 Ya Gong
Ya Gong Lu Ma
Lu Ma Zhao-Zhong Du
Zhao-Zhong Du Wei-Shuang Zheng3
Wei-Shuang Zheng3 Zong-Jun Du
Zong-Jun Du- 1Marine College, Shandong University, Weihai, Shandong, China
- 2State Key Laboratory of Microbial Technology, Shandong University, Qingdao, Shandong, China
- 3Peking University-The Hong Kong University of Science and Technology (PKU-HKUST) Shenzhen-Hong Kong Institution, Marine Institute for Bioresources and Environment, Shenzhen, Guangdong, China
Spiribacter is the most abundant bacterial genus in the intermediate-salinity zones of hypersaline environments. However, Spiribacter strains are extremely difficult to isolate in pure culture. Therefore, the characteristics, genome features, and adaption mechanisms that allow Spiribacter strains to thrive in highly saline conditions are largely unknown. Here, we show that Spiribacter is predominant in brines from marine solar salterns and sulfate-type salt lakes with intermediate to saturated salinities. Using a high-salt medium, we isolated a novel strain, Spiribacter halobius E85T, which possesses a relatively large and distinct genome. The genome of strain E85T has a length of 4.17 Mbp, twice that of other Spiribacter species genomes and the largest described genome within the family Ectothiorhodospiraceae. Comparative genomic analyses indicate that approximately 50% of E85T genes are strain-specific, endowing functional differences in its metabolic capabilities, biosynthesis of compatible solutes, and transport and pumping of solutes into the cell from the environment. Hundreds of insertion sequences result in many pseudogenes and frequent gene fragment rearrangements in the E85T genome. Dozens of genomic islands, which show a significant preference for replication, recombination and repair, and cell motility and may have been gained from other bacterial species, are scattered in the genome. This study provides important insights into the general genetic basis for the abundance of Spiribacter in hypersaline environments and the strain-specific genome evolutionary strategies of strain E85T.
Introduction
Hypersaline environments, such as saline lakes and solar salterns, are distributed worldwide (Saccò et al., 2021; Shu and Huang, 2021). The unique environments of hypersaline lakes are considered representations of the conditions on Mars and the early Earth, providing an opportunity to study the presence of life in these distinct habitats (Pontefract et al., 2017; Belilla et al., 2019). The microbial community structure and the potential underlying ecological and evolutionary processes in these extreme hypersaline environments have been extensively studied using cultivation-independent methods (Narasingarao et al., 2012; Fernández et al., 2014). The square haloarchaeon Haloquadratum walsbyi and the halophilic bacterium Salinibacter ruber predominate crystallizer ponds with the highest salinities, while surprisingly high prokaryotic diversity and abundance are observed in intermediate-salinity conditions (Ventosa et al., 2015). However, these relatively abundant halophilic microbes are commonly slow growing and extremely difficult to isolate in pure culture (Bolhuis et al., 2004; Ghai et al., 2011). Homologous recombination and horizontal gene transfer are prevalent among halophilic bacteria and archaea, enabling adaptation to extreme and sometimes highly fluctuating environmental conditions. High recombination frequencies have been observed between populations of Halobacterium sp. AUS1 (Papke et al., 2004) and between different species in the genus Haloferax (Naor et al., 2012), indicating that halophilic archaea prefer genetic exchange and recombination. In prokaryotic genomes, horizontal gene transfer is the main source of gene gain and has been observed between S. ruber and haloarchaea. However, the genomes of many taxa have not yet been sequenced, and the genome evolution of halophilic bacteria remains to be uncovered.
The assembly of metagenomic data from intermediate-salinity marine solar saltern ponds revealed that the most abundant bacteria was a novel genus, Spiribacter, which belongs to the family Ectothiorhodospiraceae and the class Gammaproteobacteria (Fernández et al., 2014; León et al., 2014). The first cultured species of this genus, Spiribacter salinus, was documented in 2013 (León et al., 2013), and two streamlined genomes were reported in 2013 (López-Pérez et al., 2013). To date, the genus Spiribacter comprises four species with validly published names: S. salinus, S. curvatus, S. roseus, and S. vilamensis, which are halophilic bacteria isolated from salterns (León et al., 2014; León et al., 2015; León et al., 2016; León et al., 2020). The novel taxon ‘S. halalkaliphilus’, which is most similar to the type strain S. salinus, was recently obtained from soda-saline lakes in Inner Mongolia, China (Xue et al., 2021). The relatively restricted metabolic versatility and osmoregulatory mechanisms of Spiribacter have been investigated based on the genomes of S. salinus, S. curvatus, and ‘S. halalkaliphilus’ (López-Pérez et al., 2013; León et al., 2018; Xue et al., 2021). These genomes are approximately 2 Mbp in length, representing the smallest genomes within Ectothiorhodospiraceae and among all halophilic bacteria. Elucidation of the genomic and phenotypic repertoire of the genus Spiribacter has been limited by the lack of pure cultured strains.
Yuncheng Salt Lake is located in China’s Shanxi province and contains sulfate. Wendeng Solar Saltern, in China’s Shandong province, is a multi-pond saltern that originated from seawater. In a recent study, we sequenced samples from five Wendeng Solar Saltern ponds and analyzed the prokaryotic community composition, including the Spiribacter genus, the abundance of which was found to be positively related to the salinity of the water samples (Song et al., 2022). In the present study, we conducted amplicon sequencing variant (ASV) analyses of brine and sediment samples with different salinities from Yuncheng Salt Lake and Wendeng Solar Saltern. A novel Spiribacter strain (E85T) was isolated, and its taxonomic status was investigated based on phenotypic, chemotaxonomic, and genotypic data. Comparative genomics analyses of all Spiribacter genomes were performed to identify the potential metabolic capabilities, high-salinity adaptation mechanisms, and expansion pathways of the relatively large and distinct genome of strain E85T.
Results and discussion
Spiribacter was highly abundant in solar salterns and salt lakes
Considering the high abundance of the genus Spiribacter in different saline environments (López-Pérez et al., 2013; Zhao et al., 2020), we performed ASV sequencing analyses of brines and sediments to gain insights into the microbial community composition of marine solar salterns and sulfate-type salt lakes. In a previous study, samples from five Wendeng Solar Saltern ponds were sequenced and operational taxonomic units were clustered with a cut-off value of 97%, and the results showed that the abundance of Spiribacter exhibited a positive relationship with salinity in the water samples (Song et al., 2022). The sequence data and physicochemical parameters of hypersaline Wendeng Solar Saltern samples were used and compared in this study, but the 16S rRNA gene sequence data (accession numbers PRJNA559148 and PRJNA799174 in the Sequence Read Archive) were re-analyzed via ASV analyses with a threshold of 100% similarity. In this study, we also collected brine and sediment samples from six Yuncheng Salt Lake ponds in Yuncheng, China (Data S1). The physicochemical properties of the Yuncheng Salt Lake and Wendeng Solar Saltern brines were distinct. The salinities of Yuncheng Salt Lake ponds ranged from 170‰ to 340‰, with pH values of 7.84–8.53, and the salinities of Wendeng Solar Saltern ponds ranged from 45‰ to 265‰, with pH values of 7.53–8.02. The sulfate and chloride concentrations of Yuncheng Salt Lake ponds ranged from 47.13 to 136.37 g/L and from 24.22 to 23.91 g/L, respectively, and the chloride concentrations of the Wendeng Solar Saltern ponds ranged from 22.59 to 118.71 g/L.
Microbial genomic DNA from all brine and sediment samples of Yuncheng Salt Lake ponds was extracted, the V4–V5 region of the bacterial 16S rRNA gene sequence was amplified and sequenced, and the raw data were deposited in the Sequence Read Archive under accession numbers PRJNA825478 and PRJNA825481. With a threshold of 100% similarity, ASVs from all Yuncheng Salt Lake and Wendeng Solar Saltern samples were defined and assigned (Data S2). More than 50% of ASVs in most brine and sediment samples were not assigned to any previously described genus (Figure 1), suggesting that more pure culture, classification, and function studies of microbes in saline environments are needed.
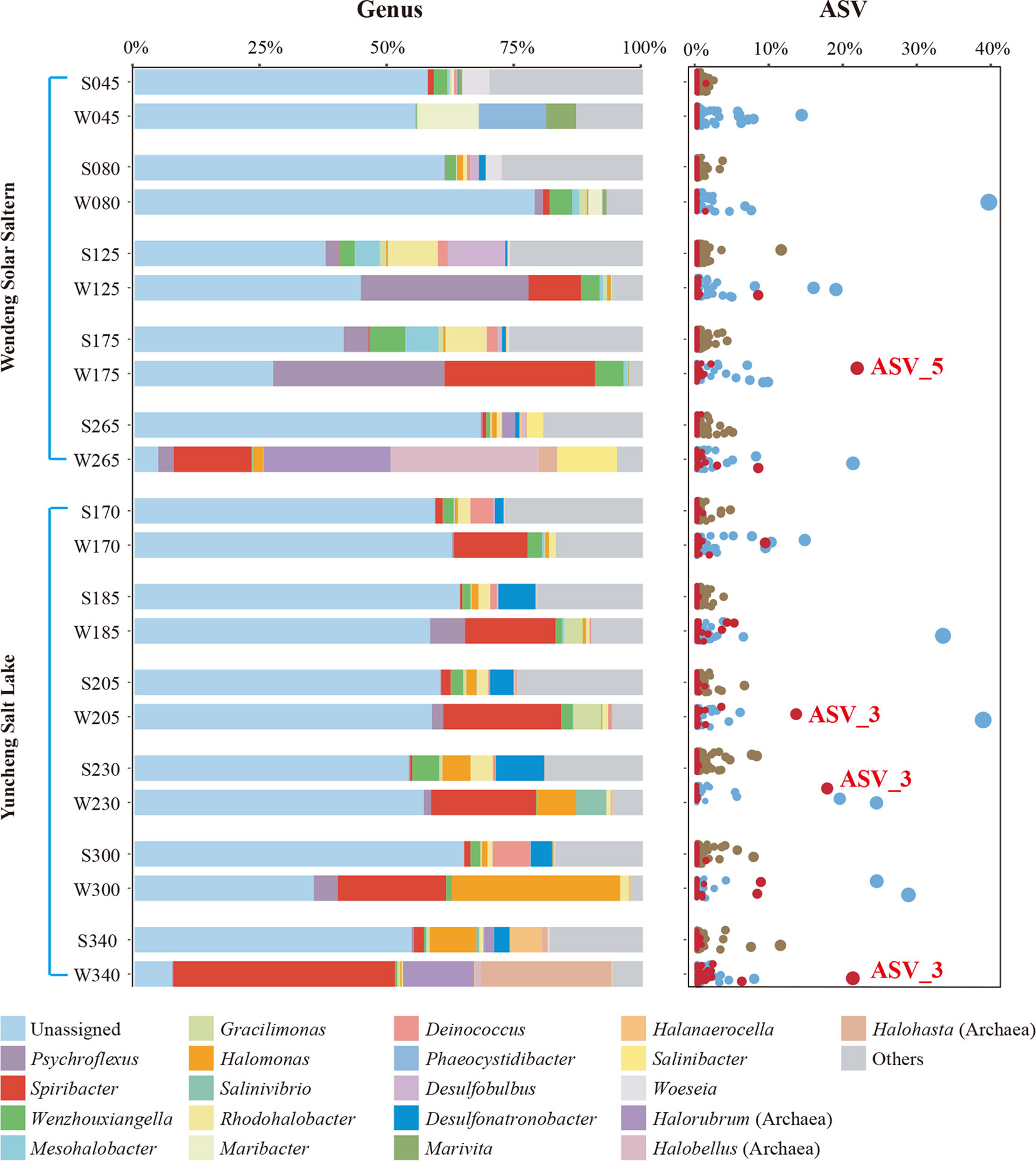
Figure 1 The prokaryotic community composition of two different saline environments along a gradient of increasing salinity at the genus and ASV levels. The top 22 most abundant genus taxa (including unassigned and others) and all ASVs are shown. In the sample names, the prefixes “S” and “W” represent sediment and water samples, respectively, and the numbers represent the salinity (‰). ASVs of the genus Spiribacter are shown in red, and the two representative ASVs are labeled.
