulate antibody circulation. Molecular and structural studies have provided insight into how the hinge and constant domains from antibodies across different species, isotypes, subclasses, and alleles are recognized by host cell receptors and complement protein C1q. The molecular details of these interactions have led to manipulation of the sequences and glycosylation of hinge and constant domains to enhance or reduce antibody effector functions and circulating half-life. This review will describe the concepts being applied to optimize the hinge and crystallizable fragment of antibodies, and it will detail how these interactions can be tuned up or down to mediate a biological function that confers a desired disease outcome.
Introduction
Since the approval of the first monoclonal antibody by the FDA in 1986 (1), there has been a rapid increase in the number of available monoclonal antibodies or antibody derivatives. In 2015 there were 44 antibodies approved for human use in the United States and Europe (2). Consistent with an expected annual approval rate of six to nine additional antibodies (3), the number of approved antibodies and antibody-like biologics in the United States has climbed to more than 70 (1, 4). It is estimated that global sales of antibody-based products approach $60–75 billion in any given year (2–8). Therefore, many pharmaceutical companies are including antibody-like molecules in their development portfolio due to their high capacity to generate revenue.
Basic science continues to discover the underlying mechanisms of genetic disorders, cancer, and infectious diseases (9, 10). Elucidation of these mechanisms fuels the development of antibody-based biologics to counteract the abnormal biologic process that is causing disease. How the antibody counteracts the biologic process can be optimized for selectivity and potency by modifying the sequence of the antibody-based molecule to enhance or abrogate its interaction with the host immune system (11, 12). This concept is the foundation of antibody optimization efforts in industry laboratories as well as academic research laboratories. While most approved biologics are traditional antibodies, optimized antibodies like Orencia® (abatacept), Soliris® (eculizumab), Nplate® (romiplostim), and Removab® (catumaxomab) have paved the way for optimized antibodies as treatment options (11).
To optimize an antibody one must understand how the antibody is constructed and the role of each of its parts. An intact full-length antibody consists of two 50 kD heavy chains and two 25 kD light chains resulting in a 150 kD full-length, soluble immunoglobulin (13). Each heavy chain associates with a light chain through disulfide bonds and non-covalent interactions to form a heterodimer (14). The two heterodimers are paired together via disulfide bonds between the heavy chains (15, 16). Each heavy and light chain heterodimer includes the antigen binding fragment (Fab) composed of the light chain paired to the variable region of the heavy chain and the CH1 domain of the heavy chain constant region (17, 18). C-terminal to the Fab is the hinge, and the crystallizable fragment (Fc) (17, 18). The hinge region can be subdivided into upper, core, and lower hinge regions (19). The Fc includes the CH2 and CH3 domains of the heavy chain constant region (14).
The constant region of antibodies also contributes to the sequence variation of the heavy chain. The variable region of the heavy chain recombines with the heavy chain constant region to produce a full-length heavy chain (20, 21). The antibody can vary in isotype depending on whether the alpha, mu, gamma, epsilon, or delta constant region gene segment is recombined with the variable region (22). Among the human gamma gene segments there are 4 different subclasses designated as gamma 1, 2, 3, and 4, which are approximately 90% identical to each other (1). In a clinical context, each subclass is important since each subclass specializes in the elimination of different types of pathogens (23). For example, there is an association between deficiency in IgG2 antibodies and infection with encapsulated bacteria (24). The molecular basis of the association may be a diminished antibody response to polysaccharide antigens in individuals lacking IgG2 antibodies (25). For antibody engineering, the different isotypes and subclasses are important for antibody optimization since the sequence variation occurs at sites that determine affinities and specificities for FcRn, Fc alpha receptor, Fc gamma receptors, and complement protein C1q (26). There are 5 Fc gamma receptors (FcγR) that activate effector cells upon binding to IgG. Among the activating receptors there are FcγRI, FcγRIIa, FcγRIIc, FcγRIIIa, and FcγRIIIb (27). There is one inhibitory Fc gamma receptor—FcγRIIb (28, 29). The FcγRs are polymorphic, where certain alleles exhibit higher affinity for Fc than others. For example Val158 allelic variants of FcγRIIIa bind with higher affinity to IgG1 Fc than the Phe158 allelic variant (30). Antibody binding to these receptors can facilitate the recruitment of effector cells to opsonized target cells or opsonized pathogens for clearance [Figure 1A; (2)]. Therefore, changes to the sequence and post-translational modification of the Fc and hinge regions of antibodies allows one to manipulate the effector functions and circulation of a given antibody or antibody-like protein (31). In addition to sequence variation, the Fc region also contains an N-linked glycosylation site at residue 297, which is important for Fc structure and function (32). Most clinically approved antibody-based products are of the gamma isotype, subclass 1 (IgG1) (2). There are currently 3 IgG2 antibodies that are approved for use in the United States (2). IgG3 has long hinge region prone to proteolytic cleavage (23), and exhibits a reduced half-life relative to other IgG subclasses (33). For these reasons it has it has not been the subclass of choice for biologics. Since most clinically-approved antibodies are of the gamma isotype (2), the optimization of antibody binding to FcγRs has been the major focus of the Fc engineering field. However, it is important to note that IgA, IgM, and IgE isotypes have Fc receptors as well, which can be exploited by Fc engineering as discussed below (34–36).
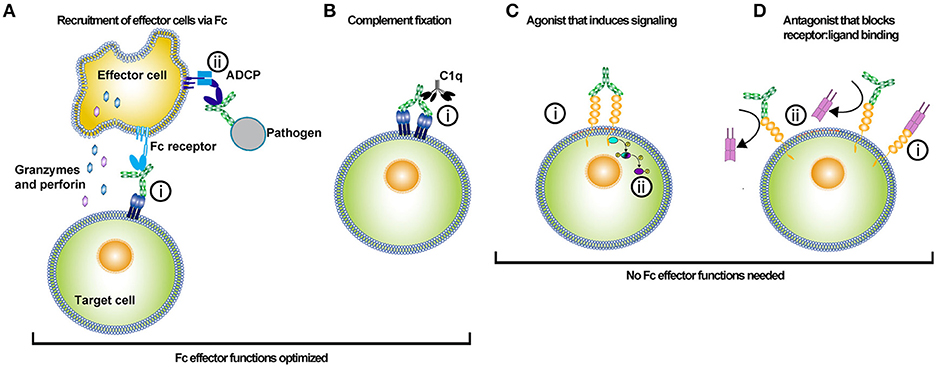
Figure 1. Uses of antibody-based biologics. The type of Fc optimization undertaken for an antibody depends on the desired function of the antibody in vivo. Antibody-based biologics are principally used as adaptor molecules for effector cell recruitment to targets, inducers of the classical complement pathway, agonists, or antagonists. (A) Antibody biologics can be used to opsonize target cells or pathogens and recruit effector cells to kill or phagocytose the target cell or pathogen. (i) Antibodies bind to the antigen via their Fabs and recruit effector cells via their Fc interacting with Fc receptors. Recruitment of the effector cell to the target cell and release of cytotoxic molecules, such as granzyme and perforin results in target cell killing. Alternatively, (ii) effector cells can bind to the Fc of an antibody that has opsonized a pathogen, and subsequently phagocytose the pathogen via antibody-dependent cellular phagocytosis (ADCP). Much of the Fc engineering effort has been focused on improving Fc affinity for various activating FcγRs. The improvement of Fc affinity for FcγRs has led to augmented ADCC and ADCP activity for the optimized antibody-based biologic. (B) Antibodies can kill target cells through the initiation of the complement pathway. The initial step in initiating the complement pathway is the binding of complement protein 1q (C1q). (i) C1q binds to the Fc of antibodies in complex with antigen. Specific mutations have been introduced into the Fc of antibodies to enhance Fc binding affinity to C1q. (C) Agonistic antibody-based molecules are designed to bind to ligand and induce signaling by crosslinking a membrane-bound receptor. (i) Binding of both Fab arms crosslinks the cell receptor and (ii) subsequent phosphorylation of intracellular signal transducers potentiates the receptor signaling. These types of antibody-based biologics do not require Fc function; hence these molecules are prime candidates for Fc optimization that silences FcγR and complement binding. (D) Antagonistic antibody-based biologics bind to a target molecule and prevent the function of that protein either directly or by blocking its ligand from binding. (i) Typically the ligand binds to its receptor, (ii) but in the presence of the antagonistic antibody the receptor binding site is competitively or allosterically blocked. The lack of ligand binding prevents intracellular signaling by the membrane-bound receptor. In this instance, Fc effector functions are not needed and thus mutations that silence Fc effector function can be employed to optimize these types of biologics.
In this review, the approaches utilized to optimize or eliminate Fc interactions with host proteins will be discussed. This review will focus on changes to Fc sequence and glycosylation as a means to modulate Fc function. While Fc optimization is presented as two distinct categories of either enhancement or abrogation of Fc binding the review will describe how a single mutation can have both effects; thus, the two categories are not mutually exclusive. Ultimately, the reader will gain knowledge of how to alter the Fc region of an antibody to change its immunologic properties.
Antibody Fc Mutations for the Improvement of Effector Functions
Enhanced FcγR Binding
Engagement of FcγRs is required for antibody effector functions, such as antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP) (37). Below, modifications that affect binding to FcγRs which result in enhanced ADCC and ADCP are described.
Point Mutations to Enhance FcγR Binding
The Fc of antibodies has been optimized using multiple approaches in attempts to increase binding affinity to selected FcγR (Table 1 and Figure 2). Guided by the 3.2 Å structure of the Fc of IgG1 (61), Shields et al. performed alanine scanning mutagenesis of the solvent exposed amino acid residues on Fc (38). Antibodies encoding Fc regions with alanine mutations were screened for their binding to FcγRI, FcγRIIa, FcγRIIb, FcγRIIIa, and FcRn. Each individual mutation was subdivided into improved or reduced binding to each FcγR and FcRn. Twenty-seven individual mutations increased binding to at least one FcγR or FcRn. In an attempt to engineer an Fc that bound strongly to FcγRIII—a receptor that mediates ADCC (62, 63)—alanine mutations at different sites were combined into one modified Fc. The combination of Ser298Ala, Glu333Ala, and Lys334Ala mutations (sometimes referred to as the AAA mutations) had an additive improvement on the affinity of IgG1 for FcγRIIIa (Table 1) (38). The improved binding to FcγRIIIa translated to 50–100-fold more potent killing in vitro of Her2+ cells by the antibody Herceptin when Ser298Ala, Glu333Ala, and Lys334Ala mutations were incorporated.
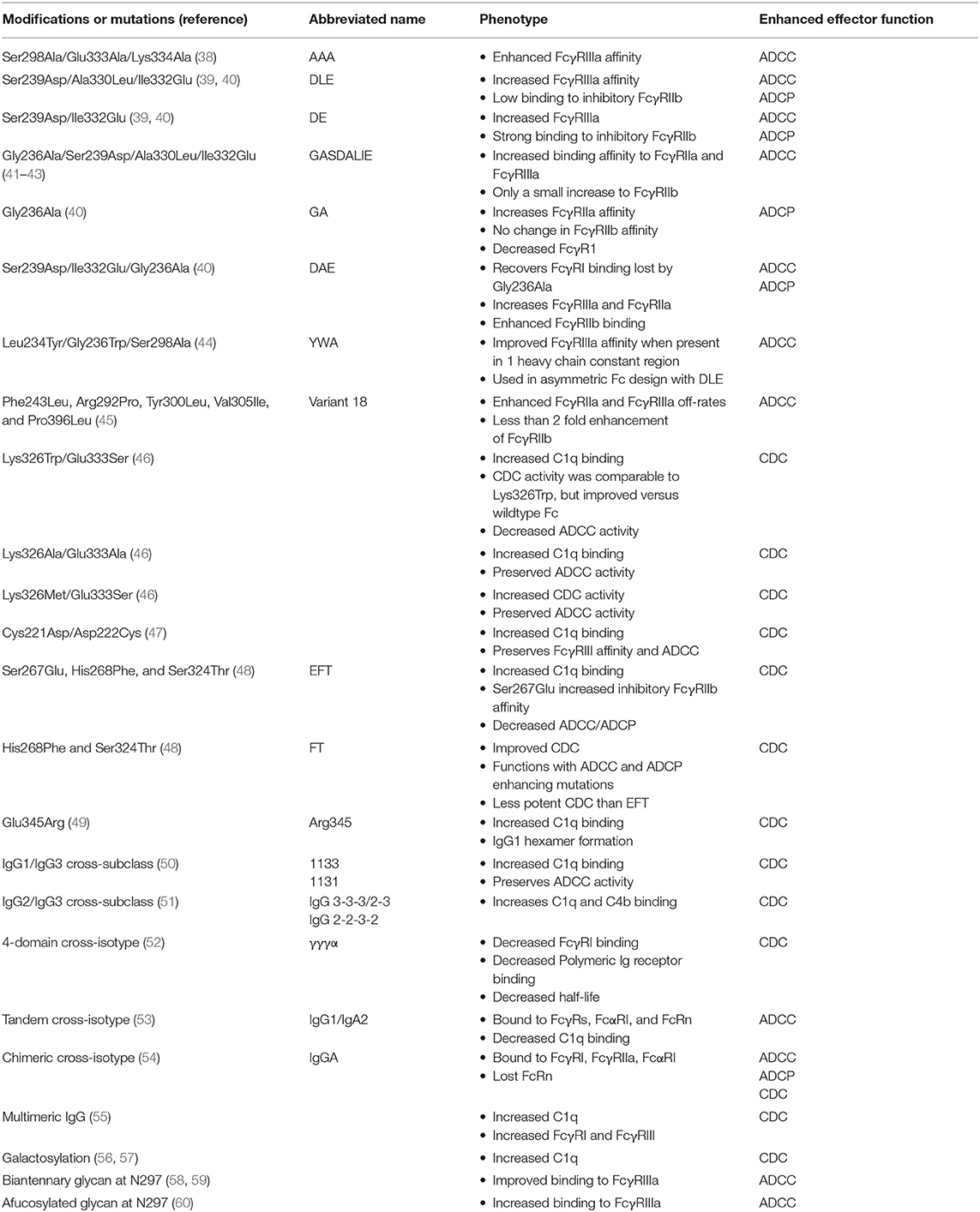
Table 1. Fc modifications to enhance antibody effector function.
