 Vanessa Arranz1,2*
Vanessa Arranz1,2* Lea Schmütsch-Molina1,2
Lea Schmütsch-Molina1,2 Robert Fernandez-Vilert1,2
Robert Fernandez-Vilert1,2 Jose Carlos Hernández3
Jose Carlos Hernández3 Rocío Pérez-Portela1,2
Rocío Pérez-Portela1,2- 1Departament de Biologia Evolutiva, Ecologia i Ciències Ambientals, Universitat de Barcelona, Barcelona, Spain
- 2Institut de Recerca de la Biodiversitat (IRBio), Universitat de Barcelona, Barcelona, Spain
- 3Departamento de Biología Animal, Edafología y Geología, Universidad de la Laguna, Tenerife, Canary Islands, Spain
Understanding holobiont dynamics is essential for unraveling the complex interactions between marine hosts and their microbiota. Sea urchins play pivotal roles in shaping benthic ecosystems, yet the functional roles of their microbial symbionts remain poorly characterized. Here, we present a comparative microbiome analysis of two sympatric echinoid species, Arbacia lixula and Paracentrotus lividus which occupy contrasting trophic niches. P. lividus is primarily herbivorous, while A. lixula exhibits omnivorous and carnivorous feeding behavior. We characterized microbial communities from coelomic fluid, coelomocytes, and egested fecal pellets, collected from two biogeographic regions, the Northeastern Atlantic Ocean and the Mediterranean Sea. Applying Next-Generation sequencing of the 16S rRNA gene (V3-V4 region) and using the FAPROTAX functional annotation database to infer microbial ecological functions, we found distinct microbial signatures shaped by host species, body compartment, and location. Notably, species-specific differences may reflect dietary preferences, with P. lividus enriched in sulfur-metabolizing and phototrophic bacteria, while A. lixula displayed functional signatures potentially linked to nitrogen cycling and microbial pathogenesis. Fecal microbiota exhibited the highest diversity and functional enrichment in carbohydrate degradation and nutrient cycling. Coelomic compartment hosted microbial assemblages with potential immune host-interaction traits, including intracellular symbiosis or parasitism. Geographic variation further shaped microbiota composition, with stronger location-dependent functional shifts observed in P. lividus. These findings reveal a high degree of spatial and functional differentiation in sea urchin microbiomes, highlighting the plastic nature of sea urchin microbiomes and their potential role in host adaptation to environmental change.
1 Introduction
Recently, greater emphasis has been placed on the concept of holobiont in marine ecosystems. This concept refers to a biological unit composed of a host and its microbiota, as they coexist and interact (Simon et al., 2019; Bordenstein and Theis, 2015). Advances in high-throughput sequencing, curated reference databases and standardized protocols have led to uncovering the complexity and functional roles of host-associated microbial communities in a wide range of organisms (Stévenne et al., 2021; Pita et al., 2018; Dittami et al., 2021). In marine ecosystems, microbial symbionts have an important effect on the host’s development (Carrier and Reitzel, 2019), immunity (Dantan et al., 2024), physiology and metabolism (Venn et al., 2008; Nicholson et al., 2012), as they can carry out processes like nitrogen fixation (Guerinot and Patriquin, 1981; Petersen et al., 2016), sulfur cycling (Enomoto et al., 2012) and micronutrients supply (Li et al., 2025), among others. Microbiomes also play a crucial role in animal survival under variable and extreme environments by enhancing the host’s ecological plasticity and resilience through dynamic shifts in composition and functionality (Bang et al., 2018; Torda et al., 2017). Microbiomes exhibit dynamic shifts during early developmental stages, influenced by feeding regimes and environmental factors, suggesting a crucial role in growth and acclimatization (Carrier and Reitzel, 2020, 2019; Buschi et al., 2023). They are also highly responsive to environmental changes, underscoring their sensitivity to external stressors and their contribution to host resilience under variable conditions (Marangon et al., 2021, 2023).
