 Laura E. Timm
Laura E. Timm Lys M. Isma
Lys M. Isma Matthew W. Johnston
Matthew W. Johnston Heather D. Bracken-Grissom
Heather D. Bracken-Grissom- 1CRUSTOMICS Laboratory, Department of Biological Sciences, Institute of Water and Environment, Florida International University, North Miami, FL, United States
- 2Evolutionary Genomics Laboratory, Department of Biochemistry and Molecular Genetics, University of Colorado-Anschutz Medical Campus, Aurora, CO, United States
- 3Oceanic Ecology Laboratory, Halmos College of Natural Sciences and Oceanography, Nova Southeastern University, Dania Beach, FL, United States
The Gulf of Mexico experiences frequent perturbations, both natural and anthropogenic. To better understand the impacts of these events, we must inventory natural variability within the ecosystem, communities, species, and populations, and contextualize these findings in relation to physical features. Here, we present an integrated study of comparative population genomics and biophysical oceanography. Targeting three species of mesopelagic shrimp common to the Gulf of Mexico midwater (Acanthephyra purpurea, Systellaspis debilis, and Robustosergia robusta), we analyzed genetic diversity and population connectivity as proxies for species health and resilience, respectively. We also simulated a range of vertical migratory behaviors for the shrimp to infer the relationship between diel vertical migration and horizontal transmission between the Gulf of Mexico and the greater Atlantic Ocean. This study aims to establish biological baselines and characterize these values in terms of the prevailing oceanographic feature of the midwater: the Gulf Loop Current. Generally, the oplophorid species (A. purpurea and S. debilis) exhibit lower genetic diversity and higher interpopulation homogeneity compared to the sergestid (R. robusta). Biophysical simulations suggest the differences in vertical migratory regimes between these two groups have important implications for horizontal transport out of the Gulf of Mexico. Because of the difference in vertical migration patterns, access to the Gulf Loop Current varies across taxa and impacts inter-basin migration. Our findings suggest a negative correlation between surface abundance and genetic diversity in these three shrimp species. We hypothesize that this correlation may be due to the relationships between surface abundance and access to the fastest moving waters of the Gulf Loop Current.
Introduction
The Gulf of Mexico experiences frequent environmental perturbations. In the past decade alone, the region has been struck by two major hurricanes: Hurricane Ike in 2008 (Kraus and Lin, 2009) and Hurricane Harvey in 2017 (van Olderborgh et al., 2017). Additionally, three major oil spills have impacted the region: the Deepwater Horizon Oil Spill in 2010 (Beyer et al., 2016), the Shell Brutus Platform Spill in 2016, and an additional pipeline rupture 40 miles south of the Louisiana coastal city of Venice in 2017 (Nelson and Grubesic, 2018). The Gulf of Mexico also hosts a hyper-diverse mesopelagic zone (Sutton et al., 2017) and is described as a unique biogeographic ecoregion, distinct from the Caribbean Sea, Sargasso Sea, and greater Atlantic Ocean (Backus et al., 1977; Gartner, 1988). The frequent perturbations, both natural and anthropogenic, may have a drastic impact on the Gulf mesopelagic given its unique biological community and connections (St. John et al., 2016). Research efforts must focus on diagnosing Gulf health, contextualizing health in relation to the Gulf's relationship to the greater Atlantic, and understanding the role(s) of major oceanographic features on inter-basin population connectivity.
Genetic diversity and genetic connectivity, common metrics targeted in population genomics, provide especially valuable information about enigmatic species, serving as established proxies for species health and resilience, respectively (Hellberg et al., 2002; Hughes and Stachowicz, 2004; Danovaro et al., 2008; Cowen and Sponaugle, 2009). Genetic diversity is measured as the number of alleles present within a population or species. A population's or species' ability to adapt to new or changing environments is closely tied to higher genetic diversity (Hughes and Stachowicz, 2004; Danovaro et al., 2008; Cowen and Sponaugle, 2009). Thus, local adaptation can be crucial to a population's maintained health in the face of environmental disturbances. The movement and distribution of genes within or between systems is described by population connectivity. Population connectivity can be characterized as inter-population gene flow or migration, or the historical demography of populations, such as recent separation or re-mixing of distinct populations and/or changes to population size (Cowen et al., 2007). The ecological implications of these population dynamics are crucial to species resilience: following a localized perturbation event, gene flow between geographically separated populations can provide a functional genetic reservoir outside the disturbed area (Hellberg et al., 2002; Cowen and Sponaugle, 2009).
This study focuses on population genomics and biophysical connectivity of three mesopelagic crustacean species in relation to the Gulf Loop Current (GLC) and associated eddies, the principal mixing features in the Gulf of Mexico. Generally, the GLC enters the Gulf of Mexico through the Yucatan Channel and exits through the Florida Straits, occupying the upper (surface to ~800 m) water column (Oey et al., 2005; Hamilton et al., 2014). The GLC is characterized by relatively warm, fast-moving water with speeds as swift as 1.7 m s−1 (Forristall et al., 1992) in surface waters (e.g., the top 100 m of the water column; Hamilton et al., 2014), decreasing to a maximum speed of 0.4 m s−1 between 100 and 200 m depth, and continuing to slow with depth. Below 1,000 m depth, water movement is generally considered to be independent of the Gulf Loop Current (Oey et al., 2005; Hamilton et al., 2014). Recent work focused on characterizing water masses in the Gulf presents evidence of distinctly structured microbial communities associated with mesoscale features (Johnston et al., 2019). Given that the Loop Current has been associated with lower biomass and abundances of pelagic organisms (Biggs, 1992; Biggs and Muller-Karger, 1994; Zimmerman and Biggs, 1999; Wells et al., 2017), it is not unrealistic to conclude the current has real, biologically significant impacts on the diversity and distribution of pelagic fauna within the Gulf.
Many midwater organisms exhibit diel vertical migratory behavior, occupying deeper water during the day and moving into epipelagic/surface water at night (Loose and Dawidowicz, 1994; Brierley, 2014). This behavior results in substantial, diel increases in surface abundances for a number of “midwater” species. However, differences in migratory behavior (ranging from “non-migratory,” wherein depth-discrete abundances remain unchanged over a solar cycle, to different degrees of migratory) can be described in terms of migratory regimes (Brierley, 2014). Recently, a population genetics/genomics study of three mesopelagic cephalopod species, representing a range of migratory regimes, found evidence for a correlation between surface abundance and inter-basin population dynamics in the Gulf of Mexico and the greater Atlantic Ocean (Timm et al., 2020). The authors posit that this putative relationship between surface abundance and inter-basin population dynamics is due to the division of these regimes into concomitant “tiers” of access to the Gulf Loop Current. Here, we seek to further investigate this trend through the addition of biophysical modeling of migration regimes and the population genomics analysis of three crustacean species.
All three species targeted in this study (Figure 1) vertically migrate to some degree, but exhibit contrasting life histories, specifically in reproductive behavior and generation time. The oplophorids Acanthephyra purpurea (Milne-Edwards, 1881a) and Systellaspis debilis (Milne-Edwards, 1881b) brood their eggs (Herring, 1967, 1974a,b) and can live multiple years (Ramirez Llodra, 2002). The sergestid species, Robustosergia robusta (Smith, 1882) reproduces by releasing fertilized eggs into the water column (Vereshchaka, 2009) and lives ~15 mo (based on studies of co-occurring sergestid species, see Genthe, 1969; Uchida and Baba, 2008). Additionally, surveys have indicated that R. robusta diel vertical migratory behavior differs geographically (Foxton, 1970; Donaldson, 1975; Froglia and Giannini, 1982; Froglia and Gramitto, 2000; Vereshchaka, 2009) and ontologically: larvae migrate into shallower waters than juveniles, which in turn migrate into shallower waters than adults (Flock and Hopkins, 1992). Such ontological shifts in diel vertical migratory behavior have also been found in A. purpurea and S. debilis (Roe, 1984; De Robertis, 2002). These insights into diel vertical migration make Gulf-specific observations of vertical migratory behavior necessary for both oplophorid species (Burdett et al., 2017) and R. robusta (Frank, pers. comm.). Combined with our increased understanding of the complexities of the Gulf Loop Current (Johnston et al., 2019) and the short lifespan of R. robusta, this behavior may have an amplified impact on population dynamics. In short, we expect this study to yield great insight into the interplay between behavior and population dynamics in the Gulf midwater.
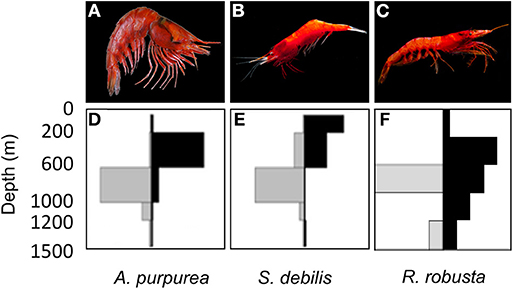
Figure 1. Three species of mesopelagic shrimp targeted in this study, including the oplophorids (A) Acanthephyra purpurea (Milne-Edwards, 1881a) (photo credit: Dr. T.-Y. Chan) and (B) Systellaspis debilis (Milne-Edwards, 1881b) (photo credit: Dr. Danté Fenolio), and the sergestid (C) Robustosergia robusta (Smith, 1882) (photo credit: Dr. Danté Fenolio). Discrete depth abundances are reported for A. purpurea and S. debilis (D,E), respectively (Burdett et al., 2017) and R. robusta (F) (Frank, pers. comm.). Relative abundances, indicated by bar length, are plotted by depth (in meters) and solar cycle (“Day” is represented by gray bars to the left; “Night” is represented by black bars to the right).
The research presented here seeks fine-scale resolution to identify differences in diversity and connectivity (the latter, both genetic and biophysical) in non-model organisms across relatively small geographic distances. To quantify genetic diversity and inter-basin gene flow with the greatest power realistically available, we utilized a powerful next-generation sequencing (NGS) method, double digest Restriction site Associated DNA sequencing (ddRADseq, as described by Peterson et al., 2012). This approach allowed us to query a representative, reproducible fraction of the genome and generate orders of magnitude more data with greater statistical power than traditional population genetics studies have done (Davey and Blaxter, 2010; Peterson et al., 2012; Reitzel et al., 2013; Catchen et al., 2017). Next, we employed a biophysical dispersal model to simulate the migration behaviors and subsequent residency within the Gulf of Mexico of both diel migrators and non-migrators. The model integrated ocean circulation in the upper 1,500 m of the water column from the Hybrid Coordinate Ocean Model (HYCOM) and passive dispersal (exclusive of diel migrations) of particles representing our study species. The goal was to emulate the overall displacement effect of swift surface waters on migrators vs. non-migrators. Integrating the results from these two approaches, genetic and biophysical, allowed us to objectively define migration regimes and test for regime effect on population genomics across species.