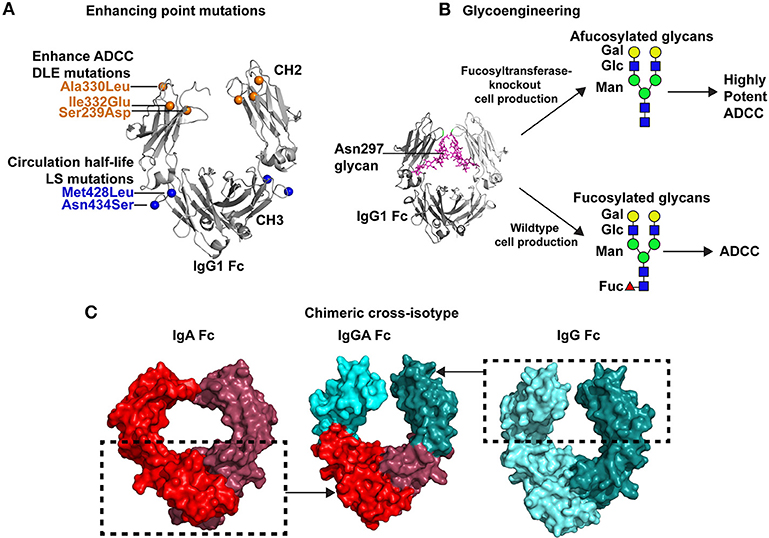
Figure 2. Strategies for improving antibody Fc-mediated effector functions. (A) Multiple point mutations have been identified that improve binding affinity of Fc for specific FcγRs. In some instances a single FcγR, such as FcγRIIIa is the receptor of interest. Directed evolution, alanine scanning, or structure-guided design have been used to identify these mutations. An example of these mutations is the DLE (Ser239Asp/Ile332Glu/Ala330Leu) set of mutations that are shown in the crystal structure of the Fc by orange spheres (PDB:3DO3; 42). These mutations improve ADCC activity. Additionally, mutations can be inserted that improve antibody circulation in vivo. The LS mutations, depicted by blue spheres, (Met428Leu/Asn434Ser) are one example of antibody half-life extension mutations. (B) Antibody effector functions can be enhanced by glycoengineering the Fc domain. The Fc domain contains a N-linked glycan at position 297. A crystal structure of the IgG1 Fc (gray) and the N297 glycan (magenta) are shown (PDB:4BYH). Expression of antibody in wildtype cells results in a fucosylated complex glycans present at N297. However, specialized cells have been created with fucosyltransferase knocked out, which results in afucoylated glycans at Asn297. Antibodies with afucosylated glycans exhibit upto 50-fold more potent ADCC than the same antibody with a fucosylated glycan at Asn297 (60). Green circles, mannose; blue squares, GlcNAc2; yellow circles, galactose; and red triangles, fucose. (C) Antibody effector functions can be improved by expanding the breadth of Fc receptors capable of interacting with Fc. To improve antibody effector function the Fc of a single antibody can be engineered to bind to Fc receptors for multiple antibody isotypes. This concept has led to the design of cross-isotype IgGA antibodies (center) where the IgG1 CH2 a1 loop residues 245–258 and the IgG1 CH3 domain (cyan) were exchanged with the structurally analogous regions of IgA (54). The regions inside the dashed box were combined to create a chimeric cross-isotype Fc. The IgG1 segments are colored light and dark cyan (right, PDB: 3DO3), and the IgA segments are colored light and dark red (left, PDB: 1OW0). The cross-isotype Fc is capable of binding to FcγRI and FcαRI, hence either of these Fc receptors can be used to recruit diverse effector cells to target cells (54).
In a directed evolution approach, Lazar et al. used a computational algorithm to calculate amino acid substitutions that would be predicted to improve the interaction between Fc and FcγRIIIa (39). They also generated a set of quality improvement mutations that would be predicted to improve stability and solubility. Ser239Asp and Ile332Glu in the CH2 domain individually improved FcγRIIIa binding affinity by one log compared to the wildtype Fc (Figure 2A). To maximize binding these two mutations were combined into one Fc construct which resulted in an approximately 2-log enhancement in binding affinity for FcγRIIIa compared to the wildtype Fc. However, an unwanted increase in binding to the inhibitory FcγRIIb was also conferred by the Ser239Asp/Ile332Glu double mutant (39). This undesired effect was partially negated by adding an Ala330Leu mutation to the Ser239Asp/Ile332Glu variant (Figure 2A). The triple mutant Ser239Asp/Ile332Glu/Ala330Leu (commonly referred to as DLE) was introduced into the Fc of anti-cancer antibody Trastuzumab. Compared to wildtype Trastuzumab, Trastuzumab with the DLE mutations had 2-log more potent ADCC killing of Her2+ cancer cell lines expressing low or high levels of cancer antigen (39). The DLE mutations also increased ADCC activity of an anti-integrin antibody, MEDI-522, against a human melanoma cell line (64). Similarly, these mutations increased ADCP by the anti-CD20 cancer antibody Rituximab (39). At the cellular level the DLE mutations function to recruit higher numbers of natural killer cells per target cell coated with the DLE-optimized antibody compared to wildtype antibody (65). Approximately 90% of the recruited NK cells kill cell the target upon first contact, and then move to a second cell for killing (65). Thus, single NK cells kill more target cells per contact and more target cells over time (65). This increase in recruitment and more efficient killing conferred by the DLE-optimized antibody was shown to be due to enhanced FcγRIII-mediated signaling as measured by ZAP70 phosphorylation (65).
The crystal structure of the Fc containing the Ser239Asp/Ile332Glu/Ala330Leu mutations was solved to understand how these mutations affect FcγRIIIa binding. The structure showed an open conformation of the Fc where the two CH2 domains were separated from each other by an additional 30 Å compared to wildtype Fc (66). Thermostability measurements suggested that the opening of the CH2 domain could be because the CH2 domain was less stable or more flexible upon addition of the Ser239Asp/Ile332Glu/Ala330Leu mutations (66). The structure of the optimized Fc was modeled interacting with FcγRIIIa, to determine whether the Ser239Asp/Ile332Glu/Ala330Leu mutations created additional interactions with the FcγRIIIa. Indeed, the structural model suggested additional hydrogen bonds between Ser239Asp/Ile332Glu in the Fc and Lys158 in the FcγRIIIa. Ala330Leu potentially created more hydrogen bonds with Ile85 in the FcγRIIIa as well (66). Additional electrostatic and hydrophobic interactions were also suggested by the structural model (66).
Mimoto et al. engineered an asymmetric Fc that combined the DLE mutations with their newly-identified Fc optimization mutations (44). In a large saturating mutagenesis screen, they examined the binding of 1,000 single Fc mutants to identify mutations that improved FcγRIIIa binding when present in only one of the heavy chains within an IgG molecule. They selected three mutations, Leu234Tyr, Gly236Trp, and Ser298Ala (termed YWA mutations), from their screen to incorporate into one heavy chain constant region. Since the DLE mutations had been shown to increase FcγRIIIa binding they incorporated these mutations into the other heavy chain constant region. Antibodies bearing YWA mutations in one heavy chain and DLE in the other mediated ADCC of tumor antigen-expressing cells in vitro more potently than symmetrical antibodies that contained only the YWA or DLE mutations (44). Thus, this asymmetric Fc design enables one to incorporate multiple optimization mutations to additively improve Fc function.
Macrophages utilize FcγRIIa to phagocytose antibody-opsonized antigens (67). To increase Fc receptor binding to FcγRIIa, Richards et al. screened 900 Fc variants for binding to FcγRIIa and identified Gly236Ala substitution alone increased the binding affinity approximately 6-fold for both His131 and Arg131 alleles of FcγRIIa (40). Unfortunately, the addition of Gly236Ala into IgG1 Fc reduced the IgG1 affinity for the activating receptor FcγRI (40). To recover the FcγRI binding, previously reported Ser239Asp/Ile332Glu mutations were introduced into the IgG1 Fc. This triple combination of mutations showed a 3-fold increase in FcγRI up to 70-fold increase in affinity for FcγRIIa, and up to a 31-fold increase in affinity for FcγRIIIa. The Ser239Asp/Ile332Glu/Gly236Ala mutations enhanced in vitro FcγRIIa-dependent phagocytosis and FcγRIII-dependent ADCC activity of an IgG1 targeting adenocarcinoma cell lines (40).
The activating receptor FcγRIIa is 90% similar to the inhibitory receptor FcγRIIb (68), and thus the increase in FcγRI and FcγRIIIa affinity for Ser239Asp/Ile332Glu/Gly236Ala was accompanied by a 13-fold enhancement in binding to FcγRIIb. To compare the binding of both the activating and inhibitory FcγRIIs the ratio of binding was determined. The ratio of binding between the activating FcγRIIa and inhibitory FcγRIIb receptors was higher for Gly236Ala and the Ser239Asp/Ile332Glu/Gly236Ala than wildtype IgG1 (40). Thus, the ratio may be most important for determining the final functional activity of antibodies encoding the Ser239Asp/Ile332Glu/Gly236Ala mutations. Smith et al. attempted to improve the binding ratio of FcγRIIa to FcγRIIb by combining related sets of mutations to generate Gly236Ala/Ser239Asp/Ala330Leu/Ile332Glu (referred to as GASDALIE) (41). This collection of mutations increased binding affinity to FcγRIIIa encoding the low affinity allele Phe158 by 30-fold, most likely because of increased electrostatic interactions between the Fc and FcγRIIIa (41, 42). Similarly, binding affinity to FcγRIIa was improved 25-fold (41). FcγRIIb binding affinity affinities were only slightly increased, which resulted in a FcγRIIa to FcγRIIb affinity ratio of 11.6 compared to 1.6 for wildtype IgG1 (41). In a second experiment, investigators attempted to optimize Fc while avoiding any mutations that increased FcγRIIb binding. Using yeast display the investigators identified mutations that increased FcγRIIIa binding and reduced FcγRIIb binding. Upon making their mutant libraries and expressing them on the surface of yeast they used bead depletion to remove antibody Fc variants that bound to FcγRIIb. After FcγRIIb-bead depletion, the library of remaining Fc regions was screened for binding to recombinant FcγRIIIa. Using two different libraries, seven single mutations appeared to lack FcγRIIb binding while improving FcγRIIIa binding. These mutations were introduced into the Fc region of IgG1 individually as well as in various combinations. Combinations of Phe243Leu, Arg292Pro, Tyr300Leu, Val305Ile, and Pro396Leu mutations slowed the off-rates of Fc binding to FcγRIIa and FcγRIIIa relative to wildtype Fc without increasing binding to the inhibitory FcγRIIb receptor. The Fc that included all five mutations—termed variant 18—had a 10-fold improvement in affinity for FcγRIIa and FcγRIIIa, and <2-fold increase in FcγRIIb affinity. The variant 18 Fc exhibited potent ADCC activity against colon, ovarian, and breast cancer cell lines in vitro for several different antibodies (45). In vivo, an IgG1 encoding Phe243Leu, Arg292Pro, Tyr300Leu, Val305Ile, and Pro396Leu mutations conferred a significant increase in survival in a lethal ovarian tumor transplantation model (45). These mutations have shown promise in treatment of cancer in humans as well. Anti-HER2 monoclonal antibody Margetuximab, which contains Phe243Leu/Arg292Pro/Tyr300Leu/Val305Ile/Pro396Leu optimization mutations, exhibits improved ADCC activity compared to the standard of care antibody trastuzumab (69). In one clinical trial 78% of the response-evaluatable patients who received Margetuximab showed a reduction in tumor size (70), highlighting the potential promise for Fc optimization to improve disease treatment.
Glycoengineering to Enhance FcγR Binding
The Fc of IgG1 contains a single N-linked glycosylation site at position 297. The glycan present at N297 typically consists of two N-acetylglucosamine (GlcNAc), three mannose, and two more GlcNAc linked to the mannose to form a biantennary complex glycan (71). The two GlcNAc are linked to mannose through either a β1,2 linkage to α-3 or α-6 of the mannose. Thus, each arm of the glycan can be distinguished as the α1,3 or α1,6 arm depending upon how the mannose and GlcNAc2 are linked (71). Additional fucose, galactose, sialic acid, and GlcNAc can be added to the core glycan structure (Figure 2B). IgG found circulating in human sera are generally fucosylated, however during recombinant IgG production the glycan composition can be altered by expressing the antibody in plant cells, knocking in or knocking out specific glycosidases, or in vitro enzymatic digestion of the glycosylated IgG (Figure 2B) (72). Since both heavy chains are glycosylated it is possible for a single IgG molecule to have significant glycan heterogeneity (71). The glycan has direct effects on FcγR binding. The Asn297 glycan on the Fc can clash with glycans on the FcγRIII protein, which results in poor engagement of effectors cells that mediate ADCC. Also, nuclear magnetic resonance studies have shown that Fc regions containing different glycans at Asn297 adopt different hinge region conformations (73). Since the hinge region is contacted by FcγRs, the glycosylation of N297 indirectly affects the ability of the Fc to interact with FcγRs. Thus, optimization of Fc glycosylation has been important for producing antibody biologics with a desired function.
To modulate antibody activity several studies have modified antibody Fc glycosylation by expressing or inhibiting enzymes in the producer cells. Expression of β(1,4)-N-acetylglucosaminyltransferase III when expressing IgG gives an antibody glycosylated at N297 that has a biantennary glycan and has better ADCC activity (58). Davies et al. produced anti-CD20 IgG1 under these conditions and found the antibody had 10–20-fold more potent FcγRIIIa-dependent killing of CD20+ cells (59). Despite these results the importance of bisecting GlcNAc is debated, and the removal of fucose has been asserted as an alternative hypothesis [Figure 2B; (74)]. Antibodies deficient in fucose have been shown to have 50-fold higher binding to FcγRIIIa and enhanced ADCC activity (60). The enhancement of afucosylated antibody binding to FcγRIIIa is higher for the high affinity Val158 allele compared to the Phe158 allele, but both alleles show an increase in binding to afucosylated IgG1 compared to fucosylated IgG1 (40, 75). The most dramatic increase in binding by afucosylated Fc is for glycosylated FcγRIIIa (76), with the removal of the Asn162 glycan in FcγRIIIa completely abrogating this enhanced binding (76). The mechanism for glycosylated FcγRIII recognition was later determined by structural studies, which showed that the afucosylated Asn297 glycan interacts with the Asn162 glycan on FcγRIII (77). The addition of fucose to the Fc glycan creates clashes with the GlcNAc2 on FcγRIIIa, providing a structural explanation for why afucosylated antibodies bind better to FcγRIIIa (77). The approval of mogamulizumab (POTELIGEO®) in Japan marked the first approval for human use of an afucosylated antibody with enhanced ADCC activity (78–80).
