 Carlos Henríquez-Castillo1,2*†
Carlos Henríquez-Castillo1,2*† Alvaro M. Plominsky3†
Alvaro M. Plominsky3† Salvador Ramírez-Flandes4,5
Salvador Ramírez-Flandes4,5 Anthony D. Bertagnolli6,7
Anthony D. Bertagnolli6,7 Frank J. Stewart6,7
Frank J. Stewart6,7 Osvaldo Ulloa4,5
Osvaldo Ulloa4,5- 1Laboratorio de Fisiología y Genética Marina, Centro de Estudios Avanzados en Zonas Áridas, Coquimbo, Chile
- 2Facultad de Ciencias del Mar, Universidad Católica del Norte, Coquimbo, Chile
- 3Marine Biology Research Division, Scripps Institution of Oceanography, University of California, San Diego, La Jolla, CA, United States
- 4Instituto Milenio de Oceanografía, Universidad de Concepción, Concepción, Chile
- 5Departamento de Oceanografía, Universidad de Concepción, Concepción, Chile
- 6Department of Microbiology and Cell Biology, Montana State University, Bozeman, MT, United States
- 7School of Biological Sciences and Center for Microbial Dynamics and Infection, Georgia Institute of Technology, Atlanta, GA, Bozeman, United States
Gammaproteobacteria of the genus Alteromonas are prominent members of pelagic marine microbial communities, playing critical roles in the aerobic degradation of particulate organic matter. Comparative genomic studies of these microorganisms have mainly focused on the metabolic and genomic plasticity of strains isolated primarily from oxygenated environments. In this study, we show that Alteromonas significantly contribute to marine microbial communities from suboxic waters ([O2] < 5 uM) in both the free-living (FL) and particle-attached (PA) fractions, but considerably decrease in abundance in the anoxic waters. The highest proportion of Alteromonas transcripts was found within the secondary fluorescence maximum (SFM) of Oxygen Minimum Zones (OMZs). This metatranscriptomic information suggests an in situ coupling of Alteromonas iron (Fe) and carbon metabolisms, and a relevant role of the glyoxylate cycle across the different layers of the OMZs. This study demonstrates that Alteromonas is an abundant and active member of the OMZ microbial communities, with a potentially significant impact on the carbon cycling in these ecosystems. These results provide valuable environmental evidence to support previous culture-based studies assessing the physiology and ecology of these ubiquitous marine heterotrophs under low-oxygen conditions.
Introduction
Bacteria of the gammaproteobacterial genus Alteromonas are ecologically relevant copiotrophs that inhabit almost every marine environment, from surface waters to bathypelagic depths (Mikhailov et al., 2006; Nayfach et al., 2016). Members of this genus are considered r-strategists that can proliferate when organic nutrients are available (Math et al., 2012) and are typically associated with particles or aggregates (López-Pérez and Rodríguez-Varela, 2016). A study from Lekunberri et al. (2013) found that the relative abundance of Alteromonas increased with depth in the tropical Atlantic. Also, Schattenhofer et al. (2009) showed that Alteromonas was more abundant in the euphotic layer than in mesopelagic waters in a latitudinal Atlantic transect. Similar results were reported by Dobal-Amador et al. (2016) off the Galician coast of Spain. In terms of activity, Alteromonas has repeatedly accounted for most of the increase in cell abundance and activity in a variety of microcosms (Jin et al., 2016; Aguayo et al., 2020).
Alteromonas ecotypes can occupy diverse niches, with such plasticity linked to the presence of genes associated with genomic islands or mobile elements (López-Pérez and Rodriguez-Valera, 2016; López-Pérez et al., 2017; Koch et al., 2020). Surface ecotypes are enriched in genes involved in degrading simple carbohydrates, amino acids, and regulatory elements (e.g., two-component systems). Contrastingly, deep ecotypes seem specialized to microaerophilic conditions and the degradation of complex organic matter while attached to sinking particles (Ivars-Martinez et al., 2008a). Regarding metabolic activity, coastal and oceanic Alteromonas have shown strain-specific responses to Fe-limitation according to their different strategies for carbon metabolism and energy acquisition (Fourquez et al., 2014).
Eastern boundary oceanic ecosystems are among the most productive on Earth. In these regions, reduced circulation and high productivity at the surface contribute to enhanced oxygen consumption and the formation of oxygen minimum zones (OMZs) at intermediate depths (Messié and Chavez, 2015). Within the OMZs from the Eastern Tropical North and South Pacific (ETNP and ETSP, respectively) and the Arabian Sea, dissolved oxygen concentrations often drop below the detection limit of the most sensitive modern sensors (< 1–10 nM) (Revsbech et al., 2009). Thus, these areas have also been called ‘Anoxic Marine Zones’ (AMZs; Ulloa et al., 2012) and can be distinguished by a peak in the accumulation of nitrite, which typically occurs within their anoxic core when O2 falls below the nanomolar detection limit (Thamdrup et al., 2012). These systems are characterized by a remarked redox gradients that profoundly impacts the composition and activities of microbial communities at the base of marine food webs (Medina et al., 2017). Notably, AMZs may have a secondary fluorescence maximum (SFM) within the lower euphotic depths that overlap with the anoxic layer (Tiano et al., 2014; Banse et al., 2017; Garcia-Robledo et al., 2017). Studies in the ETNP suggest two important aspects regarding the presence of the anoxic SFM: the covariation of oxygen in its spatial distribution, and the association between the SFM and the 26.0 kg m-3 isopycnal (Cepeda-Morales et al., 2009; Márquez-Artavia et al., 2019). This phenomenon is also linked to the presence of unique ecotypes of the cyanobacterium Prochlorococcus (Lavin et al., 2010; Ulloa et al., 2021), whose oxygenic photosynthesis drives a cryptic oxygen cycle in these anoxic waters, and provides organic matter to the microbial community (Garcia-Robledo et al., 2017, Fuchsman et al., 2019).
However, the biogeography, genomic potential, and environmental impact of Alteromonas in these marine regions has been briefly documented regarding their representatives inhabiting the oxic layers of the OMZs (Ivars-Martinez 2008a, Ivars-Martinez 2008b, López-Pérez et al., 2017; Koch et al., 2020). Moreover, there is an overall lack of information regarding the abundance, metabolic potential, and in situ transcriptional activity of Alteromonas representatives across the redox gradients of low-oxygen environments, such as OMZs. This study addressed these aspects of the Alteromonas populations inhabiting the Pacific Ocean AMZs by using various 16S rRNA gene amplicon-based ecological analysis, as well as metabolic assessments based on single-cell genome and metatranscriptomic sequencing. Through these multi-omic approaches we demonstrate that Alteromonas spp. are an abundant, and active group of the AMZ microbial communities, especially within the SFM, with potentially major impacts on carbon cycling in these ecosystems.
Material and methods
16S rRNA gene amplicon processing
Samples from the ETSP-AMZ were collected during the LowPhOx I and II cruises (November 2015 and February 2018, respectively). Additional samples corresponding to the SFM of both the ETNP and ETSP AMZs were collected during four cruises: NH1410 (May 2014) and RB1603 (April 2016) for the ETNP, and NBP1305 (June 2013) and AT2626 (January 2015) for the ETSP (Padilla et al., 2016; Padilla et al., 2017; Aldunate et al., 2020). Samples were collected based on the fluorescence and oxygen profiles of each station, sampling water from the surface, oxycline, the SFM, and the core of the anoxic layer. The collection of water samples was performed using a pump profiler system, an instrument that pumps water directly from the desired depth while profiling the water column with an attached conductivity‐temperature‐depth (CTD) system (Seabird SBE‐19 plus for ETNP and Seabird SBE‐25 for ETSP). For the LowpHOx II cruise, seawater samples were collected using 10L Niskin bottles. Seawater was sequentially filtered through 20 µm, 3 µm, and 0.2 µm pore size diameter filters (Millipore, Darmstadt, Germany). Total DNA was extracted from the 0.2 - 3 µm (Free-Living) and 3 - 20 µm (Particle-Attached) fractions with the Phenol: Chloroform protocol described by Sambrook and Russell, 2006. Additional metabarcoding samples from the ETNP-AMZ and from Golfo Dulce (GD), an anoxic tropical basin with a O2/H2S interphase, were obtained from the NCBI SRA repository. These samples corresponded to two size fractions, from 0.22 to 1.6 µm (here also considered as Free-Living) and from 1.6 to 30 µm (here also considered as Particle-Attached) for samples from the ETNP (Ganesh et al., 2015), and only for 0.22-1.6 for GD samples (Padilla et al., 2016). Accession numbers are in Table S1. All samples analyzed here were sequenced using the modified universal F515 (5’-GTGYCAGCMGCCGCGGTAA-3’, Parada et al., 2016) and 806R (5’-GGACTACNVGGGTWTCTAAT-3’, Apprill et al., 2015) primer pairs (Walters et al., 2016), which amplify the hyper-variable V4 region of the 16S rRNA gene, and the Illumina MiSeq platform. All amplicon sequence data were analyzed together using the DADA2 package (v1.11.3) (Callahan et al., 2016) implemented in R (v3.4.4). The primers were removed using cutadapt (v1.2.1) (Martin, 2011), and the sequences from each pair were trimmed to 210 and 190 bases respectively and quality filtered (truncLen=c(210, 190), maxEE=1, maxN=0, truncQ=11, rm.phix=TRUE). Amplicon Sequence Variants (ASVs) were inferred from de-replicated sequences. Chimeras were removed using the “consensus” removal method. Taxonomic assignment was performed using the Silva v132 16S rRNA gene database (Quast et al., 2013). The relative abundance of ASVs in each sample and community analysis (Observed species, alpha-diversity indices) calculated from subsampled datasets were performed in the R environment with the phyloseq package (McMurdie and Holmes, 2013). Inference of microbial interaction networks was performed through statistical co-occurrence using FlashWeave with the sensitive option (Tackmann et al., 2019).
Clustering and correlations analysis
The ASVs count table was normalized using the variance stabilization transformation through the DESeq package (Bolaños et al., 2020). Hierarchical clustering was performed using the Euclidean distance of the normalized count table, and the WardD2 method was applied for the linkage. A principal component analysis was also performed using the normalized count table. Inference of microbial interaction networks was performed through statistical co-occurrence using FlashWeave (Tackmann et al., 2019) with the sensitive option. We used the function envfit to fit environmental variables onto ordination. All these analyses were performed in the R environment.