The community compositions of brine and sediment samples differed. Some brine samples, such as W265 from Wendeng Solar Saltern (265‰ salinity) and W340 from Yuncheng Salt Lake (340‰ salinity), were enriched in the archaea genera Halorubrum, Halobellus, and Halohasta. Additionally, the genus Spiribacter was abundant in all brine samples from both Yuncheng Salt Lake and Wendeng Solar Saltern, despite the vastly different physicochemical parameters of the two sets of samples. The relative abundance of Spiribacter exceeded 10% in brine samples W175 and W265 from Wendeng Solar Saltern and all Yuncheng Salt Lake brine samples. The W340 sample had the highest relative abundance of Spiribacter, accounting for 43.60%. At the ASV level, there were some highly abundant Spiribacter ASVs; ASV_3 and ASV_5 accounted for approximately 20%. By contrast, the relative abundance of Spiribacter was below 2% in all sediment samples. These results showed that Spiribacter was predominant in the brines of marine solar salterns and sulfate-type salt lakes with intermediate and saturation salinities ranging from 125‰ to 340‰. The data confirmed that Spiribacter species are very abundant in brines, but not saline sediments, of soda-saline lakes (Zhao et al., 2020). Thus, Spiribacter was abundant in different hypersaline environments.
Isolation, identification, and characteristics of a novel Spiribacter species
Given the high abundance of this genus in high-salt conditions, we intended to isolate Spiribacter species to investigate the novel metabolic pathways and potential osmotic regulatory mechanisms enabling them to function at high salinity. Considering the halotolerance of Spiribacter, the brine and sediment samples of Yuncheng Salt Lake and Wendeng Solar Saltern were 10-fold serially diluted and plated on marine agar 2216 supplemented with 10% (w/v) NaCl. All cultured colonies were picked, and several pure colonies were identified as Spiribacter. Among them, strain E85T was identified as a novel species that grew faster than other Spiribacter strains and was thus chosen for further study.
The optimal growth of strain E85T occurred at 37–40°C (range 20–50°C), pH 7.5–8.0 (range 7.0–9.0), and 3–6% NaCl (range 0.5–16%, w/v). Distinctively, S. salinus LMG 27464T could tolerate 10–25% NaCl and showed optimal growth at 15% (León et al., 2014). E85T cells were Gram stain-negative, straight to curved rods measuring 0.2–0.3 μm × 1.2–4.5 μm in size and possessing a single polar flagellum (Figure S1). Unlike other Spiribacter strains, E85T grew under anaerobic conditions and could reduce nitrate to nitrite. Strain E85T was susceptible to ampicillin (10 μg), clindamycin (2 μg), chloromycetin (30 μg), streptomycin (10 μg), and norfloxacin (30 μg), but resistant to penicillin (10 μg), tetracycline (30 μg), neomycin (30 μg), cefotaxime sodium (30 μg), gentamycin (30 μg), sulfamethoxydiazine (10 μg), vancomycin (30 μg), and kanamycin (30 μg). In the Biolog Gen III test, strain E85T was resistant to troleandomycin, rifamycin SV, minocycline, lincomycin, and vancomycin.
The predominant respiratory quinone of strain E85T was ubiquinone-8, consistent with previous Spiribacter genus descriptions. Similarly, the major fatty acids of strain E85T were C18:1ω7c (36.8%) and C16:0 (19.2%, Table S1), but the details of the fatty acids were different; for example, strain E85T had four unique fatty acids, summed feature 2, iso-C15:0, anteiso-C15:0, and C13:1. The polar lipids of strain E85T were one phosphatidylcholine, one phosphatidylglycerol, one phosphatidylethanolamine, one diphosphatidylglycerol, one phosphoaminoglycolipid, and three phospholipids (Figure S2). S. salinus LMG 27464T and strain E85T shared four polar lipids, whereas the former exclusively contained a phosphoglycolipid and the latter harbored a phosphatidylcholine and a diphosphatidylglycerol. More differential morphological, physiological, and biochemical characteristics of strain E85T and closely related species are provided in the species description and summarized in Table S2.
Phylogenetic placement of strain E85T
An almost complete 16S rRNA gene sequence (1461 bp) was cloned from strain E85T and deposited in GenBank under the accession number KY407792.1. A sequence similarity calculation using the EzBioCloud database revealed that the closest relatives of strain E85T were S. salinus LMG 27464T (97.3%), S. curvatus CECT 8396T (96.3%), S. roseus CECT 9117T (96.2%), ‘S. halalkaliphilus’ IM2438T (96.2%), and S. vilamensis SV525T (96.0%). The phylogenetic tree constructed based on the 16S rRNA gene sequences of all cultured Spiribacter strains and the type strains of the related genera of the family Ectothiorhodospiraceae showed that strain E85T formed a separate clade within the genus Spiribacter (Figure S3). Additionally, the phylogenomic tree of 14 Spiribacter genomes, 11 genomes from pure culture strains, three metagenome-assembled genomes, and genomes from the type strains of related genera based on 120 ubiquitous bacterial single-copy genes also support the conclusion that strain E85T represents a novel lineage within the Spiribacter genus (Figure 2).
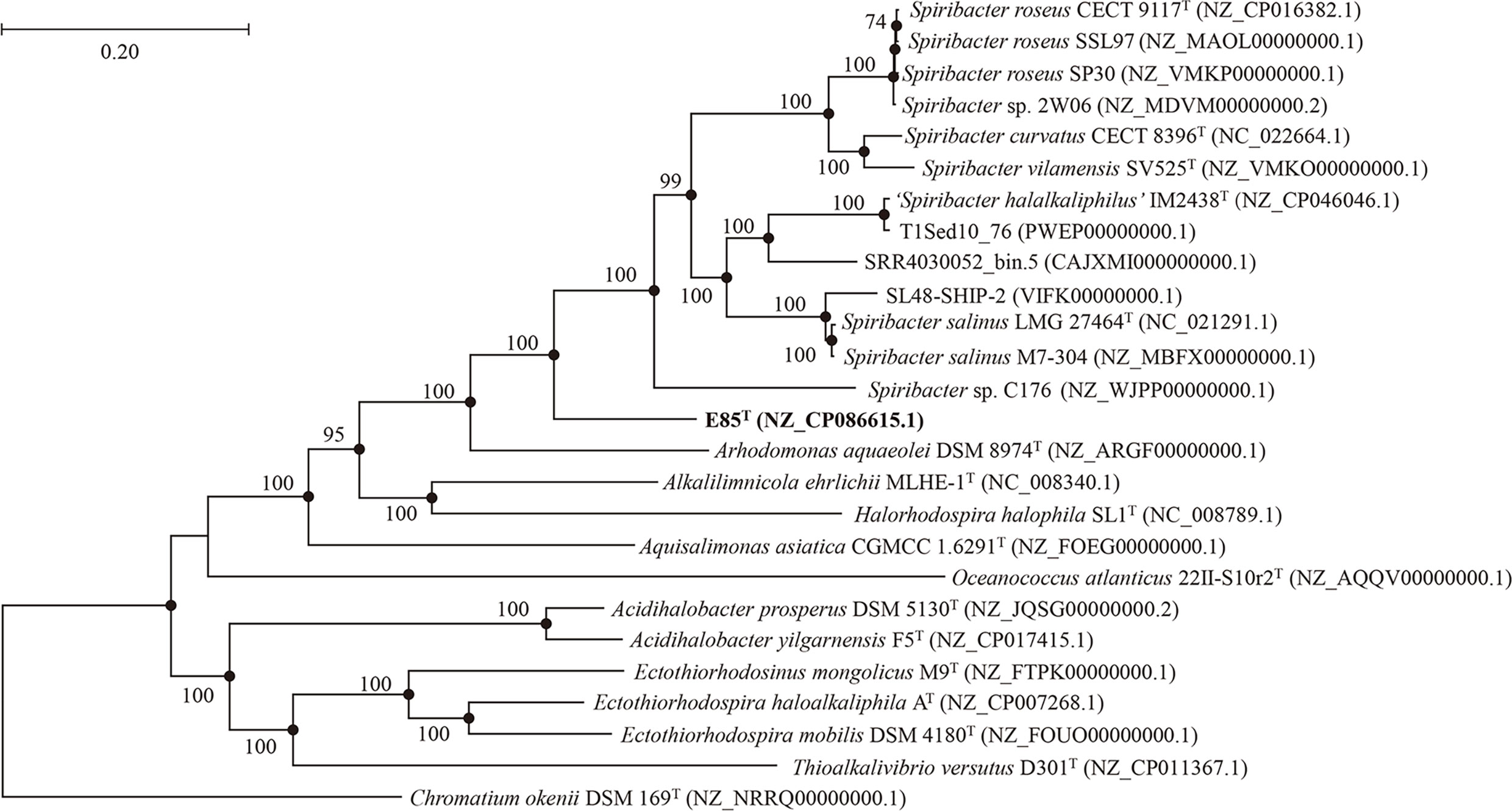
Figure 2 Phylogenetic tree generated from the alignment of 120 conserved proteins from 26 genomes of related strains, showing the taxonomic position of strain E85T among members of the genus Spiribacter. The numbers on the nodes represent bootstrap values (IQTree) based on 1,000 replications; bootstrap values higher than 70% are marked on the branches. Filled circles indicate nodes overlapping on trees reconstructed using the FastTree and IQTree algorithms. The genome sequence GenBank accession numbers are given in parentheses. The genome of Chromatium okenii DSM 169T (NRRQ00000000.1) is used as an outgroup. Bar: 0.20 substitutions per nucleotide position.
Furthermore, phylogenomic analyses of strain E85T, four validly published Spiribacter species, and the recently isolated strain ‘S. halalkaliphilus’ IM2438T were performed (Table S3). The average nucleotide identity value between the genomes of strain E85T and S. roseus CECT 9117T was 74.5%, followed by S. vilamensis SV525T (73.9%), ‘S. halalkaliphilus’ IM2438T (73.5%), S. curvatus CECT 8396T (73.4%), and S. salinus LMG 27464T (73.2%). The digital DNA-DNA hybridization value between the genomes of strain E85T and S. vilamensis SV525T was highest at 20.1%, followed by S. roseus CECT 9117T (19.4%), S. curvatus CECT 8396T (19.3%), S. salinus LMG 27464T (19.2%), and ‘S. halalkaliphilus’ IM2438T (19.1%). These values were far below the species threshold values of 95–96% for average nucleotide identity (Goris et al., 2007) and 70% for digital DNA-DNA hybridization (Auch et al., 2010). In addition, the average amino-acid identity values between genomes of strain E85T and other Spiribacter species ranged from 68.8% to 70.8%. This also suggested that strain E85T might be a novel species according to the genera boundary of 45–65% and the species boundary of 65–95% (Konstantinidis et al., 2017). Therefore, based on these phylogenetic and phylogenomic analyses, we propose that strain E85T is a novel species of the genus Spiribacter.