Enhancing FcγR Binding by Exchange of Fc Domains Across Isotypes (Cross-Isotype Antibodies)
In addition to increasing affinity for receptors by introducing point mutations or modifying glycans, the Fc can be optimized to engage a wider range of Fc receptors (Figure 2C). As stated above, Fc receptors for isotypes other than gamma exist on particular leukocytes. By creating a Fc region that can interact with multiple Fc receptors, such as FcγR and FcαRI, one creates an antibody with expanded, novel abilities to engage effector cells (2). Neutrophils are the most abundant leukocyte in the body, and they engage Fc of IgA antibodies via the FcαRI (81, 82). Single domains of IgA2 were appended to end of the gamma 1 constant region creating a four-domain constant region (CH1g-CH2g-CH3g-CH3a) (52) in an attempt to engage FcγRs and FcαRI. To make the constant region more similar to the alpha constant region the CH1 domain of gamma 1 was substituted for the alpha 1 constant region domain (CH1a-CH2g-CH3g-CH3a). These four-domain cross-isotype IgGA chimeric antibodies bound to J chain similar to natural IgA2 but had reduced transport by polymeric Ig receptor. The four-domain, cross-isotype antibodies also had a 3–5-fold decrease in FcγRI affinity, and possessed the short serum half-life of IgA2 instead of the protracted serum circulation of IgG1. Despite these shortcomings, the four-domain cross-isotype IgGA design was capable of mediating complement-dependent lysis of sheep red blood cells and appeared to be more pH-resistant than IgG1 (52). In a similar approach a second type of cross-isotype Fc has been created by fusing the gamma 1 and alpha constant regions together to create a tandem G1-A Fc region (53). In this design the hinge, CH2, and CH3 of IgA2 is fused to the C-terminus of IgG1. This tandem cross-isotype IgG/IgA design had similar expression levels, antigen binding, and thermostability as antibodies made in the IgG1 format. In vitro, the tandem cross-isotype IgG/IgA bound to FcαRI and FcγRI, FcγRII, FcγRIIIa, and FcRn with affinities similar to wildtype IgA and IgG, respectively. This binding to various FcRs translated to the antibody mediating ADCC activity with polymorphonuclear cells and NK cells. C1q binding to the tandem cross-isotype IgG/IgA was reduced 3-fold compared to the IgG1 format of the antibody. Lastly, in BALB/c mice, tandem IgG/IgA circulated with a half-life similar to that of IgG1. In a third design, Kelton et al. created a cross-isotype antibody by exchanging the CH3 domain and CH2 α1 loop residues 245–258 (PKPKDTLMISRTPE) of the gamma 1 constant region with that of the alpha constant region [Figure 2C; (54)]. The chimeric Fc possessed the ability to bind to FcγRI, FcγRIIa, and FcαRI (54). Antibodies made in this IgGA format were capable of mediating ADCC with polymorphonuclear cells, mediating ADCP with macrophages, and activating complement (54). However, this design lacked binding to neonatal Fc receptor which regulates antibody half-life (54). Thus, further optimization would be required for effective in vivo use of this design. In total, these designs show the promise of the concept of engaging a wide range of effector cells for antibody Fc optimization.
IgG Multimerization Augments FcγR Binding
Multimerizing IgG has shown promise in the treatment of autoimmune diseases (83, 84). The IgG multimers are constructed in various ways including the addition of heterologous multimerization domains such as isoleucine zippers (83), another hinge region at the N-terminus of the natural hinge, or another hinge region at the C-terminus of the CH3 domain (83). Similarly, hexamers of IgG have been created by appending the IgM tailpiece to the C-terminus of the IgG1 Fc and creating a cysteine bond at position 309 (85, 86). The multimeric IgG formed by the addition of the IgM tailpiece bound strongly to FcγRI, FcγRIIa, and FcγRIIIa and bound weakly to FcγRIIb and FcγRIIIb (85, 86). Across the various designs, multimeric IgG bound to a higher magnitude than monomeric IgG to FcγRI, FcγRIIb, and FcγRIII (83, 85). Such molecules have shown promise in preclinical models of arthritis, neuropathy, and autoimmune myasthenia gravis (83, 84, 87). Hence the multimeric IgG platform is being further optimized to fine-tune the immune receptors, such as FcRn that can interact with the multimer (55).
Enhancing Complement Fixation
Point Mutations to Increase C1q Binding and Complement-Dependent Cytotoxicity (CDC)
Antibodies can exert cytotoxic effects by engaging the complement pathway. The initial step of this process is the binding of C1q to the CH2 domain of an antibody-opsonized antigen [Figure 1B; (88)]. Alanine scanning mutagenesis of the human IgG1 Fc identified Asp270, Lys322, Pro329, and Pro331 as essential for C1q binding to the Fc (89), although Fc from different species utilize different residues for binding to C1q (90). To increase Fc binding to C1q Idusogie et al. identified Lys326 and Glu333 as proximal to the core binding site of C1q within the Fc. The importance of these residues was first tested by alanine mutagenesis. Introducing Lys326Ala and Glu333Ala increased C1q binding and CDC activity by 50%. To optimize the Fc for binding to C1q various amino acids were introduced at 326 and 333 individually and in combination. The combination of Lys326Trp and Glu333Ser increased C1q binding by 5-fold (46). However, CDC activity conferred by the Lys326Trp/Glu333Ser double mutant Fc was the same as the Lys326Trp single mutant, and the Fc lost the ability to mediate ADCC. In instances when ADCC activity is also important, mutating positions 326 and 333 to two alanines, or mutating positions 326 and 333 to methionine and serine, respectively, provided an increase in CDC without hindering ADCC (46). Similar mutagenesis experiments have been done for the hinge region to determine whether it affects C1q binding and CDC activity. The mutation of the hinge region was not intuitive since C1q binds to the CH2 domain below the hinge region. However, it was shown that in the upper hinge region substituting Trp in various combinations at positions 222, 223, 224, and 225 increased C1q binding and increased CDC activity relative to wildtype IgG1 (47). ADCC activity and FcγRIIIa binding were unchanged by these modifications (47). Cys221Asp and Asp222Cys alone or in combination with Trp substitutions also increased C1q and CDC activity (47). Thus, the hinge of human IgG1 modulated C1q binding to the CH2 domain (47).
To identify other point mutations that improve CDC activity of IgG1, Moore et al. made 38 Fc variants of an anti-CD20 antibody and screened them in vitro for their ability to mediate CDC against Raji cells (48). Among the 38 variants, three variants encoding Ser267Glu, His268Phe, and Ser324Thr (termed the EFT mutations) changes were identified as having more potent CDC activity vs. wildtype IgG1 (48). The largest improvement in CDC activity was achieved when the three mutations were combined into one Fc variant (48). Correlation analyses suggested the improvement in CDC potency was due to increased C1q binding (48). The triple EFT mutations had increased binding to the inhibitory FcγRIIb, which presumably limited its ADCC and ADCP activity. The addition of ADCC and ADCP-enhancing mutations to the EFT mutations restored ADCC and ADCP function back to wildtype IgG1 levels, but did not confer an improvement (48). The increased binding to FcγRIIb could be reduced by eliminating the Ser267Glu from the EFT mutations, however this change came a cost of reduced CDC potency. The His268Phe and Ser324Thr mutations were then capable of being combined with ADCC and ADCP-enhancing mutations to create a single Fc with improved CDC, ADCC, and ADCP activity (48). This study highlights the complicated balance between optimizing one effector function without decreasing another effector function.
Insertions and Deletions to Increase to Increase C1q Binding and Complement-Dependent Cytotoxicity (CDC)
Hinge length is important for C1q recognition of antibodies or antibody-based proteins. IgG3 has a distinct extended hinge of 62 amino acids that arises from the duplication of 3 exons that encode for part of the core hinge region (91). For IgG3 antibodies complement activation is increased by shortening its hinge region (92). While complete removal of the hinge ablates CDC function, a hinge of 15 amino acids instead of 62 amino acids exhibited 10-fold more potent CDC activity (92). More specifically, removal of the three repeats regions within the core hinge does not eliminate CDC, but instead improved CDC potency for anti-bacterial antibodies (93). This result for IgG3 is in contrast to IgG1 where two amino acid deletions in the core hinge region reduced C1q binding, CDC activity, and ADCC (47).
Cross-Subtype Antibodies to Improve C1q Binding
IgG1 is the preferred subclass for antibody biopharmaceuticals over IgG3 since the long hinge of IgG3 complicates large scale production of the antibody (2). However, IgG3 possesses the best in vitro binding to C1q (94). As a means to enhance IgG1 C1q binding, domains of IgG3 where substituted for IgG1 domains to create IgG1/G3 cross-subtype antibodies (50). These chimeras eliminated the difficulty of purifying antibodies with long hinge regions but capitalized on IgG3 effector functions. In one of the best variants, termed 1133, the CH1 and hinge region from IgG1 was fused to the Fc from IgG3 (50). Its ADCC activity and antigen binding were unchanged, while, its CDC activity and C1q binding were enhanced relative to wildtype IgG1 or IgG3 (50). Furthermore, the 1133 design allowed for CDC activity when the antigen levels were low (95). However, the 1133 Fc variant lacked protein A binding, which is important for easy purification of the antibody. Thus, an antibody with the CH1, hinge, and CH3 of IgG1 and CH2 of IgG3 was constructed since C1q binds the CH2 domain and protein A binds the CH3 domain. This variant had improved CDC activity and the ability to bind protein A (50). The molecular basis for the improved binding of the chimeras is presumed to be the amino acid differences in the CH2 domain that are proximal to the C1q binding site in the tertiary structure of the Fc. In mutagenesis experiments aiming to define the amino acids required for C1q binding to IgG1 and IgG3 K322 was found to be important for both subclasses, but dependence on other amino acids varied between subclasses. For example, P331 was required for CDC activity of IgG1 (89), but had only a modest effect on IgG3 CDC activity (96). These results indicate that C1q binding differs between IgG3 and IgG1 thus chimeric antibodies may be able to enhance binding by combining Fc:C1q interactions from both gamma subclasses. While the goal of Fc designs has been to boost IgG1 activity, cross-subclass designs have also been used to confer activity to functionally silent subclasses. IgG2 and IgG4 have very little ability to mediate CDC compared to IgG1 or IgG3 (51). However, replacing the CH2 domain of IgG2 with that of IgG3 can instill CDC activity to the otherwise IgG2 Fc (51). Similarly, IgG4 differs from IgG1 at position 331, which has been shown to be proximal to the C1q binding site (90). Changing the IgG4 residue at position 331 to match IgG1 conferred a moderate level of CDC. Therefore, if one knows the key residues for mediating an effector function they can be introduced into functionally silent Fc domains to confer specific functions.
IgG1 Hexamer Formation Boosts C1q Binding and CDC Activity
The multimerization of IgG by binding to antigen is known to enhance C1q binding substantially (97). To engender multimerization of the IgG in the absence of antigen, analysis of IgG structures identified position 345 as an amino acid that could facilitate multimerization between the Fc regions of different antibodies (49). The IgG structure suggested that the introduction of a positively charged amino acid would confer Fc:Fc interactions. Thus, a Glu345Arg mutation was introduced into the IgG1 Fc. Electron microscopy and mass spectrometry confirmed that this mutation resulted in monomeric IgG1 as well as multimeric IgG1 linked via the Fc (49). The multimeric IgG possessed higher binding to C1q and more potent lysis of a Burkitt's lymphoma cell line (49). Interestingly, the Glu345Arg mutation not only increased CDC activity by IgG1, but also IgG2, IgG3, and IgG4. Multimers of IgG1 have also been described by the addition of IgM tailpiece and Cys309 in the IgG1 Fc as stated above (see IgG multimerization augments FcγR binding). IgG hexamers created using the IgM tailpiece strategy also showed improved binding to C1q and C5b relative to wildtype IgG1 (55). Thus, multimerization of IgG is another method in addition to point mutations and IgG1/IgG3 cross-isotype antibodies to increase C1q binding affinity.
Glycoengineering to Improve Complement Binding
The Asn297 glycan within the CH2 domain of the Fc can be modified to improve CDC activity (56, 57, 98–101). In a large screen of 20 different glycoforms of anti-trinitrophenyl hapten IgG1 Fc an overabundance of galactosylation increased C1q binding and CDC activity compared to the unmodified glycoform of IgG1 (57). Galactosylation appeared to be the principal glycan residue that affected CDC activity as significant positive correlations were observed between abundance of galactosylation on Fc and CDC potency (57). In a separate study Peschke et al. confirmed the importance of galactosylation for CDC activity using a different antibody specificity and multiple IgG subclasses. Galactosylation of the IgG1 Fc improved CDC activity of the anti-CD20 antibody Rituxumab against Raji B cells in vitro (56). The improved CDC activity conferred by galactosylation of the Fc was applicable to IgG3, but was not applicable to IgG2 or IgG4 in this in vitro model (56). The improved CDC activity conferred by galactosylated Fc was not due to changes in antigen binding, but instead was associated with enhanced C1q binding (56, 57). Overabundance of galactosylation on IgG1 Fc also improved thermostability when measured by differential scanning calorimetry (99). Thus, galactosylating the Fc is one strategy for producing a stable biologic with highly potent CDC activity.
Improved Antibody Half-Life Circulation
In addition to improving antibody effector functions by increasing affinity for activating FcγR and C1q, Fc optimization efforts have also tried to improve antibody circulation in vivo. In vivo IgG catabolism is regulated by its interaction with the neonatal Fc receptor (FcRn) (102). The FcRn binds to IgG at the junction of the CH2 and CH3 domains in a pH dependent manner (102–104). IgG is endocytosed by cells where it can be shuttled to lysosomes or recycled back to the cell surface (105). Binding of IgG to FcRn at low pH (pH < 6.5) in the endosomes allows the antibody to be trafficked with the FcRn back to the cell surface (106, 107). Poor binding to FcRn at pH < 6.5 results in the antibody being trafficked to the lysosome and degraded (105). At the physiologic pH of the extracellular environment IgG has weak affinity for FcRn which results in its release from the FcRn back into circulation (105). The pH dependent binding is regulated by protonation of His310, 435, 436 in the Fc at low pH (108). The protonation creates positively charged residues that can bind to negatively charged Glu117, Glu132, and Asp137 in the FcRn (109).
Point Mutations to Increase FcRn Binding Affinity
In a global approach to increasing IgG1 half-life, alanine scanning mutagenesis of the Fc was performed. In this screen 17 amino acids that affect IgG Fc binding to FcRn were identified (38). Among the 17 amino acids Asn434 and Glu380 showed large increases in affinity when mutated to alanine (38). The Asn434Ala mutation has been useful for countering the poor FcRn affinity that can result from the introduction of FcγR affinity-optimizing mutations (38), thus Asn434Ala is typically added to Ser298Ala, Glu333Ala, and Lys334Ala to create a AAAA variant with enhanced FcγR binding and normal or slightly improved FcRn binding (Table 2) (38). Additional half-life mutations were identified by sequentially performing random and directed evolution screens of phage libraries. The phage binding was done at pH 6 to mimic endosomal pH, and elution was done at pH 7.4 to find variants that did not bind at physiologic pH. Six collections of mutations were identified that improved FcRn binding across three different assays including Glu294deletion/Thr307Pro/Asn434Tyr (termed C6A-66) and Thr256Asn/Ala378Val/Ser383Asn/Asn434Tyr (referred to as C6A-78) (116). Asn434Tyr was among the most common mutations found in each collection (114). One of the differences between these collections of mutations was the ability to bind to FcγRIIIa, thus one could extend half-life while also retaining ADCC activity or knocking out ADCC activity (114). In more recent work, the C6A-66 collection of mutations were analyzed further since it showed only a moderate increase in FcRn binding, but had the best serum half-life in vivo. The collection of mutations were studied as individual mutations to elucidate the function of the deletion of Glu294 (115). This deletion resulted in higher sialylation of the Asn297 glycan on the Fc (115). The abundance of sialic acid was necessary for the increase in antibody half-life in vivo (115). Thus, increased FcRn binding was not the only factor that contributed to increased half-life. Sialylation also has a role in regulating serum half-life (115).