Single Amplified Genomes (SAGs)
Samples were collected on 19th November, 2010, during the BiGRAPA cruise from the Secondary Fluorescence Maximum (53 m depth) of the ETSP-AMZ off the coast of Iquique, Chile (Table S3). Single-cell sorting, whole-genome amplification, 16S rRNA real-time PCR screening, and PCR product sequencing were performed at the Bigelow Laboratory for Ocean Sciences, Single Cell Genomics Center (SCGC, www.bigelow.org/scgc), as described previously (Stepanauskas et al., 2017). This procedure identified 19 SAGs affiliated to Alteromonas, based on the 16S rRNA partial sequences obtained during the SAG screening (Plominsky et al., 2018). Single cells were sequenced utilizing the HiSeq 2000 platform (Canada’s Michael Smith Genome Sciences Centre). Artifactual sequences were filtered from the raw data, and draft SAGs were assembled using SPAdes 3.5 with default parameters (Bankevich et al., 2013). SAG contigs >1Kbp in length were decontaminated using ProDeGe (Tennessen et al., 2016) and uploaded for gene prediction and annotation through the IMG/MER pipeline (Markowitz et al., 2009). SAG completeness and contamination were estimated by the presence of conserved single-copy genes using CheckM v1.0.7 (Parks et al., 2015). Average nucleotide identity (ANI) was computed using fastANI (Jain et al., 2018).
Pangenome analysis
Based on 16S rRNA and ANI comparisons, contigs from similar SAGs (>97% similarity) were analyzed as Alteromonas OMZ “populations”. The operational definition of “population” is described in Delmont and Eren (2018), as an agglomerate of naturally occurring microbial cells, genomes of which are similar enough to align to the same genomic reference with high sequence identity as defined by the read recruitment stringency. Reference Alteromonas genomes (76) were downloaded from the IMG/MER platform and the NCBI SRA repository (accession numbers available in Table S3). Pangenome analysis was performed using the Anvi’o software v5 (Eren et al., 2015) by clustering based on the presence/absence of genes in each genome with the mcl algorithm (inflation = 8) through Euclidean distance and the wardD2 linkage method. Annotation of predicted genes was performed using DIAMOND (Buchfink et al., 2015) with the sensitive option to find homologs in different databases, including the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000), Pfam (Mistry et al., 2021), the Clusters of Orthologous Groups (COGs) (Tatusov et al., 2000). The distribution of genes, COG categories, KEGG Orthologs, and Pfam modules across genomes were analyzed in the R environment. A phylogenetic tree was generated using 137 single-copy genes obtained with the Anvi’o function anvi-get-sequences-for-hmm-hits with the -return-best-hit option. The phylogenetic tree was inferred from the concatenated alignment of the single-copy genes using IQ-TREE (Nguyen et al., 2015).
Metatranscriptome read recruitments
Metatranscriptomes from OMZs were downloaded from the NCBI SRA (reported in Stewart et al., 2012; Padilla et al., 2016 and Garcia-Robledo et al., 2017; Table S6). Raw metatranscriptomic sequences were quality filtered using Trimmomatic (Bolger et al., 2014) and the protein-encoding genes were functionally predicted using the KEGG database. The Alteromonas genomes and SAGs were used to recruit reads from the metatranscriptomic dataset using DIAMOND, with a cutoff nucleotide identity of 97% over a minimum alignment length of 100 nucleotides and a bitscore >50. The relative recruitment over each Alteromonas genome was calculated based on the number of KOs detected in the metatranscriptome associated with the Alteromonas genomes and normalized to the total number of KO predicted in each Metatranscriptome multiplied by 100. Protein-coding gene abundances (those with a KO) were normalized by the fraction of KEGG assigned to Alteromonas from the total KEGG assigned in each metatranscriptome.
Results
Distribution of Alteromonas along the OMZ redox gradient and among biomass size fractions
The diversity of Alteromonas Amplicon Sequence Variants (ASVs) in the free-living (FL) and the particle-attached (PA) biomass fractions was assessed in geographically diverse locations across the ETNP and ETSP AMZs (Figure 1A). The sections sampled were classified according to their water chemistry (Oxygen and Nitrite concentrations) and collection depth as Surface (depths of 0 – 30 m); SFM (within the Secondary Fluorescence Maximum); Oxycline ([O2] > 5 uM); Suboxic ([O2] < 5 uM); Anoxic Core ([O2] < 5 uM and > 1 µM Nitrite); and Mesopelagic (depths below 500 m; [O2] > 5 uM) (Table S1).
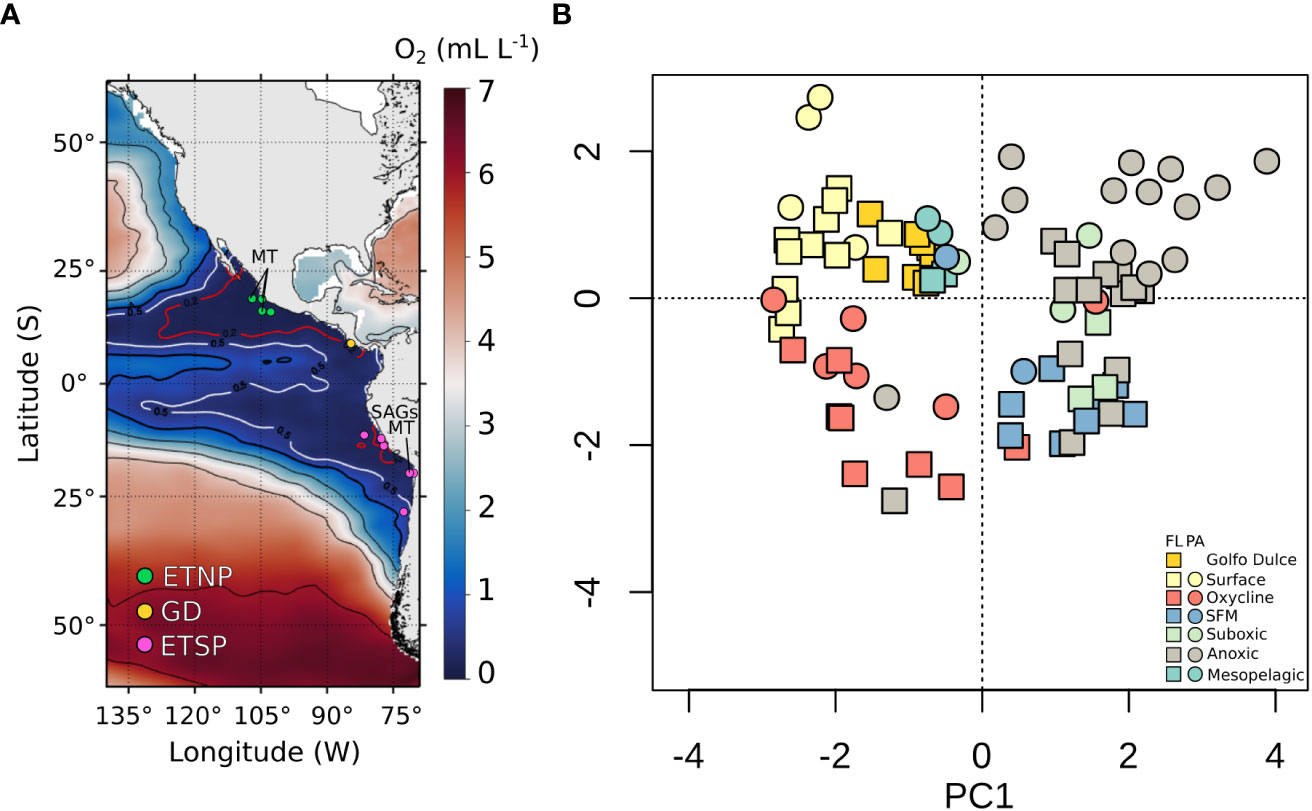
Figure 1 Geographic distribution of samples and prokaryotic community structure across OMZs of the Pacific Ocean. Geographic and depth distribution of samples across OMZs of the Pacific Ocean. (A) Map of the Eastern Pacific Ocean showing the stations utilized in this study (white filled circles). Colors and the contour lines correspond to the oxygen concentrations in mL L-1 at 325 meters depth (mean depth of the OMZ core, CARS 2009). (B) Principal component analysis of the bacterial community composition according to 16S rRNA gene metabarcoding data. The ASVs counts table was normalized using the variance-stabilizing transformation performed by DESeq. The two principal components were plotted. Samples were colored based on the water chemistry and collection depths. Surface: samples from depths of 0 -~30m; SFM Secondary Fluorescence Maximum; Oxycline: [O2] > 5 uM); Suboxic: [O2] < 5 uM); Anoxic: [O2] < 5 uM, and > 4 µM of Nitrite. ETNP, Eastern Tropical North Pacific; GD, Golfo Dulce; ETSP, Eastern Tropical South Pacific; FL, Free-Living; PA, Particle-Attached.
A PCA based on all ASVs from all study sites revealed a clear partitioning of samples (Figure 1B) based on these chemical/depth classifications (PERMANOVA, Pr(>r) = 0.001). Here, all communities from low-oxygen waters ([O2] < 5 µM) within the AMZs (i.e., “Suboxic”, “SFM”, and “Anoxic-Core”) clustered together regardless of their geographic origin and were distinct from the low-oxygen communities from the sulfidic, coastal OMZ of Golfo Dulce, Costa Rica (Hierarchical Cluster, Figure 2). Samples from the FL fraction corresponding to the SFM, Suboxic, and Anoxic Core formed distinct sub-clusters, whereas all PA communities sampled from low-oxygen waters ([O2] < 5 µM) clustered together regardless of their nitrite levels or depth (Figure 2). The FL fraction from low-oxygen waters was dominated by bacteria classified as SAR406, SUP05, SAR324, and SAR11 clades I and II (Figure S1A). In contrast, the PA fraction was dominated by bacteria classified as Alteromonas, UBA10353 marine group, NS9 Marine group, SAR406, Oleibacter, and archaea of Marine group III Euryarchaeota (Figure S1B).
