 SuHyeon Oh
SuHyeon Oh SangJoon Lee
SangJoon Lee- Department of Biological Science, Ulsan National Institute of Science and Technology (UNIST), Ulsan, Republic of Korea
Innate immunity is an important first line of defense against pathogens, including viruses. These pathogen- and damage-associated molecular patterns (PAMPs and DAMPs, respectively), resulting in the induction of inflammatory cell death, are detected by specific innate immune sensors. Recently, Z-DNA binding protein 1 (ZBP1), also called the DNA-dependent activator of IFN regulatory factor (DAI) or DLM1, is reported to regulate inflammatory cell death as a central mediator during viral infection. ZBP1 is an interferon (IFN)-inducible gene that contains two Z-form nucleic acid-binding domains (Zα1 and Zα2) in the N-terminus and two receptor-interacting protein homotypic interaction motifs (RHIM1 and RHIM2) in the middle, which interact with other proteins with the RHIM domain. By sensing the entry of viral RNA, ZBP1 induces PANoptosis, which protects host cells against viral infections, such as influenza A virus (IAV) and herpes simplex virus (HSV1). However, some viruses, particularly coronaviruses (CoVs), induce PANoptosis to hyperactivate the immune system, leading to cytokine storm, organ failure, tissue damage, and even death. In this review, we discuss the molecular mechanism of ZBP1-derived PANoptosis and pro-inflammatory cytokines that influence the double-edged sword of results in the host cell. Understanding the ZBP1-derived PANoptosis mechanism may be critical for improving therapeutic strategies.
Introduction
Innate immunity and virus
Innate immunity is the first line of defense against viral infections in host cells. The pro-inflammatory response of innate immunity induces the migration of immune cells, including macrophages and neutrophils, to remove infectious agents (1, 2). The innate immune system is activated by the viral pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) by pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), nucleotide-binding oligomerization domain (NOD)-like receptor family proteins (NLRs), absent in melanoma 2 (AIM2), and Z-DNA binding protein 1 (ZBP1). For example, several TLRs are involved in the detection of β-coronaviruses. TLR7 senses severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV) (3), and murine hepatitis virus (MHV) (4), and TLR2 senses SARS-CoV-2 (5). RIG-I senses viral RNA, including IAV (6–9) and hepatitis C virus (HCV) (10). Subsequently, TLR- and RLR-mediated signaling leads to the secretion of type 1 interferons (IFNs), that stimulate the expression of IFN-stimulated genes (ISGs) in infected and neighboring cells, thereby inducing an antiviral state. In particular, some PRRs, including NLRs and AIM2, assemble a large protein complex known as inflammasome, comprising a sensor, an adaptor, and an effector. They are assembled after sensing viral infections and activate the programmed cell death (PCD) pathway. The most well-established PCDs are pyroptosis, apoptosis, and necroptosis. These PCD pathways are activated against various viral infections to remove infected cells and suppress viral spread. Some viruses derive the crosstalk between the multiple PCD pathways known as PANoptosis, including IAV (11–14) and HSV1 (15–18) infection. PANoptosis occurs via PANoptosome, wherein the key molecules of pyroptosis, apoptosis, and necroptosis simultaneously interact with each other (14, 18–22). In this review, we summarize the molecular mechanisms of each PCD and PANoptosis against viral infection.
ZBP1
At first, ZBP1 was considered as the cytosolic DNA sensor (23, 24). However, Zbp1–/–and wild type (WT) mice displyed a similar phenotype in B-DNA-induced innate immune activation (23–25). ZBP1 comprises three parts: the N-terminal Z-DNA binding domain (ZBD), the receptor-interacting protein homotypic interaction motifs (RHIM), and the C-terminal signal domain (SD) (26–29). The N-terminal ZBD, also called the Zα1 and Zα2, binds to the left-handed helical Z-conformation nucleic acid (Z-NA) (30, 31). Zα domains, particularly the Zα2 domain, are known to play a critical role in the activation of PCDs (18, 30, 32–34). For example, the deficiency of the Zα domains or Zα2 alone limits ZBP1-RIPK3-mediated inflammatory cell death after IAV infection (30, 34). In addition to Zα domains, ZBP1 has two RHIMs that interact with other RHIMs in RIPK1 and RIPK3 (26, 30, 35, 36). The C-terminal SD of ZBP1 participates in the type 1 IFN response induced by ZBP1 (29).
ZBP1 has recently been shown to act as a central regulator of PANoptosis, by defending against viral infections, such as IAV (11, 34) and HSV1 (18). In contrast, some viruses cause cell death that severely impacts host health. For example, SARS-CoV-2, the causative virus of coronavirus disease 19 (COVID-19), activates multiple inflammatory cell death pathways and induces the hyperactivation of cytokine secretion, which results in severe symptoms (32, 37–39). Thus, the regulation of the adverse mechanisms of PCD is essential for protecting the host from death.
ZBP1-NLRP3 inflammasome
The inflammasome mediates pyroptosis by forming a large protein complex after sensing PAMPs and DAMPs. Inflammasomes induce the activation of protease enzyme families such as caspase-1, 4, 5, and 11, which process GSDMD and release the N-terminus to oligomerize and form pores in the plasma membrane after their cleavage (40–42). Activated caspase-1 cleaves the end to release IL-1β and IL-18. There are five well-known inflammasome sensors: NLRP1 (43), NLRP3 (44–46), NLRC4 (47, 48), AIM2 (49–52), and pyrin (53). Some other innate sensors have also been reported to initiate inflammasome assembly under specific conditions, such as NLRP6 (54), NLRP9 (55), NLRP12 (56), interferon-γ-inducible protein 16 (IFI16) (57), RIG-I (58), and myxovirus resistance protein A (MxA) (1). These sensors interact with apoptosis-associated speck-like protein containing CARD (ASC), an adaptor molecule, to activate caspase-1. Among them, NLRP3 has been extensively studied in various stimuli, from endogenous danger signals (44–46) to gram-positive (59) and gram-negative bacteria (60–62). In addition, NLRP3 senses RNA viruses, IAV (11, 63–66), and West Nile virus (WNV) (67), in addition to DNA viruses, HSV-1 (68, 69) to activate the antiviral immune response.
Two signals are required to activate the NLRP3 inflammasome. First, the priming signal from stimuli promotes the NF-κB and ERK pathways, which may elevate the gene expression of inflammasome components and manage post-translational modifications of NLRP3, such as ubiquitination (70), phosphorylation (71, 72), and SUMOylation (73). Subsequently, the activation signal stimulates NLRP3 activation, which may be due to specific cellular stress patterns such as K+ efflux (74, 75) and mitochondrial dysfunction (76, 77). The activated NLRP3 inflammasome contains NLRP3, caspase-1, and ASC and facilitates IL-1β maturation (78). Notably, regardless of the presence of stimuli, ASC specks can be released into the extracellular space and oligomerized in the neighboring macrophages via a prion-like mechanism in Arf6–/– macrophages (79).
Recently, the interaction between ZBP1 and NLRP3 inflammasome has been revealed in several viral infections. ZBP1 detects IAV infection and activates the NLRP3 inflammasome to induce inflammatory cell death (11, 30, 80). And NLRP3 inflammasome activation was diminished during IAV infection in Zbp1–/– bone marrow-derived macrophages (BMDMs) (11). In addition, the Zα2 domain of ZBP1 influences the activation of NLRP3 inflammasome and PANoptosis (30). ZBP1 can interact with IAV nucleoprotein (NP), polymerase subunit PB1, and IAV Z-RNA (11, 34), thereby activating the NLRP3 inflammasome via the RIPK1-RIPK3-caspase-8 axis (11). Overall, these results indicate that ZBP1 is an essential regulator of the NLRP3 inflammasome in response to viral infection.
In this review, we summarize the role of ZBP1 as an essential regulator of innate immune response and cell death during viral infection. Herein, we describe how ZBP1 senses the entry of viral Z-RNA and stimulates the inflammatory cell death pathway. We also describe a newly emerging concept of inflammatory cell death, PANoptosis, which leads to host survival when balanced or fetal symptoms and even death when exacerbated, which may be a decisive target in various viral diseases.
ZBP1 and PCDs
ZBP1 senses viral genome and plays a key role as a central mediator of PCDs
The sensing of viral elements by the innate immune system induces inflammatory cell death pathways. After ZBP1 senses viral Z-RNA, a cascade of pro-inflammatory cytokines occurs, and PCDs are induced individually or together through crosstalk for host defense (Figure 1).