Our study represents a comparative, integrative NGS and biophysical modeling investigation into the role of behavior and oceanography on population dynamics in three species of crustacean ubiquitous in the mesopelagic Gulf. This study utilizes a dual approach to infer biological resilience in the Gulf and model the role of the Gulf Loop Current in maintaining this resilience. To accomplish this goal we (1) quantify genetic diversity in each species and compare between the Gulf and Bear Seamount in the northern Atlantic; (2) characterize population connectivity between the Gulf and greater Atlantic from a hybrid population genomics-biophysical modeling perspective; (3) correlate surface abundance with diversity and connectivity; and (4) improve our understanding of crustacean health and resilience in the region, specifically in the context of species- and/or population-specific diel vertical migratory behavior and the major oceanographic feature of the region, the Gulf Loop Current.
Materials and Methods
Adult specimens of A. purpurea, S. debilis, and R. robusta were collected from the northern Gulf of Mexico during the wet (August) and dry (May) seasons of 2015 and 2016 as part of the Gulf of Mexico Research Initiative (GOMRI)-funded Deep Pelagic Nekton Dynamics of the Gulf of Mexico (DEEPEND) project on the R/V Point Sur (Figure 2). During the DEEPEND cruises, every collection site was sampled twice: a day sample (entire water column from 0 to 1,500 m depth, sampled at noon) and a night sample (0–1,500 m depth, sampled at midnight). Gulf samples were collected with a Multiple Opening/Closing Net and Environmental Sensing System (MOC-10) rigged with six 3-mm mesh nets, allowing for collected specimens to be assigned to a depth bin (0–200 m, 200–600 m, 600–1,000 m, 1,000–1,200 m, and 1,200–1,500 m; the sixth net sampled from 0 to 1,500 m). In 2016, samples of A. purpurea and S. debilis were also collected from the Florida Straits aboard the R/V Walton Smith. Maximum sampling depth in the Florida Straits was determined by water depth and trawls ran every few hours. For this cruise, specimens were collected with a 9 m2 Tucker trawl fitted with a cod-end capable of closure at-depth, allowing for discrete depth sampling. All three species were collected from Bear Seamount in the Northern Atlantic in 2014 as part of the Deepwater Systematics project, funded by the NMFS Northeast Fisheries Science Center and conducted on the R/V Pisces. Samples were collected from Bear Seamount with a modified Irish herring trawl.
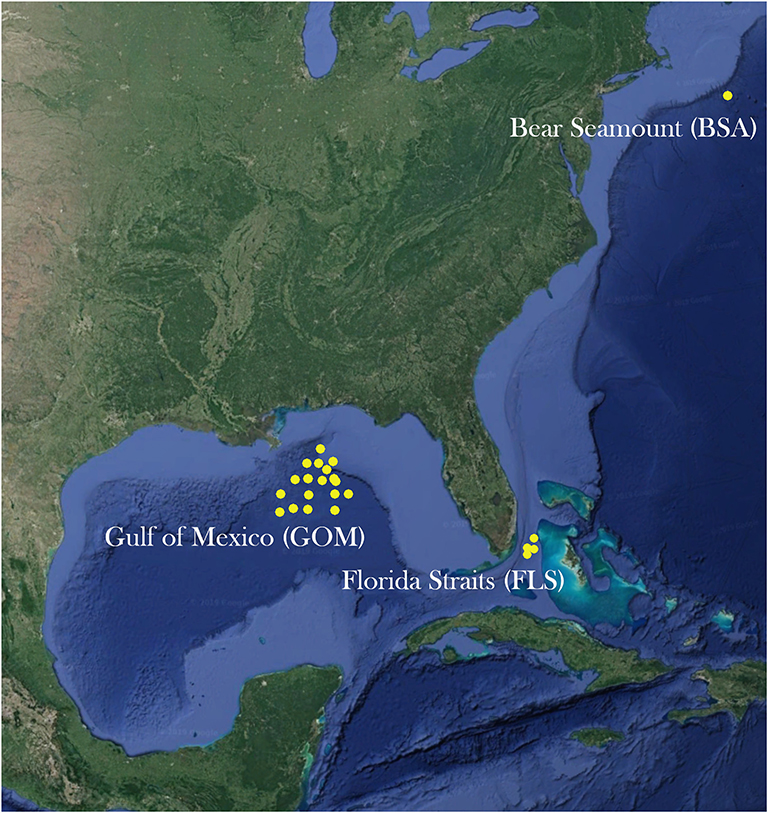
Figure 2. A map of sites sampled from the Gulf of Mexico, the Florida Straits, and Bear Seamount in the Atlantic Ocean.
All samples were identified to species and collected as whole-specimens, either in 70% EtOH or a RNA-stabilizing buffer, and stored at −20°C onboard the vessel before being transferred to a −80°C freezer in the Florida International University Crustacean Collection (FICC). Collected samples were then given a unique voucher ID in the FICC database, including all relevant collection data. Muscle tissue was plucked for each specimen and stored in 70% EtOH or a RNA-stabilizing buffer, in accordance with how the whole-specimen was originally collected, and stored in a −80°C freezer. Voucher specimens were preserved in 70% EtOH and deposited in the FICC. In total, 247 samples of A. purpurea were collected, 218 samples of S. debilis, and 95 samples of R. robusta. For each species, a subset of individuals was selected to provide adequate representation for each sampling locality (Bear Seamount, Florida Straits, and the Gulf of Mexico). These subsets and metadata associated with each specimen are included in this study are detailed in Supplemental Information 1.
DNA Extraction and Sample Barcoding
DNA was extracted with the DNeasy Blood and Tissue Kit (Qiagen), following the protocol provided by the manufacturer. Due to the high quality of DNA necessary for robust ddRADseq data, several quality control measures were taken, many of which are detailed in O'Leary et al. (2018). First, the amount of DNA was ascertained with the Qubit dsDNA High Sensitivity Assay (Thermo Fisher). Next, DNA extractions were visualized on a 2% agarose gel with GelRed (Biotium) run for 90 min at 100 V to ensure the presence of exclusively high molecular weight DNA. Samples with <500 ng DNA and/or a preponderance of degraded DNA were excluded from library preparation.
Finally, every individual eligible for ddRADseq library preparation was barcoded with the 16S ribosomal subunit, 16S (A. purpurea and S. debilis) or cytochrome oxidase subunit I, COI (R. robusta). Because these barcodes were used solely to confirm taxonomic species identification (and not for downstream analyses), genes were selected based on ease of amplification for each species (that is, universal primers were effective). Polymerase Chain Reaction (PCR) occurred in 25-μl volumes: 12.5 μl GoTaq DNA Polymerase (Promega), 1 μl of each primer, 8.5 μl of sterile distilled water, and 2 μl of template DNA. The primer combinations, sequences, and references, as well as annealing temperatures and amplicon length (in base pairs) are presented in Supplemental Information 2. All PCR products were visualized on a 1% agarose gel in the same manner as the DNA extractions.
Amplicons were cleaned and sequenced at the Genewiz sequencing facility in Newark, NJ, USA. Quality filtering of raw reads, contig assembly, ambiguity determination, primer removal, and alignment with MAFFT (Katoh and Standley, 2013) occurred in Geneious v.9.3 (Kearse et al., 2012). The alignment was visually inspected for errors in MEGA7 (Kumar et al., 2016) before determining the reading frame and codon position of COI.
Cleaned, aligned sequences were queried against the NCBI GenBank database using the Basic Local Alignment Search Tool (BLAST) for standard nucleotide. Before querying, we confirmed that all three species were present in the database for the locus we sequenced (16S or COI). A barcode was considered a match when the percent identity of the match was ≥99%. Only individuals whose taxonomic identification was supported by BLAST results were included in ddRADseq library preparation.
Next-Generation Sequencing With ddRADseq
Library Preparation
Double digest RADseq libraries were successfully prepared for 96 individuals of A. purpurea, 96 individuals of S. debilis, and 95 individuals of R. robusta. Reduced representation libraries were prepared according to the double digest RADseq (ddRADseq) method (Peterson et al., 2012). Generally, enzyme trials were completed to determine the appropriate enzyme combinations and size selection windows. DNA was digested with a combination of two enzymes (New England Biolabs) and custom barcoded adapters were synthesized and ligated to the fragments resulting from double digest. Once barcoded, samples could be pooled into sublibraries, which were size selected on a PippinPrep (Sage Science). Specific enzyme combinations, custom barcoded adapter sequences, and size selection schemes are reported in Supplemental Information 3. Size selected fragments were then amplified via PCR with Phusion Hi-Fidelity Polymerase (Thermo Scientific), which also incorporated indices (i7) and Illumina adapters into the fragments and allowed for pooling of sublibraries into the final libraries; 12 sublibraries per library and one library per species. The final libraries were quality checked on an Agilent BioAnalyzer 2100 (Agilent Technologies) before the library was sent for sequencing on an Illumina NextSeq, SE75 high output, at the Georgia Genomics Facility at the University of Georgia.
Quality Filtering and Data Assembly
Raw sequence files were processed with the STACKS v1.45 (Catchen et al., 2013) pipeline on the FIU High Performance Computing Cluster (HPCC). In process_radtags, reads were demultiplexed, cleaned (-c), and quality-filtered (-q). The ustacks program aligned identical reads within each individual, then these consensus reads were cataloged in cstacks. All putative loci were queried against this catalog with sstacks before rxstacks corrected individual genotype calls according to the accumulated population data. Here, “population” is determined by the collection location of each specimen; for example, all specimens collected from the Gulf of Mexico were labeled as members of the “Gulf” population. Finally, the populations program output a file of aligned, putatively unlinked single-nucleotide polymorphisms (SNPs). Two requirements had to be met for a given SNP to be called: first, the minimum read depth (-m = 5) had to be met; second, the SNP needed to be found in 25% of the individuals of a population (-r = 0.25) for the SNP to be called for that population. After SNPs were called according to these parameters, two additional requirements needed to be met for a given SNP to be retained: the SNP had to be present in all populations (Bear Seamount, Florida Straits, and Gulf) and, to increase the likelihood of excluding linked loci, only one random SNP was called per locus (–write_random_snp). These parameter settings were chosen to exclude reads originating from mitochondrial and ribosomal sequences (relative to nuclear sequence, these are generally considered to differ substantially in frequency, thus these are functionally removed with the stack depth parameter) and to prevent the inclusion of paralogs (also controlled with stack depth).
