 Javier Paredes1,2*
Javier Paredes1,2* Daniel Varela2,3
Daniel Varela2,3 Camila Martínez2
Camila Martínez2 Andrea Zúñiga2
Andrea Zúñiga2 Karen Correa2,4Adrián Villarroel2
Karen Correa2,4Adrián Villarroel2 Bianca Olivares2,4
Bianca Olivares2,4- 1Programa de Doctorado en Biología y Ecología Aplicada, Departamento de Biología Marina, Facultad de Ciencias del Mar, Universidad Católica del Norte, Coquimbo, Chile
- 2Centro i∼mar, Universidad de Los Lagos, Puerto Montt, Chile
- 3Centre for Biotechnology and Bioengineering, Santiago, Chile
- 4Instituto de Fomento Pesquero (IFOP), Centro de Estudios de Algas Nocivas (CREAN), Puerto Montt, Chile
In southern Chile, Alexandrium catenella is the main species generating harmful algal blooms (HABs) and over time it has expanded its range since it was first recorded in the Magallanes region in 1972. In 2016 a severe bloom of an Alexandrium species occurred, which was notable for its intensity and geographical extent, extending into new areas to the north of the Patagonian fjords including areas along the open Pacific Ocean coast. Given the exceptional nature of this event, we verified the taxonomic classification of the species that generated the bloom and evaluated the influence of the range expansion process on its genetic structure and population diversity. This was achieved by isolating clones collected in 2014 from cyst beds located at the northern limit of its then known distribution, along with clones isolated from the water samples taken during the 2016 bloom. These clones were characterized genetically with LSU rDNA and AFLPs molecular markers. Phylogenetic analyses showed that all clones were aggregated in the Group I of the A. tamarense species complex, which confirmed that A. catenella was the species responsible for the 2016 bloom. High genetic diversity was observed within populations though there were no significant differences between populations. Furthermore, genetic structure showed an isolation by distance relationship among populations, and several analyses consistently indicated a high divergence among the population groups derived from both cysts and vegetative cells. Despite this study not finding the patterns expected for species range expansion (i.e., diversity gradient and/or high population divergence), the genetic diversity and structure indicated that these were influenced by the geographic distance effect and the physical restrictions to gene flow, along with the demographic processes that occurred during the distinct phases of the life cycle of A. catenella.
Introduction
Around the world, factors such as climate change and eutrophication of coastal waters have favored the recurrence and severity of harmful algal blooms (HABs), increasing the probability that harmful species will expand their ranges to new areas (Wells et al., 2015). Species of the A. tamarense species complex are among the most prominent HAB species (Anderson et al., 2012b; Murray et al., 2015). These species, following a trend shown by other harmful algae (Anderson et al., 2012a), have over recent decades expanded their geographic ranges in both the northern and southern hemispheres (Penna et al., 2005; Persich et al., 2006).
Underlying the process of species range expansion are the colonization-extinction dynamics of the populations, whether a proportion of the migrating population will survive or not and successfully reproduce in the new area (e.g., Sexton et al., 2009). Theoretically, it is expected that the range expansion process will generate a gradient in genetic diversity and marked genetic differences between populations, due to a fraction of the migrants establishing in the new area changing due to the effects of genetic drift (Excoffier et al., 2009; Sexton et al., 2009). The genetic characterization of populations using AFLPs (Amplified fragment length polymorphisms) has permitted the study of genetic diversity and differentiation in several harmful species, with an important advantage over microsatellite markers due to the high amount of genome-wide loci observed (Rengefors et al., 2017) and its power in identifying weak genetic population differentiation (Alpermann et al., 2009). Although, AFLPs disadvantages include variation in the precision of sizing of fragments, and the assumption that bands of the same size are homologous, might not always be right, especially at higher levels of divergence (John et al., 2004; Fry et al., 2009). For instance, AFLP population characterizations have permitted the examination of the range expansion process of species such as Gonyostomum semen and Alexandrium ostenfeldii, which show no evidence of diversity gradients, but do show levels of population divergence and isolation by distance (Tahvanainen et al., 2012; Sassenhagen et al., 2015). However, in A. ostenfeldii, Brandenburg et al. (2018) recently described a contrasting genetic and phenotypic variability between a highly diverse population established in the Baltic Sea approximately at 3000 – 8000 BP and the low variability of the populations that colonized the Netherlands localities in 2012, that represent examples of established and founder population respectively. Furthermore, genetic characterization using AFLPs and microsatellites of Alexandrium populations have shown the presence of high levels of genetic diversity, which is related in turn to high phenotypic variability, factors which are relevant to their adaptive potential (Alpermann et al., 2010; John et al., 2015; Kremp et al., 2016; Brandenburg et al., 2018). In the case of A. tamarense species complex, the population genetics characterization using microsatellites and AFLPs, have revealed that the population divergence could have been generated by demographic changes that have occurred over time, in the populations of cysts in the sediments, and/or in the population of vegetative cells in the water column, and as a result of reduced gene flow. For example, it has been hypothesized that the vegetative cells generated in a bloom would come from diverse banks of cysts, formed by different cohorts, that have accumulated in the sediment over time (Alpermann et al., 2009). In addition, during the bloom the environmental conditions could favor changes in allele frequency, due to selective pressures on certain genotypes, which could then be amplified by asexual reproduction (Alpermann et al., 2009; Dia et al., 2014). Finally, the potential migration of vegetative cells could be limited by oceanographic barriers (Casabianca et al., 2012) or restricted by geographic distance (Nagai et al., 2007), generating high levels of differentiation between populations.
The taxonomic identification of species comprising the A. tamarense species complex is fundamental not just for monitoring programs and implementing management strategies (John et al., 2014), but also, in order to verify that the genetic analyses of populations are made with conspecific individuals. Over time various authors have discussed the taxonomic classification of the species of the A. tamarense species complex which occurs in southern Chile, proposing a range of nomenclatural alternatives, such as A. tamarense (Jedlicki et al., 2012), A. fundyense (John et al., 2014; Mcneill et al., 2014; Wang et al., 2014) or A. catenella (Cordova and Müller, 2002). However, this discussion was recently concluded with priority being given to the name A. catenella (Willem, 2017). This classification was based in large part on the homology of ribosomal genetic characters (rDNA) rather than morphology, as morphology can be highly plastic and variable within the species (John et al., 2014). For example, historically the capacity to form chains has been used as a character for distinguishing between the species of the A. tamarense species complex. However, evidence indicates that the formation of chains is influenced by environmental variables such as a high pH and low pCO2 (Mardones et al., 2016b). On the other hand, the phylogenetic analysis with rDNA molecular markers consistently produces five clades, where clones are separated, including those with a sympatric distribution (John et al., 2014).
In southern Chile Alexandrium catenella is the principal species causing HABs, resulting in serious ecological, social and economic impacts throughout the Patagonian fjords, where this species has expanded its range northward (Varela et al., 2012). Since 1972, when it was first identified in the Magallanes region (ca. 56°S), monitoring programs have registered the northward expansion of the species range distribution, arriving in the Aysén region in 1992, and in the waters of the southern end of Chiloé Island (ca. 43°S), Los Lagos region, in 2002 (Molinet et al., 2003; Varela et al., 2012). Until 2015 the blooms of A. catenella were restricted to the fjords and channels between Magallanes and southern Chiloé Island (Molinet et al., 2003; Mardones et al., 2010; Varela et al., 2012). However, a severe bloom of A. catenella occurred during 2016 that extended from the central-northern region of Aysén (45° 27′S) to the Chiloé inland sea, and for the first time, the bloom occurred in the open Pacific Ocean along the west coast of Chiloé Island (Figure 1; Buschmann et al., 2016).