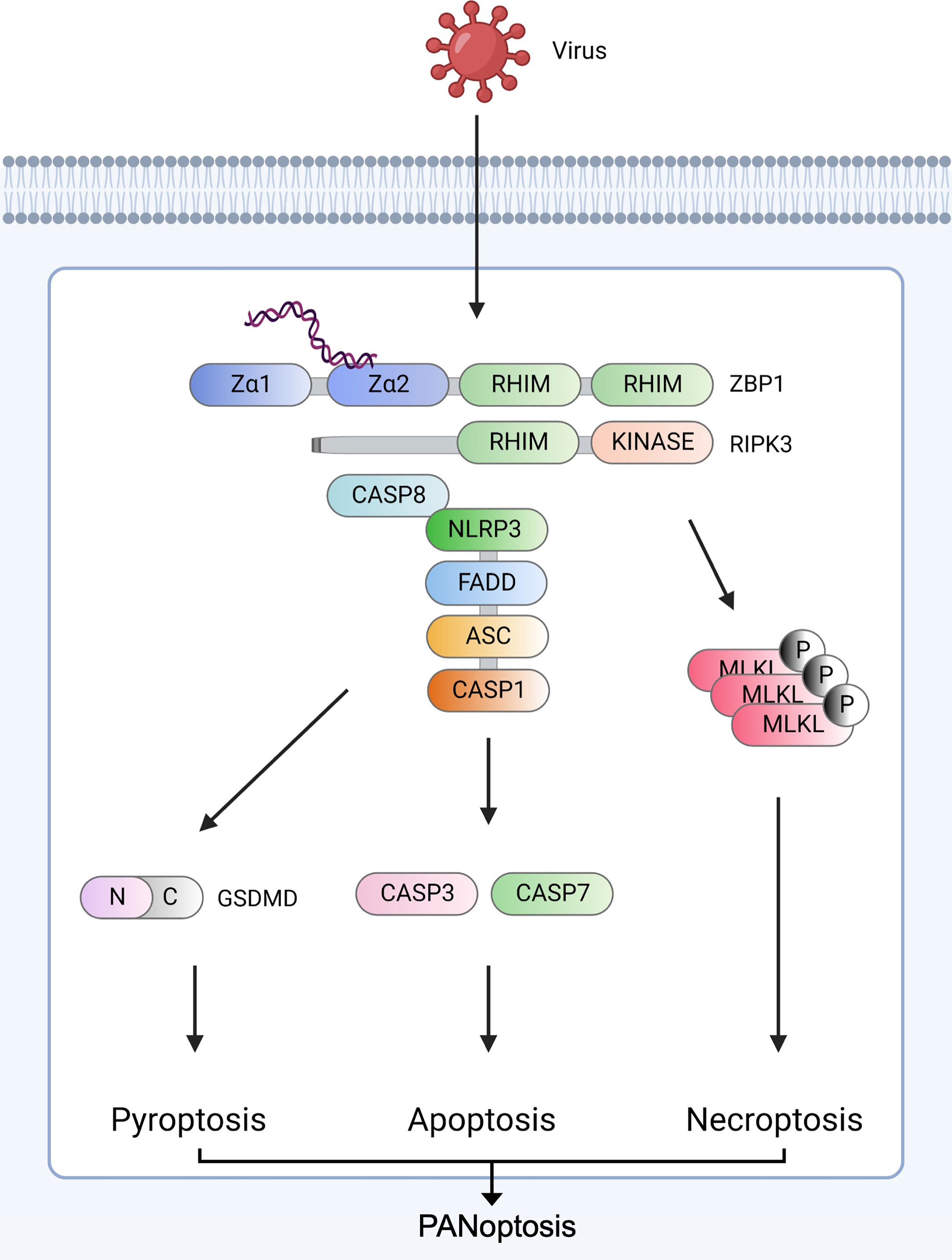
Figure 1 ZBP1-derived PANoptosis ZBP1 senses viral nucleic acids (Z-NA) and interacts with RIPK1 (not shown) and RIPK3 via RHIM domains to recruit caspase-8. The ZBP1-RIPK3-caspase-8 complex induces three major inflammatory cell death pathways: GSDMD-mediated pyroptosis, caspase-3/caspase-7-mediated apoptosis, and MLKL-mediated necroptosis. RIPK3 and caspase-8 induce NLRP3 inflammasome assembly via ASC and pro-caspase-1. After assembly, mature caspase-1 cleaves GSDMD to form pores in the plasma, leading to pyroptosis. Caspase-8 stimulates the secretion of caspase-3 and caspase-7. ZBP1-RIPK3 activates MLKL-mediated necroptosis. Created with BioRender.com.
ZBP1 induces pyroptosis during viral infection
Pyroptosis is a form of inflammatory cell death, typically mediated by inflammasomes. The term pyroptosis was first defined in 2001 semantically distinguishing pyroptosis and apoptosis (81). Canonical pyroptosis is regulated by inflammasome activation, which cleaves GSDMD and releases IL-1β and IL-18 (40, 41, 82). Gasdermin E (GSDME), another member of the gasdermin (GSMD) family, is also involved in pyroptosis via caspase-3 and -8 activation by undergoing cleavage and releasing N-terminus, thereby forming channels on the cell membrane (83, 84). The inflammasome is a multi-protein complex containing parts of a sensor, adaptor, and effector that assemble in response to the virus entry. The assembly of the inflammasome begins to sense certain stimuli through its sensor protein (85). For example, poly (dA:dT) is recognized by AIM2 (86), and NLRC4 detects Salmonella flagellin (87). Subsequently, pro-caspase-1 proteins form oligomers and activate caspase-1. Furthermore, the activated caspase-1 can proteolytically cleave the cytokines pro–IL-1β and pro–IL-18 into their bioactive forms to induce pro-inflammatory responses.
The relationship between the NLRP3 inflammasome and ZBP1 is well established. The ZBP1-NLRP3 inflammasome facilitates the maturation of pro-inflammatory cytokines, including IL-1β and IL-18, and GSDMD by activating caspase-1. IL-1β and IL-18 are processed into their active forms to upregulate the pro-inflammatory signaling pathway (88, 89). Simultaneously, GSDMD is cleaved by caspase-1 and it self-oligomerizes to form a pore in the membrane, releasing cytokines to induce inflammatory cell death through a process called pyroptosis (40–42). During IAV infection, ZBP1 activates the NLRP3 inflammasome and induces pyroptosis in BMDMs. Pyroptosis-associated cytokines, IL-1β and IL-18, were significantly reduced in Zbp1–/– BMDMs (11). In BMDMs, ZBP1-induced pyroptosis is regulated by RIG-I-MAVS and TLR signaling pathways during IAV infection (13). However, in MHV infection, Zbp1–/– mice survived more than WT mice after IFN-γ treatment (32). Similar to the ZBP1-NLRP3 inflammasome, the ZBP1-AIM2 inflammasome facilitates the expression of pyroptotic markers, such as caspase-1, GSDMD, and GSDME, and inflammasome activation is reduced in Zbp1–/– BMDMs in response to HSV-1 infection (18).
ZBP1 induces apoptosis during viral infection
Apoptosis was first structurally distinguished from cell death and found to be involved in PCD in the development of Caenorhabditis elegans (90). Apoptosis is triggered by numerous stimuli, including viruses, and mediated by successive caspase reactions. This activation occurs via the initiator caspase, which is present upstream of the effector (or executioner) caspase. The apoptotic initiator caspase contains caspases-2, -8, -9, and -10, and effector caspases contain caspase-3, -6, and -7. These effector caspases play a central role in apoptosis by catalyzing their substrates.
ZBP1-associated apoptosis is also mediated by caspase-8, caspase-3, and caspase-7. Caspase-8 activates caspase-3, which promotes the maturation of GSDME to form pores in the membrane. Additionally, caspase-7 is activated by caspase-8. In Zbp1–/– BMDMs, caspase-8, -3, and -7 are downregulated during HSV-1 and IAV infection (11, 18). Additionally, the activation of caspase-8, -3, and -7 was attenuated in Zbp1–/– and Zbp1ΔZα2/ΔZα2 BMDMs than in WT during MHV infection with IFN-β treatment (32).
ZBP1 induces necroptosis during viral infection
Necroptosis and apoptosis differ in their morphology and molecular pathways. Apoptosis is characterized by cell shrinking, nuclear fragmentation, intra-nucleosomal cleavage, and membrane blebbing (91, 92). In addition, cells exposed to apoptosis show engulfment signals that are then detected by phagocytes. Necroptosis is characterized by a bursting membrane, cell lysis, and pro-DAMP release (93–95). Necroptosis is mediated by RIPK3, which interacts with other RHIM domain-containing molecules via the RHIM domain at the C-terminus (96). Similarly, ZBP1 induces necroptosis via the activation of RIPK3, which phosphorylates MLKL via its kinase domain. Phosphorylated MLKL is then inserted into the membrane, which constitutes a necroptotic pore. During HSV-1 infection, necroptotic markers, phosphorylated RIPK3 and MLKL, are reduced in Zbp1–/– BMDMs (18). The HSV1 viral protein ICP6 induces necroptosis in RHIM-RIPK3-MLKL dependent manner in ZBP1 deficient cells (97). After sensing IAV Z-RNA, ZBP1 induces RIPK3-MLKL-dependent necroptosis (98). In murine cytomegalovirus (MCMV) infection, ZBP1 regulates necroptosis with RIPK3, independent of RIPK1 (99). The phosphorylation of MLKL and RIPK3 was downregulated in the absence of the ZBP1 and Zα2 domains of BMDMs during MHV infection with IFN-β treatment (32). The Zα2 domain of ZBP1 senses vaccinia virus (VV) and induces necroptosis (100). In a study, after SARS-CoV-2 infection, the mRNA levels of ZBP1 and MLKL were increased in mouse neurons and brains (101). The expression of ZBP1, RIPK3, and caspase-8 was found to be increased in blood samples of patients with severe COVID-19, as analyzed using expression quantitative trait loci (eQTL) (102).
ZBP1-PANoptosis: a double-edged sword
PANoptosis
PANoptosis is a unique inflammatory cell death process controlled by the PANoptosome, which reacts to specific stimuli, including viruses. The term PANoptosis was established based on studies that revealed a crosstalk between pyroptosis, apoptosis, and necroptosis. The crosstalk was first observed between pyroptosis and apoptosis (103, 104). Subsequently, the overlapping functions of caspase-8 and caspase-1/NLRP3 for pyroptotic, apoptotic, and necroptotic molecules were identified (61, 105, 106). ZBP1 (11), TAK1 (19, 107), and caspase-6 (14) were recently found to solidify the concept of PANoptosis. Additionally, the Zα2 domain of ZBP1 has been shown to be an essential component of IAV (30) and HSV1 (18)-induced PANoptosis. Furthermore, the roles of PANoptosis and cytokine storms in coronavirus infection have been studied (32, 37, 108). Overall, PANoptosis has been implicated in defense against various pathogens, including viruses (Figure 2).
