 Hannes Lindahl1,2*
Hannes Lindahl1,2* Yenan T. Bryceson1,3,4
Yenan T. Bryceson1,3,4- 1Clinical Immunology and Transfusion Medicine, Karolinska University Hospital, Stockholm, Sweden
- 2Center for Molecular Medicine, Department of Clinical Neuroscience, Karolinska Institute, Karolinska University Hospital, Stockholm, Sweden
- 3Center for Hematology and Regenerative Medicine, Department of Medicine, Karolinska Institute, Karolinska University Hospital Huddinge, Stockholm, Sweden
- 4Brogelmann Research Laboratory, Department of Clinical Sciences, University of Bergen, Bergen, Norway
The advent of high-throughput sequencing has facilitated genotype-phenotype correlations in congenital diseases. This has provided molecular diagnosis and benefited patient management but has also revealed substantial phenotypic heterogeneity. Although distinct neuroinflammatory diseases are scarce among the several thousands of established congenital diseases, elements of neuroinflammation are increasingly recognized in a substantial proportion of inborn errors of immunity, where it may even dominate the clinical picture at initial presentation. Although each disease entity is rare, they collectively can constitute a significant proportion of neuropediatric patients in tertiary care and may occasionally also explain adult neurology patients. We focus this review on the signs and symptoms of neuroinflammation that have been reported in association with established pathogenic variants in immune genes and suggest the following subdivision based on proposed underlying mechanisms: autoinflammatory disorders, tolerance defects, and immunodeficiency disorders. The large group of autoinflammatory disorders is further subdivided into IL-1β-mediated disorders, NF-κB dysregulation, type I interferonopathies, and hemophagocytic syndromes. We delineate emerging pathogenic themes underlying neuroinflammation in monogenic diseases and describe the breadth of the clinical spectrum to support decisions to screen for a genetic diagnosis and encourage further research on a neglected phenomenon.
Introduction
A monogenic disease may be suspected when onset is unusually early, there are similar manifestations in other family members, or there is consanguinity (1). With affordable high-throughput sequencing it is now feasible to pursue a genetic diagnosis in individual patients, which adds information on disease prognosis, potential treatments, and may also facilitate prenatal diagnostics.
The most well-recognized disease with a primary neuroinflammatory etiology is multiple sclerosis (MS). The pathogenesis is generally assumed to be autoimmune but the antigen specificity of the autoreactive lymphocytes that drive the disease remain undefined (2). Neuroinflammatory diseases for which an autoimmune pathogenesis is more substantiated include neuromyelitis optica (NMO) spectrum disorder and autoimmune encephalitis. Conversely, other forms of neuroinflammation appear less likely to be driven by autoreactive lymphocytes but rather dysregulated innate immune cells, such as those observed in Bechet’s disease, an idiopathic systemic vasculitis syndrome (3). Moreover, neuroinflammation can cause collateral damage in central nervous system (CNS) infection, neurodegeneration, ischemia, and trauma.
In clinical practice, it is not uncommon that a patient presents with apparent neuroinflammation, e.g. with subacute onset of CNS symptoms associated with abnormalities on magnetic resonance imaging (MRI) and cerebrospinal fluid (CSF) analysis (Figure 1), but without distinct features that justify a definite diagnosis. The patient will then typically be managed under a presumptive diagnosis until the clinical picture develops further, which often entails accumulation of neurological deficits. A prompt molecular diagnosis can thus benefit treatment. Early clinical presentation and a history consistent with a hereditary disease may suggest a genetic etiology, warranting genetic investigations. Furthermore, the increasing affordability implies that genome sequencing may soon constitute part of the routine first-line diagnostic work-up of severe neuroinflammation. Recently, McCreary and colleagues published an extensive panel of neuroinflammation-associated genes and demonstrated that it could provide a molecular diagnosis in 20% of unresolved pediatric patients with suspected genetic neuroinflammation (4). Herein, we review the literature on neuroinflammatory manifestations associated with monogenic immune-mediated diseases and discuss the different underlying immunological mechanisms. Our compilation can concretize what may constitute a monogenic neuroinflammatory clinical presentation.
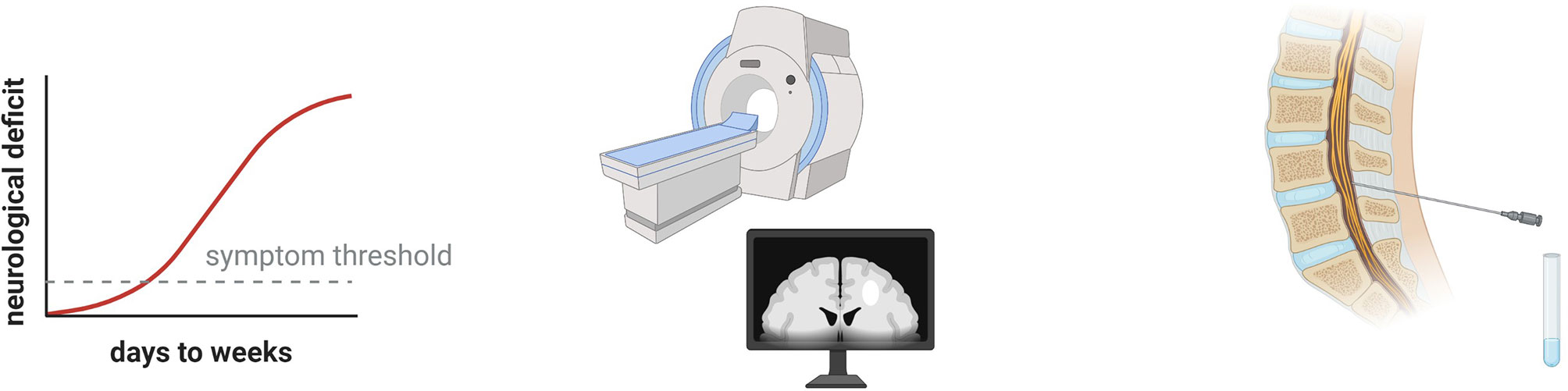
Figure 1 Clinical hallmarks of neuroinflammation. The patient may have a subacute onset of neurological symptoms that may resolve spontaneously or respond to immunomodulatory drugs. Using magnetic resonance imaging (MRI), acute lesions with blood brain barrier disruption can be visualized as contrast enhancing T1 lesions. Various pathologies can appear as T2 lesions, such as demyelination, axonal damage, and edema. Analysis of cerebrospinal fluid allows quantitative and qualitative assessment of intrathecal inflammation as well as demonstration of blood brain barrier disruption.
Autoinflammatory Disorders
Autoinflammatory disorders are clinical disorders of the innate immune system characterized by chronic or periodic inflammation without engagement of adaptive lymphocytes, i.e. autoreactive antibodies or T cells. The most recognized monogenic autoinflammatory syndromes are familial Mediterranean fever (FMF), cryopyrin-associated periodic syndrome (CAPS), hyperimmunoglobulinemia D syndrome (HIDS), and tumor necrosis factor receptor-associated periodic syndrome (TRAPS). IL-1β-driven or -biased inflammation is a common feature of these four syndromes. In disorders caused by NF-κB dysregulation the uncontrolled production of cytokines involves IL-1β as well as several other proinflammatory cytokines. By comparison, interferonopathies represent a distinct category of rare diseases characterized by increased type I IFN signaling and loss of negative regulation. And lastly, although caused by impaired lymphocyte effector functions, the most prominent feature of hemophagocytic syndromes is hyperinflammation with uncontrolled innate immune activation and they are therefore included under this heading in this review.
IL-1β-Mediated Autoinflammatory Disorders
Familial Mediterranean Fever
Familial Mediterranean fever (FMF; MIM: 249100) is classically regarded as an autosomal recessive disease caused by variants in MEFV, but one mutant allele is sufficient to cause subclinical systemic inflammation (5) (Table 1). Furthermore, autosomal dominant FMF has also been described (MIM: 134610) (55) and likely involves a distinct subset of MEFV variants (56). MEFV encodes pyrin, an intracellular pattern recognition receptor that is expressed mainly by neutrophils and monocytes/macrophages. Upon its activation, an inflammasome complex forms, leading to proteolytic maturation of IL-1β as well as IL-18 and their subsequent extracellular release through a form of programmed cell death called pyroptosis (57). MEFV variants that cause FMF represent pyrin gain-of-function variants that lower the activation threshold of inflammasome formation. Clinically, FMF is characterized by periodic fever, serositis, arthralgia, and rash. A feared complication is renal failure due to amyloidosis. Apart from headache (10%) and seizures (4%), other neurological manifestations of FMF are very rare (8). Recurrent aseptic meningitis in FMF is occasionally reported (58–61), as well as a wide range of other neuroinflammatory manifestations, however the causal relationship with FMF is uncertain (8). Moreover, elevated protein in CSF as a sign of meningeal irritation may occur without clinical evidence of meningitis (6). Whereas MEFV variants that are associated with FMF are gain-of-function variants, a subset of rare MEFV variants that result in defective binding to an inhibitory 14-3-3 protein cause a distinct autosomal dominant disease termed pyrin-associated autoinflammation with neutrophilic dermatosis (PAAND), for which neurological involvement has not been reported (62, 63). Collectively, awareness that the episodic inflammation in FMF may also engage the CNS is important.
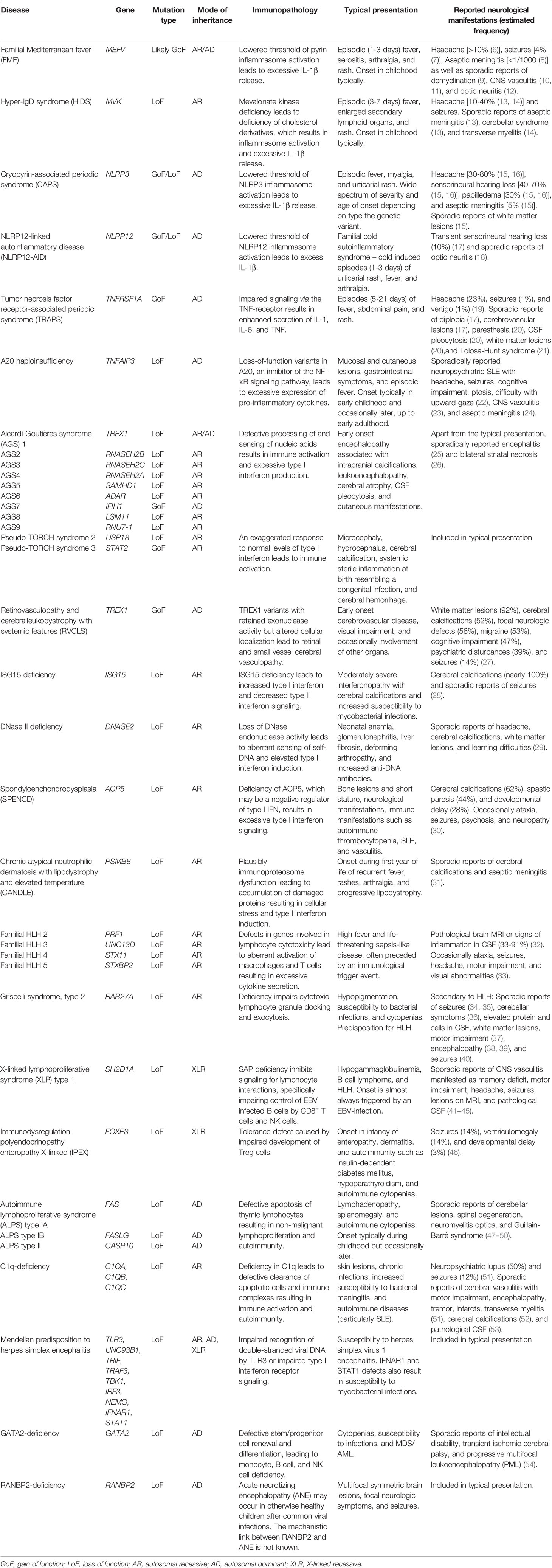
Table 1 Summary of neuroinflammatory manifestations associated with inborn errors of immunity.
Mevalonate Kinase Deficiency
Mevalonate kinase deficiency (MKD) consists of the autosomal recessive syndromes mevalonic aciduria (MVA; MIM: 610377) and hyperimmunoglobulinemia D syndrome (HIDS; MIM: 260920). Both are caused by loss-of-function variants in MVK, which encodes the cholesterol biosynthesis enzyme mevalonate kinase. MVA and HIDS represent opposite ends of a clinical spectrum associated with absent to subnormal enzyme activity, respectively (64). The exact pathogenesis is elusive but involves build-up of mevalonate and loss of pyrin inhibition resulting in inflammasome activation and excess IL-1β production (65). The mevalonate pathway produces the substrate for a form of post-translational modification, prenylation, which is involved in the regulation of TLR-induced phosphoinositide 3-kinase (PI3K) activation (66). Furthermore, impaired prenylation of RhoA, a small GTPase, inactivates RhoA, decreases pyrin phosphorylation and subsequent 14-3-3-mediated negative regulation of pyrin activity (67). Loss of prenylation in MKD thus contributes to pyrin activation and the hyperinflammatory phenotype. MVA is associated with severe developmental delay, ataxia, epilepsy, and shortened lifespan (65). HIDS is an autoinflammatory periodic fever syndrome associated with persistently elevated IgD and increased mevalonic acid in the urine during attacks (13). Common clinical manifestations of HIDS include rash, hepatosplenomegaly, and lymphadenopathy. The most common neurological manifestation is headache (10-40%), which may be present independent of the fever episodes (65). Sporadic reports of seizures, transverse myelitis, cerebellar syndrome, and aseptic meningitis exist (13, 14). Although the exact pathogenesis of FMF and HIDS are not completely understood, similarities in the affected pathways are consistent with a similar pattern of CNS engagement that mainly involves headache and only rarely overt neuroinflammation.
Cryopyrin-Associated Periodic Syndrome
Cryopyrin-associated periodic syndrome (CAPS) can be regarded as a continuum of three phenotypes that were described before the common disease-causing gene was identified – from the milder familial cold autoinflammatory syndrome (FCAS, MIM: 120100), via Muckle-Wells syndrome (MWS, MIM: 191900), to the most severe form chronic infantile neurological cutaneous and articular syndrome (CINCA, MIM: 607115). All three are caused by autosomal dominant variants in NLRP3 (68–70). Analogous to MEFV, NLRP3 encodes an intracellular pattern recognition receptor, cryopyrin, and the CAPS-associated gain-of-function variants lower the threshold of inflammasome activation leading to excessive IL-1β production and release. Additionally, NLRP3 acts as a negative regulator of NF-κB signaling and loss-of-function NLRP3 variants are also reported as the cause of autoinflammation (71). Clinically, CAPS is characterized by periodic fever, urticarial rash, conjunctivitis, and arthralgia. Neurological symptoms are common and most frequently consist of headache (30-80%), sensorineural hearing loss (40-70%), and papilledema (30%) (15, 72, 73). Less common neurological manifestations include aseptic meningitis (5%) and hydrocephalus, seizures, chorea, white matter lesions, and CSF pleocytosis have sporadically been reported (16, 74–76). A case has been described that mimicked Tolosa-Hunt syndrome with steroid-responsive periorbital pain, diplopia, and granulomatous inflammation (77). Neuroradiological manifestations are not well characterized, but a case report in which serial imaging was performed showed enhancement in leptomeninges, cochlea, and cranial nerves (78). The higher frequency of neurological symptoms in CAPS compared to the other IL-1β-mediated autoinflammatory disorders may be linked to the increasingly appreciated role of NLRP3 in microglia as the main contributor to neuroinflammation in neurodegeneration, infection, and stroke (79).
TNF Receptor-Associated Periodic Syndrome
TNF Receptor-Associated Periodic Syndrome (TRAPS; MIM: 142680) is an autosomal dominant periodic fever syndrome that is caused by gain-of-function variants in TNFRSF1A, which encodes one of the major TNF receptors (80). Despite that variants associated with TRAPS impair signaling via the TNF receptor, inflammation is enhanced with increased IL-1β, IL-6, and TNF secretion, likely due to intracellular accumulation of misfolded protein (81). Approximately 10% of cases have adult onset. The clinical presentation typically consists of fever, abdominal pain, arthralgia, myalgia, and a migratory rash. Neurological symptoms are not core features of TRAPS, but approximately 20% of patients manifest headache and to lesser extent seizures (1%), vertigo (1%), diplopia, and cerebrovascular lesions (17, 19, 21). Occasionally paresthesia, CSF pleocytosis, white matter lesions (20), and Tolosa-Hunt syndrome have been reported (21).