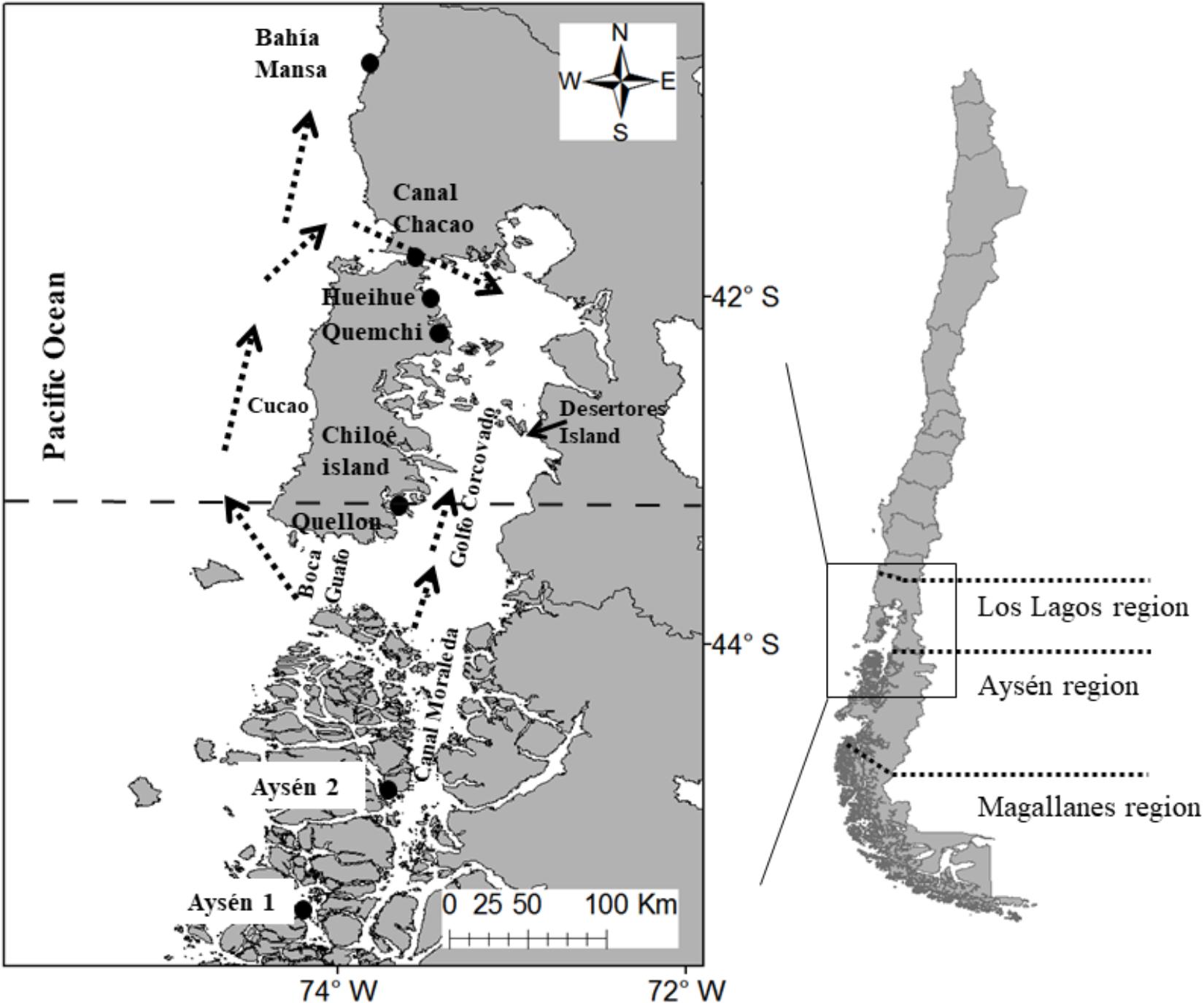
Figure 1. Localities from where the clones were isolated (black circles): Aysen 1, Aysén 2, and Quellón, the populations from northern limit of the known distribution of A. catenella up to 2015, where the clones were isolated from cysts beds in 2014. Quemchi, Hueihue, Canal Chacao, Bahía Mansa, the populations isolated from vegetative cells of the 2016 bloom, isolate from opportunistic sampling. Arrows show a hypothesized route of the spread of the 2016 bloom. Horizontal dotted lines illustrate both the political boundary of the regions (right) and the horizontal segmented line show distribution limit of the species up to 2015 (left).
The exceptional bloom of 2016 was surprising due to its wide geographical distribution, extending into new areas along the open coast of the Pacific Ocean and Chiloé inland sea (Figure 1). The event chronology indicated that the first evidence of the bloom was recorded around November–December of 2015 in the central-northern area of the Aysén region (45°23′S). Later, during January–February of 2016 the highest abundances of vegetative cells were observed in the southern area of the Chiloé inland sea (Buschmann et al., 2016). Until then, the bloom had followed the same pattern of geographical distribution observed during the severe blooms that occurred in 2002 and 2009 (Molinet et al., 2003; Mardones et al., 2010). However, from the first week of March 2016 the bloom extend northward, through the Chiloé inland sea up to Desertores Island (42° 41′ S), from where high levels of paralytic shellfish poison (PSP) were detected in mollusks (Buschmann et al., 2016). By April, the bloom declined in the Chiloé inland sea, but surprisingly the highest concentrations of PSP in mollusks were found along the exposed Pacific coast of Chiloé Island (42°37′S). This was the first evidence of the spread of the bloom into the open Pacific Ocean. Subsequently, PSP increased rapidly in mollusks living further north along the exposed coast, vegetative cells abundance also increased in areas beyond the Patagonian fjords (up to 39°42′S). Finally, by mid-May, both the cellular abundance and PSP concentrations began to decline rapidly in most areas (Buschmann et al., 2016; Hernández et al., 2016).
Given the exceptional nature of the 2016 bloom, several hypotheses have been proposed to explain the origin and dynamics of the range expansion, including doubt as to whether the species that caused the bloom were the same species that have historically affected the Patagonian fjords. Thus, the objective of the current investigation was to verify the taxonomic classification of the species that generated this exceptional bloom and evaluate the influence of the range expansion process on its genetic structure and population diversity. In order to achieve this, rDNA large subunit (LSU) and AFLPs molecular markers were used for the genetic characterization of clones. This was done with two groups of samples; the first group were vegetative cells isolated from cysts collected in 2014 from sediments located at the northern limit of its known distribution; and the second group were vegetative cells isolated from samples taken from the bloom expansion areas observed during the 2016 bloom in waters of the open Pacific Ocean coast and from the waters of the Chiloé inland sea (Figure 1).
Materials and Methods
Sampling and Cultivation of Clones
The samples used for the genetic characterization of the exceptional bloom of 2016 were collected in April, during the peak of the event, from central-northern area of its distribution, and they were compared with samples collected previously, in 2014, from sites near the origin of the same bloom event. In 2014 sediment samples containing A. catenella cysts were collected by SCUBA divers from the Aysén 1, Aysén 2 and Quellón localities (Populations from the northern limit of the then known species distribution (Figure 1 and Table 1). The sediment samples were transported to the laboratory, Centro i∼mar Universidad de Los Lagos, where the cysts were isolated using the methodology described by Varela et al. (2012). The cysts were then placed in multi-welled cultivation plates with L1 medium and exposed to conditions that facilitate germination (i.e., 12°C, 30 psu, 35 ± 5 μmol photons m-2 s-1, and a photoperiod of 16:8 h light/dark). From each well where cysts germinated, a single cell was selected and transferred to a new well (48-well plate) with the objective of generating clonal cultures. The single cell was isolated using an inverted microscope (Olympus CKX 42) by an extended Pasteur pipette (Anderson and Kawachi, 2005) and then deposited in a well with an area of 0.64 cm2 (48-well plate) with 500 μl of L1. Subsequently, the well was carefully examined using an inverted microscope to verify the successful transfer. Additionally, in April of 2016, seawater samples were collected during the A. catenella bloom at Quemchi, Hueihue, Canal Chacao, and Bahía Mansa localities (Figure 1 and Table 1). These samples were part of an opportunistic sampling that attempted to determine the extent of the 2016 bloom beyond its historic distribution. Seawater samples were also transported to the laboratory (Centro i∼mar) where vegetative cells were isolated and maintained following the same methodology and environmental conditions described above. Moreover, after 24 days of culturing, cell characteristic were determined (N = 50 per locality), i.e., viability in culture (number of well with cells alive), cell density, growth rate and the number of the cells forming chains (c.f. Paredes et al., 2016). Finally, cell isolates, both from cysts and vegetative cells, once they began to multiply, were transferred first to culture tubes, then to 250 ml conical flasks, from which cells were harvested for DNA extraction. Thus, all the strains utilized in this study were established as clonal cultures and maintained under the same laboratory conditions (i.e., 12°C, 30 psu, 35 ± 5 μmol photons m-2 s-1, photoperiod of 16:8 h light/dark, and L1 cultivation medium).
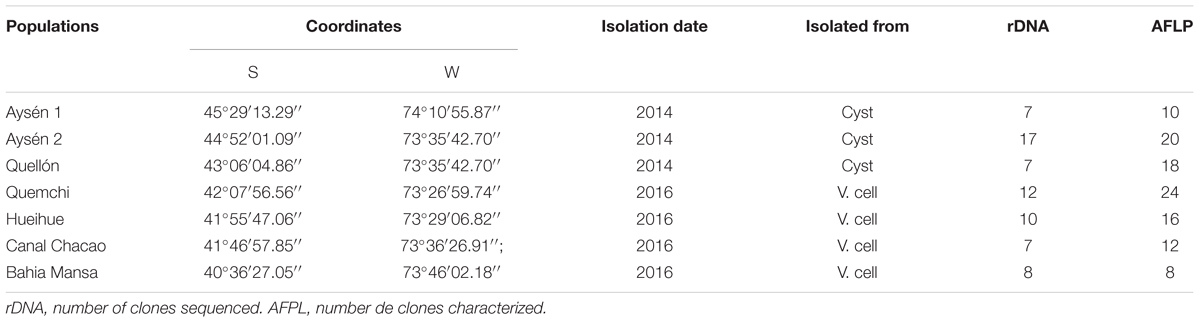
Table 1. Alexandrium catenella samples information.
Taxonomic Identification of the Clones
To verify the taxonomic classification of the clones a total of 96 clones isolated from the 2016 bloom and the cysts were characterized using rDNA LSU (D1D2) molecular marker as described by Scholin et al. (1994). DNA extractions were performed with the DNeasy plant mini Kit (Qiagen, Hilden, Germany) using 250 ml of culture harvested in the exponential phase of growth, and concentrated by centrifugation. The amplification of the DNA was performed using a thermocycler (Px2 Thermal Cycler, Thermo Electron Corporation, Waltham, MA, United States) with the following settings: one cycle of 5 min at 95°C; 30 cycles of 45 s at 95°C, 45 s at 56°C and 1 min at 72°C; and a final elongation for 10 min at 72°C. The amplifications were verified using an agarose gel (1.4%), and then, the PCR products were sent to biotechnological company Macrogen®for sequencing. The obtained sequences were evaluated visually and the homology with the sequences available in GenBank was explored. Finally, a phylogenetic reconstruction was performed using the sequences generated by John et al. (2014) which were obtained from the Supplementary Material (i.e., Supplementary Figure 1). Alignments of the SSU, ITS and LSU sequences for the phylogenetic analyses presented in this study. 1-s2.0-S1434461014001011-mmc1.doc). The reconstruction was performed with the maximum likelihood (ML) method, with a bootstrap analysis (1000 replicates) which evaluated the robustness of the nodes, and using an alignment of 826 base pair (bp). These analyses were performed with the software PAUP (Swofford, 2002). To perform the ML reconstruction, previous DNA sequences were analyzed with the MODELTEST 3.7 software which determined the most appropriate model of nucleotide substitution for the data (Possada and Crandall, 1998). Thus, the GTR + G + I model was selected with the following corrections: a base frequency of A = 0.26130, C = 0.16530, G = 0.24910, T = 0.32430; and a substitution rate matrix of [A _ G] = 2.2885, [A _ T] = 0.7838, [C _ G] = 0.7838, [C _ T] = 3.3797, [A _ C] = 1, and [G _ T] = 1; with no proportion of invariable sites and an unequal distribution of rates at variable sites (gamma shape parameter of alpha = 1.2519).