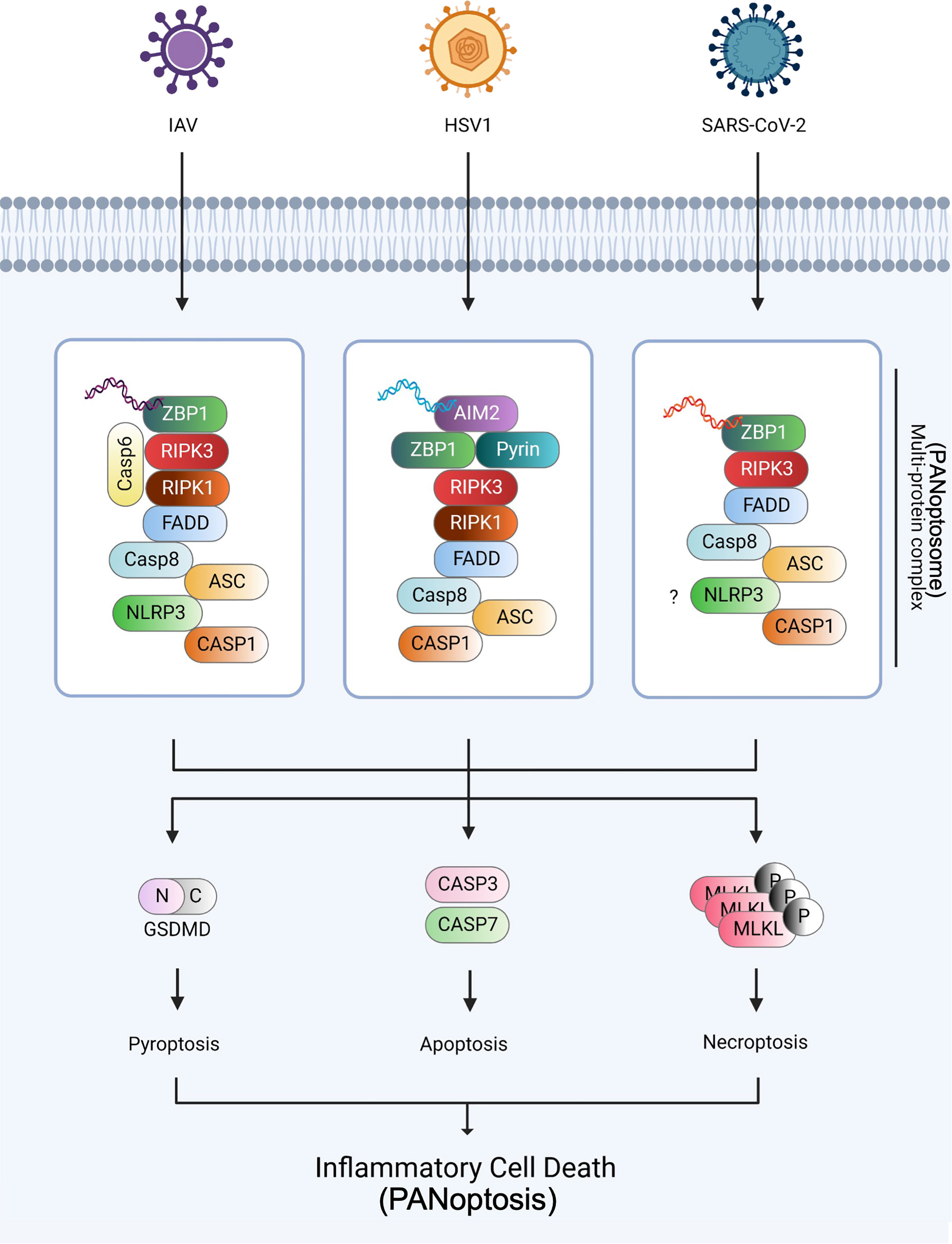
Figure 2 PANoptosome and PANoptosis against IAV, HSV1, and SARS-CoV-2 infections The three viruses, IAV, HSV1, and SARS-CoV-2, were established as the models of PANoptosis. During IAV infection, ZBP1 senses viral dsRNA and recruits RIPK3, RIPK1, caspase-6, and caspase-8 to assemble the PANoptosome, causing GSDMD-mediated pyroptosis, caspase-3 and -7-mediated apoptosis, and MLKL-mediated necroptosis. During HSV1 infection, AIM2, the dsDNA sensor, senses HSV1 dsDNA and recruits ZBP1 and pyrin during PANoptosis. During SARS-CoV-2 infection, ZBP1 interacts with the NLRP3 inflammasome via an unknown mechanism and induces PANoptosis. Created with BioRender.com.
ZBP1 regulates PANoptosis as a central upstream molecule
ZBP1 has been investigated as a necroptotic sensor; however, recently, it has also been revealed to regulate multiple inflammatory cell death processes, including PANoptosis. The first study of ZBP1-derived PANoptosis reported that ZBP1 regulates NLRP3 inflammasome activation to induce PANoptosis via RIPK1-RIPK3-caspase-8 axis during IAV infection. This study suggests that ZBP1 is a central molecule that senses IAV infection by detecting the IAV viral proteins, NP and PB1 (11). IAV Z-RNA also induces PCD by activating ZBP1 and resulting in RIPK1 recruitment and caspase-8 activation (34). Furthermore, the Zα2 domain of ZBP1 regulates PANoptosis and NLRP3 inflammasome during IAV infection (30).
ZBP1-PANoptosis defends host against viral infection
Several studies have established the crosstalk theory that explains the co-activation of pyroptosis, apoptosis, and necroptosis (PANoptosis). Since the first crosstalk between pyroptosis and apoptosis was revealed, ZBP1 showed a critical role in the crosstalk between inflammatory cell deaths. ZBP1 has been studied as a necroptotic sensor (99, 109). However, the regulatory role of ZBP1 was identified in multiple inflammatory cell death pathways. First, ZBP1 stimulates not only NLRP3 inflammasome activation but also apoptosis and necroptosis during IAV infection (11). This study suggests that ZBP1 is a key regulator of the three delegable inflammatory cell death pathways. ZBP1 was found to be highly expressed in IAV-infected WT BMDMs, while being downregulated in IAV-infected Ifnar1–/– BMDMs. IL-1β and IL-18 levels were reduced in Zbp1–/– BMDMs. The scientists observed an interaction between ZBP1 and RIPK3 using immunoprecipitation in IAV-infected WT BMDMs, which induced apoptosis and necroptosis during IAV infection (110, 111). Subsequently, the Zα2 domain of ZBP1 regulates influenza-inducible PANoptosis and NLRP3 inflammasome (30). The absence of the Zα2 domain of ZBP1 induced the downregulation of caspase-1 activation and GSDMD cleavage during IAV infection, which is a criterion for pyroptosis. The activation of caspase-8, caspase-3, and RIPK3 lacked Zbp1ΔZα2/ΔZα2 BMDMs. These results indicated that the Zα2 domain of ZBP1 is critical for the activation of PANoptosis against IAV infection. Additionally, the absence of ZBP1 increases cell death. In summary, ZBP1 is beneficial for host cell survival against IAV infection (11, 32, 34). In other viral infections, such as HSV1, ZBP1 forms a complex with AIM2 and pyrin and defends the host via the induction of PANoptosis (18). In this study, ZBP1 cooperated with pyrin in AIM2 inflammasome activation during HSV1 infection, and ZBP1 induced PANoptosis in response to HSV1 infection in an AIM2-dependent manner. Additionally, a deficiency of the Zα2 domain of ZBP1 reduced HSV1-induced cell death and Zbp1–/– BMDMs. Overall, these studies suggest that ZBP1 is a central mediator of PANoptosis against viral infections to protect host cells from viral lethality.
ZBP1-PANoptosis increases viral lethality through cytokine storm
PANoptosis is not always beneficial for host survival. Dysregulation of cytokines can cause cell death, tissue damage, and mortality due to viral infections (32, 38, 112). Immune hyperactivation occurs as an acute induction of pro-inflammatory cytokine secretion, resulting in a cytokine storm in β-coronavirus infections, including those of SARS-CoV (113–118), MERS-CoV (119–125), MHV (32, 108), and SARS-CoV-2 (32, 39, 126, 127). During SARS-CoV-2 infection, co-treatment with TNF-α and IFN-γ, which mimics the cytokine storm, induces PANoptosis in vitro and in vivo, viral lethality, and severe symptoms, such as tissue damage (37). Robust release of cytokines has been suggested to correlate with lung injury and multiple organ failure (Figure 3) (128–130). After screening a publicly available dataset, various pro-inflammatory cytokines were found to be upregulated in patients with severe COVID-19. Co-treatment with IFN-γ and TNF-α significantly induced PANoptosis in BMDMs and THP-1 cells via the STAT1-interferon regulatory factor 1 (IRF1)-inducible nitric oxide synthase (iNOS)-nitric oxide (NO) axis. NO induces apoptosis by activating caspase-8 (131, 132). Similarly, Ripk3–/–Casp8–/– BMDMs were rescued from PANoptosis induced by IFN-γ and TNF-α when compared with Ripk3–/– BMDMs. Moreover, Ripk3–/–Fadd–/– BMDMs were saved from PANoptosis induced by co-treatment with IFN-γ and TNF-α. Overall, the RIPK1-FADD-CASP8 axis induces PANoptosis by IFN-γ and TNF-α co-treatment. In the in vivo experiment, the levels of serum lactate dehydrogenase (LDH) and immune cells in the blood were reduced in STAT1–/– and RIPK3–/–Casp8–/– mice co-treated with IFN-γ and TNF-α. Blocking IFN-γ and TNF-α using neutralizing antibodies significantly increased the survival of SARS-CoV-2-infected mice when compared to that of an isotype control. Collectively, IFN-γ and TNF-α play critical roles in the induction of PANoptosis and cytokine storms during SARS-CoV-2 infection. Similarly, the Zα2 domain of ZBP1 upregulates PANoptosis and cytokine storm during β-coronavirus, SARS-CoV-2, and MHV infections with IFN treatment (32). Interestingly, delayed IFN-β release and STAT1 activation were observed in MHV infection, which mirrors the biology of human β-coronavirus. IFN-β treatment a few days after MHV and SARS-CoV-2 infection induces the activation of PANoptosis markers, including caspase-1, GSDMD, GSDME, caspase-8, -3, -7, MLKL, and RIPK3. Delayed IFN-β treatment influences the pathogenesis of MERS-CoV in a mouse model (3) and delayed IFN-α2b treatment upregulates mortality in patients with SARS-CoV-2 (133). These results indicate that delayed cytokine release favors β-coronavirus lethality. ZBP1 was then used to identify the sensing mechanism of β-coronavirus among ISGs significantly upregulated by MHV infection in immortalized BMDMs (iBMDMs). ZBP1-deficient mice showed no significant difference in the presence or absence of IFN-β, and ZBP1 was highly expressed in the lungs of mice infected with MHV following IFN-β treatment than in the lungs of MHV-infected and untreated mice. Similarly, a deficiency of the Za2 domain of ZBP1 reduces cell death during MHV infection. However, IFN-β treatment did not induce any changes in cell death during IAV infection in Zbp1–/– and Zbp1ΔZα2 mice. Moreover, Zbp1–/– and Zbp1ΔZα2 mice showed the downregulation of PANoptosis markers compared to WT mice during MHV infection with IFN-β treatment. In a very recent study, it was observed that the deletion of ZBP1 or RIPK3 reduced the secretion of inflammatory cytokines and chemokines and attenuated immune cell infiltration and lung damage during SARS-CoV-2 infection in vivo (39).
