Abstract
Introduction:
Compared to mammals and birds, sex-determining genes differ in most fish species. Largemouth bass (Micropterus Salmoides) is one of the most important cultured fish species in China, and there are growth differences between males and females. However, its sex-determining genes and mechanisms currently remain unknown.
Methods:
We explored the sex-determination mechanism by integrating whole-genome sequencing, resequencing and comparative genomics approaches.
Results:
In this study, we employed HiFi and Hi-C sequencing technologies to construct a chromosome-level haplotypic genome assembly for male largemouth bass, with a genome size of 875.69 Mb. The assembled genome contains 23 chromosomes, covering 95.31% of the complete sequences with a high scaffold N50 of 35.93 Mb. A genome-wide association study (GWAS) of sex was performed with four populations consisting of 62 males and 58 females. For the sex trait, a total of 3,838 SNP loci were identified to be significantly associated with sexual discrepancy. Interestingly, almost all these significant SNPs (3,825) were clustered on chromosome 10 (Chr10), within a 3.5-Mb sex-determination region (SDR). They were homozygous in females while heterozygous in males. We therefore speculate that largemouth bass owns a XX/XY sex determination system. By comparing genomics data and examining coverage depth of resequencing reads, we revealed a ~51-kb male-specific region (MSR) on Chr10. Gene annotation discovered a coding sequence (msy) within MSR-1, which may contribute to sex determination of largemouth bass. By differential expression analysis, two candidate sex-determining genes (ccdc103 and jockey) were predicted within the target SDR. Moreover, we applied two male-specific non-coding fragments (within MSR-2 and MSR-3) to design specific sex markers, successfully obtaining universal gender identity in examined largemouth bass.
Discussion:
Overall, our findings improve our understanding of the molecular basis for sex determination in largemouth bass, which will thereby promote the mono-sexual breeding progress in the aquaculture industry.
1 Introduction
Sex determination (SD) is an important process, by which sexually reproducing organisms begin to differentiate into males and females. In various fishes, it is fulfilled by a complex system that is triggered by certain genetic and/or environmental factors (Dan et al., 2018; Tao et al., 2021). Compared to the highly conserved sex determination systems of mammals and birds, fishes display a diversity of sex determination patterns with various sex-determining genes (Chen et al., 2012; Mei and Gui, 2015; Smith and Wootton, 2016). Interestingly, most fishes have homomorphic sex chromosomes, which cannot be distinguished by the phenotype of sex chromosomes (Devlin and Nagahama, 2002). In fact, only a few fishes own dimorphic sex chromosomes, such as in half-smooth tongue sole (Cynoglossus semilaevis) (Liao et al., 2014), which also increases the possibility for identifying any SD system and sex-determining genes.
So far, sex-determining genes in more than 30 fish species have been reported, and interestingly most of these genes belong to the transforming growth factor-beta (TGF-β) gene family (Chen et al., 2022), such as anti-Mullerian hormone (amh) in black rockfish (Sebastes schlegelii) (Song et al., 2021), Nile tilapia (Oreochromis niloticus) (Li et al., 2015), Northern pike (Esox lucius) (Pan et al., 2019), mandarin fish (Siniperca chuatsi) (Han et al., 2020) and three-spine stickleback (Gasterosteus aculeatus) (Peichel et al., 2020), and anti-Mullerian hormone receptor type 2 (amhr2) in yellow perch (Perca flavescens) (Feron et al., 2020), tiger puffer (Takifugu rubripes) (Kamiya et al., 2012), Southern catfish (Silurus meridionalis) (Zheng et al., 2022) and ayu (Plecoglossus altivelis) (Nakamoto et al., 2021). In addition, doublesex and mab-3 related transcription factor 1 (dmrt1), SRY-box transcription factor 3 (sox3) and gonadal somatic cell derived factor (gsdf) have been identified in three medaka fish species, Oryzias latipes (Nanda et al., 2002), O. dancena (Takehana et al., 2014) and O. luzonensis (Myosho et al., 2012) respectively. The interferon regulatory factor 9 (located in a male-specific genomic sequence and named as sdy), dmrt1 and breast cancer anti-estrogen resistance protein 1 (bcar1) have also been determined as sex-determining genes in rainbow trout (Oncorhynchus mykiss) (Yano et al., 2012), Siamese fighting fish (Betta splendens) (Wang et al., 2022), and channel catfish (Ictalurus punctatus) (Bao et al., 2019). Recently, some new master sex-determining genes have been validated, such as the inhibitor of DNA binding 2b (id2b) in arapaima (Arapaima gigas) (Adolfi et al., 2021), follicle stimulating hormone receptor (fshr) in Senegales sole (Solea senegalensis) (De la Herrán et al., 2023), and PDZ domain-containing gene (pfpdz1) in yellow catfish (Pelteobagrus fulvidraco) (Dan et al., 2018). As more and more sex-determining genes are identified in various fishes, it is likely that more and more complicated SD systems will be discovered.
According to published reports, fish sex-determining genes can be mainly divided into four categories, including i) a sex-specific region containing a gene (usually duplicate of a sex-determining gene that can be expressed specifically in one sex to result in sexual differentiation), such as an amhr2by gene inserted into the male specific region of yellow perch (Feron et al., 2020), and a duplicated copy of dmrt1 on the Y chromosome of medaka (Matsuda et al., 2002); ii) allelic variants in sex-determining genes of both sexes, such as amhr2 in tiger pufferfish (Kamiya et al., 2012) and fshr in Senegales sole (De la Herrán et al., 2023); iii) sex-specific expression of an isotype of the sex-determining gene at an early stage of sex differentiation (Bao et al., 2019); and iv) presence of a heteromorphic sex chromosome (such as in C. semilaevis) (Chen et al., 2014). Meanwhile, other modes of sex regulation have also been reported. For instance, epigenetic modifications (including histone modification and genomic DNA methylation) and cis-regulatory elements have been confirmed to control SD by regulating the expression of sex-determining genes in some fish species (Shao et al., 2014; Takehana et al., 2014; Wang et al., 2022).
Largemouth bass (Micropterus salmoides) is an important cultured euryhaline fish species in China (Sun et al., 2021). It exhibits an interesting dimorphism in sexual growth, with males generally living longer and becoming larger than females (Wei, 2022). Obviously, establishment of an all-male population would help to increase the aquaculture production of largemouth bass. Previous studies confirmed that the sex determination system in largemouth bass is male heterogametic (XX/XY) (Du et al., 2021). A sex-linked region was identified on Chr10 or Chr7 (from different genome data), but these researchers did not find any large region with remarkable difference between both sexes (He et al., 2022b; Wen et al., 2022). However, we validated that some previously reported molecular markers were tested with low accuracy when the examined largemouth bass samples were collected from multiple populations with different origins. Up to now, the precise sex-determining genes in largemouth bass are still unknown.
Two genome assemblies of female largemouth bass have been published (He et al., 2022a; Sun et al., 2021). Here, we constructed a high-quality genome assembly of male largemouth bass. Based on the assembled male fish genome and subsequent Genome-Wide Association Study (GWAS), we identified sex-specific regions and potential sex-determining genes. Within the sex determination region (SDR), we developed two molecular markers that can accurately determine the genetic sex in different populations, which solves the problem of low efficiency for those reported sex identification markers from other studies. Our present work has laid a good foundation for further practical research on mono-sex breeding of largemouth bass.
2 Materials and methods
2.1 Fish sampling and whole-genome resequencing of four populations
A total of 120 adult individuals (58 females: average body length of 29.6 ± 2.29 cm and average body weight of 567.44 ± 45.37 g; 62 males: average body length of 31.8 ± 5.17 cm and average body weight of 612.5 ± 40.45 g) from four populations (30 individuals per population) were collected for whole-genome resequencing. A breeding population introduced from Taiwan (abbreviated as TWL), a common breeding population in the mainland of China (TL), and Youlu (YL) and Jiadefeng (JDF) breeding populations from Guangdong province were obtained for comparison (refer to Sun et al., 2023a). These fishes were cultured for sampling at Foshan Xinrong Aquatic Co. Ltd. (Foshan city, Guangdong province, China) and Guangzhou Huaxuan Aquatic Co. Ltd. (Guangzhou city, Guangdong province, China). Their sex phenotype was confirmed by dissection and histologic observation. Caudal fins from each fish were collected for storage in 95% ethanol at – 20°C before DNA extraction.
Genomic DNA (gDNA) was isolated from collected fins using a QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA). The quality of gDNA was checked via agarose gel electrophoresis and Agilent 4200 Bioanalyser (Agilent Technologies, Palo Alto, CA, USA). Illumina DNA libraries with an average insert size of 350 bp were constructed using a Genomic DNA Sample Prep kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. These libraries were subsequently sequenced by an Illumina NovaSeq 6000 platform with a paired-end mode (PE150).
2.2 DNA collection, genome sequencing, and genome assembly for a male fish
Muscle tissue of an adult male largemouth bass (from the JDF population) was sampled from Pearl River Fisheries Research Institute (Guangzhou, China) and used for whole genome sequencing. An Illumina library with an insert size of 350 bp was constructed and then sequenced using a DNBSEQ-T7 platform (MGI, Shenzhen, China). We employed SOAPfilter v.2.2 for quality control to clean raw reads (Chen et al., 2018). A High-fidelity (HiFi) library was generated using SMRTbell Express Template Prep Kit 2.0 and sequenced on PacBio Sequel IIe platform (Pacific Biosciences, Menlo Park, USA). The chromosome conformation capture (Hi-C) library was constructed from muscle tissue for chromosome construction. DNA was cross-linked with 4% formaldehyde, digested by MboI, labeled with biotin-14-DCTP, and then linked with T4 DNA ligase. The ligated DNA was digested into 200~600-bp fragments and then sequenced on a MGI DNBSEQ-T7 platform with the PE150 module.
The male genome was assembled using Hifiasm v.0.16.0-r369 with default parameters, which can indeed perform haplotype phasing and split the total genome sequence into two haplotypes (Cheng et al., 2021). For the Hi-C assembly, clean Hi-C reads were mapped to the constructed contigs using HiC-Pro with default settings (Servant et al., 2015). Subsequently, an initial chromosome-level genome was assembled using the 3D-DNA pipeline with default parameters (Dudchenko et al., 2017). Finally, Juicebox (Durand et al., 2016) was employed to visualize before manual refinements to construct the final chromosome-scale assembly.