Echinoderms are an important group of marine organisms that host a diverse microbiome, yet their holobiont dynamics are only beginning to be disentangled, laying the groundwork for understanding the roles of microorganisms in their health, development, and physiology (e.g., Carrier and Reitzel, 2017). Representing key components of marine ecosystems, echinoderms are found across a wide range of marine environments, from coastal to deep bottoms and from tropical to polar regions, often constituting a significant portion of biomass in these habitats (Lawrence, 2020). Microbiome studies across echinoderm classes, such as sea stars (Galac et al., 2016; Nakagawa et al., 2017), brittle stars (Dong et al., 2023), sea cucumbers (Pagán-Jiménez et al., 2019; Zhang et al., 2021), and sea urchins (Faddetta et al., 2020; Schwob et al., 2020), have revealed a wide range of symbiotic functions. These include nutrient metabolism, such as the breakdown of complex organic compounds, and even indigestible products (Zhang et al., 2014; Pagán-Jiménez et al., 2019), nitrogen fixation, sulfuric oxidation, and amino acid uptake (Miller et al., 2021). In addition to digestive processes, echinoderm-associated microbiota contributes to host functions such as antibacterial, antiviral, anticoagulant and antitumoral functions, particularly studied in holothurians due to bioactive compounds such as triterpene glycosides (McCracken et al., 2023; Chludil et al., 2003; Offret et al., 2019).
Within echinoderms, sea urchins are a pivotal group playing a key role in determining the structure and function of marine ecosystems by regulating the abundance and distribution of benthic species and energy flow in marine communities (Lawrence, 2020). Through their grazing activity and habitat modifications, sea urchins influence not only the composition of benthic communities but also the microbial assemblages associated with them. These environmental shifts, in turn, shape the bacterial communities that sea urchins host, contributing to the establishment of their native microbiota. The bacteria associated with sea urchins play fundamental roles in digestion, nutrient cycling, immunity, and overall host homeostasis (Hakim et al., 2016; Brothers et al., 2018). Distinct microbial communities are associated with different compartments in sea urchins, including the digestive tract, coelomic fluid, and external surfaces, with specialized functions tied to each compartment (Faddetta et al., 2020; Masasa et al., 2023). Studies reveal that digestive microbiomes vary by species, diet, and environmental conditions, contributing to nutrient acquisition and organic matter degradation (Becker et al., 2007; Zhang et al., 2014). For example, sulfur-metabolizing bacteria in the digestive systems of certain sea urchins support nutrient cycling (Thorsen et al., 2003). Studies on Lytechinus variegatus and Strongylocentrotus purpuratus have highlighted gut compartment-specific microbial compositions, with changes in bacterial assemblages supporting their roles in digestion and host health (Hakim et al., 2016, 2019). Beyond the digestive system, the coelomic fluid of sea urchins harbors a dynamic microbiota community that plays critical roles in immunity and host health, contradicting earlier assumptions of it being a sterile environment (Nakagawa et al., 2017; Faddetta et al., 2020). This fluid contains coelomocytes, a variety of different cell populations, some of them with phagocytic and antimicrobial functions, forming a complex innate immune system responsive to pathogens and environmental challenges (Smith et al., 2018). These compartmentalized and environmentally responsive microbial communities highlight the intricate symbiotic relationships making sea urchins a valuable system for understanding host-microbe interactions in marine ecosystems, particularly in diverse environments and across species.
The sea urchins Arbacia lixula (Linnaeus, 1758) and Paracentrotus lividus (Lamark, 1816), are sympatric species dominating shallow coastal ecosystems. A. lixula is a subtropical species distributed across the Mediterranean, Atlantic, and Brazilian coasts (Bonaviri et al., 2011; Gianguzza and Bonaviri, 2013) although the Brazilian populations have been found to be a different evolutionary unit (Wangensteen et al., 2012; Pérez-Portela et al., 2019). P. lividus, is a temperate-cold species distributed through the Mediterranean Sea and northeastern Atlantic (Boudouresque and Verlaque, 2001). These key structuring species play a crucial role in shaping benthic communities, with their intense and joint grazing activity, driving barren formation that significantly reduce algae cover and benthic biodiversity (Palacín et al., 1998; Sala et al., 1998; Boudouresque et al., 2020; Bulleri et al., 1999; Klaoudatos et al., 2022; Hereu et al., 2004). Both species are capable of exploiting diverse food sources, yet they exhibit distinct dietary preferences and different enzymatic digestive profiles (Trenzado et al., 2012). A. lixula is considered omnivorous with a tendency to carnivory in the Mediterranean (Wangensteen et al., 2011), whereas P. lividus primarily feeds on fleshy macroalgae and seagrass (Privitera et al., 2011). A. lixula shows a preference for encrusting corallines and consumes newly settled organisms, often maintaining the barren state even in the absence of P. lividus in some areas of the Mediterranean (Wangensteen et al., 2011; Hereu et al., 2004).