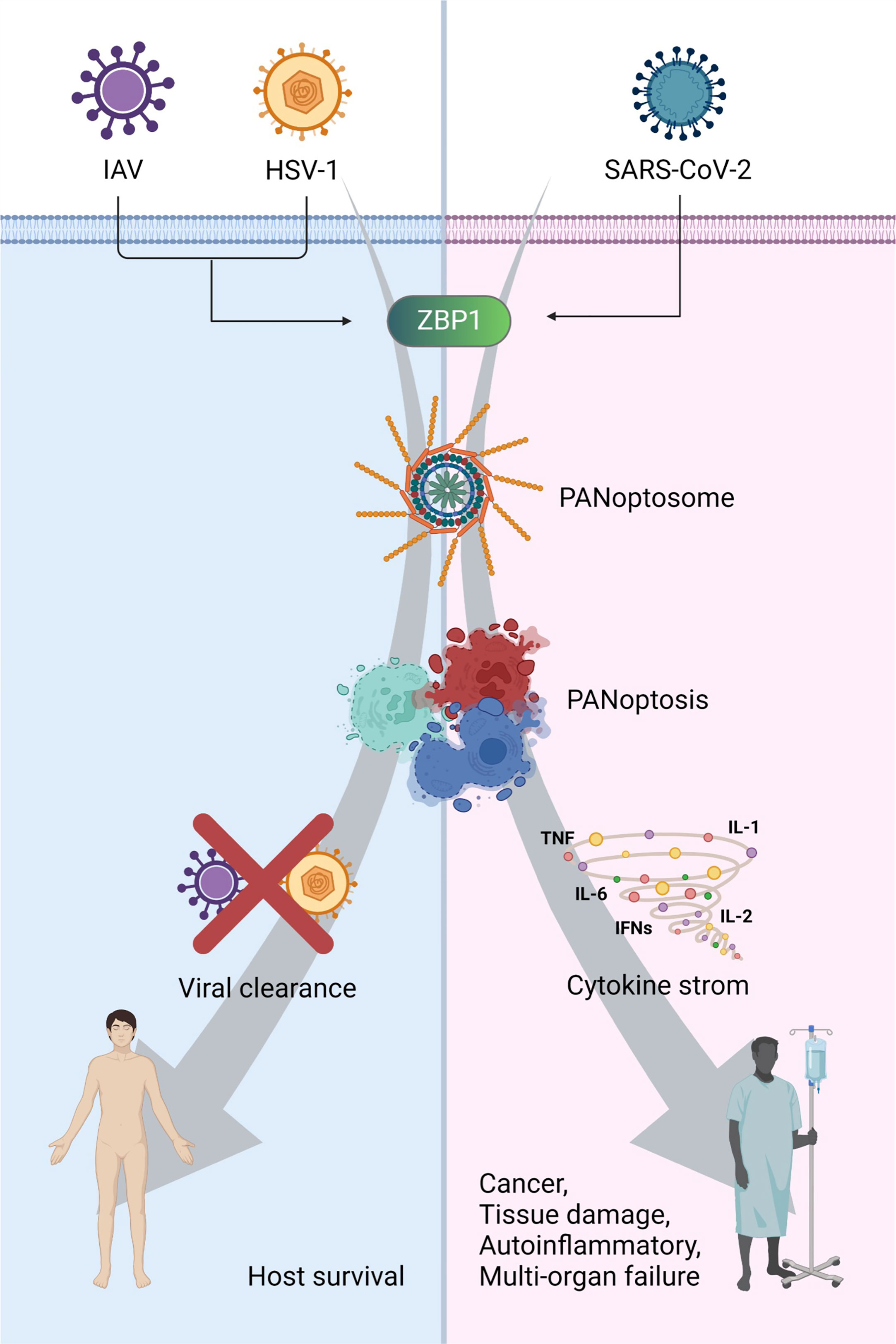
Figure 3 The consequence of PANoptosis: a double-edged sword in viral pathogenesis ZBP1-mediated PANoptosis can protect the host against viral infections, such as IAV and HSV1. However, it can trigger cytokine storms and cause cancer, tissue damage, autoinflammation, and multiple-organ failure. Created with BioRender.com.
Viral immune evasion
The virus can evade the host immune system through viral components including viral proteins. Viral immune evasion helps viruses grow, transmit, and survive in the host body to escape the host immune system, thereby causing failure in the immune response (134). The virus has diverse strategies for escaping the host immune system. They inhibit signaling pathways by targeting specific immune signaling-mediated proteins, such as inhibiting IRF9 (also known as p48) (135, 136) or blocking the phosphorylation of STAT1 (135, 137, 138). ZBP1 has also been reported as a target of viral evasion strategies (26, 99, 139, 140). An MCMV viral protein, M45, is considered to suppress the interaction of the ZBP1-RIPK1/RIPK3 and downstream signaling pathway depending on the RHIM domain (26), and ZBP1-RIPK3 interaction occurs in M45mutRHIM MCMV infection but not in WT MCMV (11, 99). This study identified ZBP1 as a target of MCMV evasion. VV viral protein, E3, which contains the Zα domain inhibits IFN and RIPK3-dependent necroptosis with ZBP1 during VV infection (140). Additional studies about viral evasion strategies are suggested from an RHIM-dependent perspective (141).
Other viral proteins may disrupt the inflammatory cell death signaling pathway. The nonstructural protein (NS1) is a well-known IAV viral protein that inhibits the transcription of antiviral genes and intracellular ISGs, including protein kinase R (PKR) (142) and 2,’ 5’-oligoadenylate synthetase (2’-5’ OAS), by binding to viral RNA to prevent detection by ISGs (143). The other IAV viral protein, PB1, particularly PB1-F2, directly interacts with MAVS (144, 145). PB1-F2 induces pyroptosis by interacting with the NLRP3 inflammasome, which causes an increased production of IL-1β (146). Additionally, the substitution of an amino acid (Asn66Ser) is known to inhibit type 1 and type 3 IFNs (147). Similarly, HSV can evade the host immune system through various mechanisms. Similar to IAV, HSV represses the IFN response through viral proteins, including ICP0, ICP27, ICP34.5, Us3, and vhs. Each protein inhibits IFN expression in diverse ways. ICP0 modifies IRF3 and IRF7 (148, 149), and ICP27 reduces IFN and cytokine expression by inhibiting IRF3 and NF-kB activation (150). Downstream molecules of the IFN signaling pathway, such as STAT1, are also targeted by HSV viral proteins. In addition, HSV viral proteins ICP4, ICP27, ICP34.5, and gJ inhibit apoptosis in various ways, such as caspase inhibition and downregulation of Fas ligand (151). Coronavirus impedes the innate immune system using viral proteins, nonstructural proteins (Nsp), and open reading frames (OFR). Nsp1 inhibits the IFN signaling pathway, particularly SARS-CoV-2 Nsp1, which suppresses the promoter activity of IFN-stimulated response elements (ISREs) (152). Nsp3, the largest protein encoded by the coronavirus genome, can bind to IRF3 and suppress the phosphorylation and nuclear translocation of IRF3, thereby leading to the inhibition of the IFN signaling pathway (153). Moreover, Nsp13 and Nsp15 can modify the viral RNA to escape from the guards of the host. The 5’-ppp moiety, a type of RIG-I ligand, is regulated by Nsp13 (154, 155), and Nsp15 removes the 5’-polyuridine (polyU) region from 5’-polyU-containing, negative-sense RNAs, which helps viral RNA to hide from cytosolic dsRNA sensors, including PKR, MDA5, and OAS/RNase L (156). The ORF family also antagonizes the host inflammatory and IFN signaling pathways (157–167). These numerous strategies allow viruses to evade the host innate immune system, particularly the IFN signaling pathway, and these strategies may evolve owing to the importance of IFN in defending the host from viruses.
Cytokine storm-related cytokines: IFNs and TNF
IFNs and TNF are essential components of the innate immune system. IFN secretion is activated when PRRs (RIG-I) sense viral components and stimulate IRF3, which induces the secretion of type 1 IFNs. Secreted IFNs are then detected by IFNAR1/2, and the STAT1 signaling pathway is induced (80, 168). ZBP1 is also a downstream molecule in the IFN pathway. Induction of IRF9 by the STAT1 pathway stimulates ZBP1 expression in Ifnar1–/–, Stat1–/–, and Irf9–/– cells, and ZBP1 activation was abolished (11). IFNs are required to induce ZBP1-derived inflammatory cell death. IFNs mediate pyroptosis (11, 169), necroptosis (170), and apoptosis (171) associated with ZBP1. Therefore, IFN is critical for PANoptosis. Cytokine storms, which compensate for severe viral lethality in the host, are also related to IFNs. One of the well-studied theories about the relationship between IFNs and cytokine storms is the delayed activation of IFN. IFN delay enhances cytokine secretion and disease during viral infections (3, 133, 172). During MERS-CoV infection, IFN-β delay enhances pro-inflammatory cytokines released in monocytes, macrophages, and neutrophils (3). Delayed type 1 IFN signaling promotes SARS-CoV infection (172). Besides, CD4+ and CD8+ T cells, which are involved in adaptive immune, are reduced in severe COVID-19 (173, 174), and Th17 CD4+ T cells, which act in a pro-inflammatory role, are increased (126). In a recent study, SARS-CoV-2 revealed that can directly infect T lymphocytes in a spike-ACE2/TMPRSS2-independent manner (175). The dysregulated immune system induces non-specific immune cells and the release of pro-inflammatory factors (176, 177). In conclusion, the IFN secretory pathway may burst during the late phase of viral infection and promote host survival.