The integrity of our genome assembly was estimated using Benchmarking Universal Single-Copy Orthologs (BUSCO) with the actinopterygii_odb9 database as the reference (Simão et al., 2015). Genomic comparison between male (this study) and female (He et al., 2022a) assemblies was fulfilled by JCVI v1.0.9 (Tang et al., 2008) and syri v1.3 (Goel et al., 2019) with default parameters.
2.3 Repeat element annotation, gene prediction, and functional annotation
Repetitive sequences were predicted by combination of homology and de novo approaches. For the homology prediction, Tandem Repeats Finder (v4.07) was applied to search for tandem repeats (Benson, 1999). Transposable elements (TEs) were identified using RepeatMasker (v4.0.6) and RepeatProteinMask (v4.0.6) (Tarailo-Graovac and Chen, 2009). For the de novo approach, RepeatModeler v1.0.8 (Abrusan et al., 2009) and LTR_FINDER v1.0.6 (Xu and Wang, 2007) were employed to generate a de novo repeat library, and then RepeatMasker was applied to annotate repeat elements against this repeat library.
We annotated gene structures by integration of de novo prediction, homology-based prediction, and transcriptome-based prediction. First, the ab inito gene prediction was performed by AUGUSTUS v3.2.1 (Stanke et al., 2006). Second, the homology-based prediction was conducted by using the GeMoMa software (Keilwagen et al., 2019). We aligned homology proteins from six other fish species, including zebrafish (Danio rerio), Asian seabass (Lates calcarifer), medaka (Oryzias latipes), spotted gar (Lepisosteus oculatus), Smallmouth bass (Micropterus dolomieu) and European bass (Dicentrarchus labrax) (downloaded from the NCBI). Third, the transcriptome (RNA-seq) data from five tissues (including muscle, spleen, gill, liver and brain) were aligned to the assembled genome using Hisat2 v2.0.13 (Kim et al., 2019), and then we applied cufflinks v2.1.1 to predict gene structures (Trapnell et al., 2012). Finally, these gene sets were integrated by the MAKER pipeline v1.0 (Elsik et al., 2007). Transposonpsi (http://transposonpsi.sourceforge.net) was employed to align this gene set to the transposon database with default parameters. Remove those genes homologous to transposons from the final gene set.
These predicted genes were aligned against five public databases, including Interpro, Kyoto Encyclopedia of Genes and Genomes (KEGG), Swiss-Prot, TrEMBL and NCBI Non-Redundant Protein Sequence (NR), using BLASTP (Balakrishnan et al., 2005). Data from the searches were concatenated as the final annotation file.
2.4 Phylogenetic tree construction and divergence time estimation
To identify gene families in the male largemouth bass genome, protein-coding sequences of spotted gar, Atlantic herring (Clupea harengus), Mexican tetra (Astyanax mexicanus), channel catfish (Ictalurus punctatus), Northern pike (Esox lucius), rainbow trout (Oncorhynchus mykiss), Atlantic cod (Gadus morhua), medaka, Nile tilapia (Oreochromis niloticus), greater amberjack (Seriola dumerili), half-smooth tongue sole (C. semilaevis), turbot (Scophthalmus maximus), three-spined stickleback (Gasterosteus aculeatus), yellow perch (Perca flavescens), big head croaker (Collichthys lucidus), Mandarin fish (Siniperca chuatsi), and smallmouth bass (Micropterus dolomieu) were downloaded from the NCBI. Subsequently, OrthoMCL v1.4 (Li et al., 2003) was executed to cluster these predicted gene families among the examined genomes. We selected single copy orthologous genes from the genomes of the representative eighteen teleost species, and applied MUSCLE v3.8.31 (Edgar, 2004) and PhyML v3.0 (Guindon et al., 2009) to construct a phylogenetic tree. Based on the phylogenetic topology, MCMCTREE in the PAML v4.9e package (Yang, 2007) was employed to estimate divergence times between largemouth bass and other examined species, with assistance of fossil records from the TimeTree (Kumar et al., 2017).
2.5 Read trimming and variant calling of whole-genome resequencing data
We employed Trimmomatic v0.32 (Bolger et al., 2014) to remove adapter sequences and low-quality reads. The high-quality clean reads were subsequently aligned to the assembled male genome using BWA v0.7.15 (Li and Durbin, 2009) with default parameters. SAMtools v 1.3.1 (Li et al., 2009) was employed to create index and retain those mapped high-quality sequences. The MarkDuplicates tool in Picard v1.54 software (https://broadinstitute.github.io/picard/) was applied to remove PCR duplicated reads.
Genome Analysis Toolkit (GATK) v4.0 (Franke and Crowgey, 2020) was employed for SNP and Indel variant detection and variant filtering, as it can identify single-base substitutions in addition to small insertions and deletions. Quality control was conducted on all the SNPs compiled by variant call format (VCF) using the VCFtools v0.1.13 software (Danecek et al., 2011) after genotyping of each sample. SNPs were filtered using the following criteria: (i) SNPs with QD < 2.0, MQ<40.0, MQRankSum < -12.5, FS > 60.0, and ReadPosRankSum < -8.0; (ii) variant missing rate < 10%; and (iii) minor allele frequency (MAF) > 0.05.
2.6 Identification of potential sex-determination regions by a genome-wide association study and fixation index (Fst)
High-quality SNPs obtained after filtering were used for GWAS. Missing data were imputed by beagle v5.0 (Browning et al., 2018). The association analysis was performed with EMMAX using the Mixed Linear Model (MLM) (Legarra et al., 2018). Manhattan plots were generated by the ggplot2 package (Ito and Murphy, 2013). Threshold for these plots, represented as the -log10(0.05/n), was set at 9.
Fst was calculated using VCFtools v0.1.13 (Danecek et al., 2011) in every 100-kb sliding window with a step size of 10-kb (-window-pi 100000 -window-pi-step 10000), and the results were visualized through R package ggplot2. The level of genetic differentiation was represented by Fst with values ranging from 0 to 1, and huge differentiation was rated as Fst > 0.25 (Choy et al., 2015).
2.7 Identification of the male-specific region
The resequencing data from both male and female largemouth bass were aligned to the assembled male genome using BWA. SAMtools and Deeptools v3.5.1 (Ramirez et al., 2014) were applied to calculate coverage depth, which was calculated using 10-kb non-overlapping sliding windows. Based on the differences between these maps, possible sex-specific regions were identified. Integrative Genomics Viewer (IGV) v2.5.2 (Robinson et al., 2011) was employed to show the coverage differences of those reads from different sexes on the reference genome.
2.8 Verification of male-specific markers
Two sex-specific primer pairs were designed based on the MSR-2 and MSR-3 sequences using Primer 6.0 (Supplementary Table S1), which were amplified in newly collected 120 male and 150 female samples from three populations (YL, TWL and JDF). gDNA of each fish was extracted as described in the section 2.1. PCR amplification was performed in a total volume of 12.5 μl, containing 0.5 μl of gDNA (100 ng/μL), 6.25 μl of 2× Premix Taq (Takara Bio Inc., Dalian, Liaoning, China), 0.5 μl of each primer (10 mM), and 4.75 μl of double distilled water.
The amplification conditions were set as follows: initial denaturation for 5 min at 94°C, denaturation at 94°C for 30 s, annealing at an optimal temperature for 30 s, extension at 72°C for 30 s, a total of 35 cycles; and the reaction was extended for 7 min at 72°C. Amplification products were assessed using 1% agarose gel and then verified by Sanger sequencing.
2.9 Expression analyses of sex-determination region genes in mature gonads
Mature testis or ovary of each fish were determined by visual observation, and subsequently three females and three males were collected for transcriptome sequencing. The cDNA library was constructed according to the manufacturer’s instructions and then sequenced on an Illumina HiSeq 4000 platform using a paired-end mode (PE150). Adaptor sequences and low-quality reads were removed using Trimmomatic (Bolger et al., 2014). The clean reads from each sample were mapped to the assembled male largemouth bass genome using Hisat2 v2.2.1 with default parameters (Kim et al., 2019). RSEM v1.2.31 (Li and Dewey, 2011) was then applied to estimate expression abundance of annotated transcripts.
3 Results
3.1 Phenotypic sexuality of largemouth bass
In order to accurately determine the phenotypic sex of largemouth bass individuals, male and female gonads were histologically sectioned. See more details of gonadal differences in Supplementary Figure S1.
3.2 A chromosome-level assembly of male genome and genomic characterization
A total of 67.73-Gb Illumina clean data were applied for a genome survey. We then combined HiFi and Hi-C data to construct a chromosome-level male genome assembly for largemouth bass. A total of 31.5-Gb HiFi long reads were assembled to produce an initial genome assembly, with 296 contigs and a contig N50 of 19.43 Mb. A total of 67.9-Gb Hi-C data were used for chromosome construction. The final chromosome-scale haplotypic genome assembly for the male largemouth bass is 875 Mb, with a scaffold N50 of 35.93 Mb (Table 1). A sum of 834-Mb (95.3%) contig sequences were anchored to 23 chromosomes (Figure 1A). The heatmap of chromosome contacts displays good completeness of this genome assembly. The assembly completeness was further evaluated using BUSCO v3.0 (Simão et al., 2015), and we predicted that 98.3% of BUSCO genes are complete in the male genome assembly.
Table 1
| Parameter | Haplotype | Haplotype 1 | Haplotype 2 |
|---|---|---|---|
| Sex | Male | Haplotype-Y | Haplotype-X |
| Contig N50 (bp) | 14,257,025 | 8,791,301 | 9,591,873 |
| Scaffold N50 (bp) | 35,933,337 | 35,897,524 | 35,724,343 |
| Scaffold number | 472 | 501 | 411 |
| Genome Size (bp) | 875,695,184 | 861,871,429 | 871,473,078 |
| GC | 40.9% | 40.9% | 40.9% |
| BUSCO | 98.3% | 97.6% | 98.2% |
| Chromosome anchor ratio | 95.3% | 95.6% | 95.7% |
| Chromosome length (bp) | 834,598,459 | 824,132,482 | 834,549,618 |
Statistics of various assemblies of largemouth bass.