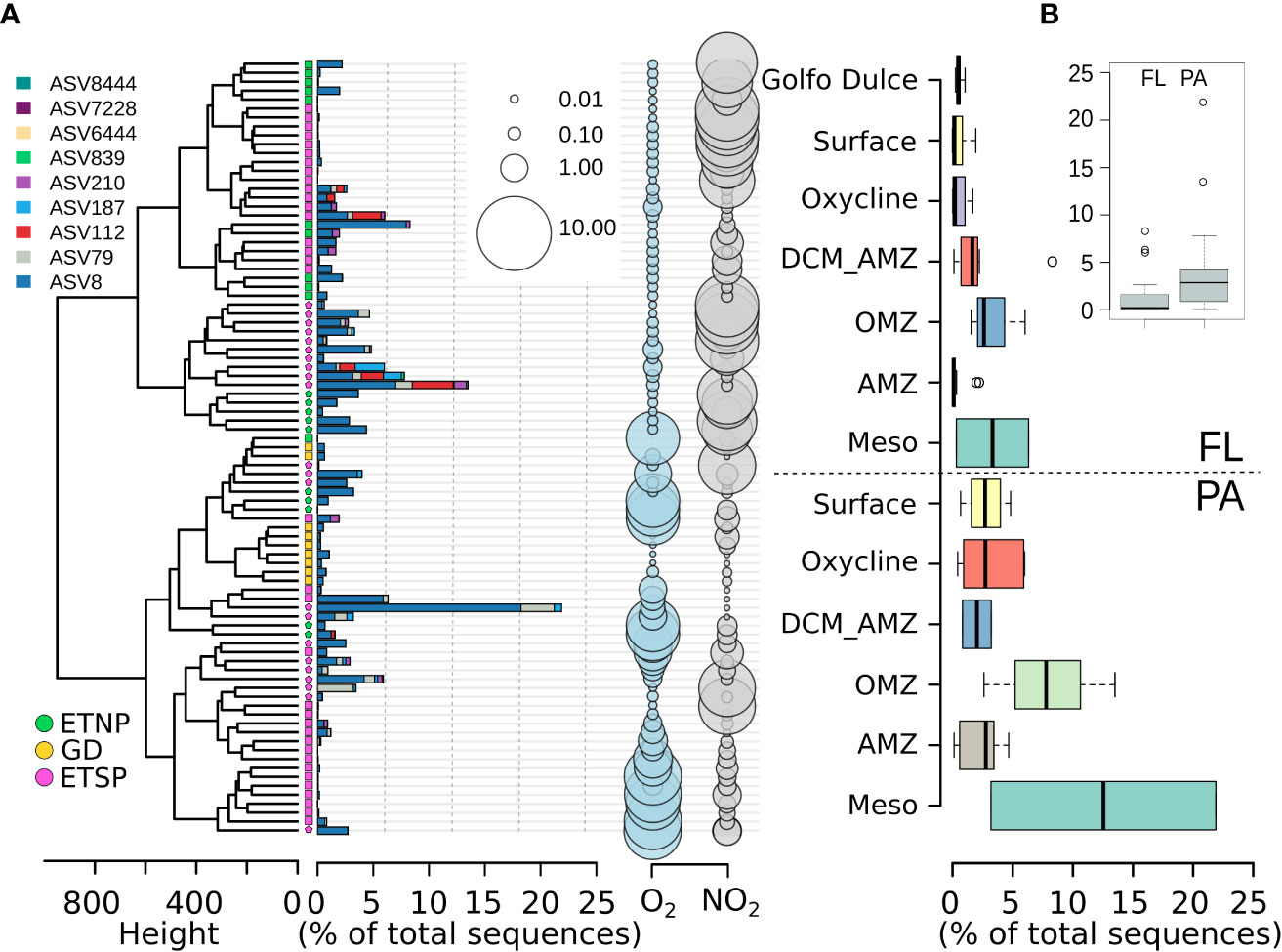
Figure 2 Alteromonas-related ASV abundances over the redox gradient of OMZ waters of the Pacific Ocean. (A) Alteromonas ASVs per sample as a percentage of the total reads in each sample. The color code corresponds to the different geographic locations. Samples from the free-living (FL) fraction are marked with squares. Particle-attached (PA) samples are marked with circles. Oxygen concentrations are presented in mL L-1, and nitrite concentrations are presented in µM. (B) Barplot corresponds to the percentage of reads classified as Alteromonas relative to the total number of reads in each sample. ETNP, Eastern Tropical North Pacific; GD, Golfo Dulce; ETSP, Eastern Tropical South Pacific; O2, Oxygen; NO2, Nitrite; SFM, Secondary Fluorescence Maximum; Meso, Mesopelagic.
ASVs assigned to the Alteromonas genus reached up to ~22% of the prokaryotic community in the sites/depths studied here (Figure 2B). A single ASV (ASV 8) represented more than the 90% of the Alteromonas-related ASVs, while three other ASVs (ASV 79, 112, and 210) were consistently detected in these sampling locations but at lower proportions (Figure 2). Alteromonas ASVs were less abundant in the FL fraction than in the PA fraction at all depths, except at the SFM, where abundances in the FL and PA fractions were similar (Figure 2B). The overall higher abundance of Alteromonas within the PA fraction here was statistically significant (p < 0.0001, Mann-Whitney test Figure 2B, inset), with the highest abundances of Alteromonas detected in mesopelagic (3.2 - 21.87%) and suboxic (2.62 - 13.5%) waters (Figure 2B) in the PA fraction. Network analysis of samples from low-oxygen waters (n = 49) revealed that while the ASV_8 was widely distributed across the oxygen gradient and size fractions, the ASV 79 was exclusively represented in the PA fraction, and the ASV 112 was restricted to hypoxic but not anoxic waters. These two ASVs co-occurred with carbon degraders as Oleibacter and Thalassolituus and host-associated bacteria including Sva0996 marine group, Absconditabacteriales_(SR1), and Margulisbacteria among others (Table S2).
Genomic potential of Alteromonas Single-cell Amplified Genomes from the ETSP OMZ.
A total of 19 Alteromonas Single-cell Amplified Genomes (SAGs) were generated from samples collected at the ETSP SFM (20.0834° S 70.7997° W, at 53 m depth, and 3.6 µM O2) (Table S3). These SAGs, with estimated completeness that ranged from 30% to 77%, corresponded to two well-defined populations based on their average nucleotide identity (ANI > 95%) (Figure S2). SAGs from AMZ population 1 (n=6) shared 100% 16S rRNA gene sequence identity with the dominant Alteromonas spp. ASV_8 and were phylogenetically related to the A. macleodii ecotype. In contrast, SAGs from AMZ population 2 (n=13) shared 100% 16S rRNA gene sequence identity with Alteromonas spp. ASV_79 were affiliated with the A. naphtalenivorans SN2 ecotype (Figure S3; Table S4).
Comparative genomics of representative Alteromonas spp. genomes (n=76) and the SAGs (two populations) revealed that the pangenome of this bacterial genus contained a core set of 3787 genes (Figure 3). This also confirmed that the SAGs from AMZ population 1 are related to the A. macleodii ecotype, with a similar gene composition to that of Alteromonas HOT1A3, EZ55, and MED G140, presenting only 34 predicted genes that were unique of this population. SAGs from AMZ population 2 were related to the SN2 ecotype and the Alteromonas stellipolaris strains PQQ42, PQQ44, LMG21856, among others, and had 224 predicted genes unique to this population. Functional analysis of the unique genes present in SAGs from AMZ population 1 revealed that those with an assigned KO function were a multidrug efflux pump and two-component systems, among others (Table S5). Instead, the unique genes with a predicted KO function from AMZ population 2 are involved in cell wall biosynthesis, processing and transport of peptides and aminoacids, and genes for stability and defense mechanisms that are also found in other organisms from the order Altermonadales, such as Colwellia sppp., and Paraglaciecola spp (Table S5).