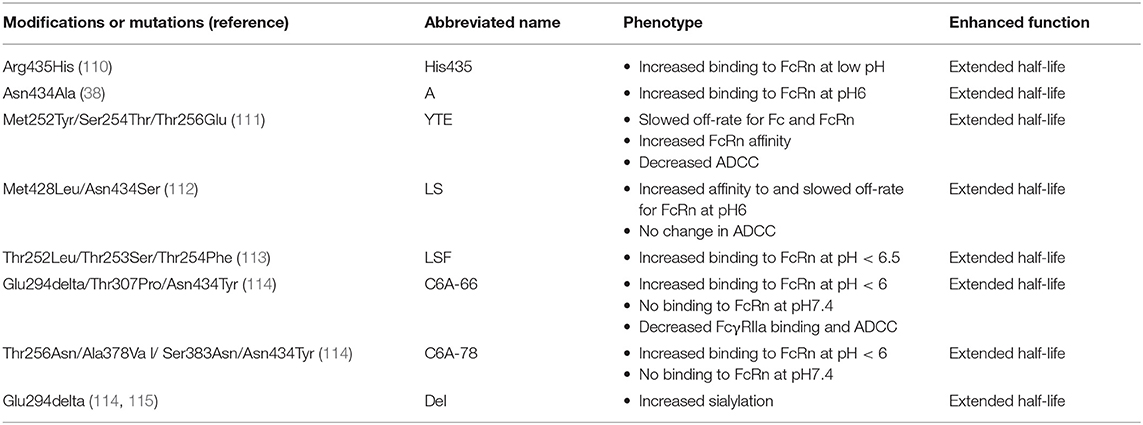
Table 2. Fc modifications to improve antibody circulation half-life.
Ghetie et al. also created large libraries of random mutations of Thr252, Thr253, and Thr254 in the mouse Fc and screened them for binding to mouse FcRn using a bacteriophage display platform (113). The three sites were chosen based on their proximity to the FcRn binding site on Fc. From the phage library the collection of Thr252Leu, Thr253Ser, and Thr254Phe was identified that had significantly longer half-life in wildtype mice. These three mutations, termed LSF, did not affect association rates of Fc with FcRn but did slow the dissociation rate of Fc from FcRn at pH6 (113). This result indicated for mouse antibodies that that position 252, 254, and 256 could be manipulated to increase antibody half-life. Hence in a later study, phage display libraries of human IgG1 were used to identify analogous mutations at positions 252, 254, and 256 (111). In the human IgG1 Met252Tyr, Ser254Thr, and Thr256Glu was observed in a high abundance among the clones isolated from the phage library. Inclusion of these 3 mutations, often called YTE, in the IgG1 Fc resulted in a 10-fold slower dissociation rate of Fc and FcRn. Overall, the YTE mutations enhanced the apparent equilibrium rate constant 3-fold for Fc binding to FcRn. To determine whether the increased binding to FcRn in vitro translated to improved pharmacokinetics in primates, wildtype IgG1 or a YTE variant were infused into cynomolgus monkeys and serum half-life was compared. The YTE antibody possessed a 4-fold increase in serum half-life as compared to the wildtype antibody (64). Additionally, there was a higher concentration of infused IgG measured in bronchioalveolar lavage fluid when the Fc included the YTE mutations (64). The pharmacokinetic profile of an IgG1 possessing the YTE mutations was determined in a phase 1, double-blind, dose escalation study. In this study the YTE-variant of IgG1 had a serum half-life of 80–112 days (117). Based on the typical serum half-life of IgG1 being 21 days, the YTE mutation seems to increase half-life 4–5-fold in humans (117). In a second study YTE was introduced into motavizumab and their half-life was directly compared in humans. Consistent with the first study, serum half-life was extended 2–4-fold compared to wildtype IgG. Remarkably, the individuals that received the YTE variant had functional therapeutic antibody present in their serum 240 days after antibody infusion (118). Thus, the YTE mutations raise the possibility of long-acting antibody-based biologics that could suppress or protect from disease in humans. While YTE improves half-life of the antibody, it also eliminates ADCC activity of the antibody (64). This pitfall can be counteracted by the addition of ADCC-enhancing mutations, such as DLE (64). Therefore, YTE mutations should be used alone only when the antibody does not need to mediate ADCC.
Another set of mutations that improve antibody half-life was discovered by Zalevsky et al. using rational protein design. Introduction of Met428Leu and Asn434Ser mutations (referred to as the LS mutations) in IgG1 Fc resulted in a decrease in the dissociation rate and an 11-fold improvement in binding affinity between Fc and human FcRn at pH6 [Figure 2A; (112)]. In contrast to YTE, LS mutations did not significantly reduce ADCC activity (119). In cynomolgus macaques, the LS mutations conferred a 3-fold increase in antibody half-life (112). A similar increase of 3–4-fold in serum antibody half-life was seen in human FcRn transgenic mice (112). The improvement in function conferred by the LS mutations was tested by engrafting tumors into the human FcRn transgenic mice and infusing wildtype or LS-mutant IgG1 (112). For two different cancer immunotherapeutic antibodies the LS mutant IgG1 inhibited tumor growth significantly better than the wildtype IgG1 (112). Since the initial description of the LS mutations, multiple groups have shown these mutations boost antibody half-life in cynomolgus macaques (120, 121) and rhesus macaques (119, 122). The LS mutations have been helpful in sustaining protection against HIV-1 infection in animal models (119, 122, 123). The incorporation of LS resulted in increased antibody concentrations at mucosal sites and prolonged serum half-life (119, 122). Together these attributes resulted in improved protection afforded by optimized IgG1 in macaque models of HIV-1 infection (119, 122, 123). Clinical trials are planned to administer anti-HIV-1 IgG1 antibodies encoding the LS mutation. The extent to which the improved pharmacokinetic profile translates from macaques to humans will be determined, and whether longer half-life improves therapeutic efficacy will be evaluated. Novel mutations to improve antibody half-life are still being pursued. Approaches to improve upon the LS mutations include finding mutations that completely eliminate Fc binding to FcRn at physiologic pH, while also enhancing binding at low pH.
Cross-Subclass Point Mutations to Enhance FcRn Binding
IgG1 makes these 3 productive Fc:FcRn contacts and has a half-life of 21 days (109). In contrast to IgG1, IgG3 alleles typically encode arginine at position 435 instead of histidine. IGHG3*17, IGHG3*18, and IGHG3*19 alleles are the exceptions, which encode histidine like IgG1. The presence of arginine vs. histidine confers a serum half-life of only 7 days (124). In vitro competition assays suggest that IgG1 with His435 outcompetes IgG3 with Arg435 for FcRn binding (110). Furthermore, Arg435 seems to increase binding of IgG3 to FcRn at physiologic pH (110), which could result in more IgG3 being absorbed to epithelial cells expressing FcRn hindering the ability of IgG3 to freely circulate in serum. To increase IgG3 half-life, position 435 was changed to histidine, which boosted FcRn binding at low pH (110). Consistent with the increased binding, serum concentrations of IgG3 are higher in individuals who express an allelic variant of IgG3 encoding His435 and infusion of IgG3 encoding His435 is more efficient (110, 125). Thus, modulation of the pH sensing ability of IgG3 Fc is one mechanism for boosting its serum half-life.
Antibody Fc Engineering for the Ablation of Effector Functions
While Fc optimization has focused heavily on gain-of-function modifications, in certain situations it can be beneficial to eliminate antibody Fc function. These situations include antibodies that are used as (1) receptor agonists to crosslink receptors and induce signaling, (2) receptor antagonists to block receptor:ligand interactions to prevent signaling, or (3) drug delivery vehicles to deliver drug to antigen-expressing target cells (Figures 1C,D). In these instances Fc engagement of receptors on effector cells or engagement of C1q is not wanted, because it can lead to undesired killing of biologically-important cells expressing the receptor or recruitment of drug-conjugated antibodies to off-target cells (126, 127). Below, strategies to eliminate FcγR binding and complement protein C1q binding are described.
Ablation of FcγR Binding
Point Mutations to Ablate FcγR Binding
One of the earliest antibodies used in humans was OKT3 to prevent transplant rejection (1). Despite humanization of the antibody, this antibody induced proinflammatory cytokine secretion, which resulted in toxicity (128, 129). The cytokine secretion was due to binding of OKT3 to CD3 followed by crosslinking of FcγRs on T cells (129). To alleviate the cytokine induction from T cells the Fc region of the antibody was mutated to eliminate FcγR binding (129). A single mutation of Leu235Glu was sufficient for knocking out binding to Fc receptors on U937 cells (129). Furthermore, the 100-fold reduction in binding to FcγR also resulted in lower T cell activation and proliferation in the presence of the Leu235Glu Fc mutant IgG1. Building upon this initial mutation it was found that the combination of Leu234Ala and Leu235Ala (commonly called LALA mutations) eliminated FcγRIIa binding [Table 3 and Figure 3A; (130, 131)]. These two mutations were later shown to eliminate detectable binding to FcγRI, IIa, and IIIa for both IgG1 and IgG4 (153). The use of LALA appears to be more effective than either Leu234Ala or Leu235Ala alone. Some groups have seen that the Leu235Glu mutation knocks out FcγRI binding (129). However, there are reports that this high affinity receptor still binds to IgG1 Fc when this mutation is present (132). Similarly, Leu234Ala single mutant Fc still possessed detectable binding to the high affinity Fc receptor FcγRI at least in some assays (131). Both the single and double mutations at position 234 and 235 reduced ADCC activity mediated by PBMCs and nearly ablated ADCC mediated by monocytes (132). Nonetheless, the LALA mutations have been tested in humans in a phase I clinical trial. Anti-CD4 antibody OKT3 encoding the LALA mutations was administered for the treatment of acute renal allograft rejection. The IgG1 with LALA mutations caused minimal adverse reactions and was able to reverse allograft rejection in 6 of 7 individuals (154). One common pairing, in addition to the LALA pairing, is Ser228Pro paired with Leu235Glu, which has been called the SPLE or PE mutations (133). The SPLE mutations have been introduced into IgG4, which has low binding to FcγR initially perhaps due to the Phe234 residue that differs from Leu234 found in IgG1 (155). This combination dramatically reduced FcγRI binding to IgG Fc to barely detectable levels by surface plasmon resonance (133), without reducing circulating half-life in rats (155). The effect of the Ser228Pro is thought to be mostly to improve stability of IgG4 (155).
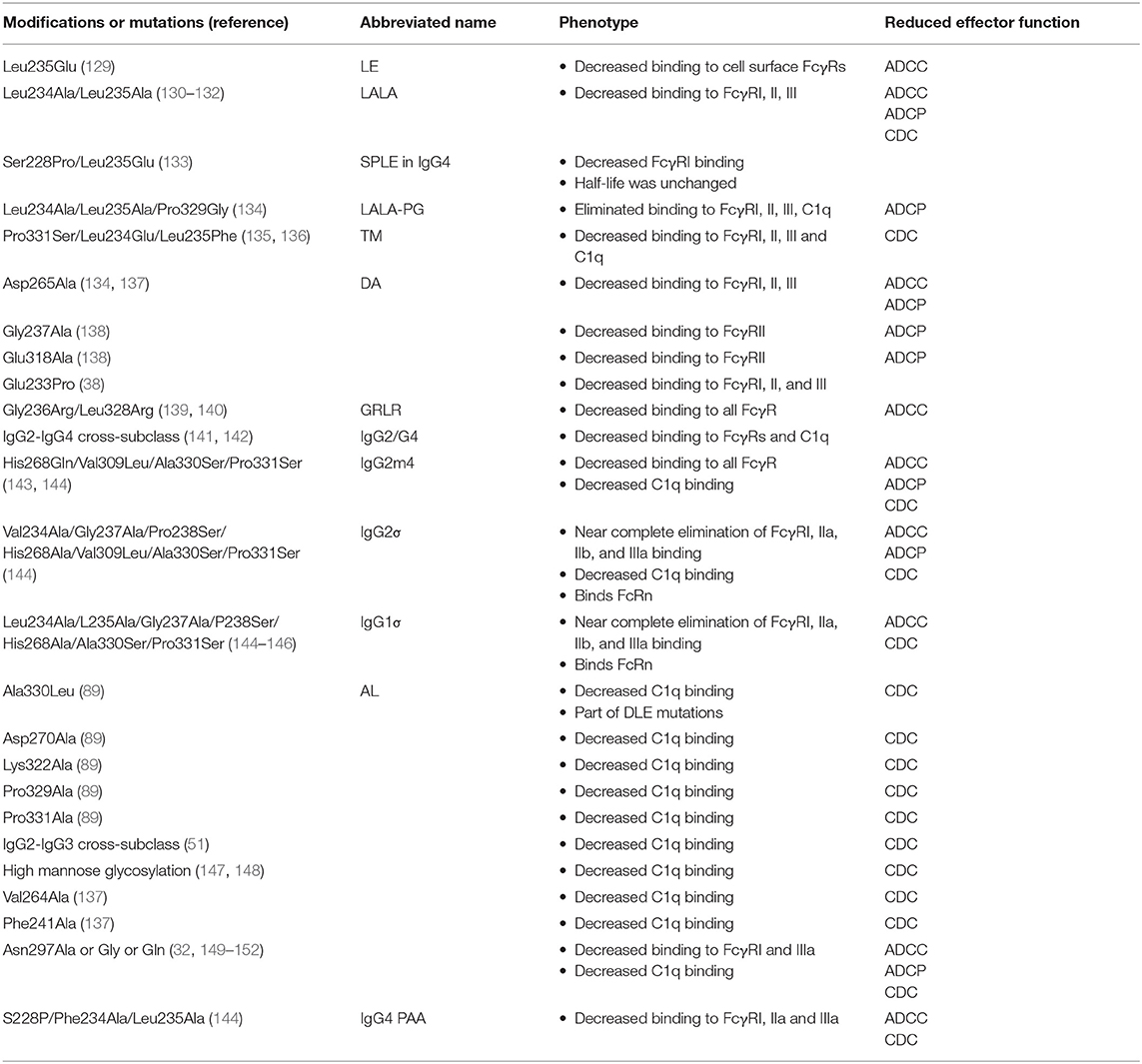
Table 3. Fc modifications to silence antibody effector function.