Genotyping Using AFLP Analyses
The genetic characterization was performed with 150 clonal strains, using the AFLPs technique and 100 ng of DNA, as suggested by Vos et al. (1995) and Mardones et al. (2016a). DNA extractions were performed with the DNeasy Plant Mini Kit (Qiagen), using 250 ml of culture harvested in the exponential phase of growth and concentrated by centrifugation. The concentration and quality of the DNA (proportion 280/260 nm) were estimated using a NanoQuant Plate (Infinite M200 PRO). The primers EcoRI- 5′ GACTGCGTACCAATTCXXX 3′ and MseI-5′ GATGAGTCCTGAGTAAXXX 3′ were used for the selective amplifications with the following combinations of primers marked with Fam dyes: EcoRIAAG x MseICTA, EcoRIAAG x MseICTT, EcoRIACC x MseICTA and, EcoRIACC x MseICTT. Selective amplifications were performed with the settings “touchdown” which included: 13 initial cycles with 30 s at 95°C, 45 s at 56°C (gradually reduced by 0.7°C per cycle), and 30 s at 72°C. This was followed by 25 cycles of 1 min at 95°C; 1 min at 56°C; 1 min at 72°C, and finally an elongation of 10 min at 72°C. The amplifications were verified in an agarose gel (1.4%) and then these were analyzed using an ABI PRISM 3500 sequencer (Applied Biosystems). Consequently, the AFLP fragments were visualized and edited with the software GeneMarker. In order to create a presence/absence matrix of co-migrating AFLPs fragment, the sequence profiles were examined visually considering a fragment detection range between 100 and 15000 relative fluorescence units (RFUs) and between 50 and 400 bp. In addition, the reproducibility of the amplifications was determined visually by repeating 20% of the samples selected at random.
Intraspecific Distance, Genetic Diversity, and Population Structure
The intraspecific distance was determined using Nei’s genetic distance (Saitou and Nei, 1987) while the genetic diversity of the populations (H) was calculated according to Nei (1973). The genetic diversity differentiation between populations was evaluated by an ANOVA, performed in a generalized linear mixed model (GLMM) framework. The genetic diversity for each locus was estimated, and the populations were considered to be a random effect to account for the lack of independence between the samples. In addition, a negative binomial residuals model distribution was used, and the null hypothesis (H0) was evaluated using a likelihood ratio test with a significance level (α) of 0.05 (Venables and Ripley, 2002). The estimation of genetic diversity per locus and population was performed with the ARLEQUIN 3.5.1.2 software (Excoffier et al., 2005) while the GLMM was performed using R software (R Core Team, 2017) and the package “lme4” (Bates et al., 2015).
The genetic structure was estimated using different analyses. Firstly, analysis of molecular variance (AMOVA) and the significance of the FST, FSC, and FCT were tested using a non-parametric analysis with 10,000 permutations. For this analysis the following sources of variation were considered: (1) between groups, composed of isolated populations in the northern limit of distribution (derived from cysts) and the populations isolated from the bloom; (2) variation between populations within groups, and (3) variation within the population. Furthermore, the level of differentiation between populations was estimated using the fixation index FST. Both analyses were performed using the software ARLEQUIN 3.5.1.2 (Excoffier et al., 2005). Subsequently, in order to visualize the similarities/dissimilarities between populations, a Principal Component Analysis (PCA) was performed using a matrix of genetic distances between populations which was estimated using Nei (1978) method. This was done using the software GENALEX 6.5 with a binary model for haploid organisms (Peakall and Smouse, 2012). The identification of genetic populations and the level of admixture of each genotype, or proportion of genotype assigned to each population, were estimated using a Bayesian cluster analysis implemented in the software STRUCTURE 2.3.4. The simulation was performed using a correlated allele frequency model and an ancestry mixed model without using location as prior information (Falush et al., 2003). The simulations were conducted using values of K populations from 1 to 10; a burning length of 100,000 and 100,000 repetitions of Monte Carlo Markov Chains (MCMC), and the results were evaluated using likelihood of K populations [L(K)]. The evaluation of K populations and the visualization of the genotype assignments were made using the web application www.pophelper.com (Francis, 2016). Finally, the software ARLEQUIN was used to conduct a Mantel analysis to evaluate isolation by distance, which included the paired values of FST (Y matrix) and the geographic distance in Km (X matrix).
Disequilibrium of the Multilocus Linkage
The index of multilocus disequilibrium (Agapow and Burt, 2001) was calculated for each population and significance was determined using 10,000 permutations. The values of the index vary between 0 and 1 indicating equilibrium and disequilibrium respectively. The analyses were performed with R software (R Core Team, 2017) and the package “Poppr” (Kamvar et al., 2014).
Results
Taxonomic Identification
Only 68 sequences of molecular marker rDNA LSU (D1D2) were used (Table 1). The remaining sequences were eliminated after visual evaluations of the chromatograms which suggested ambiguity in the assignment of some nucleotides i.e., no clear nucleotide peak or nucleotide peak overlaying. The phylogenetic analysis indicated that sequences of the clones isolated from both the 2016 bloom and the northern limit formed part of the same clade, corresponding to the Group I of the A. tamarense species complex (Lilly et al., 2007; John et al., 2014) (Figure 2). Moreover, Chi-square test did not show differences in the base frequencies among sequences used for the phylogenetic reconstruction (χ2 = 62.243, p > 0.05). The maximum genetic distance, according to maximum-likelihood distance, was observed between A._pacificum_ver v/s A._leei_ver strains (0.924). Meanwhile, among clones isolated from southern Chile the distance ranged between zero, for several pairwise clones isolated from closer and separate populations, and 0.069 between clones isolated from Bahía Mansa v/s Canal Chacao. Furthermore, in the clones isolated from the 2016 bloom, the number of cells in chains ranged from 2 to 8, cell viability from 0.396 to 0.875 probability, and growth rate from 0.140 to 0.331 div/day (c.f. Paredes et al., 2016). These responses were concordant with the characteristics of A. catenella strains previously isolated from southern Chile.
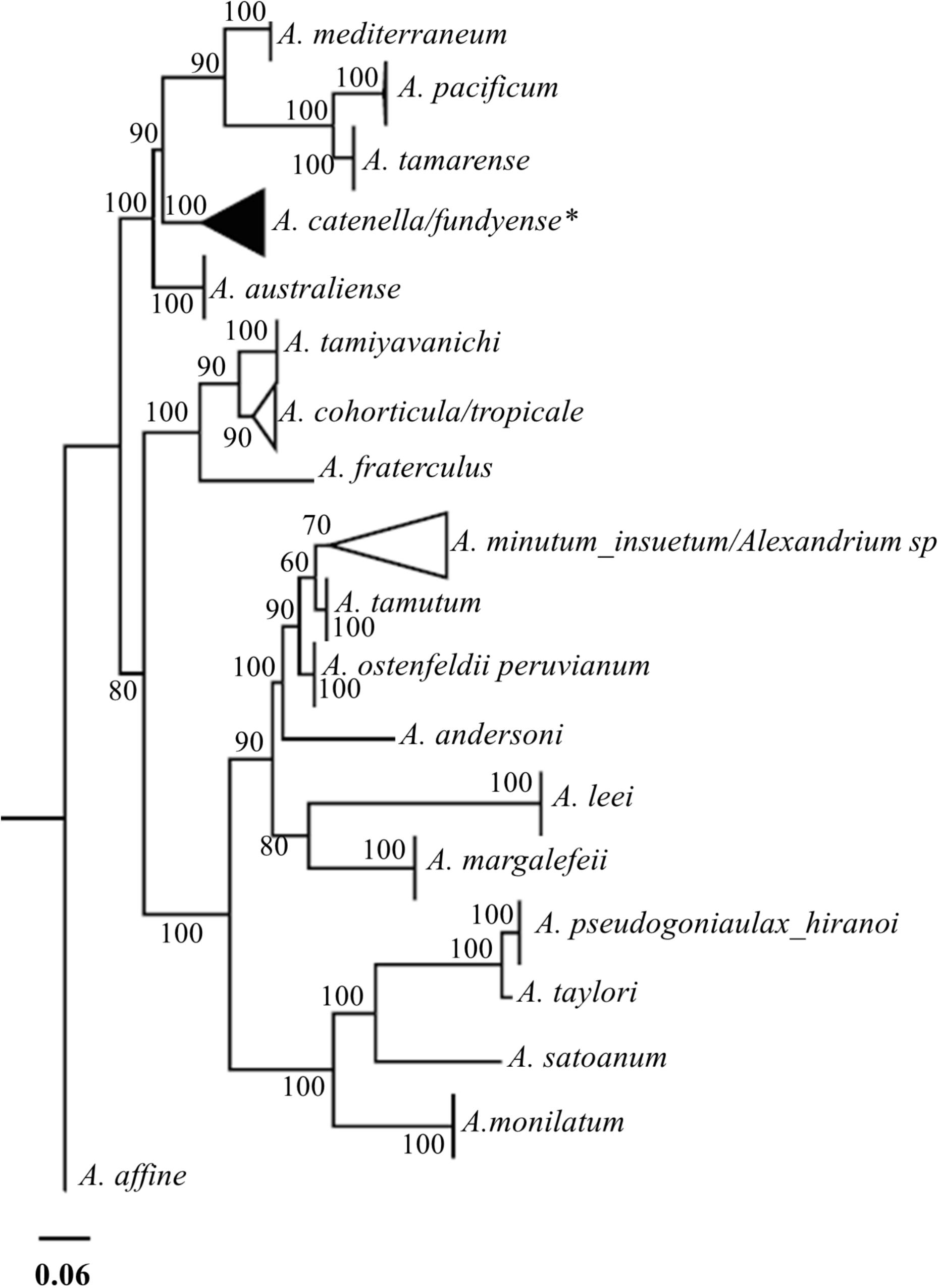
Figure 2. Maximum likelihood phylogenetic reconstruction and bootstrap values using sequences generated by John et al. (2014) and sequences of the clones isolated from the northern limit of species distribution and from the 2016 bloom. Collapsed black triangle indicates the A. catenella clade (Group 1) and aggregation of all the clone sequences in this study. ∗Sequences of the morphotype A. fundyense are also included in this clade.
