 Alexandros Popotas
Alexandros Popotas Georges Jacques Casimir
Georges Jacques Casimir Francis Corazza
Francis Corazza Nicolas Lefèvre
Nicolas Lefèvre- 1Laboratory of Pediatrics, Université Libre de Bruxelles, Brussels, Belgium
- 2Laboratory of Translational Research, Université Libre de Bruxelles, Brussels, Belgium
- 3Department of Pulmonology, Allergology and Cystic Fibrosis, Queen Fabiola Childrens University Hospital (Hôpital Universitaire des Enfants Reine Fabiola) – University Hospital of Brussels (Hôpital Universitaire de Bruxelles), Brussels, Belgium
- 4Laboratory of Immunology, Centre Hospitalier Universitaire (CHU) Brugmann, Université Libre de Bruxelles, Brussels, Belgium
An increasing number of studies have highlighted the existence of a sex-specific immune response, wherein men experience a worse prognosis in cases of acute inflammatory diseases. Initially, this sex-dependent inflammatory response was attributed to the influence of sex hormones. However, a growing body of evidence has shifted the focus toward the influence of chromosomes rather than sex hormones in shaping these inflammatory sex disparities. Notably, certain pattern recognition receptors, such as Toll-like receptors (TLRs), and their associated immune pathways have been implicated in driving the sex-specific immune response. These receptors are encoded by genes located on the X chromosome. TLRs are pivotal components of the innate immune system, playing crucial roles in responding to infectious diseases, including bacterial and viral pathogens, as well as trauma-related conditions. Importantly, the TLR-mediated inflammatory responses, as indicated by the production of specific proteins and cytokines, exhibit discernible sex-dependent patterns. In this review, we delve into the subject of sex bias in TLR activation and explore its clinical implications relatively to both the X chromosome and the hormonal environment. The overarching objective is to enhance our understanding of the fundamental mechanisms underlying these sex differences.
1 Introduction
Sex-dependent inflammatory responses are well established in the realm of adaptive immunity (1, 2). However, in the critical hours immediately following cellular insult, the rapid and robust inflammatory reaction primarily relies on innate immunity, encompassing various aspects of activation and regulation. Numerous examples illustrate the importance of innate immunity, as the breakdown of one of its pathways can lead to severe clinical consequences (3). Epidemiological evidence now corroborates disparities between men and women in the early immune response, with sex emerging as a distinct prognostic factor in acute inflammation (1). Sex-specific responses are observed in inflammatory conditions such as acute infections, trauma, or surgery. The recent coronavirus disease 2019 (COVID-19) pandemic has further highlighted these sex differences (4–7). Nevertheless, the precise mechanistic underpinnings of this sexual dimorphism remain elusive. While hormonal influence was initially attributed as the main driver, emerging evidence suggests the involvement of an alternative mechanism, potentially linked to genes residing on the sex chromosomes that play a role in the inflammatory response (8). Among the various mediators, Toll-like receptors (TLRs), which are specific receptors of the innate immune system, play a pivotal role in activating inflammatory markers associated with sex-related inflammatory responses (9). In this review, we shed light on the sex bias in TLR expression and activation, striving to elucidate the underlying mechanisms, including the influence of the hormonal environment and contributions from the X chromosome.
2 Sex differences in acute inflammatory responses
2.1 Infectious diseases
2.1.1 Bacterial infections
Substantial disparities in the incidence, prognosis, and immune response between men and women become evident in the context of acute infectious diseases. Clinical studies have consistently demonstrated a higher incidence of bacteremic infections among men, including conditions such as invasive pneumococcal disease (10), Pseudomonas aeruginosa (11), or Staphylococcus aureus (12) infections. When these infections progress toward sepsis, the male sex emerges as a significant risk factor, as higher mortality and sequelae are reported in men (12–15). In a large cohort including more than 10 million cases of sepsis, the male sex was associated with a higher incidence (16). Similarly, men admitted to intensive care units (ICUs) exhibit a heightened incidence of sepsis and shock (17). Despite these findings suggesting that the male sex is linked to an elevated risk of bacteremic infections, it is important to note that the incidence of Escherichia coli bacteremia is higher in women (18, 19). This observation, however, can be attributed to anatomical differences of the urinary tract, where E. coli urinary tract infections (UTI) predominate in women. Nevertheless, men face a greater risk of morbidity (20, 21) and mortality (22) from the complications of UTIs and require more extended treatment to eliminate the bacterial infection (23).
Sex differences are also evident in bacterial lung infections. The male sex is a risk factor in the context of community-acquired pneumonia (CAP) (24–27). Men not only experience a more unfavorable disease progression, but also exhibit a higher rate of admissions to ICUs (26, 27). The most common pathogen isolated in CAP is Streptococcus pneumoniae (28), with men being disproportionately affected by severe pneumococcal CAP (29). Tuberculosis, another prominent bacterial infection with a global impact on patient mortality, also displays sex-based disparities. Mycobacterium tuberculosis primarily afflicts men, who concurrently manifest elevated mortality rates compared with women (30, 31).
These clinical observations align with corresponding biological findings observed in sepsis and other infectious diseases. An investigation involving prepubertal children admitted to ICUs for sepsis revealed higher maximum white blood cell and neutrophil counts, lower pH, and higher C-reactive protein (CRP) levels in women (32). In the early stages of the disease, general biological markers of inflammation are more elevated in women, indicating a heightened activation of innate defense mechanisms. However, over time, a distinct kinetic pattern emerges, highlighting differences in the return to homeostasis between women and men. In contrast to these general inflammatory markers, levels of proinflammatory cytokines such as interleukin 1β (IL-1β), IL-6, and tumor necrosis factor alpha (TNF-α) are higher in men, whereas women exhibit elevated levels of anti-inflammatory mediators, notably IL-10 (13, 33). Men with CAP, who face a worse prognostic compared with women with CAP, present stronger release of TNF-α (34).
Findings from studies involving human subjects are congruent with observations in animal models. In an experimental model of sepsis induced by cecal ligation, female mice exhibit higher survival rates and maintain splenocyte functions (35), whereas male mice release higher levels of IL-6, TNF-α, and prostaglandin E2 (PGE2) (36). Among mice, there is a heightened incidence of systemic pneumococcal infection and pneumococcal respiratory infection in males, who also present higher mortality rates than their female counterparts (37). Further, male mice infected with S. pneumoniae produce significantly greater quantities of proinflammatory cytokines, including IL-6, IL-12p70, interferon gamma (IFN-γ), granulocyte colony-stimulating factor (G-CSF), and IL-17A (37). Mycobacterium bovis, which causes bovine tuberculosis, and Mycoplasma pulmonis, responsible for respiratory mycoplasmosis in mice, both display a more severe clinical outcome and higher mortality in males (31). Interestingly, M. pulmonis induces alveolar neutrophil infiltrate with edema and hemorrhage in males, resembling an acute alveolar inflammatory response. Conversely, female lungs with respiratory mycoplasmosis display macrophage infiltration with minimal neutrophils, akin to a chronic peribronchial inflammatory response (31).
Sex disparities in bacterial infections extend beyond mammals, with documented differences in defense against pathogens observed in insects and birds (38, 39).
2.1.2 Viral infections
Sexual dimorphism in clinical outcomes and immune responses is notably evident in viral infections. Throughout the COVID-19 pandemic, epidemiological reports have consistently described a better clinical course in women (4–7), with men presenting higher levels of proinflammatory cytokines (40). Similarly, during the outbreaks of severe acute respiratory syndrome (SARS-CoV) and Middle East respiratory syndrome (MERS-CoV), men showed a higher incidence and more severe disease (41, 42). Sex disparities are also apparent in Paramyxoviridae infections; boys infected with respiratory syncytial virus (RSV) or human metapneumovirus (hMPV) develop more severe clinical manifestations compared with girls (43–45).
Dengue virus (DENV) a substantial global health concern, also displays sex differences, with men experiencing a disadvantage (46).
In contrast to the aforementioned viruses, among adults infected with influenza A, women are more susceptible to an increased risk of severe clinical presentations (47), accompanied by higher IFN-γ levels compared with men (44, 48, 49).
Sex-specific disparities in viral responses have been corroborated in animal models. Male golden Syrian hamster exhibit a poorer outcome and develop more severe SARS-CoV-2 pneumonia than their female counterparts (50) and male mice infected with RSV display impaired IFN-β expression and viral control (51). Female mice infected with equal viral loads of influenza virus to males, experience a worse outcome, with greater cytokine and chemokine release in their lungs (44, 52).
2.2 Trauma and surgery
2.2.1 Trauma
Sex-specific patterns in complication rates are evident among trauma patients. As reported by Mörs et al. (53), men exhibit higher injury scores and an increased risk of developing sepsis following trauma. In polytraumatized patients, men have a higher incidence of multiple organ dysfunction syndrome and sepsis (54). Offner et al. (55) further emphasized that the male sex constitutes a significant risk factor for major post-trauma infections. Moreover, in cohorts involving trauma patients with either blunt or penetrating injuries, post-trauma complications such as pneumonia (56, 57), sepsis (58), and multiple organ failure (59) are more prevalent in men. Factors such as the length of ICU stay and ventilator use are influenced by the patient’s age and the severity of the trauma, but they predominantly impact men (60).
While disparities in trauma complications between men and women are well established, the impact of sex on survival after injury remains less clear. Mortality rates stemming from penetrating or blunt trauma, as well as post-trauma infection, exhibit a pronounced male predilection (60–63). While studies delineating mortality disparities across age cohorts manifest certain inconsistencies, discernible trends persist, albeit with variation across investigations (60, 62, 63).
Biological sex differences become apparent in trauma patients, with men producing significantly higher IL-6 levels during the initial 2 days after injury (53). Similar findings have been reported in polytraumatized patients, where men exhibit higher levels of IL-6 and IL-8 compared with women (54). Additionally, men with multiple injuries and those who develop post-trauma sepsis produce increased levels of IL-6, IL-8, and TNF-α (64).
Animal studies further emphasize the existence of inflammatory sexual dimorphism in the context of trauma, with varying survival rates in different species and trauma contexts, emphasizing the complexity of sex-related responses to trauma (65, 66). In experimental mouse trauma models, male macrophages produce higher levels of IL-6 (67, 68).
Burn injuries are another type of trauma with discrepant results regarding complications and survival in humans. Several authors have demonstrated increased length of hospital or ICU stays and higher mortality rates in women (69–72). Cumming et al. (73) identified an association between the male sex and the development of multiple organ dysfunction and severe sepsis following burn trauma. The prognosis may be influenced by the duration of the process, with worse outcomes in men during the acute phase, while chronic inflammation consequences may exacerbate the prognosis in women.
The conflicting outcomes observed in burn trauma studies may stem from variations in the timing of analyses and the involvement of cofactors such as skin loss, fluid depletion, hypovolemic shock, and secondary infections. Additionally, the inconsistencies may arise from the heterogeneous nature of the aggression, incorporating elements like fractures, hematomas, bacterial translocations. Consequently, the comparison between study populations becomes notably challenging.
2.2.2 Surgery
Surgery itself acts as an inflammatory trigger, yet there is a scarcity of research in this domain. Similarly to the studies on trauma, the presence of confounding factors such as underlying pathology or infectious complications adds complexity to research endeavors in surgery and inflammation.
Among patients who have undergone surgery, men present a higher mortality risk and are more susceptible to complications. In a study involving 512 surgical patients, men demonstrated a higher risk of developing major respiratory complication, with no sex-specific differences regarding the duration of the operation, the length of hospital stay, or mortality rates (74). Similarly, in a study on surgical intensive care patients, the authors observed a higher incidence of severe sepsis or septic shock among men (17), and a large retrospective analysis revealed a higher perioperative morbidity in male surgical patients (75).
Sex-specific cytokine profiles have been identified in surgical patients, revealing an excessive release of TNF-α and diminished levels of IFN-γ in stimulated male peripheral blood mononuclear cells (PBMCs) (76).
There is very limited information on surgery-induced inflammation and mortality in animals. In equine surgical operations for colic, some researchers have reported higher mortality rates for males (77), while others studies have not confirmed these results (78).
3 TLRs
3.1 TLR structure
Whether in humans or animals, the early inflammatory response to infection or injury is predominantly orchestrated by the innate immune system. This crucial defense mechanism identifies pathogens or damaged cells through pattern recognition receptors (PRRs). Among the well-studied PRRs, TLRs stand out. They rapidly recognize infectious agents or damaged particles, subsequently initiating signals for the elimination of microbial pathogens or the activation of adaptive immune responses.
The human innate immune system comprises 10 TLRs, each characterized by a distinct structural composition. These receptors consist of two essential domains. The N-terminal end features an extracellular leucine-rich repeat (LRR) domain that functions as a ligand binder. Conversely, the C-terminal end contains the intracellular domain known as the IL-1 receptor (Toll/IL-1 receptor [TIR]), responsible for initiating the signaling cascade (79, 80).
TLRs are expressed in diverse cellular locations; some are found on the cell membrane, while others are located in endosomes within the cell’s cytoplasm. Specifically, TLR3, TLR7, TLR8, and TLR9 are endosomal, whereas the remaining TLRs mainly function as membrane-bound receptors (79, 80). These TLRs are expressed in a wide array of cell types, including but not limited to endothelial cells; epithelial cells; parenchymal cells; synovial fibroblasts; and hematopoietic-derived cells such as dendritic cells (DCs), macrophages, neutrophils, B cells, and T cells (79, 80).
3.2 TLR signaling
The extracellular N-terminus of TLRs is responsible for ligand binding, which varies depending on the specific type of TLR. After ligand recognition, signaling cascades are enhanced through the intracellular C-terminal domain, leading to the activation of transcription factors and the subsequent induction of cytokine expression (9, 80, 81).
The intracellular domain of TLRs, known as the TIR domain, is essential for mediating signal transduction. This domain interacts with various adaptor proteins, including myeloid differentiation factor 88 (MyD88), MyD88-adapter-like (Mal), TIR domain-containing adaptor inducing IFN-β (TRIF), and TRIF-related adaptor molecule (TRAM) (9, 80–83). MyD88 is a common adaptor utilized by all TLRs, with TLR3 being a potential exception, whereas the other adaptor proteins are much more restricted in their TLR interactions. Mal is involved in signaling via TLR2 and TLR4, TRIF is a component of the signaling machinery for TLR3 and TLR4, and TRAM is specifically associated with TLR4 (Figure 1) (9, 80–83).
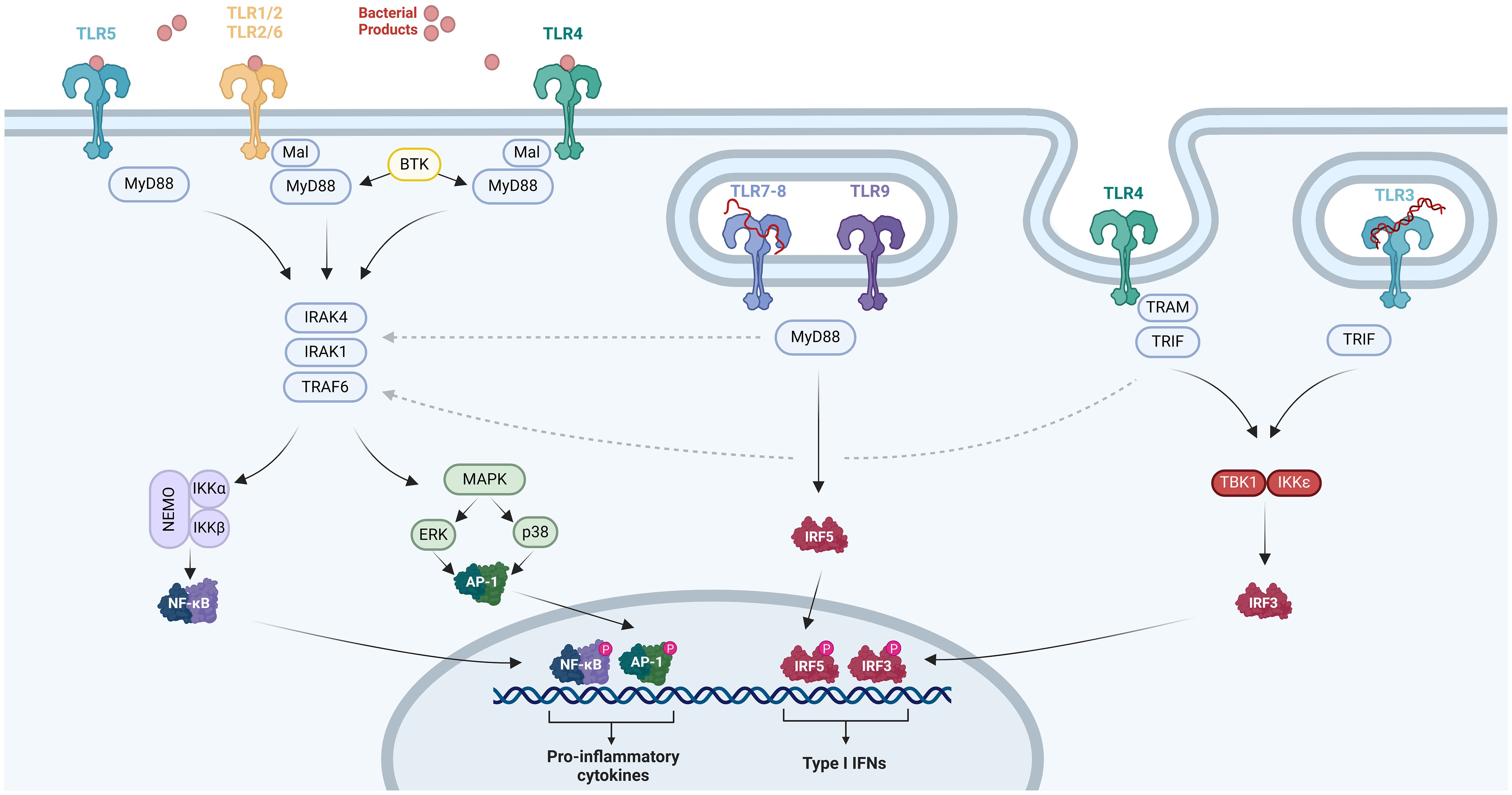
Figure 1 TLR pathways. The TIR domain within TLR2, TLR4, or TLR5 interacts with MyD88, initiating the activation of IRAK1, IRAK4, and TRAF6. This interaction triggers the phosphorylation of the NF-κB complex and instigates NF-κB-regulated factors. Additionally, it activates p38 MAPK and ERK1/2, which subsequently induce AP-1 activity. The activation of NF-κB and AP-1 prompts the production of proinflammatory cytokines. TRIF functions as an adaptor molecule for TLR3 and TLR4, facilitating NF-κB activation through its interaction with TRAF6, while also initiating TBK1 and IKKϵ activation. TBK1 and IKKϵ phosphorylate IRF3, leading to the expression of IFN-a/b. TLR7, TLR8, and TLR9 are implicated in IFN expression through TRIF-independent mechanisms, potentially involving IRF-5 or the MyD88–IRAK1–TRAF6 pathway, although the exact mechanism remains to be elucidated. Created with BioRender.com.