Each file of aligned SNPs then underwent an iterative missing data filter. Loci with >95% missing data were removed, followed by individuals with >95% missing data. This was repeated with 90% missing data, then 85%, and so on. This was repeated until only 10% missing data was allowed by locus and individual or until ~500 loci remained. This “500 SNP” rule was necessary in the case of the oplophorids A. purpurea and S. debilis, as strict filtering resulted in data sets reduced to unusably small sizes. This is likely the result of very large genome sizes: the amount of data returned from the Illumina NextSeq is relatively fixed, therefore larger genomes will yield smaller amounts of consistently reproducible reads across individuals. Finally, we used BayeScan v2.1 (Foll and Gaggiotti, 2008) to identify FST outliers within each filtered data set. Any loci identified as outliers were removed. Sample sizes for each species following quality filtering are reported in Table 1.
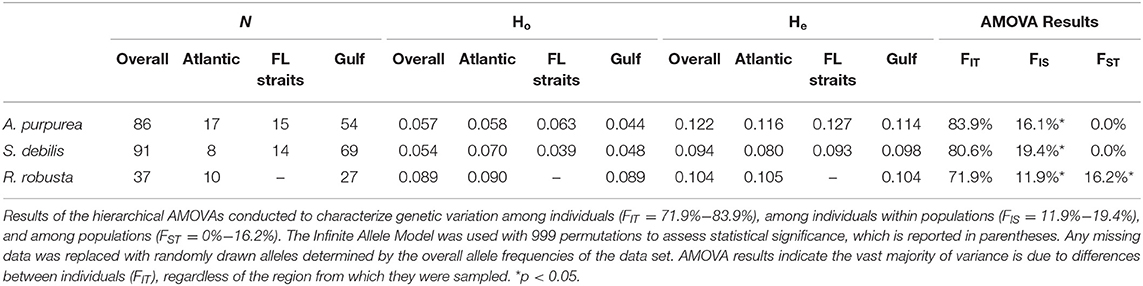
Table 1. Sample sizes and diversity indices, including observed heterozygosity (Ho) and expected heterozygosity (He), for the three targeted species: Acanthephyra purpurea, Systellaspis debilis, and Robustosergia robusta.
Molecular Data Analysis
Several genetic diversity indices were calculated in GENODIVE v2.0b23 (Meirmans and Van Tienderen, 2004), including observed heterozygosity (Ho), the inbreeding coefficient (GIS), and expected heterozygosity (He, which was calculated from the Ho and GIS values). Jackknifing over loci was used to calculate standard deviation.
GENODIVE was also used to measure population differentiation (FST) and calculate hierarchical Analyses of Molecular Variance (AMOVAs, including FIT and FIS) with the Infinite Allele Model. Both analyses were run under 999 permutations to assess significance. For the AMOVAs, missing data were replaced with randomly drawn alleles determined by overall allele frequencies.
We employed the Bayesian program STRUCTURE v2.3.4 (Pritchard et al., 2000) to test for population structure within the data. Seven K-values were tested (K = 1–7) 10 times each under the admixture model. Following a burn-in of 20,000 generations, 200,000 Markov Chain Monte Carlo generations ran. In STRUCTURE HARVESTER v0.6.94 (Earl and VonHoldt, 2012), STRUCTURE results were collated and ad hoc posterior probability models (Pritchard et al., 2000) and the Evanno method (Evanno et al., 2005) were used to infer the optimal K value. STRUCTURE HARVESTER also generated CLUster Matching and Permutation Program (CLUMPP) files for individuals and populations. These files were input into CLUMPP v1.1.2 (Jakobsson and Rosenberg, 2007), resulting in input files compatible with distruct v1.1 (Rosenberg, 2004) and facilitating the visualization of estimated membership coefficients.
Two additional, non-model based methods were also employed for inferring and visualizing population structure: multi-dimensional scaling (MDS) plots and Principle Component Analyses (PCAs) were rendered for each data set using the R packages MASS (Venables and Ripley, 2002) and adegenet (Jombart and Ahmed, 2011), respectively. These methods are very similar, however MDS preserves distance/dissimilarity between data points while PCA preserves covariance within the data.
Biophysical Oceanographic Simulations
To further assess the potential population connectivity between the Gulf of Mexico (GOM) and greater Atlantic for the three target species, R. robusta, A. purpurea, and S. debilis, we ran a suite of simulations representing both migrating and non-migrating deep-sea fauna (hereafter “particles”) using a derivation of a particle-tracking, Lagrangian biophysical model previously used to study the dispersal of marine organisms (Johnston and Bernard, 2017; Riegl et al., 2018). The purpose was to assess if strong surface circulation had an overall effect on the diffusion of diel migrators vs. non-migrators outside of the GOM (i.e., a proxy for connectivity to the greater Atlantic). Please see Supplemental Information 4 for a complete description of the model logic following the Overview, Design concepts, and Design (ODD) protocol as per (Grimm et al., 2006, 2010; Grimm and Railsback, 2005). The following is an abbreviated description of the simulations that were run, including their parameterization.
The primary “model domain” spanned 98–76.5°W longitude and 18–35°N latitude, encompassing the entire GOM and the Eastern Florida Shelf northward to 35°N. Ocean condition data for the simulations were derived from the GOM 1/25° resolution Hybrid Current Ocean Model (HYCOM). HYCOM simulation data are high resolution approximations of water flow that have been used in many previous studies that rely on particle-tracking biophysical models (e.g., Kool et al., 2010; Johnston and Purkis, 2015; Johnston and Bernard, 2017). We used three-dimensional daily snapshot (i.e., at 00:00 UTM) HYCOM data for the upper 1,500 m of the water column for the year 2015 and ran 60-day simulations, commencing on January 1, 2015. The year 2015 was chosen as it was a typical, representative year in the GOM when the Gulf Loop Current (GLC) was in an extended state and 2015 also corresponds to the start of the sampling period by the DEEPEND Consortium which provided the samples for our genetic analysis. It should be noted that during the GLC's extended state is when connectivity outside of the GOM should be at its maximum and connectivity would expectedly be lower when the GLC is in a retracted state.
At the start of each simulation, we released five particles at each of the 46 stations (total n = 230 per simulation) in the DEEPEND sample grid in the northern GOM (Figure 3), a quantity we deemed sufficient to demonstrate the potential for individual retention and/or export out of the GOM. We ran 15 simulations (see Supplemental Information 5 for a summary of all simulations) to represent non-migrating particles (hereafter the “non-migratory simulations”), with releases at 100 m water depth increments, spanning 1,500 to 100 m. These simulations emulated the dispersal of particles that do not migrate vertically and inhabit discrete depths. We next ran a suite of 105 simulations over all possible combinations of diel vertical migration patterns (hereafter the “migratory simulations”) from 1, 500 to 100 m in 100 m increments (i.e., from 1,500 to 1,400 m, from 1,500 to 1,300 m, from 1,400 to 1,300 m, from 1,400 to 1,200 m, and so on) to represent the range of diel migratory behaviors (see Supplemental Information 6 for the animation showing migrators vs. non-migrators).
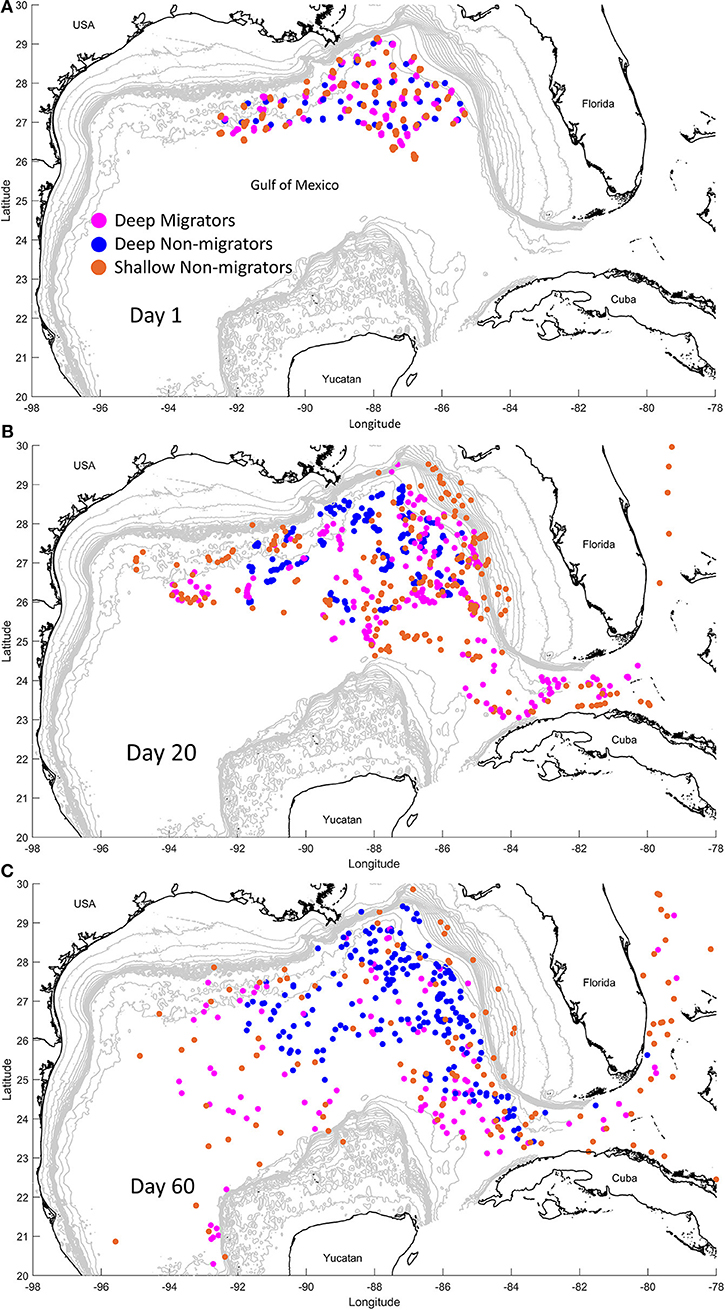
Figure 3. Snapshots of the biophysical modeling simulation at day 1 (A), day 20 (B), and day 60 (C). Particles that exhibit diel vertical migration (“Migrators”) from 900 m to 200 m are in pink. Particles that do not exhibit this behavior, instead residing at 900 m (blue, “Deep Non-migrators”) or 100 m (orange, “Shallow Non-migrators”), are also depicted.