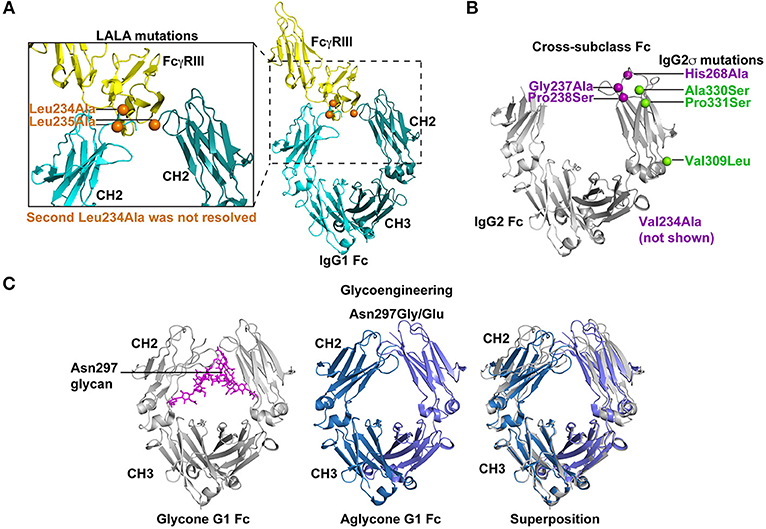
Figure 3. Strategies for silencing antibody effector functions. (A) Point mutations in the Fc have been identified that disrupt antibody effector functions. The elucidation of key amino acids in the interaction of Fc with FcγRs has led to collections of point mutations that can eliminate or drastically reduce Fc binding to specific FcγRs. The Leu234Ala/Leu235Ala (LALA) mutations are perhaps the most commonly used mutations for disrupting antibody effector function (130, 131). As shown in the co-crystal structure (PDB: 1T83) with orange spheres the LALA mutations are proximal to FcγRIII (yellow) when it binds the IgG1 Fc (light and dark teal). These mutations can be combined with other effector function silencing strategies to engineer a Fc that is devoid of any FcγR binding or C1q binding. Leu 234 was only resolved in one of the chains of the Fc region. (B) Effector functions can be disrupted by exchanging amino acids between two Fc molecules from different IgG subclasses. These cross-subclass Fc designs rationally combine mutations that knockdown binding to a given FcγR or complement protein. IgG2m4 and IgG2σ are two examples of engineered Fc regions that were generated by this approach (143, 144). IgG2σ is perhaps the most effector function silent Fc and it combines cross-subclass mutations Val309Leu, Pro331Ser, and Ala330Ser (green spheres) with four additional mutations not naturally found in human Fc sequences (purple spheres) (144). The crystal structure of the IgG2 Fc (gray) encoding these mutations (green and blue spheres) showed the CH2 domains moved farther apart from each other. Also, Asp270 and Pro329, which are essential for binding to FcγR and C1q, were repositioned (PDB:4L4J; 145). Position 234 was not visible in the crystal structure and is not shown in figure. (C) Removal of the Asn297 glycan in the IgG Fc severely reduces Fc binding to FcγRs by inducing a Fc closed conformation. Several Fc designs have removed the N-linked glycosylation site at position 297 by introducing Asn297Gly or Asn297Glu changes (151, 152). The crystal structure of glycosylated IgG1 Fc (gray) is shown with the N297 glycan (magenta; PDB:4BYH). The introduction of a Gly or Glu residue at position 297 produces an aglycone IgG1 Fc (blue and lilac; PDB:3S7G). Superposition of the glycone and aglycone Fc crystal structures shows the lilac and blue CH2 domains in the aglycone are closer in proximity than the gray CH2 domains. The altered CH2 conformation has been hypothesized to be the structural explanation for reduced FcγR binding by the aglycone Fc.
The LALA mutations have provided a foundation for the addition of other mutations or new modifications to Leu235. Building upon the LALA mutant phenotype, Oganesyan et al. mutated Leu234 and Leu235, but also added Pro331Ser to the Fc design to completely abrogate binding between Fc and FcγRs (135). The triple mutant Pro331Ser, Leu234Glu, and Leu235Phe eliminated all FcγR binding (135) without disrupting the overall conformation of the Fc (135). Similarly, Pro329Gly mutation was added to the LALA mutations, which inhibited binding to murine FcγRI, II, and III by IgG2a Fc (134, 156). The amino acid at 329 was changed, because this residue makes contact with Trp108 and Trp131 of FcγRIIIa (61). The LALA-PG was an improvement over LALA mutations alone in that they nullified Fc function in mouse and human IgG (134), whereas LALA alone still retains murine FcγRIII binding to murine IgG2a (157). The significance of the LALA-PG mutations are that observed results in murine models are expected to more accurately translate to humans since the mutations confer a similar phenotype for both murine IgG2a and human IgG1 (134).
The LALA mutations are among the most common point mutations used to disrupt Fc receptor binding, however other sites have been reported to knockout Fc receptor binding. Using a panel of 32 site-directed alanine mutations in IgG Fc Lund and colleagues showed that Gly237 and Glu318 were required for FcγRII binding (131, 138, 158). This lack of binding resulted in poor phagocytosis in vitro (138). Additional alanine scanning mutagenesis experiments determined 9 different amino acid substitutions that resulted in loss of binding to FcγRI, IIa, IIb, and IIIa. Notably, Asp265Ala and Glu233Pro mutations reduced binding to all 4 receptors by >80% (38). Ala318, Ala237, Ala265, and Pro233 represent a collection of mutations that can be used in various combinations to eliminate Fc receptor binding to Fc (134, 137, 138).
Cross-Subclass Fc Designs Eliminate FcγR Binding
To silence the effector functions of Fc, large portions of Fc regions from different subclasses have been exchanged to generate cross-subclass Fc regions (141). These designs aim to silence the Fc effector functions by combining CH domains from different subclasses that lack opposing functions. For example, IgG2 has poor FcγR binding but binds C1q, and IgG4 lacks C1q binding but reacts with FcγRs (142). Hence, combinations of IgG2 and IgG4 CH domains have been constructed that are devoid of both C1q and FcγR binding (141, 142). Typically, in the IgG2/G4 chimeras the hinge and CH1 domain originates from IgG2 and the CH2 and CH3 domains are from IgG4 (141, 142).
Using a different approach to the same concept An and colleagues compared the amino acid sequences of different IgG subclasses and introduced mutations into IgG2 that would completely eliminate FcγR binding. The aim of this approach is to introduce natural amino acids into the Fc so that the Fc would not be immunogenic. The investigators made conservative changes in the IgG2 primary sequence resulting in His268Gln/Val309Leu/Ala330Ser/Pro331Ser mutations (143). This cross-subclass design was termed IgG2m4 and lacked binding to all FcγR (143). The circulating half-life of this antibody was comparable to wildtype IgG in macaques, which suggested the transplantation of IgG4 residues did not make the IgG2 more immunogenic (143).
More recently, Vafa et al. combined many of the mutations that have been discovered over the last 25 years to create an engineered construct called G2σ (Figure 3B). This construct included Val234Ala/Gly237Ala/Pro238Ser/His268Ala/Val309Leu/Ala330Ser/Pro331Ser mutations where many of the mutations were previously established as silencing mutations and the remaining mutations were selected as cross-subclass mutations that introduced IgG4 residues into IgG2 (144). In direct comparisons with IgG1, IgG2, IgG4, and IgG2m4, IgG2σ had the most profound elimination of binding to FcγRI, IIa, and IIIa (144). However, both IgG2m4 and IgG2σ lacked in vitro ADCC activity mediated by human PBMCs effector cells and possessed very little ADCP activity against breast cancer cell lines (144). Given the success of IgG2σ at ablating Fc effector functions, the design was translated to IgG1 and IgG4 (145, 146, 159). The IgG1σ (Leu234Ala/L235Ala/Gly237Ala/P238Ser/His268Ala/Ala330Ser/Pro331Ser), IgG2σ, and an IgG4 Fc encoding S228P/Phe234Ala/Leu235Ala mutations (termed IgG4 PAA) versions of Fc were compared for binding to FcγRs from multiple species (144, 145). IgG1σ and IgG2σ lacked binding to FcγRI and III (145). For FcγRIIa and IIb extremely weak binding could be seen to IgG1σ and IgG2σ at high concentrations of antibody (145). Both IgG1σ and IgG2σ exhibited lower binding to FcγRs than IgG4 PAA (145). IgG4 PAA also showed species-specific differences in binding to FcγRs, whereas IgG1σ and IgG2σ lacked binding for human, macaque, and mouse FcγRs. Therefore, IgG1σ and IgG2σ are among the most effective mutations for knocking out Fc effector function.
Mimoto et al. sought to use FcγRIIb as a way to capture immune complexes on the surface of FcγRIIb-expressing B cells (160). Thus, they engineered the Fc to selectively bind to FcγRIIb with a 200-fold increase in affinity, and a 10-fold lower affinity for the other FcγRs (160). The improved affinity for FcγRIIb conferred the desired boost in B cell presentation of peptides to T cells in vitro (160). Since FcγRIIb is an inhibitory receptor these mutations could be used to silence effector function by changing the ratio of Fc binding to activating vs. inhibitory receptors.
Ablation of C1q Binding to Reduce Complement Dependent Cytotoxicity (CDC)
Point Mutations to Ablate Complement Binding
Inducing the complement cascade has been associated with antibody injection site adverse reactions (161, 162). Therefore, eliminating C1q binding to Fc—the initial event in the activation of antibody-dependent complement cytotoxicity (163)—has been a goal of Fc optimization. One of the benefits of the mutations engineered to eliminate FcγR binding is that many of them eliminate C1q binding too. In a structure-guided screen of Fc mutations, an Ala330Leu mutation was observed to decrease C1q binding (39). As stated above this mutation also eliminated FcγRIIb binding (39). In in vitro assays the Ala330Leu mutation reduced the ability of the antibody to mediate complement-dependent cytotoxicity (CDC) of target cells, presumably because Ala330Leu disrupted C1q binding to Fc. However, not all amino acids introduced at position 330 disrupted C1q binding, thus the effect was specific to the introduction of only certain amino acids at position 330 (39). It should be noted that Ala330Leu is one of the mutations in the set of mutations comprising the DLE mutations (Ser239Asp Ile332Glu Ala330Leu) that improve FcγR-mediated effector functions. The DLE optimization eliminates CDC activity mediated by the antibody, which can be rescued by removing the Ala330Leu mutation from the set. In instances where CDC causes adverse reactions to antibody administration the DLE mutations may be able to enhance FcγR-mediated effector functions and eliminate injection site reactions (39).
Additional amino acids that reduce C1q binding were identified by an alanine scan of the Fc. Asp270, Lys322, Pro329, and Pro331 were all implicated as sites in IgG that confer binding to C1q (89). Among these amino acid positions Asp270Ala and Pro329Ala, showed the most pronounced deficiency in complement activation and C1q binding across multiple concentrations of serum C1q (89). The Leu234Glu/Leu235Phe/Pro331Ser triple mutant Fc lacks binding to FcγRs, but also these mutations eliminate Fc binding to C1q (135). Similarly, the creation of IgG2m4 not only eliminates FcγR binding but also eliminates C1q binding (143). Vafa et al. examined the CDC activity of IgG2m4 (His268Gln/Val309Leu/Ala330Ser/Pro331Ser) and IgG2σ (Val234Ala/Gly237Ala/Pro238Ser/His268Ala/Val309Leu/Ala330Ser/Pro331Ser) formats of Rituxan (144). Neither antibody format conferred CDC against a lymphoma cell line using human serum complement (144) showing both are potential designs for eliminating complement-mediated functions. The structure of IgG2σ Fc was solved to 1.9 angstroms and showed that it is in a more open conformation meaning the CH2 domains of the Fc are spaced relatively far apart (144). Moreover, the loop containing Leu328 is repositioned compared to wildtype IgG2 Fc. Thus, it is postulated that the change in conformation results in reorientation of Asp270 and Pro329, which eliminates FcγR and C1q binding to IgG2σ (144).
Cross-Subclass Fc Regions to Ablate Complement Activation
Fundamental knowledge of how each IgG subclass interacts with complement allows for fine tuning of CDC. For example, IgG2 can have moderate to low CDC activity (51). Thus to reduce IgG3 CDC activity the CH2 domain of IgG2 can be used to replace the CH2 domain of IgG3 (51). Similarly, to ablate IgG1 CDC activity a Pro331Ser mutation was introduced based on the fact that IgG4 has Ser331 and lacks CDC activity (136). These mutations are an example of how basic science can be applied to the design of antibody-based biologics. Also many of the point mutations made to knockout C1q binding are cross-subclass mutations (Table 3).
Glycoengineering to Ablate FcγR and C1q Binding
The Fc of IgG contains an N-linked glycosylation site at position 297 [Figure 3C; (72)]. Typically the glycan present at N297 is a complex biantennary glycan (164, 165). The modification of this glycan to high mannose glycan reduced the affinity of IgG1 Fc for C1q, which in turn reduced CDC activity (147, 148). Inhibiting the incorporation of galactose or sialic acid into carbohydrate synthesis did not dramatically silence immune effector functions of the antibody Fc (147). While glycans devoid of galactose or sialic acid appear to function normally, mutations in the Fc that knockout C1q and FcγRI binding can also lead to an increase in galactosylation and sialylation of the Asn297 glycan eliminate (137). Galactosylation and sialylation-increasing mutations include Phe241Ala, Val264Ala, and Asp265Ala mutations (137). Whether the change in glycosylation profile has a role in reducing C1q binding or is an unrelated bystander effect is not clear. In one study, sialylation of the Fc reduced C1q binding 4-fold suggesting hypersialylation could directly impair CDC responses (101). However, hypersialylation reduces terminal galactosylation complicating which factor contributes to the reduced C1q binding (101).
Another common method to eliminate Fc effector function has been to completely remove the glycosylation site by substituting alanine, glutamine, or glycine at position 297 (32, 149–152). The removal of the glycosylation site dramatically reduced IgG1 binding to FcγRI and C1q [Figure 3C; (137, 152, 166)]. In the context of IgG3, the Fc lacking glycosylation—the aglycone Fc—has reduced binding to FcγRI and C1q. In vitro, the aglycone IgG3 Fc loses the ability to mediate ADCC via FcγRIIIa (151, 152). However, removal of Asn297 glycan reduces, but does not eliminate, binding to mouse Fc receptors (157). Additionally, it is thought that avidity can overcome the low binding affinity to FcγRI conferred by mutating Asn297 (144). Therefore, on monocytes and macrophages where FcγRI is expressed at a high density the Asn297Gly mutation may not be sufficient to eliminate all binding. Lo et al. combined the Asn297Gly mutation with Asp265Ala mutation to further reduce FcγR binding to Fc. As stated above each of these mutations reduces FcγR and C1q binding individually and when combined showed a further reduction in Fc binding to FcγRs and C1q (134). Combinations such as the Asn297Gly/Asp265Ala are useful for near complete knockout of binding between Fc and FcγRs or C1q.