Figure 1
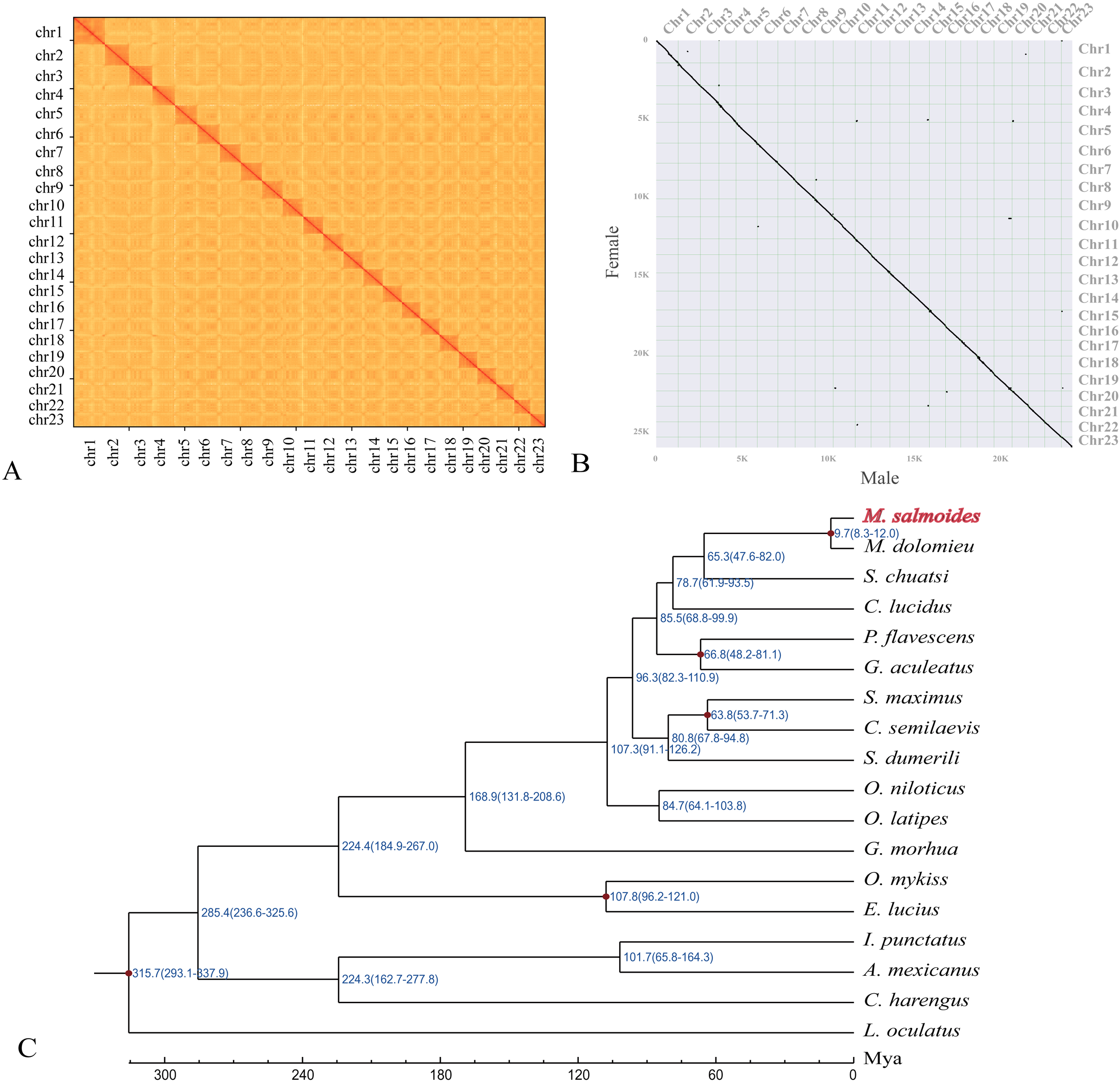
Construction of a male (XY) genome assembly of largemouth bass for comparative analysis. (A) Hi-C interactions among 23 chromosomes in the assembled XY genome. Strong to weak interactions are shown in red to yellow. (B) A synteny analysis between this XY genome and reported female genome (He et al., 2022a) assemblies using JCVI. (C) A phylogenetic tree showing relationships between largemouth bass and other representative teleost species. Blue numbers represent the divergence time (Million years ago, Mya). The purple points on five internal nodes indicate those fossil calibration times. The Latin name of largemouth bass is marked (at the top) to highlight its phylogenetic location.
Genome assemblies of the haplotype 1 (Y chromosome sequences) and haplotype 2 (X chromosome sequences) were separated, with lengths of 861 and 871 Mb and scaffold N50 values of 35.8 and 35.7 Mb, respectively. Additionally, to evaluate the quality of both assemblies, we performed a synteny analysis between male (this study) and female (GCA_022435785.1) genomes. Not surprisingly, the assembled 23 chromosomes of XY genome (Figure 1A) showed a full correspondence (Figure 1B) to previously reported XX female genome (He et al., 2022a).
Homology and de novo prediction results showed that repeated sequences accounted for 32.03% of the assembled male genome. Transposable elements (TEs) are the main category of repeat sequences. LINEs (long interspersed repeated segments) were the most abundant (18.02%), followed by DNA transposons (12.03%) and LTR (long terminal repeats, 5.09%). In total, 26,319 protein-coding genes were annotated by integration of de novo, homologous, and transcriptome-based prediction in the assembled male genome. Finally, 25,191 genes (95.71% of the total) were successfully annotated with at least one hit from the searched databases (Supplementary Table S2).
3.3 Phylogenomic analyses
A total of 746 single-copy ortholog genes were collected for the prediction of phylogenetic relationships and divergence times. Our results showed that largemouth bass is clustered with smallmouth bass within the genus Micropterus (Figure 1C). We also estimated evolutionary divergence times; it seems that Micropterus diverged from Mandarin fish ~65.3 Mya, while largemouth bass and smallmouth bass diverged ~9.7 Mya (Figure 1C).
3.4 Summary of resequencing and genomic variations
A total of 120 individuals were collected for whole-genome resequencing. They generated a total of 1.35-Tb raw data, with an average sequencing depth of 11.77× per fish. After filtering low-quality reads, a total of 1.27-Tb clean data were obtained. These clean reads were then mapped to the assembled male genome. The mapping rates varied from 95.23% to 99.66%, and the depth of effective mapped reads averaged at ~11.4× (ranged from 10.2 to 12.47 ×). We obtained a total of 4,099,996 SNPs, which were then filtered to produce a final list of 2,538,505 high-quality SNPs for further analysis.
3.5 Identification of a sex-determination region
To explore sex-determination loci in largemouth bass, both females and males were genotyped by whole-genome resequencing. Based on the significance threshold of p < 1×10-9, we detected 3,838 SNP loci in significant association with the phenotypic sex, which are distributed on Chr10 and Chr21. Interestingly, among them 3,825 loci were densely distributed on Chr10 within a 3.5-Mb region (from 32.7 to 36.2 Mb in the male genome; Figure 2A), exhibiting a clustered distribution of sex-associated signals on the Chr10. Meanwhile, our Fst results (Supplementary Figure S3) also indicate that Chr10 (32.6 - 36.0 Mb) contained more divergent variants between males and females than any other chromosomes.
Figure 2
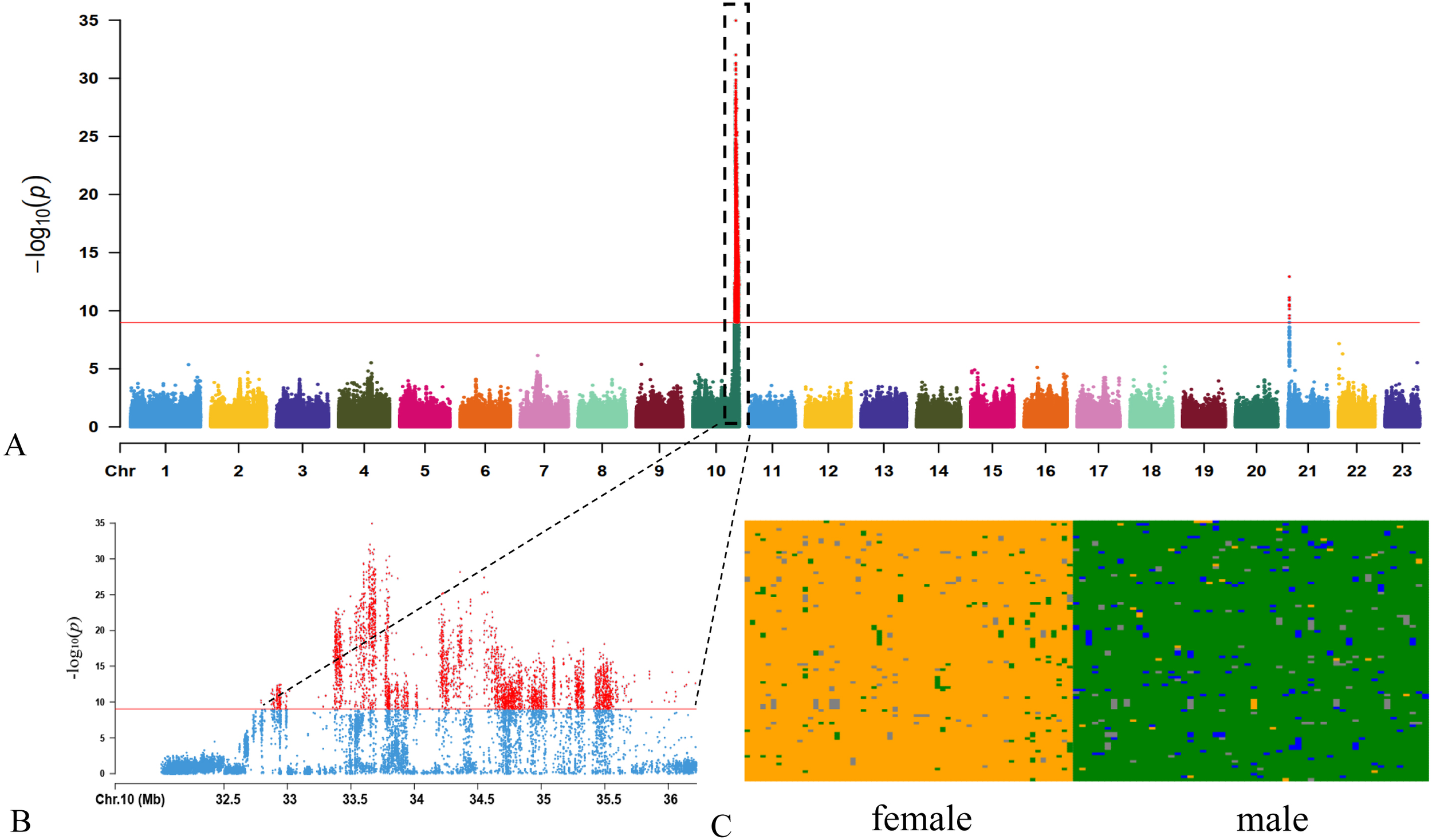
Characterization of sex chromosome and sex-related loci in female largemouth bass. (A) A Manhattan plot of the sex trait. (B) A sex significantly associated region on the Chr10 (32.7-36.2 Mb). (C) Genotypes of SNP loci significantly associated with sex (-log10(p)>25). Vertical axis of each population represents the 120 examined individuals (62 males and 58 females), and horizontal axis of each population illuminates detailed SNP allele distribution patterns among different individuals. Meanings of difference colors: orange, homozygous site of reference allele; green, heterozygous site; blue, homozygous site of alternative allele; gray, missing data.