Other IL-1β-Mediated Autoinflammatory Disorders
Variants in NLRP12 can also cause an autoinflammatory syndrome characterized by excessive IL-1β secretion (MIM: 611762). Like NLRP3, both gain-of-function and loss-of-function variants have been described that cause increased inflammasome activation and loss of NF-κB inhibition, respectively (18, 82, 83). NLRP12-associated disorders are very rare and reported symptoms primarily include inflammation in skin and bone. Neuroinflammatory manifestations such as headache and sensorineural hearing loss are occasionally observed (17) and rare reports of optic neuritis exist (18). In addition, loss-of-function mutations in IL1RN, encoding the IL-1 receptor antagonist, also cause excess IL-1β signaling. These patients primarily have autoinflammatory manifestations in skin and bone but a patient with CNS vasculitis has also been reported (84).
NF-κB Dysregulation
NF-κB constitute a ubiquitously expressed family of transcription factors that have been conserved throughout eukaryotes and that serve as key mediators of inflammatory responses (85). Upon activation, IKK phosphorylates the NF-κB inhibitor IκBα at two N-terminal serines, thereby triggering ubiquitin-dependent, proteasome-mediated IκBα degradation, resulting in rapid and transient nuclear translocation of canonical NF-κB members that can induce transcription of a variety of pro-inflammatory cytokine genes. Low penetrance variants in TNFAIP3, encoding A20, have been associated with many different complex immune mediated diseases (86), but more recently, heterozygous loss-of-function variants have established an autoinflammatory disease entity termed A20 haploinsufficiency (MIM: 616744), with similarities to Bechet’s disease and autoimmune lymphoproliferative syndrome (ALPS) (23, 87, 88). A20 is a ubiquitin editing enzyme and a potent inhibitor of NF-κB (89). Most cells express baseline amounts of A20 but expression is upregulated in pro-inflammatory settings (86). Deficiency of A20 consequently results in insufficient suppression of NF-κB and dysregulated innate immunity via overproduction of cytokines such as TNF, IL-1β, IL-18, and IL-6 (24). Sporadic reports exist of A20 haploinsufficiency with neurological symptoms, such as aseptic meningitis, and CNS vasculitis (23, 24). Furthermore, a frameshift variant in TNFAIP3 has been reported as the cause of neuropsychiatric SLE manifested as headache, seizures, cognitive impairment, ptosis, and difficulty with upward gaze (22). The pathogenesis may involve an influence on blood brain barrier (BBB) permeability from the disease associated variant (22). Although Tnfaip3-/- mice are prone to fulminant neuroinflammation involving the NLRP3 inflammasome (90), the neurological spectrum in human TNFAIP3 haploinsufficiency suggests that species differences exist or that one TNFAIP3 allele is sufficient to restrain inflammation in the CNS.
ITCH, RNF11, and TAX1BP1 comprise additional essential components of the ubiquitin-editing protein complex that normally ensures the transient nature of inflammatory NF-κB-mediated signaling pathways (85). Autosomal recessive mutations in ITCH are associated with syndromic multisystem autoimmune disease (MIM: 613385) (91). Although ITCH is highly expressed in the brain, neurological symptoms are not a prominent feature of patients with ITCH deficiency. Altogether, excessive NF-κB activation is associated with autoinflammation and a low incidence of neuroinflammation. Furthermore, loss-of-function variants in several additional components of the NF-κB signaling pathway have been described with immunodeficiency, but where neuroinflammation is not a prominent feature.
Comment on Neuroinflammation in IL-1β-Mediated Autoinflammatory Disorders
Headache is the most common neurological manifestation in the IL-1β-mediated autoinflammatory disorders. This is consistent with the emerging role for inflammasomes in migraine (92) and the association of MEFV variants with the risk of migraine (93). Moreover, seizures is another commonly reported neurological manifestation of these diseases. Importantly, both acute and chronic neuroinflammation has been linked to epileptogenesis (94) and in experimental systems TNF-blockade has reduced epileptic activity (95). Interestingly, the frequency of seizures in FMF patients (4%) appears to be higher than in the other disorders. This may be a consequence of unique aspects of the pyrin inflammasome such as sensing of bacterial modification/inactivation of Rho GTPases (96).
Pathogenic insights can be gained not only from studying cellular processes but also from clinical observations of what drugs have efficacy treating specific inborn errors of immunity. Most autoinflammatory diseases have been reported to respond to anti-IL-1 therapy (97). Moreover, first-line treatment for FMF is colchicine, which likely acts by interfering with the cytoskeletal changes required for inflammasome assembly, also implicating IL-1 (98). Colchicine-resistance is observed in approximately 10% of all FMF patients, which generally receive anti-IL-1 therapy as second-line treatment (99). Anti-IL-6 and anti-IL-18 therapy are occasionally used for autoinflammatory diseases in general (98). Anti-TNF therapy has been mainly used for TRAPS with inconsistent results. Anti-TNF may even provoke an inflammatory flair in TRAPS, akin to experience from MS (100, 101). A20 haploinsufficiency frequently responds to colchicine but a very wide range of treatments have been used in reported cases (102). Interestingly, experience from A20 haploinsufficiency suggests that patients who carry the same pathogenic mutation do not necessarily respond to the same treatment implying a role for genetic background and/or environment (102).
Most cells produce IL-1β to some degree but during neuroinflammation major contributors are T cells, infiltrating myeloid cells, and microglia (Figure 2) (103). Physiological IL-1 signaling in the CNS has a role in, for example, memory performance or neuronal survival (104) but elevated IL-1 signaling has a wide range of pathological effects (103). Which cell types in the CNS that respond to IL-1β is controversial, but sequential deletion of the IL-1 receptor in mouse CNS cells suggests that endothelial cells in the BBB as well as in cerebral ventricles are the major targets (105, 106). Microglia are activated by IL-1β in vivo but this may be an indirect effect via signaling from endothelial cells (105). For sterile neuroinflammation in general, the inciting factor that triggers inflammation is rarely understood. An inflammatory cascade that starts in the periphery and then migrates across the BBB is often hypothesized (107). In the case of IL-1β-mediated autoinflammation, a wide range of pathogen associated molecular patterns (PAMP) and damage associated molecular patterns (DAMP) can elicit IL-1β secretion with insufficient regulatory feedback mechanisms for resolution. IL-1β promotes the differentiation of IL-17 and GM-CSF-secreting T helper cells, which are implicated in the pathogenesis of MS as well as other autoimmune diseases and can cross the BBB (108–110). Once inflammation has spread to the CNS, a vicious cycle can potentially be established including IL-1β-mediated immune cell chemotaxis to the CNS via activated endothelial cells, skewing of T helper cells and astrocytes towards encephalitogenic phenotypes (111), as well as neuronal excitotoxicity and subsequent synaptopathy (Figure 2) (112).
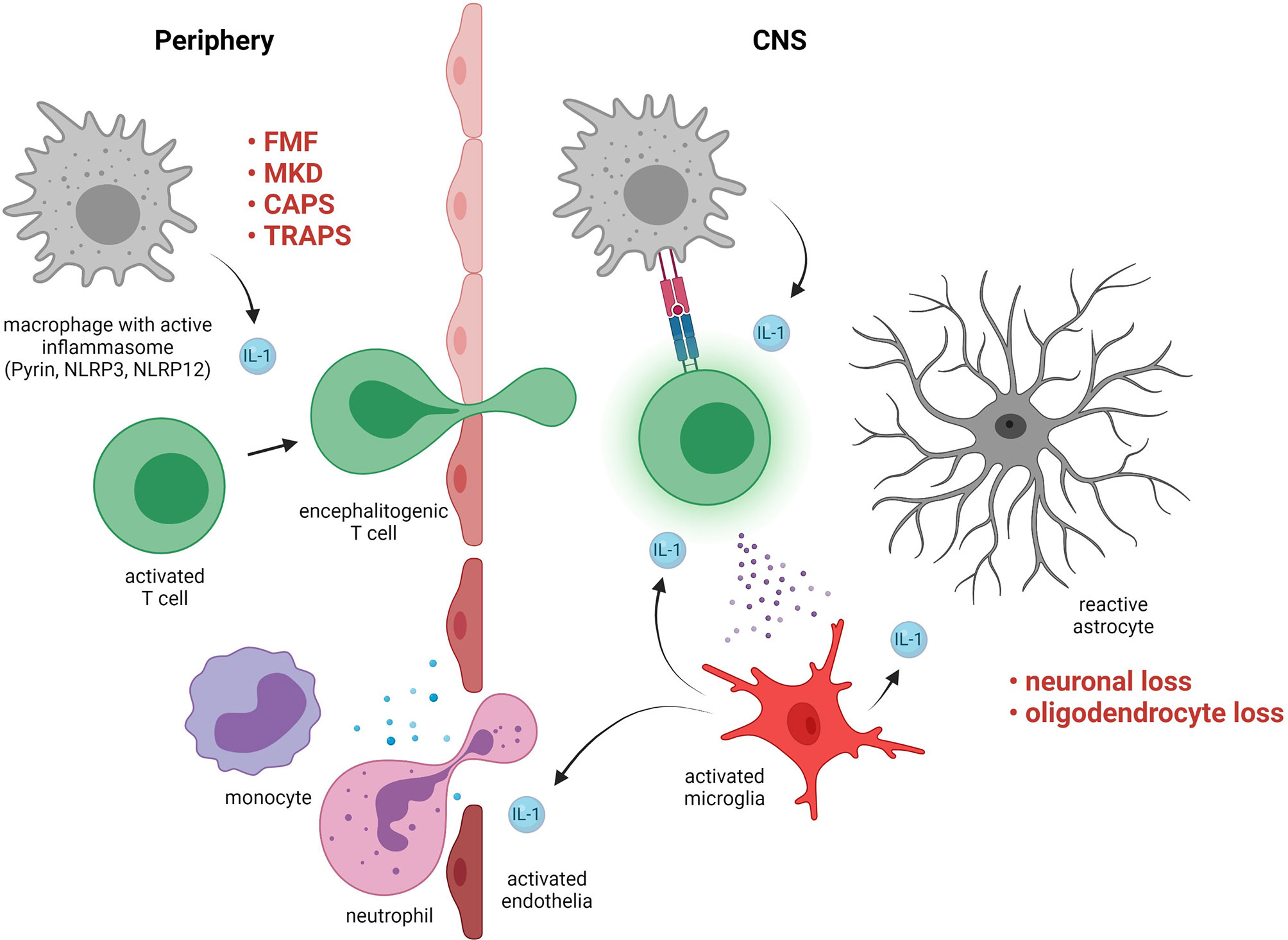
Figure 2 Pathogenic roles of interleukin (IL)-1-signaling in neuroinflammation. Examples of disorders with exaggerated IL-1-signaling include familial Mediterranean fever (FMF), mevalonate kinase deficiency (MKD), cryopyrin-associated periodic syndrome (CAPS), and TNF receptor-associated periodic syndrome (TRAPS). IL-1 in the periphery contributes to skewing of activated T cells towards an encephalitogenic phenotype. The T cells cross the blood brain barrier and in the central nervous system (CNS) they are activated by resident antigen presenting cells via MHC interactions and cytokines. The T cells activate microglia, which produce IL-1 that triggers a pro-inflammatory response in astrocytes and activates endothelia resulting in further immune cell infiltration. Maladaptive CNS-inflammation can cause irreversible damage such as loss of neurons and oligodendrocytes.
The wide range of observed neurological presentations is intriguing considering the common pathogenesis of dysregulated inflammasomes and may reflect differences in genetic background, comorbidities, or environmental exposures. Furthermore, apart from constitutional neurological symptoms such as headache, the neuroinflammatory manifestations reported in association with IL-1β-associated autoinflammatory diseases are rare and could potentially reflect separate disease entities with complex etiology for which the autoinflammatory disease gene variants act as risk-factors. Interestingly, several of the autoinflammation associated variants are commonly implicated as modifyers of complex diseases. Bechet’s disease has similarities with FMF regarding both symptomology and treatment, which in 3-30% of cases is complicated by neuroinflammation (113). Patients with neuro-Bechet’s disease and the closely related neuro-Sweet’s disease that carry mutations in MEFV have more pronounced neurological manifestations such as headache and neuroimaging findings such as white matter lesions and non-parenchymal lesions (114, 115). Notably, co-occurrence of MS in carriers of homozygous or heterozygous pathogenic MEFV variants is well described (8, 116–118). Moreover, NLRP12 has recently been reported as a candidate gene for familial MS (119) and NLRP3 as well as TNFRSF1A variants appear to modify susceptibility and/or severity of MS (118). Thus, autoinflammatory susceptibilities can likely fuel other neuroinflammatory processes, triggering or exacerbating disease.
Type I Interferonopathies
Aicardi-Goutières Syndrome
A subset of the autoinflammatory disorders called type I interferonopathies is distinguished by excess IFNα/β signaling (25). In 1984, Jean Aicardi and Françoise Goutières described a form of severe familial progressive encephalopathy in children (120). A majority of these patients have signs of upregulated type I interferon signaling, often assessed using a panel of interferon-stimulated genes as a proxy measure (121). To date, variants in at least 9 genes have been identified, which together underlie most cases of Aicardi–Goutières syndrome (AGS): TREX1 (MIM: 225750), RNASEH2A (MIM: 610333), RNASEH2B (MIM: 610181), RNASEH2C (MIM: 610329), SAMHD1 (MIM: 612952), ADAR1 (MIM: 615010), IFIH1 (MIM: 615846), LSM11 (MIM: 619486), and RNU7-1 (MIM: 619487) (122–127). Loss-of-function variants in these genes cause autosomal recessive disease inheritance, except for IFIH1 and TREX1. Gain-of-function variants in IFIH1 are associated with autosomal dominant inheritance, whereas for TREX1 both recessive and dominant inheritance has been reported. Disease mechanisms include accumulation or modification of endogenous nucleic acids, enhanced activity of nucleic acid sensor or other components in the interferon signaling pathway triggering excessive type I IFN production, and loss of negative regulation (128). TREX1 encodes an exonuclease that cleaves nucleic acids in the cytosol, thereby preventing their accumulation and triggering of a type I interferon associated inflammatory response (129), which is toxic to neurons (130). Cardinal features of AGS are early onset of a severe neurological disorder with intracranial calcification, leukoencephalopathy, cerebral atrophy, cerebrospinal pleocytosis, as well as cutaneous manifestations. As more data have been gathered, a wider range of neurological and other clinical phenotypes have been associated with TREX1 variants (26, 131). Phenotype-genotype associations exist to some extent in AGS and regarding the neurological manifestations, variants in TREX1 appear more prone to have an encephalitic presentation (25). Furthermore, ADAR1 variants have been linked to the pediatric neurodegenerative disorder bilateral striatal necrosis (BSN) (132). BSN is clinically characterized by acute or subacute onset of dystonia and radiological symmetrical abnormalities in the striatum. Onset can be as late as in adolescence, often with an infectious trigger. Although clinically distinct from AGS, BSN patients with ADAR1 variants have signs of interferonopathy such as upregulated interferon-stimulated genes and may have brain calcifications and signs of neuroinflammation such pleocytosis, elevated neopterin in CSF, and spinal cord involvement (132, 133). In summary, excessive type I interferon signaling frequently causes neuropathology but the phenotypic heterogeneity associated with these genetic variants suggests that unrecognized modifying factors have a significant influence on individual outcome.