Strain E85T possesses a relatively large and distinct genome
The draft genome of strain E85T (GenBank accession number QFFI00000000.1) was assembled, with a total sequence length of 4,155,782 bp, 90 scaffolds, 150× genome coverage, and a scaffold N50 value of 118,172 bp. The National Center for Biotechnology Information (NCBI) Prokaryotic Genome Annotation Pipeline was used to annotate 3,863 genes. Among them, 3,755 protein-coding genes and 52 RNA genes were identified. Furthermore, the complete genome of strain E85T was defined by assembling the next-generation sequencing data. A circular 4,172,350 bp sequence was obtained and deposited in the GenBank database under accession number NZ_CP086615.1 (Figure 3A). The complete genome of strain E85T contained 3,874 genes, with 3,821 coding sequences (CDSs), 3,771 protein-coding genes, three rRNAs, 46 tRNAs, four ncRNAs, and 50 pseudogenes. The similarity between the draft genome and the complete genome of strain E85T was compared based on the whole-genome alignment, and the results indicated that the gene sequences were highly similar; however, the gene arrangements differed due to the different contigs (Figure S4A). The complete genome of strain E85T was used for the genomic analyses of this study.
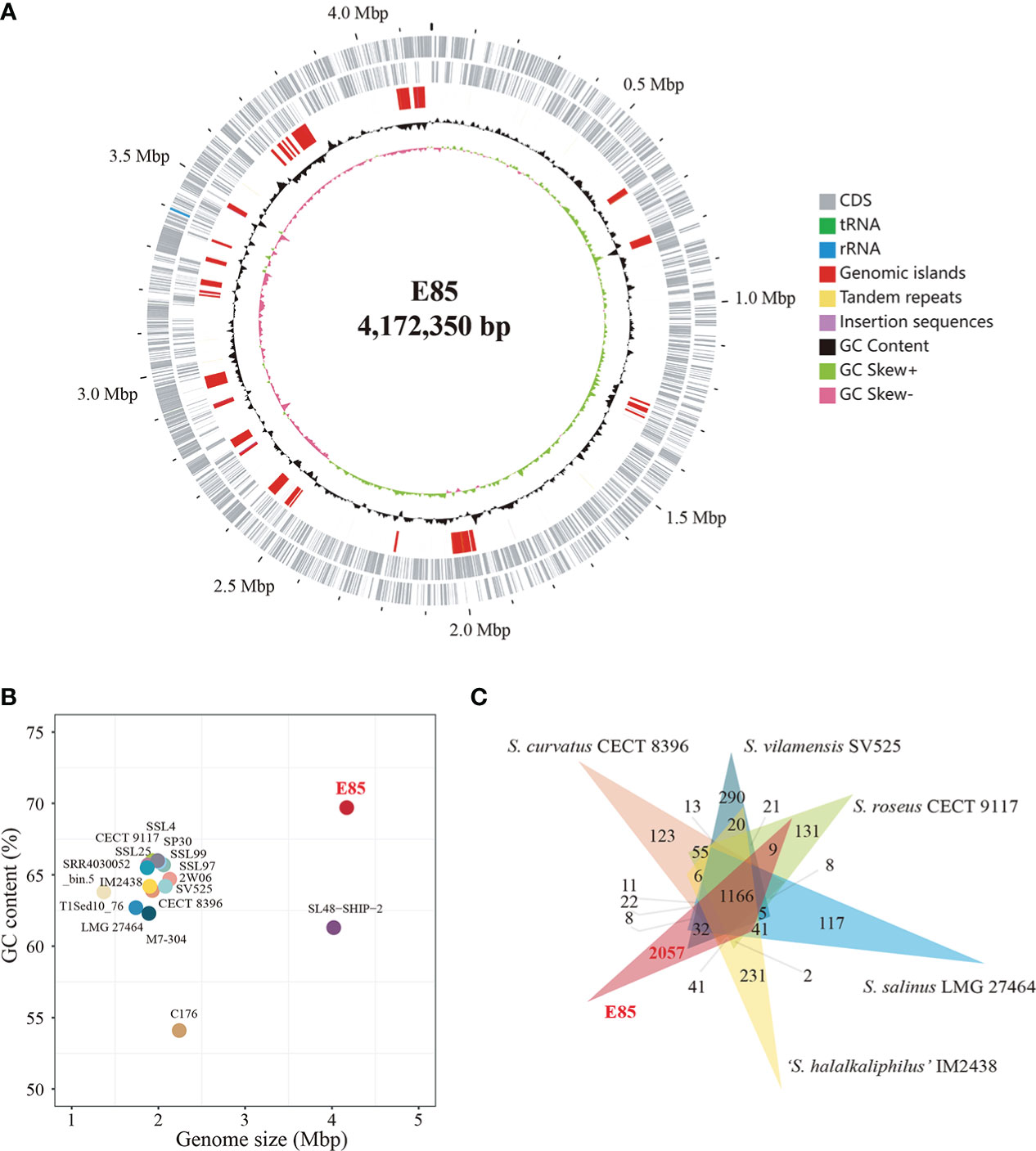
Figure 3 Genome features of strain E85 and closely related species. (A) Genome map of Strain E85. Ordered from outer ring to inner ring: 1) forward CDS, tRNA, and rRNA; 2) reverse CDS, tRNA, and rRNA; 3) genomic islands, tandem repeats, and insertion sequences; 4) GC content; 5) GC skew. (B) Genome size and GC content of all Spiribacter genomes: 14 genomes from pure culture strains (strain E85, S. roseus CECT 9117, S. roseus SSL97, S. roseus SP30, Spiribacter sp. 2W06, S. curvatus CECT 8396, S. vilamensis SV525, ‘S. halalkaliphilus’ IM2438, S. salinus LMG 27464, Spiribacter sp. C176, S. roseus SSL4, S. roseus SSL25, S. roseus M7-304, and Spiribacter sp. SSL99) and three metagenome-assembled genomes (SL48-SHIP-2, T1Sed10_76, and SRR4030052_bin.5). (C) Distribution of homologous genes in the genomes of strain E85 and five other published Spiribacter species: 1,166 core gene clusters were represented in all genomes. Strain E85, ‘S. halalkaliphilus’ IM2438, S. salinus LMG 27464, S. roseus CECT 9117, S. vilamensis SV525, and S. curvatus CECT 8396 contained 2,057, 231, 117, 131, and 290 species-specific gene clusters, respectively. The clustering results were used to generate a Venn diagram (Data S3).
The genome of strain E85T was distinctly larger than other Spiribacter genomes, 14 pure culture strain genomes (from 1.74 Mb for S. salinus LMG 27464T to 2.24 Mb for Spiribacter sp. C176) and three metagenome-assembled genomes, including SL48-SHIP-2 (4.02 Mb, Figure 3B). The E85T genome is the largest described within the family Ectothiorhodospiraceae (the detailed information of genomes included in this research is shown in Table S4). Additionally, the DNA G+C content of strain E85T was 69.7%, which corresponds to that obtained using HPLC (70.2 mol%) and is the highest among the Spiribacter genomes. The annotated gene numbers and CDS numbers of strain E85T were almost double those of other Spiribacter genomes (Table 1). Moreover, 288 tandem repeats, generally non-coding DNA, were found in the genome of strain E85T.
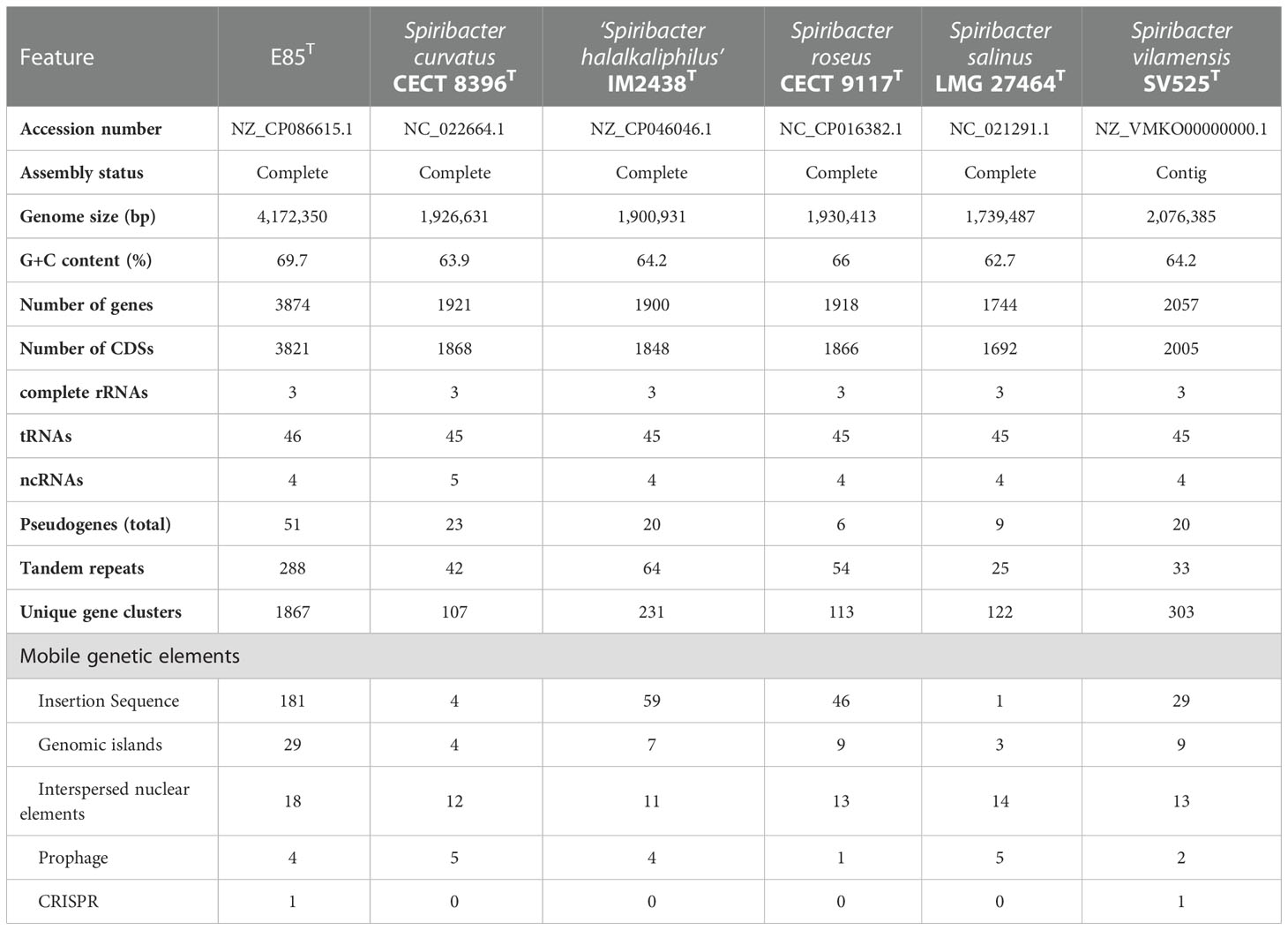
Table 1 General genome features of strain E85T and other Spiribacter species.
Further, the homologous genes of strain E85T, ‘S. halalkaliphilus’ IM2438T, S. salinus LMG 27464T, S. curvatus CECT 8396T, S. roseus CECT 9117T, and S. vilamensis SV525T were clustered using the GeneFamily Method and Pan-Genome Analysis Web Server (Chen et al., 2018) with an e-value of 1e-10, coverage of 0.5, and identity of 0.5. A total of 4,961 homologous gene clusters were identified, and 1,166 gene clusters were shared by the six genomes (Figure 3C, Data S3). Strikingly, strain E85T harbored 2,057 unique gene clusters, corresponding to 2,173 genes and accounting for approximately 56.09% of the total gene number of strain E85T. These substantial differences between the genomes were also reflected in the progressiveMauve algorithm results based on the whole-genome alignment. The homologous gene regions were matched, whereas several gene regions of strain E85T were not shared with other strains (Figure S4B). Thus, the genome of strain E85T was relatively distinct.