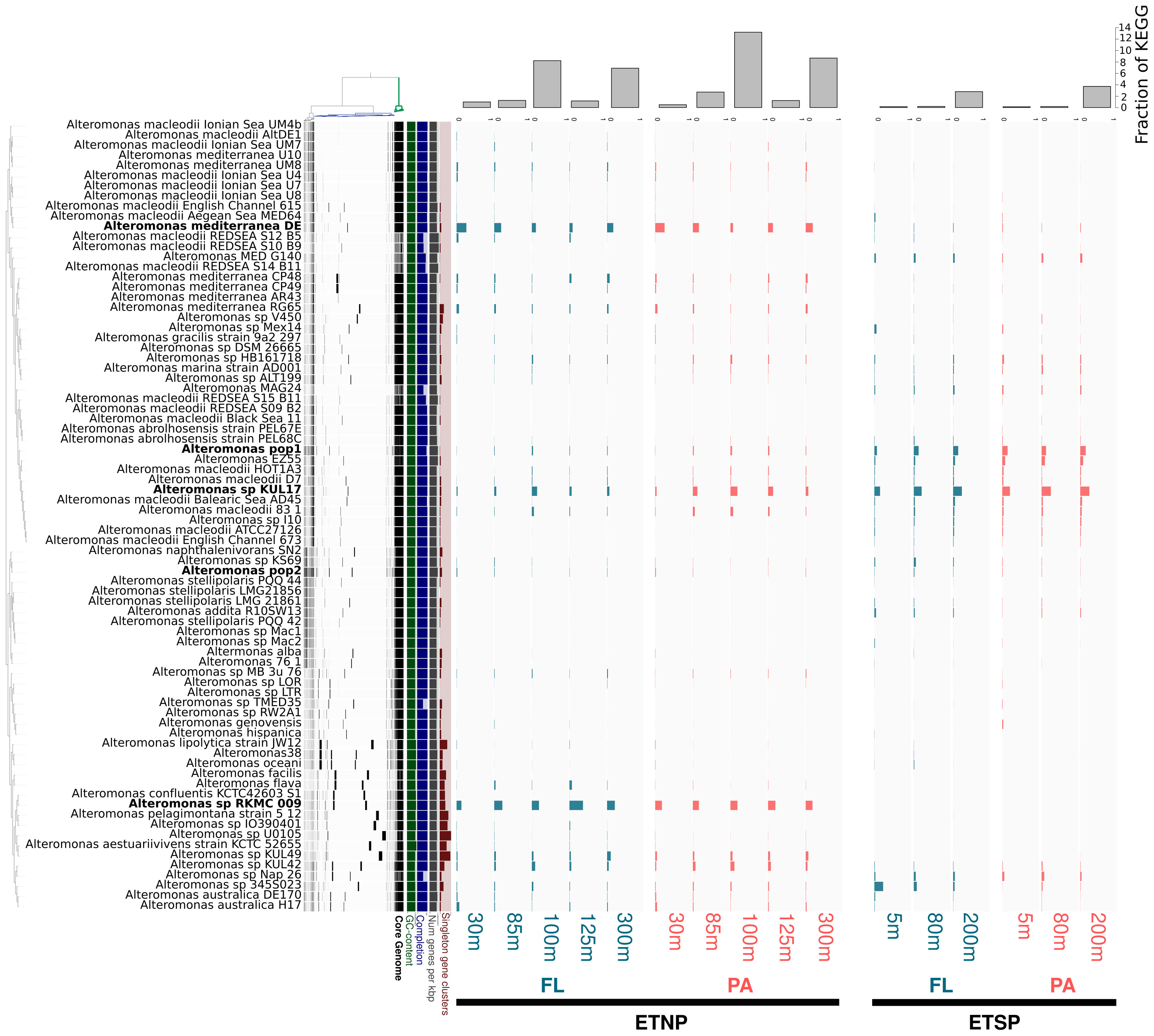
Figure 3 Pangenomic analysis of the Alteromonas genomes and percentage recruitment of OMZ metatranscriptome read fragments. Pangenomic analysis of the Alteromonas genomes and fragment recruitment of OMZ metatranscriptomes. A. The dendrogram at the left shows the clustering of 76 isolated genomes and the two SAGs populations based on the distribution of 7,385 genes (black bars) recovered from the pangenomic analysis (Euclidean distance and ward D2 clustering). The barplots corresponds to metatranscriptome reads recruited to each genome represented as a fraction of the total Alteromonas recruited reads in the metatranscriptome. ETNP, Eastern Tropical North Pacific; ETSP, Eastern Tropical South Pacific; FL, Free-Living; PA, Particle-Attached. The upper barplot corresponds to the relative recruit over all the Alteromonas genomes, calculated based on the number of KOs detected in the Metatranscriptome that are present in the Alteromonas genomes and normalized to the total number of KO predicted in each Metatranscriptome (fraction of KEGG).
Environmental transcriptomic activity of Alteromonas in OMZs
To further evaluate the in situ activity and the functional role of Alteromonas lineages across the OMZ redox gradient and between the FL and PA biomass fractions, we mapped AMZ metatranscriptomic reads against 76 reference Alteromonas genomes from public repositories in addition to the SAGs from the ETSP SFM. Overall, the proportion of Alteromonas transcripts was highest in ETNP compared to the ETSP (Table S6). The higher representation of Alteromonas was observed at 100 m depth of the ETNP (SFM) in the PA fraction where they accounted for 1.8% of the total reads and 13.23% of the reads assignable to KEGG functions.
A differential reprentation of Alteromonas ecotypes was detected among the different AMZ sites and depths (Figure 3; Table S6). Of all the genomes used as references, Alteromonas strain RKMC-009 recruited the most metatranscriptomic reads at the ETNP, especially between 85 m and 125 m depth in the FL fraction (constituting up to 37% of all the Alteromonas-recruited reads) (Figure 3), and a homogeneous representation in the PA fraction with an average recruitment of 18% of all the Alteromonas-recruited reads). Other genome that recruited a high percentage of transcripts in the ETNP corresponded to the Alteromonas strains DE, with a peak in abundance in the surface chlorophyll maxima (25% of the Alteromonas-recruited reads in both the FL and PA fraction), Alteromonas KUL17 dominated in terms of transcript abundances at the ETSP in both the FL and PA fraction (22% of the Alteromonas-recruited reads). Alteromonas SAGs from population 1 represented up to 15% of the Alteromonas-recruited reads in the ETSP, where Alteromonas sp. 345S023 was the genome that recruited the most transcripts from the surface waters at the ETSP (Figure 3).
Regarding their functional representation, the most abundant and prevalent Alteromonas transcripts in the ETNP and ETSP AMZs encoded the glucose-1-phosphate thymidylyltransferase (rfbA, rffH; K00973) and a pre-protein translocase, respectively. The iron complex outer membrane receptor protein (TC.FEV.OM; K02014) was also highly represented in both AMZs (Table S7). No differences in functional representation were detected in Alteromonas representatives between FL and PA metatranscriptomes (Table S7). The most abundant functional transcripts encoded iron complex outer membrane receptor protein, a solute:Na+ symporter, a biopolymer transport protein ExbB threonine synthase, and the glutamine Synthetase. Iron metabolism-related transcripts encoding the Fe-II oxidizing enzyme bacterioferritin (K03594) and a superoxide dismutase from the Fe-Mn family (K04564) and to a lesser extent, the periplasmic protein TonB (K03832) were also detected, coinciding with the high level of transcripts for the iron complex outer membrane receptor protein (Figure 4; Table S7).
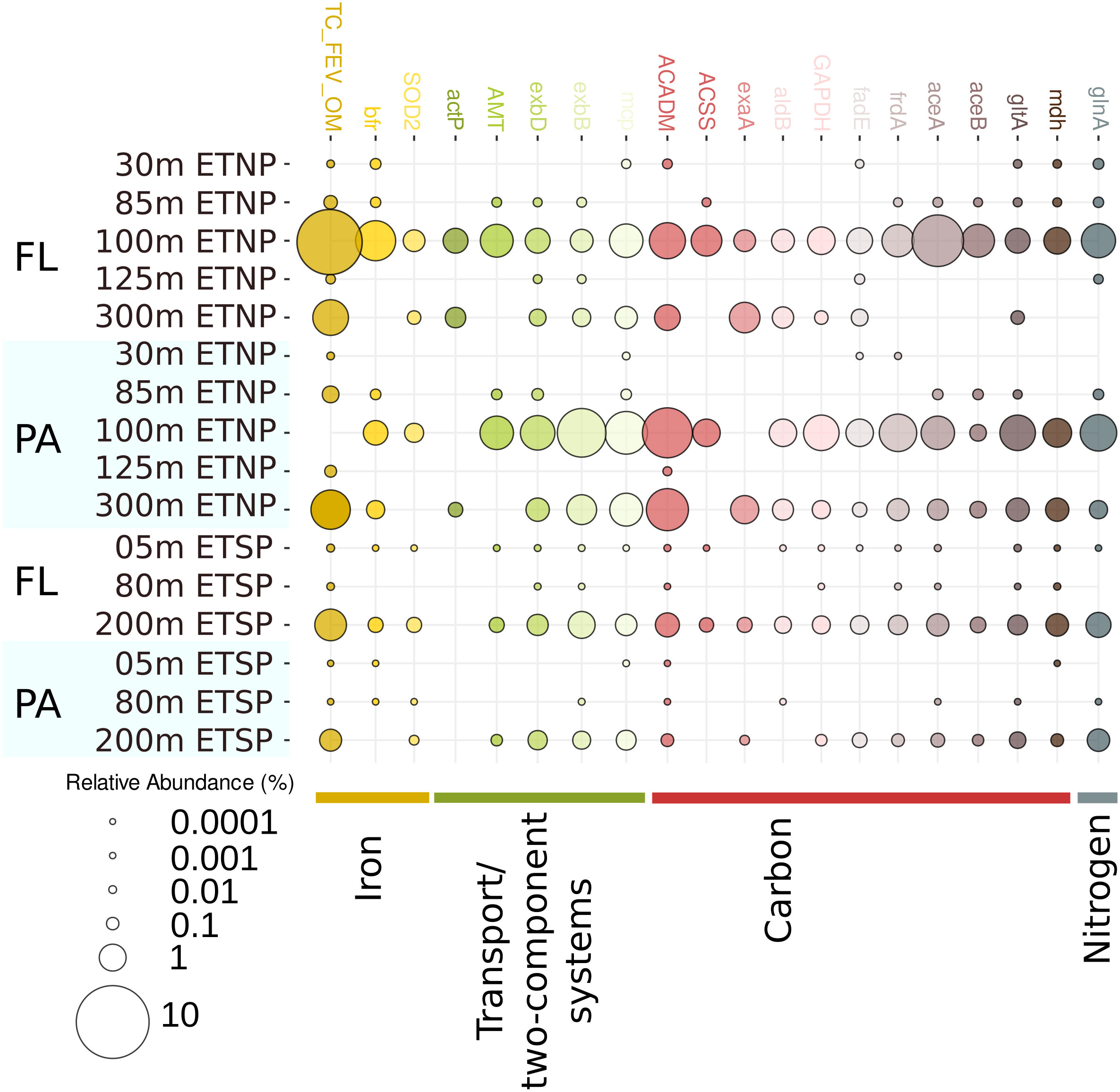
Figure 4 Percentage recruitment of ETNP and ETSP OMZs metatranscriptome read fragments to key Alteromonas functional genes. Alteromonas genomes and SAGs populations were used to recruit reads from the metatranscriptomic data from both the ETNP and ETSP using a cutoff of 97% nucleotide identity over a minimum alignment length of 100 nucleotides. the top 20 KEGG orthologs (KOs) were selected. FL, Free-living; PA, Particle-attached. Values for each KO were normalized by the fraction of KEGG assigned to Alteromonas from the total KEGG assigned in each metatranscriptome.
In addition to glucose-1-phosphate thymidylyltransferase, the most abundant Alteromonas transcripts related to carbon metabolism encoded isocitrate lyase (K01637), malate synthase (K01638), aconitate hydratase 2 (K01682), and succinate dehydrogenase/fumarate reductase (K00239) (Table S7). Levels of these transcripts were the highest within the AMZ SFM (Figure 4) from the ETNP.