We then screened for the most significant sites associated with sex (-log10(p)>25, 114 SNPs), and observed that these sites are almost all heterozygous in males while homozygous in females (Figure 2B), which strongly supports the male heterogametic (XY) sex-determination system in largemouth bass (Figure 2C). These findings indicate that the Chr10, possessing sex-linked genomic sequences, may play a sex-determination role in largemouth bass. Based on a collinear analysis, we confirmed that the Chr10 of assembled male genome is consistent with the Chr10 of female genome, and the SDR (from 32.8 to 35.7 Mb) localizes in the Chr10 of female genome.
3.6 Identification of a male specific region
The results of previous reports and the sex-link SNP genotypes in this study indicate that largemouth bass owns an XX/XY sex-determination system. To identify Y-specific regions in sex-linked chromosomes, reads mapping depth and coverage analysis were conducted using the whole-genome resequencing data. According to the depth differences of male and female mappings, we identified a relatively small region of approximately 51 kb localized on the Chr10 (from 33.623 to 33.674 Mb), which contains five male-specific regions (MSRs) with lengths of 12 kb (termed as MSR-1), 710 bp (MSR-2), 826 bp (MSR-3), 6.5 kb (MSR-4), and 750 bp (MSR-5), respectively. However, none of the female reads were mapped to these MSRs (see those black rectangles in Figure 3A) of the male genome. This coverage pattern confirms the XX/XY sex-determination system in largemouth bass, and strongly supports that Chr10 is one critical sex chromosome.
Figure 3
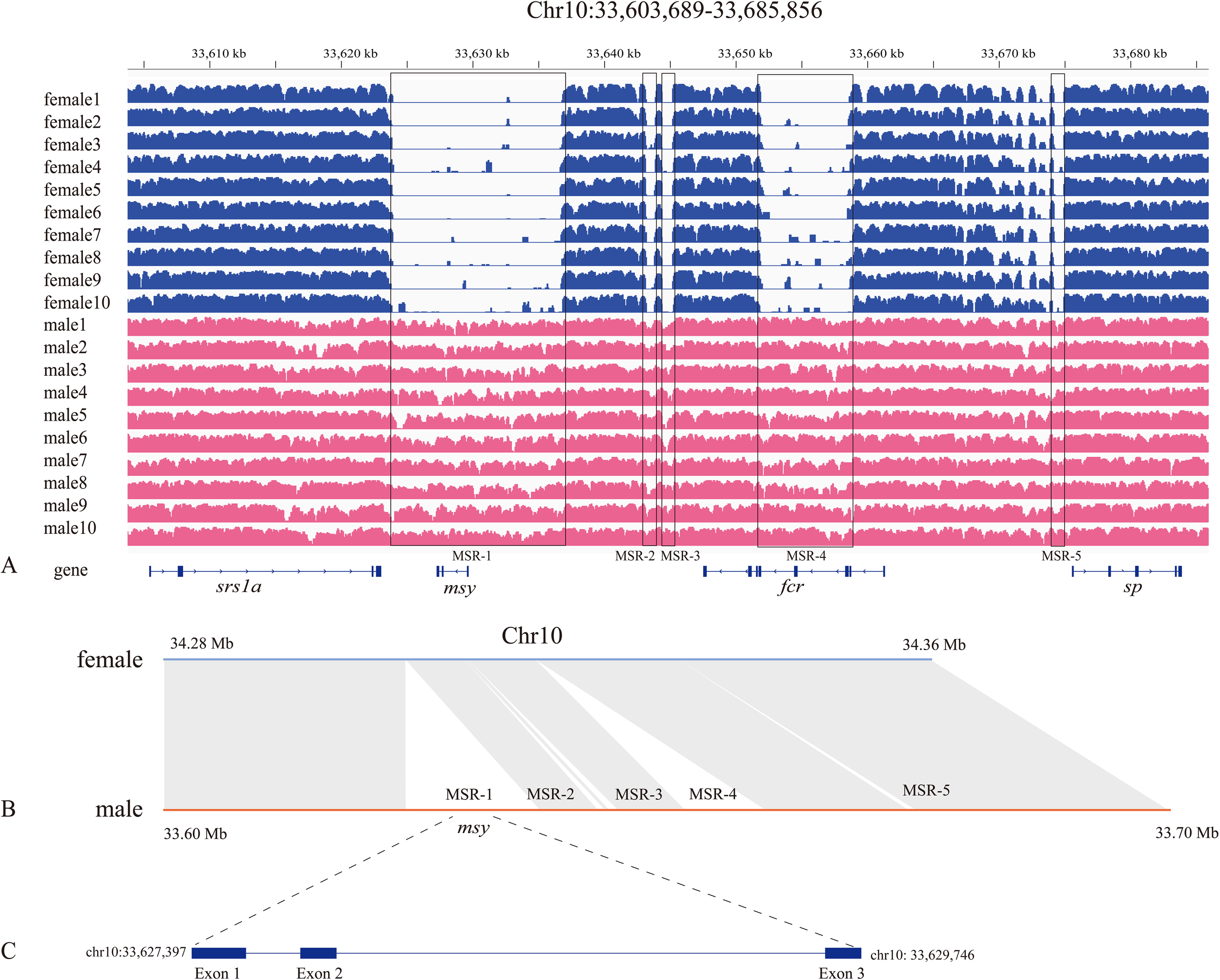
Characterization of male-specific insertions on the Chr10 of male genome. (A) Coverage depth of female and male resequencing reads in SDR (on the Chr10). Note several male specific regions (MSRs) in the male genome with a complete absence of corresponding female reads (within the black boxes). (B) Comparative alignment of the SDR between female and male genomes. (C) The predicted gene structure of msy in the MSR-1.
We then applied SYRI (Synteny and Rearrangement Identifier) to identify sequence differences between female and male genomes (He et al., 2022a), which confirmed existence of the SDR (Figure 3B). Gene structure annotation shows that the MSR-1 on Chr10 (from 33,623,700 to 33,636,800 bp) contains a coding sequence (msy) for male-specific region of the Chr10 (Y chromosome) with three exons and two introns (Figure 3C), displaying a high similarity to the C-terminal domain of putative RNA-directed DNA polymerase from transposon BS. However, in other MSRs, we could not identify any coding sequence with a high similarity to any known sex-determining gene.
3.7 Genome-wide distribution of repetitive sequences
A sex chromosome usually has a high proportion of repetitive sequences, and the emergence and evolution of sex chromosome(s) are often related to the dynamics of transposable elements (TEs) and repeats (Chalopin et al., 2015). The distribution pattern of repetitive elements among various chromosomes was plotted in the male largemouth bass genome (Figure 4A). These repeats were generally concentrated at both ends of each chromosome, except for Chr1 and Chr10. Through detailed analysis of repeat sequences, we observed that they occupied 76.9% and 60.61% of the MSRs and the SDR, respectively. These values are greater than those of the whole chromosome (32.03%) and sex chromosome (34.19% in the male genome Chr10). Those TEs within the MSRs and the SDR were mostly long interspersed elements (LINEs; 87.7% and 64.5%, respectively). This interesting accumulation of TEs in the SDR implies that the sex chromosome of largemouth bass may be still at an early stage of differentiation.