Upon activation of these adaptor proteins, members of the IL-1 receptor-associated kinase (IRAK) family, including IRAK1 and IRAK4, as well as TNF receptor-associated factor 6 (TRAF6) and inhibitors of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) kinases (IKKs), facilitate signal transmission. This activation results in the phosphorylation of the NF-κB complex, leading to the activation of NF-κB-regulated factors and, concurrently, phosphorylation of mitogen-activated protein kinases (MAPKs), including p38 MAPK and extracellular signal-regulated kinase 1/2 (ERK1/2), which induce activator protein 1 (AP-1) activation (9, 80). These transcription factors are primarily responsible for the release of proinflammatory molecules (84). Additionally, Bruton’s tyrosine kinase (BTK) can contribute to this pathway through its interactions with MyD88 and IRAK1 (Figure 1) (9, 80–83). It is noteworthy that BTK, IRAK1, and IKBKG—which encodes IKKγ, also known as NF-κB essential modulator (NEMO)—are encoded on the X chromosome at Xq21 and Xq28, respectively (85).
TRIF can directly activate NF-κB through interaction with TRAF6 and is also capable of initiating signal transduction to TRAF family member associated NF-κB activator binding kinase (TBK) 1 and IKKϵ. These kinases phosphorylate and activate IFN regulatory factor 3 (IRF3), which is essential for the induction of IFN-α/β. Therefore, TRIF plays a central role as the adaptor molecule enabling TLR3 and TLR4 to induce the expression of IFN-α/β (Figure 1) (9, 80–82). In contrast, TLR7, TLR8, and TLR9 appear to trigger IFN expression through TRIF-independent mechanisms, although the precise mechanism remains unclear. Some data suggest the involvement of IRF-5 or the MyD88–IRAK1–TRAF6 pathway (Figure 1) (9, 80–82).
The TLR4 and TLR2 pathways are two well-known pathways of the innate immune system. TLR2 usually builds heterodimer structures with TLR1 or TLR6, while homodimerization is observed for TLR4. TLR4 plays a critical role in the inflammatory response to gram-negative bacteria through recognition of lipopolysaccharide (LPS). To achieve this, TLR4 associates with the CD14 receptor and the MD-2 co-receptor, allowing for the recognition of LPS and the initiation of downstream signaling (82).
On the other hand, TLR1–TLR2 detects pathogens containing molecules of triacylated lipoproteins and gram-negative bacteria (9, 79–81, 86). Meanwhile, TLR2–TLR6 is responsible for recognizing microbes with diacylated lipoproteins, zymosan in fungi, bacterial lipoteichoic acid as seen in Staphylococcus spp., peptidoglycans in gram-positive bacteria, and released microbial heat shock proteins (HSPs) (9, 79–81, 86).
TLR3 recognizes double-stranded RNA (dsRNA) viruses and triggers antiviral immune responses by promoting the secretion of proinflammatory cytokines and type I IFN. On the other hand, TLR7 and TLR8 are specialized in detecting viral single-stranded RNA (ssRNA). TLR9 recognizes unmethylated CpG DNA motifs belonging to bacteria, fungi, viruses, and protozoa (79, 86).
In addition to infectious ligands, cellular damage from pathogens or injuries, leads to the release of molecules known as damage-associated molecular patterns (DAMPs). These DAMPs can contribute to inflammation and potentially exacerbate tissue damage or impair wound healing (87, 88). Normally confined within the intracellular compartment, DAMPs are inadvertently released into the extracellular environment following cellular damage (87, 88). Examples of DAMPs include RNA, DNA, high-mobility group box-1 protein (HMGB-1) associated with chromatin, adenosine triphosphate (ATP), degradation products like uric acid, and stress-induced molecules such as HSPs (87, 88).
TLRs have the ability to recognize DAMPs and to elicit an appropriate and fast response. Several DAMPs have been shown to induce cytokine release through the recognition of TLR2 and TLR4. Furthermore, β-defensin-2 and β-defensin-3, as danger molecules, activate TLR2 and TLR4 (89, 90). TLR1 also plays a role in recognizing β-defensin-3 (87, 88). Nucleic acids are recognized by TLR3, TLR7, and TLR8, which are activated by messenger RNA (mRNA), ssRNA, and IgG-chromatin complexes, respectively (87, 88).
3.3 The implication of TLRs in acute inflammatory diseases
3.3.1 TLRs and bacterial infections
TLR2 and TLR4 play a significant role in the recognition of these pathogens and the subsequent release of inflammatory cytokines during sepsis (84, 91, 92). The presence of a specific IRAK-1 variant haplotype, a crucial protein in the TLR2 and TLR4 pathway, is linked to increased nuclear translocation of NF-κB, contributing to a worse prognostic clinical presentation in sepsis (93). Moreover, TLR2 and TLR4 polymorphisms are associated with a higher risk of staphylococcal or gram-negative infections, further underscoring the involvement of TLRs in sepsis (94, 95). TLR2 and TLR4 mRNA upregulation is also described in monocytes from patients with sepsis (96, 97).
Multiple TLRs recognize S. pneumoniae, the most frequent bacteria in CAP: TLR2 recognizes lipoteichoic acid and lipoproteins from the pneumococcal cell wall (91), TLR4 detects pneumolysin (91, 98) and TLR9 is activated through recognition of pneumococcal bacterial DNA which contains unmethylated CpG motifs (89). All three of these receptors regulate the NF-κB pathway to enhance proinflammatory cytokine release (91).
Animal experiments further support the involvement of TLRs in sepsis and pneumococcal lung infection: mice with experimental sepsis exhibit a cytokine pattern dependent on NF-κB and MAPKs (37) and TLR2 and TLR4 knockout (KO) mice display less effective cytokine release compared with wild mice (90).
In murine models of sepsis, TLR2 and TLR4 are upregulated in hepatic and splenic macrophages (90). Similarly, mice with peritonitis show higher expression of TLR2 and TLR4 mRNA in the lungs and liver (99). The administration of endotoxin leads to the upregulation of TLR2 mRNA through TLR4- and MyD88-dependent signaling in alveolar macrophages and endothelial cells (100, 101). This crosstalk between TLR2 and TLR4 is achieved through activated polymorphonuclear neutrophils (PMNs), making their presence a determinant element in the response within TLR pathway (100, 101).
In mice with endothelial cells lacking TLR4, pulmonary neutrophil recruitment is significantly reduced after exposure to LPS, illustrating the importance of alveolar endothelial TLR4 expression in pulmonary neutrophil recruitment during sepsis (102).
In animal experiments simulating pneumococcal lung infection, TLR4 KO mice demonstrate a less vigorous immune response (103, 104). TLR2-deficient mice show decreased release of proinflammatory cytokines (105, 106), and TLR9-deficient mice exhibit weaker lung bacterial clearance compared with wild-type mice (89).
3.3.2 TLRs and viral infections
The role of TLRs in viral infections has gained significant attention in the context of the COVID-19 pandemic. Initially, information about TLR recognition of SARS-CoV-2 was extrapolated from data on SARS-CoV-1. Among the TLRs described in SARS-CoV-1 infection, TLR3 and TLR7 are the most prominent, leading to type I IFN expression through the TRIF–IRF3 pathway (83, 107, 108). There is also evidence for MyD88-dependent pathway activation by SARS-CoV-1, such as NF-κB (109, 110). Furthermore, AP-1 activation has been previously described solely in the context of SARS-CoV infections, with the spike and nucleocapsid proteins of the virus identified as the triggering factors (111, 112).
In the context of severe COVID-19 cases in adults involving a massive release of proinflammatory cytokines, numerous studies have emphasized the role of TLRs. Molecular docking investigations have revealed a strong interaction between TLR4 and the spike glycoprotein of SARS-CoV-2. Additionally, the upregulation of TLR4 and its associated pathway components (CD14, MyD88, Mal, TRAF6, IRAK1, and TRIF) have been observed in PBMCs from patients with COVID-19 (113–115). TLR2 can recognize SARS-CoV-2 by interacting with the SARS-CoV-2 E protein; this interaction amplifies intracellular immune pathways, leading to the activation of an immune response against the pathogen (116).
We previously described the relation between endothelial TLR4 and neutrophil recruitment in experimental models simulating bacterial infection (102). An intriguing observation is that COVID-19 in adults is also marked by a swift and substantial influx of neutrophils (117). This is further reflected in the elevated peripheral blood neutrophil count in patients with COVID-19, with the neutrophil-to-lymphocyte ratio (NLR) serving as an indicator of disease severity (40, 117). Nevertheless, the impact of TLRs and sex on neutrophil levels in COVID-19 remains an understudied area (40). Only one study suggests a male predominance in the NLR among patients with COVID-19, which is associated with a more unfavorable prognosis (118).
Animal models have produced similar findings regarding the implication of TLRs in Coronaviridae detection. For SARS-CoV-1, direct evidence points to the implication of TLR4, as TLR4 KO mice are more susceptible to SARS-CoV-1 than wild-type mice, exhibiting higher viral titers (115).
Another virus notorious for eliciting a robust inflammatory response, as well as notable and swift neutrophil recruitment, is RSV (117, 119). RSV activates the NF-κB pathway through TLR2 and TLR4, similarly to bacterial pathogens, to trigger cytokine release and to recruit immune cells into the airways (43). Notably, TLR4 polymorphisms are associated with severe RSV clinical presentations in children (120–123). RSV upregulates TLR4 in airway epithelial cells and monocytes among children with acute bronchiolitis (124, 125). In vitro studies on human peripheral macrophages suggest that TLR4 recognizes the RSV F protein (126), and bronchoalveolar lavage analyses have demonstrated elevated neutrophil levels in children with severe bronchiolitis (127–129). Girls with viral bronchiolitis tend to have higher levels of circulating neutrophils compared with boys (130).
Studies in mice underscore the contributions of TLR2 and TLR4 to RSV recognition, with TLR2 KO mice showing a higher viral load and impaired cytokine mediation (131) and TLR4 KO mice displaying an impaired immune response and reduced virus clearance (132). TLR4 also plays a role in recognizing human metapneumovirus and enhancing the innate immune response against this pathogen (133).
Human cytomegalovirus (HCMV) is detected by both membranous and endosomal TLRs. Glycoproteins on the HCMV surface interact with TLR2 and TLR4, resulting in the activation of the NF-κB pathway and subsequent production of proinflammatory cytokines (134). Furthermore, HCMV nucleic acids are recognized by endosomal TLR3, leading to the release of IFN-α (134). Studies investigating mutations or polymorphisms in TLR3 and TLR9 have indicated an increased risk of CMV infection, underscoring the significant role played by these receptors in the recognition of HCMV (134).
The recognition of the DENV involves activation of both the MyD88 and IRF-3 or IRF-5 pathways, dependent on the specific viral components engaged in the interaction with the host’s immune response (135). Specifically, DENV2 triggers TLR3, TLR7, and TLR8, resulting in a robust induction of IL-8 and IFN-α (136, 137). Additionally, the DENV NS1 protein induces signaling pathways associated with TLR2, TLR6, and TLR4, leading to subsequent proinflammatory cytokine production (138, 139). Individuals with TLR4 polymorphisms exhibit an increased susceptibility to DENV infection (140) and patients with dengue hemorrhagic fever demonstrate elevated TLR2 expression in comparison to those with dengue fever (141). In animal models of experimental dengue infection, mice lacking TLR6 exhibit improved survival rates following exposure to DENV2 and NS1, in contrast to wild-type mice (139). In macaques, dengue virus recognition is described through TLR3, TLR7, and TLR8 (142).
As mentioned above, influenza A exhibits a different pattern, with women being more susceptible to an increased risk of severe clinical presentations (47) and higher IFN-γ levels compared with men (44, 48, 49). The influenza virus primarily activates TLR7 and the IRF3 pathway (143) rather than the TLR4 or MyD88-dependent pathways. In rodent models of influenza infection, TRL3- and TLR7-deficient mice are unable to control viral replication and succumb to the disease (144). TLR4 contribution was not demonstrated in TLR4 KO mice with influenza infection, thereby affirming the predominant involvement of TLR3 and TLR7 activation in response to influenza virus (132).
3.3.3 TLRs and surgery or trauma
There are no studies focusing on the impact of surgical procedures on the activation of TLR pathways. In many surgical pathologies, TLRs are already stimulated by pathogens or DAMPs (145). As in humans, the literature available on the interaction between surgery and TLR responses in animals is limited. However, a notable finding comes from experiments involving mice subjected to sternotomy and treated with a TLR9 antagonist. In this context, a decrease in IL-6 production is observed, highlighting the potential significance of the TLR9 pathway in modulating the inflammatory response elicited by surgical procedures (146).
The precise involvement of TLRs in trauma remains a topic of ongoing investigation, with conflicting findings in the literature. Trauma patients with an ISS (injury severity score) over 12 display heightened expression of TLR2 and TLR4 on monocytes (147). However, in patients with an ISS exceeding 25, TLR4 expression on monocytes decreases, and TLR2 expression is similar to healthy subjects (148). Some studies report unaltered TLR4 but reduced TLR2 expression in patients with an ISS greater than 21 (149). These findings are based on limited populations, emphasizing the need to expand these studies before drawing definitive conclusions. Furthermore, thoughtful consideration should be given to the potential limitation in TLR2 and TLR4 availability resulting from their excessive engagement in the context of trauma.
Trauma patients exhibit higher serum levels of IL-6 and IL-10 (147). Previous studies have established a discernible correlation between IL-6 levels and ISS within the initial 72 hours of hospitalization (150). TLR2, -4 and -9 activation results in trauma patients results in an impaired proinflammatory response, as evidenced by lower TNF and IL-6 levels (148, 149) and increased release of IL-10 (148).
Findings from a murine model indicate that TLR4 plays a crucial role in neutrophil accumulation and well as increased TNF-α production (denoted by increased mRNA and protein levels) following experimental hemorrhage (151). Hemorrhage in mice leads to increased upregulation of p38, ERK1/2, and c-Jun N-terminal kinase (JNK) in Kupffer cells, as described by Thobe et al. (152). Furthermore, the release of proinflammatory cytokines mediated by TLR2 and TLR4 is modulated through the p38, ERK1/2, and JNK pathways (152). The inflammatory response after lung injury in mice appears to be TLR9-mediated, as TLR9-deficient mice exhibit reduced proinflammatory cytokine release (153). TLR9-dependent TNF-α and IL-6 production has been shown to rely on JNK (152).
TLRs have also been implicated in burn injuries: The levels of TLR2, TLR4, and TLR9 on DCs are higher in burn patients compared with healthy volunteers. Additionally, TLR2 protein expression is associated with the survival prospects of individuals affected by burn injuries (154). However, in the specific context of burn trauma, hemodynamic and infectious cofactors impede a thorough analysis of the specific TLR implication in thermal injuries. As highlighted in the above studies on TLR analysis in trauma patients, the constrained sample size underscores the need for further investigations in this field.
In murine models, TLR function is impaired in the context of burn injuries. Macrophages from burned mice show reduced expression of TLR3 and TLR9 mRNA compared with macrophages from their healthy counterparts (155). DCs from burned mice have lower TLR9 mRNA expression, resulting in an altered cytokine release profile characterized by an anti-inflammatory response, including elevated IL-10 levels and reduced production of IL-6, TNF-α, and IL-12p70 (156).
4 Sex differences and TLRs
4.1 Sex bias in TLRs and their pathways
As discussed above, the literature emphasizes sex-based disparities in the inflammatory response during bacterial, viral infections, and even trauma. What is particularly remarkable is the involvement of TLR pathways in these mechanisms (Figure 2), but, surprisingly, very few studies have focused on the relationship between sex and TLR expression within the immune system.