Research on the digestive microbiota of P. lividus in the Mediterranean revealed distinct microbial communities across compartments, such as the esophagus, stomach, and intestine, with functional roles in nutrient acquisition, organic matter degradation, and metabolic support (Meziti et al., 2007; Laport et al., 2018). These microbial communities are influenced by diet and environmental factors, suggesting a transient microbiota that adapts to local conditions (Faddetta et al., 2020; Liu et al., 2024). Additionally, certain bacterial strains associated with P. lividus coelomic fluid exhibit antimicrobial properties, contribute to environmental detoxification, and support the host’s immune responses, showcasing the functional versatility of its microbiome (Laport et al., 2018). For A. lixula, no comprehensive studies on its microbiota have been conducted, leaving a significant knowledge gap.
The aim of this study was to characterize the microbiomes associated with two sympatric sea urchins, and to evaluate how they vary across host- and environment-related factors. Specifically, we sought (i) to compare the microbiomes of the two species, which differ in their trophic ecology (ii) to investigate whether microbial communities differ among host body compartments (coelomic fluid, coelomocytes, and feces), and (iii) to assess whether microbial composition varies geographically between populations from the Northeastern Atlantic (La Palma) and the Mediterranean (Blanes). By addressing these objectives, our study provides the first integrated analysis of interspecific, inter-compartmental, and geographic variability in sea urchin-associated microbiota, contributing to a better understanding of host–microbe associations in marine invertebrates.
2 Materials and methods
2.1 Sample collection
Nine specimens of A. lixula and nine P. lividus were sampled from each of the sampling locations: La Palma (Canary Islands, Spain) and Cala de Sant Francesc, Blanes (Girona, Spain) in October 2023 and February 2024, respectively (Figure 1 and Supplementary Table S1). In La Palma, nine P. lividus individuals were collected from Fuencaliente, while nine A. lixula specimens were sampled from La Bajita in the eastern part of the island. These individuals were transported in specialized containers to the Observatorio Marino de Cambio Climático (OMACC) in Fuencaliente. Specimens from Blanes were transported and processed at the Universitat de Barcelona. Specimens were separated into individual tanks containing seawater collected from their respective sampling sites during the sample collection process. Once in the laboratory, coelomic fluid was extracted using a 5 mL sterile syringe through the peristomial membrane pre-filled with 2 mL of sterile anticoagulant buffer [80% Calcium/Magnesium Free Artificial Sea Water (CM-ASW), osmolarity 1200 mOsM+20% EDTA stock solution (13.53 g/L)]. The fluid was briefly centrifuged to separate coelomic fluid and coelomocytes, the latter were stored in absolute ethanol and at -20 °C. The remaining coelomic fluid was filtered using sterile Sartorius Minisart™ filters with a pore size of 0.22 µm to retain microbial cells. The filters were fixed with absolute ethanol and stored at -20 °C. Fecal pellets were collected after a 24-hour fasting period to ensure defecation, and samples were preserved in absolute ethanol at -20 °C and used as a non-invasive proxy to investigate the digestive tract-associated microbiota. In La Palma, coelomic fluid (CF), coelomocytes (C), and fecal pellets (F) were collected, whereas in Blanes only coelomic fluid samples were obtained. For La Palma, coelomocytes and feces were collected from multiple individuals, but only five samples per species were selected for sequencing. As controls, two samples of 5 mL of anticoagulant buffer used during coelomocyte extraction were filtered through 0.22 µm membranes and stored in absolute ethanol and at -20 °C. Following processing, the specimens were returned alive to their original sampling locations.