During each simulation, particles were reliant upon water flow for dispersal, with the exception of the inclusion of a small percentage of stochasticity to represent eddy diffusivity and small-scale animal movement (see SI for the specifics). Migratory particles underwent a diel migration from the depths to the surface waters to the depths over a 4-hr span in each direction. Morning migrations downwards began at 5:00 a.m. at the starting depth and ended at 9:00 a.m. at the bottom depth, as specified in Supplemental Information 5. Evening migrations started from the bottom depth at 5:00 p.m. and ended in shallow waters at 9:00 p.m. Particles were tracked for 60 days, during which we corrected their position hourly and recorded their cumulative horizontal displacement distance. For the purposes of determining connectivity outside of the GOM, we considered those particles that were transported east of −80° to be exported from the GOM and into the western Atlantic. Finally, we summed both the total particle movements for each simulation and those movements which occurred outside of the GOM to calculate retention and export percentages. We also averaged the overall cumulative distance traveled of each particle for each simulation to demonstrate the horizontal dispersal distance per scenario. Though we were primarily interested here in the outputs that represented the specific behaviors of R. robusta, A. purpurea, and S. debilis, the suite of simulations we completed may be useful in the future to study the connectivity of other diel migrating and non-migrating deep-sea fauna in the GOM.
Integrating Analyses and Comparing Migration Regimes
Biophysical oceanographic modeling (BPOM) results were used alongside discrete depth abundance data (Burdett et al., 2017; Frank, pers. comm.) to distinguish between migration regimes, based on the depths at which modeled particles were exported from the Gulf, and classify each species as shallow non-migrator, deep non-migrator, strong migrator, or weak migrator. Based on the depths at which modeled particles were exported from the Gulf, each species was classified as a shallow non-migrator, deep non-migrator, strong vertical migrator, or weak vertical migrator. Once these evidence-based regimes were identified, data from species targeted in this study, as well as those targeted in Timm et al. (2020), were classified and binned by migration regime. To test for general correlation between surface abundance and genetic diversity indices, we began by defining “surface abundance” as the percent of total day abundance found above 600 m at night, as determined by MOC-10 net abundances (Figure 1). This cutoff was informed by the BPOM results: in migrators, particle export from the GOM ceased below 500 m; in non-migrators, export ceased at 500 m (Table 2). Because we did not have a net that discretely sampled above and below 500 m, we instead used 600 m as the cutoff. We plotted each diversity index (observed and expected heterozygosity and the inbreeding coefficient) against surface abundance for each species. Data from Timm et al. (2020) was also included to increase sample size. A trendline was fit to each index and R2 was used to determine goodness-of-fit. To statistically test for correlation, we calculated Kendall's τ and Spearman's rank. We did not calculate Pearson's index because the data was not normally distributed.
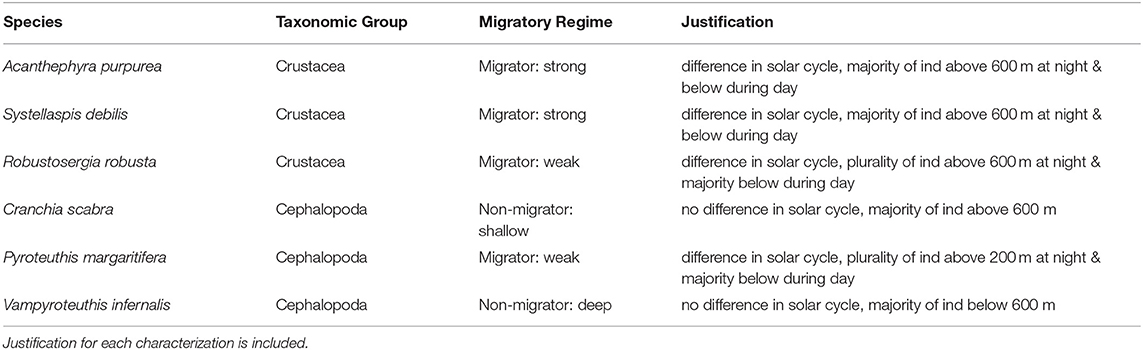
Table 2. Characterization of each species by migratory regime based on biophysical oceanographic modeling (BOM) (export ceases for migrators below 600 m and non-migrators below 500 m) and recorded diel vertical migratory behavior (difference in depth-discrete abundance by solar cycle and proportion of individuals above or below the BOM export depths).
Results
Of the 288 prepared libraries (96 individuals within each species), 279 could be aligned and assembled within STACKS (95 of A. purpurea, 95 of S. debilis, and 89 of R. robusta). The initial data sets included: 596 SNPs (A. purpurea), 652 SNPs (S. debilis), and 4,196 SNPs (R. robusta). After applying the missing data filter, the A. purpurea data set included 522 SNPs across 87 individuals, the S. debilis data set included 525 SNPs across 91 individuals, and the R. robusta data set included 1,066 SNPs across 37 individuals. Across all data sets, only the R. robusta set was found to contain FST outliers: three SNPs were identified by BAYESCAN and removed from the final data set. This information is summarized in Supplemental Information 1 and demultiplexed fastq reads have been uploaded and are publicly available through the Gulf of Mexico Research Initiative's Information & Data Cooperative (Timm et al., 2019), as well as on NCBI's SRA database under BioProject PRJNA553831. The SNP counts for each species in this data set are relatively low for a ddRADseq study, where tens of thousands of SNPs might be genotyped. We attribute the low counts to two primary causes: first, no genomes have been annotated, assembled, or even sequenced for any of the targeted species, lowering confidence in SNP calls; second, the oplophorid species, for which SNP counts were very low, are hypothesized to have large genome sizes (the only oplophorid with a genome size estimate, Hymenodora sp., has a C-val of 38.00; Dixon et al., 2001). DNA barcoding efforts confirmed taxonomic identification of 90 specimens of A. purpurea (90 de novo sequences of 16S: GenBank Accessions MN507733-MN507822) and 80 specimens of S. debilis (80 de novo sequences of 16S: GenBank Accessions MN507553-MN507632). Sanger sequencing of the COI gene in R. robusta generated 57 de novo sequences (GenBank Accessions MN510870-MN510926). However, due to a lack of archived COI sequences for R. robusta in GenBank, BLAST results identified five individuals as Robustosergia regalis, none of which were included in downstream analyses.
Population Genomics
Genetic Diversity
Values across species were very similar (Ho: 0.057–0.089; He: 0.094–0.122) with exception of the inbreeding coefficient which was highest in A. purpurea (0.534), slightly lower in S. debilis (0.425), and lowest in R. robusta (0.146). As the inbreeding coefficient reflects the relationship between Ho and He ([He-Ho]/He), it ranges from −1 to 1, with positive values indicating inbreeding or a recent decrease in population size. These results are reported in Table 1.
Observed heterozygosity is the actual, measured amount of heterozygosity found in a population and can be impacted by an excess of homozygosity. Expected heterozygosity, however, describes the theoretical amount of heterozygosity present assuming the population of interest is in Hardy-Weinberg Equilibrium. It considers the number of alleles as well as their abundance, regardless of homozygosity. These two metrics, observed and expected heterozygosity, are compared using the inbreeding coefficient, as described in the Materials and Methods section. In all species and basins studied here, expected heterozygosity was found to be higher than observed heterozygosity, with the largest difference in A. purpurea, followed by S. debilis, then R. robusta. Generally, inbreeding coefficients approaching 1 indicate decreases in population size or local purifying selection, suggesting that the oplophorids have experienced population decreases or uneven selection pressures that R. robusta has not faced.
When genetic diversity was compared by basin (Gulf of Mexico [GOM] vs. Bear Seamount in the greater Atlantic [BSA]), both A. purpurea and R. robusta exhibited slightly higher diversity in the greater Atlantic (A. purpurea BSA HE = 0.116 > GOM HE = 0.114; R. robusta BSA HE = 0.105 > GOM HE = 0.104), while S. debilis had higher diversity in the Gulf (HE BSA = 0.080 < HE GOM = 0.098). In R. robusta, the inbreeding coefficient was found to be slightly lower in the Gulf than the greater Atlantic (BSA GIS = 0.148 > GOM GIS = 0.143). The oplophorids had significantly higher GIS in the Gulf compared to the greater Atlantic (A. purpurea BSA GIS = 0.500 < GOM GIS = 0.614; S. debilis BSA GIS = 0.126; GOM GIS = 0.510). This is illustrated in Figure 4.
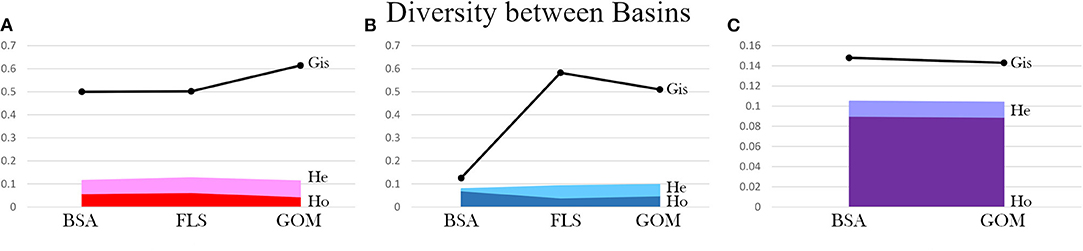
Figure 4. Diversity metrics (observed heterozygosity Ho, expected heterozygosity He, and inbreeding coefficient Gis) are compared between collection localities: Bear Seamount in the Atlantic (BSA), Florida Straits (FLS), and the Gulf of Mexico (GOM) for (A) Acanthephyra purpurea, (B) Systellaspis debilis, and (C) Robustosergia robusta (note the change in y-axis scale).
Population Differentiation and Structure
AMOVA results, reported in Table 1, indicate a lack of population differentiation between basins in the oplophorids: FIT ranged from 80.6% in S. debilis to 83.9% in A. purpurea and the rest of molecular variance was accounted for by FIS (19.4% in S. debilis and 16.1% in A. purpurea). The majority of variance in R. robusta was from FIT (71.9%), however the remainder was comprised of FIS (11.9%) and FST (16.2%), indicating statistically significant genetic differentiation between the Gulf and the Atlantic.
STRUCTURE results strongly support and aptly illustrate the AMOVA results for each species (Figure 5). For the oplophorids, optimal K was determined to be 2; for R. robusta, K = 3 was deemed optimal. In the oplophorids, the admixture of ancestral populations within each individual is nearly identical across BSA, the Florida Straits, and the GOM, while there is some variation within each sampling locality. R. robusta, however, exhibits a dramatic difference in admixture proportion between the GOM and BSA. While admixture from all three ancestral populations is present in every individual, the individuals from the Atlantic consist of nearly equal admixture from populations 1 and 2, with the majority from population 3, while individuals from the Gulf have a very small proportion of admixture from population 3, nearly identical proportions of admixture from population 1 as seen in BSA, and the vast majority of admixture from population 2.