Figure 2 TLR sex differences in humans and rodents. Created with BioRender.com.
4.1.1 TLR2 and TLR4
There have been diverse results regarding TLR2 and TLR4 expression. An in vitro study on neutrophils from healthy young adults stimulated with either LPS or IFN-γ revealed higher TLR4 expression in men (157). Conversely, higher TLR2 expression is reported in monocytes from healthy women (76). However, other researchers have not identified any sex difference in TLR4 or TLR2 expression on human immune cells after exposure to LPS (76, 158).
In some studies exploring the potential influence of sex on TLR responses, researchers have analyzed cytokine release subsequent to TLR activation. In vitro stimulation of TLR4 and TLR2 by LPS or zymosan, respectively, results in augmented proinflammatory cytokine release in whole blood or isolated leukocyte populations from men (157, 159–162). Furthermore, after LPS stimulation, men exhibit a higher percentage of IL-12-, IL-1β-, and TNF-α-producing monocytes compared with women (163).
As discussed previously, men tend to produce predominantly proinflammatory cytokines, whereas women primarily release cytokines from the IL-10 family. This distinctive sex-related pattern is further corroborated through TLR2 and TLR4 activation. When subject to whole blood endotoxin stimulation, women demonstrate elevated levels of IL-10, relative to the monocyte concentration, enhancing their ability to regulate the immune response effectively (164). Indeed, IL-10 plays an essential role in maintaining the integrity and homeostasis of the cellular environment during inflammation (165).
Regarding the downstream proteins within the TLR2–TLR4 pathway, male neutrophils present greater activation of ERK, p38, JNK, and Akt upon LPS stimulation. However, there are no discernible sex differences in either IκBα phosphorylation or degradation subsequent to endotoxin exposure (157).
The above studies reveal divergent patterns in the sex-related expression of TLR2 and TLR4 in human. However, activation through ligands primarily elicits an enhanced response in men, with increased cytokine release and higher TLR2- and TLR4-associated proteins. These differences appear to stem from the core pathway, specifically involving the proteins associated with TLRs and the mechanisms governing cytokine release, rather than the receptors themselves.
We previously described the relation between endothelial TLR4 and neutrophil recruitment in bacterial infection (102) or viral infections characterized by a significant recruitment of neutrophils, such as COVID-19 (117) and RSV bronchiolitis (43, 117, 119). The predominant function of TLR2 and TLR4 in men offers a promising avenue of research to identify the etiology of worse clinical outcomes and heightened inflammatory responses observed in men.
Research on TLR2 and TLR4 mRNA and protein expression in animal models has yielded inconsistent results. At basal state, male mice express higher levels of TLR2, while higher TLR4 levels are registered on macrophages from female mice (166). Conversely, certain studies suggest that while there are no discernible sex differences in TLR4 mRNA expression under baseline conditions, male murine macrophages exhibit higher levels of TLR4 protein on their cell surfaces (167).
These contradictory results also extend to stimulated conditions in animals. Stimulated female rodent peritoneal leukocytes show increased TLR2 mRNA and TLR4 mRNA levels (168). However, a communication by Marriot et al. (167) suggests that stimulated murine macrophages do not exhibit a sex difference in TLR4 mRNA expression. In an infectious model involving rodents infected with coxsackievirus, female mice demonstrate improved survival rates, which correspond to higher TLR2 expression on lymphocytes (169). Females also display lower TLR4 expression on lymphocytes, monocytes, and DCs (169). In the same model, male mice display an increase in TLR2 expression on monocytes and DCs (169). Furthermore, after trauma and hemorrhagic shock, and also following LPS stimulation, mouse macrophages do not display sex-dependent TLR4 expression on their surface (170).
Interestingly, when analyzing cytokine response to LPS stimulation, male subjects display elevated IL-6 levels (167, 168). Moreover, male-derived macrophages exposed to LPS exhibit higher levels of IL-1β (167). Once again, similarly to human studies, it appears that sex differences are more evident within the context of the TLR2 and TLR4 pathways than in the receptors themselves.
4.1.2 TLR3
Limited information is available regarding the TLR3 sex bias in infectious diseases. In human studies, researchers have assessed activation of the TLR3 pathway by measuring cytokine release after stimulating PBMCs with polyinosinic:polycytidylic acid (poly I:C). They have registered significant sex differences only for TNF-α, specifically higher levels in male PBMCs (171, 172). Stimulation with herpes simplex virus 1 (HSV1), a double-stranded DNA virus, induces higher IL-10 production in men, although it is essential to note that this virus also engages TLR9 (172).
Findings from animal experiments indicate greater TLR3 mRNA expression in peritoneal leukocytes from female mice, although the authors did not investigate protein expression under infectious conditions (168). In virus challenge models involving Seoul virus, female rats exhibit lower TLR3 expression compared with male rats (173).
4.1.3 TLR7 and TLR8
Higher expression of TLR7 has been registered in women (174). As described above, TLR7 activates the type I IFN signaling pathway. Consequently, increased levels of type I IFN are observed in immune cells from adult and adolescent women following exposure to TLR7 ligands (175, 176). In patients with COVID-19, women demonstrate elevated production of IFN-α2 (2).
Stimulation of TLR7 and TLR8 leads to amplified production of both type I and type II IFNs and decreased levels of IL-1β, IL-6, IL-8, IL-10, and TNF-α in female PBMCs (160, 172, 176, 177). In whole blood samples, resiquimod induces significant sex differences only in IL-1β, IL-6, and TNF-α, with higher production in men (160). Interestingly, HCMV suppresses the response to TLR7/8 stimulation specifically in women (178). In rats, greater TLR7 expression occurs in females following viral exposure (173).
Certain viruses, such as influenza mainly activates IRF3 pathway through TLR7 triggering (143). This leads to a distinct pattern where women exhibit greater susceptibility to an increased risk of severe clinical presentations (47) and elevated IFN-γ compared with men (44, 48, 49). The more pronounced TLR7 activation in women likely serves as an explanation for the differing sex-specific patterns in inflammatory responses and clinical outcomes in the case of these pathogens.
4.1.4 TLR9
In most studies, stimulation of human DCs with CpG, a TLR9 ligand, results in no sex bias in IFNα production (175, 177). However, a recent investigation using CpG-A indicates increased type I and II IFN expression in PBMCs obtained from blood samples from women (176).
When considering animal models, mouse cytomegalovirus (MCMV) infection leads to an upregulation of TLR9 expression in male immune cells compared with female cells. In MCMV-infected wild-type mice, males exhibit higher neutrophil and DC infiltration. However, in TLR9 KO mice, there are no sex differences following infection (179).
5 TLRs and sex hormones
Sex bias in TLR expression and TLR pathway–associated proteins have been progressively established. Whether the differential TLR expression in each sex is influenced by the hormonal environment or by sex chromosome–linked genes involved in the inflammatory response requires further investigation. As essential PRRs for a robust innate immune response and due to their significant involvement in infectious diseases, it is imperative to explore TLRs and their sex-specific variations. Sex hormones have been demonstrated to modulate the innate immune response. Namely, estrogens are known to be immunostimulators, whereas progesterone and androgens are considered immunosuppressors. The influence of these hormones on the expression of TLRs and TLR-mediated cytokine release may represent one of the primary factors contributing to sex-based disparities in immunity.
5.1 Estrogens and progesterone
5.1.1 TLR2 and TLR4
Under conditions of bacterial stimulation, exposure to exogenous or naturally occurring estradiol tends to have an anti-inflammatory effect, resulting in decreased production of IL-1β, IL-6, TNF-α, and GM-CSF, while increasing the release of IL-10, IL-12, IL-23, and IL-27 (180). In vitro, human PBMCs stimulated with LPS and estradiol, show suppressed TNF-α production (159).
On the other hand, a review from Bouman et al. (181), presents contentious evidence regarding the impact of estrogens and progesterone on IL-1β, IL-6, TNF-α, and IL-12 expression. Additionally, a study involving menopausal women who underwent hormone replacement therapy revealed no significant effects on IL-6, IL-1ra, IL-1α, and TNF-α production (182), while Another investigation focusing on human ectocervical epithelial cells demonstrated that following LPS stimulation, estradiol treatment increases the secretion of IL-1β, IL-6, IL-8, and IFN-γ compared with untreated cells (183). At first sight, the impact of estrogen and progesterone on the expression of inflammatory cytokines appears to be inconsistent among different studies (Figures 3, 4).

Figure 3 The influence of estrogens on TLR expression and activation in human and animal models. Created with BioRender.com.
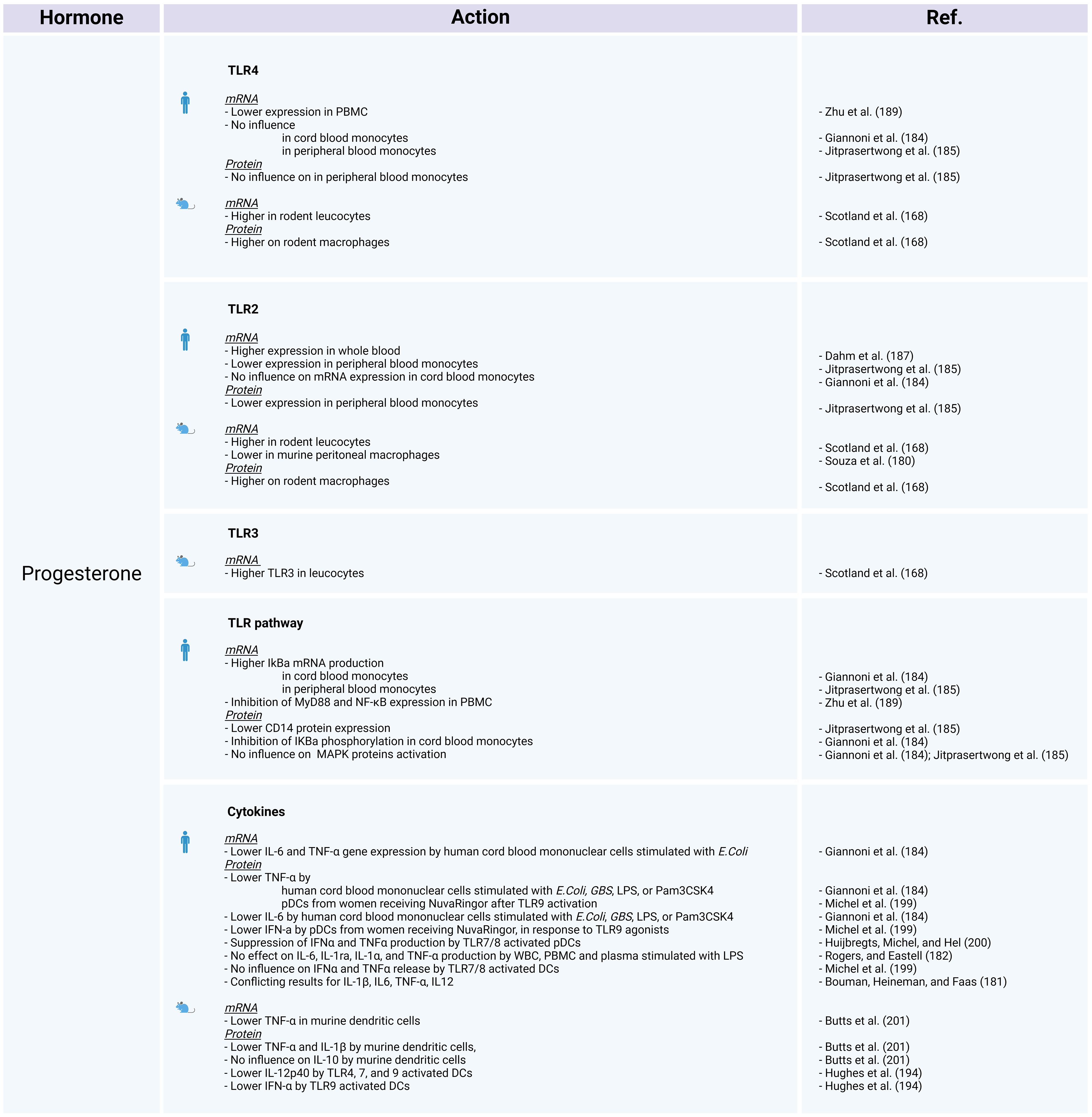
Figure 4 The influence of progesterone on TLR expression and activation in human and animal models. Created with BioRender.com.
A study investigating the impact of sex on stimulated human cord blood mononuclear cells, reveals that estradiol and progesterone decrease TNF and IL-6 protein and gene expression (184). The authors assessed TLR2 and TLR4 mRNA expression, as well as the protein levels of components within the NF-κB and MAPK pathways. Estradiol and progesterone do not influence TLR2 or TLR4 mRNA expression or the MAPK pathway, but do mediate cytokine release by inhibiting the phosphorylation of IκBα and increasing NFKBIA mRNA levels (184). The study concluded that the absence of phosphorylation prevents the ubiquitination and degradation of the IκBα protein, while the elevated mRNA levels suggest a potential increase in IκBα transcription. Under these circumstances, IκBα is unable to release the NF-κB p65 subunit, leading to inhibition of the NF-κB pathway (184). This finding could account for the observed difference in NF-κB dependent cytokine release regulation between women and men, with women showing more regulated release and men displaying higher levels of pro-inflammatory cytokines.
Jitprasertwong et al. (185) confirmed that exposure to estradiol and progesterone blocks the NF-κB pathway but does not affect TLR4 gene and protein expression or activation of MAPKs (phosphorylated forms of p38, ERK1/2, and JNK). The same authors also reported TLR2 mRNA and protein downregulation, as well as reduced CD14 protein expression on stimulated human monocytes exposed to estradiol and progesterone (185).
Reduced TLR2 mRNA expression was also observed after stimulation with S. aureus in male monocytes treated with 17β-estradiol and monocytes from women during their fertile period (180). However, conflicting findings arise again for these receptors; while some studies indicate no influence on TLR2 mRNA (186), others demonstrate its upregulation in whole blood exposed to 17β-estradiol or progesterone (norethisterone), as well as in THP-1 cells treated with 17β-estradiol (187, 188). When female PBMCs are treated with progesterone, it results in the inhibition of both responsiveness and TLR4 expression (189).
Relative to other TLR pathway–related proteins, comparison of the gene expression profiles of women during their fertile period and men has revealed several noteworthy distinctions. In stimulated female monocytes TNF, MAPK8, and other genes are downregulated. Likewise, a comparison between male immune cells exposed to 17β-estradiol and their untreated but stimulated counterparts demonstrated that estradiol negatively regulates the NFKB1, IRAK2, IL1B, IL1A, IL12A, IKBKB, and BTK genes, but does not affect the IRAK1 gene (180). Stimulated female PBMCs treated with progesterone show markedly reduced MYD88 and NFKB1 mRNA levels (189).
To delve deeper into the influence of sex steroids on immune responses, several studies have examined TLR2 and TLR4 pathways modulation in women, across the menstrual cycle or during pregnancy (Figure 5). According to some researchers, female hormones impede relative TLR2, CD14, BTK, and TNF gene expression in human peripheral blood monocyte, while higher IL-12 levers are observed in the presence of these hormones (180). On the other hand, higher cytokine responsiveness to TLR2 activation is observed during follicular phase when estrogen is predominant, and then decreases during the luteal phase marked by progesterone presence (190). Some publications suggest that TLR4 expression remains consistent in women throughout the menstrual cycle (191), while others describe increased TLR4 responsiveness during the early luteal phase, which then decreases during the late luteal phase (190). According to Ziegler et al. (192), at the beginning of pregnancy, stimulation of TLR4 in PBMCs induces the release of TNF-α, which subsequently decreases as the pregnancy progresses.

Figure 5 TLR expression and activation across the menstrual cycle. Created with BioRender.com.
Overall, the abovementioned studies have yield highly contrasting outcomes concerning the expression and activation of TLR2 and TLR4 following exposure to estrogens and progesterone (Figures 3-5). The unclear influence of these steroids challenges their role as the primary etiology of sex differences in TLR2- and TLR4-mediate inflammatory responses in humans.
In animal models, estradiol enhances the expression of TLR4 on rodent macrophages (193). In addition, ovariectomy reduces the expression of TLR2 and TLR4 in leukocytes and protein expression of TLR2 and TLR4 on macrophages (168). On the other hand, Souza et al. (180) reported that murine peritoneal macrophages from ovariectomized females express higher TLR2 mRNA levels than wild-type females. Moreover, treatment of ovariectomized females or males with estradiol decreases TLR2 expression. In mouse plasmacytoid dendritic cells (pDCs), progesterone exposure decreases the IL-12p40 induction through TLR4 (194). These findings highlight the complex interplay between sex hormones and TLR expression in animals.
5.1.2 TLR7 and TLR8
Estradiol increases TLR8 expression, both at the RNA and protein levels, in PBMCs from healthy premenopausal women. Moreover, it enhances TLR7 gene expression in PBMCs from healthy premenopausal females and pDCs from postmenopausal women (186, 195). However, in monocytes, TLR7 and TLR8 gene expression does not vary between fertile women and men or between cells treated compared to those not treated with estradiol (Figures 3, 4) (180).