The mechanism behind the aglycone reducing Fc binding to C1q and FcγRs is not fully understood. The aglycone Fc is more susceptible to protease cleavage, which suggests the structure of the glycone Fc differs from that of the aglycone (152). Nuclear magnetic resonance studies have similarly suggested structural perturbations are present in the aglycone Fc (32, 73, 151). The clearest evidence for structural changes in the agylcosylated Fc was provided by the crystal structure of mouse Fc without the N297 glycan (167). The CH3 domains appeared identical, but the CH2 domains of aglycone Fc and glycosylated Fc differed in position by 10 to 14 angstroms (167). In the aglycone Fc the CH2 domains were closer together than in the glycosylated Fc, thus the aglycone had a “closed” conformation. This closed conformation was not unique to mouse Fc. Crystal structures of both aglycosylated human IgG1 Fc (168) and IgG4 Fc (169) have shown the CH2 domains of a single Fc to undergo rigid-body movements of 10–20 angstroms to be closer in proximity to each other when aglycosylated (Figure 3C). The closed conformation of the aglycone Fc is mediated, at least in part, by the perturbed C'E loop (149, 150, 167, 168, 170). In total, eliminating Fc glycosylation induces a closed conformation that confers silencing of the Fc effector functions.
Conclusions
Many approaches including phage display, alanine scanning mutations, and structure-based design have all been successful in optimizing the Fc of antibody-based biologics (38, 39, 113). Underlying the optimization of the Fc is modulating its ability to bind to Fc receptors, C1q, and FcRn. These interactions can be modulated by the introduction of point mutations, inserting or deleting amino acids, modifying glycan composition, or appending protein domains (171). Overall, strengthening or disrupting Fc interactions with its binding partners as measured by in vitro affinity has translated to the desired outcome in vivo. The optimization of Fc for specific functions not only can improve in vivo functions, but it also provides a means to dissect the importance of specific Fc receptors (172) and downstream CDC, ADCC, phagocytosis, or circulation half-life in treating specific diseases (119, 173). While antibody-based biologics have been successful in the treatment of disease, new opportunities exist for antibody biologics as durable prevention strategies for infectious diseases (174–176). The antibody-mediated prevention (AMP) study will test the efficacy of monoclonal antibody passive infusion to prevent HIV-1 infection in 2,700 participants (NCT02716675). This phase 2b trial could provide the first proof-of-concept that neutralizing antibodies can provide protection from HIV-1 infection in humans. The VRC01 antibody that will be used contains a wildtype Fc; however, the next generation of HIV-1 antibody protection studies will likely include combinations of Fc-optimized HIV-1 neutralizing antibodies with prolonged circulation half-life (116), since longer half-life was important for protection in preclinical nonhuman primate studies (119, 122, 123). The ability to optimize the Fc region of antibodies continues to be a powerful approach for combating heritable diseases, infectious diseases, and cancer.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
KS is supported by NIAID extramural project grant R01-AI120801.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Fonseca MHG, Furtado GP, Bezerra MRL, Pontes LQ, Fernandes CFC. Boosting half-life and effector functions of therapeutic antibodies by Fc-engineering: an interaction-function review. Int J Biol Macromol. (2018) 119:306–11. doi: 10.1016/j.ijbiomac.2018.07.141
2. Brezski RJ, Georgiou G. Immunoglobulin isotype knowledge and application to Fc engineering. Curr Opin Immunol. (2016) 40:62–9. doi: 10.1016/j.coi.2016.03.002
3. Reichert JM. Antibodies to watch in 2017. MAbs. (2017) 9:167–81. doi: 10.1080/19420862.2016.1269580
4. Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov. (2018) 17:197–223. doi: 10.1038/nrd.2017.227
5. Gura T. Therapeutic antibodies: magic bullets hit the target. Nature. (2002) 417:584–6. doi: 10.1038/417584a
6. Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol. (2005) 23:1073–8. doi: 10.1038/nbt0905-1073
7. Kaplon H, Reichert JM.Antibodies to watch in 2019. MAbs. (2018) 11:219–38. doi: 10.1080/19420862.2018.1556465
8. Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs. (2015) 7:9–14. doi: 10.4161/19420862.2015.989042
9. Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. (2006) 124:823–35. doi: 10.1016/j.cell.2006.02.016
10. Sim F, Leidner R, Bell RB. Immunotherapy for head and neck cancer. Oral Maxillofac Surg Clin North Am. (2019) 31:85–100. doi: 10.1016/j.coms.2018.09.002
11. Strohl WR. Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr Opin Biotechnol. (2009) 20:685–91. doi: 10.1016/j.copbio.2009.10.011
12. Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol. (2006) 6:343–57. doi: 10.1038/nri1837
13. Padlan EA. Anatomy of the antibody molecule. Mol Immunol. (1994) 31:169–217. doi: 10.1016/0161-5890(94)90001-9
14. Huber R, Deisenhofer J, Colman PM, Matsushima M, Palm W. Crystallographic structure studies of an IgG molecule and an Fc fragment. Nature. (1976) 264:415–20. doi: 10.1038/264415a0
15. Nisonoff A, Wissler FC, Lipman LN, Woernley DL. Separation of univalent fragments from the bivalent rabbit antibody molecule by reduction of disulfide bonds. Arch Biochem Biophys. (1960) 89:230–44. doi: 10.1016/0003-9861(60)90049-7
17. Harris LJ, Larson SB, Hasel KW, Day J, Greenwood A, Mcpherson A. The three-dimensional structure of an intact monoclonal antibody for canine lymphoma. Nature. (1992) 360:369–72. doi: 10.1038/360369a0
18. Silverton EW, Navia MA, Davies DR. Three-dimensional structure of an intact human immunoglobulin. Proc Natl Acad Sci USA. (1977) 74:5140–4. doi: 10.1073/pnas.74.11.5140
19. Kim H, Matsunaga C, Yoshino A, Kato K, Arata Y. Dynamical structure of the hinge region of immunoglobulin G as studied by 13C nuclear magnetic resonance spectroscopy. J Mol Biol. (1994) 236:300–9. doi: 10.1006/jmbi.1994.1136
20. Dreyer WJ, Bennett JC. The molecular basis of antibody formation: a paradox. Proc Natl Acad Sci USA. (1965) 54:864–9. doi: 10.1073/pnas.54.3.864
21. Brack C, Hirama M, Lenhard-Schuller R, Tonegawa S. A complete immunoglobulin gene is created by somatic recombination. Cell. (1978) 15:1–14. doi: 10.1016/0092-8674(78)90078-8
22. Kataoka T, Kawakami T, Takahashi N, Honjo T. Rearrangement of immunoglobulin gamma 1-chain gene and mechanism for heavy-chain class switch. Proc Natl Acad Sci USA. (1980) 77:919–23. doi: 10.1073/pnas.77.2.919
23. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. (2014) 5:520. doi: 10.3389/fimmu.2014.00520
24. Jefferis R, Kumararatne DS. Selective IgG subclass deficiency: quantification and clinical relevance. Clin Exp Immunol. (1990) 81:357–67. doi: 10.1111/j.1365–2249.1990.tb05339.x
25. Barrett DJ, Ayoub EM. IgG2 subclass restriction of antibody to pneumococcal polysaccharides. Clin Exp Immunol. (1986) 63:127–34.
26. Woof JM, Burton DR. Human antibody-Fc receptor interactions illuminated by crystal structures. Nat Rev Immunol. (2004) 4:89–99. doi: 10.1038/nri1266
27. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. doi: 10.1038/nri2206
28. Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig MC, Ravetch JV. A 13-amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature. (1994) 369:340. doi: 10.1038/369340a0
29. Amigorena S, Bonnerot C, Drake JR, Choquet D, Hunziker W, Guillet JG, et al. Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B lymphocytes. Science. (1992) 256:1808–12. doi: 10.1126/science.1535455
30. Koene HR, Kleijer M, Algra J, Roos D, Von Dem Borne AE, De Haas M. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood. (1997) 90:1109–14. doi: 10.1016/S0165-2478(97)85823-3
31. Presta LG. Molecular engineering and design of therapeutic antibodies. Curr Opin Immunol. (2008) 20:460–70. doi: 10.1016/j.coi.2008.06.012
32. Dwek RA, Lellouch AC, Wormald MR. Glycobiology: ‘the function of sugar in the IgG molecule’. J Anat. (1995) 187 (Pt 2):279–92.
33. Morell A, Terry WD, Waldmann TA. Metabolic properties of IgG subclasses in man. J Clin Invest. (1970) 49:673–80. doi: 10.1172/JCI106279
34. Fritsche R, Spiegelberg HL. Fc receptors for IgE on normal rat lymphocytes. J Immunol. (1978) 121:471–8.
35. Lum LG, Muchmore AV, Keren D, Decker J, Koski I, Strober W, et al. A receptor for IgA on human T lymphocytes. J Immunol. (1979) 122:65–9.
36. Kubagawa H, Oka S, Kubagawa Y, Torii I, Takayama E, Kang DW, et al. Identity of the elusive IgM Fc receptor (FcmuR) in humans. J Exp Med. (2009) 206:2779–93. doi: 10.1084/jem.20091107
37. Presta LG. Engineering antibodies for therapy. Curr Pharm Biotechnol. (2002) 3:237–56. doi: 10.2174/1389201023378256
38. Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. (2001) 276:6591–604. doi: 10.1074/jbc.M009483200
39. Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci USA. (2006) 103:4005–10. doi: 10.1073/pnas.0508123103
40. Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther. (2008) 7:2517–27. doi: 10.1158/1535–7163.MCT-08-0201
41. Smith P, Dilillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcgamma receptor structural and functional diversity. Proc Natl Acad Sci USA. (2012) 109:6181–6. doi: 10.1073/pnas.1203954109
42. Ahmed AA, Keremane SR, Vielmetter J, Bjorkman PJ. Structural characterization of GASDALIE Fc bound to the activating Fc receptor FcgammaRIIIa. J Struct Biol. (2016) 194:78–89. doi: 10.1016/j.jsb.2016.02.001
43. Bournazos S, Klein F, Pietzsch J, Seaman MS, Nussenzweig MC, Ravetch JV. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell. (2014) 158:1243–53. doi: 10.1016/j.cell.2014.08.023
44. Mimoto F, Igawa T, Kuramochi T, Katada H, Kadono S, Kamikawa T, et al. Novel asymmetrically engineered antibody Fc variant with superior FcgammaR binding affinity and specificity compared with afucosylated Fc variant. MAbs. (2013) 5:229–36. doi: 10.4161/mabs.23452
45. Stavenhagen JB, Gorlatov S, Tuaillon N, Rankin CT, Li H, Burke S, et al. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcgamma receptors. Cancer Res. (2007) 67:8882–90. doi: 10.1158/0008–5472.CAN-07-0696
46. Idusogie EE, Wong PY, Presta LG, Gazzano-Santoro H, Totpal K, Ultsch M, et al. Engineered antibodies with increased activity to recruit complement. J Immunol. (2001) 166:2571–5. doi: 10.4049/jimmunol.166.4.2571
47. Dall'acqua WF, Cook KE, Damschroder MM, Woods RM, Wu H. Modulation of the effector functions of a human IgG1 through engineering of its hinge region. J Immunol. (2006) 177:1129–38. doi: 10.4049/jimmunol.177.2.1129
48. Moore GL, Chen H, Karki S, Lazar GA. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. MAbs. (2010) 2:181–9. doi: 10.4161/mabs.2.2.11158
49. Diebolder CA, Beurskens FJ, De Jong RN, Koning RI, Strumane K, Lindorfer MA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science. (2014) 343:1260–3. doi: 10.1126/science.1248943
50. Natsume A, In M, Takamura H, Nakagawa T, Shimizu Y, Kitajima K, et al. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res. (2008) 68:3863–72. doi: 10.1158/0008–5472.CAN-07-6297
51. Sensel MG, Kane LM, Morrison SL. Amino acid differences in the N-terminus of C(H)2 influence the relative abilities of IgG2 and IgG3 to activate complement. Mol Immunol. (1997) 34:1019–29. doi: 10.1016/S0161-5890(97)00112-0
52. Chintalacharuvu KR, Vuong LU, Loi LA, Larrick JW, Morrison SL. Hybrid IgA2/IgG1 antibodies with tailor-made effector functions. Clin Immunol. (2001) 101:21–31. doi: 10.1006/clim.2001.5083
53. Borrok MJ, Luheshi NM, Beyaz N, Davies GC, Legg JW, Wu H, et al. Enhancement of antibody-dependent cell-mediated cytotoxicity by endowing IgG with FcalphaRI (CD89) binding. MAbs. (2015) 7:743–51. doi: 10.1080/19420862.2015.1047570
54. Kelton W, Mehta N, Charab W, Lee J, Lee CH, Kojima T, et al. IgGA: a “cross-isotype” engineered human Fc antibody domain that displays both IgG-like and IgA-like effector functions. Chem Biol. (2014) 21:1603–9. doi: 10.1016/j.chembiol.2014.10.017
55. Czajkowsky DM, Andersen JT, Fuchs A, Wilson TJ, Mekhaiel D, Colonna M, et al. Developing the IVIG biomimetic, hexa-Fc, for drug and vaccine applications. Sci Rep. (2015) 5:9526. doi: 10.1038/srep09526
56. Peschke B, Keller CW, Weber P, Quast I, Lunemann JD. Fc-galactosylation of human immunoglobulin gamma isotypes improves C1q binding and enhances complement-dependent cytotoxicity. Front Immunol. (2017) 8:646. doi: 10.3389/fimmu.2017.00646
57. Dekkers G, Treffers L, Plomp R, Bentlage AEH, De Boer M, Koeleman CaM, et al. Decoding the human immunoglobulin G-Glycan repertoire reveals a spectrum of Fc-receptor- and complement-mediated-effector activities. Front Immunol. (2017) 8:877. doi: 10.3389/fimmu.2017.00877
58. Umana P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat Biotechnol. (1999) 17:176–80. doi: 10.1038/6179
59. Davies J, Jiang L, Pan LZ, Labarre MJ, Anderson D, Reff M. Expression of GnTIII in a recombinant anti-CD20 CHO production cell line: expression of antibodies with altered glycoforms leads to an increase in ADCC through higher affinity for FC gamma RIII. Biotechnol Bioeng. (2001) 74:288–94. doi: 10.1002/bit.1119
60. Shields RL, Lai J, Keck R, O'connell LY, Hong K, Meng YG, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. (2002) 277:26733–40. doi: 10.1074/jbc.M202069200
61. Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature. (2000) 406:267–73. doi: 10.1038/35018508
62. Erbe DV, Pfefferkorn ER, Fanger MW. Functions of the various IgG Fc receptors in mediating killing of Toxoplasma gondii. J Immunol. (1991) 146:3145–51.