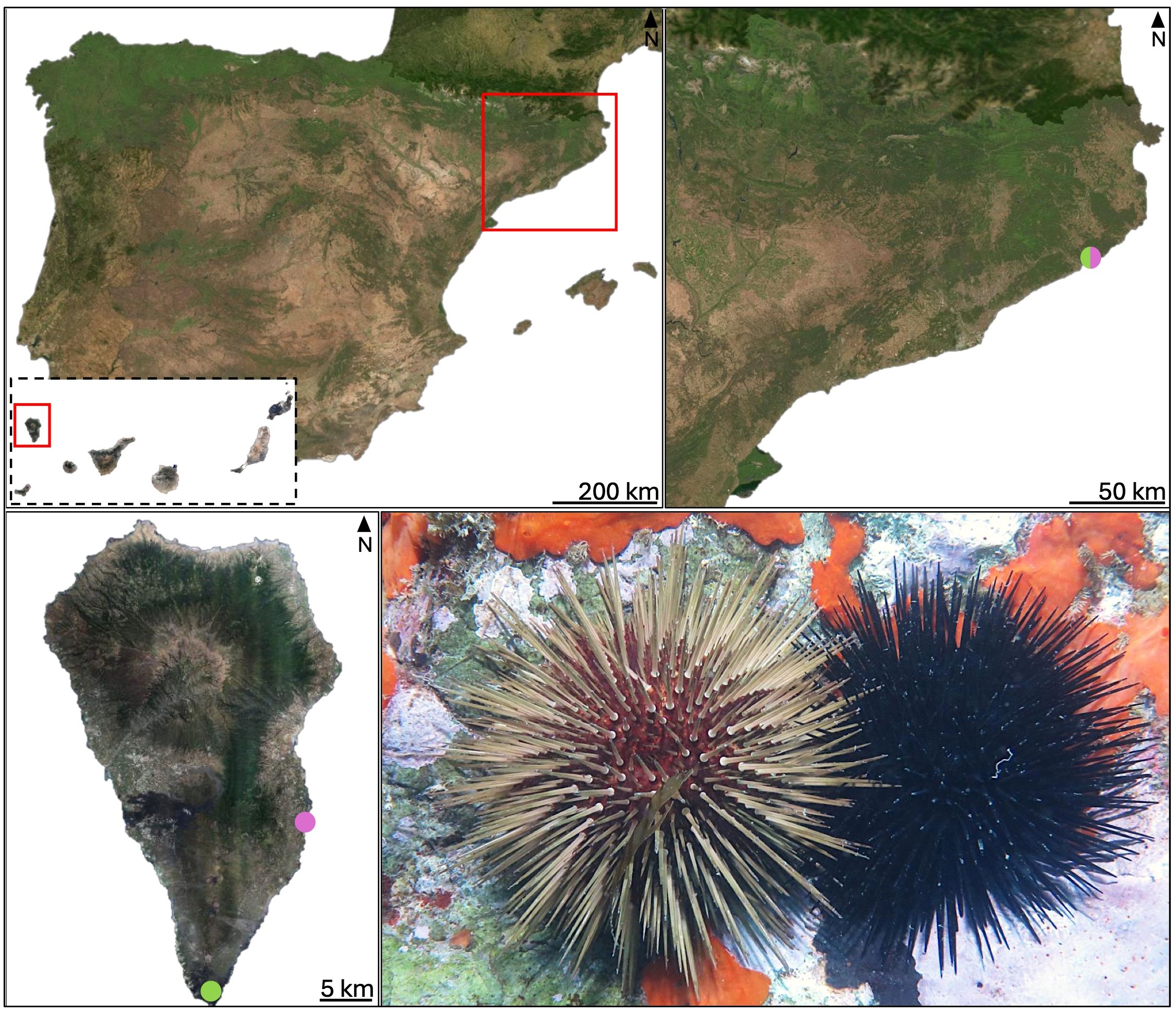
Figure 1. Map showing the sampling locations (see Supplementary Table S1). Locations where Arbacia lixula specimens were collected are marked in purple, and those where Paracentrotus lividus were collected are marked in green. Blanes is located in the Mediterranean Sea and La Palma in Northeast Atlantic ocean. The lower right image shows P. lividus (left) and A. lixula (right). Photo credit: Robert Vilert-Fernández.
2.2 DNA Extraction and sequencing
DNA was extracted from all collected samples using DNeasy™Blood & Tissue kit (QIAGEN, Maryland, USA) following a slightly modified manufacturer’s protocol. Coelomocytes and fecal pellets were briefly ethanol-dried, followed by overnight lysis in 20 µL proteinase K and 180 µL ATL buffer at 56 °C. For coelomic fluid samples, half of the filter was cut in small pieces, and an additional 40 µL ATL buffer was added to ensure complete coverage. All DNA was further purified using OneStep PCR Inhibitor removal kit (ZymoBIOMICS™, Zymo Research, Irvine, CA, USA). DNA quantification was evaluated using a Qubit dsDNA HS (High Sensitivity) Assay Kit with the Qubit® Fluorometer (Massachusetts, USA). A total of 58 samples were selected for sequencing including an artificial seawater and DNA extraction blank as negative controls. We employed Illumina MiSeq sequencing (2×300 bp paired-end reads) to analyse the V3–V4 hypervariable region of the 16S rRNA gene, which was amplified using universal primers 341F (5’-CCTACGGGNGGCWGCAG-3`) and 785R (5’-GACTACHVGGGTATCTAATCC-3`) (Klindworth et al., 2013). Library preparation and sequencing were conducted at the Centre for Genomic Research in Barcelona, Spain. The sequences are available in GenBank Bioproject PRJNA1252121 with accession numbers SRR33214878-SRR33214973.