The production of IFN-a, which relies on TLR7 activation, is reduced in PBMCs from women in the first trimester of pregnancy; however, there is an increase in stimulated pDCs during pregnancy (1, 192). Notably, the menstrual cycle does not affect TLR7 activation (190). In contrast, a decrease in responsiveness is described after TLR8 activation during the luteal phase (190). Remarkably, higher TLR7-induced IFN levels in pDCs from women compared with men (196) occur even before puberty (Figures 3–5) (197), prompting further discussion about the actual role of sex steroids in immune dimorphism and cytokine release.
Studies investigating the influence of hormonal contraception on TLR7/8 signaling have yielded mixed results. Some have described no significant difference in DC activation in women receiving depot medroxyprogesterone acetate (DMPA) or NuvaRing compared with those not using contraception (198, 199). Other studies suggest an inhibitory effect of medroxyprogesterone acetate on the production of IFN-α and TNF-α by TLR7–TLR8 activated pDCs (200). Interestingly, the presence of estrogen during the fertile period downregulates the IRF1, IFNG, IFNB1, and IFNA1 genes in monocytes, all of which are involved in TLR7/8 signaling (180).
In mice exposed to estradiol, there is an increase in TLR8 mRNA in various tissues, including the spleen, thymus, bone marrow, and lymph nodes (186). Mouse DCs exposed to estradiol display increased TNF-α release following TLR7 and TLR9 activation (195). Interestingly, ablation of the ESR1 gene (which encodes estrogen receptor α) in DCs abolishes the TLR7-mediated IFN response (195). In murine DCs, pre-treatment with progesterone impairs the ability to release TNF-α and IL-1β, as TNF gene expression is also disturbed in a dose-dependent manner (201). On the contrary, progesterone exposure does not affect the production of IL-10 (201). However, progesterone exerts a negative influence on IL-12p40 induction through TLR 7 stimulation in mouse pDCs (Figures 3, 4) (194).
5.1.3 TLR9
Researchers have reported diverse effects of estrogens on TLR9. Higher TLR9 gene expression is registered in PBMCs exposed to estradiol (186). In DCs, the presence of estradiol induces higher amounts of cytokine release upon TLR9 activation (195). Conversely, the use of hormonal contraception (such as DMPA or NuvaRing) or in vitro progesterone treatment reduce the capacity of pDCs to produce IFN-α, TNF-α, or IL-12p40 in response to TLR9 agonists (194, 199, 200). In rodents, DMPA hampers the production of IFN-α by pDCs following TLR9 activation or viral infection (Figures 3, 4) (194).
5.1.4 TLR3
There is limited information available on the influence of sex hormones on TLR3 expression and activation in humans. PBMCs treated with estradiol show enhanced TLR3 mRNA expression (186), while there are no differences in monocytes (180). In endometrial epithelial cells, estradiol does not affect TLR3 gene or protein expression, but suppresses the release of IL-6 and IL-8 upon TLR3 stimulation with poly I:C. Lesmeister et al. (202) suggest that these results may be attributed to estradiol modulating the function rather than the quantity of this receptor. Nonetheless, no variations in TLR3-induced cytokines or TLR3 mRNA expression have been identified throughout the menstrual cycle (190, 203). In mouse models, ovariectomy decreases TLR3 expression in leukocytes and TLR3 protein expression on macrophages (Figures 3–5) (168).
5.1.5 TLR5
There is also scare literature regarding whether there is a TLR5 sex bias. In monocytes treated with 17-β estradiol, there are no differences in TLR5 mRNA expression (180). Concerning TLR5-induced cytokines during the menstrual cycle, an increase in IL-1β and TNF-α release has been described from the follicular to the early luteal phase, while a decrease in IL-6 has been noted during the luteal phase (Figures 3–5) (190).
5.2 Androgens
In contrast to female sex hormones, there is very little information on the influence of androgens on the modulation of TLRs and their associated signaling pathways in human cells. Exposing PBMCs to testosterone does not affect the expression of TLR2, TLR3, TLR4, TLR7, TLR8, and TLR9 (186).
Given the clinical evidence discussed previously, which indicates that men have a more proinflammatory cytokine profile than women, it is intriguing to find that research on the impact of androgens on cytokines suggests a suppressive effect. In fact, there is a correlation between elevated concentrations of androgens and increased production IL-10 via TLR9 stimulation in male PBMCs (172). Observations in PBMCs and whole blood from patients with androgen deficiencies have revealed increased production of proinflammatory cytokines, specifically IL-1β and TNF-α, both at baseline and following exposure to LPS (204). Remarkably, exogenous testosterone administration negatively affects proinflammatory cytokine release (191). This inverse relationship between exogenous testosterone levels and TNF-α and IL-1β production has been confirmed in studies investigating diseases characterized by low androgen levels and hormone replacement therapy (205, 206).
In cultured human umbilical vein endothelial cells, exposure to testosterone inhibits the NF-κB DNA-binding activity (207). Correspondingly in umbilical cord serum, testosterone levels are negatively associated with TNF-α, while IL-10 is increased (208). These findings suggest that testosterone may mitigate the production of proinflammatory cytokines by downregulating NF-κB activity, although it does not appear to exert a direct influence on the expression of TLR genes or proteins in humans.
In animal models, testosterone upregulates IL-10 expression and reduces TNF production in LPS-treated murine macrophages (209). Similarly, in castrated rats, exogenous testosterone decreases the expression of TNF-α in a dose-dependent manner (210) within the prostate. In contrast to findings in human diseases, the absence of endogenous testosterone following orchidectomy, has been associated with an increased TLR4 expression on murine macrophages as well as the susceptibility to endotoxin shock. In contrast, control mice or those subjected to orchidectomy but subsequently treated with exogenous testosterone replacement exhibit reduced TLR4 expression and a corresponding decrease in endotoxin-induced severity scores (211).
Based on the insights gained from both animal and human studies, it appears that testosterone tends to have anti-inflammatory properties, in contrast to the proinflammatory patterns often observed in clinical settings in men. This intriguing paradox might be elucidated by examining potential variations in receptor kinetics or considering the role of hormones, which may not be as critical in underpinning sex-specific differences in immune responses as previously thought.
Indeed, according to the above evidence, estrogens, progesterone, and testosterone exert a discernible influence on the innate immune response, and, among other functions, the expression or function of TLRs. However, the emergence of sex-related inflammatory responses in prepubertal children, along with discernible TLR sex disparities prior to the onset of puberty, suggest an additional mechanistic layer at play, potentially involving genetic factors such as chromosomal genes (32, 197, 212, 213). Notably, genetic polymorphisms located on the X chromosome contribute to the immune sex bias, manifesting as differences in susceptibility to bacterial and viral infections between men and women (214).Therefore, determining the influence of sex chromosomes on TLR-mediated responses might enhance our understanding of the intricacies underlying the immune sex bias.
6 TLRs and the X chromosome
Women carry a pair of X chromosomes, while in men, the X chromosome is paired with the Y chromosome. The X chromosome harbors more than 1,000 genes, contrary to the Y chromosome, on which less than 100 genes have been identified. Several of these X-linked genes encode proteins integral to the functioning of the innate immune response (215–218). Female cells undergo random inactivation of the X chromosome, also referred to as silencing. This process leads to the aleatory expression of only one of the parental X chromosomes in each cell. The chromosome that will be inactivated is coated with the long noncoding RNA Xist, located on the X-inactivation center, which triggers a biological process resulting in chromosome condensation by heterochromatin modifications (2, 219–221).
Recent studies have described a more complicated procedure of X methylation in women. At least 23% of X-linked genes escape inactivation, inducing their overexpression. Thus, X-chromosome inactivation (XCI), designed to balance gene expression in women, may be incomplete, and some genes are expressed from both X chromosomes (2, 217, 222, 223). In addition to XCI, another mechanism leading to imbalanced gene expression in women is skewed XCI. In this case, X-chromosome silencing does not occur randomly; rather, one of the parental X chromosomes is preferentially expressed in nearly all cells (2, 217, 222, 223). Of note, the presence of only one X chromosome in men, in addition to the occurrence of skewed XCI in women, explain why the majority of pathologies related to X-linked genes predominantly affect boys and men. This imbalance in the expression of X-linked genes between men and women may contribute to immune sex differences and, particularly with respect to TLR disparities, may offer a more diversified immune response in women (2, 217, 222, 223).
In one study, researchers assessed individuals with aneuploidies, comparing XY men and XX women, to evaluate the respective contribution of the X chromosome and sex hormones (Figure 6) (158). The authors measured the activation of some TLRs and found significant disparities in the innate immune responses between men and women. Indeed, whole blood stimulation, employing TLR2, TLR4, and TLR7/8 ligands, induces augmented release of proinflammatory cytokines in men (158). Individuals with Klinefelter syndrome, who exhibit a male phenotype despite the presence of an extra X chromosome, display a cytokine release profile similar to that of XX women (158). However, their estradiol levels do not significantly differ from those observed in women (158). These findings underscore the influential role of sex chromosomes over sex steroids. Moreover, the production of inflammatory cytokines in response to TLR ligands does not differ significantly according to the level of 17-β estradiol, reinforcing the less pronounced role of estrogens in these immune responses (160). Further investigation, conducted on purified monocytes, demonstrated lower levels of inflammatory cytokine production in women compared with men upon TLR activation (160).
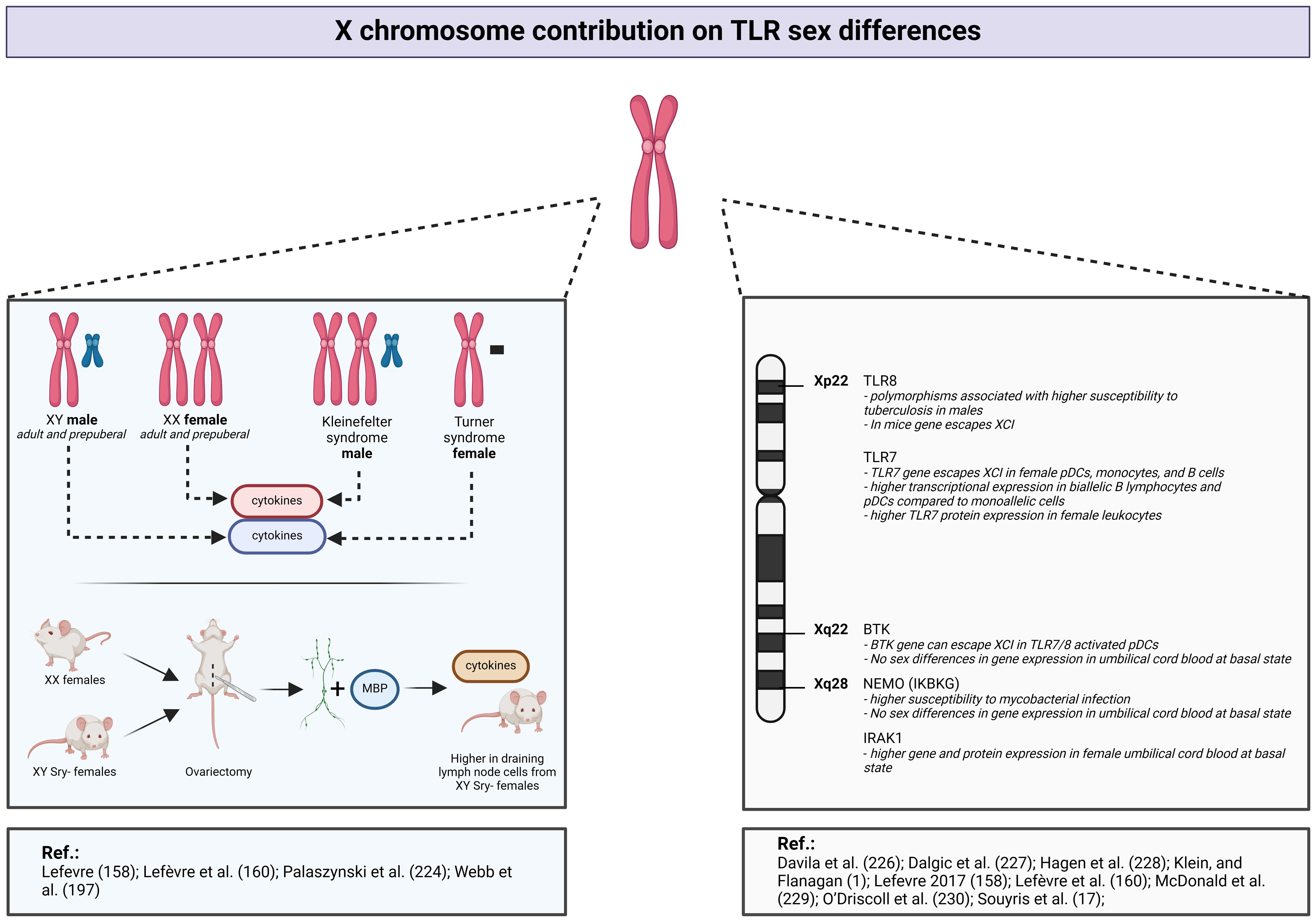
Figure 6 X chromosome contribution to TLR sex differences. Created with BioRender.com.
An animal model employing XX female mice and XY-Sry deficient mice has provided additional information regarding sex-related differences in immune responses (Figure 6) (224). The SRY gene is a critical determinant in male fetal development. The absence of this gene leads to suppression of male differentiation, and thus the above mice are XY females. After ovariectomy in all groups of mice, the researchers collected draining lymph nodes cells and stimulated them with myelin basic protein (224). Despite the absence of gonadal hormonal influences and similar phenotypes, sex differences in proinflammatory cytokine release persist, depending on the number of X chromosomes (224).
Crucial components of the TLR-mediated innate immune response are under the genetic control of loci located on the X chromosome. Specifically, X-linked genes play a pivotal role in the regulation of TLR7, TLR8, as well as key constituents of the NF-κB pathway, including IRAK-1, NEMO, and BTK (Figure 6) (174, 216–218, 225).
Several studies have reported a relation between TLR8 polymorphisms and tuberculosis susceptibility, with a more pronounced effect observed in men (226, 227). These findings imply that mutations in the TLR8 gene, located on the X chromosome, will have a greater impact in men who carry only one parental chromosome.
As discussed previously, TLR7 stimulation results in higher IFN production in women (196), even before the onset of puberty (197). Notably, according to a study including adults, prepubertal children, transgender men and women, and individuals with Turner syndrome, the IFN-1 production from TLR7-activated DCs is associated with the number of X chromosomes (197). More precisely, regardless of the sex steroid concentrations, the production of IFN production by pDCs following TLR7 stimulation is higher in subjects with a karyotype including two X chromosomes (prepubertal girls, adult women, and individuals assigned female at birth who identify as transgender) compared with subjects with a single X chromosome (prepubertal boys, adult men, individuals assigned male at birth who identify as transgender, and patients with Turner syndrome) (197). Transcriptional analyses conducted before and after vaccination in human subjects or following virus challenge in animal models show higher amounts of TLR-pathway and proinflammatory genes expression, such as TLR7 or NF-κB in female PBMCs and tissues (1).
Souyris et al. (174) and more recently Hagen et al. (228) have demonstrated that the TLR7 gene, located on the X chromosome, escapes X inactivation in pDCs, monocytes, and B cells. This phenomenon leads in higher TLR7 expression in women. Indeed, female biallelic B cells and pDCs express higher TLR7 mRNA levels than monoallelic cells, and female leukocytes have greater TLR7 protein levels (174, 228). Similar pattern of TLR7 X-inactivation escape was observed in patients with Klinefelter syndrome, who possess an extra X chromosome (174). Thus, the differential expression and function of TLR7 showing a female predomination is mainly attributed to the X chromosome (Figure 6).
In mouse models, TLR8 also seems to escape from XCI; however, this phenomenon has not been proven in a human model (Figure 6) (229).
TLR1/2, TLR2/6, and TLR4 mainly activate the NF-κB pathway, which includes the X-related proteins IRAK-1, NEMO, and BTK. A recent study showed that the BTK gene can escape XCI, offering a potential explanation for certain sex-based variations in the TLR signaling pathway (Figure 6) (228). However, this observation was established in cells stimulated with a TLR7/8 agonist and not a TLR2 or TLR4 ligand. In neonatal PBMCs at the basal state, there are no sex-based differences in BTK or NEMO expression (Figure 6) (230). In contrast, IRAK1 gene and protein expression is higher in female PBMCs (Figure 6) (230). Nevertheless, this model was not evaluated under inflammatory conditions. Both clinical studies and in vitro experiments involving TLR2 and TLR4 ligands, show higher proinflammatory cytokine release in men (32, 160). However, the authors did not observe sex differences in TLR2 and TLR4 expression or the phosphorylated forms of NF-κB or MAPK-mediated pathway proteins, suggesting that cytokine sex bias is potentially induced at the transcriptional level (32, 160). These TLR2 and TLR4 signaling pathway differences have not yet been firmly established in ex vivo models.
The X chromosome also contains several microRNAs (miRNAs), which regulate protein synthesis by targeting specific mRNAs, thereby modulating gene expression through translational repression or enhancement. It has been suggested that X-linked miRNAs also undergo silencing and could contribute to the sex-specific immune response and TLR sex differences. Among miRNAs located on the X chromosome, interactions with TLRs have been described for miRNA-98, miRNA-223, and miRNA-105. miRNA-98, in synergy with let-7i, appears to regulate the TLR4 pathway, influencing SNAP3 and IL-10 expression. However, miRNA-98 itself is modulated by TLR4, as its expression is downregulated after LPS exposure (231–233). In contrast, miRNA-223 exhibits upregulation in response to TLR4 signaling and is implicated by modulation of neutrophil differentiation from myeloid precursors (233, 234). miRNA-105 has a negative regulatory effect on TLR2 mRNA in human gingival keratinocytes: Higher levels are associated with oral keratinocytes from patients exhibiting a diminished TLR2 response and cytokine production (235).