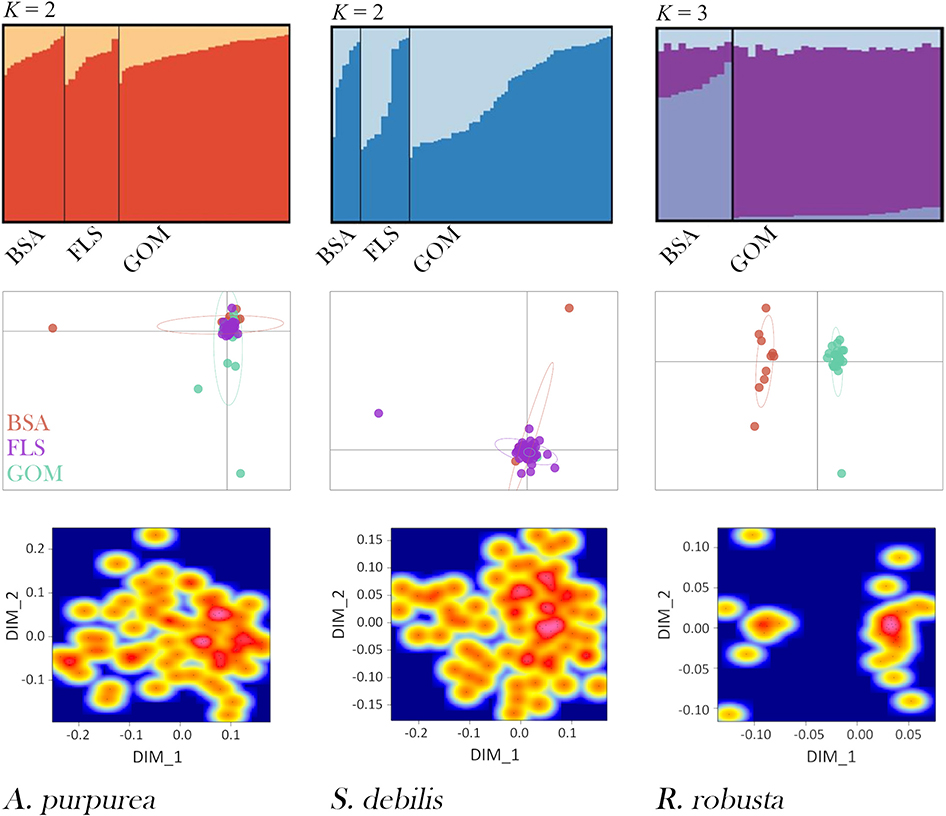
Figure 5. DISTRUCT plots (top), Principal Component Analyses (PCAs; middle), and multidimensional scaling (MDS) heat maps (bottom) for Acanthephyra purpurea, Systellaspis debilis, and Robustosergia robusta. Collection localities are denoted “BSA” for Bear Seamount in the north Atlantic, “FLS” for the Florida Straits, and “GOM” for the Gulf of Mexico., The first two principal components shown for each species are as follows: A. purpurea PC1 = 3.5%, PC2 = 3.1%, S. debilis PC1 = 2.7%, PC2 = 2.5%, and R. robusta PC1 = 5.9%, PC2 = 3.6%.
The PCAs and MDSs present these results another way: both oplophorid species have all individuals fall into a single cluster, regardless of collection locality. Conversely, the population differentiation seen in the AMOVA results for R. robusta, as well as the STRUCTURE analysis, is made further evident in the PCA and MDS: both plots show two distinct clusters, one containing individuals from Bear Seamount in the northern Atlantic and the other containing Gulf specimens. Results from PCA and MDS are depicted in Figure 5.
Biophysical Oceanographic Simulations
In the non-migratory simulations, dispersal out of the GOM (and inferred external connectivity to the greater Atlantic) primarily occurred in particles that were resident in water depths of 600 m or shallower (Table 2 and Figure 3). The percentage range of particle movements outside of the GOM was a minimum of 0.14% for those residing at 600 m to a maximum of 15.72% for those found at 100 m water depth. Average horizontal dispersal distance for the non-migrating particles ranged from 422.03 km (1,500 m residents) to 2,558.25 km (100 m residents), demonstrating that those residing at shallower depths were dispersed much greater distances than those inhabiting the deep.
For the migratory simulations, dispersal out of the GOM was associated with those migrations that occurred from the deepest depths (e.g., 1,500–1,000 m) to a minimum of 500 m water depth, with increases in both the percentage of export and horizontal dispersal distance in depths shallower than 500 m. When migrating from midwater depths (e.g., 900–200 m), increases in the percentage of export and horizontal distance were again seen with shallower migrations, however almost all midwater simulations showed some level of export from the GOM. The maximum export percentage measured was 14.94% and the maximum horizontal displacement was 3,824.88 km, both in particles that migrated from 200 to 100 m water depth on a diel cycle.
Integrating Analyses and Comparing Migration Regimes
BPOM identified minimum depths for export out of the Gulf of Mexico for both migrators (500 m) and non-migrators (600 m). These values, along with discrete depth abundances calculated from MOC-10 capture, were used to characterize each of the six species (Table 2): the three species of mesopelagic shrimp and three species of mesopelagic cephalopod included from Timm et al. (2020). Generally, a negative correlation between surface abundance and genetic diversity was statistically supported (Figure 6). Across analyses, correlation was strongest between surface abundance and observed heterozygosity (R2 = 0.868, rs = −0.942, τ statistically significant; Table 3). Correlation between surface abundance and expected heterozygosity was weaker (R2 = 0.494, rs = −0.543, τ not statistically significant; Table 3). Inbreeding coefficient was not found to be correlated to surface abundance (R2 = 0.073, rs = −0.543, τ not statistically significant; Table 3).
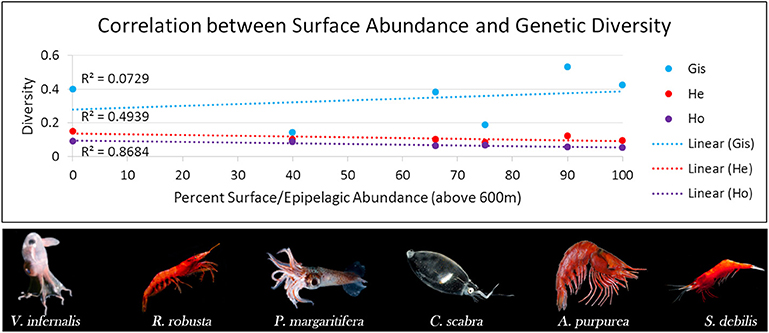
Figure 6. Graph relating genetic diversity (inbreeding coefficient [GIS] in blue, expected heterozygosity [He] in red, and observed heterozygosity [Ho] in purple) to abundance in the surface/epipelagic (here, we define this as above 600 m) across midwater invertebrate species with differing diel vertical migratory behaviors. We find an indirect relationship, with diversity decreasing as the percent of individuals found in the surface/epipelagic increases. This correlation is strongest in Ho (R2 = 0.87) compared to He (R2 = 0.49) and GIS (R2 = 0.073). Robustosergia robusta, Pyroteuthis margaritifera, Cranchia scabra, and Systellaspis debilis photo credit: Dr. Danté Fenolio. Vampyroteuthis infernalis photo credit: David Wrobel. Acanthephyra purpurea photo credit: Dr. T.-Y. Chan.
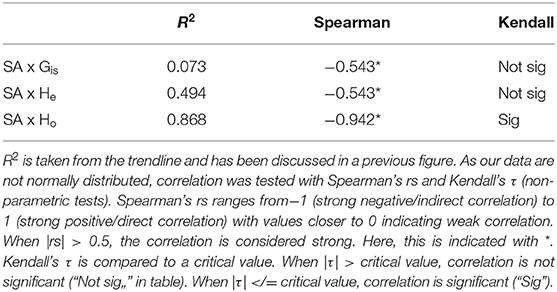
Table 3. Results of testing for correlation between surface/epipelagic abundance (“SA,” here defined as above 600 m) and three diversity metrics: inbreeding coefficient (GIS), expected heterozygosity (He), and observed heterozygosity (Ho).
Discussion
Through the integrated analysis of genomic proxies, namely diversity and connectivity, and biophysical models, we are beginning to address a persistent data gap in the mesopelagic Gulf by establishing biological baselines. We investigated how genetic diversity is organized between the Gulf of Mexico and the greater Atlantic, including the Florida Straits. Between basins, expected and observed heterozygosity paralleled each other well in each species, with the exception of S. debilis in the north Atlantic, wherein the two were nearly equal, greatly decreasing the inbreeding coefficient. In the oplophorids, inbreeding was lower in samples collected from Bear Seamount in the greater Atlantic compared to the Gulf, with the Florida Straits being nearly equal to Bear Seamount (in the case of A. purpurea) or significantly higher than the Gulf (in the case of S. debilis). This may be indicative of Gulf-localized perturbation or purifying selection affecting the oplophorids. However, the low inbreeding coefficient, high diversity, and small inter-basin diversity differences seen in R. robusta suggest quite different population dynamics.
To better understand the processes maintaining these contrasting dynamics, we investigated how this inter-basin organization is maintained through population structure and genetic connectivity and also modeled physical connectivity. Here again, we found a notable difference between the oplophorids and R. robusta. The oplophorids exhibited high population connectivity, indicating historical and current gene flow. Results of population structure analyses indicate each oplophorid species consists of a single population spanning the Gulf, Florida Straits, and the north Atlantic. Individuals from these populations are comprised of admixture from two ancestral populations of each species. R. robusta, however, exhibits significant population differentiation between basins. Analyses of population structure indicate this is coupled with different patterns of admixture from three ancestral populations, forming two distinct genetic signatures. Both of these results were echoed in our biophysical model results: the strong migrators (i.e., the oplophorids) were flushed from the Gulf while the weak migrators (i.e., R. robusta) were retained in the Gulf over the simulation timeframe (Table 2 and Figure 3).