63. Shen L, Graziano RF, Fanger MW. The functional properties of Fc gamma RI, II and III on myeloid cells: a comparative study of killing of erythrocytes and tumor cells mediated through the different Fc receptors. Mol Immunol. (1989) 26:959–69. doi: 10.1016/0161-5890(89)90114-4
64. Dall'acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem. (2006) 281:23514–24. doi: 10.1074/jbc.M604292200
65. Romain G, Senyukov V, Rey-Villamizar N, Merouane A, Kelton W, Liadi I, et al. Antibody Fc engineering improves frequency and promotes kinetic boosting of serial killing mediated by NK cells. Blood. (2014) 124:3241–9. doi: 10.1182/blood-2014-04-569061
66. Oganesyan V, Damschroder MM, Leach W, Wu H, Dall'acqua WF. Structural characterization of a mutated, ADCC-enhanced human Fc fragment. Mol Immunol. (2008) 45:1872–82. doi: 10.1016/j.molimm.2007.10.042
67. Jung ST, Kelton W, Kang TH, Ng DT, Andersen JT, Sandlie I, et al. Effective phagocytosis of low Her2 tumor cell lines with engineered, aglycosylated IgG displaying high FcgammaRIIa affinity and selectivity. ACS Chem Biol. (2013) 8:368–75. doi: 10.1021/cb300455f
68. Rankin CT, Veri MC, Gorlatov S, Tuaillon N, Burke S, Huang L, et al. CD32B, the human inhibitory Fc-gamma receptor IIB, as a target for monoclonal antibody therapy of B-cell lymphoma. Blood. (2006) 108:2384–91. doi: 10.1182/blood-2006-05-020602
69. Nordstrom JL, Gorlatov S, Zhang W, Yang Y, Huang L, Burke S, et al. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcgamma receptor binding properties. Breast Cancer Res. (2011) 13:R123. doi: 10.1186/bcr3069
70. Bang YJ, Giaccone G, Im SA, Oh DY, Bauer TM, Nordstrom JL, et al. First-in-human phase 1 study of margetuximab (MGAH22), an Fc-modified chimeric monoclonal antibody, in patients with HER2-positive advanced solid tumors. Ann Oncol. (2017) 28:855–861. doi: 10.1093/annonc/mdx002
71. Liu L. Antibody glycosylation and its impact on the pharmacokinetics and pharmacodynamics of monoclonal antibodies and Fc-fusion proteins. J Pharm Sci. (2015) 104:1866–84. doi: 10.1002/jps.24444
72. Jefferis R. Glycosylation of antibody therapeutics: optimisation for purpose. Methods Mol Biol. (2009) 483:223–38. doi: 10.1007/978-1-59745-407-0_13
73. Yamaguchi Y, Nishimura M, Nagano M, Yagi H, Sasakawa H, Uchida K, et al. Glycoform-dependent conformational alteration of the Fc region of human immunoglobulin G1 as revealed by NMR spectroscopy. Biochim Biophys Acta. (2006) 1760:693–700. doi: 10.1016/j.bbagen.2005.10.002
74. Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. (2003) 278:3466–73. doi: 10.1074/jbc.M210665200
75. Niwa R, Hatanaka S, Shoji-Hosaka E, Sakurada M, Kobayashi Y, Uehara A, et al. Enhancement of the antibody-dependent cellular cytotoxicity of low-fucose IgG1 is independent of FcgammaRIIIa functional polymorphism. Clin Cancer Res. (2004) 10:6248–55. doi: 10.1158/1078–0432.CCR-04-0850
76. Ferrara C, Stuart F, Sondermann P, Brunker P, Umana P. The carbohydrate at FcgammaRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J Biol Chem. (2006) 281:5032–6. doi: 10.1074/jbc.M510171200
77. Ferrara C, Grau S, Jager C, Sondermann P, Brunker P, Waldhauer I, et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc Natl Acad Sci USA. (2011) 108:12669–74. doi: 10.1073/pnas.1108455108
78. Beck A, Reichert JM. Marketing approval of mogamulizumab: a triumph for glyco-engineering. MAbs. (2012) 4:419–25. doi: 10.4161/mabs.20996
79. Ishii T, Ishida T, Utsunomiya A, Inagaki A, Yano H, Komatsu H, et al. Defucosylated humanized anti-CCR4 monoclonal antibody KW-0761 as a novel immunotherapeutic agent for adult T-cell leukemia/lymphoma. Clin Cancer Res. (2010) 16:1520–31. doi: 10.1158/1078–0432.CCR-09-2697
80. Ito A, Ishida T, Yano H, Inagaki A, Suzuki S, Sato F, et al. Defucosylated anti-CCR4 monoclonal antibody exercises potent ADCC-mediated antitumor effect in the novel tumor-bearing humanized NOD/Shi-scid, IL-2Rgamma(null) mouse model. Cancer Immunol Immunother. (2009) 58:1195–206. doi: 10.1007/s00262-008-0632-0
81. Braster R, O'toole T, Van Egmond M. Myeloid cells as effector cells for monoclonal antibody therapy of cancer. Methods. (2014) 65:28–37. doi: 10.1016/j.ymeth.2013.06.020
82. Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res. (2011) 71:2411–6. doi: 10.1158/0008–5472.CAN-10-2583
83. Jain A, Olsen HS, Vyzasatya R, Burch E, Sakoda Y, Merigeon EY, et al. Fully recombinant IgG2a Fc multimers (stradomers) effectively treat collagen-induced arthritis and prevent idiopathic thrombocytopenic purpura in mice. Arthritis Res Ther. (2012) 14:R192. doi: 10.1186/ar4024
84. Niknami M, Wang MX, Nguyen T, Pollard JD. Beneficial effect of a multimerized immunoglobulin Fc in an animal model of inflammatory neuropathy (experimental autoimmune neuritis). J Peripher Nerv Syst. (2013) 18:141–52. doi: 10.1111/jns5.12022
85. Blundell PA, Le NPL, Allen J, Watanabe Y, Pleass RJ. Engineering the fragment crystallizable (Fc) region of human IgG1 multimers and monomers to fine-tune interactions with sialic acid-dependent receptors. J Biol Chem. (2017) 292:12994–3007. doi: 10.1074/jbc.M117.795047
86. Mekhaiel DN, Czajkowsky DM, Andersen JT, Shi J, El-Faham M, Doenhoff M, et al. Polymeric human Fc-fusion proteins with modified effector functions. Sci Rep. (2011) 1:124. doi: 10.1038/srep00124
87. Thiruppathi M, Sheng JR, Li L, Prabhakar BS, Meriggioli MN. Recombinant IgG2a Fc (M045) multimers effectively suppress experimental autoimmune myasthenia gravis. J Autoimmun. (2014) 52:64–73. doi: 10.1016/j.jaut.2013.12.014
88. Hughes-Jones NC, Gardner B. Reaction between the isolated globular sub-units of the complement component C1q and IgG-complexes. Mol Immunol. (1979) 16:697–701. doi: 10.1016/0161-5890(79)90010-5
89. Idusogie EE, Presta LG, Gazzano-Santoro H, Totpal K, Wong PY, Ultsch M, et al. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J Immunol. (2000) 164:4178–84. doi: 10.4049/jimmunol.164.8.4178
90. Duncan AR, Winter G. The binding site for C1q on IgG. Nature. (1988) 332:738–40. doi: 10.1038/332738a0
91. Huck S, Fort P, Crawford DH, Lefranc MP, Lefranc G. Sequence of a human immunoglobulin gamma 3 heavy chain constant region gene: comparison with the other human C gamma genes. Nucleic Acids Res. (1986) 14:1779–89. doi: 10.1093/nar/14.4.1779
92. Michaelsen TE, Aase A, Westby C, Sandlie I. Enhancement of complement activation and cytolysis of human IgG3 by deletion of hinge exons. Scand J Immunol. (1990) 32:517–28. doi: 10.1111/j.1365–3083.1990.tb03192.x
93. Giuntini S, Granoff DM, Beernink PT, Ihle O, Bratlie D, Michaelsen TE. Human IgG1, IgG3, and IgG3 hinge-truncated mutants show different protection capabilities against meningococci depending on the target antigen and epitope specificity. Clin Vaccine Immunol. (2016) 23:698–706. doi: 10.1128/CVI.00193-16
94. Bruggemann M, Williams GT, Bindon CI, Clark MR, Walker MR, Jefferis R, et al. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med. (1987) 166:1351–61. doi: 10.1084/jem.166.5.1351
95. Natsume A, Shimizu-Yokoyama Y, Satoh M, Shitara K, Niwa R. Engineered anti-CD20 antibodies with enhanced complement-activating capacity mediate potent anti-lymphoma activity. Cancer Sci. (2009) 100:2411–8. doi: 10.1111/j.1349–7006.2009.01327.x
96. Thommesen JE, Michaelsen TE, Loset GA, Sandlie I, Brekke OH. Lysine 322 in the human IgG3 C(H)2 domain is crucial for antibody dependent complement activation. Mol Immunol. (2000) 37:995–1004. doi: 10.1016/S0161-5890(01)00010-4
97. Gaboriaud C, Thielens NM, Gregory LA, Rossi V, Fontecilla-Camps JC, Arlaud GJ. Structure and activation of the C1 complex of complement: unraveling the puzzle. Trends Immunol. (2004) 25:368–73. doi: 10.1016/j.it.2004.04.008
98. Aoyama M, Hashii N, Tsukimura W, Osumi K, Harazono A, Tada M, et al. Effects of terminal galactose residues in mannose alpha1-6 arm of Fc-glycan on the effector functions of therapeutic monoclonal antibodies. MAbs. (2019) doi: 10.1080/19420862.2019.1608143. [Epub ahead of print].
99. Wada R, Matsui M, Kawasaki N. Influence of N-glycosylation on effector functions and thermal stability of glycoengineered IgG1 monoclonal antibody with homogeneous glycoforms. MAbs. (2019) 11:350–72. doi: 10.1080/19420862.2018.1551044
100. Raju TS. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr Opin Immunol. (2008) 20:471–8. doi: 10.1016/j.coi.2008.06.007
101. Quast I, Keller CW, Maurer MA, Giddens JP, Tackenberg B, Wang LX, et al. Sialylation of IgG Fc domain impairs complement-dependent cytotoxicity. J Clin Invest. (2015) 125:4160–70. doi: 10.1172/JCI82695
102. Sockolosky JT, Szoka FC. The neonatal Fc receptor, FcRn, as a target for drug delivery and therapy. Adv Drug Deliv Rev. (2015) 91:109–24. doi: 10.1016/j.addr.2015.02.005
103. Oganesyan V, Damschroder MM, Cook KE, Li Q, Gao C, Wu H, et al. Structural insights into neonatal Fc receptor-based recycling mechanisms. J Biol Chem. (2014) 289:7812–24. doi: 10.1074/jbc.M113.537563
104. Burmeister WP, Huber AH, Bjorkman PJ. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature. (1994) 372:379–83. doi: 10.1038/372379a0
105. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. (2007) 7:715–25. doi: 10.1038/nri2155
106. Ober RJ, Martinez C, Vaccaro C, Zhou J, Ward ES. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. J Immunol. (2004) 172:2021–9. doi: 10.4049/jimmunol.172.4.2021
107. Rodewald R. pH-dependent binding of immunoglobulins to intestinal cells of the neonatal rat. J Cell Biol. (1976) 71:666–9. doi: 10.1083/jcb.71.2.666
108. Raghavan M, Bonagura VR, Morrison SL, Bjorkman PJ. Analysis of the pH dependence of the neonatal Fc receptor/immunoglobulin G interaction using antibody and receptor variants. Biochemistry. (1995) 34:14649–57. doi: 10.1021/bi00045a005
109. Martin WL, West AP Jr, Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell. (2001) 7:867–77. doi: 10.1016/S1097-2765(01)00230-1
110. Stapleton NM, Andersen JT, Stemerding AM, Bjarnarson SP, Verheul RC, Gerritsen J, et al. Competition for FcRn-mediated transport gives rise to short half-life of human IgG3 and offers therapeutic potential. Nat Commun. (2011) 2:599. doi: 10.1038/ncomms1608
111. Dall'acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, et al. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol. (2002) 169:5171–80. doi: 10.4049/jimmunol.169.9.5171
112. Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, et al. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. (2010) 28:157–9. doi: 10.1038/nbt.1601
113. Ghetie V, Popov S, Borvak J, Radu C, Matesoi D, Medesan C, et al. Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat Biotechnol. (1997) 15:637–40. doi: 10.1038/nbt0797-637
114. Monnet C, Jorieux S, Souyris N, Zaki O, Jacquet A, Fournier N, et al. Combined glyco- and protein-Fc engineering simultaneously enhance cytotoxicity and half-life of a therapeutic antibody. MAbs. (2014) 6:422–36. doi: 10.4161/mabs.27854
115. Bas M, Terrier A, Jacque E, Dehenne A, Pochet-Beghin V, Beghin C, et al. Fc sialylation prolongs serum half-life of therapeutic antibodies. J Immunol. (2019) 202:1582–94. doi: 10.4049/jimmunol.1800896
116. Mendoza P, Gruell H, Nogueira L, Pai JA, Butler AL, Millard K, et al. Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature. (2018) 561:479–84. doi: 10.1038/s41586-018-0531-2
117. Yu XQ, Robbie GJ, Wu Y, Esser MT, Jensen K, Schwartz HI, et al. Safety, tolerability, and pharmacokinetics of MEDI4893, an investigational, extended-half-life, anti-staphylococcus aureus alpha-toxin human monoclonal antibody, in healthy adults. Antimicrob Agents Chemother. (2017) 61:e01020-16. doi: 10.1128/AAC.01020-16
118. Robbie GJ, Criste R, Dall'acqua WF, Jensen K, Patel NK, Losonsky GA, et al. A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob Agents Chemother. (2013) 57:6147–53. doi: 10.1128/AAC.01285-13
119. Ko SY, Pegu A, Rudicell RS, Yang ZY, Joyce MG, Chen X, et al. Enhanced neonatal Fc receptor function improves protection against primate SHIV infection. Nature. (2014) 514:642–5. doi: 10.1038/nature13612
120. Nnane IP, Han C, Jiao Q, Tam SH, Davis HM, Xu Z. Modification of the Fc region of a human anti-oncostatin M monoclonal antibody for higher affinity to FcRn receptor and extension of half-life in cynomolgus monkeys. Basic Clin Pharmacol Toxicol. (2017) 121:13–21. doi: 10.1111/bcpt.12761
121. Shen Y, Li H, Zhao L, Li G, Chen B, Guo Q, et al. Increased half-life and enhanced potency of Fc-modified human PCSK9 monoclonal antibodies in primates. PLoS ONE. (2017) 12:e0183326. doi: 10.1371/journal.pone.0183326
122. Saunders KO, Pegu A, Georgiev IS, Zeng M, Joyce MG, Yang ZY, et al. Sustained delivery of a broadly neutralizing antibody in nonhuman primates confers long-term protection against simian/human immunodeficiency virus infection. J Virol. (2015) 89:5895–903. doi: 10.1128/JVI.00210-15
123. Gautam R, Nishimura Y, Gaughan N, Gazumyan A, Schoofs T, Buckler-White A, et al. A single injection of crystallizable fragment domain-modified antibodies elicits durable protection from SHIV infection. Nat Med. (2018) 24:610–6. doi: 10.1038/s41591-018-0001-2
124. West APJr, Bjorkman PJ. Crystal structure and immunoglobulin G binding properties of the human major histocompatibility complex-related Fc receptor(,). Biochemistry. (2000) 39:9698–708. doi: 10.1021/bi000749m
125. Einarsdottir H, Ji Y, Visser R, Mo C, Luo G, Scherjon S, et al. H435-containing immunoglobulin G3 allotypes are transported efficiently across the human placenta: implications for alloantibody-mediated diseases of the newborn. Transfusion. (2014) 54:665–71. doi: 10.1111/trf.12334
126. Mcdonagh CF, Kim KM, Turcott E, Brown LL, Westendorf L, Feist T, et al. Engineered anti-CD70 antibody-drug conjugate with increased therapeutic index. Mol Cancer Ther. (2008) 7:2913–23. doi: 10.1158/1535–7163.MCT-08-0295
127. Uppal H, Doudement E, Mahapatra K, Darbonne WC, Bumbaca D, Shen BQ, et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin Cancer Res. (2015) 21:123–33. doi: 10.1158/1078–0432.CCR-14-2093
128. Richards J, Auger J, Peace D, Gale D, Michel J, Koons A, et al. Phase I evaluation of humanized OKT3: toxicity and immunomodulatory effects of hOKT3gamma4. Cancer Res. (1999) 59:2096–101.