Figure 4
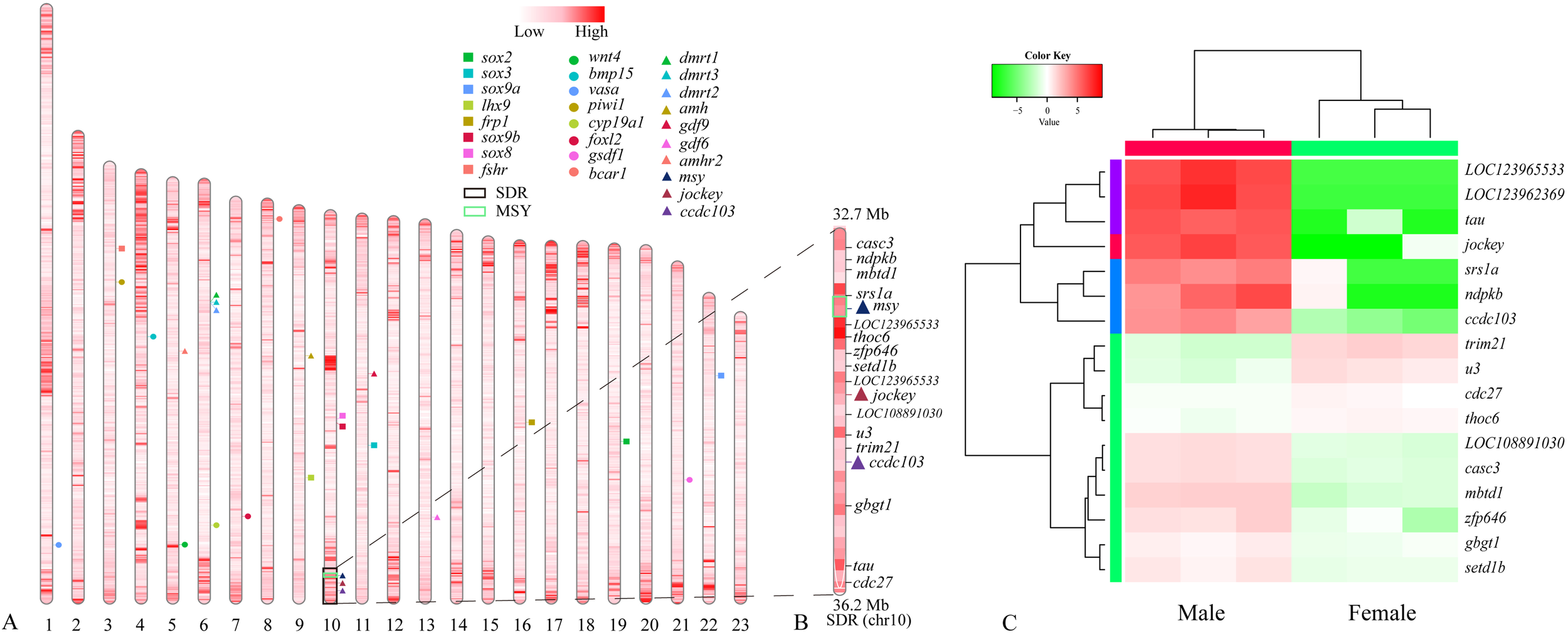
Genome-wide distribution of repetitive elements and identification of candidate sex-determining genes in largemouth bass. (A) Localization of repetitive elements and candidate sex-determining genes. The top color bar represents the density of repetitive elements (number per 100 kb) along these chromosomes, and 26 key genes for potential teleost sex-determination (from previous studies) were localized on different chromosomes. (B) Localization of differentially expressed genes (DEGs) in the SDR. (C) A clustering heat map of these DEGs.
3.8 Identification of potential sex-determining genes
We applied gonad transcriptome data to explore expression of those genes in the SDR. In adult gonads, seventeen sex-biased genes were identified (Figures 4B, C), and some of them were predominantly transcribed in the testis, while almost absent in the ovary. Coiled-coil domain-containing protein 103 (ccdc103) and RNA-directed DNA polymerase from mobile element jockey-like (jockey) are two examples (see more details in Figure 4C and Supplementary Table S3). However, the msy in the MSR-1 (Figure 3C) was not detectable in adult testis transcriptome (although few transcriptome reads were aligned to this coding sequence). Meanwhile, based on comparison of homologous sequences (known genes associated with sex in other vertebrates), we identified 23 candidate key genes for sex determination in the assembled male genome (Figure 4A).
3.9 Development of a PCR-based genotyping method for genetic sex identification
Based on the identified male-specific insertion sequences (Figure 3A), we developed two pairs of sex-specific primers that can effectively identify genetic females (XX) and males (XY) of largemouth bass. We applied both primer pairs for preliminary validation of 120 female and 150 male largemouth bass from 3 different cultured populations (TWL, YL and JDF). Some electropherogram images were provided (Figure 5).
Figure 5
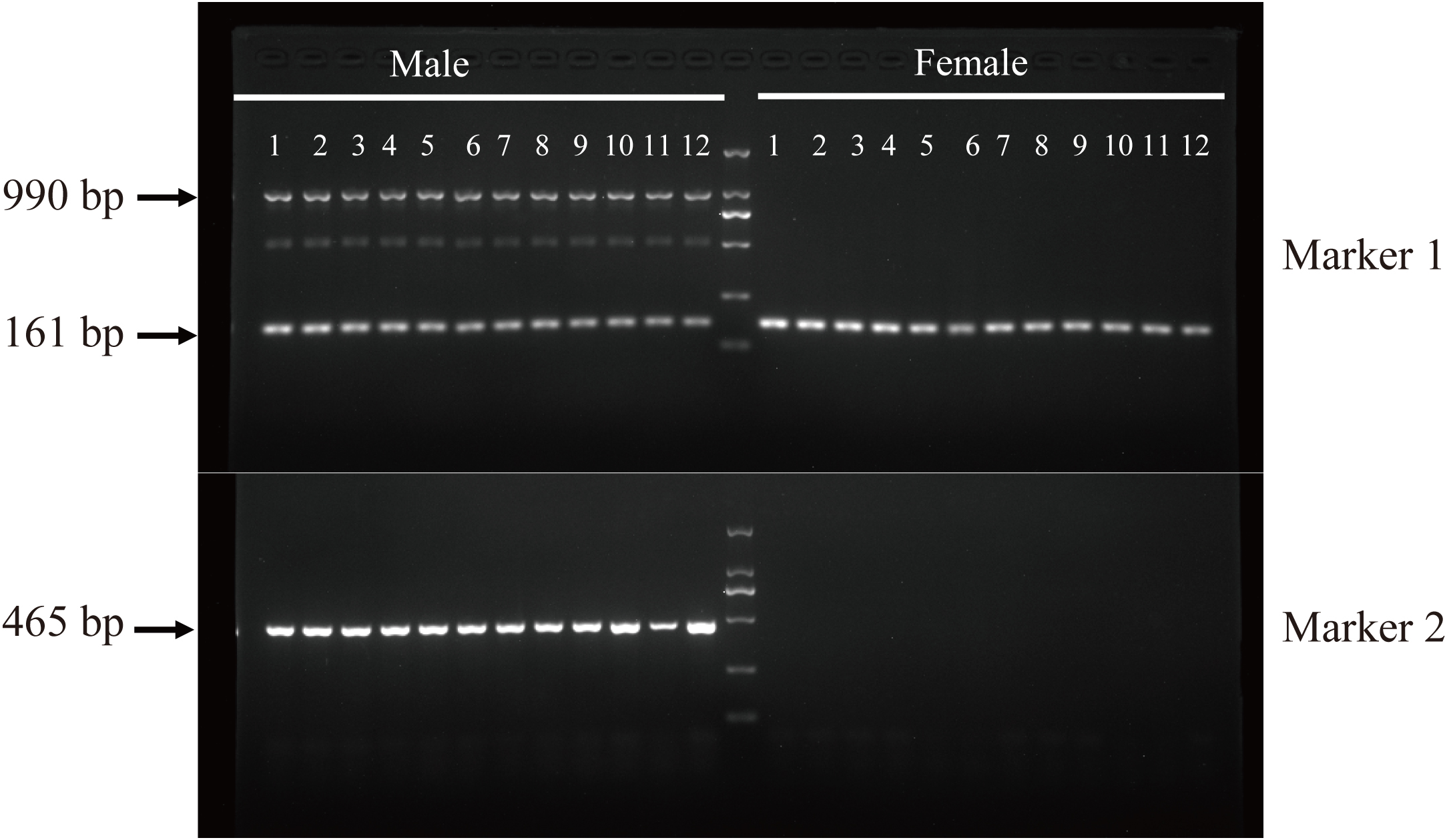
PCR detection of universal sex-specific markers in largemouth bass. In the male samples Marker 1 amplified three bands (including two target bands and the middle artificial band), whereas only one amplicon was detected in female samples. Marker 2 produced single amplicon for male samples, while no band was detectable in female samples.
One pair of primers (named as Marker 1; located in the MSR-3) amplified two target bands (161 and 990 bp, respectively) and the middle artificial band (between the two target bands; possibly generated by a repeat sequence in other locations of the male genome) in male samples, while only one single amplicon was detected in female samples (top panels in Figure 5). Another pair of primers (Marker 2; located in the MSR-2) produced a single amplicon (465 bp) in male samples, while no band was detectable in female samples (below panels in Figure 5). Encouragingly, the identification rate between males and females reached 100% when integrating both male-specific markers. Overall, these findings validate high efficiency and accuracy of our sex-specific primers, which may benefit for practical development of mono-sex populations.
4 Discussion
In our present study, a chromosome-scale genome assembly for male largemouth bass was obtained by integration of HiFi and Hi-C sequencing technologies. The quality of this assembled genome was assessed by a variety of evaluation metrics. Firstly, the size of our assembled genome (875 Mb) is similar to the previous report (GCA_019677235.1; He et al., 2022a), and it also matches the estimate of our genome survey (901 Mb; Supplementary Figure S2). However, it is much shorter than the genome assembly of a female largemouth bass (the same fish species; 963Mb from the GCA_014851395.1) that was recently reported (Sun et al., 2021). Interestingly, through a comparative analysis, we found that the genome data of this female contained a large number of redundant sequences (Supplementary Figure S4).
Sexual dimorphisms, especially reflected in growth rate and body shape, have been reported in a considerable number of aquatic animals, such as yellow catfish (Pelteobagrus fulvidraco) (Zhang et al., 2016), half-smooth tongue sole (Sun et al., 2010), giant freshwater prawns (Macrobrachium Rosenbergii) (Jiang et al., 2019) and tilapia (Oreochromis mossambicus) (Wan et al., 2019). Largemouth bass has been one of the most economically valuable fish species in China, and presents a sexual growth dimorphism (Wen et al., 2022). Therefore, understanding their sex determination mechanisms can not only improve the basic research of vertebrate sex chromosome evolution, but also promote the practical mono-sex breeding of largemouth bass, thus greatly increasing profits of the largemouth bass industry. Both GWAS and Fst have been widely used for fish sex determination studies (Xu et al., 2024; Zhang et al., 2024). In our present study, we detected a SD genetic mechanism of largemouth bass using GWAS, Fst and comparative genomic approaches, due to identification of a big region of 3.5 Mb at the end of Chr10 as the SDR of largemouth bass. Additionally, sex-linked SNPs exhibited homozygous patterns in females while being heterozygous in males, suggesting a XX/XY SD system in largemouth bass that is consistent with a previous report (He et al., 2022b).