Potential metabolic capabilities and high-salinity adaptation mechanisms
Bioinformatics analyses revealed that 3,197 CDSs were devoted to a clear Clusters of Orthologous Genes (COG) functional category, accounting for 83.67% of the total CDSs in the strain E85T genome (Table S5). Meanwhile, the predicted results showed that, on average, 93.31% of the total CDSs in the genomes of the other five Spiribacter species were assigned to a functional category, with the functions of COG-J (translation, ribosomal structure, and biogenesis) and COG-M (cell wall/membrane/envelope biogenesis) as the top terms (Figure 4A). However, CDSs with the functions of COG-K (transcription), COG-L (replication, recombination, and repair), COG-T (signal transduction mechanisms), COG-E (amino acid transport and metabolism), COG-C (energy production and conversion), COG-P (inorganic ion transport and metabolism), COG-G (carbohydrate transport and metabolism), COG-I (lipid transport and metabolism), and COG-Q (secondary metabolite biosynthesis, transport, and catabolism) were abundant in the genome of strain E85T, suggesting that they may play a role in survival strategies. These functions enriched in strain E85T were consistent with the previous finding that larger genomes preferentially accumulate genes related to secondary metabolism and energy conversion (Konstantinidis and Tiedje, 2004).
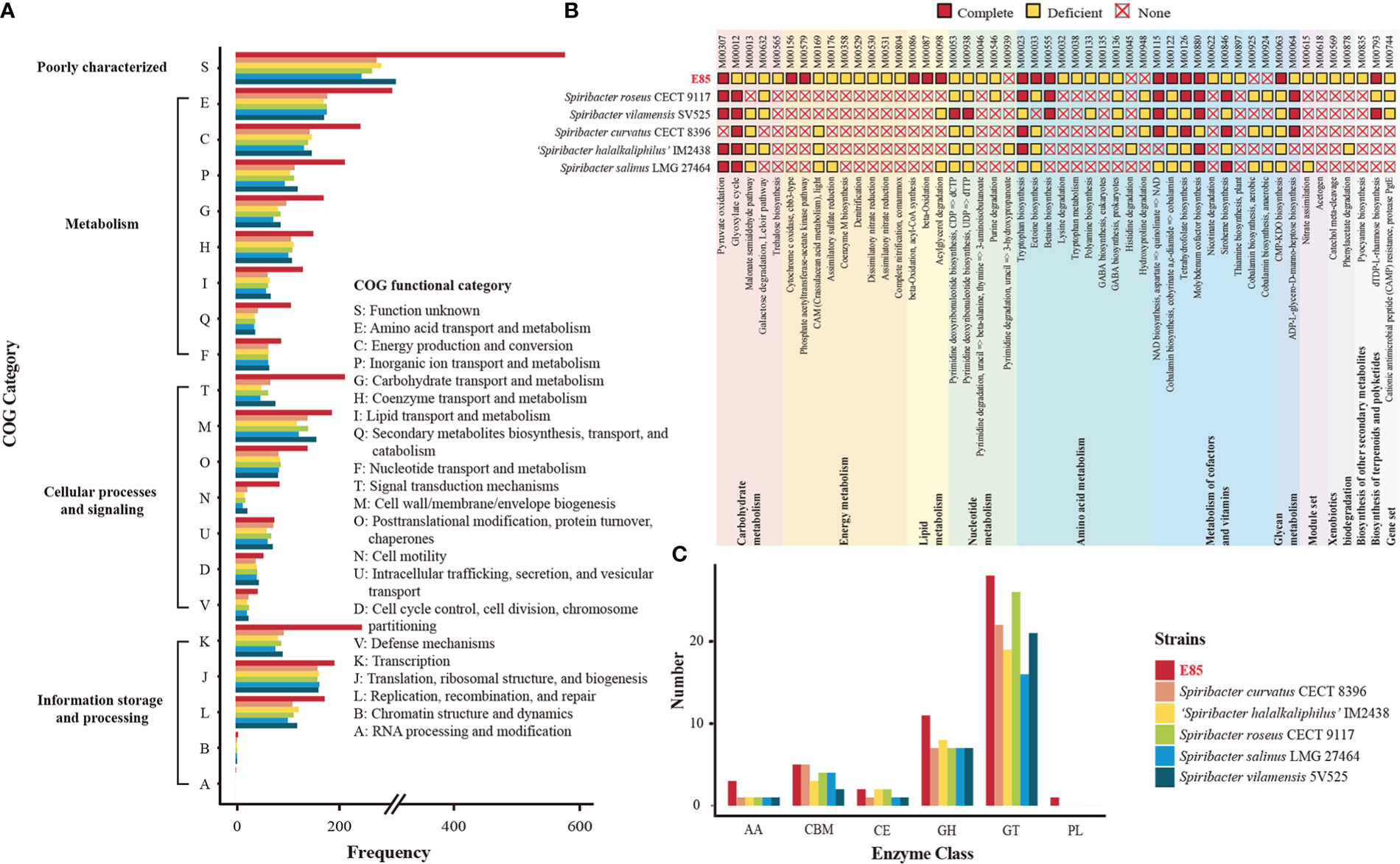
Figure 4 Comparison of potential function between the six Spiribacter genomes. (A) Functional distribution based on the COG classifications. The COG features are divided into four broad categories: information storage and processing, cellular processes and signaling, metabolism, and poorly characterized. (B) Inferred metabolism of six Spiribacter strains based on the KEGG database. Modules were manually categorized as complete, deficient (one block missing, two blocks missing, and incomplete), and none. (C) Quantification of carbohydrate-active enzyme genes in Spiribacter genomes. GH, glycoside hydrolases; GT, glycosyl transferases; PL, polysaccharide lyases; CE, carbohydrate esterases; CBM, carbohydrate-binding modules; AA, auxiliary activities; Annotation details are provided in Table S5.
To determine the possible ecological benefits possessed within the large genome of strain E85T, the KEGG database was used to identify metabolic pathways. The KEGG annotation results revealed that 50 pathway modules differed between the genomes of strain E85T and the other five Spiribacter species (Figure 4B). Among them, several pathways were only complete in the genome of strain E85T. For example, strain E85T had two complete energy metabolism pathways, M00156 (cytochrome c oxidase, cbb3-type) and M00579 (phosphate acetyltransferase-acetate kinase pathway), whereas other Spiribacter strains lacked these pathways. The cbb3 cytochrome c oxidases contain four-subunit integral membrane complexes and use c-type cytochromes as electron donors with high oxygen affinity, functioning in O2 reduction, proton pumping across the inner membrane, and the initiation of other metabolic pathways required for growth under microaerobic conditions (Durand et al., 2018). The bacterial enzymes acetate kinase and phosphotransacetylase form key pathways for synthesis of the central metabolic intermediate acetyl coenzyme A from acetate or ATP from excess acetyl coenzyme A, playing an important role in physiology and metabolism (Ingram-Smith et al., 2006). These two complete energy metabolism pathways might be beneficial for the growth of strain E85T, which grew faster than the other Spiribacter strains. The colony diameter of strain E85T reached 0.5–1.5 mm after 3 days of incubation at 37°C, while S. salinus LMG 27464T, S. curvatus CECT 8396T, and S. roseus CECT 9117T reached colony diameters of 0.5–1.5 mm, 0.2–0.5 mm, and 0.5–1.0 mm, respectively, after 10 days of incubation at 37°C (León et al., 2014; León et al., 2015; León et al., 2016). The colony diameter of S. vilamensis SV525T was pinpoint (< 0.5 mm) after 5 days of incubation at 30°C, and that of ‘S. halalkaliphilus’ IM2438T was 1.0–2.0 mm after 3–5 days of incubation at 37°C (León et al., 2014; León et al., 2015; León et al., 2016; León et al., 2020; Xue et al., 2021).
Considering the halotolerance of Spiribacter strains, the biosynthesis of compatible solutes and transport pumping solutes into the cell from the environment were investigated. Water-soluble organic molecules, including sugars, alcohols, and amino acid derivatives, are typically compatible solutes with the amino acids ectoine, glycine betaine, and proline, which are widespread in prokaryotes (Gunde-Cimerman et al., 2018). The genome analyses showed that strain E85T harbored three complete amino acid metabolism pathways, M00555 (betaine biosynthesis, choline → betaine), M00033 (ectoine biosynthesis, aspartate → ectoine), and M00015 (proline biosynthesis, glutamate → proline), to synthesize these compatible solutes. Among the other five Spiribacter species, all strains possessed the complete proline biosynthesis pathway, and S. roseus CECT 9117T and S. vilamensis SV525T had the complete betaine biosynthesis pathway. In addition, the compatible solute transporters, betaine/carnitine transporter, choline/glycine/proline betaine transport protein, and glycine betaine/proline transport system (glycine betaine/proline transport system ATP-binding protein [EC:7.6.2.9]; glycine betaine/proline transport system permease protein; glycine betaine/proline transport system substrate-binding protein), were identified in the Spiribacter genomes, functioning in the uptake of compatible solutes (Table S5). All six Spiribacter strains harbored betaine/carnitine transporter and the glycine betaine/proline transport system, but strain E85T lacked choline/glycine/proline betaine transport protein. Therefore, the adaptation strategies of Spiribacter to high-salt environments might differ between species. Strain E85T produces diverse compatible solutes, whereas other Spiribacter species might depend on an enhanced uptake capacity for compatible solute accumulation in cells. The ability of S. salinus LMG 27464T and ‘S. halalkaliphilus’ IM2438T to import and synthesize compatible solutes has been documented (López-Pérez et al., 2013; León et al., 2018; Xue et al., 2021). S. salinus LMG 27464T produces ectoine and trehalose and possesses several uptake systems, and some marker genes for ectoine, glycine betaine, and trehalose were annotated in the genome of ‘S. halalkaliphilus’ IM2438T, indicating that these compatible solutes might not be synthesized de novo for these metabolism pathways in these strains.
Other unique pathway modules of strain E85T were lipid metabolism M00086 (beta-oxidation, acyl-CoA synthesis), M00087 (beta-oxidation), and M00098 (acylglycerol degradation); metabolism of cofactors and vitamins M00122 (cobalamin biosynthesis); and glycan metabolism M00063 (CMP-KDO biosynthesis). These pathways might be beneficial to the growth of strain E85T in saline environments. The pathway modules of strain E85T and related strains identified by KEGG are listed in Table S5. Many pathway modules involving carbohydrate metabolism (glycolysis, citrate cycle, pentose phosphate pathway, PRPP biosynthesis, and the Entner–Doudoroff pathway), energy metabolism (NADH: quinone oxidoreductase, succinate dehydrogenase, cytochrome bc1 complex respiratory unit, cytochrome c oxidase, and F-type ATPase), and amino acid biosynthesis were shared by all Spiribacter strains. To analyze the potential ability of Spiribacter strains to degrade and metabolize complex carbohydrates, carbohydrate-active enzymes were identified by dbCAN2 annotation. The results showed that strain E85T contained more glycoside hydrolases than other Spiribacter strains, which contained more glycosyltransferases (Figure 4C). Glycoside hydrolases usually catalyze glycosidic bond breakage, which suggests that strain E85T may utilize organic compounds as carbon and energy sources.