Intraspecific Genetic Distance and Population Diversity
Based on the evaluation of the banding patterns, following the RFU criteria and amplification reproducibility, 264 loci were used for the genetic evaluation, from a total of 108 clones distributed across seven populations (Table 1). Similar genotypes were not observed between clones. The genetic distance between clones varied between 0.100 and 0.708, with smallest distances between Hueihue and Canal Chacao; and the greatest distances observed between the clones sampled in Quemchi and Hueihue. On the other hand, the mean genetic diversity (H) had values that varied between 0.289 and 0.415, for Aysén 2 and Bahía Mansa populations respectively (Figure 3). Although, significant differences in the genetic diversity (p > 0.05, GLMM) were not observed among populations.
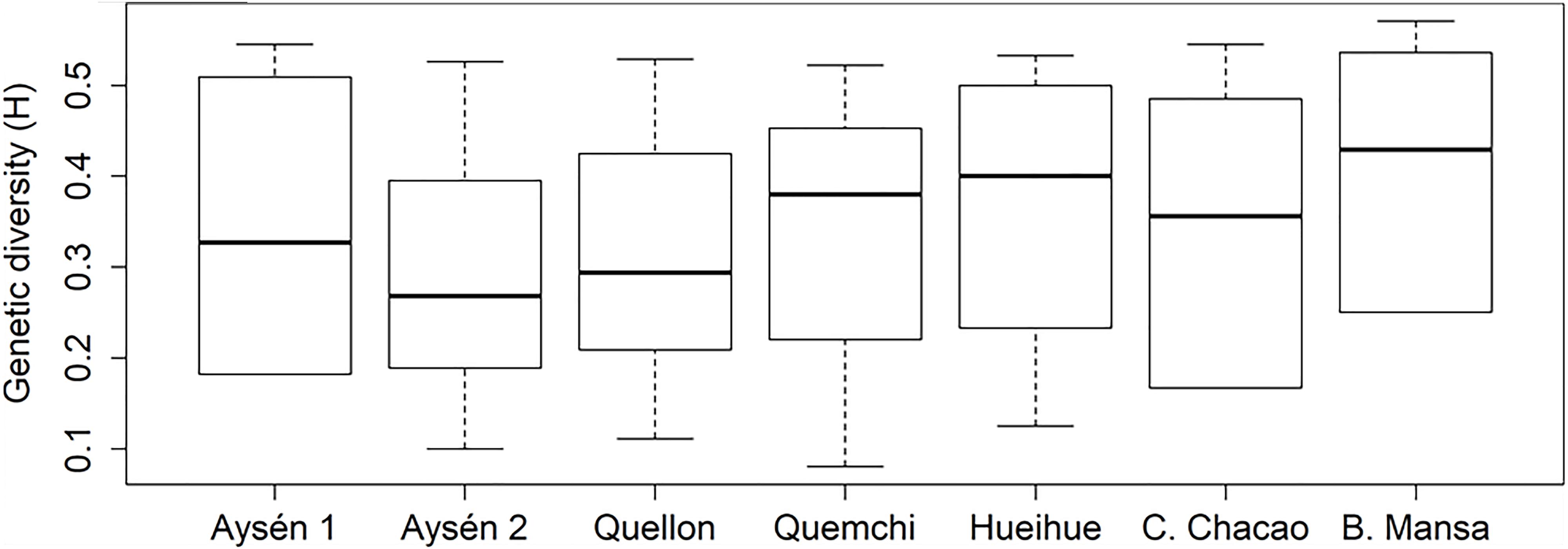
Figure 3. Genetic diversity boxplot of A. catenella populations (GLMM, p > 0.05).
Genetic Structure
The genetic variability of A. catenella was significantly structured (AMOVA, p < 0.05, for each of the fixation indices; Table 2). Although most of the genetic variability was explained by variation within populations (90.450%), the genetic differences were significant between groups, explaining 6.730% of the genetic variation, and between populations within groups, which explained 2.820% of the variation (Table 2). The paired index of fixation FST was significant for the majority of comparisons between populations, with the highest values being observed for the comparisons made between the populations derived from cysts and those derived from vegetative cells collected in the 2016 bloom (Table 3). Of the comparisons, the population in Quemchi had the greatest differences compared with each of the three populations derived from cysts (FST = 0.120 – 0.122), these were followed by Bahía Mansa (FST = 0.104 – 0.109), Hueihue (FST = 0.101 – 0.103) and the population in the Canal Chacao (FST = 0.076 – 0.093). Among the populations derived from cysts the genetic differences were relatively similar, with the largest value observed between populations of Quellón and Aysén 2 (FST = 0.032). The paired values of FST between populations derived from the bloom were more heterogeneous. There were no significant differences in the values of FST between Bahía Mansa and the populations of the Canal Chacao and Hueihue, the same was true for the comparison between the Canal Chacao and Quemchi. However, there were significant differences in the values of FST for the comparisons between Quemchi and Hueihue (FST = 0.048), Quemchi and Bahía Mansa (FST = 0.054), and between Hueihue and Canal Chacao (FST = 0.031). These differences were relatively higher than those found for the comparisons between populations derived from cysts. PCA indicated segregation of the populations in at least three groups. Thus, the first coordinate explained 65.76% of the variation and showed evident genetic similarities between the populations isolated from cysts beds in 2014 (Aysén 1, Aysén 2 and Quellon) and populations isolated from 2016 bloom. Moreover, among these last populations the second coordinates, which explained 18.35% of the variation, showed genetic similarities between Bahía Mansa and Hueihue; and between Canal Chacao and Quemchi (Figure 4).
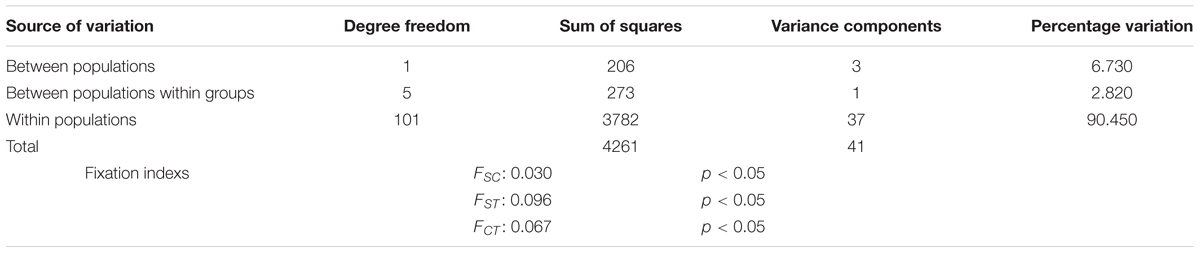
Table 2. Analysis of molecular variance (AMOVA).
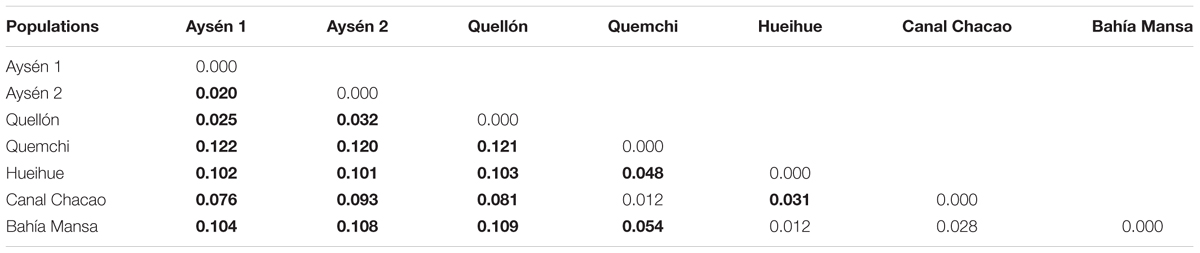
Table 3. Pair-wise FST parameters between populations (bold numbers = p < 0.05).