High connectivity and little population structure in oplophorids, evidenced by high FIT, low FST, and results of structure analyses, may constrain genetic diversity through purifying selection: in both species, a single population must contend with two very different basins and environments (Backus et al., 1977; Gartner, 1988; Sutton et al., 2017). Any potential local or basin-specific adaptations must also be fit for the other basin. Additionally, in the case of S. debilis, it seems the entire inter-basin population is impacted by local perturbations: a localized die-off in the Gulf of Mexico can be seen in the overall population (Gulf and northwestern Atlantic, see Table 1 FIS results). R. robusta, however, exhibits the highest diversity and lowest inbreeding of species included in this study. This may be attributable to a larger number of ancestral populations (three, instead of two in the oplophorids) or potentially local adaptation to the Gulf of Mexico and the Atlantic Ocean, relatively independently. Random genetic drift within each basin may also explain the results we see. Relatively high, statistically significant FST, indicating population differentiation between basins, could suggest local adaptation following the recent separation and isolation of two distinct subspecies. However, more work is needed to fully address this, specifically a comprehensive phylogeny of sergestids.
Previous work investigating genetic connectivity between the Gulf of Mexico and the greater Atlantic has largely focused on shark species with high potential dispersal distances. Research into population connectivity in Atlantic sharpnose sharks and sandbar sharks (conducted with mitochondrial RFLP and allozymes) found a single continuous population in both cases (Heist et al., 1995, 1996; Heist and Gold, 1999). A study of blacktip sharks (incorporating microsatellite data as well as the mitochondrial control region) identified population structure between the basins and largely attributed this to female shark preference for their own natal nursery grounds for parturition (Keeney et al., 2005). A study of genetic connectivity of the coral Montastraea cavernosa collected from across the Atlantic identified three populations; one of which (the Caribbean-North Atlantic) spans Bear Seamount and the Gulf of Mexico (Nunes et al., 2009). The authors attribute this connectivity to larval dispersal across long distances, while acknowledging the difficulties of modeling dispersal purely in terms of current-mediated transport. They cite larval lifespan, predation, micro-environmental fluctuations, and active swimming behavior as complicating variables in modeling larval dispersal via currents; all of which may also apply to the shrimp species targeted in this study.
Across the analyses presented here, results exhibited fairly clear distinctions between two taxonomic groups that represent distant evolutionary histories: the oplophorids A. purpurea and S. debilis, and the sergestid R. robusta. These two groups differ in many ways, including reproductive behavior and strength of diel vertical migration. Brooding behavior, exhibited by the oplophorids, may contribute greatly to connectivity between basins by facilitating inter-basin migration: while fecundity may differ by reproductive strategy (Ramirez Llodra, 2002), brooded young tend to have a better chance of survivorship (MacIntosh et al., 2014). Moreover, a survey of R. robusta, which releases fertilized eggs without brooding, from 1992 describes an ontological shift in diel vertical migration strength, with juvenile shrimp exhibiting stronger migration behavior than adults (Flock and Hopkins, 1992). As such, though larvae of R. robusta may have better access to the fastest moving waters of the Gulf Loop Current, they may also be less likely to survive and contribute to the effective population. The authors have noted this anecdotally: on research cruises to the Florida Straits, adults of A. purpurea, S. debilis, and sergestids known to exhibit strong diel vertical migration (Flock and Hopkins, 1992) were quite abundant, but adults of R. robusta were functionally absent and non-migrating sergestid larvae were neither collected nor noted. However, as mentioned, this requires confirmation. Statistical analysis of size distributions along the depth gradient is needed to clarify the role of larvae as migrants connecting the Gulf and Atlantic. While larvae can be critical for population connectivity in marine species (Palumbi, 2003; Gaines et al., 2007; Cowen and Sponaugle, 2009), there is also strong evidence that potential dispersal is rarely correlated with realized dispersal (Shanks, 2009).
Despite the potentially confounding variables identified in determining dispersal through current-mediated transport (e.g., disparity between potential and realized dispersal, oversimplifying active swimming behaviors, and ignoring the importance of rare individuals dispersing long distances; see Shanks, 2009 for a more thorough discussion), biophysical modeling can be used in concert with genetic evidence to improve our understanding of the dynamic relationships between marine organisms and their environment (Liggins et al., 2013). This integrative approach has been used to differentiate between broad-ranged natural populations and exotic introduced populations in the globally-distributed moon jellyfish genus, Aurelia (Dawson et al., 2005). Combining thorough empirical genetic sampling with biophysical modeling of dispersal has also proved valuable in explaining population structure in the highly dispersive spiny lobster, Panulirus argus (Truelove et al., 2017).
Our study particularly focused on diel vertical migration of adults, resultant surface/epipelagic abundance and transport on swift surface currents, and population dynamics. Including data from Timm et al. (2020), we find a trend of high surface abundance associated with low (if not absent) population differentiation between basins. However, this relationship appears to be binary. More stringent, statistical testing, across as many species as possible is needed to properly investigate this putative relationship. Genetic diversity shows much higher variability, allowing for statistical testing of correlation. Generally, an indirect/negative correlation was found, with higher surface abundance associated with lower genetic diversity. This relationship was clearest in observed heterozygosity, though still present in expected heterozygosity. It was nearly absent in the inbreeding coefficient. In the context of our simulation results, we suspect species with higher surface abundance have better access to the Gulf Loop Current, promoting inter-basin migration and homogenizing the population.
Testing for an effect of migration regime, informed by discrete depth abundance observations combined with oceanographic modeling, provides compelling evidence that vertical migration behavior alone is not sufficient to explain differences in genetic diversity across these species. Generally, modeling indicated an increase in export from the Gulf of Mexico into the greater Atlantic and an increase in dispersal distance as simulated particles reached shallower depths. Indeed, we find that minimum depth reached by each species during a diel cycle may be particularly indicative of access to the Gulf Loop Current and ability to migrate between basins.
In many ways, this study only begins to uncover the mechanisms driving and maintaining natural variability in the mesopelagic species inhabiting the Gulf of Mexico and between the Gulf and the greater Atlantic. The establishment of baselines for genetic diversity and connectivity is crucial to understanding the Gulf and for future appraisal of damages following disturbance events. We hypothesize that specific differences in population dynamics may be explained by access to the Gulf Loop Current: populations with higher abundance in the surface or epipelagic potentially have greater access to the fastest moving waters of the Gulf Loop Current. It can be logically reasoned that this access could maintain a single population spanning the Gulf and Atlantic in the strong vertical migrators, homogenizing if not functionally preventing local adaptation and population differentiation.
The results presented here, contextualized in terms of environment (the Gulf Loop Current) and life history (reproductive strategy and diel vertical migratory behavior), serve as the first glimpse of the natural variability present in the Gulf midwater and begin to describe potential drivers of this variability. First, we find that genetically, the oplophorids included in this study, A. purpurea and S. debilis, each form a single population spanning the eastern Gulf of Mexico and the northwest Atlantic. While this is associated with lower diversity, suggesting a lack of natural variability within each population and raising some concern over these species' health, it also indicates unimpeded gene flow between basins, a result also indicated in our model simulations. This is a good prognosis for genetic rescue potential and resilience in the Gulf. Robustosergia robusta, however, shows an opposite trend: high diversity, indicative of natural variability and species health, and genetic population differentiation between basins with low physical connectivity suggests lower potential for genetic rescue—a strategy for replenishing lost genetic diversity following a localized environmental perturbation (Mussmann et al., 2017). The unique genetic signatures of each basin may mean that, despite gene flow between basins, diversity lost within one basin may not be easily replaced through inter-basin migration.
Future Directions
In light of the immense difficulties associated with deep-sea specimen collection (especially of deep, non-migrating species), we recognize that continued collection efforts are needed to increase sample sizes. Additionally, before attempts to model surface abundance-genetic diversity correlation are undertaken, the correlation should be tested in more species. As fishes represent a major proportion of the mesopelagic biomass and are generally better studied, a similar study to the one presented here, focused on fish species, could substantially improve our understanding of the state and flux of genetic diversity in the mesopelagic Gulf of Mexico. When model testing begins, pervasive depth-dependent environmental variables (i.e., salinity, temperature, hydrostatic pressure, dissolved oxygen concentration, and chlorophyll concentration) should be considered as well as physical oceanographic parameters, such as water velocity and direction in relation to the Florida Straits, and biological traits such as active retention within the GOM via directional swimming during diel vertical migration.
Data Availability Statement
All data are publicly available through the Gulf of Mexico Research Initiative Information & Data Cooperative (GRIIDC) at https://data.gulfresearchinitiative.org (doi: 10.7266/n7-3p3y-g470) and from NCBI (BioProject PRJNA553831).
Author Contributions
LT and HB-G contributed conception and design of the study. Population genomics data generation and analysis was conducted by LT and LI. MJ carried out the biophysical modeling component of the study. MJ and LT worked together to integrate the results from population genomics and biophysical modeling. LT wrote the manuscript, with the exception of the biophysical modeling sections, which were written by MJ. All authors contributed to the revision process, have read, and approve this manuscript.
Funding
This research was made possible in part by a grant from The Gulf of Mexico Research Initiative to the Deep Pelagic Nekton Dynamics of the Gulf of Mexico (DEEPEND) Consortium, as well as a Division of Environmental Biology National Science Foundation (NSF) grant awarded to HB-G (DEB#1556059). Samples from Bear Seamount were collected through the Deepwater Systematics project, funded by the NMFS Northeast Fisheries Science Center. Funding was also provided by the Florida International University (FIU) Presidential Fellowship, the FIU Doctoral Evidence Acquisition Fellowship, and the FIU Dissertation Year Fellowship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AL declared a past co-authorship with one of the authors MJ to the handling editor.
Acknowledgments
We would like to specially thank Dr. Tammy Frank and Dr. Rosanna Milligan for their support in sample collection and statistical analysis, as well as Mr. Jorge Perez-Moreno and Mr. Charles Golightly for support in initial sample processing. We also thank LT's Ph.D. committee: Dr. José Eirin-Lopez, Dr. Mauricio Rodriguez-Lanetty, Dr. Eric von Wettberg, and Dr. Wensong Wu. We especially thank Dr. Emily Warschefsky, Dr. Joseph Ahrens, and Mr. Jordon Rahaman for advice and feedback on ddRADseq library prep and data assembly. Finally, we thank the reviewers for feedback on earlier versions of this manuscript. The work presented here is part of LT's doctoral dissertation (Timm, 2018) and is publicly available online. This is contribution #174 from the Coastlines and Oceans Division of the Institute of Environment at Florida International University.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.00019/full#supplementary-material
Supplemental Information 1. Metadata for all samples included in this study, including: the Illumina i7 index and custom barcode combination, listed under “Idx-BC,” sample ID (HBG number), species, date and basin of collection, as well as the Station ID and coordinates for the collection site, and the depth range from which the sample was collected. The gene targeted for Sanger sequencing, to be used for DNA barcoding to confirm taxonomic identification, was either the 16S small ribosomal subunit (16S) or cytochrome c oxidase subunit I (COI). This is reported under “Gene” and the associated GenBank Accession number is also listed.