The strain E85T genome contains many insertion sequences and DNA fragment rearrangements
Mobile genetic elements were analyzed to explore the genetic basis for the apparent differences in gene contents and metabolic capabilities between strain E85T and other Spiribacter strains. Mobile genetic elements are DNA sequences, such as transposons, integrons, and genomic islands, that can mediate horizontal gene transfer between bacterial strains and species, facilitating genome expansion, rapid evolution, and environmental adaptation (Frost et al., 2005). Compared with other Spiribacter genomes, the genome of strain E85T was enriched in insertion sequences (181, Table 1, Figure 3A). Insertion sequences are small transposable elements widespread in bacterial genomes, representing a dynamic evolutionary force. In bacterial genomes, copy number increases and subsequent insertion sequence losses can have functional consequences such as gene inactivation, pseudogene formation, mediation of intervening sequence deletion between two copies of the insertion sequence, and genome rearrangements (Siguier et al., 2014). The strain E85T genome contained 51 pseudogenes (Table 1), and the S. curvatus CECT 8396T, S. vilamensis SV525T, ‘S. halalkaliphilus’ IM2438T, S. salinus LMG 27464T, and S. roseus CECT 9117T genomes contained 21, 18, 15, 9, and 7 pseudogenes, respectively. To determine whether gene fragment rearrangement occurred in strain E85T, synteny between the genomes of strain E85T and the other five Spiribacter strains was analyzed. Obvious genome synteny existed between S. salinus LMG 27464T, ‘S. halalkaliphilus’ IM2438T, and S. curvatus CECT 8396T, but no genome synteny was observed between strain E85T and the other five Spiribacter strains (Figure 5). Although there was genome synteny between S. vilamensis SV525T and S. salinus LMG 27464T, ‘S. halalkaliphilus’ IM2438T, S. curvatus CECT 8396T, and S. roseus CECT 9117T, the rearrangement of a large DNA fragment containing approximately half of the genome was found in S. vilamensis SV525T. A small DNA fragment, approximately one-sixth of the genome, was also rearranged in the genome of S. roseus CECT 9117T. These results indicate that genome mutation caused by gene fragment rearrangement frequently occurs in Spiribacter, especially in strain E85T.
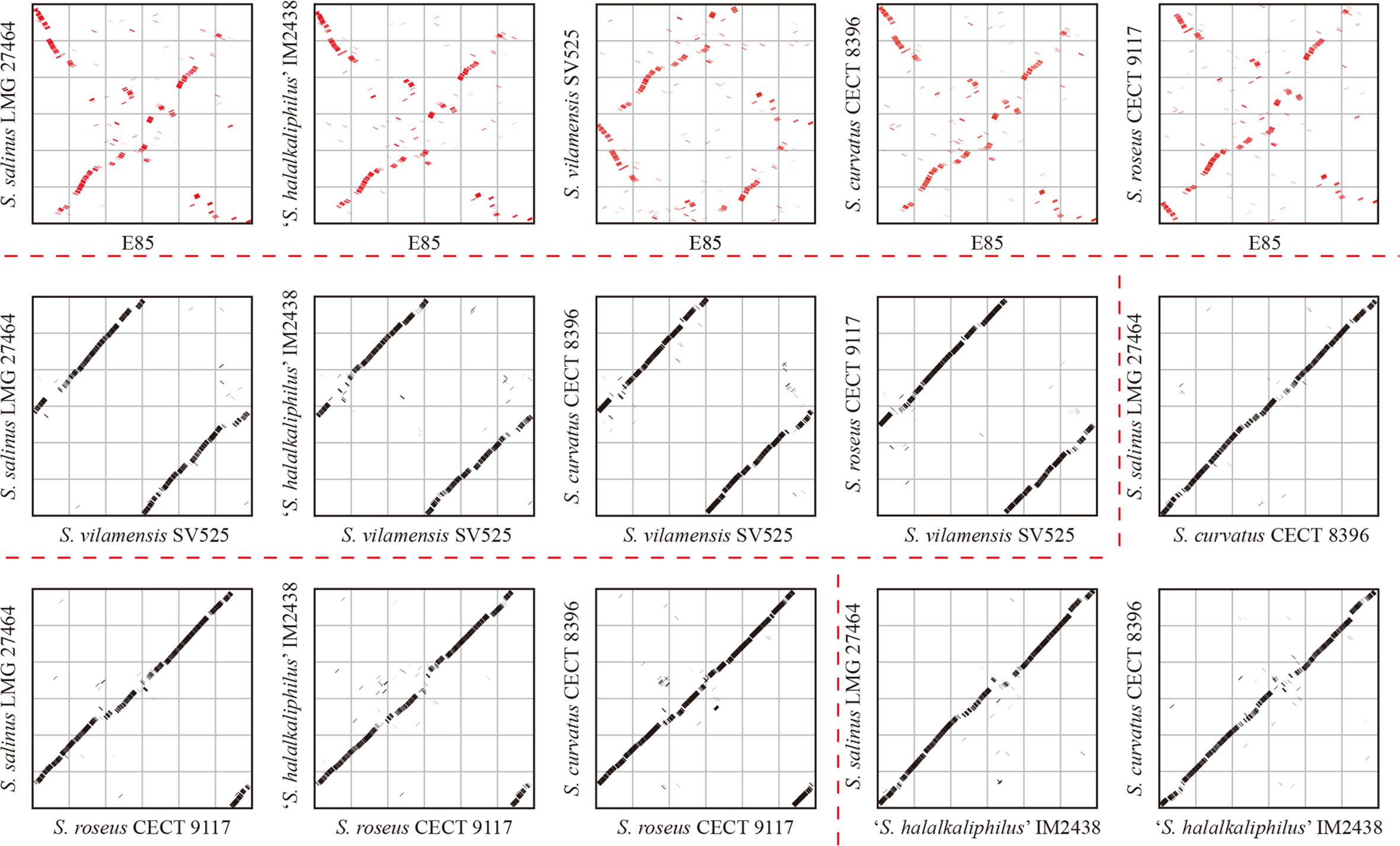
Figure 5 Genome synteny analyses of six Spiribacter species genomes. Significant synteny between genomes is represented by a clear line on the graph, whereas no synteny is represented by scattered points. Obvious synteny is observed between the genomes of S. salinus LMG 27464, ‘S. halalkaliphilus’ IM2438, and S. curvatus CECT 8396, but no synteny is observed between strain E85 and the other five Spiribacter strains (marked in red).
The genome of strain E85T was enriched in genomic islands, which might be from different sources
Genomic islands (GIs) are, in essence, discrete DNA segments that differ between closely related bacterial strains to which some past or present mobility is attributable. Horizontal gene transfer facilitated by GIs is widely recognized to play a crucial role in the evolution of bacterial species (Juhas et al., 2009). In these six Spiribacter genomes, GIs were recognized using IslandViewer 4 (Bertelli et al., 2017). The results showed that 29 GIs were scattered in the genome of strain E85T, and relatively few GIs were present in other Spiribacter genomes (Table 1). Other than strain E85T, S. roseus CECT 9117T and S. vilamensis SV525T harbored the highest number of GIs (nine each). S. salinus LMG 27464T contained the fewest GIs (three), which might be a consequence of having the smallest genome among the species examined (López-Pérez et al., 2013). Therefore, the genome of strain E85T was enriched in GIs.
To determine the potential function of GIs in strain E85T, the gene features and functional differences were analyzed. The 29 GIs were distributed throughout the genome, with sequence lengths ranging from 4,684 to 48,684 bp (Data S4). Among them, eight large GIs were longer than 20 Kbp, and 13 small GIs were shorter than 10 Kbp. The total length of all 29 GIs was 484,275 bp, accounting for 11.61% of the strain E85T genome length. The GIs harbored 481 genes, representing 12.42% of the total number of genes. According to the COG functional annotation, 313 CDSs were classified into COG functional categories, accounting for 65.07% of the total CDSs in the 29 GIs, far less than the proportion of functional CDSs (83.67%) in the genome of strain E85T. Based on the COG categories, the functional preferences of CDSs encoded by GIs and the complete genome were analyzed (Figure 6A). The proportions of CDSs encoded by the genome of strain E85T in COG-J (translation, ribosomal structure, and biogenesis), COG-C (energy production and conversion), COG-E (amino acid transport and metabolism), COG-F (nucleotide transport and metabolism), COG-H (coenzyme transport and metabolism), and COG-P (inorganic ion transport and metabolism) were 5.97, 7.54, 9.45, 2.75, 4.69, and 6.60%, respectively, more than twice that in the GIs. However, the GIs were enriched in COG-L (replication, recombination, and repair) and COG-N (cell motility) functional CDSs, accounting for 16.61 and 14.80% of total COG functional CDSs in the 29 GIs, respectively. These two proportions were very different from those in the genome of strain E85T, which corresponded to 5.38 and 2.66%, respectively. These results indicate that core cell functions, such as the translation process, energy metabolism, and transport and metabolism of amino acids, nucleic acids, coenzymes, and inorganic ions, were concentrated in the genome of strain E85T, while the CDSs encoded by the 29 GIs were related to replication, recombination and repair, and cell motility functions. More importantly, the GIs encoded plenty of transposases, integrases, and phage-related proteins, reflecting the potential for horizontal gene transfer and recombination. In addition, the GIs were genomic regions with GC skew, and the GC content of GIs was often low (Figure 2A), suggesting that the GIs might have been gained from the genomes of other strains or bacterial species.
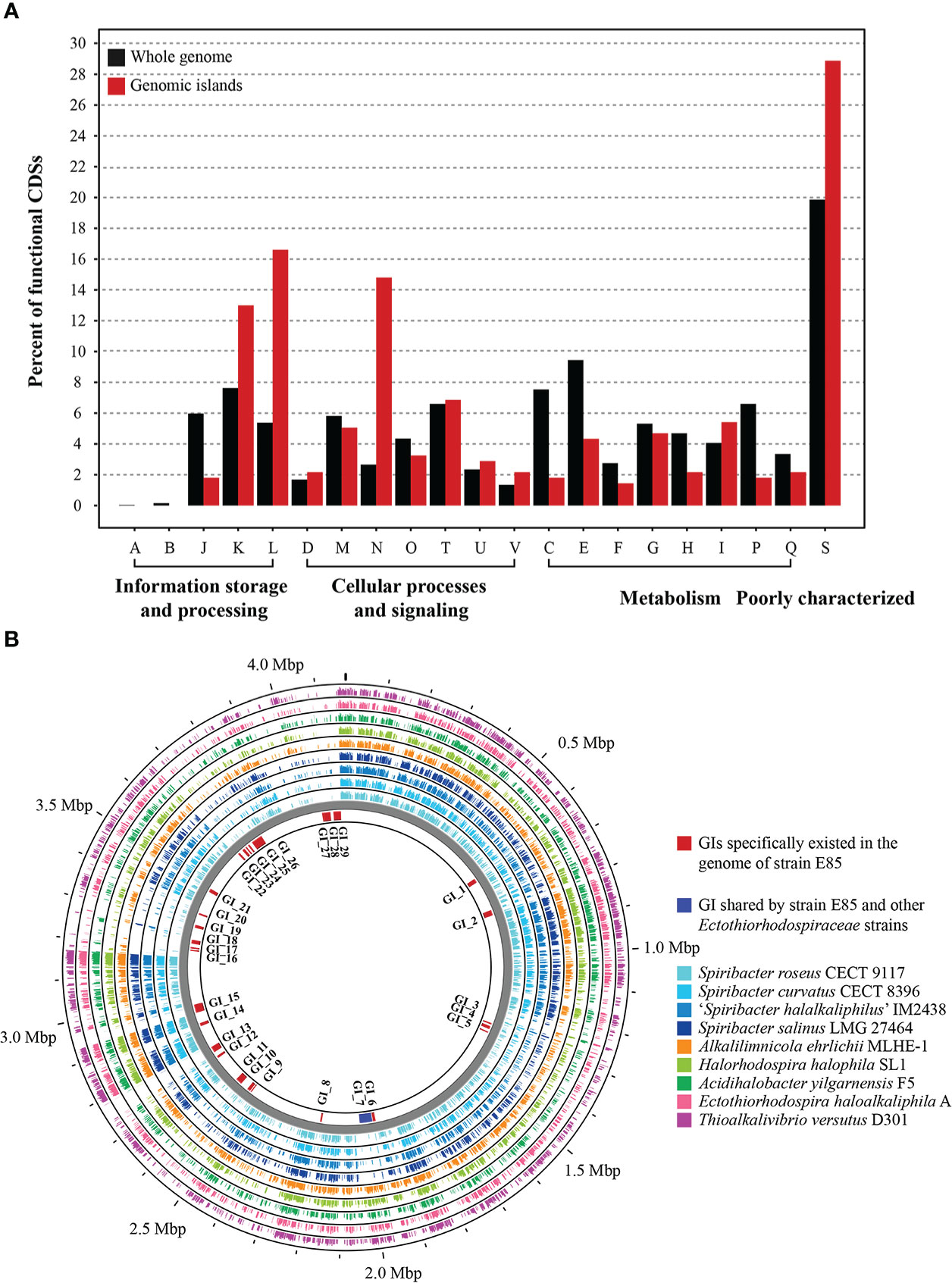
Figure 6 Genomic islands in the genome of strain E85. (A) Functional differences between the genomic islands and the whole genome of strain E85. The percentages of CDSs belonging to each COG are presented, and the function of each COG is shown in Figure 4. Compared with the functional CDSs in the whole genome of strain E85, the genomic islands harbor more than twice the proportions of COG-L and COG-N CDSs. (B) Whole-genome comparison of strain E85 and the complete genomes of Spiribacter and related genera within the Ectothiorhodospiraceae family. Genomic islands are shown inside the backbone and scattered in the genome of strain E85. Circular plots were generated using the BLASTn Proksee tools. Genomes are ordered from the inner ring to the outer ring: 1) S. roseus CECT 9117; 2) S. curvatus CECT 8396; 3) ‘S. halalkaliphilus’ IM2438; 4) S. salinus LMG 27464; 5) Alkalilimnicola ehrlichii MLHE-1; 6) Halorhodospira halophila SL1; 7) Acidihalobacter yilgarnensis F5; 8) Ectothiorhodospira haloalkaliphila A; 9) Thioalkalivibrio versutus D301. The BLASTn results of each CDS in the genomic islands of strain E85 are displayed in Data S4.