Other enzymes related to aminoacid metabolism and catabolism, including glutamine synthetase (K01915), ketol-acid reductoisomerase (K00053), and acyl-CoA dehydrogenase (K06445), were also highly transcribed in the SFM in the ETNP. The most abundant nitrogen metabolism-related transcript encoded an ammonium transporter (amt, K03320). Transcripts encoding type I and type III cytochrome c oxidases peaked in abundance in the SFM and at the anoxic core in the coastal station (Table S7). Accordingly, transcripts for a major ammonium assimilation pathway for (the glutamine synthetase/glutamate synthase [GS/GOGAT] pathway) were also highly abundant at the ETNP (Figure 4, Table S7).
Finally, transcripts encoding alcohol dehydrogenase (K00114), a cold-shock protein (K03704) and a methyl-accepting chemotaxis protein (K03406) were also abundant at anoxic depths (Figure 4).
Discussion
Alteromonas bacteria constitute a relevant fraction of microbial communities inhabiting the Pacific Ocean AMZ
Although Alteromonas is one of the most prevalent bacterial taxa in the oceans (Nayfach et al., 2016), there is still a lack of knowledge regarding the distribution, diversity, and ecology of these bacteria in oxygen-deficient marine environments. A high representation of Alteromonas have been reported in the Sansha Yongle Blue Hole, an O2 - deficient environment in the South China (He et al., 2020), and in the Arabian Sea OMZ (Bandekar et al., 2018). This study reports a field-based analysis of the diversity and distribution of Alteromonas lineages across various redox gradients and depths in the Pacific Ocean AMZs. Based on 16S rRNA gene amplicon data from both free-living (FL) and particle-attached (PA) biomass fractions, we show that Alteromonas is a quantitatively relevant member of the microbial community across all the AMZ waters of the eastern boundary of the Pacific Ocean, especially within the PA fraction where it can reach more than 20% of total prokaryotic 16S rRNA gene sequences (Figure 2).
A single ASV, related to Alteromonas AMZ population 1 SAGs and the A. macleodii ecotype, dominated in all the oxygen-deficient marine environments included in this study (Figure 1A). The abundance of Alteromonas in the FL fraction of the SFM and suboxic waters was significantly higher than in the anoxic core of the AMZ (Figure 2B). The absence of detectable concentrations of nitrite in both the SFM and Suboxic samples suggests the presence of low, though undetectable, amounts of oxygen. A cryptic oxygen cycle linked to oxygenic cyanobacterial lineages has been demonstrated to occur within SFM depths of the Eastern Pacific AMZs (Lavin et al., 2012; Garcia-Robledo et al., 2017). Accordingly, the abundance of genes and transcripts of aerobic respiration and nitrification spike in the Prochlorococcus-enriched AMZ-SFM (Bertagnolli and Stewart, 2018). Cryptic oxygen supply by the AMZ Prochlorococcus lineages, can potentially allow Alteromonas and other heterotrophs to degrade complex carbohydrates fueled by aerobic respiration and/or supplemented by facultative anaerobic respiration of nitrate, nitrite, or sulfur compounds in these scarcely oxygenated waters.
The Alteromonas populations support their higher abundance in the Particle-Attached fraction
The genomic versatility among and within Alteromonas ecotypes presumably contributes to the success of these bacteria under diverse environmental conditions (Math et al., 2012; López-Pérez and Rodriguez-Valera, 2016; Koch et al., 2020). Most Alteromonas ecotypes, including those explored in our study, contain genes supporting aerobic respiration and the degradation of organic matter under low-oxygen conditions (Figure S4). These genes are assembled in a cluster flanked by a universal stress protein-coding gene and encode an anaerobic transcriptional regulator, the cbb3-type cytochrome c oxidase, a bacterioferritin a P-type Cu+2 transporting ATPase, and glutamate synthase (NADPH) large chain subunit (Figure S4). Most of these genes are transcribed in the oxygen-deficient-waters studied here (Table S7; Figure 4). The decrease in the representation of Alteromonas ASVs in the anoxic core, especially in the FL fraction, coincides with the drop of the oxygen concentrations below the detection limit of current sensors ( <10 nM oxygen) and where anaerobic processes largely replace oxygenic respiration, as shown by the accumulation of nitrite (Figure 2).
The relatively high proportional abundance of Alteromonas in the PA fraction at the anoxic core is more challenging to explain. This taxonomic signal may be somewhat refractory, representing organisms that were active on particles at oxic depths but are relatively inactive when those particles sink into the anoxic core of the AMZ. Alternatively, since the Alteromonas transcripts from low-oxygen waters PA fractions (Figure 4 and Table S6) included a high-affinity terminal oxidases and a nitrite reductases, among other transcribed genes, these populations may be utilizing the undetectable traces of oxygen or conducting anaerobic metabolism. Finally, the higher abundances of Alteromonas in both the FL and PA fraction in the mesopelagic waters below the AMZ indicate an abundance of usable organic matter at these depths. Previously sediment trap data indicated reduced degradation of sinking particles in AMZs (Keil et al., 2016; Cram et al., 2022), which would allow these particles to be a source of energy for aerobic heterotrophs such as Alteromonas in these deeper waters.
Our results show that in these low oxygen waters, Alteromonas ecotypes have higher abundances in association with particles or cell-cell aggregates. Indeed, a high abundance of traditionally biofilm-associated marine bacteria (i.e., Oleibacter Marinus and Marinobacter hydrocarbonoclasticus) was found in the OMZ PA fraction (Table S2). Such microorganisms specialize in degrading hydrophobic organic compounds that can represent a significant fraction of organic matter in marine waters (Mounier et al., 2014). It remains unclear if AMZ Alteromonas are filling a similar metabolic niche on particles or are potentially interacting with these or other PA-associated taxa. In surface waters a single Alteromonas strain is able to consume as much DOC as diverse free-living microbial communities (Pedler et al., 2014). Here, the enrichment of Alteromonas in the SFM is consistent with a potential association between Alteromonas and picocyanobacteria (Aharonovich and Sher, 2016; Hennon et al., 2018). In such associations, Alteromonas has been shown to alleviate oxidative stress and nutrient limitation during extended periods of darkness (Coe et al., 2016; Biller et al., 2018). For example, Alteromonas may secrete asparagine and glutamine, which has been shown to coincide with the up-regulation of related importers by the co-cultured Prochlorococcus (Koch et al., 2020).
Alteromonas bacteria are transcriptionally active members of the AMZ microbial communities
A differential representation of Alteromonas species was observed between the ETSP and ETNP AMZ transcripts. Although both AMZs often present similar oxygen and nitrate profiles through depth (Tiano et al., 2014), the differences in nitrite and ammonium between the two AMZs at the sampling time could have an influence over the representation of the different ecotypes, and the abundance of the gene transcripts (Table S7). AMZ Alteromonas transcripts were dominated by sequences most closely related to those of Alteromonas strain RKMC-009, isolated from the sponge Xestospongia muta (MacIntyre et al., 2019) and of the Alteromonas DE, with a differential representation between the ETSP and the ETNP (Figure 3). Interestingly, the unique KOs and COGs assigned to strains RKMC-009 and DE were only sparsely represented in the metatranscriptomic dataset analyzed. A similar trend was observed for all the prevalent Alteromonas genotypes, suggesting a disconnect between their representation and their unique genomic functions (Tables S5, S6). The genome of RKMC-009 contains the highest amount of carbohydrate-active enzymes among the Alteromonas genomes analyzed here (CAZymes, n = 142, with 31 unique genes related to carbohydrate transport and metabolism), in addition to unique genes associated with inorganic ion transport and metabolism. This genomic characteristic suggests that the ability of RKMC-009 to degrade diverse polysaccharides may extend to its AMZ Alteromonas relatives and underlie their success in these environments. The Alteromonas DE ecotype, described initially from deep-sea sites in the Mediterranean (Ivars-Martinez et al., 2008a) was transcriptionally active in all the samples analyzed. This “deep” strain is adapted to live on large particles that sink to meso- and bathy-pelagic depths. However, Alteromonas DE relatives are also present in surface habitats (Lopez-Pérez et al., 2012), as also demonstrated here, and are not an obligate bathytype (Ivars-Martinez, et al., 2008). The SAGs from AMZ population 1 recruited their highest proportion of reads from the ETSP AMZ transcriptomes. These SAGs are closely related to Alteromonas KUL17, which is one of the most represented genomes in the transcriptomes. This bacterium was isolated from a mollusk and can utilize diverse carbon sources for growth, including ulvan and L-rhamnose (He et al., 2017). The SAGs from AMZ population 2 were phylogenetically related to both Alteromonas SN2 (isolated from a crude oil-contaminated marine tidal flat; Jin et al., 2012) and Alteromonas 76-1 (isolated from an alginate-supplemented microcosm at the Patagonian continental shelf; Koch et al., 2019) and had low transcriptional representation in the samples studied here (Figure 3). The low recruitment by AMZ Alteromonas SAGs, particularly from the population 2 might be explained by the low overall representation of Alteromonas in metatranscriptomic samples from the ETSP, were the SAGs were collected, but also by the lack of metatranscriptomic samples from the SFM in this area. This remains to be explored in future research.
Metabolic contributions of Alteromonas bacteria to the AMZ microbial communities
Regarding carbon metabolism, two key glyoxylate shunt genes were found at high proportional abundance in the Alteromonas-associated transcript pool, particularly in the SFM (Figure 4). These encode the enzymes isocitrate lyase (which utilizes isocitrate from the Citric Acid Cycle and catalyzes its transformation into succinate and glyoxylate) and malate synthase (which catalyzes the formation of malate from glyoxylate and Acetyl-CoA). Along with citrate synthase, aconitase, and phosphoenolpyruvate carboxykinase, these enzymes compose the glyoxylate shunt. This pathway is likely a relevant carbon acquisition pathway for marine heterotrophic bacteria including other prevalent marine bacteria, including Photobacterium spp. and Alphaproteobacterial of the SAR11 group (Beier et al., 2015; Koedooder et al., 2018).
With respect to the nitrogen metabolism, the high abundance of genes for the GS/GOGAT pathway in the ETNP (Figure 4, Table S7) is similar to results described by Jin et al. (2016) when exposing the Alteromonas SN2 marine ecotype to different environmental conditions. Accordingly, the aforementioned study reported a higher transcription of genes related to glyoxylate metabolism and glutamate synthesis (glutamate dehydrogenase) in a tidal flat habitat that experiences low-oxygen conditions.