Supplemental Information 2. The primer pairs and annealing temperatures associated with PCR amplification of two mitochondrial genes targeted for DNA barcoding of samples included in the ddRADseq library preparations.
Supplemental Information 3. Details of ddRADseq protocol for each species, including enzymes, custom-made barcoded-adapter sequences, and size selection schemes. Both strands of each adapter are given (1.1 and 1.2) in the 5′ to 3′ direction. These strands are annealed prior to ligation to the ddRADseq fragments. The barcode section of the adapter is underlined. Note that the overhang in the 1.1 strands differs between the “oplo” and the “flex” adapters. Illumina i7 adapters were also used, specifically index 1, 3, 7, 12, 13, 16, 21, 24, 29, 37, 42, and 43.
Supplemental Information 4. This model description follows the Overview, Design concepts, and Details (ODD) protocol for describing individual- and agent-based models (Grimm and Railsback, 2005; Grimm et al., 2006) and consists of seven elements. The first three elements provide an overview of the model, the fourth element explains general concepts underlying the model's design, and the remaining three elements provide details of the model processes.
Supplemental Information 5. Summary of the dispersal of migrators and non-migrators within and outside of the GOM.
Supplemental Information 6. Three-dimensional animation of the dispersal of migrating species vs. non-migrating species spanning 80 days. Blue dots represent non-migrating animals at 900 m depth and pink represent animals with a diel vertical migration range of 200 m to 900 m depth.
References
Backus, R. H., Craddock, J. E., Haedrich, R. L., and Robison, B. H. (1977). Atlantic mesopelagic zoogeography. Fish. Western N. Atlantic 7, 266–287 doi: 10.1575/1912/4359
Beyer, J., Trannum, H. C., Bakke, T., Hodson, P. V., and Collier, T. K. (2016). Environmental effects of the Deepwater Horizon oil spill: a review. Mar. Pollut. Bull. 110, 28–51. doi: 10.1016/j.marpolbul.2016.06.027
Biggs, D. C. (1992). Nutrients, plankton, and productivity in a warm-core ring in the western Gulf of Mexico. J. Geophys. Res. 2, 2143–2154. doi: 10.1029/90JC02020
Biggs, D. C., and Muller-Karger, F. E. (1994). Ship and satellite observations of chlorophyll stocks in interacting cyclone-anticyclone eddy pairs in the western Gulf of Mexico. J. Geophys. Res.4, 7371–7384. doi: 10.1029/93JC02153
Brierley, A. S. (2014). Diel vertical migration. Curr. Biol. 24, R1074–R1076. doi: 10.1016/j.cub.2014.08.054
Burdett, E. A., Fine, C. D., Sutton, T. T., Cook, A. B., and Frank, T. M. (2017). Geographic and depth distributions, ontogeny, and reproductive seasonality of decapod shrimps (Caridea: Oplophoridae) from the northeastern Gulf of Mexico. Bull. Mar. Sci. 93, 743–767. doi: 10.5343/bms.2016.1083
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A., and Cresko, W. A. (2013). Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140. doi: 10.1111/mec.12354
Catchen, J. M., Hohenlohe, P. A., Bernatchez, L., Funk, W. C., Andrews, K. R., and Allendorf, F. W. (2017). Unbroken: RADseq remains a powerful tool for understanding the genetics of adaptation in natural populations. Mol. Ecol. Res. 17, 362–365. doi: 10.1111/1755-0998.12669
Cowen, R. K., Gawarkiewicz, G., Pineda, J., Thorrold, S. R., and Werner, F. (2007). Population connectivity in marine systems. Oceanography 20, 14–21. doi: 10.5670/oceanog.2007.26
Cowen, R. K., and Sponaugle, S. (2009). Larval dispersal and marine population connectivity. Ann. Rev. Mar. Sci. 1, 443–466. doi: 10.1146/annurev.marine.010908.163757
Danovaro, R., Gambi, C., Dell'Anno, A., Corinaldesi, C., Fraschetti, S., Vanreusel, A., et al. (2008). Exponential decline of deep-sea ecosystem functioning linked to benthic biodiversity loss. Curr. Biol. 18, 1–8. doi: 10.1016/j.cub.2007.11.056
Davey, J. L., and Blaxter, M. W. (2010). RADseq: next-generation population genetics. Brief. Funct. Genom. 9, 416–423. doi: 10.1093/bfgp/elq031
Dawson, M. N., Gupta, A. S., and England, M. H. (2005). Coupled biophysical global ocean model and molecular genetic analyses identify multiple introductions of cryptogenic species. Proc. Natl. Acad. Sci. U.S.A. 102, 11968–11973. doi: 10.1073/pnas.0503811102
De Robertis, A. (2002). Size-dependent visual predation risk and the timing of vertical migration: an optimization model. Limnol. Oceanogr. 47, 925–933. doi: 10.4319/lo.2002.47.4.0925
Dixon, D. R., Dixon, L. R. J., Wilson, J. T., and Pascoe, P. L. (2001). Chromosomal and nuclear characteristics of deep-sea hydrothermal-vent organisms: correlates of increased growth rate. Mar. Biol. 139, 251–255. doi: 10.1007/s002270100564
Donaldson, H. A. (1975). Vertical distribution and feeding of sergestid shrimps (Decapoda: Natantia) collected near Bermuda. Mar. Biol. 31, 37–50. doi: 10.1007/BF00390646
Earl, D. A., and VonHoldt, B. M. (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Gene. Res. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Flock, M. E., and Hopkins, T. L. (1992). Species composition, vertical distribution, and food habits of the sergestid shrimp. J. Crustacean Biol. 12, 210–223. doi: 10.2307/1549076
Foll, M., and Gaggiotti, O. (2008). A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180, 977–993. doi: 10.1534/genetics.108.092221
Forristall, G. Z., Schaudt, K. J., and Cooper, C. K. (1992). Evolution and kinematics of a loop current eddy in the Gulf of Mexico during 1985. J. Geophys. Res. 97, 2173–2184. doi: 10.1029/91JC02905
Foxton, P. (1970). The vertical distribution of pelagic decapods (Crustacea: Natantia) collected on the Sond cruise 1965 II. the Penaeidea and general discussion. J. Mar. Biol. Assoc. U.K. 50, 961–1000. doi: 10.1017/S0025315400005907
Froglia, C., and Giannini, S. (1982). Osservazioni sugli spostamenti verticali nictemerali di Sergestes arcticus kroyer e Sergia robusta (Smith) (Crustacea, Decapoda, Sergestidae) nel Mediterraneo occidentale. Atti Del Convegno Delle Unità Operative Afferenti Ai Sottoprogetti Risorse Biologiche e Inquinamento Marino. 1, 311–319.
Froglia, C., and Gramitto, M. E. (2000). A new pelagic shrimp of the genus Sergia (Decapoda, Sergestidae) from the Atlantic Ocean. J. Crustacean Biol. 20, 71–77. doi: 10.1163/1937240X-90000009
Gaines, S., Gaylord, B., Gerber, L., Hastings, A., and Kinlan, B. (2007). Connecting places: the ecological consequences of dispersal in the sea. Oceanography 20, 90–99. doi: 10.5670/oceanog.2007.32
Gartner, J. V. (1988). The lanternfishes (Pisces: Myctophidae) of the eastern Gulf of Mexico. Fish. Bull. 85, 81–98.
Genthe, H. C. (1969). The reproductive biology of Sergestes similis (Decapoda, Natantia). Mar. Biol. 2, 203–217. doi: 10.1007/BF00351142
Grimm, V., Berger, U., Bastiansen, F., Eliassen, S., Ginot, V., Giske, J., et al. (2006). A standard protocol for describing individual-based and agent-based models. Ecol. Model. 198, 115–126. doi: 10.1016/j.ecolmodel.2006.04.023
Grimm, V., Berger, U., DeAngelis, D. L., Polhill, J. G., Giske, J., and Railsback, S. F. (2010). The ODD protocol: a review and first update. Ecol. Model. 221, 2760–2768. doi: 10.1016/j.ecolmodel.2010.08.019
Grimm, V., and Railsback, S. F. (2005). Individual-Based Modeling Ecology. Princeton, NJ: Princeton Unviersity Press.
Hamilton, P., Donohue, K., Hall, C., Leben, R., Quian, H., Sheinbaum, J., et al. (2014). Observations and Dynamics of the Loop Current. OCS Study BOEM 5015-006. New Orleans, LA.
Heist, E. J., and Gold, J. R. (1999). Microsatellite DNA variation in sandbar sharks (Carcharhinus plumbeus) from the Gulf of Mexico and Mid-Atlantic Bight. Copeia 1999, 182–186. doi: 10.2307/1447399
Heist, E. J., Graves, J. E., and Musick, J. A. (1995). Population genetics of the sandbar shark (Carcharhinus plumbeus) in the Gulf of Mexico and Mid-Atlantic Bight. Copeia 1995, 555–562. doi: 10.2307/1446752
Heist, E. J., Musick, J., and Graves, J. E. (1996). Mitochondrial DNA diversity and divergence among sharpnose sharks, Rhizoprionodon terraenovae, from the Gulf of Mexico and Mid-Atlantic Bight. Fish. Bull. 94, 664–668.
Hellberg, M. E., Burton, R. S., Neigel, J. E., and Palumbi, S. R. (2002). Genetic assessment of connectivity among marine populations. Bull. Mar. Sci. 70, 273–290.