Pseudo-TORCH
An early-onset syndrome caused by severe congenital infections goes under the acronym TORCH, which stands for Toxoplasmosis, Other agents, Rubella, Cytomegalovirus, and Herpes simplex. The term pseudo-TORCH is used for the same clinical picture when no infection is detected. Three genetically defined forms of pseudo-TORCH exist to date caused by recessive variants in OCLN (MIM: 251290), USP18 (MIM: 617397), and STAT2 (MIM: 618886), respectively, of which the latter two are type I interferonopathies. All three disorders cause brain malformations, intracranial calcifications, and severe developmental delay. Most reported cases have died at an early age from cerebral hemorrhage. AGS is a common differential diagnosis. Features that are more consistent with pseudo-TORCH are microcephaly, thrombocytopenia, liver dysfunction, and hepatosplenomegaly. OCLN codes for occludin, a tight junction protein. Occludin deficiency may cause BBB dysfunction but the pathophysiology of this disease remains unclear (134). The clinical features suggest elevated type I interferon signaling but this has not been demonstrated. USP18 is a STAT2-dependent negative regulator of type I interferon signaling and lack of USP18 or a STAT2 variant that is unable to interact with USP18 consequently result in prolonged signaling downstream of the type I interferon receptor (135–137). In the initial report of STAT2 variant pseudo-TORCH, a severe multisystemic inflammatory disorder developed at the age of 8 months, which included marked neuroinflammation in the form of brain calcifications as well as white matter and cerebellar abnormalities (137). Pseudo-TORCH therefore encompasses genetic diagnoses with considerable clinical overlap with AGS and that may present with neuroinflammation.
Vasculopathies
Pathologic type I interferon signaling is associated with cerebrovascular disease, both in monogenic interferonopathies and typically more complex genetic diseases such as SLE (138, 139). In addition to AGS, TREX1 variants have been associated with the adult onset disorder retinovasculopathy and cerebral leukodystrophy with systemic features (RVCLS, MIM: 192315) (140). However, unlike AGS, TREX1 variants associated with RVCLS do not disrupt exonuclease activity but rather manifest as gain-of-function variants with dominant inheritance. The underlying pathology of RVCLS can be vasculitis, thrombotic microangiopathy, or aneurysmal dilatation and occurs in both small and large vessels (141, 142). Moreover, TREX1 variants have been suggested as the genetic basis of patients with cerebral small vessel disease with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL)-like phenotype (143) and monogenic SLE.
Gain-of-function variants in STING1 cause the type I interferonopathy STING-associated vasculopathy with onset in infancy (SAVI, MIM: 615934) (144, 145). Core manifestations of SAVI are early-onset systemic inflammation, skin vasculopathy, and interstitial lung disease. Consistent with other interferonopathies cerebral calcifications are observed as a sign of CNS involvement. Occasionally CSF pleocytosis or overt neurological symptoms such as spastic paresis and seizures have been reported (27, 146).
Other Interferonopathies
Autosomal recessive loss-of-function variants in ISG15 cause a syndrome termed ISG15-deficiency (MIM: 616126)103. ISG15 is an intracellular ubiquitin-like protein induced by type I interferon that in humans stabilizes USP18 in a feedback inhibition loop. ISG15-deficiency consequently leads to an interferonopathy but results in a milder neurological phenotype compared to pseudo-TORCH (28, 147). Moreover, ISG15 acts as an extracellular inducer of type II interferon and its deficiency is consequently also associated with increased susceptibility to mycobacterial disease. All described ISG15-deficient patients have cerebral calcifications, a sign of elevated type I interferon signaling, and seizures have frequently been reported. DNase II deficiency underlies a distinct clinical syndrome of autoinflammation and elevated type I interferon signaling. Neurological manifestations include cerebral calcifications and white matter hyperintensities (29). Loss-of- function variants in ACP5 has been identified as a cause of spondyloenchondrodysplasia with immune dysregulation (SPENCDI, MIM: 607944), a rare autosomal recessive syndrome characterized by skeletal and immune abnormalities (30, 148). ACP5 encodes an enzyme that regulates the activity of osteopontin and is involved in non-nucleic acid dependent type I interferon induction. A wide spectrum of immune dysregulation associated with SPENCDI has been described, including development of SLE. Neurological manifestations include developmental delay, spastic paresis, and intracranial calcifications (149). Mutations in PSMB8 is a cause of a disorder previously called chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE). The disease now belongs to a subcategory of interferonopathies called proteasome-related autoinflammation (PRAAS, MIM: 256040) and is called PRAAS1. Like other interferonopathies cerebral calcifications have been reported but also aseptic meningitis (31, 150, 151). Thus, disease-causing variants in an increasing spectrum of genes are being linked to interferonopathies with neuroinflammatory manifestations.
Comment on Neuroinflammation in Type I Interferonopathies
Unlike the IL-1β-mediated autoinflammatory diseases where conspicuous neuroinflammation is rare, most interferonopathies have signs of CNS involvement. Assuming that the intracranial calcifications observed in virtually all interferonopathies are caused by type I interferon, this can either be a result of direct interferon signaling in cells of the brain or of signals transduced from CNS cells that are in contact with the peripheral circulation. Notably, in homeostatic conditions, neither IFN-α nor IFN-β readily cross the BBB and reach the brain parenchyma (152–154) but as the barrier opens in inflammatory settings, cytokines and cells can gain access to the CNS. Although all somatic cells can produce type I interferons, the major contributors are the plasmacytoid dendritic cells (DC) (155, 156). In a mouse model of AGS, Trex1 deletion restricted to the hematopoietic compartment was sufficient for full disease, whereas deletion restricted to DCs yielded less pronounced manifestations (157). On the other hand, transfer of wild type bone marrow into irradiated Trex1-/- mice did not rescue inflammation, implying that non-hematopoietic cells also are sufficient to trigger the type I IFN-mediated pathology (158). In inflammatory settings, CNS cells that produce type I interferon include microglia, astrocytes, and neurons. During homeostasis, microglia present in white matter receive a constant interferon signal, which may be necessary to increase housekeeping functions like phagocytosis of myelin debris (159). Furthermore, constitutive interferon signaling is required for the homeostasis of neurons in mice demonstrated by the development of age associated neurodegeneration with similarities to Parkinson’s disease and Lewy body dementia in Ifnb–/– mice, with a likely contribution from defective autophagy (160). Humans that lack type I interferon signaling are rare and reported examples of IFNAR1 and IFNAR2 deficiencies have all been in children (161–163) or young adults not specifically assessed for signs of neurodegenerative disease (164).
In inflammatory settings, most CNS cells including infiltrating immune cells have the potential to respond to type I interferon (165). In a mouse model of MS, USP18 expression specifically in microglia has an important role in dampening excessive interferon input that otherwise would result in pathological microglia activation, which reflects the human interferonopathy caused by USP18 deficiency (135). In contrast, IFN-β is a well-established therapy for MS (166) and has been reported to reduce the severity of mouse models of the disease (167). Notably, not all MS patients respond to IFN-β, and exacerbations have been described (168, 169). Moreover, NMO, as well as several autoimmune diseases that do not primarily affect the CNS, are likely worsened by type I interferon and it has been hypothesized that IFN-β is an effective treatment in Th1-biased but not in Th17-biased diseases or disease subphenotypes (170). Although it is not definitively established that type I interferons are driving the disease manifestations of the interferonopathies it is at least likely. AGS and MS are immunologically and clinically very different and the proposed T helper cell polarizing effect of IFN-β in MS is probably not a dominant feature in the type I interferonopathies.
Signal transduction downstream of the type I interferon receptor involves activation of JAK1 and TYK2 followed by activation of STAT1 and STAT2, which together with IRF9 translocate to the nucleus to initiate transcription of interferon stimulated genes (ISG). Increasing experience suggests that treatment with JAK inhibitors is efficacious in some patients with interferonopathies (171). However, in one study, treatment with JAK inhibition before onset of symptoms did not prevent development of AGS (172). Additionally, based on the hypothesis that endogenous retroelements drive interferon signaling in AGS, a small (n=8) open-label pilot study of reverse transcriptase inhibitor therapy has been conducted with promising results (173).
In summary, interferon-related autoinflammation is qualitatively different from the classical autoinflammatory disorders with prominent IL-1β-biased inflammation. The neuroinflammation observed in interferonopathies is likely less dependent on recruitment of innate or adaptive immune cells. Although the pathogenesis is clearly related to excess type I interferon signaling, what part of this complex system is appropriate to target pharmacologically for these disorders is not well understood.
Hemophagocytic Syndromes
Familial Hemophagocytic Lymphohistiocytosis
Hemophagocytic lymphohistiocytosis (HLH) is a rare hyperinflammatory syndrome characterized by fever, cytopenias, and splenomegaly, which often leads to a sepsis-like condition with multiple organ dysfunction. HLH can be divided into familial HLH (FHL) caused typically by recessive mutations and secondary non-Mendelian forms. FHL is most frequently caused by loss-of-function variants in genes required for lymphocyte cytotoxicity, including genes that are required for CD8+ T cell and NK cell exocytosis. To date, mutations in PRF1 (MIM: 603553), UNC13D (MIM: 608898), STX11 (MIM: 603552), STXBP2 (MIM: 613101), and RHOG have been linked to FHL (174–178). Both FHL and secondary HLH often have an apparent triggering event, for example an infection or malignancy, leading to uncontrolled immune stimulation. The defective killing of target cells makes the host unable to clear the antigenic stimulus and control activated immune cells. This results in persistence and amplification of the immune response with excessive secretion of pro-inflammatory cytokines. In turn, the pro-inflammatory cytokines activate macrophages and mediate much of the tissue damage. Diagnostic HLH criteria have been established and a diagnosis is made based on the fulfillment of 5/8 of the criteria (179). Neuroinflammatory manifestations occur at any time during the disease course in FHL or secondary HLH but are not part of the diagnostic criteria. In pediatric HLH, neurological symptoms or signs have been reported in 13-73% of patients and pathological MRI or CSF findings in 33-91% (32). Importantly, when the disease presents as an isolated neuroinflammatory disorder, perhaps without the diagnostic criterion cytopenia, HLH may not be included as a differential diagnosis (180, 181). An international survey and literature review of confirmed FHL cases that presented as an isolated neuroinflammatory disease (CNS-HLH) has been reported (33). Common symptoms at presentation in this case series were ataxia, seizures, headache, motor impairment, and visual abnormalities. Brain MRI findings were non-specific and included a range of multifocal bilateral abnormalities including demyelination in the hemispheres, basal ganglia, cerebellum, or brain stem. Occasionally MRI may reveal a single mass-like lesion (182) or may mimic septic embolization (183). MRI abnormalities have been reported in FHL due to PRF1 variants even in the absence of neurological symptoms (184). Therefore, CNS-HLH is frequently initially diagnosed as acute demyelinating encephalomyelitis (ADEM), CNS vasculitis, meningitis, leukodystrophy, or MS (38, 185–188). The mean time from onset of symptoms of CNS-HLH to confirmed molecular diagnosis is more than two years in published cases and it is recognized that a fully developed HLH disease can arise several years after onset of CNS-HLH (33). Two patients with cerebellar involvement on MRI and consistent symptoms in the form of ataxia and dysarthria were shown to have had a FHL-associated biallelic PRF1 mutations without developing FHL before dying at the age of 1 and 5 years, respectively (189). Conversely, when in remission and off treatment for typical FHL, a relapse may present as isolated CNS-HLH (190). Importantly, compared to most of the other disorders reviewed herein, FHL is prone to rapidly develop into a life-threatening inflammatory condition and vigilance is required regarding early symptoms, which may be from the CNS.
Familial Hemophagocytic Lymphohistiocytosis-Related Disorders
An additional set of inherited disorders also frequently cause HLH. These include Griscelli syndrome type 2 (GS2, MIM: 607624), Chediak-Higashi syndrome (CHS, MIM: 214500), X-linked lymphoproliferative syndrome (XLP) type 1 (XLP1; MIM: 308240) and XLP type 2 (XLP2; MIM: 300635).
A distinct feature of GS2 and CHS is hypopigmentation because of defective granule transport in melanocytes. GS2 is an autosomal recessive disease caused by loss-of-function variants in RAB27A, which encodes a protein that facilitates secretory lysosome trafficking and exocytosis in several cell types (191). The impaired cytotoxic activity in T and NK cells can result in increased susceptibility to infections as well as failure to control immune responses and development of HLH. Microglial dependence on Rab27a for migration towards sites of injury in the CNS suggests a possible mechanism for neuroinflammation in the setting of Rab27a deficiency (192). A GS2 patient with isolated CNS-HLH presenting with developmental regression, seizures, and eventually status epilepticus has been described (193). Other cases of GS2 have been reported with neurological involvement in the form of encephalopathy, headache and tonsillar herniation, focal seizures, multifocal or diffuse white matter lesions, and pathological CSF (38–40). An atypical case of GS2 without hypopigmentation describes a 14-year old male that presented with myoclonus, dysmetria, dysarthria, ataxia, fever, and pancytopenia (37). The neuroinflammatory manifestations resolved within 3 months despite conservative management but returned after 2 months with hemiparesis, abducens nerve palsy, fever, and pancytopenia. Chediak-Higashi syndrome may have neurological manifestations, but these are almost always neurodegenerative, and MRI typically reveal atrophy but no focal lesions (194, 195).
Loss-of-function variants in SH2D1A and XIAP cause XLP type 1 and 2, respectively, both with X-linked recessive inheritance. SH2D1A codes for an adaptor protein expressed in T cells but its role in the pathogenesis of XLP1 is not fully established (196). Deletion of SH2D1A may result in mild immunodeficiency early in life in males but can also present as early-onset HLH. Clinical manifestations of XLP1 are typically triggered by Epstein-Barr virus (EBV) infection, resulting in deficient humoral immunity, polyclonal proliferation of B and T cells, lymphoma, and HLH. A case of Burkitt lymphoma in a 14-year old male with hypogammaglobulinemia followed by fatal CNS vasculitis has been reported (41). The CNS vasculitis manifested as short-term memory deficit and MRI revealed multiple edematous and hemorrhagic lesions. The symptomatology of other reported cases of XLP and CNS vasculitis includes headache, blurred vision, seizures, memory impairment, motor deficits. CSF analysis may reveal elevated white cells and protein. MRI often show aneurysms, T2 hyperintensities, and hemorrhagic lesions (42–45). Thus, severe neuroinflammatory symptoms are not isolated to FHL patients with defective lymphocyte cytotoxicity.
Comment on Neuroinflammation in Hemophagocytic Syndromes
Although all characterized gene variants that underlie FHL result in impaired lymphocyte cytotoxicity, the tendency for CNS involvement in FHL subtypes likely differ. Neuroinflammation in FHL due to PRF1 variants was observed in 36% of 124 cases in a multi-center study (197). The corresponding proportion associated with UNC13D variants was estimated at 60% (198, 199) and with STXBP2 variants at approximately 50% (34). Estimates were less certain for other FHL types but a study suggests that CNS involvement is less frequent in FHL due to STX11 variants (200). Furthermore, it is becoming increasingly recognized that hypomorphic variants with some residual function, often cause FHL with later onset and atypical clinical presentations, for example isolated CNS involvement (201–206).
The immunopathogenesis of HLH is described as a cytokine storm. Several genetic defects that predispose to HLH impair the delivery of cytotoxic granules from CD8+ T cells and NK cells, which is essential both for clearance of pathogens but also of activated antigen presenting cells. A feed-forward loop is initiated with increasing tissue infiltration and activation of immune cells that secrete a plethora of pro-inflammatory cytokines, most notably IFN-γ, TNF, IL-1, IL-2, IL-6, IL-10, IL-12, IL-18, and GM-CSF. Consequently, the established treatments for HLH involve broad immunosuppression to achieve remission while hematopoietic stem cell transplantation is needed to cure primary HLH (207). Animal models of FHL imply that IFN-γ derived from CD8+ T cells drive the pathogenesis (208, 209). However, HLH-like disease in the absence of IFN-γ has also been reported recently (210). There is a large body of work on the role of IFN-γ in neuroinflammation. Although protective effects have been suggested, most data point towards a pathogenic role. An early trial in MS patients showed that administration of IFN-γ worsened disease (211). Briefly, in experimental conditions, low doses of IFN-γ generally exert protective effects on neurons, microglia and oligodendrocytes whereas high doses appear to have the opposite effect (211). Although neutralization of specific cytokines such as TNF and IL-1 have been used as second line treatment for HLH, their efficacy is unknown. Novel treatment strategies that are being pursued include anti-IFN-γ and JAK-inhibition (212, 213).
In summary, the CNS is vulnerable to the hyperinflammation associated with FHL and related disorders, but even severe cases can have a good outcome if diagnosed early so that appropriate treatment can be initiated in parallel with arrangements for a hematopoietic stem cell transplantation.