2.3 Bioinformatic analysis
DNA sequences pre-processing and bioinformatic analyses were performed using the software environment Quantitative Insights into Microbial Ecology version 2019.4 (Bolyen et al., 2019). QIIME2 provides a software environment, data standards, and tool wrappers allowing seamless interoperability between tools used for microbial community analysis. The bioinformatic pipeline used is available at https://github.com/leaschmutsch/microbiota_characterization. Primers were removed without mismatch tolerance using the Cutadapt plugin (Martin, 2011). We used the DADA2 plugin (Callahan et al., 2016) for paired-end merging, trimming parameters were set at 260bp for forward and reverse reads, chimera removal (using consensus method) and clustering into Amplicon Sequence Variants (ASVs). To create a phylogenetic tree, we used the ‘align-to-tree-mafft-fasttree’ pipeline from the ‘q2-phylogeny plugin’. This step consists of doing a multiple-sequence alignment, then filtering the alignment to remove highly variable positions and applying FastTree to generate a phylogenetic tree from the masked alignment. Taxonomic assignment of ASVs was performed against the SILVA v138.1 reference database (Quast et al., 2012). Singletons and ASVs taxonomically assigned to eukaryotes were discarded for statistical analysis in R programming environment (R Core Team, 2024).
2.4 Statistical analysis
Potential contaminants were removed with the decontam R package v1.4 (Davis et al., 2018) using the prevalence method, and the extraction and PCR negative controls. The ASV table was used to calculate rarefaction curves using phyloseq R package v1.46 (McMurdie and Holmes, 2013). We optimized the rarefaction thresholds to balance sequencing depth and sample retention, selecting 7,000 sequences per sample to support robust statistical comparisons. Relative abundance barplots were generated at the family, order and phylum level using the plot_composition function in the phyloseq R package.
We then estimated alpha diversity (taxonomic richness) and beta diversity (community composition) based on pairwise dissimilarity. To test differences, we applied a planned comparisons framework, focusing on a set of predefined, biologically meaningful hypotheses. Seven comparisons were defined a priori, including differences: (i) between A. lixula and P. lividus within the same body compartment; (ii) among compartments within a species; and (iii) between geographic locations for the same species and compartment (see Supplementary Table S2 for details). These comparisons were encoded in a contrast matrix, assigning weights (−1, 0, + 1) to each level depending on the comparison of interest (Crawley, 2012). Each column of the contrast matrix corresponds to a particular comparison. Because our experimental design was partially unbalanced (species × body compartment × location), we set up a dummy variable to uniquely identified each factor combination, and each sample was then assigned to one of these levels. For example, a sample of coelomic fluid of A. lixula from la Palma would have been coded as “Arb.CF.Palma”. This resulted in eight levels for the 56 samples considered, excluding the negative controls. This ensured that all planned comparisons could be tested directly without unnecessary subsetting or multiple testing of the same data. As some contrasts were non-orthogonal (analogous to collinear predictors), we used the inverse of the transposed contrast matrix to calculate the fixed-effects design matrix. By implementing planned contrasts through contrast matrices, and by explicitly modelling fixed effects (species, compartments, locations) and random effects (sample identity), we efficiently used the degrees of freedom in our design and tested only the comparisons of biological interest. Importantly, because all planned contrasts were defined within the models, corrections for multiple testing were not required. This approach provides robust and interpretable estimates while accounting for the hierarchical structure of the sampling design.