Concluding remarks
In this review, we summarize the molecular-based mechanism of inflammatory cell death by focusing on viruses that cause all three major cell deaths. Considering IAV and HSV1 infections, ZBP1-derived PANoptosis plays an important role in host survival. However, side effects such as a cytokine storm in coronavirus infections lead to systemic inflammation, organ failure, and even death of the host. Therefore, virus-induced cell death must be controlled to reduce the hyperactivation of the immune response by conducting virus-specific studies. There are differences in the viral infection method, viral life cycle, and immune evasion strategies for each virus. In addition, various viral proteins help viruses evade and cause confusion in the host immune system. Further studies should be conducted to elucidate the exact mechanism through which the cytokine storm occurs.
The regulation of cytokines and signaling pathways is important for controlling the pathogenesis of the hyperactivated immune system. In particular, TNF and IFN-γ synergize cytokine storms by generating a feedback loop in coronavirus infection. Thus, the neutralization of TNF and IFN-γ may be valuable to rescue excessive cytokine secretion (178). This process is induced by the caspase-8-JAK1/2-STAT1 axis (178). Inhibitors of molecules that participate in this signaling pathway, such as STAT1 or JAK1/2 inhibitors, would be effective. In the case of JAK1/2 inhibitors, baricitinib received an emergency use authorization to cure COVID-19 in 2020. Collectively, pro-inflammatory cytokines must be regulated to rescue the host from the cytokine storm loop and subsequent lethal symptoms, such as systemic inflammation.
Other strategies that target inflammasome components, including sensors, ZBP1, NLRP3, and downstream molecules, caspases, RIPK1, RIPK3, and ASC may be helpful. In caspase-8 and MLKL double-knockout (DKO) mice, weight loss induced by SARS-CoV-2 infection was abolished, although the viral burden did not change (179). Casp8–/–Ripk3–/– DKO mice were rescued from viral lethality induced by TNF and IFN-γ co-treatment but not Ripk3–/– mice (37). These results indicate that the components of PANoptosis play an important role in fatal cytokine storms. If we can directly control ZBP1 and NLRP3, this may be an efficient method. One of the possible strategies is by using ADAR1. ADAR1 acts as a repressor of the ZBP1-NLRP3 inflammasome and causes multiple inflammatory cell deaths (33). Additionally, there are other studies on the capacity of ADAR1 for suppressing ZBP1-mediated PCD (180–183). Collectively, we can overcome ZBP1-derived multiple inflammatory cell death and severe signs, such as cytokine storms, using molecular-based therapeutic strategies.
Furthermore, IFN delay causes hyperactivated secretion of cytokine (3, 133, 172). Thus, IFN therapy, which is used for the treatment of viral infection, would be harmful to patients with COVID-19 by eliciting excessive activation of cytokines. Therefore, treatment should be administered with caution after further research. In summary, the unresolved questions need to be addressed to develop a strong defense strategy against viral infections and to control multiple inflammatory cell deaths.
Author contributions
SO and SL conceived the manuscript. SO and SL wrote the manuscript. SO and SL critically revised and approved the final version of the manuscript. All authors have contributed to the manuscript and approved the submitted version.
Funding
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2022R1C1C1007544 to SL), by grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare (HV22C015600 to SL), by research grant from the Korea National Institute of Health funded by Korea Disease Control and Prevention Agency (KDCA) (2.221151.01 to SL), by research grant from Korea Virus Research Institute (KVRI), Institute for Basic Science (IBS) (2.230625.01 to SL), by research fund from Ulsan National Institute of Science & Technology (UNIST) (1.220112.01, 1.220107.01 to SL). This study also received funding from Yuhan Corporation (2.220961.01 to SL). The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Acknowledgments
We thank the members of the Lee Lab (Viral Immunology Lab) for their helpful comments and suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lee S, Ishitsuka A, Noguchi M, Hirohama M, Fujiyasu Y, Petric PP, et al. Influenza restriction factor MxA functions as inflammasome sensor in the respiratory epithelium. Sci Immunol (2019) 4(40). doi: 10.1126/sciimmunol.aau4643
2. Lee S, Hirohama M, Noguchi M, Nagata K, Kawaguchi A. Influenza a virus infection triggers pyroptosis and apoptosis of respiratory epithelial cells through the?type I interferon signaling pathway in a mutually exclusive manner. J Virol (2018) 92(14). doi: 10.1128/JVI.00396-18
3. Channappanavar R, Fehr AR, Zheng J, Wohlford-Lenane C, Abrahante JE, Mack M, et al. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest (2019) 129:3625–39. doi: 10.1172/JCI126363
4. Cervantes-Barragan L, Züst R, Weber F, Spiegel M, Lang KS, Akira S, et al. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood (2007) 109:1131–7. doi: 10.1182/blood-2006-05-023770
5. Zheng M, Karki R, Williams EP, Yang D, Fitzpatrick E, Vogel P, et al. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat Immunol (2021) 22:829–38. doi: 10.1038/s41590-021-00937-x
6. Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell (2010) 140:397–408. doi: 10.1016/j.cell.2010.01.020
7. Baum A, Sachidanandam R, García-Sastre A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci U.S.A. (2010) 107:16303–8. doi: 10.1073/pnas.1005077107
8. Weber M, Gawanbacht A, Habjan M, Rang A, Borner C, Schmidt AM, et al. Incoming RNA virus nucleocapsids containing a 5'-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe (2013) 13:336–46. doi: 10.1016/j.chom.2013.01.012
9. Liu G, Park HS, Pyo HM, Liu Q, Zhou Y. Influenza a virus panhandle structure is directly involved in RIG-I activation and interferon induction. J Virol (2015) 89:6067–79. doi: 10.1128/JVI.00232-15
10. Sumpter R, Omma C,jr., et al. Regulating intracellular antiviral defense and permissiveness to hepatitis c virus RNA replication through a cellular RNA helicase, RIG-I. J Virol (2005) 79:2689–99. doi: 10.1128/JVI.79.5.2689-2699.2005
11. Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol (2016) 1(2). doi: 10.1126/sciimmunol.aag2045
12. Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH, 3rd Ingram JP, et al. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza a virus. Cell Host Microbe (2016) 20:13–24. doi: 10.1016/j.chom.2016.05.011
13. Kesavardhana S, Kuriakose T, Guy CS, Samir P, Malireddi RKS, Mishra A, et al. ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J Exp Med (2017) 214:2217–29. doi: 10.1084/jem.20170550
14. Zheng M, Karki R, Vogel P, Kanneganti TD. Caspase-6 is a key regulator of innate immunity, inflammasome activation, and host defense. Cell (2020) 181:674–687.e613. doi: 10.1016/j.cell.2020.03.040
15. Pham TH, Kwon KM, Kim YE, Kim KK, Ahn JH. DNA Sensing-independent inhibition of herpes simplex virus 1 replication by DAI/ZBP1. J Virol (2013) 87:3076–86. doi: 10.1128/JVI.02860-12
16. Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, et al. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe (2015) 17:243–51. doi: 10.1016/j.chom.2015.01.003
17. Hayes CK, Wilcox DR, Yang Y, Coleman GK, Brown MA, Longnecker R. ASC-dependent inflammasomes contribute to immunopathology and mortality in herpes simplex encephalitis. PloS Pathog (2021) 17:e1009285. doi: 10.1371/journal.ppat.1009285
18. Lee S, Karki R, Wang Y, Nguyen LN, Kalathur RC, Kanneganti TD. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature (2021) 597:415–9. doi: 10.1038/s41586-021-03875-8
19. Malireddi RKS, Gurung P, Kesavardhana S, Samir P, Burton A, Mummareddy H, et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J Exp Med (2020) 217(3). doi: 10.1084/jem.20191644
20. Malireddi RKS, Kesavardhana S, Karki R, Kancharana B, Burton AR, Kanneganti TD. RIPK1 distinctly regulates yersinia-induced inflammatory cell death, PANoptosis. Immunohorizons (2020) 4:789–96. doi: 10.4049/immunohorizons.2000097
21. Christgen S, Zheng M, Kesavardhana S, Karki R, Malireddi RKS, Banoth B, et al. Identification of the PANoptosome: a molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol (2020) 10:237. doi: 10.3389/fcimb.2020.00237
22. Wang Y, Kanneganti TD. From pyroptosis, apoptosis and necroptosis to PANoptosis: a mechanistic compendium of programmed cell death pathways. Comput Struct Biotechnol J (2021) 19:4641–57. doi: 10.1016/j.csbj.2021.07.038
23. Fu Y, Comella N, Tognazzi K, Brown LF, Dvorak HF, Kocher O. Cloning of DLM-1, a novel gene that is up-regulated in activated macrophages, using RNA differential display. Gene (1999) 240:157–63. doi: 10.1016/S0378-1119(99)00419-9
24. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature (2007) 448:501–5. doi: 10.1038/nature06013
25. Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature (2008) 451:725–9. doi: 10.1038/nature06537
26. Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep (2009) 10:916–22. doi: 10.1038/embor.2009.109
27. Schwartz T, Behlke J, Lowenhaupt K, Heinemann U, Rich A. Structure of the DLM-1-Z-DNA complex reveals a conserved family of z-DNA-binding proteins. Nat Struct Biol (2001) 8:761–5. doi: 10.1038/nsb0901-761
28. Rothenburg S, Schwartz T, Koch-Nolte F, Haag F. Complex regulation of the human gene for the z-DNA binding protein DLM-1. Nucleic Acids Res (2002) 30:993–1000. doi: 10.1093/nar/30.4.993