Tolerance Defects
Loss-of-function variants in FOXP3, a key transcription factor in the development of regulatory T (Treg) cells, is the cause of X-linked recessive immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX, MIM: 304790). The Treg cell defect leads to loss of peripheral tolerance manifested as enteropathy, dermatitis, type I diabetes mellitus, hypoparathyroidism, autoimmune cytopenias, and other autoimmune diseases. IPEX-like syndromes may also arise due to defects in other Treg cell related genes such as loss-of-function variants in IL2RA2, CTLA4, and LRBA, or gain-of-function variants in STAT1 and STAT3 (214). A cohort of 173 patients with IPEX or IPEX-like syndrome have been reported and approximately 25% in both groups had neurological symptoms, which included seizures, ventriculomegaly, and developmental delay (46).
ALPS (MIM: 601859) is an autosomal dominant disease characterized by abnormal lymphocyte survival resulting in lymphoproliferation, autoimmunity, and secondary malignancies. Loss-of-function variants in Fas is the most common cause followed by FasL, and caspase-10. Autoimmunity typically manifests as autoimmune cytopenia but essentially any organ can be affected including the nervous system (47). ALPS without a specified genetic basis has been associated with NMO and Guillain Barré syndrome (47, 48).
Loss-of-function variants in CTLA4, a negative regulator of T cell activation has been described as a cause of ALPS (ALPS type V, MIM: 616100) but has more recently been defined as a disorder of its own. CTLA4 haploinsufficiency may cause a range of autoimmune and lymphoproliferative manifestations. In a series of 133 patients with CTLA-4 insufficiency, a penetrance of 67% was observed and upper range of age of onset was 50 years (215). Among the affected, 29% had neurological features. A wide range of neurological syndromes were observed including autoimmune encephalitis, demyelination, increased intracranial pressure, ischemic or hemorrhagic lesions. Others have described cerebellar lesions and spinal cord degeneration (49, 50). Altogether, neuroinflammatory manifestations are thus a feature in up to a third of patients with defects in immune tolerance.
Immunodeficiency Disorders
The complement system consists of approximately 30 different components and deficiencies have been described for all of them. The clinical consequences depend on which component is affected, but the spectrum broadly involves susceptibility to infections, autoimmunity, cardiovascular disease. Loss-of-function variants in any of the three subcomponent genes for C1q, i.e. C1QA, C1QB, or C1QC, cause the autosomal recessive condition C1q-deficiency (MIM: 613652), which stands out among the complement component deficiencies for being strongly linked to development of SLE. One study found that neurological symptoms at disease presentation occur to a similar extent in C1q-deficient and C1q-sufficient SLE/SLE-like patients (216) but C1q-deficient patients with SLE appear to relatively often present with neuropsychiatric symptoms (51). Seizures are a common manifestation of neuropsychiatric-SLE but encephalopathy, infarcts, and transverse myelitis have also been described (217). Neuroimaging findings include calcifications or ischemic lesions in the basal ganglia, vasculitis, and brain atrophy. Intracranial calcifications, a cardinal feature of the interferonopathies, allude to increased type I interferon also having an influence on the CNS in this SLE subset, possibly explained by the observed inhibitory role of C1q on type I interferon induction (52, 53, 218). C1q is expressed by neurons and opsonizes unwanted synapses for removal by microglia (219). Plausible indirect mechanisms for the observed neurological involvement include C1q-dependent removal of apoptotic cells or immune complexes. Most complement deficiencies are associated with increased susceptibility to bacterial meningitis due to the important role of complement for the elimination of encapsulated bacteria, such as meningococci (220). Aseptic meningitis has also been associated with several of the complement deficiencies, which often manifests as neutrophilic inflammation, making the distinction from septic meningitis difficult (51, 221–223).
Variants in TLR3 as well as TRIF, TRAF3, TBK1, IRF3, and NEMO that code for mediators downstream of TLR3, and UNC93B1 that is involved in TLR3 trafficking, all reduce type I interferon signaling and are reported genetic causes of herpes simplex encephalitis (HSE) (224–229). Disease penetrance is incomplete and very few HSE multiplex families have been described. Interestingly, even complete absence of TLR3 signaling does not appear to increase susceptibility to other infections than herpes simplex (230). Autoimmune encephalitis relatively often occurs after HSE, likely due to bystander activation and breaking of tolerance in the CNS (231). Thus, one can speculate that the risk of CNS autoimmunity is elevated in patients with type I interferon deficiencies.
Heterozygous loss-of-function variants in GATA2 (MIM: 614172) cause a wide range of defects including cytopenias, particularly affecting monocytes, B cells and NK cells, resulting in susceptibility to infections in addition to a risk of developing myelodysplastic syndrome/acute myeloid leukemia (232). GATA2 is an essential transcription factor for hematopoietic stem/progenitor cell renewal and differentiation (233). In a survey with 79 patients, the median age of onset was early adulthood and 8% were symptom-free at the age of 40 (54). Three of these patients (4%) presented with neurological symptoms in the form of intellectual disability, transient ischemic cerebral palsy, and progressive multifocal leukoencephalopathy (PML), respectively. PML is an often fatal neuroinflammatory consequence of reactivation of latent JC-virus, which is observed in patients with immunodeficiency, primary or secondary to a wide range of immunosuppressive drugs (234).
COPA encodes a protein involved in endoplasmic reticulum (ER)-Golgi transport. Variants in the gene cause the rare COPA-syndrome (MIM: 616414), mostly associated with manifestations in the lungs, kidneys, and joints. However, a case of COPA-syndrome and NMO has been described (235). The patient had bacterial meningitis at the age of 2 and onset of NMO at the age of 6. Immunological findings associated with loss of function COPA-variants include Th17 polarization and humoral autoimmunity consistent with the immunopathology of NMO (236, 237).
Loss-of-function variants in RANBP2 are associated with the autosomal dominant post-infectious and often fatal complication acute necrotizing encephalopathy type 1 (ANE1, MIM: 608033) (238, 239). Many different viruses and some bacteria have been implicated but no association appears to exist to severity of ANE1, suggesting that genetic or other factors are more influential after the initial trigger. RANBP2 encodes the nuclear pore protein RAN binding protein 2 that is ubiquitously expressed and has several described cellular functions. Hypothesized pathogenic mechanisms include metabolic and mitochondrial dysfunction as well as induction of a cytokine storm. TNF and IL-6 are elevated in the serum and CSF of ANE1 patients, but elevated cells in the CSF is not typically observed (239). Taken together, the rarity of these immunodeficiency disorders makes a causal relationship to any observed CNS involvement difficult to establish but for several of them there are plausible neuropathological mechanisms that lend some support to the presence of etiological links.
Comment on Neuroinflammation in Tolerance Defects and Immunodeficiency Disorders
Several of the neuroinflammatory manifestations described in these sections result from immune deficiencies leading to defective tolerance mechanisms and autoimmunity. IPEX and ALPS are prototypical autoimmune syndromes caused by defective Treg cell formation and lymphocyte apoptosis, respectively. The complement system contributes to clearance of potential autoantigens and type I interferon defects lead to fulminant infections, possibly resulting in bystander activation of autoreactive lymphocytes. However, these and other autoimmune syndromes are primarily associated with involvement of other organs than the CNS. Similarly, the observation that autoimmune diseases tend to cluster in families does not apply to CNS autoimmunity such as MS to the same extent (240). Together this implies that distinct requirements are needed for tolerance in the CNS to break. Although CNS surveillance of adaptive immune cells is well-described the threshold of activation is higher inside the BBB (241). Moreover, activation of innate immune cells locally by PAMPs or DAMPs does not elicit infiltration of neutrophils or monocytes, as is the case in most other tissues (242).
The mechanisms that underlie neuroinflammation in COPA or RANBP2 deficiency are unknown. These genes are expressed in most tissues throughout the body including the CNS (gtexportal.org) and therefore a neuroinflammatory cascade that originates within the CNS may be hypothesized, in contrast to most other gene defects described herein that clearly have major direct consequences on immune cells, thus making an inflammatory reaction that is initiated in the periphery and then spills over to the CNS a more likely scenario.
In brief, the pathogenic mechanisms that underlie neuroinflammation in disorders of immune tolerance or defense are elusive and likely heterogeneous. There is not much evidence that supports the involvement of actual autoreactive encephalitogenic lymphocytes in neuroinflammation related to tolerance defect disorders or any other disease category reviewed herein. New single cell methodologies can potentially shed light on pathophysiologic mechanisms underlying these sets of immune disorders.
Conclusion
With the growing use of high-throughput exome and genome sequencing in clinical practice, it has become increasingly recognized that adult onset of monogenic disease is not necessarily uncommon (243). Moreover, genetic variants associated with early and late onset Mendelian diseases may not be the same and adjustment of filtering criteria may be needed when analyzing the sequencing data (244). As genome sequencing is increasingly applied for clinical diagnostics, more disease-causing variants will be identified. The task of filtering through the large number of variants that likely have no clinical significance will continue to be a challenge. One important consideration when assessing the possible causative role of disease-associated variants is whether they are consistent with the patient’s phenotype. Herein we have summarized reported neuroinflammatory phenotypes observed in association with monogenic diseases. These CNS-manifestations are often not part of the typical disease description and therefore relevant pathogenic variants may be overlooked when tasked with matching sequencing results with a clinical neuroinflammatory presentation.
Based on available reports, some conclusions can be drawn regarding the distinguishing features of neuroinflammation in relation to the underlying immunopathogenesis. For example: IL-1β driven autoinflammation is often associated with headache and to a lesser extent seizures and aseptic meningitis, interferonopathies with chronic cerebral abnormalities such as calcifications and developmental defects, and HLH with prominent but unspecific signs neuroinflammation on MRI and in CSF. As awareness increases regarding the potential CNS involvement in inborn errors of immunity, the clinical characterization of reported cases will improve. This will help better define what is a typical neuroinflammatory manifestation in relation to the various types of immune defects, which in turn will facilitate the process of making a genetic diagnosis.
Author Contributions
HL reviewed the literature and drafted the manuscript. YTB revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful for funding from the Swedish Research Council, Swedish Cancer Foundation, Swedish Childhood Cancer Foundation, Knut and Alice Wallenberg Foundation, and Göran Gustafsson Foundation (to YTB). All figures were created with BioRender.com.
Glossary
References
1. Matricardi S, Farello G, Savasta S, Verrotti A. Understanding Childhood Neuroimmune Diseases of the Central Nervous System. Front Pediatr (2019) 7:511. doi: 10.3389/fped.2019.00511
2. Villoslada P, Zamvil SS. The Immune Signatures of Multiple Sclerosis: Lessons From Twin Studies. Proc Natl Acad Sci USA (2020) 117:24013–5. doi: 10.1073/pnas.2016711117
3. Akman-Demir G, Serdaroglu P, Tasçi B. Clinical Patterns of Neurological Involvement in Behçet’s Disease: Evaluation of 200 Patients. The Neuro-Behçet Study Group. Brain (1999) 122(Pt 11):2171–82. doi: 10.1093/brain/122.11.2171
4. McCreary D, Omoyinmi E, Hong Y, Mulhern C, Papadopoulou C, Casimir M, et al. Development and Validation of a Targeted Next-Generation Sequencing Gene Panel for Children With Neuroinflammation. JAMA Netw Open (2019) 2:1–13. doi: 10.1001/jamanetworkopen.2019.14274
5. Çakan M, Aktay Ayaz N, Keskindemirci G, Karadağ ŞG, Tanatar A, Sönmez HE. Serum Amyloid A as a Biomarker in Differentiating Attacks of Familial Mediterranean Fever From Acute Febrile Infections. Clin Rheumatol (2020) 39:249–53. doi: 10.1007/s10067-019-04765-1
6. Gedalia A, Zamir S. Neurologic Manifestations in Familial Mediterranean Fever. Pediatr Neurol (1993) 9:301–2. doi: 10.1016/0887-8994(93)90068-n
7. Ertekin V, Selimoğlu MA, Pirim I. Familial Mediterranean Fever in a Childhood Population in Eastern Turkey. Pediatr Int (2005) 47:640–4. doi: 10.1111/j.1442-200x.2005.02140.x
8. Feld O, Yahalom G, Livneh A. Neurologic and Other Systemic Manifestations in FMF: Published and Own Experience. Best Pract Res Clin Rheumatol (2012) 26:119–33. doi: 10.1016/j.berh.2012.01.004
9. Ugurlu S, Bolayir E, Candan F, Gumus C. Familial Mediterranean Fever and Multiple Sclerosis–A Case Report. Acta Reumatol Port (2009) 34:117–9.
10. Ozkaya O, Bek K, Alaca N, Ceyhan M, Açikgöz Y, Taşdemir HA. Cerebral Vasculitis in a Child With Henoch-Schönlein Purpura and Familial Mediterranean Fever. Clin Rheumatol (2007) 26:1729–32. doi: 10.1007/s10067-006-0485-x
11. Finsterer J, Stöllberger C, Shinar Y. Cranial Nerve Lesions and Abnormal Visually Evoked Potentials Associated With the M694V Mutation in Familial Mediterranean Fever. Clin Rheumatol (2002) 21:317–21. doi: 10.1007/s100670200083
12. Lossos A, Eliashiv S, Ben-Chetrit E, Reches A. Optic Neuritis Associated With Familial Mediterranean Fever. J Clin Neuroophthalmol (1993) 13:141–3.