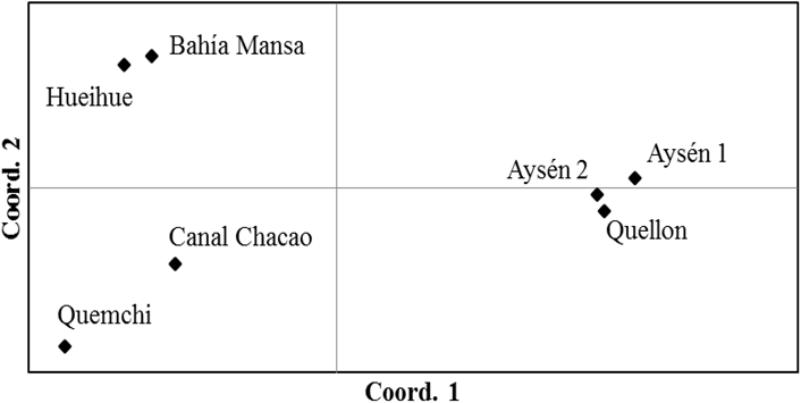
Figure 4. Principal components analysis of A. catenella populations.
A similar pattern described with PCA was observed when K = 3 (Supplementary Figure 1) or K = 5 Bayesian cluster analysis was plotted, distinguishing clearly between the strains from cysts and those from the bloom population, and recognizing higher variability within the group of isolated from the bloom. However, the Bayesian cluster analyses suggested a K = 5 as the more probable number of clusters [L(K) = -13490, Supplementary Figure 2]. The strains derived from cysts, in the main, were assigned more or less in the same proportions in two subgroups (pink and light blue, Figure 5). In the case of the strains derived from the bloom, despite the higher degree of mixing in their assignment, it was also possible to distinguish between geographic populations. Thus, the majority of the strains from Quemchi and Canal Chacao presented a high proportion of assignment to a third subgroup (yellow, Figure 5). Meanwhile, the strains from Hueihue and Bahía Mansa presented a higher degree of heterogeneity, with individuals assigned to different subgroups (blue, red or pink, Figure 5) and others assigned more or less equally to the same subpopulations.
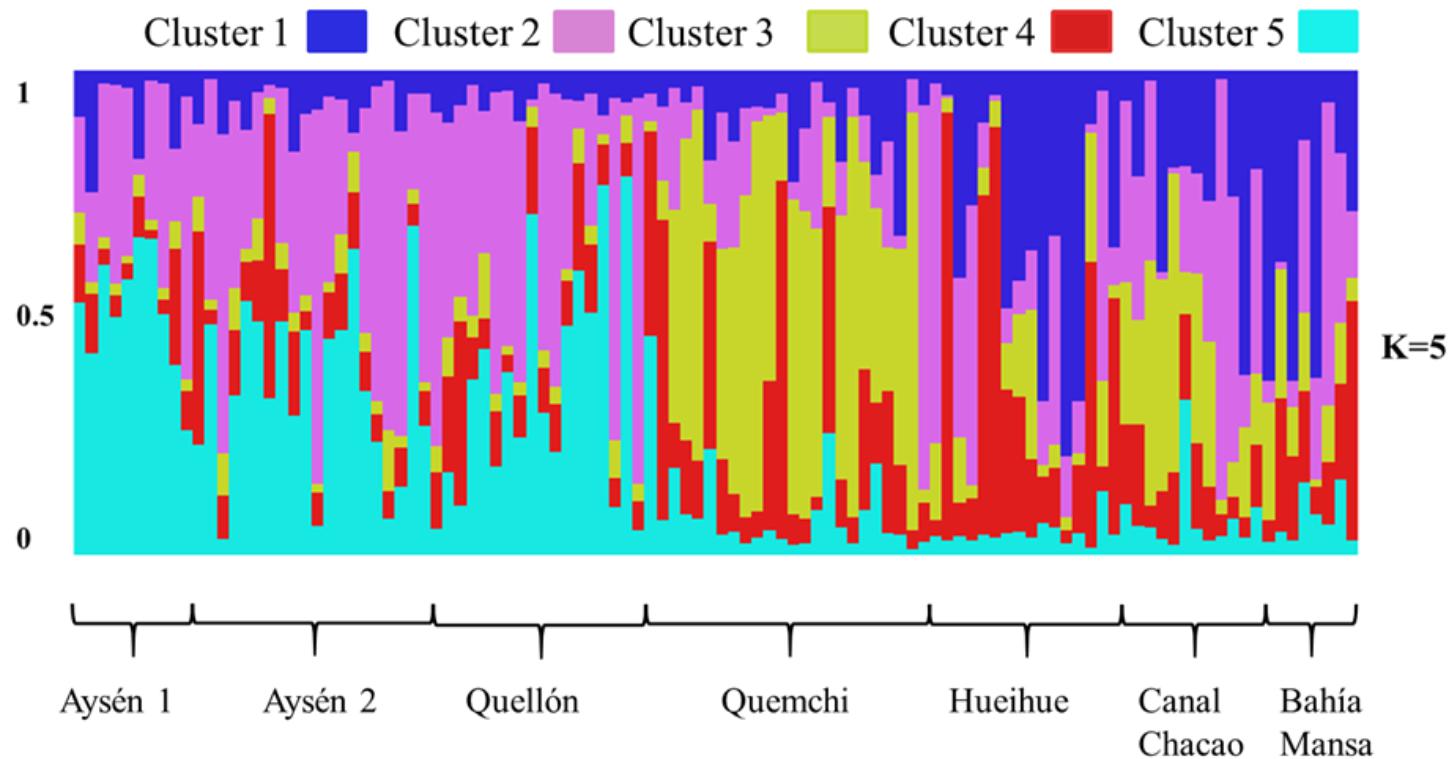
Figure 5. Bayesian cluster analysis. The proportion of genotype membership to K population is illustrated by a bar plot with different colors. Below the graph are the names of the populations from which the clones were isolated.
Isolation by Distance
There was a significant positive relationship between the genetic and geographic distances based on the results of the Mantel test (p < 0.01), with a correlation coefficient of 0.612 and determination of y by x of 0.375.
Disequilibrium of the Multilocus Linkage
The index of multilocus disequilibrium gave significant values (p < 0.05) for all the populations (Table 4). The highest and lowest values were observed in the populations derived from the 2016 bloom, and ranged between 0.021 and 0.007 for Hueihue and Quemchi respectively. Between the populations derived from cysts intermediate values were observed, ranged between 0.010 and 0.015.

Table 4. Multilocus linkage disequilibrium index per population and significance value (p).
Discussion
Phylogeny and Genetic Diversity
The phylogenetic reconstruction based on rDNA LSU sequences indicated that all the clones analyzed from the populations associated with the 2016 bloom, as well as those isolated from the northern limit of distribution, were aggregated in the Group I of the A. tamarense species complex, verifying that the species being studied was A. catenella (Lilly et al., 2007; John et al., 2014; Willem, 2017). The current taxonomic revision of A. tamarense species complex, supported more by ribosomal phylogeny than morphological characters (John et al., 2014), has merged into A. catenella species group some toxic strains previously referred to as A. fundyense or A. tamarense (John et al., 2014; Willem, 2017). As a result, the studies made by John et al. (2004), Nagai et al. (2007), Alpermann et al. (2009, 2010), Erdner et al. (2011), and Richlen et al. (2012) with the toxic strains of A. tamarense and A. fundyense now correspond to A. catenella, with A. tamarense and A. fundyense considered as morphotypes (c.f. John et al., 2014). In turn, the A. tamarense species is the only known non-toxic species among the species comprising the A. tamarense species complex, which segregate in Group 3 (John et al., 2014). Similar to our results, clones isolated from the Patagonian fjords between 1994 and 2016 have been placed in the same Group I (Cordova and Müller, 2002; Aguilera-Belmonte et al., 2011; Varela et al., 2012; Mardones et al., 2016a). Moreover, several studies have consistently found PSP toxins in the clones characterized and in the shellfish after blooms where A. catenella was the trigger (Krock et al., 2007; Molinet et al., 2010; Aguilera-Belmonte et al., 2011; Varela et al., 2012; Buschmann et al., 2016). These facts reduce the possibility that other relevant species comprising the A. tamarense species complex are present in the Patagonian fjords, as has been suggested by other authors (e.g., Jedlicki et al., 2012), and they confirm that the diversity and variation in genetic populations observed in the present study are intraspecific. However, taking account the huge geographical extent of the 2016 bloom and the impossibility of sampling many localities in different times, we cannot discount the presence of the A. ostenfeldii. This species has been identified before in the Patagonian fjords in low densities in the same zones where the 2016 bloom initiated, and on some localities within the northward expansion of the bloom (e.g., Salgado et al., 2015; Pizarro et al., 2018).
In this study, the high genetic diversity observed among A. catenella populations, isolated from both cysts and vegetative cells, are consistent with the high genetic diversity reported in A. catenella populations in the northern hemisphere (Alpermann et al., 2010; Erdner et al., 2011). Mutation is the fundamental mechanism generating genetic variation in both the asexual and sexual phase of all organisms (Rengefors et al., 2017). In Alexandrium species the mutation can be produced during asexual division of vegetative cells in the water column, and also during genetic recombination of distinct cells (i.e., heterothallic mating; Mardones et al., 2016a) that generate cysts that sink to the seafloor (Brandenburg et al., 2018). In the growth season, these cysts hatch continuously and thereby supply new genotypes to the water column, a mechanism by which they contribute to the genetic diversity (Brandenburg et al., 2018). Possibly, the high genetic diversity observed within the A. catenella populations is an attribute that confers to the species the capacity to adapt to the variable and heterogeneous environmental conditions observed across the Patagonian fjords. There, latitudinal gradients in the physical-chemical variables explain the presence of ecoregion, bioclimatic region, and biogeographic patterns (Camus, 2001; Spalding et al., 2007). However, further studies are needed to understand if the genetic diversity of A. catenella is coupled with phenotypic variability, as some authors have observed in the northern hemisphere (e.g., Alpermann et al., 2010; Brandenburg et al., 2018).