We searched for homologs of the strain E85T GIs and discovered that several homologous genes were gained from the genomes of other Spiribacter species and other taxa within the Ectothiorhodospiraceae family. Therefore, the genome of strain E85T was compared with the complete genomes of Spiribacter and related genera within the family Ectothiorhodospiraceae to confirm the sources of these GIs. The BLASTn results indicated that most GIs were unique to the genome of strain E85T, but the flagellar gene cluster GI_7 was shared by other Ectothiorhodospiraceae strains, suggesting that this GI might have been lost in other Spiribacter strains with smaller genomes (Figure 6B, Data S4). This result is consistent with the cell micrograph of strain E85T, which showed a single polar flagellum that was not observed in other Spiribacter strains. The functions of the E85T strain-specific GIs were diverse and included phage-related GI_1, GI_6, GI_13, and GI_15, terpene biosynthetic gene cluster GI_2, NRPS-like biosynthetic gene clusters GI_4 and GI_5, and antitoxin/toxin GI_16, GI_17, and GI_18. These GIs might have been gained through other sources. Transposases existed in GI_3, GI_4, GI_7, GI_12, GI_14, GI_15, GI_17, GI_18, GI_21, GI_26, GI_28, and GI_29, and integrases were found in GI_1, GI_2, GI_6, GI_8, GI_11, and GI_26, which might be involved in gene shuffling. The nucleic acid of a temperate phage integrated into the host genome is called a prophage (Freifelder and Meselson, 1970). The prediction results identified four prophages in the genome of strain E85T in the gene regions of 1,879,172–1,885,707, 2,751,299–2,767,792, 2,780,026–2,791,106, and 2,931,958–2,968,527 bp. The strain-specific phage-related GI_13 and GI_15 were prophages in the regions of 2,751,299–2,767,792 and 2,780,026–2,791,106 bp, respectively. The homologous proteins of these strain-specific GIs were searched using BLASTp in the standard NCBI nr database, and five proteins in GI_2 were similar to proteins from Halomonas rituensis, with approximately 50% amino acid sequence similarity (Figure S5). In GI_22, three neighboring genes, LMH63_RS17140, LMH63_RS17145, and LMH63_RS17150, corresponding to the proteins WP_109680169.1, WP_109680170.1, and WP_146205287.1, respectively, showed the highest amino acid sequence similarity with proteins WP_199257896.1 (65.98%), WP_199257897.1 (48.61%), and WP_199257898.1 (44.96%) encoded by three neighboring genes from the α-proteobacteria Paracoccus binzhouensis. In GI_29, similar results were gained. However, the sources of this strain-specific GI could not be determined due to low amino acid sequence similarity, indicating that additional detailed studies are needed to identify the source.
Description of Spiribacter halobiussp. nov.
Spiribacter halobius (ha.lo′bi.us. Gr. n. halos, salt; Gr. n. bios, life; M. L. masc. adj. halobius, living on salt).
The cells are Gram stain-negative, facultatively anaerobic, motile, straight to curved rods approximately 0.2–0.3 μm in width and 1.2–4.5 μm in length. The colonies are circular, smooth, convex, entire, cream-white, and 0.5–1.5 mm in diameter after incubation at 37°C for 3 days. Growth occurs at 20–50°C (optimum 37–40°C), pH 7.0–9.0 (optimum 7.5–8.0), and 0.5–16% (w/v) NaCl (optimum 3–6%). Nitrate can be reduced to nitrite. The cells cannot produce indole or H2S. Gelatin is weakly hydrolyzed. Starch; Tween 20, 40, 60, and 80; alginate; agar; and cellulose are not hydrolyzed. The cells are positive for citrate utilization and negative for the Voges–Proskauer reaction. Myoinositol, glycerol, glucuronamide, l-lactic acid, d-malic acid, l-malic acid, β-hydroxy-d, and l-butyric acid are oxidized. Acids are produced from mannose, d-fructose, inositol, 2-keto-potassium gluconate, and 5-keto-potassium gluconate. The cells are positive for oxidase, catalase, alkaline phosphatase, esterase lipase (C8), leucine arylamidase, valine arylamidase, naphthol-AS-BI-phosphohydrolase, α-glucosidase arginine, dihydrolase, and urease; negative for esterase (C4), lipase (C14), cystine arylamidase, trypsin, α-chymotrypsin, acid phosphatase, α-galactosidase, β-galactosidase, β-glucuronidase, β-glucosidase, N-acetyl-glucosaminidase, α-mannosidase, o-nitrophenyl-beta-d-galactopyranoside, lysine decarboxylase, ornithine decarboxylase, and tryptophan deaminase. The major respiratory quinone is ubiquinone-8. The predominant cellular fatty acids are C18:1ω7c and C16:0. The polar lipids are one hosphatidylcholine, one phosphatidylglycerol, one phosphatidylethanolamine, one diphosphatidylglycerol, one phosphoaminoglycolipid, and three phospholipids.
The type strain is E85T (= KCTC 52699T = MCCC 1H00230T) isolated from sediment collected from a marine solar saltern in Weihai, China. The DNA G+C content is 69.7% (data from genome sequence).
Materials and methods
Sampling, DNA extraction, and sequencing
Samples were collected in June 2019 from six Yuncheng Salt Lake ponds with salinities of 170, 185, 205, 230, 300, and 340‰ (Figure S6). Brine and sediment samples were collected in triplicate from each pond. After collection, samples were kept at 4°C during transport to the laboratory and −80°C after treatment. Brine samples were filtered through 0.22-μm polycarbonate membrane filters (Millipore, MA, USA). A FastDNA Spin Kit for soil (MP Biomedical, France) was used to extract DNA from all samples following the manufacturer’s protocol. The V4–V5 region of the bacterial 16S rRNA gene was amplified using the specific barcoded primer set 515F (5´barcode-GTGCCAGCMGCCGCGG-3´) and 907R (5´-CCGTCAATTCMTTTRAGTTT-3´). Sequencing libraries were generated following Illumina’s genomic DNA library preparation procedure using an Illumina MiSeq PE300 platform at Shanghai Majorbio Bio-pharm Technology Co., Ltd. (Shanghai, China). The raw reads were deposited into the NCBI Sequence Read Archive database (Bioproject Numbers: PRJNA825478 and PRJNA825481).
Sequence analyses
The pipeline of VSEARCH v2.15.2 (Rognes et al., 2016) was used for quality filtering and dereplicating sequences. The unique sequences among the remaining reads were used to define ASVs using the UNOISE3 pipeline (Edgar and Flyvbjerg, 2015) in USEARCH (version 10.0.240, http://drive5.com/usearch/) with a threshold of 100% similarity. VSEARCH was used to analyze the phylogenetic affiliation of each 16S rRNA gene sequence against the Ribosomal Database Project (v18) 16S rRNA database (Cole et al., 2013) with a confidence threshold of 60%. Chloroplast and mitochondrial sequences were removed in downstream analyses. The data statistics of each sample are listed in Table S6. The rarefication curves of each group based on the richness and Shannon index (Figure S7) indicated that the sequencing depth was sufficient to cover most of the prokaryotic sequences in the sample, which could be used for subsequent analyses.
Measurement of physicochemical factors
The water salinity and pH values of each pond were measured in situ. The sediment samples were weighed in a Petri dish and then dried for 6 h at 105°C. The sediment weights before and after drying were used to calculate the moisture content. The pH was measured through extracts by combining the dried sediment samples and water in a 1:2.5 ratio. Dried sediment samples (5 g) were mixed with 25 mL of water in a 50-mL centrifuge tube. The mixture was shaken for 3 min to obtain a 5:1 water–sediment extract. The water–sediment extracts and water samples from the salt lakes were filtered through 0.22-μm polyether sulfone membranes (Millipore, MA, USA). The solution ion concentrations of sediment samples and other ions (Cl−, Br−, , Na+, , K+, Mg2+, and Ca2+) in brine and sediment samples were measured using an ICS-1100 (Thermo, Sunnyvale, CA, USA). The dissolved organic and inorganic carbon were measured using an ISO TOC CUBE-ISOPRIME 100 (Elementar, German) and a Delta V Advantage (Thermo, Sunnyvale, CA, USA), respectively. The total nitrogen and total phosphorus contents were determined using an alkaline potassium persulphate digestion-UV spectrophotometric method (Baird et al., 2017).
Bacterial isolation and cultivation
Strain E85T was isolated from sediment of Wendeng Solar Saltern using the standard dilution plating technique on marine agar 2216 (MA; Becton Dickinson) supplemented with 10% (w/v) NaCl. The effects of different NaCl concentrations (0, 0.5, 1, 2, 4, 6, 8, 10, 12, 14, 16, 18, and 20%, w/v) on growth were assessed using a medium composed of yeast extract (0.1%), peptone (0.5%), and agar (1.8%) and prepared with artificial seawater (0.32% MgSO4, 0.23% MgCl2, 0.12% CaCl2, 0.07% KCl, and 0.02% NaHCO3, w/v). Strain E85T was cultured on modified MA plates (MA 2216 supplemented with 3% NaCl), and modified marine broth (MB 2216 supplemented with 3% NaCl) was used as the liquid medium. Cell cultures were maintained at −80°C in sterile modified MB supplemented with 20% (v/v) glycerol. The type strains S. salinus LMG 27464T from the Belgian Coordinated Collections of Microorganisms and S. curvatus CECT 8396T and S. roseus CECT 9117T from the Spanish Type Culture Collection were used as related type strains for comparative purposes in this study.