13. ter Haar NM, Jeyaratnam J, Lachmann HJ, Simon A, Brogan PA, Doglio M, et al. The Phenotype and Genotype of Mevalonate Kinase Deficiency: A Series of 114 Cases From the Eurofever Registry. Arthritis Rheumatol (2016) 68:2795–805. doi: 10.1002/art.39763
14. Bader-Meunier B, Florkin B, Sibilia J, Acquaviva C, Hachulla E, Grateau G, et al. Mevalonate Kinase Deficiency: A Survey of 50 Patients. Pediatrics (2011) 128(1):e152–9. doi: 10.1542/peds.2010-3639
15. Parker T, Keddie S, Kidd D, Lane T, Maviki M, Hawkins PN, et al. Neurology of the Cryopyrin-Associated Periodic Fever Syndrome. Eur J Neurol (2016) 23:1145–51. doi: 10.1111/ene.12965
16. Levy R, Gérard L, Kuemmerle-Deschner J, Lachmann HJ, Koné-Paut I, Cantarini L, et al. Phenotypic and Genotypic Characteristics of Cryopyrin-Associated Periodic Syndrome: A Series of 136 Patients From the Eurofever Registry. Ann Rheum Dis (2015) 74:2043–9. doi: 10.1136/annrheumdis-2013-204991
17. Qin W, Wu D, Luo Y, Zhao M, Wang Y, Shi X, et al. Neurological Manifestations of Autoinflammatory Diseases in Chinese Adult Patients. Semin Arthritis Rheum (2020) 50:1500–6. doi: 10.1016/j.semarthrit.2019.12.003
18. Borghini S, Tassi S, Chiesa S, Caroli F, Carta S, Caorsi R, et al. Clinical Presentation and Pathogenesis of Cold-Induced Autoinflammatory Disease in a Family With Recurrence of an NLRP12 Mutation. Arthritis Rheum (2011) 63:830–9. doi: 10.1002/art.30170
19. Lachmann HJ, Papa R, Gerhold K, Obici L, Touitou I, Cantarini L, et al. The Phenotype of TNF Receptor-Associated Autoinflammatory Syndrome (TRAPS) at Presentation: A Series of 158 Cases From the Eurofever/EUROTRAPS International Registry. Ann Rheum Dis (2014) 73:2160–7. doi: 10.1136/annrheumdis-2013-204184
20. Minden K. Tumour Necrosis Factor Receptor Associated Periodic Syndrome (TRAPS) With Central Nervous System Involvement. Ann Rheum Dis (2004) 63:1356–7. doi: 10.1136/ard.2003.016006
21. Ueda N, Ida H, Washio M, Miyahara H, Tokunaga S, Tanaka F, et al. Clinical and Genetic Features of Patients With TNFRSF1A Variants in Japan: Findings of a Nationwide Survey. Arthritis Rheumatol (2016) 68:2760–71. doi: 10.1002/art.39793
22. Duan R, Liu Q, Li J, Bian X, Yuan Q, Li Y, et al. A De Novo Frameshift Mutation in TNFAIP3 Impairs A20 Deubiquitination Function to Cause Neuropsychiatric Systemic Lupus Erythematosus. J Clin Immunol (2019) 39:795–804. doi: 10.1007/s10875-019-00695-4
23. Aeschlimann FA, Batu ED, Canna SW, Go E, Gül A, Hoffmann P, et al. A20 Haploinsufficiency (HA20): Clinical Phenotypes and Disease Course of Patients With a Newly Recognised NF-kB-Mediated Autoinflammatory Disease. Ann Rheum Dis (2018) 77:728–35. doi: 10.1136/annrheumdis-2017-212403
24. Kadowaki T, Ohnishi H, Kawamoto N, Hori T, Nishimura K, Kobayashi C, et al. Haploinsufficiency of A20 Causes Autoinflammatory and Autoimmune Disorders. J Allergy Clin Immunol (2018) 141:1485–8.e11. doi: 10.1016/j.jaci.2017.10.039
25. Crow YJ, Black DN, Ali M, Bond J, Jackson AP, Lefson M, et al. Cree Encephalitis Is Allelic With Aicardi-Goutières Syndrome: Implications for the Pathogenesis of Disorders of Interferon Alpha Metabolism. J Med Genet (2003) 40:183–7. doi: 10.1136/jmg.40.3.183
26. Schmelzer L, Smitka M, Wolf C, Lucas N, Tüngler V, Hahn G, et al. Variable Clinical Phenotype in Two Siblings With Aicardi-Goutières Syndrome Type 6 and a Novel Mutation in the ADAR Gene. Eur J Paediatr Neurol (2018) 22:186–9. doi: 10.1016/j.ejpn.2017.11.003
27. Stam AH, Kothari PH, Shaikh A, Gschwendter A, Jen JC, Hodgkinson S, et al. Retinal Vasculopathy With Cerebral Leukoencephalopathy and Systemic Manifestations. Brain (2016) 139:2909–22. doi: 10.1093/brain/aww217
28. Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, et al. Human Intracellular ISG15 Prevents Interferon-α/β Over-Amplification and Auto-Inflammation. Nature (2015) 517:89–93. doi: 10.1038/nature13801
29. Rodero MP, Tesser A, Bartok E, Rice GI, Della Mina E, Depp M, et al. Type I Interferon-Mediated Autoinflammation Due to DNase II Deficiency. Nat Commun (2017) 8(1):2176. doi: 10.1038/s41467-017-01932-3
30. Briggs TA, Rice GI, Daly S, Urquhart J, Gornall H, Bader-meunier B, et al. Tartrate-Resistant Acid Phosphatase Deficiency Causes a Bone Dysplasia With Autoimmunity and a Type I Interferon Expression Signature. Nat Genet (2011) 43:127–32. doi: 10.1038/ng.748
31. Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, et al. Mutations in Proteasome Subunit β Type 8 Cause Chronic Atypical Neutrophilic Dermatosis With Lipodystrophy and Elevated Temperature With Evidence of Genetic and Phenotypic Heterogeneity. Arthritis Rheum (2012) 64:895–907. doi: 10.1002/art.33368
32. Horne AC, Wickström R, Jordan MB, Yeh EA, Naqvi A, Henter J-II, et al. How to Treat Involvement of the Central Nervous System in Hemophagocytic Lymphohistiocytosis? Curr Treat Options Neurol (2017) 19:3. doi: 10.1007/s11940-017-0439-4
33. Blincoe A, Heeg M, Campbell PK, Hines M, Khojah A, Klein-Gitelman M, et al. Neuroinflammatory Disease as an Isolated Manifestation of Hemophagocytic Lymphohistiocytosis. J Clin Immunol (2020) 40:901–16. doi: 10.1007/s10875-020-00814-6
34. Meeths M, Entesarian M, Al-Herz W, Chiang SCC, Wood SM, Al-Ateeqi W, et al. Spectrum of Clinical Presentations in Familial Hemophagocytic Lymphohistiocytosis Type 5 Patients With Mutations in STXBP2. Blood (2010) 116:2635–43. doi: 10.1182/blood-2010-05-282541
35. Benson LA, Li H, Henderson LA, Solomon IH, Soldatos A, Murphy J, et al. Pediatric CNS-Isolated Hemophagocytic Lymphohistiocytosis. Neurol Neuroimmunol Neuroinflamm (2019) 6(3):e560. doi: 10.1212/NXI.0000000000000560
36. Işikay S. Cerebellar Involvement of Griscelli Syndrome Type 2. BMJ Case Rep (2014) 2014:1–2. doi: 10.1136/bcr-2014-206703
37. Woodward KE, Shah RM, Benseler S, Wei XC, Ng D, Grossman J, et al. Considering Immunologic and Genetic Evaluation for HLH in Neuroinflammation: A Case of Griscelli Syndrome Type 2 With Neurological Symptoms and a Lack of Albinism. Pediatr Blood Cancer (2020) 67:3–5. doi: 10.1002/pbc.28312
38. Tesi B, Rascon J, Chiang SCC, Burnyte B, Löfstedt A, Fasth A, et al. A RAB27A 5′ Untranslated Region Structural Variant Associated With Late-Onset Hemophagocytic Lymphohistiocytosis and Normal Pigmentation. J Allergy Clin Immunol (2018) 142:317–21.e8. doi: 10.1016/j.jaci.2018.02.031
39. Zhang Q, Zhao YZ, Ma HH, Wang D, Zhang N, Li ZG, et al. Successful Rescue of a Lethal Griscelli Syndrome Type 2 Presenting With Neurological Involvement and Hemophagocytic Lymphohistiocytosis: A Case Report. BMC Pediatr (2021) 21:1–7. doi: 10.1186/s12887-021-02720-1
40. Castaño-Jaramillo LM, Lugo-Reyes SO, Cruz Muñoz ME, Scheffler-Mendoza SC, Duran McKinster C, Yamazaki-Nakashimada MA, et al. Saez-De-Ocariz Gutierrez M Del M. Diagnostic and Therapeutic Caveats in Griscelli Syndrome. Scand J Immunol (2021) 93:1–7. doi: 10.1111/sji.13034
41. Neves JF, Raga LT, Chiang SCC, Tesi B, Vieira JP, Cordeiro AI, et al. Fatal Central Nervous System Lymphocytic Vasculitis After Treatment for Burkitt Lymphoma in a Patient With a Sh2d1a Mutation. Pediatr Infect Dis J (2019) 38:e29–31. doi: 10.1097/INF.0000000000002154
42. Talaat KR, Rothman JA, Cohen JI, Santi M, Choi JK, Guzman M, et al. Lymphocytic Vasculitis Involving the Central Nervous System Occurs in Patients With X-Linked Lymphoproliferative Disease in the Absence of Epstein-Barr Virus Infection. Pediatr Blood Cancer (2009) 53:1120–3. doi: 10.1002/pbc.22185
43. Weeks JK, Helton KJ, Conley ME, Onciu M, Khan RB. Diffuse CNS Vasculopathy With Chronic Epstein-Barr Virus Infection in X-Linked Lymphoproliferative Disease. Am J Neuroradiol (2006) 27:884–6.
44. Verhelst H, Van Coster R, Bockaert N, Laureys G, Latour S, Fischer A, et al. Limbic Encephalitis as Presentation of a SAP Deficiency. Neurology (2007) 69:218–9. doi: 10.1212/01.wnl.0000265597.56202.6c
45. Blackburn PR, Lin WL, Miller DA, Lorenzo-Betancor O, Edwards ES, Zimmermann MT, et al. X-Linked Lymphoproliferative Syndrome Presenting as Adult-Onset Multi-Infarct Dementia. J Neuropathol Exp Neurol (2019) 78:460–6. doi: 10.1093/jnen/nlz018
46. Gambineri E, Mannurita SC, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, et al. Patients With the Phenotype of Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) Syndrome. Front Immunol (2018) 9:2411. doi: 10.3389/fimmu.2018.02411
47. Teachey DT. New Advances in the Diagnosis and Treatment of Autoimmune Lymphoproliferative Syndrome. Curr Opin Pediatr (2012) 24:1–8. doi: 10.1097/MOP.0b013e32834ea739
48. Cooper N, Te Water Naude J, Connor PP. Neuromyelitis Optica Complicating Autoimmune Lymphoproliferative Syndrome in a 4-Year-Old Girl. Br J Haematol (2011) 152:247–7. doi: 10.1111/j.1365-2141.2010.08460.x
49. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal Dominant Immune Dysregulation Syndrome in Humans With CTLA4 Mutations. Nat Med (2014) 20:1410–6. doi: 10.1038/nm.3746
50. Long H, Chen Z, Xiao B, Tang B, Jiang H. Myeleterosis in an ALPS5 Patient With Primary Immune Dysregulation Syndrome. CNS Neurosci Ther (2020) 26:773–5. doi: 10.1111/cns.13301
51. van Schaarenburg RA, Magro-Checa C, Bakker JA, Teng YKOO, Bajema IM, Huizinga TW, et al. C1q Deficiency and Neuropsychiatric Systemic Lupus Erythematosus. Front Immunol (2016) 7:647. doi: 10.3389/fimmu.2016.00647
52. Troedson C, Wong M, Dalby-Payne J, Wilson M, Dexter M, Rice GI, et al. Systemic Lupus Erythematosus Due to C1q Deficiency With Progressive Encephalopathy, Intracranial Calcification and Acquired Moyamoya Cerebral Vasculopathy. Lupus (2013) 22:639–43. doi: 10.1177/0961203313486950
53. Lubbers R, Beaart-van de Voorde LJJ, van Leeuwen K, de Boer M, Gelderman KA, van den Berg MJ, et al. Complex Medical History of a Patient With a Compound Heterozygous Mutation in C1QC. Lupus (2019) 28:1255–60. doi: 10.1177/0961203319865029
54. Donadieu J, Lamant M, Fieschi C, De Fontbrune FS, Caye A, Ouachee M, et al. Natural History of GATA2 Deficiency in a Survey of 79 French and Belgian Patients. Haematologica (2018) 103:1278–87. doi: 10.3324/haematol.2017.181909
55. Aldea A, Campistol JM, Arostegui JI, Rius J, Maso M, Vives J, et al. A Severe Autosomal-Dominant Periodic Inflammatory Disorder With Renal AA Amyloidosis and Colchicine Resistance Associated to the MEFV H478Y Variant in a Spanish Kindred: An Unusual Familial Mediterranean Fever Phenotype or Another MEFV-Associated Periodic. Am J Med Genet (2004) 124 A:67–73. doi: 10.1002/ajmg.a.20296
56. Rowczenio DM, Iancu DS, Trojer H, Gilbertson JA, Gillmore JD, Wechalekar AD, et al. Autosomal Dominant Familial Mediterranean Fever in Northern European Caucasians Associated With Deletion of P.M694 Residue-a Case Series and Genetic Exploration. Rheumatol (Oxford) (2017) 56:209–13. doi: 10.1093/rheumatology/kew058
57. Heilig R, Broz P. Function and Mechanism of the Pyrin Inflammasome. Eur J Immunol (2018) 48:230–8. doi: 10.1002/eji.201746947
58. Sugie M, Ouchi T, Kishida D, Yasaki S. Atypical Type of Familial Mediterranean Fever: An Underdiagnosed Cause of Chronic Aseptic Meningitis. Neurol Clin Neurosci (2018) 6:191–3. doi: 10.1111/ncn3.12232
59. Kimura K, Mizooka M, Migita K, Ishida R, Matsumoto M, Yamasaki S, et al. Five Cases of Familial Mediterranean Fever in Japan: The Relationship With MEFV Mutations. Intern Med (2018) 57:2425–9. doi: 10.2169/internalmedicine.0057-17
60. Hosoi T, Ishii K, Tozaka N, Kishida D, Sekijima Y, Tamaoka A. Familial Mediterranean Fever Is Important in the Differential Diagnosis of Recurrent Aseptic Meningitis in Japan. Intern Med (2020) 59:125–8. doi: 10.2169/internalmedicine.3432-19
61. Vilaseca J, Tor J, Guardia J, Bacardi R. Periodic Meningitis and Familial Mediterranean Fever. Arch Intern Med (1982) 142:378–9.