The sex chromosomes of mammals and most birds have diverged significantly (Zhou et al., 2014). In mammals, the Y chromosome seems to be highly degraded (when compared to the X counterpart), while contains more repetitive sequences (Graves, 2006). In teleost fishes, there is little difference in the morphology of sex chromosomes in most species. Few species are indeed morphologically differentiated in sex chromosomes, although only 10% of species have been identified (176 species out of 1,700 species) with karyological studies (Devlin and Nagahama, 2002; Ezaz et al., 2006). However, this does not mean that these morphologically undifferentiated sex chromosomes could not have a differentiated SDR at the molecular level. In fact, a relatively sex-specific region was reported in multiple fish species with morphologically undifferentiated sex chromosomes. For example, in ayu a duplicate copy of the amhr2by was identified within the male-specific region (approximately 70 kb), which can be expressed specifically in the undifferentiated gonads of males and thus may participate in sex determination. In arapaima (Arapaima gigas), id2bby is a duplicated copy of id2b, and as a candidate male sex-determining gene it was identified in a small male-specific region (9,656 bp) on the Chr9 (Adolfi et al., 2021). However, sex-specific regions of some fish species do not contain any sex-determining gene. For instance, a 4–5 kb male-specific fragment (insertion) in both Channa argus and C. maculate was validated, but this region does not contain any protein-coding sequences (Sun et al., 2023b). In O. dancena, a male-specific region consists of highly repetitive non-coding sequences, while it has a cis-regulatory element that upregulates neighboring sox3 expression in the developing gonad (Takehana et al., 2014). In largemouth bass, previous studies have identified large sex-linked regions, but not large male-specific insertions (He et al., 2022b; Wen et al., 2022). In our current study, the male-specific fragment of approximately 51 kb was identified in the assembled genome. The male-specific region (MSR) contains a coding sequence (msy) that displays similarity to the C-terminal domain of RNA-directed DNA polymerase from transposon BS (bs). This bs belongs to the reverse transcriptase (RT) family, which was reported with expression in the maturing testis of Scallop Nodipecten subnodosus (Llera-Herrera et al., 2013). These RTs have transcriptase activity for RNA-directed DNA polymerization, which is required prior to reintegration into the target genome (Chang et al., 2011). Unfortunately, we couldn’t reveal transcription of this coding sequence in the transcriptome data of adult gonads, although we still speculate that this coding sequence in the male-specific fragments of largemouth bass may potentially contribute to certain biological processes such as sex determination or sex differentiation (possibly at an early stage). In the coming future, we will apply other methods to analyze and verify this coding sequence.
Based on comparative transcriptomics, two male sex-biased genes (ccdc103 and jockey) were identified in the SDR of largemouth bass. ccdc103 belongs to the coiled-coil domain-containing protein family, members of which play a variety of roles in male reproduction (Priyanka and Yenugu, 2021). The expression of ccdc38 mRNA is testicle-specific in mice, and its encoded protein is mainly confined to the nuclei of spermatogonia and spermatocytes (Lin et al., 2016). Compared to wild-type mice, ccdc136 knockout male mice were completely infertile and produced predominant round headed sperms; ccdc136 is involved in fertilization and acrosome formation (Geng et al., 2016). ccdc70, ccdc78 and ccdc189 regulate spermatogenesis, sperm maturation, and sperm motility and fertilization (Majczenko et al., 2012; Chen et al., 2016; Iso-Touru et al., 2019). jockey is a typical LINE element (non-LTR retrotransposon), and it was identified in the SDR of Nile tilapia, rainbow trout and Chinook salmon (Faber-Hammond et al., 2015).
Transposons have been shown to play a key role in sex determination by causing an insertion or replication and at the initial stage of sex chromosome differentiation and evolution (Natri et al., 2013; Chalopin et al., 2015; Ding et al., 2021). For example, three salmon species have a homologous sequence of about 4.1 kb, which contains the genetic material required for masculinization, along with transposable elements (Faber-Hammond et al., 2015). The eye stalk is an important organ of sex regulation in various arthropods, and jockey is also significantly differentially expressed in the eyestalk transcriptomes between male and female kuruma prawn (Marsupenaeus japonicus) (Toyota et al., 2023). Therefore, it is reasonable to propose that ccdc103 or jockey genes may be new candidates of master sex-determination genes in largemouth bass, but their roles and regulatory mechanisms in the SD system should be investigated by gene editing experiments.
Meanwhile, in this study we have developed two universal sex-specific PCR primer pairs that can effectively distinguish the sexuality of largemouth bass, which will promote the practical development of sex-controlled breeding strategy in largemouth bass.
5 Conclusions
In summary, the chromosome-level genome assembly of male largemouth bass was obtained by integration of PacBio HiFi and Hi-C sequencing techniques. Based on a GWAS analysis, the Chr10 of largemouth bass was validated as a sex chromosome. Three potential sex-determining genes (msy, ccdc103 and jockey) were identified in this sex determination region. A male-specific genomic insertion within the sex-linked region was detected through a genomic comparison of whole-genome resequencing data, which enabled us to generate two universal sex-specific PCR primer pairs to distinguish the fish sexuality effectively. Anyway, this study provides new insights into the genetic architecture of sex determination in largemouth bass, and the identification of male-specific markers provides a powerful tool for our on-going breeding of all-male varieties. Taken together, this male genome assembly for largemouth bass will become a valuable resource for a variety of in-depth theoretical research and practical applications. The identified MSR in largemouth bass is of great significance not only for its ecological protection and aquaculture industry, but also for elucidation of detailed molecular mechanisms of sex determination in various teleost species.
Statements
Data availability statement
The raw reads of whole-genome sequencing, resequencing, and transcriptome sequencing were deposited at the USA National Center for Biotechnology Information (NCBI) BioProject database under accession number PRJNA1012887. The assembled male genome has been deposited at GeneBank under the accession GCA_036785525.1.
Ethics statement
The animal studies were approved by Animal Care and Use Committee (IACUC) of the Pearl River Fisheries Research Institute, Chinese Academy of Fishery Sciences (CAFS), China. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
XZ: Data curation, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. JD: Formal analysis, Investigation, Methodology, Writing – review & editing. ZR: Data curation, Methodology, Visualization, Writing – review & editing. FG:. WZ: Investigation, Resources, Writing – review & editing. XXY: Data curation, Formal analysis, Investigation, Writing – review & editing. JC: Data curation, Resources, Visualization, Writing – review & editing. QS: Funding acquisition, Project administration, Supervision, Writing – review & editing. XY: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing. CS: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The research was supported by the China Agriculture Research System of MOF and MARA (no. CARS-46), Research and development projects in key areas of Guangdong Province (No. 2021B0202020001), Central Public-interest Scientific Institution Basal Research Fund (no. CAFS (2023TD95), a special fund for Science Technology Innovation and Industrial Development at Shenzhen Dapeng New District (no. KJYF202101-02) and Shenzhen Natural Science Foundation (no. JCYJ20241202124511016).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2025.1586534/full#supplementary-material
References
1
Abrusan G. Grundmann N. DeMester L. Makalowski W. (2009). TEclass–a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics25, 1329–1330. doi: 10.1093/bioinformatics/btp084
2
Adolfi M. C. Du K. Kneitz S. Cabau C. Zahm M. Klopp C. et al . (2021). A duplicated copy of id2b is an unusual sex-determining candidate gene on the Y chromosome of arapaima (Arapaima gigas). Sci. Rep.11, 21544. doi: 10.1038/s41598-021-01066-z
3
Balakrishnan R. Christie K. R. Costanzo M. C. Dolinski K. Dwight S. S. Engel S. R. et al . (2005). Fungal BLAST and Model Organism BLASTP Best Hits: new comparison resources at the Saccharomyces Genome Database (SGD). Nucleic Acids Res.33, D374–D377. doi: 10.1093/nar/gki023
4
Bao L. Tian C. Liu S. Zhang Y. Elaswad A. Yuan Z. et al . (2019). The Y chromosome sequence of the channel catfish suggests novel sex determination mechanisms in teleost fish. BMC Biol.17, 1–16. doi: 10.1186/s12915-019-0627-7
5
Benson G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res.27, 573–580. doi: 10.1093/nar/27.2.573
6
Bolger A. M. Lohse M. Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics30, 2114–2120. doi: 10.1093/bioinformatics/btu170
7
Browning B. L. Zhou Y. Browning S. R. (2018). A one-penny imputed genome from next-generation reference panels. Am. J. Hu. Genet.103, 338–348. doi: 10.1016/j.ajhg.2018.07.015
8
Chalopin D. Volff J. N. Galiana D. Anderson J. L. Schartl M. (2015). Transposable elements and early evolution of sex chromosomes in fish. Chromosome Res.23, 545–560. doi: 10.1007/s10577-015-9490-8
9
Chang P. L. Dunham J. P. Nuzhdin S. V. Arbeitman M. N. (2011). Somatic sex-specific transcriptome differences in Drosophila revealed by whole transcriptome sequencing. BMC Genom.12, 364. doi: 10.1186/1471-2164-12-364
10
Chen Y. Chen Y. Shi C. Huang Z. Zhang Y. Li S. et al . (2018). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience7, 1–6. doi: 10.1093/gigascience/gix120
11
Chen J. Hu W. Zhu Z. (2012). Progress in studies of fish reproductive development regulation. Chin. Sci. Bull.58, 7–16. doi: 10.1007/s11434-012-5577-1
12
Chen S. Zhang G. Shao C. Huang Q. Liu G. Zhang P. et al . (2014). Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nat. Genet.46, 253–260. doi: 10.1038/ng.2890
13
Chen J. B. Zheng W. Z. Li Y. C. Lin S. R. Zhang Z. Wu Y. et al . (2016). Expression characteristics of the ccdc70 gene in the mouse testis during spermatogenesis. Zhonghua nan ke xue= Natl. J. andrology22, 12–16.