29. Wang Z, Choi MK, Ban T, Yanai H, Negishi H, Lu Y, et al. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc Natl Acad Sci U.S.A. (2008) 105:5477–82. doi:?10.1073/pnas.0801295105
30. Kesavardhana S, Malireddi RKS, Burton AR, Porter SN, Vogel P, Pruett-Miller SM, et al. The Zα2 domain of ZBP1 is a molecular switch regulating influenza-induced PANoptosis and perinatal lethality during development. J Biol Chem (2020) 295:8325–30. doi: 10.1074/jbc.RA120.013752
31. Ha SC, Van Quyen D, Hwang HY, Oh DB, Brown BA, 2nd Lee SM, et al. Biochemical characterization and preliminary X-ray crystallographic study of the domains of human ZBP1 bound to left-handed z-DNA. Biochim Biophys Acta (2006) 1764:320–3. doi: 10.1016/j.bbapap.2005.12.012
32. Karki R, Lee S, Mall R, Pandian N, Wang Y, Sharma BR, et al. ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci Immunol (2022) 7(74):eabo6294. doi: 10.1126/sciimmunol.abo6294
33. Karki R, Sundaram B, Sharma BR, Lee S, Malireddi RKS, Nguyen LN, et al. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep (2021) 37:109858. doi: 10.1016/j.celrep.2021.109858
34. Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, et al. DAI senses influenza a virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe (2016) 20:674–81. doi: 10.1016/j.chom.2016.09.014
35. Kaiser WJ, Upton JW, Mocarski ES. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa b activation via the DNA-dependent activator of IFN regulatory factors. J Immunol (2008) 181:6427–34. doi: 10.4049/jimmunol.181.9.6427
36. Muendlein HI, Connolly WM, Magri Z, Smirnova I, Ilyukha V, Gautam A, et al. ZBP1 promotes LPS-induced cell death and IL-1β release via RHIM-mediated interactions with RIPK1. Nat Commun (2021) 12:86. doi: 10.1038/s41467-020-20357-z
37. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell (2021) 184:149–168.e117. doi: 10.1016/j.cell.2020.11.025
38. Lee S, Channappanavar R, Kanneganti TD. Coronaviruses: innate immunity, inflammasome activation, inflammatory cell death, and cytokines. Trends Immunol (2020) 41:1083–99. doi: 10.1016/j.it.2020.10.005
39. Li S, Zhang Y, Guan Z, Ye M, Li H, You M, et al. SARS-CoV-2 z-RNA activates the ZBP1-RIPK3 pathway to promote virus-induced inflammatory responses. Cell Res (2023) 33(3):1–14. doi: 10.1038/s41422-022-00775-y
40. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
41. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin d is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res (2015) 25:1285–98. doi: 10.1038/cr.2015.139
42. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin d for non-canonical inflammasome signalling. Nature (2015) 526:666–71. doi: 10.1038/nature15541
43. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
44. Kanneganti TD, Ozören N, Body-Malapel M, Amer A, Park JH, Franchi L, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature (2006) 440:233–6. doi: 10.1038/nature04517
45. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature (2006) 440:228–32. doi: 10.1038/nature04515
46. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature (2006) 440:237–41. doi: 10.1038/nature04516
47. Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozören N, Jagirdar R, et al. Cytosolic flagellin requires ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol (2006) 7:576–82. doi: 10.1038/ni1346
48. Ren T, Zamboni DS, Roy CR, Dietrich WF, Vance RE. Flagellin-deficient legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PloS Pathog (2006) 2:e18. doi: 10.1371/journal.ppat.0020018
49. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458:509–13. doi: 10.1038/nature07710
50. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458:514–8. doi: 10.1038/nature07725
51. Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science (2009) 323:1057–60. doi: 10.1126/science.1169841
52. Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol (2009) 10:266–72. doi: 10.1038/ni.1702
53. Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate immune sensing of bacterial modifications of rho GTPases by the pyrin inflammasome. Nature (2014) 513:237–41. doi: 10.1038/nature13449
54. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145:745–57. doi: 10.1016/j.cell.2011.04.022
55. Zhu S, Ding S, Wang P, Wei Z, Pan W, Palm NW, et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature (2017) 546:667–70. doi: 10.1038/nature22967
56. Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, et al. The NLRP12 inflammasome recognizes yersinia pestis. Immunity (2012) 37:96–107. doi: 10.1016/j.immuni.2012.07.006
57. Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe (2011) 9:363–75. doi: 10.1016/j.chom.2011.04.008
58. Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol (2010) 11:63–9. doi: 10.1038/ni.1824
59. Mathur A, Feng S, Hayward JA, Ngo C, Fox D, Atmosukarto II, et al. A multicomponent toxin from bacillus cereus incites inflammation and shapes host outcome via the NLRP3 inflammasome. Nat Microbiol (2019) 4:362–74. doi: 10.1038/s41564-018-0318-0
60. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479:117–21. doi: 10.1038/nature10558
61. Gurung P, Anand PK, Malireddi RK, Vande Walle L, Van Opdenbosch N, Dillon CP, et al. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol (2014) 192:1835–46. doi: 10.4049/jimmunol.1302839
62. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514:187–92. doi: 10.1038/nature13683
63. Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem (2006) 281:36560–8. doi: 10.1074/jbc.M607594200
64. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza a virus through recognition of viral RNA. Immunity (2009) 30:556–65. doi: 10.1016/j.immuni.2009.02.005
65. Thomas PG, Dash P, Aldridge JR, Jr. , Ellebedy AH, Reynolds C, Funk AJ, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza a virus via the regulation of caspase-1. Immunity (2009) 30:566–75. doi: 10.1016/j.immuni.2009.02.006
66. Balachandran S, Mocarski ES. Viral z-RNA triggers ZBP1-dependent cell death. Curr Opin Virol (2021) 51:134–40. doi: 10.1016/j.coviro.2021.10.004
67. Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, et al. IL-1β signaling promotes CNS-intrinsic immune control of West Nile virus infection. PloS Pathog (2012) 8:e1003039. doi: 10.1371/journal.ppat.1003039
68. Gimenez F, Bhela S, Dogra P, Harvey L, Varanasi SK, Jaggi U, et al. The inflammasome NLRP3 plays a protective role against a viral immunopathological lesion. J Leukoc Biol (2016) 99:647–57. doi: 10.1189/jlb.3HI0715-321R
69. Johnson KE, Chikoti L, Chandran B. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J Virol (2013) 87:5005–18. doi: 10.1128/JVI.00082-13
70. Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell (2013) 49:331–8. doi: 10.1016/j.molcel.2012.11.009
71. Song N, Liu ZS, Xue W, Bai ZF, Wang QY, Dai J, et al. NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol Cell (2017) 68:185–197.e186. doi: 10.1016/j.molcel.2017.08.017
72. Stutz A, Kolbe CC, Stahl R, Horvath GL, Franklin BS, van Ray O, et al. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J Exp Med (2017) 214:1725–36. doi: 10.1084/jem.20160933
73. Barry R, John SW, Liccardi G, Tenev T, Jaco I, Chen CH, et al. SUMO-mediated regulation of NLRP3 modulates inflammasome activity. Nat Commun (2018) 9:3001. doi: 10.1038/s41467-018-05321-2
74. Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ (2007) 14:1583–9. doi: 10.1038/sj.cdd.4402195
75. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
76. Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science (2008) 320:674–7. doi: 10.1126/science.1156995
77. Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem (2007) 282:2871–9. doi: 10.1074/jbc.M608083200
78. Place DE, Kanneganti TD. Recent advances in inflammasome biology. Curr Opin Immunol (2018) 50:32–8. doi: 10.1016/j.coi.2017.10.011
79. Lee S, Ishitsuka A, Kuroki T, Lin YH, Shibuya A, Hongu T, et al. Arf6 exacerbates allergic asthma through cell-to-cell transmission of ASC inflammasomes. JCI Insight (2021) 6(16). doi: 10.1172/jci.insight.139190
80. Kuriakose T, Zheng M, Neale G, Kanneganti TD. IRF1 is a transcriptional regulator of ZBP1 promoting NLRP3 inflammasome activation and cell death during influenza virus infection. J Immunol (2018) 200:1489–95. doi: 10.4049/jimmunol.1701538
81. D'Souza CA, Heitman J. Dismantling the cryptococcus coat. Trends Microbiol (2001) 9:112–3. doi: 10.1016/S0966-842X(00)01945-4
82. Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol (2017) 27:673–84. doi: 10.1016/j.tcb.2017.05.005
83. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun (2017) 8:14128. doi: 10.1038/ncomms14128
84. Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, et al. Caspase-8 induces cleavage of gasdermin d to elicit pyroptosis during yersinia infection. Proc Natl Acad Sci U.S.A. (2018) 115:E10888–e10897. doi: 10.1073/pnas.1809548115
85. Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol (2015) 25:308–15. doi: 10.1016/j.tcb.2014.12.009
86. Li XQ, Yu Q, Fang B, Zhang ZL, Ma H. Knockdown of the AIM2 molecule attenuates ischemia-reperfusion-induced spinal neuronal pyroptosis by inhibiting AIM2 inflammasome activation and subsequent release of cleaved caspase-1 and IL-1β. Neuropharmacology (2019) 160:107661. doi: 10.1016/j.neuropharm.2019.05.038
87. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477:596–600. doi: 10.1038/nature10510
88. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
89. Man SM, Kanneganti TD. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol (2016) 16:7–21. doi: 10.1038/nri.2015.7
90. Horvitz HR. Genetic control of programmed cell death in the nematode caenorhabditis elegans. Cancer Res (1999) 59(7 Suppl):1701s–6s.
91. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer (1972) 26:239–57. doi: 10.1038/bjc.1972.33
92. Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature (1998) 391:43–50. doi: 10.1038/34112
93. Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U.S.A. (2012) 109:5322–7. doi: 10.1073/pnas.1200012109
94. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol (2014) 16:55–65. doi: 10.1038/ncb2883
95. Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res (2014) 24:105–21. doi: 10.1038/cr.2013.171
96. Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem (2002) 277:9505–11. doi: 10.1074/jbc.M109488200
97. Omoto S, Guo H, Talekar GR, Roback L, Kaiser WJ, Mocarski ES. Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J Biol Chem (2015) 290:11635–48. doi: 10.1074/jbc.M115.646042
98. Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Shubina M, et al. Influenza virus z-RNAs induce ZBP1-mediated necroptosis. Cell (2020) 180:1115–1129.e1113. doi: 10.1016/j.cell.2020.02.050
99. Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe (2012) 11:290–7. doi: 10.1016/j.chom.2012.01.016
100. Koehler H, Cotsmire S, Zhang T, Balachandran S, Upton JW, Langland J, et al. Vaccinia virus E3 prevents sensing of z-RNA to block ZBP1-dependent necroptosis. Cell Host Microbe (2021) 29:1266–1276.e1265. doi: 10.1016/j.chom.2021.05.009
101. Rothan HA, Kumari P, Stone S, Natekar JP, Arora K, Auroni TT, et al. SARS-CoV-2 infects primary neurons from human ACE2 expressing mice and upregulates genes involved in the inflammatory and necroptotic pathways. Pathogens (2022) 11(2). doi: 10.3390/pathogens11020257
102. Junqueira C, Crespo Ã, Ranjbar S, Lewandrowski M, Ingber J, de Lacerda LB, et al. SARS-CoV-2 infects blood monocytes to activate NLRP3 and AIM2 inflammasomes, pyroptosis and cytokine release. Res Sq (2021). doi: 10.1101/2021.03.06.21252796
103. Malireddi RK, Ippagunta S, Lamkanfi M, Kanneganti TD. Cutting edge: proteolytic inactivation of poly(ADP-ribose) polymerase 1 by the Nlrp3 and Nlrc4 inflammasomes. J Immunol (2010) 185:3127–30. doi: 10.4049/jimmunol.1001512
104. Lamkanfi M, Kanneganti TD, Van Damme P, Vanden Berghe T, Vanoverberghe I, Vandekerckhove J, et al. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics (2008) 7:2350–63. doi: 10.1074/mcp.M800132-MCP200
105. Lukens JR, Gurung P, Vogel P, Johnson GR, Carter RA, McGoldrick DJ, et al. Dietary modulation of the microbiome affects autoinflammatory disease. Nature (2014) 516:246–9. doi: 10.1038/nature13788
106. Gurung P, Burton A, Kanneganti TD. NLRP3 inflammasome plays a redundant role with caspase 8 to promote IL-1β-mediated osteomyelitis. Proc Natl Acad Sci U.S.A. (2016) 113:4452–7. doi: 10.1073/pnas.1601636113
107. Malireddi RKS, Gurung P, Mavuluri J, Dasari TK, Klco JM, Chi H, et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med (2018) 215:1023–34. doi: 10.1084/jem.20171922
108. Zheng M, Williams EP, Malireddi RKS, Karki R, Banoth B, Burton A, et al. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J Biol Chem (2020) 295:14040–52. doi: 10.1074/jbc.RA120.015036
109. Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature (2020) 580:391–5. doi: 10.1038/s41586-020-2129-8
110. Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science (2014) 343:1357–60. doi: 10.1126/science.1249361
111. Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell (2014) 56:481–95. doi: 10.1016/j.molcel.2014.10.021
112. Place DE, Lee S, Kanneganti TD. PANoptosis in microbial infection. Curr Opin Microbiol (2021) 59:42–9. doi: 10.1016/j.mib.2020.07.012
113. Yen YT, Liao F, Hsiao CH, Kao CL, Chen YC, Wu-Hsieh BA. Modeling the early events of severe acute respiratory syndrome coronavirus infection in vitro. J Virol (2006) 80:2684–93. doi: 10.1128/JVI.80.6.2684-2693.2006
114. Cheung CY, Poon LL, Ng IH, Luk W, Sia SF, Wu MH, et al. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J Virol (2005) 79:7819–26. doi: 10.1128/JVI.79.12.7819-7826.2005
115. Law HK, Cheung CY, Ng HY, Sia SF, Chan YO, Luk W, et al. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood (2005) 106:2366–74. doi: 10.1182/blood-2004-10-4166
116. Chien JY, Hsueh PR, Cheng WC, Yu CJ, Yang PC. Temporal changes in cytokine/chemokine profiles and pulmonary involvement in severe acute respiratory syndrome. Respirology (2006) 11:715–22. doi: 10.1111/j.1440-1843.2006.00942.x
117. Wang CH, Liu CY, Wan YL, Chou CL, Huang KH, Lin HC, et al. Persistence of lung inflammation and lung cytokines with high-resolution CT abnormalities during recovery from SARS. Respir Res (2005) 6:42. doi: 10.1186/1465-9921-6-42
118. Wong CK, Lam CWK, Wu AKL, Ip WK, Lee NLS, Chan IHS, et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol (2004) 136:95–103. doi: 10.1111/j.1365-2249.2004.02415.x
119. Lau SKP, Lau CCY, Chan KH, Li CPY, Chen H, Jin DY, et al. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel middle East respiratory syndrome coronavirus: implications for pathogenesis and treatment. J Gen Virol (2013) 94:2679–90. doi: 10.1099/vir.0.055533-0
120. Zhou J, Chu H, Li C, Wong BH, Cheng ZS, Poon VK, et al. Active replication of middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J Infect Dis (2014) 209:1331–42. doi: 10.1093/infdis/jit504
121. Tynell J, Westenius V, Rönkkö E, Munster VJ, Melén K, Österlund P, et al. Middle East respiratory syndrome coronavirus shows poor replication but significant induction of antiviral responses in human monocyte-derived macrophages and dendritic cells. J Gen Virol (2016) 97:344–55. doi: 10.1099/jgv.0.000351
122. Chu H, Zhou J, Wong BH, Li C, Chan JF, Cheng ZS, et al. Middle East respiratory syndrome coronavirus efficiently infects human primary T lymphocytes and activates the extrinsic and intrinsic apoptosis pathways. J Infect Dis (2016) 213:904–14. doi: 10.1093/infdis/jiv380
123. Scheuplein VA, Seifried J, Malczyk AH, Miller L, Höcker L, Vergara-Alert J, et al. High secretion of interferons by human plasmacytoid dendritic cells upon recognition of middle East respiratory syndrome coronavirus. J Virol (2015) 89:3859–69. doi: 10.1128/JVI.03607-14
124. Min CK, Cheon S, Ha NY, Sohn KM, Kim Y, Aigerim A, et al. Comparative and kinetic analysis of viral shedding and immunological responses in MERS patients representing a broad spectrum of disease severity. Sci Rep (2016) 6:25359. doi: 10.1038/srep25359
125. Kim ES, Choe PG, Park WB, Oh HS, Kim EJ, Nam EY, et al. Clinical progression and cytokine profiles of middle East respiratory syndrome coronavirus infection. J Korean Med Sci (2016) 31:1717–25. doi: 10.3346/jkms.2016.31.11.1717
126. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in wuhan, China. Lancet (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
127. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature (2020) 584:463–9. doi: 10.1038/s41586-020-2588-y
128. Ragab D, Salah Eldin H, Taeimah M, Khattab R, Salem R. The COVID-19 cytokine storm; what we know so far. Front Immunol (2020) 11:1446. doi: 10.3389/fimmu.2020.01446
129. Jose RJ, Manuel A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir Med (2020) 8:e46–7. doi: 10.1016/S2213-2600(20)30216-2
130. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet (2020) 395:1033–4. doi: 10.1016/S0140-6736(20)30628-0
131. Du C, Guan Q, Diao H, Yin Z, Jevnikar AM. Nitric oxide induces apoptosis in renal tubular epithelial cells through activation of caspase-8. Am J Physiol Renal Physiol (2006) 290:F1044–1054. doi: 10.1152/ajprenal.00341.2005
132. Dubey M, Nagarkoti S, Awasthi D, Singh AK, Chandra T, Kumaravelu J, et al. Nitric oxide-mediated apoptosis of neutrophils through caspase-8 and caspase-3-dependent mechanism. Cell Death Dis (2016) 7:e2348. doi: 10.1038/cddis.2016.248
133. Wang N, Zhan Y, Zhu L, Hou Z, Liu F, Song P, et al. Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID-19 patients. Cell Host Microbe (2020) 28:455–464.e452. doi: 10.1016/j.chom.2020.07.005
134. Lee S, Ryu JH. Influenza viruses: innate immunity and mRNA vaccines. Front Immunol (2021) 12:710647. doi: 10.3389/fimmu.2021.710647
135. Morrison TE, Mauser A, Wong A, Ting JP, Kenney SC. Inhibition of IFN-gamma signaling by an Epstein-Barr virus immediate-early protein. Immunity (2001) 15:787–99. doi: 10.1016/S1074-7613(01)00226-6
136. Leonard GT, Sen GC. Effects of adenovirus E1A protein on interferon-signaling. Virology (1996) 224:25–33. doi: 10.1006/viro.1996.0503
137. Gotoh B, Komatsu T, Takeuchi K, Yokoo J. Paramyxovirus strategies for evading the interferon response. Rev Med Virol (2002) 12:337–57. doi: 10.1002/rmv.357
138. Audsley MD, Moseley GW. Paramyxovirus evasion of innate immunity: diverse strategies for common targets. World J Virol (2013) 2:57–70. doi: 10.5501/wjv.v2.i2.57
139. Kuriakose T, Kanneganti TD. ZBP1: innate sensor regulating cell death and inflammation. Trends Immunol (2018) 39:123–34. doi: 10.1016/j.it.2017.11.002