62. Chae JJ, Cho Y, Lee G, Cheng J, Liu PP, Feigenbaum L, et al. Article Gain-Of-Function Pyrin Mutations Induce NLRP3 Protein - Independent Interleukin-1 B Activation and Severe Autoinflammation in Mice. Immunity (2011) 34:755–68. doi: 10.1016/j.immuni.2011.02.020
63. Masters SL, Lagou V, Jéru I, Baker PJ, Van Eyck L, Parry DA, et al. Familial Autoinflammation With Neutrophilic Dermatosis Reveals a Regulatory Mechanism of Pyrin Activation. Sci Transl Med (2016) 8:1–10. doi: 10.1126/scitranslmed.aaf1471
64. Simon A, Kremer HPH, Wevers RA, Scheffer H, De Jong JG, van der Meer JWM, et al. Mevalonate Kinase Deficiency: Evidence for a Phenotypic Continuum. Neurology (2004) 62:994–7. doi: 10.1212/01.WNL.0000115390.33405.F7
65. Brennenstuhl H, Nashawi M, Schröter J, Baronio F, Beedgen L, Gleich F, et al. Phenotypic Diversity, Disease Progression, and Pathogenicity of MVK Missense Variants in Mevalonic Aciduria. J Inherit Metab Dis (2021) 44(5):1272–87. doi: 10.1002/jimd.12412
66. Akula MK, Shi M, Jiang Z, Foster CE, Miao D, Li AS, et al. Control of the Innate Immune Response by the Mevalonate Pathway. Nat Immunol (2016) 17:922–9. doi: 10.1038/ni.3487
67. Park YH, Wood G, Kastner DL, Chae JJ. Pyrin Inflammasome Activation and RhoA Signaling in the Autoinflammatory Diseases FMF and HIDS. Nat Immunol (2016) 17:914–21. doi: 10.1038/ni.3457
68. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a New Gene Encoding a Putative Pyrin-Like Protein Causes Familial Cold Autoinflammatory Syndrome and Muckle-Wells Syndrome. Nat Genet (2001) 29:301–5. doi: 10.1038/ng756
69. Feldmann J, Prieur A-M, Quartier P, Berquin P, Certain S, Cortis E, et al. Chronic Infantile Neurological Cutaneous and Articular Syndrome is Caused by Mutations in CIAS1, A Gene Highly Expressed in Polymorphonuclear Cells and Chondrocytes. Am J Hum Genet (2002) 71:198–203. doi: 10.1086/341357
70. Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, et al. De Novo CIAS1 Mutations, Cytokine Activation, and Evidence for Genetic Heterogeneity in Patients With Neonatal-Onset Multisystem Inflammatory Disease (NOMID): A New Member of the Expanding Family of Pyrin-Associated Autoinflammatory Diseases. Arthritis Rheum (2002) 46:3340–8. doi: 10.1002/art.10688
71. Jéru I, Hayrapetyan H, Duquesnoy P, Sarkisian T, Amselem S. PYPAF1 Nonsense Mutation in a Patient With an Unusual Autoinflammatory Syndrome: Role of PYPAF1 in Inflammation. Arthritis Rheum (2006) 54:508–14. doi: 10.1002/art.21618
72. Kitley JL, Lachmann HJ, Pinto A, Ginsberg L. Neurologic Manifestations of the Cryopyrin-Associated Periodic Syndrome. Neurology (2010) 74:1267–70. doi: 10.1212/WNL.0b013e3181d9ed69
73. Schuh E, Lohse P, Ertl-Wagner B, Witt M, Krumbholz M, Frankenberger M, et al. Expanding Spectrum of Neurologic Manifestations in Patients With NLRP3 Low-Penetrance Mutations. Neurol Neuroimmunol Neuroinflamm (2015) 2(4):e109. doi: 10.1212/NXI.0000000000000109
74. Schwarzbach CJ, Schmitt WH, Szabo K, Bäzner H, Hennerici MG, Blahak C. Chorea in a Patient With Cryopyrin-Associated Periodic Syndrome. Neurology (2016) 86:241–4. doi: 10.1212/WNL.0000000000002300
75. Mamoudjy N, Maurey H, Marie I, Koné-Paut I, Deiva K. Neurological Outcome of Patients With Cryopyrin-Associated Periodic Syndrome (CAPS). Orphanet J Rare Dis (2017) 12:1–7. doi: 10.1186/s13023-017-0589-1
76. Kilic H, Sahin S, Duman C, Adrovic A, Barut K, Turanli ET, et al. Spectrum of the Neurologic Manifestations in Childhood-Onset Cryopyrin-Associated Periodic Syndrome. Eur J Paediatr Neurol (2019) 23:466–72. doi: 10.1016/j.ejpn.2019.03.006
77. Höhne C, Schuh E, Kümpfel T, Straube A. Cryopyrin-Associated Periodic Fever Syndrome Manifesting as Tolosa-Hunt Syndrome. Cephalalgia (2016) 36:1392–6. doi: 10.1177/0333102416629239
78. Behringer J, Ryan M, Miller M, Jaju A. Magnetic Resonance Imaging Findings in a Patient With Cryopyrin-Associated Periodic Syndrome: A Rare Hereditary Multi-System Inflammatory Disorder. Neuroradiol J (2019) 32:420–5. doi: 10.1177/1971400919863712
79. Luo Y, Reis C, Chen S. NLRP3 Inflammasome in the Pathophysiology of Hemorrhagic Stroke: A Review. Curr Neuropharmacol (2019) 17:582–9. doi: 10.2174/1570159X17666181227170053
80. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline Mutations in the Extracellular Domains of the 55 kDa TNF Receptor, TNFR1, Define a Family of Dominantly Inherited Autoinflammatory Syndromes. Cell (1999) 97:133–44. doi: 10.1016/s0092-8674(00)80721-7
81. Lobito AA, Kimberley FC, Muppidi JR, Komarow H, Jackson AJ, Hull KM, et al. Abnormal Disulfide-Linked Oligomerization Results in ER Retention and Altered Signaling by TNFR1 Mutants in TNFR1-Associated Periodic Fever Syndrome (TRAPS). Blood (2006) 108:1320–7. doi: 10.1182/blood-2005-11-006783
82. Jéru I, Duquesnoy P, Fernandes-Alnemri T, Cochet E, Yu JW, Lackmy-Port-Lis M, et al. Mutations in NALP12 Cause Hereditary Periodic Fever Syndromes. Proc Natl Acad Sci USA (2008) 105:1614–9. doi: 10.1073/pnas.0708616105
83. Jéru I, Le Borgne G, Cochet E, Hayrapetyan H, Duquesnoy P, Grateau G, et al. Identification and Functional Consequences of a Recurrent NLRP12 Missense Mutation in Periodic Fever Syndromes. Arthritis Rheum (2011) 63:1459–64. doi: 10.1002/art.30241
84. Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An Autoinflammatory Disease With Deficiency of the Interleukin-1–Receptor Antagonist. N Engl J Med (2009) 360:2426–37. doi: 10.1056/nejmoa0807865
85. Liu T, Zhang L, Joo D, Sun S-C. NF-κb Signaling in Inflammation. Signal Transduct Target Ther 2:17023. doi: 10.1038/sigtrans.2017.23
86. Malynn BA, Ma A. A20: A Multifunctional Tool for Regulating Immunity and Preventing Disease. Cell Immunol (2019) 340:103914. doi: 10.1016/j.cellimm.2019.04.002
87. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-Of-Function Mutations in TNFAIP3 Leading to A20 Haploinsufficiency Cause an Early-Onset Autoinflammatory Disease. Nat Genet (2016) 48:67–73. doi: 10.1038/ng.3459
88. Takagi M, Ogata S, Ueno H, Yoshida K, Yeh T, Hoshino A, et al. Haploinsufficiency of TNFAIP3 (A20) by Germline Mutation Is Involved in Autoimmune Lymphoproliferative Syndrome. J Allergy Clin Immunol (2017) 139:1914–22. doi: 10.1016/j.jaci.2016.09.038
89. Martens A, van Loo G. A20 at the Crossroads of Cell Death, Inflammation, and Autoimmunity. Cold Spring Harb Perspect Biol (2020) 12:a036418. doi: 10.1101/cshperspect.a036418
90. Voet S, Mc Guire C, Hagemeyer N, Martens A, Schroeder A, Wieghofer P, et al. A20 Critically Controls Microglia Activation and Inhibits Inflammasome-Dependent Neuroinflammation. Nat Commun (2018) 9:2036. doi: 10.1038/s41467-018-04376-5
91. Lohr NJ, Molleston JP, Strauss KA, Torres-Martinez W, Sherman EA, Squires RH, et al. Human ITCH E3 Ubiquitin Ligase Deficiency Causes Syndromic Multisystem Autoimmune Disease. Am J Hum Genet (2010) 86:447–53. doi: 10.1016/j.ajhg.2010.01.028
92. Kursun O, Yemisci M, van den Maagdenberg AMJM, Karatas H. Migraine and Neuroinflammation: The Inflammasome Perspective. J Headache Pain (2021) 22:55. doi: 10.1186/s10194-021-01271-1
93. Coşkun S, Varol S, Özdemir HH, Çelik SB, Balduz M, Camkurt MA, et al. Association Between Sequence Variations of the Mediterranean Fever Gene and the Risk of Migraine: A Case-Control Study. Neuropsychiatr Dis Treat (2016) 12:2225–32. doi: 10.2147/NDT.S109414
94. Mukherjee S, Arisi GM, Mims K, Hollingsworth G, O’Neil K, Shapiro LA. Neuroinflammatory Mechanisms of Post-Traumatic Epilepsy. J Neuroinflamm (2020) 17:193. doi: 10.1186/s12974-020-01854-w
95. Chong S-A, Balosso S, Vandenplas C, Szczesny G, Hanon E, Claes K, et al. Intrinsic Inflammation Is a Potential Anti-Epileptogenic Target in the Organotypic Hippocampal Slice Model. Neurotherapeutics (2018) 15:470–88. doi: 10.1007/s13311-018-0607-6
96. Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate Immune Sensing of Bacterial Modifications of Rho GTPases by the Pyrin Inflammasome. Nature (2014) 513:237–41. doi: 10.1038/nature13449
97. Stefania S, Colia R, Cinzia R, Corrado A, Cantatore FP. Off-Label Use of Anti-IL-1 Drugs in Rheumatic Diseases. Int J Immunopathol Pharmacol (2021) 35:20587384211006584. doi: 10.1177/20587384211006584
98. Savic S, Caseley EA, McDermott MF. Moving Towards a Systems-Based Classification of Innate Immune-Mediated Diseases. Nat Rev Rheumatol (2020) 16:222–37. doi: 10.1038/s41584-020-0377-5
99. Hentgen V, Vinit C, Fayand A, Georgin-Lavialle S. The Use of Interleukine-1 Inhibitors in Familial Mediterranean Fever Patients: A Narrative Review. Front Immunol (2020) 11:971. doi: 10.3389/fimmu.2020.00971
100. Nedjai B, Hitman GA, Quillinan N, Coughlan RJ, Church L, McDermott MF, et al. Proinflammatory Action of the Antiinflammatory Drug Infliximab in Tumor Necrosis Factor Receptor-Associated Periodic Syndrome. Arthritis Rheum (2009) 60:619–25. doi: 10.1002/art.24294
101. TNF Neutralization in MS: Results of a Randomized, Placebo-Controlled Multicenter Study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology (1999) 53(3):457–65.
102. Yu M-P, Xu X-S, Zhou Q, Deuitch N, Lu M-P. Haploinsufficiency of A20 (HA20): Updates on the Genetics, Phenotype, Pathogenesis and Treatment. World J Pediatr (2020) 16:575–84. doi: 10.1007/s12519-019-00288-6
103. Musella A, Fresegna D, Rizzo FR, Gentile A, De Vito F, Caioli S, et al. ‘Prototypical’ Proinflammatory Cytokine (IL-1) in Multiple Sclerosis: Role in Pathogenesis and Therapeutic Targeting. Expert Opin Ther Targets (2020) 24:37–46. doi: 10.1080/14728222.2020.1709823
104. Depino AM, Alonso M, Ferrari C, del Rey A, Anthony D, Besedovsky H, et al. Learning Modulation by Endogenous Hippocampal IL-1: Blockade of Endogenous IL-1 Facilitates Memory Formation. Hippocampus (2004) 14:526–35. doi: 10.1002/hipo.10164
105. Liu X, Nemeth DP, McKim DB, Zhu L, DiSabato DJ, Berdysz O, et al. Cell-Type-Specific Interleukin 1 Receptor 1 Signaling in the Brain Regulates Distinct Neuroimmune Activities. Immunity (2019) 50:317–33.e6. doi: 10.1016/j.immuni.2018.12.012
106. Hauptmann J, Johann L, Marini F, Kitic M, Colombo E, Mufazalov IA, et al. Interleukin-1 Promotes Autoimmune Neuroinflammation by Suppressing Endothelial Heme Oxygenase-1 at the Blood–Brain Barrier. Acta Neuropathol (2020) 140:549–67. doi: 10.1007/s00401-020-02187-x
107. Brandão WN, De Oliveira MG, Andreoni RT, Nakaya H, Farias AS, Peron JPS. Neuroinflammation at Single Cell Level: What Is New? J Leukoc Biol (2020) 108:1129–37. doi: 10.1002/JLB.3MR0620-035R
108. Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, et al. Critical Regulation of Early Th17 Cell Differentiation by Interleukin-1 Signaling. Immunity (2009) 30:576–87. doi: 10.1016/j.immuni.2009.02.007
109. Sha Y, Markovic-Plese S. Activated IL-1ri Signaling Pathway Induces Th17 Cell Differentiation via Interferon Regulatory Factor 4 Signaling in Patients With Relapsing-Remitting Multiple Sclerosis. Front Immunol (2016) 7:543. doi: 10.3389/fimmu.2016.00543
110. Galli E, Hartmann FJ, Schreiner B, Ingelfinger F, Arvaniti E, Diebold M, et al. GM-CSF and CXCR4 Define a T Helper Cell Signature in Multiple Sclerosis. Nat Med (2019) 25:1290–300. doi: 10.1038/s41591-019-0521-4
111. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature (2017) 541:481–7. doi: 10.1038/nature21029
112. Mandolesi G, Gentile A, Musella A, Fresegna D, De Vito F, Bullitta S, et al. Synaptopathy Connects Inflammation and Neurodegeneration in Multiple Sclerosis. Nat Rev Neurol (2015) 11:711–24. doi: 10.1038/nrneurol.2015.222
113. Borhani-Haghighi A, Kardeh B, Banerjee S, Yadollahikhales G, Safari A, Sahraian MA, et al. Neuro-Behcet’s Disease: An Update on Diagnosis, Differential Diagnoses, and Treatment. Mult Scler Relat Disord (2020) 39:101906. doi: 10.1016/j.msard.2019.101906
114. Ishikawa H, Shindo A, Ii Y, Kishida D, Niwa A, Nishiguchi Y, et al. MEFV Gene Mutations in Neuro-Behçet’s Disease and Neuro-Sweet Disease. Ann Clin Transl Neurol (2019) 6:2595–600. doi: 10.1002/acn3.50937
115. Ishikawa H, Shindo A, Ii Y, Niwa A, Matsuura K, Kishida D, et al. Mediterranean Fever Gene Mutations in Patients With Possible Neuro-Sweet Disease: A Case Series. J Neurol Neurosurg Psychiatry (2018) 89:1119–21. doi: 10.1136/jnnp-2017-316667
116. Korkmaz C, Üsküdar Cansu D, Kabay SC. Familial Coexistence of Demyelinating Diseases and Familial Mediterranean Fever. Rheumatol Int (2021) 13. doi: 10.1007/s00296-021-04821-7
117. Vidmar L, Maver A, Drulović J, Sepčić J, Novaković I, Ristič S, et al. Multiple Sclerosis Patients Carry an Increased Burden of Exceedingly Rare Genetic Variants in the Inflammasome Regulatory Genes. Sci Rep (2019) 9:1–10. doi: 10.1038/s41598-019-45598-x
118. Mulazzani E, Wagner D, Havla J, Schlüter M, Meinl I, Gerdes LA, et al. Neurological Phenotypes in Patients With NLRP3-, MEFV-, and TNFRSF1A Low-Penetrance Variants. J Neuroinflamm (2020) 17:1–13. doi: 10.1186/s12974-020-01867-5
119. Vilariño-Güell C, Zimprich A, Martinelli-Boneschi F, Herculano B, Wang Z, Matesanz F, et al. Exome Sequencing in Multiple Sclerosis Families Identifies 12 Candidate Genes and Nominates Biological Pathways for the Genesis of Disease. (2019) 15(6):e1008180. doi: 10.1371/journal.pgen.1008180
120. Aicardi J, Goutières F. A Progressive Familial Encephalopathy in Infancy With Calcifications of the Basal Ganglia and Chronic Cerebrospinal Fluid Lymphocytosis. Ann Neurol (1984) 15:49–54. doi: 10.1002/ana.410150109
121. Rice GI, Forte GMA, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, et al. Assessment of Interferon-Related Biomarkers in Aicardi-Goutières Syndrome Associated With Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: A Case-Control Study. Lancet Neurol (2013) 12:1159–69. doi: 10.1016/S1474-4422(13)70258-8
122. Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the Gene Encoding the 3’-5’ DNA Exonuclease TREX1 Cause Aicardi-Goutières Syndrome at the AGS1 Locus. Nat Genet (2006) 38:917–20. doi: 10.1038/ng1845
123. Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, et al. Mutations in Genes Encoding Ribonuclease H2 Subunits Cause Aicardi-Goutières Syndrome and Mimic Congenital Viral Brain Infection. Nat Genet (2006) 38:910–6. doi: 10.1038/ng1842
124. Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, et al. Mutations Involved in Aicardi-Goutières Syndrome Implicate SAMHD1 as Regulator of the Innate Immune Response. Nat Genet (2009) 41:829–32. doi: 10.1038/ng.373
125. Rice GI, Kasher PR, Forte GMA, Mannion NM, Greenwood SM, Szynkiewicz M, et al. Mutations in ADAR1 Cause Aicardi-Goutières Syndrome Associated With a Type I Interferon Signature. Nat Genet (2012) 44:1243–8. doi: 10.1038/ng.2414
126. Rice GI, Del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, et al. Gain-Of-Function Mutations in IFIH1 Cause a Spectrum of Human Disease Phenotypes Associated With Upregulated Type I Interferon Signaling. Nat Genet (2014) 46:503–9. doi: 10.1038/ng.2933
127. Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El-Daher MT, et al. cGAS-Mediated Induction of Type I Interferon Due to Inborn Errors of Histone pre-mRNA Processing. Nat Genet (2020) 52:1364–72. doi: 10.1038/s41588-020-00737-3
128. Crow YJ. Type I Interferonopathies: Mendelian Type I Interferon Up-Regulation. Curr Opin Immunol (2015) 32:7–12. doi: 10.1016/j.coi.2014.10.005
129. Mazur DJ, Perrino FW. Identification and Expression of the TREX1 and TREX2 cDNA Sequences Encoding Mammalian 3’–>5’ Exonucleases. J Biol Chem (1999) 274:19655–60. doi: 10.1074/jbc.274.28.19655
130. Thomas CA, Tejwani L, Trujillo CA, Negraes PD, Herai RH, Mesci P, et al. Modeling of TREX1-Dependent Autoimmune Disease Using Human Stem Cells Highlights L1 Accumulation as a Source of Neuroinflammation. Cell Stem Cell (2017) 21:319–31.e8. doi: 10.1016/j.stem.2017.07.009
131. Livingston JH, Crow YJ. Neurologic Phenotypes Associated With Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutières Syndrome and Beyond. Neuropediatrics (2016) 47:355–60. doi: 10.1055/s-0036-1592307
132. Livingston JH, Lin JP, Dale RC, Gill D, Brogan P, Munnich A, et al. A Type I Interferon Signature Identifies Bilateral Striatal Necrosis Due to Mutations in ADAR1. J Med Genet (2014) 51:76–82. doi: 10.1136/jmedgenet-2013-102038
133. Samanta D, Ramakrishnaiah R. Recurrent Encephalopathy With Spinal Cord Involvement: An Atypical Manifestation of Aicardi-Goutières Syndrome. Ann Indian Acad Neurol (2019) 22:111–5. doi: 10.4103/aian.AIAN_12_18
134. O’Driscoll MC, Daly SB, Urquhart JE, Black GCM, Pilz DT, Brockmann K, et al. Recessive Mutations in the Gene Encoding the Tight Junction Protein Occludin Cause Band-Like Calcification With Simplified Gyration and Polymicrogyria. Am J Hum Genet (2010) 87:354–64. doi: 10.1016/j.ajhg.2010.07.012
135. Meuwissen MEC, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, et al. Human USP18 Deficiency Underlies Type 1 Interferonopathy Leading to Severe Pseudo-TORCH Syndrome. J Exp Med (2016) 213:1163–74. doi: 10.1084/jem.20151529
136. Alsohime F, Martin-Fernandez M, Temsah M-H, Alabdulhafid M, Le Voyer T, Alghamdi M, et al. JAK Inhibitor Therapy in a Child With Inherited USP18 Deficiency. N Engl J Med (2020) 382:256–65. doi: 10.1056/nejmoa1905633
137. Duncan CJA, Thompson BJ, Chen R, Rice GI, Gothe F, Young DF, et al. Severe Type I Interferonopathy and Unrestrained Interferon Signaling Due to a Homozygous Germline Mutation in STAT2. Sci Immunol (2019) 4(42):eaav7501. doi: 10.1126/sciimmunol.aav7501
138. Barth PG, Walter A, Van Gelderen I. Aicardi-Goutieres Syndrome: A Genetic Microangiopathy? Acta Neuropathol (1999) 98:212–6. doi: 10.1007/s004010051071
139. Cohen D, Rijnink EC, Nabuurs RJA, Steup-Beekman GM, Versluis MJ, Emmer BJ, et al. Brain Histopathology in Patients With Systemic Lupus Erythematosus: Identification of Lesions Associated With Clinical Neuropsychiatric Lupus Syndromes and the Role of Complement. Rheumatol (Oxford) (2017) 56:77–86. doi: 10.1093/rheumatology/kew341
140. Richards A, Van Den Maagdenberg AMJM, Jen JC, Kavanagh D, Bertram P, Spitzer D, et al. C-Terminal Truncations in Human 3′-5′ DNA Exonuclease TREX1 Cause Autosomal Dominant Retinal Vasculopathy With Cerebral Leukodystrophy. Nat Genet (2007) 39:1068–70. doi: 10.1038/ng2082
141. Ramesh V, Bernardi B, Stafa A, Garone C, Franzoni E, Abinun M, et al. Intracerebral Large Artery Disease in Aicardi-Goutières Syndrome Implicates SAMHD1 in Vascular Homeostasis. Dev Med Child Neurol (2010) 52:725–32. doi: 10.1111/j.1469-8749.2010.03727.x
142. Rasmussen M, Skullerud K, Bakke SJ, Lebon P, Jahnsen FL. Cerebral Thrombotic Microangiopathy and Antiphospholipid Antibodies in Aicardi-Goutieres Syndrome - Report of Two Sisters. Neuropediatrics (2005) 36:40–4. doi: 10.1055/s-2004-830532
143. Pelzer N, de Vries B, Boon EMJJ, Kruit MC, Haan J, Ferrari MD, et al. Heterozygous TREX1 Mutations in Early-Onset Cerebrovascular Disease. J Neurol (2013) 260:2188–90. doi: 10.1007/s00415-013-7050-8
144. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, et al. Activated STING in a Vascular and Pulmonary Syndrome. N Engl J Med (2014) 371:507–18. doi: 10.1056/nejmoa1312625
145. Frémond M-L, Hadchouel A, Berteloot L, Melki I, Bresson V, Barnabei L, et al. Overview of STING-Associated Vasculopathy With Onset in Infancy (SAVI) Among 21 Patients. J Allergy Clin Immunol Pract (2021) 9:803–18.e11. doi: 10.1016/j.jaip.2020.11.007
146. Frémond M-L, Crow YJ. STING-Mediated Lung Inflammation and Beyond. J Clin Immunol (2021) 41:501–14. doi: 10.1007/s10875-021-00974-z
147. Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, et al. Mycobacterial Disease and Impaired IFN-γ Immunity in Humans With Inherited ISG15 Deficiency. Science (2012) 337:1684–8. doi: 10.1126/science.1224026
148. Lausch E, Janecke A, Bros M, Trojandt S, Alanay Y, Laet C, et al. Genetic Deficiency of Tartrate-Resistant Acid Phosphatase Associated With Skeletal Dysplasia, Cerebral Calcifications and Autoimmunity. Nat Genet (2011) 43(2):132–7. doi: 10.1038/ng.749
149. Kara B, Ekinci Z, Sahin S, Gungor M, Gunes AS, Ozturk K, et al. Monogenic Lupus Due to Spondyloenchondrodysplasia With Spastic Paraparesis and Intracranial Calcification: Case-Based Review. Rheumatol Int (2020) 40:1903–10. doi: 10.1007/s00296-020-04653-x
150. Wang H, Das L, Tiong JTH, Vasanwala RF, Arkachaisri T. Candle Syndrome: An Extended Clinical Spectrum. Rheumatol (United Kingdom) (2014) 53:2119–20. doi: 10.1093/rheumatology/keu298
151. Jia T, Zheng Y, Feng C, Yang T, Geng S, Yang T, et al. A Chinese Case of Nakajo-Nishimura Syndrome With Novel Compound Heterozygous Mutations of the PSMB8 Gene. BMC Med Genet (2020) 21:1–4. doi: 10.1186/s12881-020-01060-8
152. Smith RA, Norris F, Palmer D, Bernhardt L, Wills RJ. Distribution of Alpha Interferon in Serum and Cerebrospinal Fluid After Systemic Administration. Clin Pharmacol Ther (1985) 37:85–8. doi: 10.1038/clpt.1985.16
153. Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, et al. Aging. Aging-Induced Type I Interferon Response at the Choroid Plexus Negatively Affects Brain Function. Science (2014) 346:89–93. doi: 10.1126/science.1252945
154. Blank T, Detje CN, Spieß A, Hagemeyer N, Brendecke SM, Wolfart J, et al. Brain Endothelial- and Epithelial-Specific Interferon Receptor Chain 1 Drives Virus-Induced Sickness Behavior and Cognitive Impairment. Immunity (2016) 44:901–12. doi: 10.1016/j.immuni.2016.04.005
155. Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, et al. Plasmacytoid Monocytes Migrate to Inflamed Lymph Nodes and Produce Large Amounts of Type I Interferon. Nat Med (1999) 5:919–23. doi: 10.1038/11360
156. Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, et al. The Nature of the Principal Type 1 Interferon-Producing Cells in Human Blood. Science (1999) 284:1835–7. doi: 10.1126/science.284.5421.1835
157. Peschke K, Achleitner M, Frenzel K, Gerbaulet A, Ada SR, Zeller N, et al. Loss of Trex1 in Dendritic Cells Is Sufficient To Trigger Systemic Autoimmunity. J Immunol (2016) 197:2157–66. doi: 10.4049/jimmunol.1600722
158. Gall A, Treuting P, Elkon KB, Loo YM, Gale M, Barber GN, et al. Autoimmunity Initiates in Nonhematopoietic Cells and Progresses via Lymphocytes in an Interferon-Dependent Autoimmune Disease. Immunity (2012) 36:120–31. doi: 10.1016/j.immuni.2011.11.018
159. Goldmann T, Zeller N, Raasch J, Kierdorf K, Frenzel K, Ketscher L, et al. USP 18 Lack in Microglia Causes Destructive Interferonopathy of the Mouse Brain. EMBO J (2015) 34:1612–29. doi: 10.15252/embj.201490791
160. Ejlerskov P, Hultberg JG, Wang JY, Carlsson R, Ambjørn M, Kuss M, et al. Lack of Neuronal IFN-β-IFNAR Causes Lewy Body- and Parkinson’s Disease-Like Dementia. Cell (2015) 163:324–39. doi: 10.1016/j.cell.2015.08.069
161. Hernandez N, Bucciol G, Moens L, Le Pen J, Shahrooei M, Goudouris E, et al. Inherited IFNAR1 Deficiency in Otherwise Healthy Patients With Adverse Reaction to Measles and Yellow Fever Live Vaccines. J Exp Med (2019) 216:2057–70. doi: 10.1084/jem.20182295
162. Bastard P, Manry J, Chen J, Rosain J, Seeleuthner Y, AbuZaitun O, et al. Herpes Simplex Encephalitis in a Patient With a Distinctive Form of Inherited IFNAR1 Deficiency. J Clin Invest (2021) 131(1):e139980. doi: 10.1172/JCI139980
163. Duncan CJA, Mohamad SMB, Young DF, Skelton AJ, Leahy TR, Munday DC, et al. Human IFNAR2 Deficiency: Lessons for Antiviral Immunity. Sci Transl Med (2015) 7:307ra154. doi: 10.1126/scitranslmed.aac4227
164. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn Errors of Type I IFN Immunity in Patients With Life-Threatening COVID-19. Science (2020) 370(6515):eabd4570. doi: 10.1126/science.abd4570
165. Blank T, Prinz M. Type I Interferon Pathway in CNS Homeostasis and Neurological Disorders. Glia (2017) 65:1397–406. doi: 10.1002/glia.23154
166. Cohan SL, Hendin BA, Reder AT, Smoot K, Avila R, Mendoza JP, et al. Interferons and Multiple Sclerosis: Lessons From 25 Years of Clinical and Real-World Experience With Intramuscular Interferon Beta-1a (Avonex). CNS Drugs (2021) 35:743–67. doi: 10.1007/s40263-021-00822-z
167. Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, et al. T Helper Type 1 and 17 Cells Determine Efficacy of Interferon-Beta in Multiple Sclerosis and Experimental Encephalomyelitis. Nat Med (2010) 16:406–12. doi: 10.1038/nm.2110
168. Río J, Nos C, Tintoré M, Téllez N, Galán I, Pelayo R, et al. Defining the Response to Interferon-Beta in Relapsing-Remitting Multiple Sclerosis Patients. Ann Neurol (2006) 59:344–52. doi: 10.1002/ana.20740
169. Wang A-G, Lin Y-C, Wang S-J, Tsai C-P, Yen M-Y. Early Relapse in Multiple Sclerosis-Associated Optic Neuritis Following the Use of Interferon Beta-1a in Chinese Patients. Jpn J Ophthalmol (2006) 50:537–42. doi: 10.1007/s10384-006-0359-4
170. Axtell RC, Raman C, Steinman L. Type I Interferons: Beneficial in Th1 and Detrimental in Th17 Autoimmunity. Clin Rev Allergy Immunol (2013) 44:114–20. doi: 10.1007/s12016-011-8296-5
171. Montealegre Sanchez GA, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 Inhibition With Baricitinib in the Treatment of Autoinflammatory Interferonopathies. J Clin Invest (2018) 128:3041–52. doi: 10.1172/JCI98814
172. Neven B, Al Adba B, Hully M, Desguerre I, Pressiat C, Boddaert N, et al. JAK Inhibition in the Aicardi-Goutières Syndrome. N Engl J Med (2020) 383:2190–1. doi: 10.1056/NEJMc2031081
173. Rice GI, Meyzer C, Bouazza N, Hully M, Boddaert N, Semeraro M, et al. Reverse-Transcriptase Inhibitors in the Aicardi–Goutières Syndrome. N Engl J Med (2018) 379:2275–7. doi: 10.1056/NEJMc1810983
174. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin Gene Defects in Familial Hemophagocytic Lymphohistiocytosis. Science (1999) 286:1957–9. doi: 10.1126/science.286.5446.1957
175. Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 Is Essential for Cytolytic Granules Fusion and Is Mutated in a Form of Familial Hemophagocytic Lymphohistiocytosis (FHL3). Cell (2003) 115:461–73. doi: 10.1016/s0092-8674(03)00855-9
176. zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter J-I, et al. Linkage of Familial Hemophagocytic Lymphohistiocytosis (FHL) Type-4 to Chromosome 6q24 and Identification of Mutations in Syntaxin 11. Hum Mol Genet (2005) 14:827–34. doi: 10.1093/hmg/ddi076
177. zur Stadt U, Rohr J, Seifert W, Koch F, Grieve S, Pagel J, et al. Familial Hemophagocytic Lymphohistiocytosis Type 5 (FHL-5) Is Caused by Mutations in Munc18-2 and Impaired Binding to Syntaxin 11. Am J Hum Genet (2009) 85:482–92. doi: 10.1016/j.ajhg.2009.09.005
178. Kalinichenko A, Perinetti Casoni G, Dupré L, Trotta L, Huemer J, Galgano D, et al. RhoG Deficiency Abrogates Cytotoxicity of Human Lymphocytes and Causes Hemophagocytic Lymphohistiocytosis. Blood (2021) 137:2033–45. doi: 10.1182/blood.2020008738
179. Henter J-I, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and Therapeutic Guidelines for Hemophagocytic Lymphohistiocytosis. Pediatr Blood Cancer (2007) 48:124–31. doi: 10.1002/pbc.21039
180. Murphy C, Nanthapisal S, Gilmour K, Laurent S, D’Arco F, Hemingway C, et al. Progressive Neurologic Disorder: Initial Manifestation of Hemophagocytic Lymphohistiocytosis. Neurology (2016) 86:2109–11. doi: 10.1212/WNL.0000000000002729
181. Giardino G, De Luca M, Cirillo E, Palma P, Romano R, Valeriani M, et al. Two Brothers With Atypical UNC13D-Related Hemophagocytic Lymphohistiocytosis Characterized by Massive Lung and Brain Involvement. Front Immunol (2017) 8:1892. doi: 10.3389/fimmu.2017.01892
182. Shinoda J, Murase S, Takenaka K, Sakai N. Isolated Central Nervous System Hemophagocytic Lymphohistiocytosis: Case Report. Neurosurgery (2005) 56:187.