14
Chen J. Zhu Z. Hu W. (2022). Progress in research on fish sex determining genes. Water Biol. Secur.1, 100008. doi: 10.1016/j.watbs.2022.100008
15
Cheng H. Concepcion G. Feng X. Zhang H. Li H. (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods18, 170–175. doi: 10.1038/s41592-020-01056-5
16
Choy S. H. Mahdy M. A. Al-Mekhlafi H. M. Low V. L. Surin J. (2015). Population expansion and gene flow in Giardia duodenalis as revealed by triosephosphate isomerase gene. Parasitol. Vector.8, 454. doi: 10.1186/s13071-015-1084-y
17
Dan C. Lin Q. Gong G. Yang T. Xiong S. Xiong Y. et al . (2018). A novel PDZ domain-containing gene is essential for male sex differentiation and maintenance in yellow catfish (Pelteobagrus fulvidraco). Sci. Bull.63, 1420–1430. doi: 10.1016/j.scib.2018.08.012
18
Danecek P. Auton A. Abecasis G. Albers C. A. Banks E. DePristo M. A. et al . (2011). The variant call format and VCFtools. Bioinformatics27, 2156–2158. doi: 10.1093/bioinformatics/btr330
19
De la Herrán R. Hermida M. Rubiolo J. A. Gómez-Garrido J. Cruz F. Robles F. et al . (2023). A chromosome-level genome assembly enables the identification of the follicule stimulating hormone receptor as the master sex-determining gene in the flatfish Solea Senegalensis. Mol. Ecol. Resour.23, 886–904. doi: 10.1111/1755-0998.13750
20
Devlin R. H. Nagahama Y. (2002). Sex determination and sex differentiation in fish: an overview of genetic, physiological, and environmental influences. Aquaculture208, 191–364. doi: 10.1016/S0044-8486(02)00057-1
21
Ding M. Li X. Y. Zhu. Z. X. Chen J. H. Lu M. Shi Q. et al . (2021). Genomic anatomy of male-specific microchromosomes in a gynogenetic fish. PLoS Genet.17, e1009760. doi: 10.1371/journal.pgen.1009760
22
Du J. Zhou J. Li S. Shao J. Jiang P. Dong C. et al . (2021). A PCR-based method for genetic sex identification and evidence of the XX/XY sex determination system in largemouth bass (Micropterus salmoides L.). Aquaculture545, 737220. doi: 10.1016/j.aquaculture.2021.737220
23
Dudchenko O. Batra S. S. Omer A. D. Nyquist S. K. Hoeger M. Durand N. C. et al . (2017). De novo assembly of the Aedes aEgypti genome using Hi-C yields chromosome-length scaffolds. Science356, 92–95. doi: 10.1126/science.aal3327
24
Durand N. C. Robinson J. T. Shamim M. S. Machol I. Mesirov J. P. Lander E. S. et al . (2016). Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst.3, 99–101. doi: 10.1016/j.cels.2015.07.012
25
Edgar R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res.32, 1792–1797. doi: 10.1093/nar/gkh340
26
Elsik C. G. Mackey A. J. Reese J. T. Milshina N. V. Roos D. S. Weinstock G. M. (2007). Creating a honey bee consensus gene set. Genome Biol.8, R13. doi: 10.1186/gb-2007-8-1-r13
27
Ezaz T. Stiglec R. Veyrunes F. Graves J. A. M. (2006). Relationships between vertebrate ZW and XY sex chromosome systems. Curr. Biol.16, R736–R743. doi: 10.1016/j.cub.2006.08.021
28
Faber-Hammond J. J. Phillips R. B. Brown K. H. (2015). Comparative analysis of the shared sex-determination region (SDR) among salmonid fishes. Genome Biol. Evol.7, 1972–1987. doi: 10.1093/gbe/evv123
29
Feron R. Zahm M. Cabau C. Klopp C. Roques C. Bouchez O. et al . (2020). Characterization of a Y-specific duplication/insertion of the anti-Mullerian hormone type II receptor gene based on a chromosome-scale genome assembly of yellow perch, Perca flavescens. Mol. Ecol. Resour.20, 531–543. doi: 10.1111/1755-0998.13133
30
Franke K. R. Crowgey E. L. (2020). Accelerating next generation sequencing data analysis: an evaluation of optimized best practices for Genome Analysis Toolkit algorithms. Genomics Inf.18, e10. doi: 10.5808/GI.2020.18.1.e10
31
Geng Q. Ni L. Ouyang B. Hu Y. Zhao Y. Guo J. (2016). A novel testis-specific gene, ccdc136, is required for acrosome formation and fertilization in mice. Reprod. Sci.23, 1387–1396. doi: 10.1177/1933719116641762
32
Goel M. Sun H. Jiao W. B. Schneeberger K. (2019). SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol.20, 1–13. doi: 10.1186/s13059-019-1911-0
33
Graves J. A. (2006). Sex chromosome specialization and degeneration in mammals. Cell124, 901–914. doi: 10.1016/j.cell.2006.02.024
34
Guindon S. Delsuc F. Dufayard J. F. Gascuel O. (2009). Estimating maximum likelihood phylogenies with PhyML. Bioinf. DNA Sequence Anal.537, 113–137. doi: 10.1007/978-1-59745-251-9_6
35
Han C. Zhu Q. Lu H. Wang C. Zhou X. Peng C. et al . (2020). Screening and characterization of sex-specific markers developed by a simple NGS method in mandarin fish (Siniperca chuatsi). Aquaculture527, 735495. doi: 10.1016/j.aquaculture.2020.735495
36
He Q. Ye K. Han W. Yekefenhazi D. Sun S. Xu X. et al . (2022b). Mapping sex-determination region and screening DNA markers for genetic sex identification in largemouth bass (Micropterus salmoides). Aquaculture559, 738450. doi: 10.1016/j.aquaculture.2022.738450
37
He K. Zhao L. Yuan Z. Canario A. Liu Q. Chen S. et al . (2022a). Chromosome-level genome assembly of largemouth bass (Micropterus salmoides) using PacBio and Hi-C technologies. Sci. Data9, 482. doi: 10.1038/s41597-022-01601-1
38
Iso-Touru T. Wurmser C. Venhoranta H. Hiltpold M. Savolainen T. Sironen A. et al . (2019). A splice donor variant in ccdc189 is associated with asthenospermia in Nordic Red dairy cattle. BMC Genomics20, 1–13. doi: 10.1186/s12864-019-5628-y
39
Ito K. Murphy D. (2013). Application of ggplot2 to pharmacometric graphics. CPT: Pharmacomet. Syst.2, 1–16. doi: 10.1038/psp.2013.56
40
Jiang J. Yuan X. Qiu Q. Huang G. Jiang Q. Fu P. et al . (2019). Comparative transcriptome analysis of gonads for the identification of sex-related genes in giant freshwater prawns (Macrobrachium rosenbergii) using RNA sequencing. Genes10, 1035. doi: 10.3390/genes10121035
41
Kamiya T. Kai W. Tasumi S. Oka A. Matsunaga T. Mizuno N. et al . (2012). A trans-species missense SNP in amhr2 is associated with sex determination in the tiger pufferfish, Takifugu rubripes (fugu). PLoS Genet.8, e1002798. doi: 10.1371/journal.pgen.1002798
42
Keilwagen J. Hartung F. Grau J. (2019). GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data. Gene prediction: Methods Protoc.1962, 161–177. doi: 10.1007/978-1-4939-9173-0_9
43
Kim D. Paggi J. M. Park C. Bennett C. Salzberg S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol.37, 907–915. doi: 10.1038/s41587-019-0201-4
44
Kumar S. Stecher G. Suleski M. Hedges S. B. (2017). TimeTree: a resource for Timelines, Timetrees, and divergence times. Mol. Biol. Evol.34, 1812–1819. doi: 10.1093/molbev/msx116
45
Legarra A. Ricard A. Varona L. (2018). GWAS by GBLUP: single and multimarker EMMAX and Bayes factors, with an example in detection of a major gene for horse gait. G3-Genes Genom. Genet.8, 2301–2308. doi: 10.1534/g3.118.200336
46
Li B. Dewey C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf.12, 1–16. doi: 10.1186/1471-2105-12-323
47
Li H. Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics25, 1754–1760. doi: 10.1093/bioinformatics/btp324
48
Li H. Handsaker B. Wysoker A. Fennell T. Ruan J. Homer N. et al . (2009). The sequence alignment/map format and SAMtools. Bioinformatics25, 2078–2079. doi: 10.1093/bioinformatics/btp352
49
Li L. Stoeckert C. J. Roos D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res.13, 2178–2189. doi: 10.1101/gr.1224503
50
Li M. Sun Y. Zhao J. Shi H. Zeng S. Ye K. et al . (2015). A tandem duplicate of anti-Mullerian hormone with a missense SNP on the Y chromosome is essential for male sex determination in Nile tilapia, Oreochromis niloticus. PLoS Genet.11, e1005678. doi: 10.1371/journal.pgen.1005678
51
Liao X. Xu G. Chen S. L. (2014). Molecular method for sex identification of half-smooth tongue sole (Cynoglossus semilaevis) using a novel sex-linked microsatellite marker. Int. J. Mol. Sci.15, 12952–12958. doi: 10.3390/ijms150712952
52
Lin S. R. Li Y. C. Luo M. L. Guo H. Wang T. T. Chen J. B. et al . (2016). Identification and characteristics of the testes-specific gene, ccdc38, in mice. Mol. Med. Rep.14, 1290–1296. doi: 10.3892/mmr.2016.5360
53
Llera-Herrera R. Garcia-Gasca A. Abreu-Goodger C. Huvet A. Ibarra A. M. (2013). Identification of male gametogenesis expressed genes from the scallop Nodipecten subnodosus by suppressive subtraction hybridization and pyrosequencing. PLoS One8, e73176. doi: 10.1371/journal.pone.0073176
54
Majczenko K. Davidson A. E. Camelo-Piragua S. Agrawal P. B. Manfready R. A. Li X. et al . (2012). Dominant mutation of ccdc78 in a unique congenital myopathy with prominent internal nuclei and atypical cores. Am. J. Hum. Genet.91, 365–371. doi: 10.1016/j.ajhg.2012.06.012
55
Matsuda M. Nagahama Y. Shinomiya A. Sato T. Matsuda C. Kobayashi T. et al . (2002). DMY is a Y-specific DM-domain gene required for male development in the medaka fish. Nature417, 559–563. doi: 10.1038/nature751
56
Mei J. Gui J. F. (2015). Genetic basis and biotechnological manipulation of sexual dimorphism and sex determination in fish. Sci. China Life Sci.58, 124–136. doi: 10.1007/s11427-014-4797-9