140. Koehler H, Cotsmire S, Langland J, Kibler KV, Kalman D, Upton JW, et al. Inhibition of DAI-dependent necroptosis by the z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc Natl Acad Sci U.S.A. (2017) 114:11506–11. doi: 10.1073/pnas.1700999114
141. Basavaraju S, Mishra S, Jindal R, Kesavardhana S. Emerging role of ZBP1 in z-RNA sensing, influenza virus-induced cell death, and pulmonary inflammation. mBio (2022) 13:e0040122. doi: 10.1128/mbio.00401-22
142. Lu Y, Wambach M, Katze MG, Krug RM. Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the elF-2 translation initiation factor. Virology (1995) 214:222–8. doi: 10.1006/viro.1995.9937
143. Hale BG, Randall RE, Ortín J, Jackson D. The multifunctional NS1 protein of influenza a viruses. J Gen Virol (2008) 89:2359–76. doi: 10.1099/vir.0.2008/004606-0
144. Varga ZT, Grant A, Manicassamy B, Palese P. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J Virol (2012) 86:8359–66. doi: 10.1128/JVI.01122-12
145. Varga ZT, Ramos I, Hai R, Schmolke M, García-Sastre A, Fernandez-Sesma A, et al. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PloS Pathog (2011) 7:e1002067. doi: 10.1371/journal.ppat.1002067
146. Cheung PH, Ye ZW, Lee TT, Chen H, Chan CP, Jin DY. PB1-F2 protein of highly pathogenic influenza a (H7N9) virus selectively suppresses RNA-induced NLRP3 inflammasome activation through inhibition of MAVS-NLRP3 interaction. J Leukoc Biol (2020) 108:1655–63. doi: 10.1002/JLB.4AB0420-694R
147. Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza a viruses contributes to increased virulence. PloS Pathog (2007) 3:1414–21. doi: 10.1371/journal.ppat.0030141
148. Mossman KL, Saffran HA, Smiley JR. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J Virol (2000) 74:2052–6. doi: 10.1128/JVI.74.4.2052-2056.2000
149. Lin R, Noyce RS, Collins SE, Everett RD, Mossman KL. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J Virol (2004) 78:1675–84. doi: 10.1128/JVI.78.4.1675-1684.2004
150. Melchjorsen J, Sirén J, Julkunen I, Paludan SR, Matikainen S. Induction of cytokine expression by herpes simplex virus in human monocyte-derived macrophages and dendritic cells is dependent on virus replication and is counteracted by ICP27 targeting NF-kappaB and IRF-3. J Gen Virol (2006) 87:1099–108. doi: 10.1099/vir.0.81541-0
151. Sieg S, Yildirim Z, Smith D, Kayagaki N, Yagita H, Huang Y, et al. Herpes simplex virus type 2 inhibition of fas ligand expression. J Virol (1996) 70:8747–51. doi: 10.1128/jvi.70.12.8747-8751.1996
152. Lokugamage KG, Narayanan K, Huang C, Makino S. Severe acute respiratory syndrome coronavirus protein nsp1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J Virol (2012) 86:13598–608. doi: 10.1128/JVI.01958-12
153. Devaraj SG, Wang N, Chen Z, Chen Z, Tseng M, Barretto N, et al. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J Biol Chem (2007) 282:32208–21. doi: 10.1074/jbc.M704870200
154. Ivanov KA, Ziebuhr J. Human coronavirus 229E nonstructural protein 13: characterization of duplex-unwinding, nucleoside triphosphatase, and RNA 5'-triphosphatase activities. J Virol (2004) 78:7833–8. doi: 10.1128/JVI.78.14.7833-7838.2004
155. Shu T, Huang M, Wu D, Ren Y, Zhang X, Han Y, et al. SARS-Coronavirus-2 Nsp13 possesses NTPase and RNA helicase activities that can be inhibited by bismuth salts. Virol Sin (2020) 35:321–9. doi: 10.1007/s12250-020-00242-1
156. Hackbart M, Deng X, Baker SC. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc Natl Acad Sci U.S.A. (2020) 117:8094–103. doi: 10.1073/pnas.1921485117
157. Jiang HW, Zhang HN, Meng QF, Xie J, Li Y, Chen H, et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol Immunol (2020) 17:998–1000. doi: 10.1038/s41423-020-0514-8
158. Shi CS, Qi HY, Boularan C, Huang NN, Abu-Asab M, Shelhamer JH, et al. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol (2014) 193:3080–9. doi: 10.4049/jimmunol.1303196
159. Kopecky-Bromberg SA, Martinez-Sobrido L, Palese P. 7a protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J Virol (2006) 80:785–93. doi: 10.1128/JVI.80.2.785-793.2006
160. Tan YX, Tan TH, Lee MJ, Tham PY, Gunalan V, Druce J, et al. Induction of apoptosis by the severe acute respiratory syndrome coronavirus 7a protein is dependent on its interaction with the bcl-XL protein. J Virol (2007) 81:6346–55. doi: 10.1128/JVI.00090-07
161. Yuan X, Wu J, Shan Y, Yao Z, Dong B, Chen B, et al. SARS coronavirus 7a protein blocks cell cycle progression at G0/G1 phase via the cyclin D3/pRb pathway. Virology (2006) 346:74–85. doi: 10.1016/j.virol.2005.10.015
162. Rabouw HH, Langereis MA, Knaap RC, Dalebout TJ, Canton J, Sola I, et al. Middle East respiratory coronavirus accessory protein 4a inhibits PKR-mediated antiviral stress responses. PloS Pathog (2016) 12:e1005982. doi: 10.1371/journal.ppat.1005982
163. Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep (2020) 32:108185. doi: 10.1016/j.celrep.2020.108185
164. Freundt EC, Yu L, Park E, Lenardo MJ, Xu XN. Molecular determinants for subcellular localization of the severe acute respiratory syndrome coronavirus open reading frame 3b protein. J Virol (2009) 83:6631–40. doi: 10.1128/JVI.00367-09
165. Siu KL, Yuen KS, Castaño-Rodriguez C, Ye ZW, Yeung ML, Fung SY, et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J (2019) 33:8865–77. doi: 10.1096/fj.201802418R
166. Ren Y, Shu T, Wu D, Mu J, Wang C, Huang M, et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol Immunol (2020) 17:881–3. doi: 10.1038/s41423-020-0485-9
167. Freundt EC, Yu L, Goldsmith CS, Welsh S, Cheng A, Yount B, et al. The open reading frame 3a protein of severe acute respiratory syndrome-associated coronavirus promotes membrane rearrangement and cell death. J Virol (2010) 84:1097–109. doi: 10.1128/JVI.01662-09
168. Fensterl V, Sen GC. Interferons and viral infections. Biofactors (2009) 35:14–20. doi: 10.1002/biof.6
169. Li Y, Guo X, Hu C, Du Y, Guo C, Di W, et al. Type I IFN operates pyroptosis and necroptosis during multidrug-resistant a. baumannii infection. Cell Death Differ (2018) 25:1304–18. doi: 10.1038/s41418-017-0041-z
170. Yang D, Liang Y, Zhao S, Ding Y, Zhuang Q, Shi Q, et al. ZBP1 mediates interferon-induced necroptosis. Cell Mol Immunol (2020) 17:356–68. doi: 10.1038/s41423-019-0237-x
171. Ingram JP, Thapa RJ, Fisher A, Tummers B, Zhang T, Yin C, et al. ZBP1/DAI drives RIPK3-mediated cell death induced by IFNs in the absence of RIPK1. J Immunol (2019) 203:1348–55. doi: 10.4049/jimmunol.1900216
172. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-Infected mice. Cell Host Microbe (2016) 19:181–93. doi: 10.1016/j.chom.2016.01.007
173. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
174. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in wuhan, China: a retrospective cohort study. Lancet (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
175. Shen XR, Geng R, Li Q, Chen Y, Li SF, Wang Q, et al. ACE2-independent infection of T lymphocytes by SARS-CoV-2. Signal Transduct Target Ther (2022) 7:83. doi: 10.1038/s41392-022-00919-x
176. Liu PP, Blet A, Smyth D, Li H. The science underlying COVID-19: implications for the cardiovascular system. Circulation (2020) 142:68–78. doi: 10.1161/CIRCULATIONAHA.120.047549
177. Chen R, Lan Z, Ye J, Pang L, Liu Y, Wu W, et al. Cytokine storm: the primary determinant for the pathophysiological evolution of COVID-19 deterioration. Front Immunol (2021) 12:589095. doi: 10.3389/fimmu.2021.589095
178. Woznicki JA, Saini N, Flood P, Rajaram S, Lee CM, Stamou P, et al. TNF-α synergises with IFN-γ to induce caspase-8-JAK1/2-STAT1-dependent death of intestinal epithelial cells. Cell Death Dis (2021) 12:864. doi: 10.1038/s41419-021-04151-3
179. Simpson DS, Pang J, Weir A, Kong IY, Fritsch M, Rashidi M, et al. Interferon-γ primes macrophages for pathogen ligand-induced killing via a caspase-8 and mitochondrial cell death pathway. Immunity (2022) 55:423–441.e429. doi: 10.1016/j.immuni.2022.01.003
180. Hubbard NW, Ames JM, Maurano M, Chu LH, Somfleth KY, Gokhale NS, et al. ADAR1 mutation causes ZBP1-dependent immunopathology. Nature (2022) 607:769–75. doi: 10.1038/s41586-022-04896-7
181. Jiao H, Wachsmuth L, Wolf S, Lohmann J, Nagata M, Kaya GG, et al. ADAR1 averts fatal type I interferon induction by ZBP1. Nature (2022) 607:776–83. doi: 10.1038/s41586-022-04878-9
182. Zhang T, Yin C, Fedorov A, Qiao L, Bao H, Beknazarov N, et al. ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature (2022) 606:594–602. doi: 10.1038/s41586-022-04753-7
Keywords: interferon, ZBP1, inflammasome, virus, pyroptosis, apoptosis, necroptosis, PANoptosis
Citation: Oh S and Lee S (2023) Recent advances in ZBP1-derived PANoptosis against viral infections. Front. Immunol. 14:1148727. doi: 10.3389/fimmu.2023.1148727
Received: 20 January 2023; Accepted: 03 May 2023;
Published: 16 May 2023.
Edited by:
Fajian Hou, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Sannula Kesavardhana, Indian Institute of Science (IISc), IndiaUjjaldeep Jaggi, Cedars Sinai Medical Center, United States
Copyright © 2023 Oh and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: SangJoon Lee, sangjoon.lee@unist.ac.kr