Although research concerning the role of the X chromosome in TLR-related sex differences has been relatively limited compared with the extensive examination of sex steroids, emerging findings strongly emphasize the pivotal involvement of X-linked genes. The intricate influence of XCI mechanisms on gene expression introduces a source of diversity in the women’s immune response. The imbalanced expression of X-linked genes between men and women carries substantial implications for shaping immune responses and is implied to underlie the well-documented phenomenon of immune sex differences.
7 Conclusion and future perspectives
TLR expression is one variable that influences the sex-related innate immune response. In this review, we have summarized the TLR sex bias and have attempted to identify, based on the literature, the exact etiology. Studies concerning sex bias in TLR mRNA and protein expression have mostly concentrated on TLR2, TLR4, TLR7, and TLR8, as they are highly implicated in infectious diseases. Sex steroids influence the innate immune response and, among other functions, the expression and function of TLRs. Nonetheless, the exact effects of sex hormones on TLR4 and TLR7 remain ambiguous. On the other hand in studies with postmenopausal patients receiving a hormone replacement therapy, the cytokine proinflammatory response is not affected, suggesting an additional mechanistic layer at play, potentially involving genetic factors (32, 182, 197, 212, 213). Genetic polymorphisms located on the X chromosome contribute to the immune sex bias, manifesting as differences in susceptibility to bacterial and viral infections between men and women (214). In light of these findings, we strongly recommend further investigation of genes located on the sex chromosomes to understand sex disparities in the immune system and response.
As described above, some X chromosome genes are methylated and thus escape inactivation in women. This phenomenon has been confirmed for TLR7, an X-related protein, in pDCs, monocytes, and B cells, resulting in higher expression levels of this receptor in women. Based on these observations, we should also evaluate the expression of other X-linked proteins involved in the TLR pathways.
There are divergent responses regarding the sex-related expression of TLR2 and TLR4. However, their activation through ligands mainly shows an enhanced response in men, as denoted by cytokine release. The variability in these results demands further investigation into these receptors in humans to determine whether there are sex-dependent differences in TLR2 and TLR4 expression. If there are no sex differences, and considering the uncertain impact of sex steroids, attention must be drawn to proteins of the pathways that induce cytokine release. Notably, some of the NF-κB pathway proteins such as IRAK-1, NEMO, and BTK are encoded by genes located on the X chromosome (216–218). Findings regarding the contribution of X-linked genes to TLR4 sex differences have been extrapolated from studies focusing on NF-κB-mediated cytokine release. Additional research is needed to explore IRAK-1, NEMO, and BTK gene and protein expression, as well as the phosphorylation of downstream proteins in the pathway, such as NF-κB, p38, and ERK. This approach should clarify the mechanisms underlying sex differences from TLR activation to cytokine release.
The compelling engagement of TLR2 and TLR4, resembling their roles in bacterial infections, in cases of viral infections, characterized by a significant influx of neutrophils, warrants further exploration. The involvement of PMNs in TLR pathway responses has already been suggested in studies on TLR2 and TLR4 (100, 101). The dynamic interplay between neutrophils and TLRs should be subjected to close scrutiny, with a particular focus on unravelling the implications of this interaction in the context of sexual dimorphism.
The X chromosome also harbors miRNAs that could also escape XCI inactivation and modulate TLR expression. While some research has explored the relationship between miRNAs and TLRs, the question of causality remains. Indeed, there is more robust evidence that TLRs regulate miRNAs than for the reverse situation (236). Further investigation in this area is warranted.
Most of the information about TLR sex differences has been derived from animal models. However, these data are a poor reflection of human biology (237). Furthermore, different miRNAs have been described in mice and humans, drawing attention to the thoroughness required in this type of research. The human X chromosome encompasses 76 miRNAs and the mouse X chromosome contains 65, and there are only 37 miRNAs common to both species (233). Hence, while animal research undoubtedly contributes to scientific progress, subsequent investigations involving humans are essential to validate any findings.
Moreover, in humans, isolated cells do not adequately represent the complexity of whole blood (238–240). Isolated cell populations lack the necessary context of cell communication and the surrounding environment, which can lead to unintended activation, stability issues, and biased results. Whole blood studies offer a more comprehensive analysis of immune function. Thus, research on TLRs should integrate insights from animal models and be conducted in human whole blood to establish a more physiologically relevant model.
Uncovering the cell signaling pathways underpinning the inflammatory distinctions between women and men holds the promise of identifying specific prognostic markers for individuals with inflammatory diseases or acute infections. It may also reveal new therapeutic targets to modulate the inflammatory response based on the patient’s sex. Accordingly, gaining a deeper understanding of TLR mechanisms involved in acute inflammatory processes, such as sepsis, could prove instrumental in mitigating tissue damage and, thus, be of great benefit for patients.
To summarize, the emergence of sex-related inflammatory responses in prepubertal children, along with discernible TLR sex disparities prior to the onset of puberty, provides ample support for the assertion that genes situated on the X chromosome could be responsible for some of the differences between men and women in TLR expression and activation. Future clinical trials should identify the fundamental mechanisms of the inflammatory sex differences through TLRs. Research on immune sex differences might offer novel prognostic and therapeutic targets that will help advance personalized medicine in inflammatory diseases.
Author contributions
AP: Writing – original draft, Writing – review & editing. GC: Writing – review & editing. FC: Writing – review & editing. NL: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors would like to thank the Belgian Kids’ Fund for their valuable support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. (2016) 16:626–38. doi: 10.1038/nri.2016.90
2. Wilkinson NM, Chen HC, Lechner MG, Su MA. Sex differences in immunity. Annu Rev Immunol. (2022) 40:null. doi: 10.1146/annurev-immunol-101320-125133
3. Akar-Ghibril N. Defects of the innate immune system and related immune deficiencies. Clinic Rev Allerg Immunol. (2022) 63:36–54. doi: 10.1007/s12016-021-08885-y
4. Vahidy FS, Pan AP, Ahnstedt H, Munshi Y, Choi HA, Tiruneh Y, et al. Sex differences in susceptibility, severity, and outcomes of coronavirus disease 2019: Cross-sectional analysis from a diverse US metropolitan area. PloS One. (2021) 16:e0245556. doi: 10.1371/journal.pone.0245556
5. Alwani M, Yassin A, Al-Zoubi RM, Aboumarzouk OM, Nettleship J, Kelly D, et al. Sex-based differences in severity and mortality in COVID-19. Rev Med Virol. (2021) 31:e2223. doi: 10.1002/rmv.2223
6. Abate BB, Kassie AM, Kassaw MW, Aragie TG, Masresha SA. Sex difference in coronavirus disease (COVID-19): a systematic review and meta-analysis. BMJ Open. (2020) 10:e040129. doi: 10.1136/bmjopen-2020-040129
7. Sha J, Qie G, Yao Q, Sun W, Wang C, Zhang Z, et al. Sex differences on clinical characteristics, severity, and mortality in adult patients with COVID-19: A multicentre retrospective study. Front Med (Lausanne). (2021) 8:607059. doi: 10.3389/fmed.2021.607059
8. Casimir GJ, Lefèvre N, Corazza F, Duchateau J, Chamekh M. The acid-base balance and gender in inflammation: A mini-review. Front Immunol. (2018) 9:475. doi: 10.3389/fimmu.2018.00475
9. Takeda K, Akira S. TLR signaling pathways. Semin Immunol. (2004) 16:3–9. doi: 10.1016/j.smim.2003.10.003
10. Wagenvoort GHJ, Sanders EAM, Vlaminckx BJ, Melker HEde, Ende Avd, Knol MJ. Sex differences in invasive pneumococcal disease and the impact of pneumococcal conjugate vaccination in the Netherlands, 2004 to 2015. Eurosurveillance. (2017) 22:30481. doi: 10.2807/1560-7917.ES.2017.22.10.30481
11. Al-Hasan MN, Wilson JW, Lahr BD, Eckel-Passow JE, Baddour LM. Incidence of pseudomonas aeruginosa bacteremia: A population-based study. Am J Med. (2008) 121:702–8. doi: 10.1016/j.amjmed.2008.03.029
12. ohus RM, Gustad LT, Furberg AS, Moen MK, Liyanarachi KV, Askim Å, et al. Explaining sex differences in risk of bloodstream infections using mediation analysis in the population-based HUNT study in Norway. Sci Rep. (2022) 12:8436. doi: 10.1038/s41598-022-12569-8
13. Schröder J, Kahlke V, Staubach KH, Zabel P, Stüber F. Gender differences in human sepsis. Arch Surg. (1998) 133:1200–5. doi: 10.1001/archsurg.133.11.1200
14. Schoeneberg C, Kauther MD, Hussmann B, Keitel J, Schmitz D, Lendemans S. Gender-specific differences in severely injured patients between 2002 and 2011: data analysis with matched-pair analysis. Crit Care. (2013) 17:R277. doi: 10.1186/cc13132
15. McClelland EE, Smith JM. Gender specific differences in the immune response to infection. Arch Immunol Ther Exp. (2011) 59:203–13. doi: 10.1007/s00005-011-0124-3
16. Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. (2003) 348:1546–54. doi: 10.1056/NEJMoa022139
17. Wichmann MW, Inthorn D, Andress HJ, Schildberg FW. Incidence and mortality of severe sepsis in surgical intensive care patients: the influence of patient gender on disease process and outcome. Intensive Care Med. (2000) 26:167–72. doi: 10.1007/s001340050041
18. Bou-Antoun S, Davies J, Guy R, Johnson AP, Sheridan EA, Hope RJ. Descriptive epidemiology of Escherichia coli bacteraemia in England, April 2012 to March 2014. Euro Surveill. (2016) 21:30329. doi: 10.2807/1560-7917.ES.2016.21.35.30329
19. Fischer J, Jung N, Robinson N, Lehmann C. Sex differences in immune responses to infectious diseases. Infection. (2015) 43:399–403. doi: 10.1007/s15010-015-0791-9
20. Olson P. Sex Differences in Uropathogenic Escherichia coli Urinary Tract Infection. Arts & Sciences Electronic Theses and Dissertations. Washington University in St. Louis. (2018). Available at: https://openscholarship.wustl.edu/art_sci_etds/1564.
21. Bergström T. Sex differences in childhood urinary tract infection. Arch Dis Child. (1972) 47:227–32. doi: 10.1136/adc.47.252.227
22. Abernethy JK, Johnson AP, Guy R, Hinton N, Sheridan EA, Hope RJ. Thirty day all-cause mortality in patients with Escherichia coli bacteraemia in England. Clin Microbiol Infection. (2015) 21:251.e1–8. doi: 10.1016/j.cmi.2015.01.001
23. Tandan M, Duane S, Cormican M, Murphy AW, Vellinga A. Reconsultation and antimicrobial treatment of urinary tract infection in male and female patients in general practice. Antibiotics (Basel). (2016) 5:31. doi: 10.3390/antibiotics5030031
24. Falagas ME, Mourtzoukou EG, Vardakas KZ. Sex differences in the incidence and severity of respiratory tract infections. Respir Med. (2007) 101:1845–63. doi: 10.1016/j.rmed.2007.04.011
25. Gutiérrez F, Masiá M, Mirete C, Soldán B, Rodríguez JC, Padilla S, et al. The influence of age and gender on the population-based incidence of community-acquired pneumonia caused by different microbial pathogens. J Infection. (2006) 53:166–74. doi: 10.1016/j.jinf.2005.11.006
26. López-de-Andrés A, Albaladejo-Vicente R, de Miguel-Diez J, Hernández-Barrera V, Ji Z, Zamorano-León JJ, et al. Gender differences in incidence and in-hospital outcomes of community-acquired, ventilator-associated and nonventilator hospital-acquired pneumonia in Spain. Int J Clin Practice. (2021) 75:e13762. doi: 10.1093/eurpub/ckz019
27. Reade MC, Yende S, D’Angelo G, Kong L, Kellum JA, Barnato AE, et al. DIFFERENCES IN IMMUNE RESPONSE MAY EXPLAIN LOWER SURVIVAL AMONG OLDER MEN WITH PNEUMONIA. Crit Care Med. (2009) 37:1655–62. doi: 10.1097/CCM.0b013e31819da853
28. Brown JS. Community-acquired pneumonia. Clin Med (Lond). (2012) 12:538–43. doi: 10.7861/clinmedicine.12-6-538
29. Georges H, Leroy O, Vandenbussche C, Guery B, Alfandari S, Tronchon L, et al. Epidemiological features and prognosis of severe community-acquired pneumococcal pneumonia. Intensive Care Med. (1999) 25:198–206. doi: 10.1007/s001340050816
30. Nhamoyebonde S, Leslie A. Biological differences between the sexes and susceptibility to tuberculosis. J Infect Dis. (2014) 209:S100–6. doi: 10.1093/infdis/jiu147
31. Vázquez-Martínez ER, García-Gómez E, Camacho-Arroyo I, González-Pedrajo B. Sexual dimorphism in bacterial infections. Biol Sex Differences. (2018) 9:27. doi: 10.1186/s13293-018-0187-5
32. Lefèvre N, Noyon B, Biarent D, Corazza F, Duchateau J, Casimir G. Sex differences in inflammatory response and acid-base balance in prepubertal children with severe sepsis. Shock. (2017) 47:422–8. doi: 10.1097/SHK.0000000000000773
33. Angele MK, Pratschke S, Hubbard WJ, Chaudry IH. Gender differences in sepsis. Virulence. (2014) 5:12–9. doi: 10.4161/viru.26982
34. Bacci MR, Leme RCP, Zing NPC, Murad N, Adami F, Hinnig PF, et al. IL-6 and TNF-α serum levels are associated with early death in community-acquired pneumonia patients. Braz J Med Biol Res. (2015) 48:427–32. doi: 10.1590/1414-431x20144402
35. Zellweger R, Wichmann MW, Ayala A, Stein S, DeMaso CM, Chaudry IH. Females in proestrus state maintain splenic immune functions and tolerate sepsis better than males. Crit Care Med. (1997) 25:106–10. doi: 10.1097/00003246-199701000-00021
36. Diodato MD, Knöferl MW, Schwacha MG, Bland KI, Chaudry IH. GENDER DIFFERENCES IN THE INFLAMMATORY RESPONSE AND SURVIVAL FOLLOWING HAEMORRHAGE AND SUBSEQUENT SEPSIS. Cytokine. (2001) 14:162–9. doi: 10.1006/cyto.2001.0861
37. Kadioglu A, Cuppone AM, Trappetti C, List T, Spreafico A, Pozzi G, et al. Sex-based differences in susceptibility to respiratory and systemic pneumococcal disease in mice. J Infect Dis. (2020) 204(12):1971–9. doi: 10.1093/infdis/jir657
38. Belousova I, Pavlushin S, Subbotina A, Rudneva N, Martemyanov V. Sex specificity in innate immunity of insect larvae. J Insect Science. (2021) 21:15. doi: 10.1093/jisesa/ieab097
39. Leitner G, Dan Heller E, Friedman A. Sex-related differences in immune response and survival rate of broiler chickens. Veterinary Immunol Immunopathology. (1989) 21:249–60. doi: 10.1016/0165-2427(89)90035-4
40. Haitao T, Vermunt JV, Abeykoon J, Ghamrawi R, Gunaratne M, Jayachandran M, et al. COVID-19 and sex differences: mechanisms and biomarkers. Mayo Clinic Proc. (2020) 95:2189–203. doi: 10.1016/j.mayocp.2020.07.024