Interestingly, a high proportion of transcripts were found to encode for an extracellular iron-complex binding protein, bacterioferritin, a superoxide dismutase from the Fe-Mn family (Figure 4), and, to a lesser extent, TonB-dependent transporters. This could be due to their high representation in the Alteromonas RKMC-009 genome but also with the over-expression observed in cultures of Alteromonas macleodii (Fourquez et al., 2014), Candidatus Pelagibacter ubique (SAR11 clade), and Photobacterium angustum under iron-limitation (Fourquez et al., 2014; Koedooder et al., 2018; Manck et al., 2020). Iron-related TonB-dependent transporters and extracellular iron-complex binding proteins are responsible for iron and carbon acquisition, while ferritin can be used to store iron (Andrews et al., 2003; Manck et al., 2020).
Iron is tightly linked to carbon metabolism due to its central role as a cofactor in the citric acid cycle and respiratory chain enzymes. Dissolved iron concentrations in the ocean are generally low (Moore and Braucher, 2007) and can significantly vary across OMZs (Kondo and Moffett, 2015; Vedamati et al., 2014). In low-oxygen water columns, dissolved iron concentration depends on diverse factors, potentially including conditions in the underlying sediment and in some cases connectivity with the continental shelf (Schlosser et al., 2018; Croot et al., 2019). At both the ETSP and ETNP OMZs analyzed here, total dissolved Fe and labile inorganic Fe complexes concentrations increase with depth at all stations, coincident with an increase in genes encoding Fe-proteins involved in dissimilatory nitrogen metabolisms (Glass et al., 2015). Over the ETSP continental shelf, dissolved iron concentrations increase markedly at ~100 m depth, and are almost entirely composed of reduced Fe(II) species (Heller et al., 2017). A minimum in dissolved iron concentration (dFe) that overlaps with the deep chlorophyll maximum (DCM) have been reported throughout the open ocean, which marks the lower limit of the euphotic zone (Hawco et al., 2021). However, measurements of iron concentration and speciation in the SFM from anoxic waters remain relatively sparse. Additional studies of iron and other trace metal concentrations in SFM from AMZs are needed given their potential relevance in the C-metabolism of highly abundant heterotrophs, such as Alteromonas. The seemingly coupled transcription of Alteromonas carbon and iron metabolism genes in our data supports the aforementioned culture-based physiological and genomic studies linking iron-depletion stress under anaerobic conditions and the carbon metabolism of these ubiquitous marine heterotrophs. Since heterotrophic bacteria play a significant role in the incorporation of new iron into the marine environment (Manck et al., 2022), our results raise the hypothesis that natural Alteromonas populations may overcome iron fluctuations in coastal low-oxygen environments (such as AMZs) by the uptake of larger organically complex iron substrates.
Conclusions
Our results reveal a significant representation of Alteromonas in the SFM of marine low-oxygen environments in both the free-living and particle-associated microbial communities. These Alteromonas populations co-occur with other critical microbial players in the carbon cycle, including complex-carbon degraders (i.e., Marinobacter) and primary producers (i.e., Prochlorococcus). In contrast to our current knowledge of the nitrogen, sulfur, and oxygen cycles in OMZs (Ganesh et al., 2014; Ganesh et al., 2015; Glass et al., 2015; Garcia-Robledo et al., 2016; Garcia-Robledo et al., 2017; Fuchsman et al., 2017; Saunders et al., 2019; Raven et al., 2021), the carbon cycle has been poorly characterized in these low-oxygen environments. Recently couplings between carbon, sulfur, and nitrogen metabolisms have been proposed (Plominsky et al., 2018), along with the partitioning of different carbon fixation pathways along the AMZ redox gradient (Ruiz-Fernández et al., 2020). Considering all the caveats regarding metatranscriptomic fragment recruitment, the data presented here suggests an in situ coupling of Alteromonas iron and carbon metabolisms and the potential importance of its glyoxylate cycle in the AMZs. Also, the peak transcription of iron complex outer membrane receptors, SOD2 and glutamine synthase at the SFM suggest a possible interplay between AMZ Alteromonas and co-occurring Prochloroccocus populations in subsurface waters, similar to the described for surface ecotypes when co-cultured. Our results indicate that Alteromonas may contribute significantly to the carbon metabolism in AMZs, particularly in the SFM in coastal areas, where similar functional features were transcribed in both the FL and the PA size fractions. These findings set the foundation for further studies assessing the Alteromonas ecotypes from other OMZ systems and their connections with the vertical gradients of carbon, nitrogen, oxygen, and trace metals.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
CH-C, OU, and SR-F conceived the research. CH-C, AMP, and SR-F perform the analysis. CH-C and AMP wrote the manuscript with contribution from all the authors. All authors approved the final submitted manuscript
Funding
This work was supported by the Chilean Agency for Research and Development (grant FONDECYT-POSTDOCTORADO 3180724 and ICN12_019-IMO).
Acknowledgments
The authors want to thank the crew of the R/V Cabo de Hornos from the Chilean Navy and to the IMO staff, particularly to Eduardo Navarro and Cristian Venegas for their support in the sampling during the lowphox I & II cruises. We also thank Montserrat Aldunate for their invaluable contribution, collecting the samples during the cruises: NH1410, RB1603, NBP1305 and AT2626 and to Matías Pizarro-Koch for his support with the oceanographic data.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.993667/full#supplementary-material
References
Aguayo P., Campos V. L., Henríquez C., Olivares F., De Ia Iglesia R., Ulloa O., et al. (2020). The influence of pCO2-driven ocean acidification on open ocean bacterial communities during a short-term microcosm experiment in the Eastern tropical south pacific (ETSP) off northern Chile. Microorganisms 8 (12), 1924. doi: 10.3390/microorganisms8121924
Aharonovich D., Sher D. (2016). Transcriptional response of Prochlorococcus to co-culture with a marine alteromonas: Differences between strains and the involvement of putative infochemicals. ISME J. 10 (12), 2892. doi: 10.1038/ismej.2016.70
Aldunate M., Henríquez-Castillo C., Ji Q., Lueders-Dumont J., Mulholland M. R., Ward B. B., et al. (2020). Nitrogen assimilation in picocyanobacteria inhabiting the oxygen-deficient waters of the eastern tropical north and south pacific. Limnol. Oceanogr. 65 (2), 437–453. doi: 10.1002/lno.11315
Andrews S. C., Robinson A. K., Rodríguez-Quiñones F. (2003). Bacterial iron homeostasis. FEMS Microbiol. Rev. 27 (2-3), 215–237. doi: 10.1016/S0168-6445(03)00055-X
Apprill A., McNally S., Parsons R., Weber L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 75 (2), 129–137. doi: 10.3354/ame01753
Bandekar M., Ramaiah N., Jain A., Meena R. M. (2018). Seasonal and depth-wise variations in bacterial and archaeal groups in the Arabian Sea oxygen minimum zone. Deep Sea Res. Part II Top. Stud. Oceanogr. 156, 4–18. doi: 10.1016/j.dsr2.2017.12.015
Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19 (5), 455–477. doi: 10.1089/cmb.2012.0021
Banse K., Naqvi S. W. A., Postel J. R. (2017). A zona incognita surrounds the secondary nitrite maximum in open-ocean oxygen minimum zones. Deep Sea Res. Part I Oceanogr. Res. Pap. 127, 111–113.
Beier S., Gálvez M. J., Molina V., Sarthou G., Quéroué F., Blain S., et al. (2015). The transcriptional regulation of the glyoxylate cycle in SAR11 in response to iron fertilization in the southern ocean. Environ. Microbiol. Rep. 7, 427–434. doi: 10.1111/1758-2229.12267
Bertagnolli A. D., Stewart F. J. (2018). Microbial niches in marine oxygen minimum zones. Nat. Rev. Microbiol. 16 (12), 723–729. doi: 10.1038/s41579-018-0087-z
Biller S. J., Coe A., Roggensack S. E., Chisholm S. W. (2018). Heterotroph interactions alter Prochlorococcus transcriptome dynamics during extended periods of darkness. Msystems 3 (3). doi: 10.1128/mSystems.00040-18
Bolaños L. M., Karp-Boss L., Choi C. J., Worden A. Z., Graff J. R., Haëntjens N., et al. (2020). Small phytoplankton dominate western north Atlantic biomass. ISME J., 1–12.
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Bruland K. W., Rue E. L., Smith G. J., DiTullio G. R. (2005). Iron, macronutrients and diatom blooms in the Peru upwelling regime: Brown and blue waters of Peru. Mar. Chem. 93 (2-4), 81–103. doi: 10.1016/j.marchem.2004.06.011
Buchfink B., Xie C., Huson D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P. (2016). DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods 13 (7), 581–583. doi: 10.1038/nmeth.3869
Cepeda-Morales J., Beier E., Gaxiola-Castro G., Lavín M. F., Godínez V.M (2009) Effect of the oxygen minimum zone on the second chlorophyll maximum. Ciencias Marinas, 35 (4), 389–403.