The genetic diversity observed between the populations did not indicate a process of recent colonization. Even though the reduction in diversity and segregation are expected results for a species during range expansion (Excoffier et al., 2009; Sexton et al., 2009), in this study the high diversity detected using AFLP does not differ significantly among populations (p < 0.05, GLMM) and it was distributed mainly within them (90.450%, AMOVA). Similarly, in some HAB species that have expanded their distribution, gradients of genetic diversity between established and founder populations have not been found (Tahvanainen et al., 2012; Lebret et al., 2013). Indeed, the genetic diversity of the A. catenella populations off the coasts of Japan, Korea, and the United States of America, estimated from samples taken in different months, years, localities, and life cycle phases, consistently show high values, even when population genetic divergence was found (Nagai et al., 2007; Erdner et al., 2011; Richlen et al., 2012). Although, when genetic differences in diversity and/or variability levels have been observed, they have occurred among populations separated by large geographic distances (Masseret et al., 2009) and when the colonization events have been separated by considerable periods of time (Brandenburg et al., 2018). In these latter cases, the gene flow limitation (Masseret et al., 2009) and the extended time to population diversification (Brandenburg et al., 2018) may have favored the maintenance of the diversity/variability gradient among populations. In a rapidly growing organism with a short generation time, it has been suggested that the huge population sizes could be considered as virtually infinite and genotypic diversity could thus be maintained during the bloom events, even if the organism primarily reproduces clonally (Dia et al., 2014). Thus, the lack of a genetic diversity gradient among A. catenella populations may be related to the potential observed in the genus Alexandrium for maintaining high genetic diversity even in a bloom, and the considerable gene flow among the populations, as is suggesting by the low and moderate pairwise FST values.
Population Structure
The observations that described the progressive expansion of A catenella indicate that the presence of this species in the Aysén region dates from 1992, but it was only in 2002 when an intense bloom that started in this region, reached further north up to Quellon on the south coast of Chiloé Island (Molinet et al., 2003). Considering the local oceanographic currents between the populations of Aysén and those around Quellón the dispersion among populations should be restricted (e.g., Sievers and Silva, 2003). Two strong surface (0–30 m) counter currents meet in the Golfo Corvocado and from here flow out through the Boca Guafo toward the Pacific Ocean (Sievers and Silva, 2003): one current flows south from Chiloé inland sea and, the other, flows north from the Canal Moraleda (Figure 1). Taking into account these currents and that the abundances of vegetative cells are mainly found in the surface waters, down to about 15 m (Molinet et al., 2003), it is thought that the gene flow between populations from both sides of the Boca Guafo should be limited, generating high levels of genetic differentiation. However, the lowest levels of differentiation (FST = 0.020–0.032) and similar patterns of genotype assignment to genetic population (Bayesian aggregation analysis) were observed between these populations, indicating that the current systems are not a barrier to gene flow. Indeed, the meteorological conditions that prevail during spring and summer, with the predominance of southerly winds, could be enough to push the vegetative cells northward, particularly during intense bloom events, such as those that occurred in 1998, 2002, and 2009 (Molinet et al., 2003; Mardones et al., 2010). The descriptions of these events indicated that they started in the central-northern areas of the Aysén region and then expanded to the north (Molinet et al., 2003; Mardones et al., 2010). Thus, successive bloom events could have favored the dispersal of the vegetative cells from Aysén toward the north, increasing gene flow and obscuring the evidence of an initial founder effect between the populations.
AMOVA, PCA, and Bayesian cluster analysis (K3 genetic population) indicated that the segregation of the genetic variance was important at least in two levels: between the populations coming from both the previous northern limit of the distribution and from the bloom of 2016, and also between the populations inside of each of these two groups. The differences between the former two groups of populations explained 6.730% of the variance in the AMOVA or 65.76% of the variance explained by the first axes (PCA), it also accounted for the greatest genetic differences (FST = 0.076–0.122). Within each of the two groups, smaller differences were observed between populations collected during the 2016 bloom (FST = 0.031–0.054) and the smallest differences were observed between the populations derived from cysts (FST = 0.020–0.032). In A. catenella populations significant values of FST have been observed in different geographical contexts: higher values comparing different geographical regions (FST: 0.281–0.308; John et al., 2004); or from moderate to high when comparing isolated subpopulations within the same region (FST: 0.07–0.36; Alpermann et al., 2009). Thus, the differentiation observed both between populations in the north of the distribution and those from the 2016 bloom, and within each of these groups, could be considered as subpopulations rather than clearly divergent populations.
The significant correlation between genetic and geographic distances of the populations (p < 0.05, Mantel Test) seems to support the assertion that differences observed between the populations from the northern limit of the distribution and those from the bloom of 2016 can be attributed, in part, to restrictions of the gene flow due to the limited dispersion of vegetative cells through the marine currents. Similar observations have been made for A. catenella in Japan, where the genetic structuring was correlated with geographic distance (Nagai et al., 2007), and also for A. minutum, where differentiation among samples from different locations in the Mediterranean Sea were attributed to the complex regional geography and hydrodynamics (Casabianca et al., 2012). Along over 300 km of exposed coast over which the great bloom of 2016 occurred (Buschmann et al., 2016), only some of the vegetative cells could have been advected by the unusual southern winds that were recorded throughout the month of March (Buschmann et al., 2016), limiting the dispersion to the other fraction of the population, and as a consequence, generating the observed differentiation.
Additionally, the differentiation and the changes in the genotype proportions assigned to ancestral populations, observed in the Bayesian aggregation analysis (K5 genetic population), between populations derived from cysts and those isolated from the bloom, could be the result of distinct population dynamics at each stage in the life cycle from which the samples came. The life cycle of A. catenella is characterized by alternating between a planktonic population of haploid cells that reproduce asexually and a benthic population of resting cysts that results from the occasional sexual reproduction of planktonic populations (Anderson et al., 2012b). The heterothallic behavior of the gametes (Mardones et al., 2016a), and the implicit recombination during sexual reproduction (Anderson et al., 2012b), along with the capacity of cysts to remain viable in the sediments over long periods of time (years in some cases), means that banks of cysts in the sediments act as reservoirs of genetic diversity (Alpermann et al., 2009; Anderson et al., 2012b). This could explain, in part, the relatively homogeneous assignment of the genotypes within the subpopulations derived from cysts, containing individuals with allelic proportions of the five clusters from detected ancestral subpopulations. This level of genotypic admixture and the dominance of two clusters could be the result of the allelic recombination of ancestral populations, probably of different geographic origins. However, in these subpopulations there is no genetic evidence of successive cohorts of temporally differentiated cysts, as has been observed by Alpermann et al. (2009). Despite the few studies undertaken in this area, low concentrations of cysts are observed in the Patagonian fjords (Mardones et al., 2016a). Furthermore, rapid reductions in cysts density have been observed (Diaz et al., 2014), probably due to the relatively short period of mandatory dominance (Mardones et al., 2016a) and the germination potential over the year (Varela, personal observation). This all indicates that the banks of cysts are a dynamic reservoir, without the accumulation of many cohorts over time.
Within the populations from the 2016 bloom, the heterogeneity of assignment proportions of the genotypes in genetic subpopulations (Bayesian aggregation analysis) and the high levels of the linkage disequilibrium index provided evidence of the incidence of demographic factors on the population genetic divergence. The Bayesian aggregation analysis revealed a relative dominance of individuals with a greater proportion of alleles from one of the ancestral subpopulations, with the insertion of some individuals that could be considered migrants between populations. At one extreme of the differentiation was the population from Quemchi, with a greater number of individuals with a dominant allelic proportion (yellow cluster), than the other populations which appear to be in a state of transition. This apparent process of rapid genetic spatial differentiation over the development of the bloom has also been observed in A. minutum (Dia et al., 2014) and in A. catenella (Erdner et al., 2011); differentiation which could also be temporal (Richlen et al., 2012; Dia et al., 2014). In these cases, it has been argued that these rapid genetic changes may reflect selective effects that are nevertheless not strong enough to reduce the genetic diversity (Dia et al., 2014). On the other hand, if encystment occurs during the bloom process (Brosnahan et al., 2016), and it is induced by adverse environmental conditions, this could magnify the selective effects on the proportions of different genotypes, reducing or eliminating lineages which are less successful under the prevailing conditions (Erdner et al., 2011). Both combined effects, the rate of reproduction and the differential encystment, could potentially reduce gene flow between subpopulations, generating differentiation between populations whilst maintaining the diversity of the regional population (Erdner et al., 2011).
Conclusion
In this study we did not observe the patterns generated by a range expansion process i.e., diversity gradients and/or high inter-population divergence. However, the genetic diversity and structure indicated that these were influenced by the geographic distance effects and the physical restrictions to gene flow, along with the demographic processes that occur during the distinct phases of the life cycle. Thus, the characteristics of asexual and sexual reproduction (e.g., heterothallism), the participation of ancestral populations in the allele proportions of individuals, as well as the selective value of some clonal lines amplified by asexual reproduction or by differential encystment, could be account of the genetic diversity and genetic structure observed in A. catenella populations in southern Chile.
Author Contributions
JP was responsible for the laboratory and statistical analyses, and generation of the manuscript. DV was responsible for the generation of the manuscript. JP, CM, AZ, KC, AV, and BO were responsible for the sampling collection, isolation of strains, and clones maintaining.