41. Karlberg J, Chong DSY, Lai WYY. Do men have a higher case fatality rate of severe acute respiratory syndrome than women do? Am J Epidemiol. 159(3):229–31. doi: 10.1093/aje/kwh056
42. Alghamdi IG, Hussain II, Almalki SS, Alghamdi MS, Alghamdi MM, El-Sheemy MA. The pattern of Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive epidemiological analysis of data from the Saudi Ministry of Health. IJGM. (2014) 7:417–23. doi: 10.2147/IJGM
43. Wainwright C. Acute viral bronchiolitis in children- a very common condition with few therapeutic options. Paediatr Respir Rev. (2010) 11:39–45. doi: 10.1016/j.prrv.2009.10.001
44. Ursin RL, Klein SL. Sex differences in respiratory viral pathogenesis and treatments. Annu Rev Virology. (2021) 8:393–414. doi: 10.1146/annurev-virology-091919-092720
45. Mansbach JM, Emond JA, Camargo CA. Bronchiolitis in US emergency departments 1992 to 2000: epidemiology and practice variation. Pediatr Emerg Care. (2005) 21:242–7. doi: 10.1097/01.pec.0000161469.19841.86
46. Gay L, Melenotte C, Lakbar I, Mezouar S, Devaux C, Raoult D, et al. Sexual dimorphism and gender in infectious diseases. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.698121
47. Cannon MJ, Schmid DS, Hyde TB. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev Med Virology. (2010) 20:202–13. doi: 10.1002/rmv.655
48. Abramson M, Kutin JJ, Raven J, Lanigan A, Czarny D, Walters EH. Risk factors for asthma among young adults in Melbourne, Australia. Respirology. (1996) 1:291–7. doi: 10.1111/j.1440-1843.1996.tb00045.x
49. Villacres MC, Longmate J, Auge C, Diamond DJ. Predominant type 1 CMV-specific memory T-helper response in humans: evidence for gender differences in cytokine secretion. Hum Immunol. (2004) 65:476–85. doi: 10.1016/j.humimm.2004.02.021
50. Dhakal S, Ruiz-Bedoya CA, Zhou R, Creisher PS, Villano JS, Littlefield K, et al. Sex differences in lung imaging and SARS-coV-2 antibody responses in a COVID-19 golden Syrian hamster model. mBio. (2021) 12(4):10.1128/mbio.00974-21. doi: 10.1128/mbio.00974-21
51. Malinczak CA, Fonseca W, Rasky AJ, Ptaschinski C, Morris S, Ziegler SF, et al. Sex-associated TSLP-induced immune alterations following early-life RSV infection leads to enhanced allergic disease. Mucosal Immunol. (2019) 12:969–79. doi: 10.1038/s41385-019-0171-3
52. Klein SL, Hodgson A, Robinson DP. Mechanisms of sex disparities in influenza pathogenesis. J Leukocyte Biol. (2012) 92:67–73. doi: 10.1189/jlb.0811427
53. Mörs K, Braun O, Wagner N, Auner B, Voth M, Störmann P, et al. Influence of gender on systemic IL-6 levels, complication rates and outcome after major trauma. Immunobiology. (2016) 221:904–10. doi: 10.1016/j.imbio.2016.03.005
54. Frink M, Pape HC, van Griensven M, Krettek C, Chaudry IH, Hildebrand F. INFLUENCE OF SEX AND AGE ON MODS AND CYTOKINES AFTER MULTIPLE INJURIES. Shock. (2007) 27:151–6. doi: 10.1097/01.shk.0000239767.64786.de
55. Offner PJ, Moore EE, Biffl WL. Male gender is a risk factor for major infections after surgery. Arch Surgery. (1999) 134:935–40. doi: 10.1001/archsurg.134.9.935
56. Gannon CJ, Pasquale M, Tracy JK, McCarter RJ, Napolitano LM. MALE GENDER IS ASSOCIATED WITH INCREASED RISK FOR POSTINJURY PNEUMONIA. Shock. (2004) 21:410–4. doi: 10.1097/00024382-200405000-00003
57. Napolitano LM, Greco ME, Rodriguez A, Kufera JA, West RS, Scalea and TM. Gender differences in adverse outcomes after blunt trauma. J Trauma Acute Care Surgery. (2001) 50:274–80. doi: 10.1097/00005373-200102000-00013
58. Osborn TM, Tracy JK, Dunne JR, Pasquale M, Napolitano LM. Epidemiology of sepsis in patients with traumatic injury. Crit Care Med. (2004) 32:2234–40. doi: 10.1097/01.CCM.0000145586.23276.0F
59. Sperry JL, Nathens AB, Frankel HL, Vanek SL, Moore EE, Maier RV, et al. Characterization of the gender dimorphism after injury and hemorrhagic shock: Are hormonal differences responsible?*. Crit Care Med. (2008) 36:1838–45. doi: 10.1097/CCM.0b013e3181760c14
60. Zhu Z, Shang X, Qi P, Ma S. Sex-based differences in outcomes after severe injury: an analysis of blunt trauma patients in China. Scandinavian J Trauma Resuscitation Emergency Med. (2017) 25:47. doi: 10.1186/s13049-017-0389-6
61. Kondo Y, Miyazato A, Okamoto K, Tanaka H. Impact of sex differences on mortality in patients with sepsis after trauma: A nationwide cohort study. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.678156
62. George RL, McGwin GJ, Windham ST, Melton SM, Metzger J, Chaudry IH, et al. Age-related gender differential in outcome after blunt or penetrating trauma. Shock. (2003) 19:28–32. doi: 10.1097/00024382-200301000-00006
63. Wohltmann CD, Franklin GA, Boaz PW, Luchette FA, Kearney PA, Richardson JD, et al. A multicenter evaluation of whether gender dimorphism affects survival after trauma. Am J Surgery. (2001) 181:297–300. doi: 10.1016/S0002-9610(01)00582-7
64. Majetschak M, Christensen B, Obertacke U, Waydhas C, Schindler AE, Nast-Kolb D, et al. Sex differences in posttraumatic cytokine release of endotoxin-stimulated whole blood: relationship to the development of severe sepsis. J Trauma Acute Care Surgery. (2000) 48:832–40. doi: 10.1097/00005373-200005000-00006
65. Altura BM. Sex and estrogens in protection against circulatory stress reactions. Am J Physiology-Legacy Content. (1976) 231(3):842–7. doi: 10.1152/ajplegacy.1976.231.3.842
66. Fontes GS, McCarthy RJ, Kutzler MA, Zitek-Morrison E. The effects of sex and neuter status on trauma survival in dogs: A Veterinary Committee on Trauma registry study. J Veterinary Emergency Crit Care 32(6):756–63. doi: 10.1111/vec.13210
67. Kovacs EJ, Messingham KAN, Gregory MS. Estrogen regulation of immune responses after injury. Mol Cell Endocrinology. (2002) 193:129–35. doi: 10.1016/S0303-7207(02)00106-5
68. Gregory MS, Faunce DE, Duffner LA, Kovacs EJ. Gender difference in cell-mediated immunity after thermal injury is mediated, in part, by elevated levels of interleukin-6. J Leukocyte Biol. (2000) 67:319–26. doi: 10.1002/jlb.67.3.319
69. Kerby JD, McGwin G Jr, George RL, Cross JA, Chaudry IH, Rue LW. Sex differences in mortality after burn injury: results of analysis of the national burn repository of the american burn association. J Burn Care Res. (2006) 27:452–6. doi: 10.1097/01.BCR.0000225957.01854.EE
70. Mohammadi AA, Pakyari MR, Seyed Jafari SM, Tavakkolian AR, Tolide-ie HR, Moradi Z, et al. Effect of burn sites (Upper and lower body parts) and gender on extensive burns’ Mortality. Iran J Med Sci. (2015) 40:166–9.
71. McGwin GJ, George RL, Cross JM, Reiff DA, Chaudry IH, Rue LWI. GENDER DIFFERENCES IN MORTALITY FOLLOWING BURN INJURY. Shock. (2002) 18:311–5. doi: 10.1097/00024382-200210000-00004
72. Lam NN, Hung NT, Duc NM. Influence of gender difference on outcomes of adult burn patients in a developing country. Ann Burns Fire Disasters. (2019) 32:175–8.
73. Cumming J, Purdue GF, Hunt JL, O’Keefe and GE. Objective estimates of the incidence and consequences of multiple organ dysfunction and sepsis after burn trauma. J Trauma Acute Care Surgery. (2001) 50:510–5. doi: 10.1097/00005373-200103000-00016
74. Rucker D, Warkentin LM, Huynh H, Khadaroo RG. Sex differences in the treatment and outcome of emergency general surgery. PloS One. (2019) 14:e0224278. doi: 10.1371/journal.pone.0224278
75. Al-Taki M, Sukkarieh HG, Hoballah JJ, Jamali SF, Habbal M, Masrouha KZ, et al. Effect of gender on postoperative morbidity and mortality outcomes: A retrospective cohort study. Am Surgeon. (2018) 84:377–86. doi: 10.1177/000313481808400321
76. Ono S, Tsujimoto H, Hiraki Si, Takahata R, Kinoshita M, Mochizuki H. Sex differences in cytokine production and surface antigen expression of peripheral blood mononuclear cells after surgery. Am J Surgery. (2005) 190:439–44. doi: 10.1016/j.amjsurg.2005.03.031
77. Pascoe PJ, McDonell WN, Trim CM, Van Gorder J. Mortality rates and associated factors in equine colic operations — A retrospective study of 341 operations. Can Vet J. (1983) 24:76–85.
78. Muñoz E, Argüelles D, Areste L, San Miguel L, Prades M. Retrospective analysis of exploratory laparotomies in 192 Andalusian horses and 276 horses of other breeds. Veterinary Rec. (2008) 162:303–6. doi: 10.1136/vr.162.10.303
79. Mokhtari Y, Pourbagheri-Sigaroodi A, Zafari P, Bagheri N, Ghaffari SH, Bashash D. Toll-like receptors (TLRs): An old family of immune receptors with a new face in cancer pathogenesis. J Cell Mol Med. (2021) 25:639–51. doi: 10.1111/jcmm.16214
80. Asami J, Shimizu T. Structural and functional understanding of the toll-like receptors. Protein Science: Publ Protein Soc. (2021) 30(4):761–72. doi: 10.1002/pro.4043
81. Mogensen TH, Paludan SR. Reading the viral signature by Toll-like receptors and other pattern recognition receptors. J Mol Med. (2005) 83:180–92. doi: 10.1007/s00109-004-0620-6
82. Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. (2008) 42:145–51. doi: 10.1016/j.cyto.2008.01.006
83. Chen DY, Lin CC, Chen YM, Lan JL, Hung WT, Chen HH, et al. Involvement of TLR7 MyD88-dependent signaling pathway in the pathogenesis of adult-onset Still’s disease. Arthritis Res Ther. (2013) 15:R39. doi: 10.1186/ar4193
85. Libert C, Dejager L, Pinheiro I. The X chromosome in immune functions: when a chromosome makes the difference. Nat Rev Immunol. (2010) 10:594–604. doi: 10.1038/nri2815
86. Behzadi E, Behzadi P. The role of toll-like receptors (TLRs) in urinary tract infections (UTIs). Cent Eur J Urol. (2016) 69:404–10. doi: 10.5173/ceju
87. Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. (2010) 2010:672395. doi: 10.1155/2010/672395
88. D’Arpa P, Leung KP. Toll-like receptor signaling in burn wound healing and scarring. Adv Wound Care. (2017) 6:330–43. doi: 10.1089/wound.2017.0733
89. Albiger B, Dahlberg S, Sandgren A, Wartha F, Beiter K, Katsuragi H, et al. Toll-like receptor 9 acts at an early stage in host defence against pneumococcal infection. Cell Microbiol. (2007) 9:633–44. doi: 10.1111/j.1462-5822.2006.00814.x
90. Tsujimoto H, Ono S, Efron PA, Scumpia PO, Moldawer LL, Mochizuki H. ROLE OF TOLL-LIKE RECEPTORS IN THE DEVELOPMENT OF SEPSIS. Shock. (2008) 29:315–21. doi: 10.1097/SHK.0b013e318157ee55
91. Koppe U, Suttorp N, Opitz B. Recognition of Streptococcus pneumoniae by the innate immune system. Cell Microbiol. (2012) 14:460–6. doi: 10.1111/j.1462-5822.2011.01746.x
92. de Oliviera Nascimento L, Massari P, Wetzler L. The role of TLR2 in infection and immunity. Front Immunol. (2012) 3. doi: 10.3389/fimmu.2012.00079
93. Arcaroli J, Silva E, Maloney JP, He Q, Svetkauskaite D, Murphy JR, et al. Variant IRAK-1 haplotype is associated with increased nuclear factor–κB activation and worse outcomes in sepsis. Am J Respir Crit Care Med. (2006) 173:1335–41. doi: 10.1164/rccm.200603-341OC
94. Agnese DM, Calvano JE, Hahm SJ, Coyle SM, Corbett SA, Calvano SE, et al. Human toll-like receptor 4 mutations but not CD14 polymorphisms are associated with an increased risk of gram-negative infections. J Infect Diseases. (2002) 186:1522–5. doi: 10.1086/344893
95. Lorenz E, Mira JP, Cornish KL, Arbour NC, Schwartz DA. A novel polymorphism in the toll-like receptor 2 gene and its potential association with staphylococcal infection. Infect Immun. (2000) 68:6398–401. doi: 10.1128/IAI.68.11.6398-6401.2000
96. Armstrong L, Medford AR, Hunter KJ, Uppington KM, Millar AB. Differential expression of Toll-like receptor (TLR)-2 and TLR-4 on monocytes in human sepsis. Clinical and Experimental Immunology. (2004) 136(2):312–9. doi: 10.1111/j.1365-2249.2004.02433.x
97. Tsujimoto H, Ono S, Hiraki S, Majima T, Kawarabayashi N, Sugasawa H, et al. Hemoperfusion with polymyxin B-immobilized fibers reduced the number of CD16+ CD14+ monocytes in patients with septic shock. J Endotoxin Res. (2004) 10:229–37. doi: 10.1177/09680519040100040501
98. Dessing MC, Hirst RA, Vos AFde, Poll Tvd. Role of toll-like receptors 2 and 4 in pulmonary inflammation and injury induced by pneumolysin in mice. PloS One. (2009) 4:e7993. doi: 10.1371/journal.pone.0007993
99. Williams DL, Ha T, Li C, Kalbfleisch JH, Schweitzer J, Vogt W, et al. Modulation of tissue Toll-like receptor 2 and 4 during the early phases of polymicrobial sepsis correlates with mortality*. Crit Care Med. (2003) 31:1808–18. doi: 10.1097/01.CCM.0000069343.27691.F3
100. Fan J, Li Y, Vodovotz Y, Billiar TR, Wilson MA. Hemorrhagic shock-activated neutrophils augment TLR4 signaling-induced TLR2 upregulation in alveolar macrophages: role in hemorrhage-primed lung inflammation. Am J Physiology-Lung Cell Mol Physiol. (2006) 290:L738–46. doi: 10.1152/ajplung.00280.2005
101. Fan J, Frey RS, Malik AB. TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. J Clin Invest. (2003) 112:1234–43. doi: 10.1172/JCI18696
102. Andonegui G, Bonder CS, Green F, Mullaly SC, Zbytnuik L, Raharjo E, et al. Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J Clin Invest. (2003) 111(7):1011–20. doi: 10.1172/JCI16510
103. Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, et al. Recognition of pneumolysin by toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci United States America. (2003) 100:1966–71. doi: 10.1073/pnas.0435928100
104. Srivastava A, Henneke P, Visintin A, Morse SC, Martin V, Watkins C, et al. The apoptotic response to pneumolysin is toll-like receptor 4 dependent and protects against pneumococcal disease. Infection Immun. (2005) 73(10):6479–87. doi: 10.1128/IAI.73.10.6479-6487.2005
105. Dessing MC, Florquin S, Paton JC, van der Poll T. Toll-like receptor 2 contributes to antibacterial defence against pneumolysin-deficient pneumococci. Cell Microbiol. (2008) 10:237–46. doi: 10.1111/j.1462-5822.2007.01035.x
106. Knapp S, Wieland CW, Veer Cv’t, Takeuchi O, Akira S, Florquin S, et al. Toll-like receptor 2 plays a role in the early inflammatory response to murine pneumococcal pneumonia but does not contribute to antibacterial defense. J Immunol. (2004) 172:3132–8. doi: 10.4049/jimmunol.172.5.3132
107. Frieman M, Heise M, Baric R. SARS coronavirus and innate immunity. Virus Research. (2008) 133(1):101–12. doi: 10.1016/j.virusres.2007.03.015
108. Totura AL, Whitmore A, Agnihothram S, Schäfer A, Katze MG, Heise MT, et al. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio. (2015) 6. doi: 10.1128/mBio.00638-15
109. Guo YR, Cao QD, Hong ZS, Tan YY, Chen SD, Jin HJ, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak – an update on the status. Mil Med Res. (2020) 7. doi: 10.1186/s40779-020-00240-0
110. Gemmati D, Bramanti B, Serino ML, Secchiero P, Zauli G, Tisato V. COVID-19 and individual genetic susceptibility/receptivity: role of ACE1/ACE2 genes, immunity, inflammation and coagulation. Might the double X-chromosome in females be protective against SARS-coV-2 compared to the single X-chromosome in males? Int J Mol Sci. (2020) 21:3474. doi: 10.3390/ijms21103474
111. He R, Leeson A, Andonov A, Li Y, Bastien N, Cao J, et al. Activation of AP-1 signal transduction pathway by SARS coronavirus nucleocapsid protein. Biochem Biophys Res Commun. (2003) 311:870. doi: 10.1016/j.bbrc.2003.10.075