129. Alegre ML, Collins AM, Pulito VL, Brosius RA, Olson WC, Zivin RA, et al. Effect of a single amino acid mutation on the activating and immunosuppressive properties of a “humanized” OKT3 monoclonal antibody. J Immunol. (1992) 148:3461–8.
130. Wines BD, Powell MS, Parren PW, Barnes N, Hogarth PM. The IgG Fc contains distinct Fc receptor (FcR) binding sites: the leukocyte receptors Fc gamma RI and Fc gamma RIIa bind to a region in the Fc distinct from that recognized by neonatal FcR and protein A. J Immunol. (2000) 164:5313–8. doi: 10.4049/jimmunol.164.10.5313
131. Lund J, Winter G, Jones PT, Pound JD, Tanaka T, Walker MR, et al. Human Fc gamma RI and Fc gamma RII interact with distinct but overlapping sites on human IgG. J Immunol. (1991) 147:2657–62.
132. Hezareh M, Hessell AJ, Jensen RC, Van De Winkel JG, Parren PW. Effector function activities of a panel of mutants of a broadly neutralizing antibody against human immunodeficiency virus type 1. J Virol. (2001) 75:12161–8. doi: 10.1128/JVI.75.24.12161–12168.2001
133. Wessels U, Poehler A, Moheysen-Zadeh M, Zadak M, Staack RF, Umana P, et al. Detection of antidrug antibodies against human therapeutic antibodies lacking Fc-effector functions by usage of soluble Fcγ receptor I. BIOANALYSIS. (2016) 8:2135–45. doi: 10.4155/bio-2016-0182
134. Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes JM, Zhang Y, et al. Effector-attenuating substitutions that maintain antibody stability and reduce toxicity in mice. J Biol Chem. (2017) 292:3900–8. doi: 10.1074/jbc.M116.767749
135. Oganesyan V, Gao C, Shirinian L, Wu H, Dall'acqua WF. Structural characterization of a human Fc fragment engineered for lack of effector functions. Acta Crystallogr D Biol Crystallogr. (2008) 64:700–4. doi: 10.1107/S0907444908007877
136. Xu Y, Oomen R, Klein MH. Residue at position 331 in the IgG1 and IgG4 CH2 domains contributes to their differential ability to bind and activate complement. J Biol Chem. (1994) 269:3469–74.
137. Lund J, Takahashi N, Pound JD, Goodall M, Jefferis R. Multiple interactions of IgG with its core oligosaccharide can modulate recognition by complement and human Fc gamma receptor I and influence the synthesis of its oligosaccharide chains. J Immunol. (1996) 157:4963–9.
138. Lund J, Pound JD, Jones PT, Duncan AR, Bentley T, Goodall M, et al. Multiple binding sites on the CH2 domain of IgG for mouse Fc gamma R11. Mol Immunol. (1992) 29:53–9. doi: 10.1016/0161-5890(92)90156-R
139. Horton HM, Bernett MJ, Peipp M, Pong E, Karki S, Chu SY, et al. Fc-engineered anti-CD40 antibody enhances multiple effector functions and exhibits potent in vitro and in vivo antitumor activity against hematologic malignancies. Blood. (2010) 116:3004–12. doi: 10.1182/blood-2010-01-265280
140. Horton HM, Bernett MJ, Pong E, Peipp M, Karki S, Chu SY, et al. Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia. Cancer Res. (2008) 68:8049–57. doi: 10.1158/0008-5472.CAN-08-2268
141. Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. (2007) 25:1256–64. doi: 10.1038/nbt1344
142. Mueller JP, Giannoni MA, Hartman SL, Elliott EA, Squinto SP, Matis LA, et al. Humanized porcine VCAM-specific monoclonal antibodies with chimeric IgG2/G4 constant regions block human leukocyte binding to porcine endothelial cells. Mol Immunol. (1997) 34:441–52. doi: 10.1016/S0161-5890(97)00042-4
143. An Z, Forrest G, Moore R, Cukan M, Haytko P, Huang L, et al. IgG2m4, an engineered antibody isotype with reduced Fc function. MAbs. (2009) 1:572–9. doi: 10.4161/mabs.1.6.10185
144. Vafa O, Gilliland GL, Brezski RJ, Strake B, Wilkinson T, Lacy ER, et al. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods. (2014) 65:114–26. doi: 10.1016/j.ymeth.2013.06.035
145. Derebe MG, Nanjunda RK, Gilliland GL, Lacy ER, Chiu ML. Human IgG subclass cross-species reactivity to mouse and cynomolgus monkey Fcgamma receptors. Immunol Lett. (2018) 197:1–8. doi: 10.1016/j.imlet.2018.02.006
146. Mccarthy SG, Scallon B, Tam SH, Strohl W, Vafa O. Antibody Fc Mutants With Ablated Effector Functions. Alexandria, VA: US Patent Office (2015).
147. Wright A, Morrison SL. Effect of C2-associated carbohydrate structure on Ig effector function: studies with chimeric mouse-human IgG1 antibodies in glycosylation mutants of Chinese hamster ovary cells. J Immunol. (1998) 160:3393–402.
148. Wright A, Morrison SL. Effect of altered CH2-associated carbohydrate structure on the functional properties and in vivo fate of chimeric mouse-human immunoglobulin G1. J Exp Med. (1994) 180:1087–96. doi: 10.1084/jem.180.3.1087
149. Mimura Y, Sondermann P, Ghirlando R, Lund J, Young SP, Goodall M, et al. Role of oligosaccharide residues of IgG1-Fc in Fc gamma RIIb binding. J Biol Chem. (2001) 276:45539–47. doi: 10.1074/jbc.M107478200
150. Mimura Y, Church S, Ghirlando R, Ashton PR, Dong S, Goodall M, et al. The influence of glycosylation on the thermal stability and effector function expression of human IgG1-Fc: properties of a series of truncated glycoforms. Mol Immunol. (2000) 37:697–706. doi: 10.1016/S0161-5890(00)00105-X
151. Lund J, Tanaka T, Takahashi N, Sarmay G, Arata Y, Jefferis R. A protein structural change in aglycosylated IgG3 correlates with loss of huFc gamma R1 and huFc gamma R111 binding and/or activation. Mol Immunol. (1990) 27:1145–53. doi: 10.1016/0161-5890(90)90103-7
152. Tao MH, Morrison SL. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J Immunol. (1989) 143:2595–601.
153. Xu D, Alegre ML, Varga SS, Rothermel AL, Collins AM, Pulito VL, et al. In vitro characterization of five humanized OKT3 effector function variant antibodies. Cell Immunol. (2000) 200:16–26. doi: 10.1006/cimm.2000.1617
154. Woodle ES, Bluestone JA, Zivin RA, Jolliffe LK, Auger J, Xu D, et al. Humanized, nonmitogenic OKT3 antibody, huOKT3 gamma(Ala-Ala): initial clinical experience. Transplant Proc. (1998) 30:1369–70. doi: 10.1016/S0041-1345(98)00278-4
155. Newman R, Hariharan K, Reff M, Anderson DR, Braslawsky G, Santoro D, et al. Modification of the Fc region of a primatized IgG antibody to human CD4 retains its ability to modulate CD4 receptors but does not deplete CD4(+) T cells in chimpanzees. Clin Immunol. (2001) 98:164–74. doi: 10.1006/clim.2000.4975
156. Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, et al. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel. (2016) 29:457–66. doi: 10.1093/protein/gzw040
157. Arduin E, Arora S, Bamert PR, Kuiper T, Popp S, Geisse S, et al. Highly reduced binding to high and low affinity mouse Fc gamma receptors by L234A/L235A and N297A Fc mutations engineered into mouse IgG2a. Mol Immunol. (2015) 63:456–63. doi: 10.1016/j.molimm.2014.09.017
158. Sarmay G, Lund J, Rozsnyay Z, Gergely J, Jefferis R. Mapping and comparison of the interaction sites on the Fc region of IgG responsible for triggering antibody dependent cellular cytotoxicity (ADCC) through different types of human Fc gamma receptor. Mol Immunol. (1992) 29:633–9. doi: 10.1016/0161-5890(92)90200-H
159. Tam SH, Mccarthy SG, Armstrong AA, Somani S, Wu S-J, Liu X, et al. Functional, biophysical, and structural characterization of human IgG1 and IgG4 Fc variants with ablated immune functionality. Antibodies. (2017) 6:12. doi: 10.3390/antib6030012
160. Mimoto F, Katada H, Kadono S, Igawa T, Kuramochi T, Muraoka M, et al. Engineered antibody Fc variant with selectively enhanced FcgammaRIIb binding over both FcgammaRIIa(R131) and FcgammaRIIa(H131). Protein Eng Des Sel. (2013) 26:589–98. doi: 10.1093/protein/gzt022
161. Georgakopoulos T, Tatford OC, Gurevich V, Bertolini J. C1q aggregate binding for the determination of anti-complementary activity of immunoglobulin products. Biologicals. (2011) 39:38–42. doi: 10.1016/j.biologicals.2010.11.002
162. Tawara T, Hasegawa K, Sugiura Y, Harada K, Miura T, Hayashi S, et al. Complement activation plays a key role in antibody-induced infusion toxicity in monkeys and rats. J Immunol. (2008) 180:2294–8. doi: 10.4049/jimmunol.180.4.2294
163. Kishore U, Ghai R, Greenhough TJ, Shrive AK, Bonifati DM, Gadjeva MG, et al. Structural and functional anatomy of the globular domain of complement protein C1q. Immunol Lett. (2004) 95:113–28. doi: 10.1016/j.imlet.2004.06.015
164. Lund J, Takahashi N, Nakagawa H, Goodall M, Bentley T, Hindley SA, et al. Control of IgG/Fc glycosylation: a comparison of oligosaccharides from chimeric human/mouse and mouse subclass immunoglobulin Gs. Mol Immunol. (1993) 30:741–8. doi: 10.1016/0161-5890(93)90145-2
165. Rademacher TW, Homans SW, Parekh RB, Dwek RA. Immunoglobulin G as a glycoprotein. Biochem Soc Symp. (1986) 51:131–48.
166. Kanda Y, Yamada T, Mori K, Okazaki A, Inoue M, Kitajima-Miyama K, et al. Comparison of biological activity among nonfucosylated therapeutic IgG1 antibodies with three different N-linked Fc oligosaccharides: the high-mannose, hybrid, and complex types. Glycobiology. (2007) 17:104–18. doi: 10.1093/glycob/cwl057
167. Feige MJ, Nath S, Catharino SR, Weinfurtner D, Steinbacher S, Buchner J. Structure of the murine unglycosylated IgG1 Fc fragment. J Mol Biol. (2009) 391:599–608. doi: 10.1016/j.jmb.2009.06.048
168. Borrok MJ, Jung ST, Kang TH, Monzingo AF, Georgiou G. Revisiting the role of glycosylation in the structure of human IgG Fc. ACS Chem Biol. (2012) 7:1596–602. doi: 10.1021/cb300130k
169. Davies AM, Jefferis R, Sutton BJ. Crystal structure of deglycosylated human IgG4-Fc. Mol Immunol. (2014) 62:46–53. doi: 10.1016/j.molimm.2014.05.015
170. Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J Mol Biol. (2003) 325:979–89. doi: 10.1016/S0022-2836(02)01250-0
171. Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell. (2018) 9:63–73. doi: 10.1007/s13238-017-0473-8
172. Kang TH, Lee CH, Delidakis G, Jung J, Richard-Le Goff O, Lee J, et al. An engineered human fc variant with exquisite selectivity for FcgammaRIIIaV158 reveals that ligation of FcgammaRIIIa mediates potent antibody dependent cellular phagocytosis with GM-CSF-differentiated macrophages. Front Immunol. (2019) 10:562. doi: 10.3389/fimmu.2019.00562
173. Hessell AJ, Hangartner L, Hunter M, Havenith CE, Beurskens FJ, Bakker JM, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. (2007) 449:101–4. doi: 10.1038/nature06106
174. Caskey M, Klein F, Nussenzweig MC. Broadly neutralizing anti-HIV-1 monoclonal antibodies in the clinic. Nat Med. (2019) 25:547–53. doi: 10.1038/s41591-019-0412-8
175. Paes B, Kim D, Saleem M, Wong S, Mitchell I, Lanctot KL, et al. Respiratory syncytial virus prophylaxis in infants with congenital airway anomalies compared to standard indications and complex medical disorders. Eur J Pediatr. (2019) 178:377–85. doi: 10.1007/s00431-018-03308-1
176. Griffin MP, Khan AA, Esser MT, Jensen K, Takas T, Kankam MK, et al. Safety, tolerability, and pharmacokinetics of MEDI8897, the respiratory syncytial virus prefusion F-targeting monoclonal antibody with an extended half-life, in healthy adults. Antimicrob Agents Chemother. (2017) 61:e01714–16. doi: 10.1128/AAC.01714-16
Keywords: antibody engineering, Fc optimization, therapeutic antibodies, biologics, passive immunity, immunotherapy
Citation: Saunders KO (2019) Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front. Immunol. 10:1296. doi: 10.3389/fimmu.2019.01296
Received: 20 January 2019; Accepted: 21 May 2019;
Published: 07 June 2019.
Edited by:
Gabriella Scarlatti, San Raffaele Hospital (IRCCS), ItalyReviewed by:
Simone Irene Richardson, National Institute of Communicable Diseases (NICD), South AfricaKevin R. Maisey, Universidad de Santiago de Chile, Chile
Copyright © 2019 Saunders. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevin O. Saunders, kevin.saunders@dm.duke.edu