Phenotypic, physiological, and biochemical characteristics
The influence of temperature on the growth of visible colonies on modified MA plates was tested at 15, 20, 25, 28, 30, 33, 37, 42, 45, 50, and 55°C for approximately 7 days. To test the effects of pH on growth, the pH of modified MB was adjusted by adding buffer solutions: MES (pH 5.5 and 6.0), PIPES (pH 6.5 and 7.0), HEPES (pH 7.5 and 8.0), tricine (pH 8.5), and CAPSO (pH 9.0 and 9.5). After incubation at 37°C for 3 days, the OD600 values of the cultures were measured, and the morphological and physiological features of E85T on modified MA plates or in modified MB were examined. All tests were independently conducted twice with three replicates. Cell size and morphology were observed using a transmission electron microscope (JEM-1200EX, JEOL). Motility was examined according to the hanging-drop method using a light microscope (E600, Nikon), and gliding motility was determined according to the method of (Bowman, 2000). Gram staining was performed as described by (Smibert and Krieg, 1994).
Anaerobic growth was determined by placing inoculated plates containing modified MA with or without 0.1% (w/v) KNO3 in an anaerobic jar. Modified MB supplemented with 0.1% (v/v) nitrate in test tubes was used to assess nitrate reduction. The inoculated test tubes were placed in aerobic and anaerobic conditions for cultivation. Uninoculated test tubes served as controls. Catalase activity was detected by the production of bubbles after the addition of a drop of 3% (v/v) H2O2, and oxidase activity was determined using a bioMérieux oxidase reagent kit. Starch, cellulose, lipid, and alginate hydrolysis were tested on modified MA plates supplemented with 0.2% (w/v) soluble starch, 0.5% (w/v) CM-cellulose, 1% (v/v) Tweens (20, 40, 60, and 80), and 2% (w/v) sodium alginate, respectively (Smibert and Krieg, 1994). Tests for other physiological and biochemical characteristics were determined with API 20E and API ZYM (bioMérieux), and the ability to oxidize different compounds was tested using Biolog GEN III. Acid production from carbohydrates was tested using an API 50CHB fermentation kit (bioMérieux). All API and Biolog tests were conducted according to the manufacturers’ instructions, replacing the suspension with sterilized 6% (w/v) NaCl solution. The results were recorded every 12 h after incubation at 37°C for up to 7 days. All API and Biolog tests were also performed in duplicate on the related type strains S. salinus LMG 27464T, S. curvatus CECT 8396T, and S. roseus CECT 9117T. Antibiotic sensitivity was assessed as described by the Clinical and Laboratory Standards Institute (CLSI, 2022). Briefly, a cell suspension (McFarland standard 0.5) was swabbed over the surface of modified MA plates to create a uniform lawn, and antibiotic discs were aseptically placed onto the agar surface. Inoculated plates were incubated at 37°C for up to 7 days.
Chemotaxonomic characterization
To determine the respiratory quinone, cellular fatty acid composition, and polar lipid profile, cultures shaken at 37°C for 3 days were harvested and freeze-dried. Polar lipids analysis was performed by the Identification Service of DSMZ (Braunschweig, Germany). Respiratory quinone was extracted, purified, and analyzed by HPLC (Kroppenstedt, 1982). Fatty acids were extracted, methylated, and analyzed according to the standard protocol of MIDI (Sherlock Microbial Identification System, version 6.1; (Sasser, 1990).
16S rRNA gene sequence analyses
The 16S rRNA gene of strain E85T was amplified by PCR using the universal primers 27F and 1492R as previously described (Liu et al., 2014). The PCR products were purified, ligated to the pMD18-T vector (Takara), and cloned according to the manufacturer’s instructions. Sequencing was performed by Life Biotechnology (Shanghai). The 16S rRNA gene sequence for strain E85T was submitted to the NCBI GenBank database. Gene similarities were calculated using the EzBioCloud Database (Yoon et al., 2017a). 16S rRNA gene sequence alignment was performed using the CLUSTAL_X program (Thompson et al., 1997), and positions with insertions or deletions were excluded during calculations. Phylogenetic trees were constructed using the neighbor-joining (Saitou and Nei, 1987), maximum-parsimony (Fitch, 1971), and maximum-likelihood (Felsenstein, 1981) methods with MEGA X (Kumar et al., 2018). The reliability of relationships was ensured by performing bootstrap analyses based on 1,000 replications.
Genome sequencing, assembly, and annotation
Genomic DNA of strain E85T was obtained using a genomic DNA extraction kit (Takara), and the DNA G+C content was determined by HPLC (Mesbah et al., 1989). The genome was sequenced by the Shanghai Personal Biotechnology Co., China, using the Illumina HiSeq platform and Pacbio platform. The Illumina raw sequencing data were assembled using SOAPdenovo (Li et al., 2008), and the PacBio genomes were assembled using SMRT Link (v5.0.1). The phylogenetic relationships based on amino acid sequences were analyzed via GTDB-Tk, and the phylogenetic trees were reconstructed using FastTree with JTT+CAT parameters and IQTree with the LG+F+I+G4 model and 1,000 bootstrap replicates based on the 26 genomes. The average nucleotide identity was calculated using the EzGenome web service (Yoon et al., 2017b). The Genome-to-Genome Distance Calculator (GGDC 2.0, http://ggdc.dsmz.de, (Meier-Kolthoff et al., 2013) was used to calculate digital DNA-DNA hybridization. The average amino-acid identity values were calculated based on BLAST algorithm (Konstantinidis and Tiedje, 2005).
All genomes were annotated using NCBI Prokaryote Genome Automatic Annotation Pipeline (Tatusova et al., 2016). For potential function analysis, the draft genome was annotated using KofamKOALA (https://www.genome.jp/tools/kofamkoala/; (Aramaki et al., 2020), eggNOG (http://eggnog-mapper.embl.de/; (Huerta-Cepas et al., 2019), and dbCAN2 meta server (http://bcb.unl.edu/dbCAN2; (Zhang et al., 2018). Potential prophage sequences in the genome were identified and categorized as intact, incomplete, or questionable using the PHASTER website (http://phaster.ca/; (Arndt et al., 2016). CRISPRCasFinder was used to predict CRISPR in the genomes (https://crispr.i2bc.paris-saclay.fr/Server/; (Grissa et al., 2007). Insertion sequences located within the genome were identified using the ISfinder website (https://wwwis.biotoul.fr/; (Siguier et al., 2006). Interspersed repetitive sequences and tandem repetitive sequences were identified using the RepeatMasker Web Server (http://www.repeatmasker.org/cgi-bin/WEBRepeatMasker/) and Tandem Repeats Finder (https://tandem.bu.edu/trf/trf.html; (Benson, 1999), respectively. Genomic islands were identified using IslandViewer 4 (https://www.pathogenomics.sfu.ca/islandviewer; (Bertelli et al., 2017). Ortholog clusters were identified using the pan-genomes analysis pipeline (http://pgaweb.vlcc.cn; (Zhao et al., 2012). The genome map was generated using Proksee (https://proksee.ca; (Grant and Stothard, 2008), and the Venn diagram was created using the jvenn platform (http://jvenn.toulouse.inra.fr; (Bardou et al., 2014).
Data availability statement
The data presented in the study are deposited in the GenBank and Sequence Read Archive. The accession number for the 16S rRNA gene sequence of strain E85T is KY407792.1. The accession number of draft genome and complete genome are QFFI00000000.1 and NZ_CP086615.1, respectively. The accession number of 16S rRNA gene sequence raw data from all brine and sediment samples of Yuncheng Salt Lake are PRJNA825478 and PRJNA825481.
Author contributions
D-CL isolated the strain E85T. YG, LM, Z-ZD, and W-SZ performed material preparation, experimental operation, data collection, and analyses. YG and LM wrote the manuscript. YG and Z-JD performed project guidance and critical revision of manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Science & Technology Fundamental Resources Investigation Program (Grant No. 2019FY100700) and the National Natural Science Foundation of China (Grant No. 32070002, 31900091).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1028967/full#supplementary-material
References
Aramaki T., Blanc-Mathieu R., Endo H., Ohkubo K., Kanehisa M., Goto S., et al. (2020). KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinf. (Oxford England) 36 (7), 2251–2252. doi: 10.1093/bioinformatics/btz859
Arndt D., Grant J. R., Marcu A., Sajed T., Pon A., Liang Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44 (W1), W16–W21. doi: 10.1093/nar/gkw387
Auch A. F., von Jan M., Klenk H.-P., Göker M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. genomic Sci. 2 (1), 117–134. doi: 10.4056/sigs.531120
Baird R. B., Eaton A. D., Rice E. W. (2017). Standard methods for the examination of water and wastewater. 23rd ed (Washington DC: American Water Works Association).
Bardou P., Mariette J., Escudié F., Djemiel C., Klopp C. (2014). Jvenn: an interactive Venn diagram viewer. BMC Bioinf. 15 (1), 293. doi: 10.1186/1471-2105-15-293
Belilla J., Moreira D., Jardillier L., Reboul G., Benzerara K., Lopez-Garcia J. M., et al. (2019). Hyperdiverse archaea near life limits at the polyextreme geothermal dallol area. Nat. Ecol. Evol. 3 (11), 1552–1561. doi: 10.1038/s41559-019-1005-0
Benson G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27 (2), 573–580. doi: 10.1093/nar/27.2.573
Bertelli C., Laird M. R., Williams K. P., Lau B. Y., Hoad G., Winsor G. L., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45 (W1), W30–W35. doi: 10.1093/nar/gkx343
Bolhuis H., Poele E. M., Rodriguez-Valera F. (2004). Isolation and cultivation of walsby’s square archaeon. Environ. Microbiol. 6 (12), 1287–1291. doi: 10.1111/j.1462-2920.2004.00692.x
Bowman J. P. (2000). Description of cellulophaga algicola sp. nov., isolated from the surfaces of Antarctic algae, and reclassification of Cytophaga uliginosa (ZoBell and upham 1944) reichenbach 1989 as Cellulophaga uliginosa comb. nov. Int. J. Syst. evol. Microbiol. 50 Pt 5, 1861–1868. doi: 10.1099/00207713-50-5-1861
Chen X., Zhang Y., Zhang Z., Zhao Y., Sun C., Yang M., et al. (2018). PGAweb: A web server for bacterial pan-genome analysis. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.01910
CLSI (2022). Performance standards for antimicrobial susceptibility testing, 32nd ed. Malvern, PA: Clinical and Laboratory Standards Institute.