112. Chen Iy, Chang Sc, Wu Hy, Yu Tc, Wei Wc, Lin S, et al. Upregulation of the chemokine (C-C motif) ligand 2 via a severe acute respiratory syndrome coronavirus spike-ACE2 signaling pathway. J Virol. (2010) 84. doi: 10.1128/JVI.02560-09
113. Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol 92(10):2105–13. doi: 10.1002/jmv.25987
114. Conti P, Ronconi G, Caraffa A, Gallenga C, Ross R, Frydas I, et al. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents. (2020) 34(2):327–31. doi: 10.23812/CONTI-E
115. Aboudounya MM, Heads RJ. COVID-19 and toll-like receptor 4 (TLR4): SARS-coV-2 may bind and activate TLR4 to increase ACE2 expression, facilitating entry and causing hyperinflammation. Mediators Inflammation. (2021) 2021:e8874339. doi: 10.1155/2021/8874339
116. Liu ZM, Yang MH, Yu K, Lian ZX, Deng SL. Toll-like receptor (TLRs) agonists and antagonists for COVID-19 treatments. Front Pharmacol. (2022) 13:989664. doi: 10.3389/fphar.2022.989664
117. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in wuhan, China. Clin Infect Dis 71(15):762–8. doi: 10.1093/cid/ciaa248/5803306
118. Meng Y, Wu P, Lu W, Liu K, Ma K, Huang L, et al. Sex-specific clinical characteristics and prognosis of coronavirus disease-19 infection in Wuhan, China: A retrospective study of 168 severe patients. PloS Pathogens. (2020) 16:e1008520. doi: 10.1371/journal.ppat.1008520
119. Sebina I, Phipps S. The contribution of neutrophils to the pathogenesis of RSV bronchiolitis. Viruses. (2020) 12:808. doi: 10.3390/v12080808
120. Tal G, Mandelberg A, Dalal I, Cesar K, Somekh E, Tal A, et al. Association between common toll-like receptor 4 mutations and severe respiratory syncytial virus disease. J Infect Diseases. (2004) 189:2057–63. doi: 10.1086/420830
121. Goutak M, Haidopoulou K, Pappa S, Tsakipjdis P, Frydas E, Eboriadou M, et al. The role of TLR4 and CD14 polymorphisms in the pathogenesis of respiratory syncytial virus bronchiolitis in greek infants. Int J Immunopathol Pharmacol. (2014) 27:563–72. doi: 10.1177/039463201402700412
122. Awomoyi AA, Rallabhandi P, Pollin TI, Lorenz E, Sztein MB, Boukhvalova MS, et al. Association of TLR4 polymorphisms with symptomatic respiratory syncytial virus infection in high-risk infants and young children. J Immunol. (2007) 179:3171–7. doi: 10.4049/jimmunol.179.5.3171
123. Tulic MK, Hurrelbrink RJ, Prêle CM, Laing IA, Upham JW, Souef PL, et al. TLR4 polymorphisms mediate impaired responses to respiratory syncytial virus and lipopolysaccharide. J Immunol. (2007) 179:132–40. doi: 10.4049/jimmunol.179.1.132
124. Monick MM, Yarovinsky TO, Powers LS, Butler NS, Carter AB, Gudmundsson G, et al. Respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin *. J Biol Chem. (2003) 278:53035–44. doi: 10.1074/jbc.M308093200
125. Gagro A, Tominac M, Kršulović-Hrešić V, Baće A, Matić M, Draženović V, et al. Increased Toll-like receptor 4 expression in infants with respiratory syncytial virus bronchiolitis. Clin Exp Immunol. (2004) 135:267–72. doi: 10.1111/j.1365-2249.2004.02364.x
126. Halfhide CP, Flanagan BF, Brearey SP, Hunt JA, Fonceca AM, McNamara PS, et al. Respiratory syncytial virus binds and undergoes transcription in neutrophils from the blood and airways of infants with severe bronchiolitis. J Infect Dis. (2011) 204:451–8. doi: 10.1093/infdis/jir280
127. McNamara P, Ritson P, Selby A, Hart C, Smyth R. Bronchoalveolar lavage cellularity in infants with severe respiratory syncytial virus bronchiolitis. Arch Dis Child. (2003) 88:922–6. doi: 10.1136/adc.88.10.922
128. Everard ML, Swarbrick A, Wrightham M, McIntyre J, Dunkley C, James PD, et al. Analysis of cells obtained by bronchial lavage of infants with respiratory syncytial virus infection. Arch Dis Child. (1994) 71:428–32. doi: 10.1136/adc.71.5.428
129. Cavallaro EC, Liang KK, Lawrence MD, Forsyth KD, Dixon DL. Neutrophil infiltration and activation in bronchiolitic airways are independent of viral etiology. Pediatr Pulmonology. (2017) 52:238–46. doi: 10.1002/ppul.23514
130. Casimir GJA, Mulier S, Hanssens L, Zylberberg K, Duchateau J. Gender differences in inflammatory markers in children. Shock. (2010) 33:258–62. doi: 10.1097/SHK.0b013e3181b2b36b
131. Murawski MR, Bowen GN, Cerny AM, Anderson LJ, Haynes LM, Tripp RA, et al. Respiratory syncytial virus activates innate immunity through toll-like receptor 2. J Virol. (2009) 83:1492–500. doi: 10.1128/JVI.00671-08
132. Haynes LM, Moore DD, Kurt-Jones EA, Finberg RW, Anderson LJ, Tripp RA. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J Virol. (2001) 75(22):10730–37. doi: 10.1128/JVI.75.22.10730-10737.2001
133. Velayutham TS, Kolli D, Ivanciuc T, Garofalo RP, Casola A. Critical role of TLR4 in human metapneumovirus mediated innate immune responses and disease pathogenesis. PloS One. (2013) 8:e78849. doi: 10.1371/journal.pone.0078849
134. Frascaroli G, Rossini G, Maltoni V, Bartoletti M, Ortolani P, Gredmark-Russ S, et al. Genetic and functional characterization of toll-like receptor responses in immunocompetent patients with CMV mononucleosis. Front Cell Infection Microbiol. (2020) 10:386. doi: 10.3389/fcimb.2020.00386
135. Kayesh MEH, Kohara M, Tsukiyama-Kohara K. Recent insights into the molecular mechanism of toll-like receptor response to dengue virus infection. Front Microbiol. (2021) 12:744233. doi: 10.3389/fmicb.2021.744233
136. Tsai YT, Chang SY, Lee CN, Kao CL. Human TLR3 recognizes dengue virus and modulates viral replication in vitro. Cell Microbiol. (2009) 11:604–15. doi: 10.1111/cmi.2009.11.issue-4
137. Sun P, Fernandez S, Marovich MA, Palmer DR, Celluzzi CM, Boonnak K, et al. Functional characterization of ex vivo blood myeloid and plasmacytoid dendritic cells after infection with dengue virus. Virology. (2009) 383:207–15. doi: 10.1016/j.virol.2008.10.022
138. Modhiran N, Watterson D, Muller DA, Panetta AK, Sester DP, Liu L, et al. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci Transl Med. (2015) 7:304ra142. doi: 10.1126/scitranslmed.aaa3863
139. Chen J, Ng MML, Chu JJH. Activation of TLR2 and TLR6 by dengue NS1 protein and its implications in the immunopathogenesis of dengue virus infection. PloS Pathog. (2015) 11:e1005053. doi: 10.1371/journal.ppat.1005053
140. Sharma S, Singh SK, Kakkar K, Nyari N, Dhole TN, Kashyap R, et al. Analysis of TLR4 (Asp299Gly and Thr399Ile) gene polymorphisms and mRNA level in patients with dengue infection: A case-control study. Infection Genet Evolution. (2016) 43:412–7. doi: 10.1016/j.meegid.2016.06.027
141. Torres S, Hernández JC, Giraldo D, Arboleda M, Rojas M, Smit JM, et al. Differential expression of toll-like receptors in dendritic cells of patients with dengue during early and late acute phases of the disease. PloS Negl Trop Dis. (2013) 7:e2060. doi: 10.1371/journal.pntd.0002060
142. Sariol CA, Martínez MI, Rivera F, Rodríguez IV, Pantoja P, Abel K, et al. Decreased dengue replication and an increased anti-viral humoral response with the use of combined toll-like receptor 3 and 7/8 agonists in macaques. PloS One. (2011) 6:e19323. doi: 10.1371/journal.pone.0019323
143. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. (2014) 14:315–28. doi: 10.1038/nri3665
144. Seo SU, Kwon HJ, Song JH, Byun YH, Seong BL, Kawai T, et al. MyD88 signaling is indispensable for primary influenza A virus infection but dispensable for secondary infection. J Virology. (2010) 84:12713–22. doi: 10.1128/JVI.01675-10
145. Romics L Jr, Szabo G, Coffey JC, Wang JH, Redmond HP. The emerging role of toll-like receptor pathways in surgical diseases. Arch Surgery. (2006) 141:595–601. doi: 10.1001/archsurg.141.6.595
146. Naase H, Harling L, Kidher E, Sepehripour A, Nguyen B, Kapelouzou A, et al. Toll-like receptor 9 and the inflammatory response to surgical trauma and cardiopulmonary bypass. J Cardiothoracic Surgery. (2020) 15:137. doi: 10.1186/s13019-020-01179-y
147. Pérez-Bárcena J, Regueiro V, Crespí C, Pierola J, Oliver A, Llompart-Pou JA, et al. Expression of toll-like receptors 2 and 4 is upregulated during hospital admission in traumatic patients: lack of correlation with blunted innate immune responses. Ann Surgery. (2010) 251:521–7. doi: 10.1097/SLA.0b013e3181cc8f84
148. Adib-Conquy M, Moine P, Asehnoune K, Edouard A, Espevik T, Miyake K, et al. Toll-like receptor-mediated tumor necrosis factor and interleukin-10 production differ during systemic inflammation. Am J Respir Crit Care Med. (2003) 168:158–64. doi: 10.1164/rccm.200209-1077OC
149. Lendemans S, Kreuzfelder E, Rani M, Bayeeh E, SChade FU, Flohé SB, et al. Toll-like receptor 2 and 4 expression after severe injury is not involved in the dysregulation of the innate immune system. J Trauma Acute Care Surgery. (2007) 63:740–6. doi: 10.1097/01.ta.0000240451.42238.d1
150. Giannoudis PV, Smith MR, Evans RT, Bellamy MC, Guillou PJ. Serum CRP and IL-6 levels after trauma: Not predictive of septic complications in 31 patients. Acta Orthopaedica Scandinavica. (1998) 69:184–8. doi: 10.3109/17453679809117625
151. Barsness KA, Arcaroli J, Harken AH, Abraham E, Banerjee A, Reznikov L, et al. Hemorrhage-induced acute lung injury is TLR-4 dependent. Am J Physiology-Regulatory Integr Comp Physiol. (2004) 287:R592–9. doi: 10.1152/ajpregu.00412.2003
152. Thobe BM, Frink M, Hildebrand F, Schwacha MG, Hubbard WJ, Choudhry MA, et al. The role of MAPK in Kupffer cell toll-like receptor (TLR) 2-, TLR4-, and TLR9-mediated signaling following trauma-hemorrhage. J Cell Physiol. (2007) 210:667–75. doi: 10.1002/jcp.20860
153. Suresh MV, Thomas B, Dolgachev VA, Sherman MA, Rebecca G, Mark J, et al. Toll like receptor-9 (TLR9) is requisite for acute inflammatory response and injury following lung contusion. Shock. (2016) 46:412–9. doi: 10.1097/SHK.0000000000000601
154. Zhang X, Li N, Meng Y, Zhang R, Bian J, Yao Y, et al. High-level expression of toll-like receptors on dendritic cells in adult patients with burns on ≥90% of total body surface area (TBSA). Med Sci Monit. (2016) 22:3493–9. doi: 10.12659/MSM.897433
155. Kobayashi M, Yoshida T, Herndon DN, Suzuki F. TLR3 and TLR9 mRNA expression is impaired in macrophages from severely burned mice (44.31). J Immunol. (2007) 178:S54–4. doi: 10.4049/jimmunol.178.Supp.44.31
156. Shen H, de Almeida PE, Kang KH, Yao P, Chan CW. Burn injury triggered dysfunction in dendritic cell response to TLR9 activation and resulted in skewed T cell functions. PloS One. (2012) 7:e50238. doi: 10.1371/journal.pone.0050238
157. Aomatsu M, Kato T, Kasahara E, Kitagawa S. Gender difference in tumor necrosis factor-α production in human neutrophils stimulated by lipopolysaccharide and interferon-γ. Biochem Biophys Res Commun. (2013) 441:220–5. doi: 10.1016/j.bbrc.2013.10.042
158. Lefevre N. Study of the role of the X chromosome in sex differences in pediatric inflammatory diseases (2017). Available online at: http://hdl.handle.net/2013/ULB-DIPOT:oai:dipot.ulb.ac.be:2013/260080.
159. Asai K, Hiki N, Mimura Y, Ogawa T, Unou K, Kaminishi M. GENDER DIFFERENCES IN CYTOKINE SECRETION BY HUMAN PERIPHERAL BLOOD MONONUCLEAR CELLS: ROLE OF ESTROGEN IN MODULATING LPS-INDUCED CYTOKINE SECRETION IN AN EX VIVO SEPTIC MODEL. Shock. (2001) 16:340–3. doi: 10.1097/00024382-200116050-00003
160. Lefèvre N, Corazza F, Valsamis J, Delbaere A, De Maertelaer V, Duchateau J, et al. The number of X chromosomes influences inflammatory cytokine production following toll-like receptor stimulation. Front Immunol. (2019) 10. doi: 10.3389/fimmu.2019.01052
161. Campesi I, Montella A, Franconi F. Human monocytes respond to lipopolysaccharide (LPS) stimulation in a sex-dependent manner. J Cell Physiol. (2022) 237:580–8. doi: 10.1002/jcp.30503
162. Aulock SV, Deininger S, Draing C, Gueinzius K, Dehus O, Hermann C. Gender difference in cytokine secretion on immune stimulation with LPS and LTA. J Interferon Cytokine Res. (2006) 26:887–92. doi: 10.1089/jir.2006.26.887
163. Bouman A, Schipper M, Heineman MJ, Faas MM. Gender difference in the non-specific and specific immune response in humans. Am J Reprod Immunol. (2004) 52:19–26. doi: 10.1111/j.1600-0897.2004.00177.x
164. Beenakker KGM, Westendorp RGJ, de Craen AJM, Chen S, Raz Y, Ballieux BEPB, et al. Men Have a Stronger Monocyte-Derived Cytokine Production Response upon Stimulation with the Gram-Negative Stimulus Lipopolysaccharide than Women: A Pooled Analysis Including 15 Study Populations. JIN. (2020) 12:142–53. doi: 10.1159/000499840
165. Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol. (2011) 29:71–109. doi: 10.1146/annurev-immunol-031210-101312
166. Jabeen S, Landazuri J, Nagvenkar S, Czuj B, Maghsoudi A, Javdan M, et al. TLR4 sex dimorphism correlates with sex dimorphic phagocytosis in primary macrophages. Ital J Gender-Specific Med. (2020) 6:100–6. doi: 10.1723/3432.34214
167. Marriott I, Bost KL, Huet-Hudson YM. Sexual dimorphism in expression of receptors for bacterial lipopolysaccharides in murine macrophages: A possible mechanism for gender-based differences in endotoxic shock susceptibility. J Reprod Immunol. (2006) 71:12–27. doi: 10.1016/j.jri.2006.01.004
168. Scotland RS, Stables MJ, Madalli S, Watson P, Gilroy DW. Sex-differences in resident immune cell phenotype underlies more efficient acute inflammatory responses in female mice. Blood. (2011) 118:5918–27. doi: 10.1182/blood-2011-03-340281
169. Roberts BJ, Dragon JA, Moussawi M, Huber SA. Sex-specific signaling through Toll-Like Receptors 2 and 4 contributes to survival outcome of Coxsackievirus B3 infection in C57Bl/6 mice. Biol Sex Differences. (2012) 3:25. doi: 10.1186/2042-6410-3-25
170. Eisenmenger SJ, Wichmann MW, Angele P, Faist E, Hatz R, Chaudry IH, et al. Differences in the expression of LPS-receptors are not responsible for the sex-specific immune response after trauma and hemorrhagic shock. Cell Immunol. (2004) 230:17–22. doi: 10.1016/j.cellimm.2004.08.002
171. Khan N, Summers CW, Helbert MR, Arkwright PD. Effects of age, gender, and immunosuppressive agents on in vivo toll-like receptor pathway responses. Hum Immunol. (2010) 71:372–6. doi: 10.1016/j.humimm.2010.01.018
172. Torcia MG, Nencioni L, Clemente AM, Civitelli L, Celestino I, Limongi D, et al. Sex differences in the response to viral infections: TLR8 and TLR9 ligand stimulation induce higher IL10 production in males. PloS One. (2012) 7(6):e39853. doi: 10.1371/journal.pone.0039853
173. Hannah MF, Bajic VB, Klein SL. Sex differences in the recognition of and innate antiviral responses to Seoul virus in Norway rats. Brain Behavior Immunity. (2008) 22:503–16. doi: 10.1016/j.bbi.2007.10.005
174. Souyris M, Cenac C, Azar P, Daviaud D, Canivet A, Grunenwald S, et al. TLR7 escapes X chromosome inactivation in immune cells. Sci Immunol. (2018) 3:eaap8855. doi: 10.1126/sciimmunol.aap8855
175. Berghöfer B, Frommer T, Haley G, Fink L, Bein G, Hackstein H. TLR7 ligands induce higher IFN-α Production in females. J Immunol. (2006) 177:2088–96. doi: 10.4049/jimmunol.177.4.2088