57
Myosho T. Otake H. Masuyama H. Matsuda M. Kuroki Y. Fujiyama A. et al . (2012). Tracing the emergence of a novel sex-determining gene in medaka, Oryzias luzonensis. Genetics191, 163–170. doi: 10.1534/genetics.111.137497
58
Nakamoto M. Uchino T. Koshimizu E. Kuchiishi Y. Sekiguchi R. Wang L. et al . (2021). A Y-linked anti-Mullerian hormone type-II receptor is the sex-determining gene in ayu, Plecoglossus altivelis. PLoS Genet.17, e1009705. doi: 10.1371/journal.pgen.1009705
59
Nanda I. Kondo M. Hornung U. Asakawa S. Winkler C. Shimizu A. et al . (2002). A duplicated copy of dmrt1 in the sex-determining region of the Y chromosome of the medaka, Oryzias latipes. P. Natl. Acad. Sci. U.S.A.99, 11778–11783. doi: 10.1073/pnas.182314699
60
Natri H. M. Shikano T. Merila J. (2013). Progressive recombination suppression and differentiation in recently evolved neo-sex chromosomes. Mol. Biol. Evol.30, 1131–1144. doi: 10.1093/molbev/mst035
61
Pan Q. Feron R. Yano A. Darras H. Herpin A. Koop B. et al . (2019). Identification of the master sex determining gene in Northern pike (Esox lucius) reveals restricted sex chromosome differentiation. PLoS Genet.15, e1008013. doi: 10.1371/journal.pgen.1008013
62
Peichel C. L. McCann S. R. Ross J. A. Naftaly A. F. Urton J. R. Cech J. N. et al . (2020). Assembly of the threespine stickleback Y chromosome reveals convergent signatures of sex chromosome evolution. Genome Biol.21, 1–31. doi: 10.1186/s13059-020-02097-x
63
Priyanka P. P. Yenugu S. (2021). Coiled-Coil Domain-Containing (ccdc) proteins: functional roles in general and male reproductive physiology. Reprod. Sci.28, 2725–2734. doi: 10.1007/s43032-021-00595-2
64
Ramirez F. Dundar F. Diehl S. Grüning B. A. Manke T. (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res.42, W187–W191. doi: 10.1093/nar/gku365
65
Robinson J. T. Thorvaldsdottir H. Winckler W. Guttman M. Lander E. S. Getz G. et al . (2011). Integrative genomics viewer. Nat. Biotechnol.29, 24–26. doi: 10.1038/nbt.1754
66
Servant N. Varoquaux N. Lajoie B. R. Viara E. Chen C. J. Vert J. P. et al . (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol.16, 1–11. doi: 10.1186/s13059-015-0831-x
67
Shao C. Li Q. Chen S. Zhang P. Lian J. Hu Q. et al . (2014). Epigenetic modification and inheritance in sexual reversal of fish. Genome Res.24, 604–615. doi: 10.1101/gr.162172.113
68
Simão F. A. Waterhouse R. M. Ioannidis P. Kriventseva E. V. Zdobnov E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics31, 3210–3212. doi: 10.1093/bioinformatics/btv351
69
Smith C. Wootton R. J. (2016). The remarkable reproductive diversity of teleost fishes. Fish Fish.17, 1208–1215. doi: 10.1111/faf.2016.17.issue-4
70
Song W. Xie Y. Sun M. Li X. Fitzpatrick C. K. Vaux F. et al . (2021). A duplicated amh is the master sex-determining gene for Sebastes rockfish in the Northwest Pacific. Open Biol.11, 210063. doi: 10.1098/rsob.210063
71
Stanke M. Schoffmann O. Morgenstern B. Waack S. (2006). Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinf.7, 1–11. doi: 10.1186/1471-2105-7-62
72
Sun C. Li J. Dong J. Niu Y. Hu J. Lian J. et al . (2021). Chromosome-level genome assembly for the largemouth bass Micropterus salmoides provides insights into adaptation to fresh and brackish water. Mol. Ecol. Resour.21, 301–315. doi: 10.1111/1755-0998.13256
73
Sun D. Wen H. Qi X. Li C. Sun C. Wang L. et al . (2023b). Comparative study of candidate sex determination regions in snakeheads (Channa argus and C. maculata) and development of novel sex markers. Aquaculture575, 739771. doi: 10.1016/j.aquaculture.2023.739771
74
Sun C. F. Zhang X. H. Dong J. J. You X. X. Tian Y. Y. Gao F. Y. et al . (2023a). Whole-genome resequencing reveals recent signatures of selection in five populations of largemouth bass (Micropterus salmoides). Zool. Res.44, 78. doi: 10.24272/j.issn.2095-8137.2022.274
75
Sun Y. Zhang Q. Qi J. Chen Y. Zhong Q. Li C. et al . (2010). Identification of differential genes in the ovary relative to the testis and their expression patterns in half-smooth tongue sole (Cynoglossus semilaevis). J. Genet. Genomics37, 137–145. doi: 10.1016/S1673-8527(09)60032-1
76
Takehana Y. Matsuda M. Myosho T. Suster M. L. Kawakami K. Shin-i T. et al . (2014). Co-option of sox3 as the male-determining factor on the Y chromosome in the fish Oryzias dancena. Nat. Commun.5, 4157. doi: 10.1038/ncomms5157
77
Tang H. Bowers J. E. Wang X. Ming R. Alam M. Paterson A. H. (2008). Synteny and collinearity in plant genomes. Science320, 486–488. doi: 10.1126/science.1153917
78
Tao W. Cao J. Xiao H. Zhu X. Dong J. Kocher T. D. et al . (2021). A chromosome-level genome assembly of Mozambique tilapia (Oreochromis mossambicus) reveals the structure of sex determining regions. Front. Genet.12, 796211. doi: 10.3389/fgene.2021.796211
79
Tarailo-Graovac M. Chen N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinf.5, 1–14. doi: 10.1002/0471250953.2009.25.issue-1
80
Toyota K. Mekuchi M. Akashi H. Miyagawa S. Ohira T. (2023). Sexual dimorphic eyestalk transcriptome of kuruma prawn Marsupenaeus japonicus. Gene885, 147700. doi: 10.1016/j.gene.2023.147700
81
Trapnell C. Roberts A. Goff L. Pertea G. Kim D. Kelley D. R. et al . (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc.7, 562–578. doi: 10.1038/nprot.2012.016
82
Wan Z. Y. Lin G. Yue G. (2019). Genes for sexual body size dimorphism in hybrid tilapia (Oreochromis sp. x Oreochromis mossambicus). Aquaculture Fisheries4, 231–238. doi: 10.1016/j.aaf.2019.05.003
83
Wang L. Sun F. Wan Z. Y. Yang Z. Tay Y. X. Lee M. et al . (2022). Transposon-induced epigenetic silencing in the X chromosome as a novel form of dmrt1 expression regulation during sex determination in the fighting fish. BMC Biol.20, 1–16. doi: 10.1186/s12915-021-01205-y
84
Wei Y. F. (2022). Sexual morphology, growth differences and development of gender molecular markers in Micropterus salmoides. Wuhan, China: Huazhong Agricultural University.
85
Wen M. Zhang Y. Wang S. Hu F. Tang C. Li Q. et al . (2022). Sex locus and sex markers identification using whole genome pool-sequencing approach in the largemouth bass (Micropterus Salmoides L.). Aquaculture559, 738375. doi: 10.1016/j.aquaculture.2022.738375
86
Xu Z. Wang H. (2007). LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res.35, W265–W268. doi: 10.1093/nar/gkm286
87
Xu X. Yu J. Ge J. Yi S. Weng X. Guan W. et al . (2024). A male-specific insert of Opsariichthys bidens identified based on genome-wide association analyses and comparative genomics. Aquacult. Rep.35, p.101982. doi: 10.1016/j.aqrep.2024.101982
88
Yang Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol.24, 1586–1591. doi: 10.1093/molbev/msm088
89
Yano A. Guyomard R. Nicol B. Jouanno E. Quillet E. Klopp C. et al . (2012). An immune-related gene evolved into the master sex-determining gene in rainbow trout, Oncorhynchus mykiss. Curr. Biol.22, 1423–1428. doi: 10.1016/j.cub.2012.05.045
90
Zhang K. Huang X. Wang C. Xu X. Xu X. Dong X. et al . (2024). Unveiling potential sex-determining genes and sex-specific markers in autotetraploid Carassius auratus. Sci. China Life Sci.67(11), 2444–2502. doi: 10.1007/s11427-023-2480-4
91
Zhang J. Ma W. He Y. Wu J. Dawar F. U. Ren F. et al . (2016). Sex biased expression of ghrelin and GHSR associated with sexual size dimorphism in yellow catfish. Gene578, 169–176. doi: 10.1016/j.gene.2015.12.017
92
Zheng S. Tao W. Yang H. Kocher T. D. Wang Z. Peng Z. et al . (2022). Identification of sex chromosome and sex-determining gene of southern catfish (Silurus meridionalis) based on XX, XY and YY genome sequencing. P. R. Soc B-Biol. Sci.289, 20212645. doi: 10.1098/rspb.2021.2645
93
Zhou Q. Zhang J. Bachtrog D. An N. Huang Q. Jarvis E. D. et al . (2014). Complex evolutionary trajectories of sex chromosomes across bird taxa. Science346, 1246338. doi: 10.1126/science.1246338
Summary
Keywords
largemouth bass (Micropterus salmoides), chromosome-level genome assembly, GWAS, male-specific region, sex markers
Citation
Zhang X, Dong J, Ruan Z, Gao F, Zhou W, You X, Chen J, Shi Q, Ye X and Sun C (2025) Identification and characterization of a male-specific region in largemouth bass (Micropterus Salmoides) by whole-genome sequencing, resequencing and genomics comparison. Front. Mar. Sci. 12:1586534. doi: 10.3389/fmars.2025.1586534
Received
03 March 2025
Accepted
08 May 2025
Published
30 May 2025
Volume
12 - 2025
Edited by
Seyyed Morteza Hoseini, Iranian Fisheries Science Research Institute (IFSRI), Iran
Reviewed by
Liang Guo, Hunan Normal University, China
Morteza Yousefi, Peoples’ Friendship University of Russia, Russia
Updates
Copyright
© 2025 Zhang, Dong, Ruan, Gao, Zhou, You, Chen, Shi, Ye and Sun.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chengfei Sun, scf@prfri.ac.cn; Xing Ye, gzyexing@163.com; Qiong Shi, shiqiong@szu.edu.cn
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.