Cole J. R., Wang Q., Fish J. A., Chai B., McGarrell D. M., Sun Y., et al. (2013). Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42 (D1), D633–D642. doi: 10.1093/nar/gkt1244
Durand A., Bourbon M.-L., Steunou A.-S., Khalfaoui-Hassani B., Legrand C., Guitton A., et al. (2018). Biogenesis of the bacterial cytochrome oxidase: Active subcomplexes support a sequential assembly model. J. Biol. Chem. 293 (3), 808–818. doi: 10.1074/jbc.M117.805184
Edgar R. C., Flyvbjerg H. (2015). Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinf. (Oxford England) 31 (21), 3476–3482. doi: 10.1093/bioinformatics/btv401
Felsenstein J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17 (6), 368–376. doi: 10.1007/BF01734359
Fernández A. B., Ghai R., Martin-Cuadrado A.-B., Sánchez-Porro C., Rodriguez-Valera F., Ventosa A. (2014). Prokaryotic taxonomic and metabolic diversity of an intermediate salinity hypersaline habitat assessed by metagenomics. FEMS Microbiol. Ecol. 88 (3), 623–635. doi: 10.1111/1574-6941.12329
Fitch W. M. (1971). Toward defining the course of evolution: minimum change for a specific tree topology. Syst. Biol. 20 (4), 406–416. doi: 10.1093/sysbio/20.4.406
Freifelder D., Meselson M. (1970). Topological relationship of prophage λ to the bacterial chromosome in lysogenic cells. Proc. Natl. Acad. Sci. United States America 65 (1), 200–205. doi: 10.1073/pnas.65.1.200
Frost L. S., Leplae R., Summers A. O., Toussaint A. (2005). Mobile genetic elements: the agents of open source evolution. Nat. Rev. Microbiol. 3 (9), 722–732. doi: 10.1038/nrmicro1235
Ghai R., Pašić L., Fernández A. B., Martin-Cuadrado A.-B., Mizuno C. M., McMahon K. D., et al. (2011). New abundant microbial groups in aquatic hypersaline environments. Sci. Rep. 1, 135. doi: 10.1038/srep00135
Goris J., Konstantinidis K. T., Klappenbach J. A., Coenye T., Vandamme P., Tiedje J. M. (2007). DNA-DNA Hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57 (Pt 1), 81–91. doi: 10.1099/ijs.0.64483-0
Grant J. R., Stothard P. (2008). The CGView server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36 (Web Server issue), W181–W184. doi: 10.1093/nar/gkn179
Grissa I., Vergnaud G., Pourcel C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35 (Web Server issue), W52–W57. doi: 10.1093/nar/gkm360
Gunde-Cimerman N., Plemenitaš A., Oren A. (2018). Strategies of adaptation of microorganisms of the three domains of life to high salt concentrations. FEMS Microbiol. Rev. 42 (3), 353–375. doi: 10.1093/femsre/fuy009
Huerta-Cepas J., Szklarczyk D., Heller D., Hernández-Plaza A., Forslund S. K., Cook H., et al. (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47 (D1), D309–D314. doi: 10.1093/nar/gky1085
Ingram-Smith C., Martin S. R., Smith K. S. (2006). Acetate kinase: not just a bacterial enzyme. Trends Microbiol. 14 (6), 249–253. doi: 10.1016/j.tim.2006.04.001
Juhas M., van der Meer J. R., Gaillard M., Harding R. M., Hood D. W., Crook D. W. (2009). Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33 (2), 376–393. doi: 10.1111/j.1574-6976.2008.00136.x
Konstantinidis K. T., Rosselló-Móra R., Amann R. (2017). Uncultivated microbes in need of their own taxonomy. ISME J. 11 (11), 2399–2406. doi: 10.1038/ismej.2017.113
Konstantinidis K. T., Tiedje J. M. (2004). Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl. Acad. Sci. U S A 101 (9), 3160–3165. doi: 10.1073/pnas.0308653100
Konstantinidis K. T., Tiedje J. M. (2005). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187 (18), 6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005
Kroppenstedt R. M. (1982). Separation of bacterial menaquinones by HPLC using reverse phase (RP18) and a silver loaded ion exchanger as stationary phases. J. Liquid Chromatogr. 5 (12), 2359–2367. doi: 10.1080/01483918208067640
Kumar S., Stecher G., Li M., Knyaz C., Tamura K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35 (6), 1547–1549. doi: 10.1093/molbev/msy096
León M. J., Fernández A. B., Ghai R., Sánchez-Porro C., Rodriguez-Valera F., Ventosa A. (2014). From metagenomics to pure culture: isolation and characterization of the moderately halophilic bacterium Spiribacter salinus gen. nov., sp. nov. Appl. Environ. Microbiol. 80 (13), 3850–3857. doi: 10.1128/AEM.00430-14
León M. J., Galisteo C., Ventosa A., Sánchez-Porro C. (2020). Spiribacter aquaticus Leon et al. 2017 is a later heterotypic synonym of Spiribacter roseus Leon et al. 2016. Reclassification of Halopeptonella vilamensis Menes et al. 2016 as Spiribacter vilamensis comb. nov. Int. J. Syst. evol. Microbiol. 70 (4), 2873–2878. doi: 10.1099/ijsem.0.004113
León M. J., Ghai R., Fernandez A. B., Sanchez-Porro C., Rodriguez-Valera F., Ventosa A. (2013). Draft genome of Spiribacter salinus M19-40, an abundant gammaproteobacterium in aquatic hypersaline environments. Genome Announc. 1 (1),e00179-12. doi: 10.1128/genomeA.00179-12
León M. J., Hoffmann T., Sanchez-Porro C., Heider J., Ventosa A., Bremer E. (2018). Compatible solute synthesis and import by the moderate halophile Spiribacter salinus: Physiology and genomics. –. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00108
León M. J., Rodríguez-Olmos Á., Sánchez-Porro C., López-Pérez M., Rodríguez-Valera F., Soliveri J., et al. (2015). Spiribacter curvatus sp. nov., a moderately halophilic bacterium isolated from a saltern. Int. J. Syst. Evol. Microbiol. 65 (12), 4638–4643. doi: 10.1099/ijsem.0.000621
León M. J., Vera-Gargallo B., Sánchez-Porro C., Ventosa A. (2016). Spiribacter roseus sp. nov., a moderately halophilic species of the genus Spiribacter from salterns. Int. J. Syst. evol. Microbiol. 66 (10), 4218–4224. doi: 10.1099/ijsem.0.001338
Li R., Li Y., Kristiansen K., Wang J. (2008). SOAP: short oligonucleotide alignment program. Bioinformatics 24 (5), 713–714. doi: 10.1093/bioinformatics/btn025
Liu Q.-Q., Li X.-L., Rooney A. P., Du Z.-J., Chen G.-J. (2014). Tangfeifania diversioriginum gen. nov., sp. nov., a representative of the family Draconibacteriaceae. Int. J. Syst. evol. Microbiol. 64 (Pt 10), 3473–3477. doi: 10.1099/ijs.0.066902-0
López-Pérez M., Ghai R., Leon M. J., Rodríguez-Olmos Á., Copa-Patiño J. L., Soliveri J., et al. (2013). Genomes of “Spiribacter”, a streamlined, successful halophilic bacterium. BMC Genomics 14, 787. doi: 10.1186/1471-2164-14-787
Meier-Kolthoff J. P., Auch A. F., Klenk H.-P., Göker M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf. 14, 60. doi: 10.1186/1471-2105-14-60
Mesbah M., Premachandran U., Whitman W. B. (1989). Precise measurement of the g+ c content of deoxyribonucleic acid by high-performance liquid chromatography. Int. J. Syst. Evol. Microbiol. 39 (2), 159–167. doi: 10.1099/00207713-39-2-159
Naor A., Lapierre P., Mevarech M., Papke R. T., Gophna U. (2012). Low species barriers in halophilic archaea and the formation of recombinant hybrids. Curr. Biol. 22 (15), 1444–1448. doi: 10.1016/j.cub.2012.05.056
Narasingarao P., Podell S., Ugalde J. A., Brochier-Armanet C., Emerson J. B., Brocks J. J., et al. (2012). De novo metagenomic assembly reveals abundant novel major lineage of archaea in hypersaline microbial communities. ISME J. 6 (1), 81–93. doi: 10.1038/ismej.2011.78
Papke R. T., Koenig J. E., Rodríguez-Valera F., Doolittle W. F. (2004). Frequent recombination in a saltern population of Halorubrum. Science 306 (5703), 1928–1929. doi: 10.1126/science.1103289
Pontefract A., Zhu T. F., Walker V. K., Hepburn H., Lui C., Zuber M. T., et al. (2017). Microbial diversity in a hypersaline sulfate lake: A terrestrial analog of ancient mars. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.01819
Rognes T., Flouri T., Nichols B., Quince C., Mahé F. (2016). VSEARCH: A versatile open source tool for metagenomics. PeerJ 4, e2584. doi: 10.7717/peerj.2584
Saccò M., White N. E., Harrod C., Salazar G., Aguilar P., Cubillos C. F., et al. (2021). Salt to conserve: a review on the ecology and preservation of hypersaline ecosystems. Biol. Rev. Camb. Philos. Soc. 96 (6), 2828–2850. doi: 10.1111/brv.12780
Saitou N., Nei M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4 (4), 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Sasser M. (1990). Identification of bacteria by gas chromatography of cellular fatty acids. USFCC Newsl 20, 1–6.
Shu W.-S., Huang L.-N. (2021). Microbial diversity in extreme environments. Nat. Rev. Microbiol. 20 (4), 219–235. doi: 10.1038/s41579-021-00648-y
Siguier P., Gourbeyre E., Chandler M. (2014). Bacterial insertion sequences: their genomic impact and diversity. FEMS Microbiol. Rev. 38 (5), 865–891. doi: 10.1111/1574-6976.12067
Siguier P., Perochon J., Lestrade L., Mahillon J., Chandler M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34 (Database issue), D32–D36. doi: 10.1093/nar/gkj014
Smibert R. M., Krieg N. R. (1994). Phenotypic characterization (Washington DC: Society for Microbiology).
Song T., Liang Q., Du Z., Wang X., Chen G., Du Z., et al. (2022). Salinity gradient controls microbial community structure and assembly in coastal solar salterns. Genes 13 (2), 385. doi: 10.3390/genes13020385
Tatusova T., DiCuccio M., Badretdin A., Chetvernin V., Nawrocki E. P., Zaslavsky L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44 (14), 6614–6624. doi: 10.1093/nar/gkw569
Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F., Higgins D. G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25 (24), 4876–4882. doi: 10.1093/nar/25.24.4876
Ventosa A., de la Haba R. R., Sánchez-Porro C., Papke R. T. (2015). Microbial diversity of hypersaline environments: a metagenomic approach. Curr. Opin. Microbiol. 25, 80–87. doi: 10.1016/j.mib.2015.05.002
Xue Q., Zhao D., Zhang S., Zhou H., Zuo Z., Zhou J., et al. (2021). Highly integrated adaptive mechanisms in Spiribacter halalkaliphilus, a bacterium abundant in Chinese soda-saline lakes. Environ. Microbiol. 23 (11), 6463–6482. doi: 10.1111/1462-2920.15794
Yoon S.-H., Ha S.-M., Kwon S., Lim J., Kim Y., Seo H., et al. (2017a). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. evol. Microbiol. 67 (5), 1613–1617. doi: 10.1099/ijsem.0.001755
Yoon S. H., Ha S. M., Lim J., Kwon S., Chun J. (2017b). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110 (10), 1281–1286. doi: 10.1007/s10482-017-0844-4
Zhang H., Yohe T., Huang L., Entwistle S., Wu P., Yang Z., et al. (2018). dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46 (W1), W95–W101. doi: 10.1093/nar/gky418
Zhao Y., Wu J., Yang J., Sun S., Xiao J., Yu J. (2012). PGAP: Pan-genomes analysis pipeline. Bioinf. (Oxford England) 28 (3), 416–418. doi: 10.1093/bioinformatics/btr655
Keywords: marine bacteria, Spiribacter, insertion sequences, genomic islands, hypersaline environments
Citation: Gong Y, Ma L, Du Z-Z, Zheng W-S, Lu D-C and Du Z-J (2022) Spiribacter halobius sp. nov., a novel halophilic Gammaproteobacterium with a relatively large genome. Front. Mar. Sci. 9:1028967. doi: 10.3389/fmars.2022.1028967
Received: 26 August 2022; Accepted: 07 December 2022;
Published: 23 December 2022.
Edited by:
Martin W. Hahn, University of Innsbruck, AustriaReviewed by:
Huahua Jian, Shanghai Jiao Tong University, ChinaHeng-Lin Cui, Jiangsu University, China
Copyright © 2022 Gong, Ma, Du, Zheng, Lu and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zong-Jun Du, ZHV6b25nanVuQHNkdS5lZHUuY24=
†ORCID: Zong-Jun Du, orcid.org/0000-0002-7886-5667