183. Turtzo LC, Lin DDM, Hartung H, Barker PB, Arceci R, Yohay K. A Neurologic Presentation of Familial Hemophagocytic Lymphohistiocytosis Which Mimicked Septic Emboli to the Brain. J Child Neurol (2007) 22:863–8. doi: 10.1177/0883073807304203
184. Ueda I, Kurokawa Y, Koike K, Ito S, Sakata A, Matsumora T, et al. Late-Onset Cases of Familial Hemophagocytic Lymphohistiocytosis With Missense Perforin Gene Mutations. Am J Hematol (2007) 82:427–32. doi: 10.1002/ajh.20878
185. Feng W-XX, Yang X-YY, Li J-WW, Gong S, Wu Y, Zhang W-HH, et al. Neurologic Manifestations as Initial Clinical Presentation of Familial Hemophagocytic Lymphohistiocytosis Type2 Due to PRF1 Mutation in Chinese Pediatric Patients. Front Genet (2020) 11:126. doi: 10.3389/fgene.2020.00126
186. Moshous D, Feyen O, Lankisch P, Schwarz K, Schaper J, Schneider M, et al. Primary Necrotizing Lymphocytic Central Nervous System Vasculitis Due to Perforin Deficiency in a Four-Year-Old Girl. Arthritis Rheum (2007) 56:995–9. doi: 10.1002/art.22442
187. Wang J, Tuo H, Wu L, Wang X, Wang Z. Neurological Symptoms of Familial Hemophagocytic Lymphohistiocytosis Type 2. J Integr Neurosci (2020) 19:131–5. doi: 10.31083/J.JIN.2020.01.1250
188. Beaty AD, Weller C, Levy B, Vogler C, Ferguson WS, Bicknese A, et al. A Teenage Boy With Late Onset Hemophagocytic Lymphohistiocytosis With Predominant Neurologic Disease and Perforin Deficiency. Pediatr Blood Cancer (2008) 50:1070–2. doi: 10.1002/pbc.21438
189. Dias C, McDonald A, Sincan M, Rupps R, Markello T, Salvarinova R, et al. Recurrent Subacute Post-Viral Onset of Ataxia Associated With a PRF1 Mutation. Eur J Hum Genet (2013) 21:1232–9. doi: 10.1038/ejhg.2013.20
190. Madkaikar M, Gupta M, Dixit A, Patil V. Predominant Neurologic Manifestations Seen in a Patient With a Biallelic Perforin1 Mutation (PRF1; P.R225W). J Pediatr Hematol Oncol (2017) 39:143–6. doi: 10.1097/MPH.0000000000000597
191. Ménasché G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, et al. Mutations in RAB27A Cause Griscelli Syndrome Associated With Haemophagocytic Syndrome. Nat Genet (2000) 25:173–6. doi: 10.1038/76024
192. Dou Y, Wu HJ, Li HQ, Qin S, Wang YE, Li J, et al. Microglial Migration Mediated by ATP-Induced ATP Release From Lysosomes. Cell Res (2012) 22:1022–33. doi: 10.1038/cr.2012.10
193. Panigrahi I, Suthar R, Rawat A, Behera B. Seizure as the Presenting Manifestation in Griscelli Syndrome Type 2. Pediatr Neurol (2015) 52:535–8. doi: 10.1016/j.pediatrneurol.2015.01.010
194. Introne WJ, Westbroek W, Cullinane AR, Groden CA, Bhambhani V, Golas GA, et al. Neurologic Involvement in Patients With Atypical Chediak-Higashi Disease. Neurology (2016) 86:1320–8. doi: 10.1212/WNL.0000000000002551
195. Tardieu M, Lacroix C, Neven B, Bordigoni P, De Saint Basile G, Blanche S, et al. Progressive Neurologic Dysfunctions 20 Years After Allogeneic Bone Marrow Transplantation for Chediak-Higashi Syndrome. Blood (2005) 106:40–2. doi: 10.1182/blood-2005-01-0319
196. Gartshteyn Y, Askanase AD, Mor A. SLAM Associated Protein Signaling in T Cells: Tilting the Balance Toward Autoimmunity. Front Immunol (2021) 12:654839. doi: 10.3389/fimmu.2021.654839
197. Trizzino A, Zur Stadt U, Ueda I, Risma K, Janka G, Ishii E, et al. Genotype-Phenotype Study of Familial Haemophagocytic Lymphohistiocytosis Due to Perforin Mutations. J Med Genet (2008) 45:15–21. doi: 10.1136/jmg.2007.052670
198. Santoro A, Cannella S, Bossi G, Gallo F, Trizzino A, Pende D, et al. Novel Munc13-4 Mutations in Children and Young Adult Patients With Haemophagocytic Lymphohistiocytosis. J Med Genet (2006) 43:953–60. doi: 10.1136/jmg.2006.041863
199. Sieni E, Cetica V, Santoro A, Beutel K, Mastrodicasa E, Meeths M, et al. Genotype-Phenotype Study of Familial Haemophagocytic Lymphohistiocytosis Type 3. J Med Genet (2011) 48:343–52. doi: 10.1136/jmg.2010.085456
200. Horne A, Ramme KG, Rudd E, Zheng C, Wali Y, Al-Lamki Z, et al. Characterization of PRF1, STX11 and UNC13D Genotype-Phenotype Correlations in Familial Hemophagocytic Lymphohistiocytosis. Br J Haematol (2008) 143:75–83. doi: 10.1111/j.1365-2141.2008.07315.x
201. Feldmann J, Ménasché G, Callebaut I, Minard-Colin V, Bader-Meunier B, Le Clainche L, et al. Severe and Progressive Encephalitis as a Presenting Manifestation of a Novel Missense Perforin Mutation and Impaired Cytolytic Activity. Blood (2005) 105:2658–63. doi: 10.1182/blood-2004-09-3590
202. Rudd E, Bryceson YT, Zheng C, Edner J, Wood SM, Ramme K, et al. Spectrum, and Clinical and Functional Implications of UNC13D Mutations in Familial Haemophagocytic Lymphohistiocytosis. J Med Genet (2008) 45:134–41. doi: 10.1136/jmg.2007.054288
203. Gray PE, Shadur B, Russell S, Mitchell R, Buckley M, Gallagher K, et al. Late-Onset Non-HLH Presentations of Growth Arrest, Inflammatory Arachnoiditis, and Severe Infectious Mononucleosis, in Siblings With Hypomorphic Defects in UNC13D. Front Immunol (2017) 8:944. doi: 10.3389/fimmu.2017.00944
204. Hussein AA, Hamadah T, Qandeel M, Sughayer M, Amarin R, Mansour A, et al. Hematopoietic Stem Cell Transplantation of an Adolescent With Neurological Manifestations of Homozygous Missense PRF1 Mutation. Pediatr Blood Cancer (2014) 61:2313–5. doi: 10.1002/pbc.25166
205. Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic Mutations in PRF1, MUNC13-4, and STXBP2 Are Associated With Adult-Onset Familial HLH. Blood (2011) 118(22):5794–8. doi: 10.1182/blood-2011-07-370148.An
206. Clementi R, Emmi L, Maccario R, Liotta F, Moretta L, Danesino C, et al. Adult Onset and Atypical Presentation of Hemophagocytic Lymphohistiocytosis in Siblings Carrying PRF1 Mutations. Blood (2002) 100:2266–7. doi: 10.1182/blood-2002-04-1030
207. Bergsten E, Horne AC, Aricó M, Astigarraga I, Egeler RM, Filipovich AH, et al. Confirmed Efficacy of Etoposide and Dexamethasone in HLH Treatment: Long-Term Results of the Cooperative HLH-2004 Study. Blood (2017) 130:2728–38. doi: 10.1182/blood-2017-06-788349
208. Schmid JP, Ho CH, Chrétien F, Lefebure JM, Pivert G, Kosco-Vilbois M, et al. Neutralization of Ifnγ Defeats Haemophagocytosis in LCMV-Infected Perforin - and Rab27a-Deficient Mice. EMBO Mol Med (2009) 1:112–24. doi: 10.1002/emmm.200900009
209. Jordan MB, Hildeman D, Kappler J, Marrack P. An Animal Model of Hemophagocytic Lymphohistiocytosis (HLH): CD8+ T Cells and Interferon Gamma are Essential for the Disorder. Blood (2004) 104:735–43. doi: 10.1182/blood-2003-10-3413
210. Burn TN, Weaver L, Rood JE, Chu N, Bodansky A, Kreiger PA, et al. Genetic Deficiency of Interferon-γ Reveals Interferon-γ–Independent Manifestations of Murine Hemophagocytic Lymphohistiocytosis. Arthritis Rheumatol (2020) 72:335–47. doi: 10.1002/art.41076
211. Ottum PA, Arellano G, Reyes LI, Iruretagoyena M, Naves R. Opposing Roles of Interferon-Gamma on Cells of the Central Nervous System in Autoimmune Neuroinflammation. Front Immunol (2015) 6:539. doi: 10.3389/fimmu.2015.00539
212. Merli P, Algeri M, Gaspari S, Locatelli F. Novel Therapeutic Approaches to Familial HLH (Emapalumab in FHL). Front Immunol (2020) 11:608492. doi: 10.3389/fimmu.2020.608492
213. Keenan C, Nichols KE, Albeituni S. Use of the JAK Inhibitor Ruxolitinib in the Treatment of Hemophagocytic Lymphohistiocytosis. Front Immunol (2021) 12:614704. doi: 10.3389/fimmu.2021.614704
214. Barzaghi F, Passerini L. IPEX Syndrome: Improved Knowledge of Immune Pathogenesis Empowers Diagnosis. Front Pediatr (2021) 9:612760. doi: 10.3389/fped.2021.612760
215. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, Penetrance, and Treatment of 133 Cytotoxic T-Lymphocyte Antigen 4–Insufficient Subjects. J Allergy Clin Immunol (2018) 142:1932–46. doi: 10.1016/j.jaci.2018.02.055
216. Stegert M, Bock M, Trendelenburg M. Clinical Presentation of Human C1q Deficiency: How Much of a Lupus? Mol Immunol (2015) 67:3–11. doi: 10.1016/j.molimm.2015.03.007
217. Kampylafka EI, Alexopoulos H, Kosmidis ML, Panagiotakos DB, Vlachoyiannopoulos PG, Dalakas MC, et al. Incidence and Prevalence of Major Central Nervous System Involvement in Systemic Lupus Erythematosus: A 3-Year Prospective Study of 370 Patients. PloS One (2013) 8:1–8. doi: 10.1371/journal.pone.0055843
218. Postal M, Vivaldo JF, Fernandez-Ruiz R, Paredes JL, Appenzeller S, Niewold TB. Type I Interferon in the Pathogenesis of Systemic Lupus Erythematosus. Curr Opin Immunol (2020) 67:87–94. doi: 10.1016/j.coi.2020.10.014
219. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
220. Hodeib S, Herberg JA, Levin M, Sancho-Shimizu V. Human Genetics of Meningococcal Infections. Hum Genet (2020) 139:961–80. doi: 10.1007/s00439-020-02128-4
221. Corvini M, Randolph C, Aronin SI. Complement C7 Deficiency Presenting as Recurrent Aseptic Meningitis. Ann Allergy Asthma Immunol (2004) 93:200–5. doi: 10.1016/S1081-1206(10)61476-7
222. González-Rubio C, Ferreira-Cerdán A, Ponce IM, Arpa J, Fontán G, López-Trascasa M. Complement Factor I Deficiency Associated With Recurrent Meningitis Coinciding With Menstruation. Arch Neurol (2001) 58:1923–8. doi: 10.1001/archneur.58.11.1923
223. Haerynck F, Stordeur P, Vandewalle J, Van Coster R, Bordon V, De Baets F, et al. Complete Factor I Deficiency Due to Dysfunctional Factor I With Recurrent Aseptic Meningo-Encephalitis. J Clin Immunol (2013) 33:1293–301. doi: 10.1007/s10875-013-9944-8
224. Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 Deficiency in Patients With Herpes Simplex Encephalitis. Sci (80- ) (2007) 317:1522–7. doi: 10.1126/science.1139522
225. Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, et al. Herpes Simplex Virus Encephalitis in Human UNC-93B Deficiency. Science (80- ) (2006) 314:308–12. doi: 10.1126/science.1128346
226. Sancho-shimizu V, De Diego RP, Lorenzo L, Halwani R, Alangari A, Israelsson E, et al. Recessive and Dominant TRIF Deficiency Herpes Simplex Encephalitis in Children With Autosomal Recessive and Dominant TRIF Deficiency. J Clin Invest (2011) 121(12):4889–902. doi: 10.1172/JCI59259.during
227. Pérez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, et al. Human TRAF3 Adaptor Molecule Deficiency Leads to Impaired Toll-Like Receptor 3 Response and Susceptibility to Herpes Simplex Encephalitis. Immunity (2010) 33:400–11. doi: 10.1016/j.immuni.2010.08.014
228. Herman M, Ciancanelli M, Ou YH, Lorenzo L, Klaudel-Dreszler M, Pauwels E, et al. Heterozygous TBK1 Mutations Impair TLR3 Immunity and Underlie Herpes Simplex Encephalitis of Childhood. J Exp Med (2012) 209:1567–82. doi: 10.1084/jem.20111316
229. Andersen LL, Mørk N, Reinert LS, Kofod-Olsen E, Narita R, Jørgensen SE, et al. Functional IRF3 Deficiency in a Patient With Herpes Simplex Encephalitis. J Exp Med (2015) 212:1371–9. doi: 10.1084/jem.20142274
230. Guo Y, Audry M, Ciancanelli M, Alsina L, Azevedo J, Herman M, et al. Herpes Simplex Virus Encephalitis in a Patient With Complete TLR3 Deficiency : TLR3 Is Otherwise Redundant in Protective Immunity. (2011) 208(10):2083–98. doi: 10.1084/jem.20101568
231. Armangue T, Spatola M, Vlagea A, Mattozzi S, Cárceles-Cordon M, Martinez-Heras E, et al. Frequency, Symptoms, Risk Factors, and Outcomes of Autoimmune Encephalitis After Herpes Simplex Encephalitis: A Prospective Observational Study and Retrospective Analysis. Lancet Neurol (2018) 17:760–72. doi: 10.1016/S1474-4422(18)30244-8
232. Sahoo SS, Kozyra EJ, Wlodarski MW. Germline Predisposition in Myeloid Neoplasms: Unique Genetic and Clinical Features of GATA2 Deficiency and SAMD9/SAMD9L Syndromes. Best Pract Res Clin Haematol (2020) 33:101197. doi: 10.1016/j.beha.2020.101197
233. Tsai FY, Keller G, Kuo FC, Weiss M, Chen J, Rosenblatt M, et al. An Early Haematopoietic Defect in Mice Lacking the Transcription Factor GATA-2. Nature (1994) 371:221–6. doi: 10.1038/371221a0
234. Pruitt AA. Central Nervous System Infections in Immunocompromised Patients. Curr Neurol Neurosci Rep (2021) 21(7):37. doi: 10.1007/s11910-021-01119-w
235. Taveira-Dasilva AM, Markello TC, Kleiner DE, Jones AM, Groden C, MacNamara E, et al. Expanding the Phenotype of COPA Syndrome: A Kindred With Typical and Atypical Features. J Med Genet (2018) 56(11):778–82. doi: 10.1136/jmedgenet-2018-105560
236. Watkin LB, Jessen B, Wiszniewski W, Vece TJ, Jan M, Sha Y, et al. COPA Mutations Impair ER-Golgi Transport and Cause Hereditary Autoimmune-Mediated Lung Disease and Arthritis. Nat Genet (2015) 47:654–60. doi: 10.1038/ng.3279
237. Lin J, Li X, Xia J. Th17 Cells in Neuromyelitis Optica Spectrum Disorder: A Review. Int J Neurosci (2016) 126:1051–60. doi: 10.3109/00207454.2016.1163550
238. Neilson DE, Adams MD, Orr CMD, Schelling DK, Eiben RM, Kerr DS, et al. Infection-Triggered Familial or Recurrent Cases of Acute Necrotizing Encephalopathy Caused by Mutations in a Component of the Nuclear Pore, Ranbp2. Am J Hum Genet (2009) 84:44–51. doi: 10.1016/j.ajhg.2008.12.009
239. Levine JM, Ahsan N, Ho E, Santoro JD. Genetic Acute Necrotizing Encephalopathy Associated With RANBP2: Clinical and Therapeutic Implications in Pediatrics. Mult Scler Relat Disord (2020) 43:102194. doi: 10.1016/j.msard.2020.102194
240. Roshanisefat H, Bahmanyar S, Hillert J, Olsson T, Montgomery S. Shared Genetic Factors May Not Explain the Raised Risk of Comorbid Inflammatory Diseases in Multiple Sclerosis. Mult Scler (2012) 18:1430–6. doi: 10.1177/1352458512438240
241. Forrester JV, McMenamin PG, Dando SJ. CNS Infection and Immune Privilege. Nat Rev Neurosci (2018) 19:655–71. doi: 10.1038/s41583-018-0070-8
242. Marchetti L, Engelhardt B. Immune Cell Trafficking Across the Blood-Brain Barrier in the Absence and Presence of Neuroinflammation. Vasc Biol (Bristol England) (2020) 2:H1–H18. doi: 10.1530/VB-19-0033
243. Donath X, Saint-Martin C, Dubois-Laforgue D, Rajasingham R, Mifsud F, Ciangura C, et al. Next-Generation Sequencing Identifies Monogenic Diabetes in 16% of Patients With Late Adolescence/Adult-Onset Diabetes Selected on a Clinical Basis: A Cross-Sectional Analysis. BMC Med (2019) 17:1–10. doi: 10.1186/s12916-019-1363-0
Keywords: interferonopathies, primary immunodeficiencies, neuroinflammation, familial hemophagocytic lymphohistiocytosis (FHL), Mendelian genetic diseases, interleukin-1, type I interferon, autoinflammatory disorders
Citation: Lindahl H and Bryceson YT (2022) Neuroinflammation Associated With Inborn Errors of Immunity. Front. Immunol. 12:827815. doi: 10.3389/fimmu.2021.827815
Received: 02 December 2021; Accepted: 27 December 2021;
Published: 19 January 2022.
Edited by:
Hideyuki Takeuchi, Yokohama City University, JapanReviewed by:
Tatsusada Okuno, Osaka University, JapanYoshifumi Sonobe, University of Chicago Medical Center, United States
Copyright © 2022 Lindahl and Bryceson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hannes Lindahl, Hannes.lindahl@ki.se