Coe A., Ghizzoni J., LeGault K., Biller S., Roggensack S. E., Chisholm S. W. (2016). Survival of Prochlorococcus in extended darkness. Limnol. Oceanogr. 61 (4), 1375–1388. doi: 10.1002/lno.10302
Cram J. A., Fuchsman C. A., Duffy M. E., Pretty J. L., Lekanoff R. M., Neibauer J. A., et al. (2022). Slow particle remineralization, rather than suppressed disaggregation, drives efficient flux transfer through the Eastern tropical north pacific oxygen deficient zone. Global Biogeochem. Cycles 36, e2021GB007080. doi: 10.1029/2021GB007080
Croot P. L., Heller M. I., Wuttig K. (2019). Redox processes impacting the flux of iron (II) from shelf sediments to the OMZ along the Peruvian shelf. ACS Earth Space Chem. 3 (4), 537–549. doi: 10.1021/acsearthspacechem.8b00203
Delmont T. O., Eren A. M. (2018). Linking pangenomes and metagenomes: the prochlorococcus metapangenome. PeerJ 6, e4320. doi: 10.7717/peerj.4320
Dobal-Amador V., Nieto-Cid M., Guerrero-Feijoo E., Hernando-Morales V., Teira E., Varela M. M. (2016). Vertical stratification of bacterial communities driven by multiple environmental factors in the waters (0–5000 m) off the Galician coast (NW Iberian margin). Deep-Sea Res. Pt I 114, 1–11. doi: 10.1016/j.dsr.2016.04.009
Eren A. M., Esen Ö.C., Quince C., Vineis J. H., Morrison H. G., Sogin M. L., et al. (2015). Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3, e1319. doi: 10.7717/peerj.1319
Fourquez M., Devez A., Schaumann A., Guéneuguès A., Jouenne T., Obernosterer I., et al. (2014). Effects of iron limitation on growth and carbon metabolism in oceanic and coastal heterotrophic bacteria. Limnol. Oceanogr. 59 (2), 349–360. doi: 10.4319/lo.2014.59.2.0349
Fuchsman C. A., Devol A. H., Saunders J. K., McKay C., Rocap G. (2017). Niche partitioning of the n cycling microbial community of an offshore oxygen deficient zone. Front. Microbiol. 8, 2384. doi: 10.3389/fmicb.2017.02384
Fuchsman C. A., Palevsky H. I., Widner B., Duffy M., Carlson M. C., Neibauer J. A, et al. (2019). Cyanobacteria and cyanophage contributions to carbon and nitrogen cycling in an oligotrophic oxygen-deficient zone. ISME 13 (11), 2714–2726
Ganesh S., Bristow L. A., Larsen M., Sarode N., Thamdrup B., Stewart F. J. (2015). Size-fraction partitioning of community gene transcription and nitrogen metabolism in a marine oxygen minimum zone. ISME J. 9, 2682–2696. doi: 10.1038/ismej.2015.44
Ganesh S., Parris D. J., DeLong E. F., Stewart F. J. (2014). Metagenomic analysis of size-fractionated picoplankton in a marine oxygen minimum zone. ISME J. 8, 187–211. doi: 10.1038/ismej.2013.144
Garcia-Robledo E., Borisov S., Klimant I., Revsbech N. P. (2016). Determination of respiration rates in water with sub-micromolar oxygen concentrations. Front. Mar. Sci. 3, 1–13. doi: 10.3389/fmars.2016.00244
Garcia-Robledo E., Padilla C. C., Aldunate M., Stewart F. J., Ulloa O., Paulmier A., et al. (2017). Cryptic oxygen cycling in anoxic marine zones. P Natl. Acad. Sci. U.S.A. 114 (31), 8319–8324. doi: 10.1073/pnas.1619844114
Glass J. B., Kretz C. B., Ganesh S., Ranjan P., Seston S. L., Buck K. N., et al. (2015). Meta-omic signatures of microbial metal and nitrogen cycling in marine oxygen minimum zones. Front. Microbiol. 6, 998. doi: 10.3389/fmicb.2015.00998
Hawco N. J., Fu F., Yang N., Hutchins D. A., John S. G. (2021). Independent iron and light limitation in a low-light-adapted Prochlorococcus from the deep chlorophyll maximum. ISME J. 15 (1), 359–362. doi: 10.1038/s41396-020-00776-y
He C., Muramatsu H., Kato S. I., Ohnishi K. (2017). Characterization of an Alteromonas long-type ulvan lyase involved in the degradation of ulvan extracted from ulva ohnoi. Biosci. Biotech. Bioch. 81 (11), 2145–2151. doi: 10.1080/09168451.2017.1379352
He P., Xie L., Zhang X., Li J., Lin X., Pu X, et al. (2020). Microbial diversity and metabolic potential in the stratified Sansha Yongle Blue Hole in the South China Sea. Scientific rep 10 (1), 1–17. doi: 10.1038/s41598-020-62411-2
Hennon G. M., Morris J. J., Haley S. T., Zinser E. R., Durrant A. R., Entwistle E., et al. (2018). The impact of elevated CO2 on Prochlorococcus and microbial interactions with ‘helper’ bacterium Alteromonas. ISME J. 12 (2), 520. doi: 10.1038/ismej.2017.189
Heller M. I., Lam P. J., Moffett J. W., Till C. P., Lee J. M., Toner B. M, et al. (2017). Accumulation of Fe oxyhydroxides in the Peruvian oxygen deficient zone implies non-oxygen dependent Fe oxidation. Geochimica et Cosmochimica Acta 211, 193. doi: 10.1016/j.gca.2017.05.019
Ivars-Martinez E., D'auria G., Rodriguez-Valera F., Sánchez-Porro C., Ventosa A., Joint I., et al. (2008b). Biogeography of the ubiquitous marine bacterium Alteromonas macleodii determined by multilocus sequence analysis. Mol. Ecol. 17 (18), 4092–4106. doi: 10.1111/j.1365-294X.2008.03883.x
Ivars-Martinez E., Martin-Cuadrado A. B., D'auria G., Mira A., Ferriera S., Johnson J., et al. (2008a). Comparative genomics of two ecotypes of the marine planktonic copiotroph Alteromonas macleodii suggests alternative lifestyles associated with different kinds of particulate organic matter. ISME J. 2 (12), 1194. doi: 10.1038/ismej.2008.74
Jain C., Rodriguez-R L. M., Phillippy A. M., Konstantinidis K. T., Aluru S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9 (1), 1–8. doi: 10.1038/s41467-018-07641-9
Jin H. M., Im Jeong H., Kim K. H., Hahn Y., Madsen E. L., Jeon C. O. (2016). Genome-wide transcriptional responses of Alteromonas naphthalenivorans SN2 to contaminated seawater and marine tidal flat sediment. Sci. Rep. 6, 21796. doi: 10.1038/srep21796
Jin H. M., Kim J. M., Lee H. J., Madsen E. L., Jeon C. O. (2012). Alteromonas as a key agent of polycyclic aromatic hydrocarbon biodegradation in crude oil-contaminated coastal sediment. Environ. Sci. Technol. 46, 7731–7740. doi: 10.1021/es3018545
Kanehisa M., Goto S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30. doi: 10.1093/nar/28.1.27
Keil R. G., Neibauer J. A., Biladeau C., van der Elst K., Devol A. H. (2016). A multiproxy approach to understanding the “enhanced” flux of organic matter through the oxygen-deficient waters of the Arabian Sea. Biogeosciences 13, 2077–2092. doi: 10.5194/bg-13-2077-2016
Koch H., Freese H. M., Hahnke R. L., Simon M., Wietz M. (2019). Adaptations of Alteromonas sp. 76-1 to polysaccharide degradation: A CAZyme plasmid for ulvan degradation and two alginolytic systems. Front. Microbiol. 10, 504.
Koch H., Germscheid N., Freese H. M., Noriega-Ortega B., Lücking D., Berger M., et al. (2020). Genomic, metabolic and phenotypic variability shapes ecological differentiation and intraspecies interactions of Alteromonas macleodii. Sci. Rep. 10 (1), 1–4. doi: 10.1038/s41598-020-57526-5
Kondo Y., Moffett J.W. (2015). Iron redox cycling and subsurface offshore transport in the eastern tropical South Pacific oxygen minimum zone. Marine Chemistry 168, 95–103. doi: 10.1016/j.marchem.2014.11.007
Koedooder C., Guéneuguès A., Van Geersdaële R., Vergé V., Bouget F.-Y., Labreuche Y. (2018). The role of the glyoxylate shunt in the acclimation to iron limitation in marine heterotrophic bacteria. Front. Mar. Sci. 5, 435. doi: 10.3389/fmars.2018.00435
Lavin P., González B., Santibáñez J. F., Scanlan D. J., Ulloa O. (2010). Novel lineages of Prochlorococcus thrive within the oxygen minimum zone of the eastern tropical south pacific. Env. Microbiol. Rep. 2 (6), 728–738. doi: 10.1111/j.1758-2229.2010.00167.x
Lekunberri I., Sintes E., De Corte D., Yokokawa T., Herndl G. J. (2013). Spatial patterns of bacterial and archaeal communities along the romanche fracture zone (tropical Atlantic). FEMS Microbiol. Ecol. 85 (3), 537–552. doi: 10.1111/1574-6941.12142
López-Pérez M., Rodriguez-Valera F The Family Alteromon-adaceae, in: The Prokaryotes: Gammaproteobacteria, edited by:Rosenberg E., DeLong E. F., Lory S., Stackebrandt E., Thompson F. Berlin, Heidelberg:Springer Berlin Heidelberg
López-Pérez M., Gonzaga A., Martin-Cuadrado A. B., Onyshchenko O., Ghavidel A., Ghai R. (2012). Genomes of surface isolates of Alteromonas macleodii: The life of a widespread marine opportunistic copiotroph. Scientific reports 2 (1), 11–11. doi: 10.1038/srep00696
López-Pérez M., Ramon-Marco N., Rodriguez-Valera F. (2017). Networking in microbes: Conjugative elements and plasmids in the genus Alteromonas. BMC Genomics 18 (1), 36. doi: 10.1186/s12864-016-3461-0
López-Pérez M., Rodriguez-Valera F. (2016). Pangenome evolution in the marine bacterium Alteromonas. Genome Biol. Evol. 8 (5), 1556–1570. doi: 10.1093/gbe/evw098
MacIntyre L. W., Haltli B. A., Kerr R. G. (2019). Draft genome sequence of alteromonas sp. strain RKMC-009, isolated from xestospongia muta via in situ culturing using an isolation chip diffusion chamber. Microbiol. Resour. Announc. 8 (25), e00508–e00519.
Manck L. E., Espinoza J. L., Dupont C. L., Barbeau K. A. (2020). Transcriptomic study of substrate-specific transport mechanisms for iron and carbon in the marine copiotroph Alteromonas macleodii. mSystems 5 (2). doi: 10.1128/mSystems.00070-20
Manck L. E., Park J., Tully B. J., Poire A. M., Bundy R. M., Dupont C. L., et al. (2022). Petrobactin, a siderophore produced by Alteromonas, mediates community iron acquisition in the global ocean. ISME J. 16 (2), 358–369. doi: 10.1038/s41396-021-01065-y
Markowitz V. M., Mavromatis K., Ivanova N. N., Chen I. M. A., Chu K., Kyrpides N. C. (2009). IMG ER: A system for microbial genome annotation expert review and curation. Bioinformatics 25 (17), 2271–2278. doi: 10.1093/bioinformatics/btp393
Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17 (1), 10–12. doi: 10.14806/ej.17.1.200
Márquez-Artavia A., Sánchez-Velasco L., Barton E. D., Paulmier A., Santamarı́a-Del-Ángel E., Beier E., et al. (2019). A suboxic chlorophyll-a maximum persists within the Pacific oxygen minimum zone off Mexico. Deep Sea Res. Part II: Topical Studies in Oceanography 169 (1), 104686.