Funding
Financial support was provided by the Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT N° 1080548 and 1130954) and Chilean scholar fellowship Doctorado Nacional 2012 from the Comisión Nacional de Investigación Científica y Tecnológica (CONICYT), and also by Proyecto Interno FNI02/16 ULA CR8520.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Special thanks to Dr. Federico Winkler and reviewer for significant comments to manuscript improvement, and to Dr. Matthiew Lee for English review. Moreover, thanks to Ma. Andrea Pérez Ríos from CTI Team (Competencias Transversales en Inglés) and Convenio Marco FID 1758 (Formación de Profesores 2017–2020, Universidad de Los Lagos) for the written English course imparted, which helped to substantially to the manuscript redaction.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2018.00532/full#supplementary-material
References
Agapow, P.-M., and Burt, A. (2001). Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes 1, 101–102. doi: 10.1046/j.1471-8278.2000.00014.x
Aguilera-Belmonte, A., Inostroza, I., Franco, J. M., Riobó, P., and Gómez, P. I. (2011). The growth, toxicity and genetic characterization of seven strains of Alexandrium catenella (Whedon and Kofoid) Balech 1985 (Dinophyceae) isolated during the 2009 summer outbreak in southern Chile. Harmful Algae 12, 105–112. doi: 10.1016/j.hal.2011.09.006
Alpermann, T. J., Beszteri, B., John, U., Tillmann, U., and Cembella, A. D. (2009). Implications of life-history transitions on the population genetic structure of the toxigenic marine dinoflagellate Alexandrium tamarense. Mol. Ecol. 18, 2122–2133. doi: 10.1111/j.1365-294X.2009.04165.x
Alpermann, T. J., Tillmann, U., Beszteri, B., Cembella, A. D., and John, U. (2010). Phenotypic variation and genotypic diversity in a planktonic population of the toxigenic marine Dinoflagellate Alexandrium tamarense (Dinophyceae). J. Phycol. 46, 18–32. doi: 10.1111/j.1529-8817.2009.00767.x
Anderson, D. M., Cembella, A. D., and Hallegraeff, G. M. (2012a). Progress in understanding harmful algal blooms: paradigm shifts and new technologies for research, monitoring, and management. Annu. Rev. Mar. Sci. 4, 143–176. doi: 10.1146/annurev-marine-120308-081121
Anderson, D. M., Alpermann, T. J., Cembella, A. D., Collos, Y., Masseret, E., and Montresor, M. (2012b). The globally distributed genus Alexandrium: multifaceted roles in marine ecosystems and impacts on human health. Harmful Algae 14, 10–35. doi: 10.1016/j.hal.2011.10.012
Anderson, R., and Kawachi, M. (2005). “Traditional Microalgae isolation techniques,” in Algal Culturing Techniques, ed. R. A. Andersen (Cambridge, MA: Academic Press), 83–100. doi: 10.1016/B978-012088426-1/50007-X
Bates, D., Mächler, M., Bolker, B., and Walker, S. (2015). Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48. doi: 10.18637/jss.v067.i01
Brandenburg, K. M., Wohlrab, S., John, U., Kremp, A., Jerney, A., Krock, B., et al. (2018). Intraspecific trait variation and trade-offs within and across populations of a toxic dinoflagellate. Ecol. Lett. 21, 1561–1571. doi: 10.1111/ele.13138
Brosnahan, M. L., Velo-suárez, L., Ralston, D. K., Fox, S. E., Sehein, T. R., Shalapyonok, A., et al. (2016). Rapid growth and concerted sexual transitions by a bloom of the harmful dinoflagellate Alexandrium fundyense (Dinophyceae). Limnol. Oceanogr. 60, 2059–2078. doi: 10.1002/lno.10155
Buschmann, A., Farías, L., Tapia, F., Varela, D., and Vásquez, M. (2016). Informe Final: Comisión Marea Roja (CMR). Alphen aan den Rijn: Wolters Kluwer.
Camus, P. A. (2001). Marine biogeography of continental Chile. Rev. Chil. Hist. Nat. 74, 587–617. doi: 10.4067/S0716-078X2001000300008
Casabianca, S., Penna, A., Pecchioli, E., Jordi, A., Basterretxea, G., and Vernesi, C. (2012). Population genetic structure and connectivity of the harmful dinoflagellate Alexandrium minutum in the Mediterranean Sea. Proc. Biol. Sci. 279, 129–138. doi: 10.1098/rspb.2011.0708
Cordova, J., and Müller, I. (2002). Use of PCR and partial sequencing of the large-subunit rRNA gene to identify Alexandrium catenella (Dinophyceae) from the South of Chile. Harmful Algae 1, 343–350. doi: 10.1016/S1568-9883(02)00066-5
Dia, A., Guillou, L., Mauger, S., Bigeard, E., Marie, D., Valero, M., et al. (2014). Spatiotemporal changes in the genetic diversity of harmful algal blooms caused by the toxic dinoflagellate Alexandrium minutum. Mol. Ecol. 23, 549–560. doi: 10.1111/mec.12617
Diaz, P. A., Molinet, C., Seguel, M., Diaz, M., Labra, G., and Figueroa, R. I. (2014). Coupling planktonic and benthic shifts during a bloom of Alexandrium catenella in southern Chile: implications for bloom dynamics and recurrence. Harmful Algae 40, 9–22. doi: 10.1016/j.hal.2014.10.001
Erdner, D. L., Richlen, M., McCauley, L. A. R., and Anderson, D. M. (2011). Diversity and dynamics of a widespread bloom of the toxic dinoflagellate Alexandrium fundyense. PLoS One 6:22965. doi: 10.1371/journal.pone.0022965
Excoffier, L., Foll, M., and Petit, R. J. (2009). Genetic consequences of range expansions. Annu. Rev. Ecol. Evol. Syst. 40, 481–501. doi: 10.1146/annurev.ecolsys.39.110707.173414
Excoffier, L., Laval, G., and Schneider, S. (2005). Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol. Bioinform. 1, 47–50. doi: 10.1177/117693430500100003
Falush, D., Stephens, M., and Pritchard, J. K. (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567–1587.
Francis, R. M. (2016). POPHELPER: an R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 17, 27–32. doi: 10.1111/1755-0998.12509
Fry, N., Savelkoul, P., and Visca, P. (2009). Amplified fragment length polymorphism analysis. Methods Mol. Biol. 551, 89–104. doi: 10.1007/978-1-60327-999-4_8
Hernández, C., Díaz, P. A., Molinet, C., and Seguel, M. (2016). Exceptional climate anomalies and north- wards expansion of paralytic shellfish poisoning outbreaks in Southern Chile. Harmful Algae News 54, 1–2.
Jedlicki, A., Fernández, G., Astorga, M., Oyarzún, P., Toro, J. E., Navarro, J. M., et al. (2012). Molecular detection and species identification of Alexandrium (Dinophyceae) causing harmful algal blooms along the Chilean coastline. AoB Plants 2012:ls033. doi: 10.1093/aobpla/pls033
John, U., Groben, R., Beszteri, B., and Medlin, L. (2004). Utility of Amplified Fragment Length Polymorphisms (AFLP) to analyse genetic species complex. Protist 155, 169–179. doi: 10.1078/143446104774199574
John, U., Litaker, R. W., Montresor, M., Murray, S., Brosnahan, M. L., and Anderson, D. M. (2014). Formal revision of the Alexandrium tamarense species complex (Dinophyceae) taxonomy: the introduction of five species with emphasis on molecular-based (rDNA) classification. Protist 165, 779–804. doi: 10.1016/j.protis.2014.10.001
John, U., Tillmann, U., Hülskötter, J., Alpermann, T. J., Wohlrab, S., and Van de Waal, D. B. (2015). Intraspecific facilitation by allelochemical mediated grazing protection within a toxigenic dinoflagellate population. Proc. R. Soc. B Biol. Sci. 282, 1–9. doi: 10.1098/rspb.2014.1268
Kamvar, Z. N., Tabima, J. F., and Gr, N. J. (2014). Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and /or sexual reproduction. PeerJ 2:e281. doi: 10.7717/peerj.281
Kremp, A., Oja, J., Letortorec, A. H., Hakanen, P., Tahvanainen, P., Tuimala, J., et al. (2016). Diverse seed banks favour adaptation of microalgal populations to future climate conditions. Environ. Microbiol. 18, 679–691. doi: 10.1111/1462-2920.13070
Krock, B., Seguel, C. G., and Cembella, A. D. (2007). Toxin profile of Alexandrium catenella from the Chilean coast as determined by liquid chromatography with fluorescence detection and liquid chromatography coupled with tandem mass spectrometry. Harmful Algae 6, 734–744. doi: 10.1016/j.hal.2007.02.005
Lebret, K., Kritzberg, E. S., and Rengefors, K. (2013). Population genetic structure of a microalgal species under expansion. PLoS One 8:e82510. doi: 10.1371/journal.pone.0082510
Lilly, E. L., Halanych, K. M., and Anderson, D. M. (2007). Species boundaries and global biogeography of the Alexandrium tamarense complex (Dinophyceae). J. Phycol. 43, 1329–1338. doi: 10.1111/j.1529-8817.2007.00420.x
Mardones, J. I., Bolch, C., Guzmán, L., Paredes, J., Varela, D., and Hallegraeff, G. M. (2016a). Role of resting cysts in Chilean Alexandrium catenella dinoflagellate blooms revisited. Harmful Algae 55, 238–249. doi: 10.1016/j.hal.2016.03.020
Mardones, J. I., Mu, M. N., and Hallegraeff, G. M. (2016b). Toxic dinoflagellate blooms of Alexandrium catenella in Chilean fjords: a resilient winner from climate change. ICES J. Mar. Sci. 74, 988–995. doi: 10.1093/icesjms/fsw164
Mardones, J., Clément, A., and Aparicio, C. (2010). Alexandrium catenella during 2009 in Chilean waters, and recent expansion to coastal ocean. Harmful Algae News 41, 8–9.