176. Regis E, Fontanella S, Lin L, Howard R, Haider S, Curtin JA, et al. Sex differences in innate anti-viral immune responses to respiratory viruses and in their clinical outcomes in a birth cohort study. Sci Rep. (2021) 11:23741. doi: 10.1038/s41598-021-03044-x
177. Meier A, Chang JJ, Chan ES, Pollard RB, Sidhu HK, Kulkarni S, et al. Sex differences in the Toll-like receptor–mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med. (2009) 15:955–9. doi: 10.1038/nm.2004
178. Cox M, Adetifa JU, Noho-Konteh F, Sanyang LC, Drammeh A, Plebanski M, et al. Sex-differential impact of human cytomegalovirus infection on in vitro reactivity to toll-like receptor 2, 4 and 7/8 stimulation in Gambian infants. Vaccines. (2020) 8:407. doi: 10.3390/vaccines8030407
179. Demaria O, Chasson L, Serra F, Desnues B, Alexopoulou L. Sex bias in susceptibility to MCMV infection: implication of TLR9. PloS One. (2012) 7:e45171. doi: 10.1371/journal.pone.0045171
180. Souza CLSe, Barbosa CD, Coelho HILN, Santos Júnior MN, Barbosa EN, Queiroz ÉC, et al. Effects of 17β-estradiol on monocyte/macrophage response to staphylococcus aureus: an in vitro study. Front Cell Infection Microbiol. (2021) 11:701391. doi: 10.3389/fcimb.2021.701391
181. Bouman A, Heineman MJ, Faas MM. Sex hormones and the immune response in humans. Hum Reprod Update. (2005) 11:411–23. doi: 10.1093/humupd/dmi008
182. Rogers A, Eastell R. Effects of estrogen therapy of postmenopausal women on cytokines measured in peripheral blood. J Bone Mineral Res. (1998) 13:1577–86. doi: 10.1359/jbmr.1998.13.10.1577
183. Lashkari B S, Anumba DOC. Estradiol alters the immune-responsiveness of cervical epithelial cells stimulated with ligands of Toll-like receptors 2 and 4. PloS One. (2017) 12:e0173646. doi: 10.1371/journal.pone.0173646
184. Giannoni E, Guignard L, Knaup Reymond M, Perreau M, Roth-Kleiner M, Calandra T, et al. Estradiol and progesterone strongly inhibit the innate immune response of mononuclear cells in newborns ▿. Infect Immun. (2011) 79:2690–8. doi: 10.1128/IAI.00076-11
185. Jitprasertwong P, Charadram N, Kumphune S, Pongcharoen S, Sirisinha S. Female sex hormones modulate Porphyromonas gingivalis lipopolysaccharide-induced Toll-like receptor signaling in primary human monocytes. J Periodontal Res. (2016) 51:395–406. doi: 10.1111/jre.12320
186. Young NA, Wu LC, Burd CJ, Friedman AK, Kaffenberger BH, Rajaram MVS, et al. Estrogen modulation of endosome-associated toll-like receptor 8: an IFNα-independent mechanism of sex-bias in systemic lupus erythematosus. Clin Immunol. (2014) 151:66–77. doi: 10.1016/j.clim.2014.01.006
187. Dahm AEA, Eilertsen AL, Goeman J, Olstad OK, Øvstebø R, Kierulf P, et al. A microarray study on the effect of four hormone therapy regimens on gene transcription in whole blood from healthy postmenopausal women. Thromb Res. (2012) 130:45–51. doi: 10.1016/j.thromres.2011.12.009
188. Li X, Li M, Bai X. Upregulation of TLR2 expression is induced by estrogen via an estrogen-response element (ERE). Arch Biochem Biophysics. (2014) 549:26–31. doi: 10.1016/j.abb.2014.01.028
189. Zhu Y, Wu M, Wu Cy, Xia Gq. Role of progesterone in TLR4-MyD88-dependent signaling pathway in pre-eclampsia. J Huazhong Univ Sci Technol. (2013) 33:730–4. doi: 10.1007/s11596-013-1188-6
190. Dennison U, McKernan DP, Scully P, Clarke G, Cryan J, Dinan T. Menstrual cycle influences toll-like receptor responses. NIM. (2012) 19:171–9. doi: 10.1159/000331424
191. Malkin CJ, Pugh PJ, Jones RD, Kapoor D, Channer KS, Jones TH. The effect of testosterone replacement on endogenous inflammatory cytokines and lipid profiles in hypogonadal men. J Clin Endocrinol Metab. (2004) 89:3313–8. doi: 10.1210/jc.2003-031069
192. Ziegler SM, Feldmann CN, Hagen SH, Richert L, Barkhausen T, Goletzke J, et al. Innate immune responses to toll-like receptor stimulation are altered during the course of pregnancy. J Reprod Immunol. (2018) 128:30–7. doi: 10.1016/j.jri.2018.05.009
193. Rettew JA, Huet YM, Marriott I. Estrogens augment cell surface TLR4 expression on murine macrophages and regulate sepsis susceptibility in vivo. Endocrinology. (2009) 150:3877–84. doi: 10.1210/en.2009-0098
194. Hughes GC, Thomas S, Li C, Kaja MK, Clark EA. Cutting edge: progesterone regulates IFN-α Production by plasmacytoid dendritic cells. J Immunol. (2008) 180:2029–33. doi: 10.4049/jimmunol.180.4.2029
195. Seillet C, Laffont S, Trémollières F, Rouquié N, Ribot C, Arnal JF, et al. The TLR-mediated response of plasmacytoid dendritic cells is positively regulated by estradiol in vivo through cell-intrinsic estrogen receptor α signaling. Blood. (2012) 119:454–64. doi: 10.1182/blood-2011-08-371831
196. Ziegler SM, Beisel C, Sutter K, Griesbeck M, Hildebrandt H, Hagen SH, et al. Human pDCs display sex-specific differences in type I interferon subtypes and interferon α/β receptor expression. Eur J Immunol. (2017) 47:251–6. doi: 10.1002/eji.201646725
197. Webb K, Peckham H, Radziszewska A, Menon M, Oliveri P, Simpson F, et al. Sex and pubertal differences in the type 1 interferon pathway associate with both X chromosome number and serum sex hormone concentration. Front Immunol. (2019) 9:3167. doi: 10.3389/fimmu.2018.03167
198. Shey MS, Maharaj N, Archary D, Ngcapu S, Garrett N, Abdool Karim S, et al. Modulation of female genital tract-derived dendritic cell migration and activation in response to inflammatory cytokines and toll-like receptor agonists. PloS One. (2016) 11:e0155668. doi: 10.1371/journal.pone.0155668
199. Michel KG, Huijbregts RPH, Gleason JL, Richter HE, Hel Z. Effect of hormonal contraception on the function of plasmacytoid dendritic cells and distribution of immune cell populations in the female reproductive tract. J Acquir Immune Defic Syndr. (2015) 68:511–8. doi: 10.1097/QAI.0000000000000531
200. Huijbregts RPH, Michel KG, Hel Z. Effect of progestins on immunity: medroxyprogesterone but not norethisterone or levonorgestrel suppresses the function of T cells and pDCs. Contraception. (2014) 90:123–9. doi: 10.1016/j.contraception.2014.02.006
201. Butts CL, Shukair SA, Duncan KM, Bowers E, Horn C, Belyavskaya E, et al. Progesterone inhibits mature rat dendritic cells in a receptor-mediated fashion. Int Immunol. (2007) 19:287–96. doi: 10.1093/intimm/dxl145
202. Lesmeister MJ, Jorgenson RL, Young SL, Misfeldt ML. 17beta-estradiol suppresses TLR3-induced cytokine and chemokine production in endometrial epithelial cells. Reprod Biol Endocrinology. (2005) 3:74. doi: 10.1186/1477-7827-3-74
203. Allhorn S, Böing C, Koch AA, Kimmig R, Gashaw I. TLR3 and TLR4 expression in healthy and diseased human endometrium. Reprod Biol Endocrinol. (2008) :6:40. doi: 10.1186/1477-7827-6-40
204. Musabak U, Bolu E, Ozata M, Oktenli C, Sengul A, Inal A, et al. Gonadotropin treatment restores in vitro interleukin-1β and tumour necrosis factor-α production by stimulated peripheral blood mononuclear cells from patients with idiopathic hypogonadotropic hypogonadism. Clin Exp Immunol. (2003) 132:265–70. doi: 10.1046/j.1365-2249.2003.02141.x
205. Kalinchenko SY, Tishova YA, Mskhalaya GJ, Gooren LJG, Giltay EJ, Saad F. Effects of testosterone supplementation on markers of the metabolic syndrome and inflammation in hypogonadal men with the metabolic syndrome: the double-blinded placebo-controlled Moscow study. Clin Endocrinology. (2010) 73:602–12. doi: 10.1111/j.1365-2265.2010.03845.x
206. Bobjer J, Katrinaki M, Tsatsanis C, Lundberg Giwercman Y, Giwercman A. Negative association between testosterone concentration and inflammatory markers in young men: A nested cross-sectional study. PloS One. (2013) 8:e61466. doi: 10.1371/journal.pone.0061466
207. Jin H, Qiu WB, Mei YF, Wang DM, Li YG, Tan XR. Testosterone alleviates tumor necrosis factor-alpha-mediated tissue factor pathway inhibitor downregulation via suppression of nuclear factor-kappa B in endothelial cells. Asian J Androl. (2009) 11:266–71. doi: 10.1038/aja.2008.12
208. Olmos-Ortiz A, García-Quiroz J, Halhali A, Avila E, Zaga-Clavellina V, Chavira-Ramírez R, et al. Negative correlation between testosterone and TNF-α in umbilical cord serum favors a weakened immune milieu in the human male fetoplacental unit. J Steroid Biochem Mol Biol. (2019) 186:154–60. doi: 10.1016/j.jsbmb.2018.10.009
209. D’agostino P, Milano S, Barbera C, Di Bella G, La Rosa M, Ferlazzo V, et al. Sex hormones modulate inflammatory mediators produced by macrophagesa. Ann New York Acad Sci. (1999) 876:426–9. doi: 10.1111/j.1749-6632.1999.tb07667.x
210. Jia Yl, Liu X, Yan Jy, Chong Lm, Li L, Ma Ac, et al. The alteration of inflammatory markers and apoptosis on chronic prostatitis induced by estrogen and androgen. Int Urol Nephrol. (2015) 47:39–46. doi: 10.1007/s11255-014-0845-4
211. Rettew JA, Huet-Hudson YM, Marriott I. Testosterone reduces macrophage expression in the mouse of toll-like receptor 4, a trigger for inflammation and innate immunity. Biol Reproduction. (2008) 78:432–7. doi: 10.1095/biolreprod.107.063545
212. Casimir GJ, Heldenbergh F, Hanssens L, Mulier S, Heinrichs C, Lefevre N, et al. Gender differences and inflammation: an in vitro model of blood cells stimulation in prepubescent children. J Inflammation (Lond). (2010) 7:28. doi: 10.1186/1476-9255-7-28
213. Casimir GJ, Lefèvre N, Corazza F, Duchateau J. Sex and inflammation in respiratory diseases: a clinical viewpoint. Biol Sex Differ. (2013) 4:16. doi: 10.1186/2042-6410-4-16
214. Klein SL. Immune cells have sex and so should journal articles. Endocrinology. (2012) 153:2544–50. doi: 10.1210/en.2011-2120
215. Johnston CM, Lovell FL, Leongamornlert DA, Stranger BE, Dermitzakis ET, Ross MT. Large-scale population study of human cell lines indicates that dosage compensation is virtually complete. PloS Genet. (2008) 4(1):e9. doi: 10.1371/journal.pgen.0040009
216. Lefèvre N, Corazza F, Duchateau J, Desir J, Casimir G. Sex differences in inflammatory cytokines and CD99 expression following in vitro lipopolysaccharide stimulation. Shock. (2012) 38:37–42. doi: 10.1097/SHK.0b013e3182571e46
217. Spolarics Z. THE X-FILES OF INFLAMMATION. Shock. (2007) 27:597–604. doi: 10.1097/SHK.0b013e31802e40bd
218. Li Q, Verma IM. Erratum: NF-κB regulation in the immune system. Nat Rev Immunol. (2002) 2:725–34. doi: 10.1038/nri910
219. Plath K, Mlynarczyk-Evans S, Nusinow DA, Panning B. Xist RNA and the mechanism of X chromosome inactivation. Annu Rev Genet. (2002) 36:233–78. doi: 10.1146/annurev.genet.36.042902.092433
220. Golden LC, Itoh Y, Itoh N, Iyengar S, Coit P, Salama Y, et al. Parent-of-origin differences in DNA methylation of X chromosome genes in T lymphocytes. PNAS. (2019) 116:26779–87. doi: 10.1073/pnas.1910072116
221. Sharp AJ, Stathaki E, Migliavacca E, Brahmachary M, Montgomery SB, Dupre Y, et al. DNA methylation profiles of human active and inactive X chromosomes. Genome Res. (2011) 21:1592–600. doi: 10.1101/gr.112680.110
222. Migeon BR. The role of X inactivation and cellular mosaicism in women’s health and sex-specific diseases. JAMA. (2006) 295:1428–33. doi: 10.1001/jama.295.12.1428
223. Migeon BR. Why females are mosaics, x-chromosome inactivation, and sex differences in disease. Gender Med. (2007) 4:97–105. doi: 10.1016/S1550-8579(07)80024-6
224. Palaszynski KM, Smith DL, Kamrava S, Burgoyne PS, Arnold AP, Voskuhl RR. A yin-yang effect between sex chromosome complement and sex hormones on the immune response. Endocrinology. (2005) 146:3280–5. doi: 10.1210/en.2005-0284
225. Kawai T, Akira S. TLR signaling. Cell Death Differ. (2006) 13:816–25. doi: 10.1038/sj.cdd.4401850
226. Davila S, Hibberd ML, Dass RH, Wong HEE, Sahiratmadja E, Bonnard C, et al. Genetic association and expression studies indicate a role of toll-like receptor 8 in pulmonary tuberculosis. PloS Genet. (2008) 4:e1000218. doi: 10.1371/journal.pgen.1000218
227. Dalgic N, Tekin D, Kayaalti Z, Cakir E, Soylemezoglu T, Sancar M. Relationship between toll-like receptor 8 gene polymorphisms and pediatric pulmonary tuberculosis. Dis Markers. (2011) 31:33–8. doi: 10.1155/2011/545972
228. Hagen SH, Henseling F, Hennesen J, Savel H, Delahaye S, Richert L, et al. Heterogeneous Escape from X Chromosome Inactivation Results in Sex Differences in Type I IFN Responses at the Single Human pDC Level. Cell Rep. (2020) 33:108485. doi: 10.1016/j.celrep.2020.108485
229. McDonald G, Cabal N, Vannier A, Umiker B, Yin RH, Orjalo AV, et al. Female bias in systemic lupus erythematosus is associated with the differential expression of X-linked toll-like receptor 8. Front Immunol. 6:457. doi: 10.3389/fimmu.2015.00457
230. O’Driscoll DN, De Santi C, McKiernan PJ, McEneaney V, Molloy EJ, Greene CM. Expression of X-linked Toll-like receptor 4 signaling genes in female vs. male neonates. Pediatr Res. (2017) 81:831–7. doi: 10.1038/pr.2017.2
231. Liu Y, Chen Q, Song Y, Lai L, Wang J, Yu H, et al. MicroRNA-98 negatively regulates IL-10 production and endotoxin tolerance in macrophages after LPS stimulation. FEBS Letters. (2011) 585:1963–8. doi: 10.1016/j.febslet.2011.05.029
232. Bayraktar R, Bertilaccio MTS, Calin GA. The interaction between two worlds: microRNAs and toll-like receptors. Front Immunol. (2019) 10:1053. doi: 10.3389/fimmu.2019.01053
233. Pinheiro I, Dejager L, Libert C. X-chromosome-located microRNAs in immunity: Might they explain male/female differences? BioEssays. (2011) 33:791–802. doi: 10.1002/bies.201100047
234. Lindsay MA. microRNAs and the immune response. Trends Immunol. (2008) 29:343–51. doi: 10.1016/j.it.2008.04.004
235. Benakanakere MR, Li Q, Eskan MA, Singh AV, Zhao J, Galicia JC, et al. Modulation of TLR2 protein expression by miR-105 in human oral keratinocytes. J Biol Chem. (2009) 284:23107–15. doi: 10.1074/jbc.M109.013862
236. O’Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol. (2011) 11:163–75. doi: 10.1038/nri2957
237. Bracken MB. Why animal studies are often poor predictors of human reactions to exposure. J R Soc Med. (2009) 102:120–2. doi: 10.1258/jrsm.2008.08k033
238. He D, Yang CX, Sahin B, Singh A, Shannon CP, Oliveria JP, et al. Whole blood vs PBMC: compartmental differences in gene expression profiling exemplified in asthma. Allergy Asthma Clin Immunol. (2019) 15:67. doi: 10.1186/s13223-019-0382-x
239. Kasempimolporn S, Narakorn N, Saikasem A, Sitprija V. Comparison of separated-lymphocyte and whole-blood lymphocyte proliferation assays for evaluating rabies-induced cellular immunity. Southeast Asian J Trop Med Public Health. (1996) 27:715–8.
Keywords: TLR-Toll-like receptor, innate immunity, sex, X chromosome, hormones, sepsis, infection, trauma
Citation: Popotas A, Casimir GJ, Corazza F and Lefèvre N (2024) Sex-related immunity: could Toll-like receptors be the answer in acute inflammatory response? Front. Immunol. 15:1379754. doi: 10.3389/fimmu.2024.1379754
Received: 31 January 2024; Accepted: 06 May 2024;
Published: 21 May 2024.
Edited by:
Antonietta Rossi, University of Naples Federico II, ItalyReviewed by:
Nick Gay, University of Cambridge, United KingdomMaria Grazia Morgese, University of Foggia, Italy
Copyright © 2024 Popotas, Casimir, Corazza and Lefèvre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandros Popotas, alexandros.popotas@ulb.be