Herring, P. J. (1967). Observations on the early larvae of three species of Acanthephyra (Crustacea, Decapoda, Caridea). Deep Sea Res. Oceanogr. 14, 325–329. doi: 10.1016/0011-7471(67)90075-7
Herring, P. J. (1974a). Observations on the embryonic development of some deep-living decapod crustaceans, with particular reference to species of Acanthephyra. Mar. Biol. 25, 25–33. doi: 10.1007/BF00395105
Herring, P. J. (1974b). Size, density and lipid content of some decapod eggs. Deep-Sea Res. Oceanograp. Abst. 21, 91–94. doi: 10.1016/0011-7471(74)90023-0
Hughes, A. R., and Stachowicz, J. J. (2004). Genetic diversity enhances the resistance of a seagrass ecosystem to disturbance. Proc. Natl. Acad. Sci. U.S.A. 101, 8998–9002. doi: 10.1073/pnas.0402642101
Jakobsson, M., and Rosenberg, N. A. (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806. doi: 10.1093/bioinformatics/btm233
Johnston, M. W., and Bernard, A. M. (2017). A bank divided: quantifying a spatial and temporal connectivity break between the Campeche Bank and the northeastern Gulf of Mexico. Mar. Biol. 164:12. doi: 10.1007/s00227-016-3038-0
Johnston, M. W., Milligan, R. J., Easson, C. G., DeRada, S., English, D. C., Penta, B., et al. (2019). An empirically validated method for characterizing pelagic habitats in the Gulf of Mexico using ocean model data. Limnol. Oceanograp. 17, 363–375. doi: 10.1002/lom3.10319
Johnston, M. W., and Purkis, S. J. (2015). A coordinated and sustained international strategy is required to turn the tide on the Atlantic lionfish invasion. Mar. Ecol. Prog. Ser. 533, 219–235. doi: 10.3354/meps11399
Jombart, T., and Ahmed, I. (2011). Adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071. doi: 10.1093/bioinformatics/btr521
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Keeney, D. B., Heupel, M. R., Hueter, R. E., and Heist, E. J. (2005). Microsatellite and mitochondrial DNA analyses of the genetic structure of blacktip shark (Carcharhinus limbatus) nurseries in the northwestern Atlantic, Gulf of Mexico, and Caribbean Sea. Mol. Ecol. 14, 1911–1923. doi: 10.1111/j.1365-294X.2005.02549.x
Kool, J. T., Paris, C. B., Andréfouët, S., and Cowen, R. K. (2010). Complex migration and the development of genetic structure in subdivided populations: an example from Caribbean coral reef ecosystems. Ecography 33, 597–606. doi: 10.1111/j.1600-0587.2009.06012.x
Kraus, N. C., and Lin, L. (2009). Hurricane Ike along the upper Texas coast: an introduction. Shore Beach. 77, 3–8.
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Liggins, L., Treml, E. A., and Riginos, C. (2013). Taking the plunge: an introduction to undertaking seascape genetic studies and using biophysical models. Geo. Comp. 7, 173–196. doi: 10.1111/gec3.12031
Loose, C. J., and Dawidowicz, P. (1994). Trade-offs in diel vertical migration by zooplankton: the costs of predator avoidance. Ecology 75, 2255–2263. doi: 10.2307/1940881
MacIntosh, H., de Nys, R., and Whalan, S. (2014). Contrasting life histories in shipworms: growth, reproductive development and fecundity. J. Exp. Mar. Biol. Ecol. 459, 80–86. doi: 10.1016/j.jembe.2014.05.015
Meirmans, P. G., and Van Tienderen, P. H. (2004). GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol. Ecol. Notes 4, 792–794. doi: 10.1111/j.1471-8286.2004.00770.x
Milne-Edwards, A. (1881a). “Compte rendu sommaire d'une exploration zoologique faite dans l'Atlantique, a bord du navire le Travailleu,” in Comptes Rendus Hebdomadaires de Séances de l'Académie des Sciences, Vol. 93 (Paris), 931–936.
Milne-Edwards, A. (1881b). Description de quelques crustacés macroures provenant des grandes profondeurs de la Mer des Antilles. Ann. Sci. Nat. 6, 1–15.
Mussmann, S. M., Douglas, M. R., Anthonysamy, W. J. B., Davis, M. A., Simpson, S. A., Louis, W., et al. (2017). Genetic rescue, the greater prairie chicken and the problem of conservation reliance in the anthropocene. R. Soc. Open Sci. 4:160736. doi: 10.1098/rsos.160736
Nelson, J. R., and Grubesic, T. H. (2018). The implications of oil exploration off the Gulf Coast of Florida. J. Mar. Sci. Eng. 6, 1–21. doi: 10.3390/jmse6020030
Nunes, F., Norris, R. D., and Knowlton, N. (2009). Implications of isolation and low genetic diversity in peripheral populations of an amphi-Atlantic coral. Mol. Ecol. 18, 4283–4297. doi: 10.1111/j.1365-294X.2009.04347.x
Oey, L.-Y., Ezer, T., and Lee, H.-C. (2005). “Loop current, rings, and related circulation in the Gulf of Mexico: a review of numerical models and future challenges,” in Circulation in the Gulf of Mexico: Observations and Models, eds W. Sturges and A. Lugo-Fernandez (Washington, DC: American Geophysical Union), 31–56.
O'Leary, S. J., Puritz, J. B., Willis, S. C., Hollenbeck, C. M., and Portnoy, D. S. (2018). These aren't the loci you're looking for: principles of effective SNP filtering for molecular ecologists. Mol. Ecol. 27, 3193–3206. doi: 10.1111/mec.14792
Palumbi, S. R. (2003). Population genetics, demographic connectivity, and the design of marine reserves. Ecol. Appl. 13, S146–S158. doi: 10.1890/1051-0761(2003)013<0146:PGDCAT>2.0.CO;2
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S., and Hoekstra, H. E. (2012). Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7:e37135. doi: 10.1371/journal.pone.0037135
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Ramirez Llodra, E. (2002). Fecundity and life-history strategies in marine invertebrates. Adv. Mar. Biol. 43, 87–170. doi: 10.1016/S0065-2881(02)43004-0
Reitzel, A. M., Herrera, S., Layden, M. J., Martindale, M. Q., and Shank, T. M. (2013). Going where traditional markers have not gone before: utility of and promise for RAD sequencing in marine invertebrate phylogeography and population genomics. Mol. Ecol. 22, 2953–2970. doi: 10.1111/mec.12228
Riegl, B., Johnston, M., Purkis, S., Howells, E., Burt, J., Steiner, S. C. C., et al. (2018). Population collapse dynamics in Aropora downingi, an Arabian/Persian Gulf ecosystem-engineering coral, linked to rising temperature. Glob. Change Biol. 24, 2447–2462. doi: 10.1111/gcb.14114
Roe, H. S. J. (1984). The diel migrations and distributions within a mesopelagic community in the north east Atlantic (2): vertical migration and feeding of mysids and decapod Crustacea. Prog. Oceanogr. 13, 269–318. doi: 10.1016/0079-6611(84)90011-9
Rosenberg, N. A. (2004). DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138. doi: 10.1046/j.1471-8286.2003.00566.x
Shanks, A. L. (2009). Pelagic larval duration and dispersal distance revisited. Biol. Bull. 216, 373–385. doi: 10.1086/BBLv216n3p373
Smith, S.I. (1882). Reports on the Results of Dredging Under the Supervisión of Alexander Agassiz, on the East Coast of the United States During the Summer of 1880, by the U.S. Coast Survey Steamer “Blake”, Commander J.R. Bartlett, U.S.N., Commanding. Bulletin of the Museum of comparative Zoology at Harvard College, Vol. 10, 1–108.
John, M. A., Borja, A., Chust, G., Heath, M., Grigorov, I., Mariani, P., et al. (2016). A dark hole in our understanding of marine ecosystems and their services: perspectives from the mesopelagic community. Front. Mar. Sci. 3:31. doi: 10.3389/fmars.2016.00031
Sutton, T. T., Clark, M. R., Dunn, D. C., Halpin, P. N., Rogers, A. D., Guinotte, J., et al. (2017). A global biogeographic classification of the mesopelagic zone. Deep-Sea Res. Part I Oceanogr. Res. Pap. 126, 85–102. doi: 10.1016/j.dsr.2017.05.006
Timm, L. E. (2018). Evolutionary and population dynamics of crustaceans in the Gulf of Mexico (doctoral dissertation). Miami, FL: Florida International University.
Timm, L. E., Bracken-Grissom, H. D., Sosnowski, A., Breitbart, M., Vecchione, M., and Judkins, H. (2020). Population connectivity of three deep-sea cephalopod species between the Gulf of Mexico and northwestern Atlantic Ocean. Deep Sea Res. I. 103222. doi: 10.1016/j.dsr.2020.103222
Timm, L. E., Isma, L. M., and Bracken-Grissom, H. D. (2019). Single-locus barcodes and demultiplexed ddRADseq data for comparative population genomic analysis of the mesopelagic shrimp species Acanthephyra purpurea, Systellaspis debilis, and Robustosergia robusta. Gulf of Mexico Research Initiative Information and Data Cooperative (GRIIDC), Harte Research Institute, Texas A&M University – Corpus Christi. doi: 10.7266/n7-3p3y-g470
Truelove, N. K., Kough, A. S., Behringer, D. C., Paris, C. B., Box, S. J., Preziosi, R. F., et al. (2017). Biophysical connectivity explains population genetic structure in a highly dispersive marine species. Coral Reefs 36, 233–244. doi: 10.1007/s00338-016-1516-y
Uchida, H., and Baba, O. (2008). Fishery management and the pooling arrangement in the Sakuraebi Fishery in Japan. Case Studies Fish. Self Governance 504, 175–190.
van Olderborgh, G. J., van der Wiel, K., Sebastian, A., Singh, R., Arrighi, J., Otto, F., et al. (2017). Attribution of extreme rainfall from Hurricane Harvey, August 2017. Environ. Res. Lett. 12:124009. doi: 10.1088/1748-9326/aa9ef2
Venables, W. N., and Ripley, B. D. (2002). Modern Applied Statistics With S, 4th Edn. New York, NY: Springer.
Vereshchaka, A. L. (2009). Revision of the genus Sergestes (Decapoda: Dendrobranchiata: Sergestidae): taxonomy and distribution. Galathea Rep. 22, 7–104.
Wells, R. J. D., Rooker, J. R., Quigg, A., and Wissel, B. (2017). Influence of mesoscale oceanographic features on pelagic food webs in the Gulf of Mexico. Mar. Biol. 164, 1–11. doi: 10.1007/s00227-017-3122-0
Keywords: genetic diversity, connectivity, biophysical oceanographic modeling, diel vertical migration, midwater shrimp, Gulf Loop Current, Gulf of Mexico, Bear Seamount
Citation: Timm LE, Isma LM, Johnston MW and Bracken-Grissom HD (2020) Comparative Population Genomics and Biophysical Modeling of Shrimp Migration in the Gulf of Mexico Reveals Current-Mediated Connectivity. Front. Mar. Sci. 7:19. doi: 10.3389/fmars.2020.00019
Received: 31 July 2019; Accepted: 13 January 2020;
Published: 07 February 2020.
Edited by:
Tracey T. Sutton, Nova Southeastern University, United StatesReviewed by:
Alastair Brown, University of Southampton, United KingdomAnn I. Larsson, University of Gothenburg, Sweden
Copyright © 2020 Timm, Isma, Johnston and Bracken-Grissom. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura E. Timm, bGF1cmEudGltbUBjdWFuc2NodXR6LmVkdQ==