Masseret, E., Grzebyk, D., Nagai, S., Genovesi, B., Lasserre, B., Laabir, M., et al. (2009). Unexpected genetic diversity among and within populations of the toxic dinoflagellate Alexandrium catenella as revealed by nuclear microsatellite markers. Appl. Environ. Microbiol. 75, 2037–2045. doi: 10.1128/AEM.01686-08
Mcneill, J., Redhead, S. A., Wiersema, J. H., John, U., Litaker, W., Montresor, M., et al. (2014). (2302) proposal to reject the name Gonyaulax catenella (Alexandrium catenella) (Dinophyceae). Taxon 63, 932–933. doi: 10.12705/634.21
Molinet, C., Lafon, A., Lembeye, G., and Moreno, C. (2003). Spatial and temporal distribution patterns of blooms of Alexandrium catenella (Whedon & Kofoid) Balech 1985, on inland seas of northwest Patagonia, Chile. Rev. Chil. Hist. Nat. 76, 681–698. doi: 10.4067/S0716-078X2003000400011
Molinet, C., Niklitschek, E., and Seguel, M. (2010). Trends of natural accumulation and detoxification of paralytic shellfish poison in two bivalves from the Northwest Patagonian inland sea. Rev. Biol. Mar. Oceanogr. 45, 195–204. doi: 10.4067/S0718-19572010000200001
Murray, S., Uwe, J., and Kremp, A. (2015). “Alexandrium spp.: genetic and ecological factors influencing saxitoxin production and proliferation,” in Climate Change and Marine and Freshwater Toxins, eds L. M. Botana, L. Carmen, and N. Vilariño (Berlin: Walter De Gruyter Incorporated), 125–154.
Nagai, S., Lian, C., Yamaguchi, S., Hamaguchi, M., Matsuyama, Y., Itakura, S., et al. (2007). Microsatellite markers reveal population genetic structure of the toxic dinoflagellate Alexandrium tamarense (Dinophyceae) in Japanese coastal waters. J. Phycol. 43, 43–54. doi: 10.1111/j.1529-8817.2006.00304.x
Nei, M. (1973). Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. U.S.A. 70, 3321–3323. doi: 10.1073/pnas.70.12.3321
Nei, M. (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89, 583–590.
Paredes, J., Andrea, Z., Bianca, O., Camila, M., and Daniel, V. (2016). “Vegetative cells of Alexandrium catenella from the first bloom reported in southern Chile along Pacific Ocean coast: growth attributes and genetic identification,” in Proceedings of the 17th International Conference on Harmful Algae Brazil, (Florianópolis: ICHA).
Peakall, R., and Smouse, P. E. (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28, 2537–2539. doi: 10.1093/bioinformatics/bts460
Penna, A., Garcés, E., Vila, M., Giacobbe, M. G., Fraga, S., Lugliè, A., et al. (2005). Alexandrium catenella (Dinophyceae), a toxic ribotype expanding in the NW Mediterranean sea. Mar. Biol. 148, 13–23. doi: 10.1007/s00227-005-0067-5
Persich, G. R., Kulis, D. M., Lilly, E. L., Anderson, D. M., and Garcia, V. M. T. (2006). Probable origin and toxin profile of Alexandrium tamarense (Lebour) Balech from southern Brazil. Harmful Algae 5, 36–44. doi: 10.1016/j.hal.2005.04.002
Pizarro, G., Paz, B., Alarcón, C., Toro, C., Frangópulos, M., Salgado, P., et al. (2018). Winter distribution of toxic, potentially toxic phytoplankton, and shellfish toxins in fjords and channels of the Aysén region, Chile. Lat. Am. J. Aquat. Res. 46, 120–139. doi: 10.3856/vol46-issue1-fulltext-13
Possada, D., and Crandall, K. (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818. doi: 10.1093/bioinformatics/14.9.817
R Core Team (2017). R: a Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rengefors, K., Kremp, A., Reusch, T. B. H., and Wood, A. M. (2017). Genetic diversity and evolution in eukaryotic phytoplankton: revelations from population genetic studies. J. Plankton Res. 39, 165–179. doi: 10.1093/plankt/fbw098
Richlen, M. L., Erdner, D. L., McCauley, L. A. R., Libera, K., and Anderson, D. M. (2012). Extensive genetic diversity and rapid population differentiation during blooms of Alexandrium fundyense (Dinophyceae) in an isolated salt pond on Cape Cod, MA, USA. Ecol. Evol. 2, 2588–2599. doi: 10.1002/ece3.373
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Salgado, P., Riobó, P., Rodríguez, F., and Bravo, I. (2015). Toxicon differences in the toxin pro fi les of Alexandrium ostenfeldii (Dinophyceae) strains isolated from different geographic origins: evidence of paralytic toxin, spirolide, and gymnodimine. Toxicon 103, 85–98. doi: 10.1016/j.toxicon.2015.06.015
Sassenhagen, I., Sefbom, J., Säll, T., Godhe, A., and Rengefors, K. (2015). Freshwater protists do not go with the flow: population structure in Gonyostomum semen independent of connectivity among lakes. Environ. Microbiol. 17, 5063–5072. doi: 10.1111/1462-2920.12987
Scholin, C. A., Herzog, M., Sogin, M., and Anderson, D. M. (1994). Identification of group-and strain-specific genetic markers for globally distributed Alexandrium (Dinophyceae). II. Sequence analysis of a fragment of the LSU rRNA gene. J. Phycol. 30, 999–1011. doi: 10.1111/j.0022-3646.1994.00999.x
Sexton, J. P., McIntyre, P. J., Angert, A. L., and Rice, K. J. (2009). Evolution and ecology of species range limits. Annu. Rev. Ecol. Evol. Syst. 40, 415–436. doi: 10.1146/annurev.ecolsys.110308.120317
Sievers, H. A., and Silva, N. (2003). “Masas de agua y circulación en los canales y fiordos australes,” in Avances en el Conocimiento Oceanográfico de las Aguas Interiores Chilenas, Puerto Montt a Cabo de Hornos, eds N. Silva and S. Palma (Valparaíso: Comité Oceanográfico Nacional).
Spalding, M. D., Fox, H. E., Allen, G. R., Davidson, N., Ferdaña, Z. A., Finlayson, M. A. X., et al. (2007). Marine ecoregions of the world: a bioregionalization of coastal and shelf areas. Bioscience 57, 573–583. doi: 10.1641/B570707
Swofford, D. (2002). PAUP: Phylogenetic Analysis Using Parsimony. Available at: http://phylosolutions.com/paup-test
Tahvanainen, P., Alpermann, T. J., Figueroa, R. I., John, U., Hakanen, P., Nagai, S., et al. (2012). Patterns of post-glacial genetic differentiation in marginal populations of a marine microalga. PLoS One 7:53602. doi: 10.1371/journal.pone.0053602
Varela, D., Paredes, J., Alves-de-Souza, C., Seguel, M., Sfeir, A., and Frangópulos, M. (2012). Intraregional variation among Alexandrium catenella (Dinophyceae) strains from southern Chile: morphological, toxicological and genetic diversity. Harmful Algae 15, 8–18. doi: 10.1016/j.hal.2011.10.029
Venables, W. N., and Ripley, B. D. (2002). Modern Applied Statistics with S. Berlin: Springer. doi: 10.1007/978-0-387-21706-2
Vos, P., Hogers, R., Bleeker, M., Reijans, M., van de Lee, T., Hornes, M., et al. (1995). AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 23, 4407–4414. doi: 10.1093/nar/23.21.4407
Wang, L., Zhuang, Y., Zhang, H., Lin, X., and Lin, S. (2014). DNA barcoding species in Alexandrium tamarense complex using ITS and proposing designation of five species. Harmful Algae 31, 100–113. doi: 10.1016/j.hal.2013.10.013
Wells, M. L., Trainer, V. L., Smayda, T. J., Karlson, B. S. O., Trick, C. G., Kudela, R. M., et al. (2015). Harmful algal blooms and climate change: learning from the past and present to forecast the future. Harmful Algae 49, 68–93. doi: 10.1016/j.hal.2015.07.009
Keywords: Alexandrium catenella, south Chile, range expansion, genetic population, bloom, Patagonian fjords, Pacific Ocean coast
Citation: Paredes J, Varela D, Martínez C, Zúñiga A, Correa K, Villarroel A and Olivares B (2019) Population Genetic Structure at the Northern Edge of the Distribution of Alexandrium catenella in the Patagonian Fjords and Its Expansion Along the Open Pacific Ocean Coast. Front. Mar. Sci. 5:532. doi: 10.3389/fmars.2018.00532
Received: 01 August 2018; Accepted: 31 December 2018;
Published: 17 January 2019.
Edited by:
Juan Jose Dorantes-Aranda, Institute for Marine and Antarctic Studies (IMAS), AustraliaReviewed by:
Uwe John, Alfred-Wegener-Institut Helmholtz-Zentrum für Polar- und Meeresforschung, Helmholtz-Gemeinschaft Deutscher Forschungszentren (HZ), GermanyShauna Murray, University of Technology Sydney, Australia
Copyright © 2019 Paredes, Varela, Martínez, Zúñiga, Correa, Villarroel and Olivares. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Javier Paredes, amFlbHBhbWVAZ21haWwuY29t; amF2aWVyLnBhcmVkZXNAdWxhZ29zLmNs