Math R. K., Jin H. M., Kim J. M., Hahn Y., Park W., Madsen E. L., et al. (2012). Comparative genomics reveals adaptation by Alteromonas sp. SN2 to marine tidal-flat conditions: cold tolerance and aromatic hydrocarbon metabolism. PloS One 7 (4), e35784.
McMurdie P. J., Holmes S. (2013). Phyloseq: An r package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8 (4), e61217. doi: 10.1371/journal.pone.0061217
Medina L. E., Taylor C. D., Pachiadaki M. G., Henríquez-Castillo C., Ulloa O., Edgcomb V. P. (2017). A review of protist grazing below the photic zone emphasizing studies of oxygen-depleted water columns and recent applications of in situ approaches. Front. Mar. Sci. 4, 105. doi: 10.3389/fmars.2017.00105
Messié M., Chavez F. P. (2015). Seasonal regulation of primary production in eastern boundary upwelling systems. Prog. Oceanogr. 134, 1–18. doi: 10.1016/j.pocean.2014.10.011
Mikhailov V. V., Romanenko L. A., Ivanova E. P. (2006). The genus alteromonas and related proteobacteria. the prokaryotes: A handbook on the biology of bacteria Vol. 6 (New York, USA: Springer-Verlag), 597–645.
Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G. A., Sonnhammer E. L., et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49 (D1), D412–D419. doi: 10.1093/nar/gkaa913
Moore J. K., Braucher O. (2007). Observations of dissolved iron concentrations in the world ocean: Implications and constraints for ocean biogeochemical models. Biogeosciences 4 (2), 1241–1277. doi: 10.5194/bgd-4-1241-2007
Mounier J., Camus A., Mitteau I., Vaysse P. J., Goulas P., Grimaud R., et al. (2014). The marine bacterium marinobacter hydrocarbonoclasticus SP17 degrades a wide range of lipids and hydrocarbons through the formation of oleolytic biofilms with distinct gene expression profiles. FEMS Microbiol. Ecol. 90 (3), 816–831. doi: 10.1111/1574-6941.12439
Nayfach S., Rodriguez-Mueller B., Garud N., Pollard K. S. (2016). An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res. 26 (11), 1612–1625. doi: 10.1101/gr.201863.115
Nguyen L. T., Schmidt H. A., Von Haeseler A., Minh B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi: 10.1093/molbev/msu300
Padilla C. C., Bertagnolli A. D., Bristow L. A., Sarode N., Glass J. B., Thamdrup B., et al. (2017). Metagenomic binning recovers a transcriptionally active gammaproteobacterium linking methanotrophy to partial denitrification in an anoxic oxygen minimum zone. Front. Mar. Sci. 4, 23. doi: 10.3389/fmars.2017.00023
Padilla C. C., Bristow L. A., Sarode N., Garcia-Robledo E., Ramírez E. G., Benson C. R., et al. (2016). NC10 bacteria in marine oxygen minimum zones. ISME J. 10 (8), 2067. doi: 10.1038/ismej.2015.262
Parada A. E., Needham D. M., Fuhrman J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18 (5), 1403–1414. doi: 10.1111/1462-2920.13023
Parks D. H., Imelfort M., Skennerton C. T., Hugenholtz P., Tyson G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25 (7), 1043–1055. doi: 10.1101/gr.186072.114
Pedler B. E., Aluwihare L. I., Azam F. (2014). Single bacterial strain capable of significant contribution to carbon cycling in the surface ocean. P Natl. Acad. Sci. U.S.A. 111 (20), 7202–7207. doi: 10.1073/pnas.1401887111
Plominsky A. M., Trefault N., Podell S., Blanton J. M., de la Iglesia R., Allen E. E., et al. (2018). Metabolic potential and in situ transcriptomic profiles of previously uncharacterized key microbial groups involved in coupled carbon, nitrogen and sulfur cycling in anoxic marine zones. Environ. Microbiol. 20 (8), 2727–2742. doi: 10.1111/1462-2920.14109
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucl. Acids Res. 41 (D1), D590–D596.
Raven M. R., Keil R. G., Webb S. M. (2021). Microbial sulfate reduction and organic sulfur formation in sinking marine particles. Science 3, 178–181. doi: 10.1126/science.abc6035
Revsbech N. P., Larsen L. H., Gundersen J., Dalsgaard T., Ulloa O., Thamdrup B. (2009). Determination of ultra-low oxygen concentrations in oxygen minimum zones by the STOX sensor. Limnol. Oceanogr-Meth. 7 (5), 371–381. doi: 10.4319/lom.2009.7.371
Ruiz-Fernández P., Ramírez-Flandes S., Rodríguez-León E., Ulloa O. (2020). Autotrophic carbon fixation pathways along the redox gradient in oxygen-depleted oceanic waters. Environ. Microbiol. Rep. 12 (3), 334–341. doi: 10.1111/1758-2229.12837
Sambrook J., Russell D. W. (2006). Purification of nucleic acids by extraction with phenol: chloroform. Cold Spring Harbor Protoc. 2006 (1), pdb–prot4455.
Saunders J. K., Fuchsman C. A., Mckay C., Rocap G. (2019). Complete arsenic-based respiratory cycle in the marine microbial communities of pelagic oxygen-deficient zones. Proc. Natl. Acad. Sci. U.S.A. 116, 9925–9930. doi: 10.1073/pnas.1818349116
Schattenhofer M., Fuchs B. M., Amann R., Zubkov M. V., Tarran G. A., Pernthaler J. (2009). Latitudinal distribution of prokaryotic picoplankton populations in the Atlantic ocean. Environ. Microbiol. 11 (8), 2078–2093. doi: 10.1111/j.1462-2920.2009.01929.x
Schlosser C., Streu P., Frank M., Lavik G., Croot P. L., Dengler M., et al. (2018). H2S events in the Peruvian oxygen minimum zone facilitate enhanced dissolved fe concentrations. Sci. Rep. 8 (1), 12642.
Stepanauskas R., Fergusson E. A., Brown J., Poulton N. J., Tupper B., Labonté J. M., et al. (2017). Improved genome recovery and integrated cell-size analyses of individual uncultured microbial cells and viral particles. Nat. Commun. 8 (1), 1–10.
Stewart F. J., Ulloa O., DeLong E. F. (2012). Microbial metatranscriptomics in a permanent marine oxygen minimum zone. Environ. Microbiol. 14 (1), 23–40. doi: 10.1111/j.1462-2920.2010.02400.x
Tackmann J., Rodrigues J. F. M., von Mering C. (2019). Rapid inference of direct interactions in large-scale ecological networks from heterogeneous microbial sequencing data. Cell Syst. 9 (3), 286–296. doi: 10.1016/j.cels.2019.08.002
Tatusov R. L., Galperin M. Y., Natale D. A., Koonin E. V. (2000). The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28 (1), 33–36. doi: 10.1093/nar/28.1.33
Tennessen K., Andersen E., Clingenpeel S., Rinke C., Lundberg D. S., Han J., et al. (2016). ProDeGe: A computational protocol for fully automated decontamination of genomes. ISME J. 10 (1), 269–272. doi: 10.1038/ismej.2015.100
Thamdrup B., Dalsgaard T., Revsbech N.P. (2012). Widespread functional anoxia in the oxygen minimum zone of the Eastern South Pacific. Deep Sea Res. Part I Oceanogr. Res. Pap. 65, 36–45
Tiano L., Garcia-Robledo E., Dalsgaard T., Devol A. H., Ward B. B., Ulloa O., et al. (2014). Oxygen distribution and aerobic respiration in the north and south eastern tropical pacific oxygen minimum zones. Deep Sea Res. Part I Oceanogr. Res. Pap. 94, 173–183. doi: 10.1016/j.dsr.2014.10.001
Ulloa O., Canfield D. E., DeLong E. F., Letelier R. M., Stewart F. J. (2012). Microbial oceanography of anoxic oxygen minimum zones. P Natl. Acad. Sci. U.S.A. 109 (40), 15996–16003. doi: 10.1073/pnas.1205009109
Ulloa O., Henríquez-Castillo C., Ramírez-Flandes S., Plominsky A. M., Murillo A. A., Morgan-Lang C., et al. (2021). The cyanobacterium prochlorococcus has divergent light-harvesting antennae and may have evolved in a low-oxygen ocean. P Natl. Acad. Sci. U.S.A. 118 (11), e2025638118. doi: 10.1073/pnas.2025638118
Vedamati J., Goepfert T., Moffett J. W. (2014). Iron speciation in the eastern tropical south pacific oxygen minimum zone off peru. Limnol. Oceanogr. 59, 1945–1957. doi: 10.4319/lo.2014.59.6.1945
Keywords: Oxygen Minimum Zones, environmental genomics, biogeochemical cycles, single-cell genomics, Alteromonas
Citation: Henríquez-Castillo C, Plominsky AM, Ramírez-Flandes S, Bertagnolli AD, Stewart FJ and Ulloa O (2022) Metaomics unveils the contribution of Alteromonas bacteria to carbon cycling in marine oxygen minimum zones. Front. Mar. Sci. 9:993667. doi: 10.3389/fmars.2022.993667
Received: 13 July 2022; Accepted: 30 August 2022;
Published: 20 September 2022.
Edited by:
Amal Jayakumar, Princeton University, United StatesReviewed by:
Clara A. Fuchsman, University of Maryland, College Park, United StatesRam Murti Meena, National Institute of Oceanography, India
J. Michael Beman, University of California, Merced, United States
Copyright © 2022 Henríquez-Castillo, Plominsky, Ramírez-Flandes, Bertagnolli, Stewart and Ulloa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos Henríquez-Castillo, Y2FybG9zLmhlbnJpcXVlekBjZWF6YS5jbA==
†These authors share first authorship