Alpha diversity metrics of bacterial communities associated with the sampled sea urchins were estimated using Observed ASVs and Shannon diversity index with microbiome R package v1.24 (Lahti and Shetty, 2018) and Faith’s PD index (phylogenetic diversity) with picante R package v1.8 (Kembel et al., 2010). Statistical comparisons of alpha diversity were conducted using a permutational linear model (PLM), implemented via the lmp() function from the lmPerm R package v2.1.0 (Wheeler et al., 2025), which performs analysis of variance through permutation of residuals. In these models, species, body compartment, and location were treated as fixed effects, while sample identity was included as a random effect whenever multiple compartments were measured from the same sea urchin. This explicitly accounts for repeated measures and avoids pseudoreplication. This method was chosen as it accommodates complex factorial designs while relaxing the assumptions of parametric ANOVA, which are often violated in microbiome datasets.
For the beta diversity analysis of community composition, Unweighted UniFrac, a presence/absence-based metric sensitive to phylogenetic relationships, and Weighted UniFrac, which incorporates abundance data, were calculated using the phyloseq package. For data visualization a Principal Coordinates Analysis (PCoA) was used with the UniFrac distance matrices. Permutational multivariate analysis of variance (PERMANOVA) (Anderson, 2005), was employed to assess differences in microbial community, using both UniFrac distance matrices. Statistical significance was determined with 999 permutations implemented via adonis2 function in the vegan v2.6 package (Oksanen et al., 2007). When repeated measures from the same sea urchin were present, permutations were constrained within individuals to avoid inflating type I error rates. We assessed the assumption of homogeneity of dispersions and analyzed multivariate beta diversity patterns among samples by calculating the average distance to group centroids using the betadisper function from the vegan R package.
Unique and shared taxa between species and between sample types of each species were counted at multiple taxonomic levels and visualized using Venn diagrams with ggvenn R package. Differential abundance analysis was conducted using Analysis of Composition of Microbiomes with Bias Correction (ANCOM-BC) from the Ancombc R package (Lin and Peddada, 2020; Nearing et al., 2022) on unrarefied ASV tables and taxonomic assignments were reported at genus, family and phylum level. Log fold changes (LFCs) in taxa abundances between samples were computed, with p-values adjusted using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995) to control for false discovery rates (FDR). Results were visualized using ggplot2 R package (Wickham, 2016) as heatmaps showing taxa-specific LFCs when three factors were compared (i.e sample types within each species) and as waterfall plots when two factors were compared (i.e species and locations), highlighting significant differences between sample groups.
To infer the potential functionality of microbial taxa, the Functional Annotation of Prokaryotic Taxa (FAPROTAX) software (Louca et al., 2016) was used, which maps prokaryotic taxa to ecological functions based on curated literature. For each functional group in a given sample, the count reflects the cumulative occurrences of the number of ASVs associated with that group. The functional table was normalized to relative proportions, and filtered to remove functional groups with zero abundance. Mean and standard error of functional abundances were calculated for each factor of the desired comparison, and visualized with stacked bar plots comparing functional groups with abundances ≥0.01%. To test for significant differences in predicted functional profiles, we applied PERMANOVA using Jaccard dissimilarity and 999 permutations to test for the effects of species, sample types or location as fixed factors depending of the comparison. When repeated measures from the same sea urchin were present, permutations were constrained within individuals. The functional profile table, derived from FAPROTAX annotations, was normalized to relative abundances prior to analysis. To identify the most influential functions driving observed differences, we performed a SIMPER (Similarity Percentage) analysis with vegan package in R, which estimates the contribution of individual functional groups to dissimilarities between groups.
3 Results
A total of 11,718,926 raw reads were obtained across all 58 samples. After quality filtering, 5,348,831 reads remained. Following denoising, merging and chimera removal, 4,145,444 of high-quality, non-chimeric reads were retained for downstream analysis (Supplementary Table S3). After clustering into ASVs, singleton and putative contaminants removal, we obtained 3,693 ASVs taxonomically assigned to bacteria for the whole dataset (Supplementary Table S4). Rarefaction curves indicated that most of the subsamples approached an asymptote in ASVs richness, indicating that sampling effort was sufficient to produce a representative estimate of the biodiversity in the sampled community (Supplementary Figure S1).
3.1 Comparison of microbiota associated to A. lixula and P. lividus.
To study the differences in microbial composition between the two species, all sample types of each species from La Palma were pooled, to capture the overall microbial profile of each species (Supplementary Figure S2). Alpha diversity comparisons between A. lixula and P. lividus revealed significant species-level differences for Observed ASVs and Shannon diversity. P. lividus harbored a significantly higher number of observed ASVs and Shannon diversity compared to A. lixula. In contrast, Faith’s phylogenetic diversity did not differ significantly between species (Figure 2A; Supplementary Table S5).
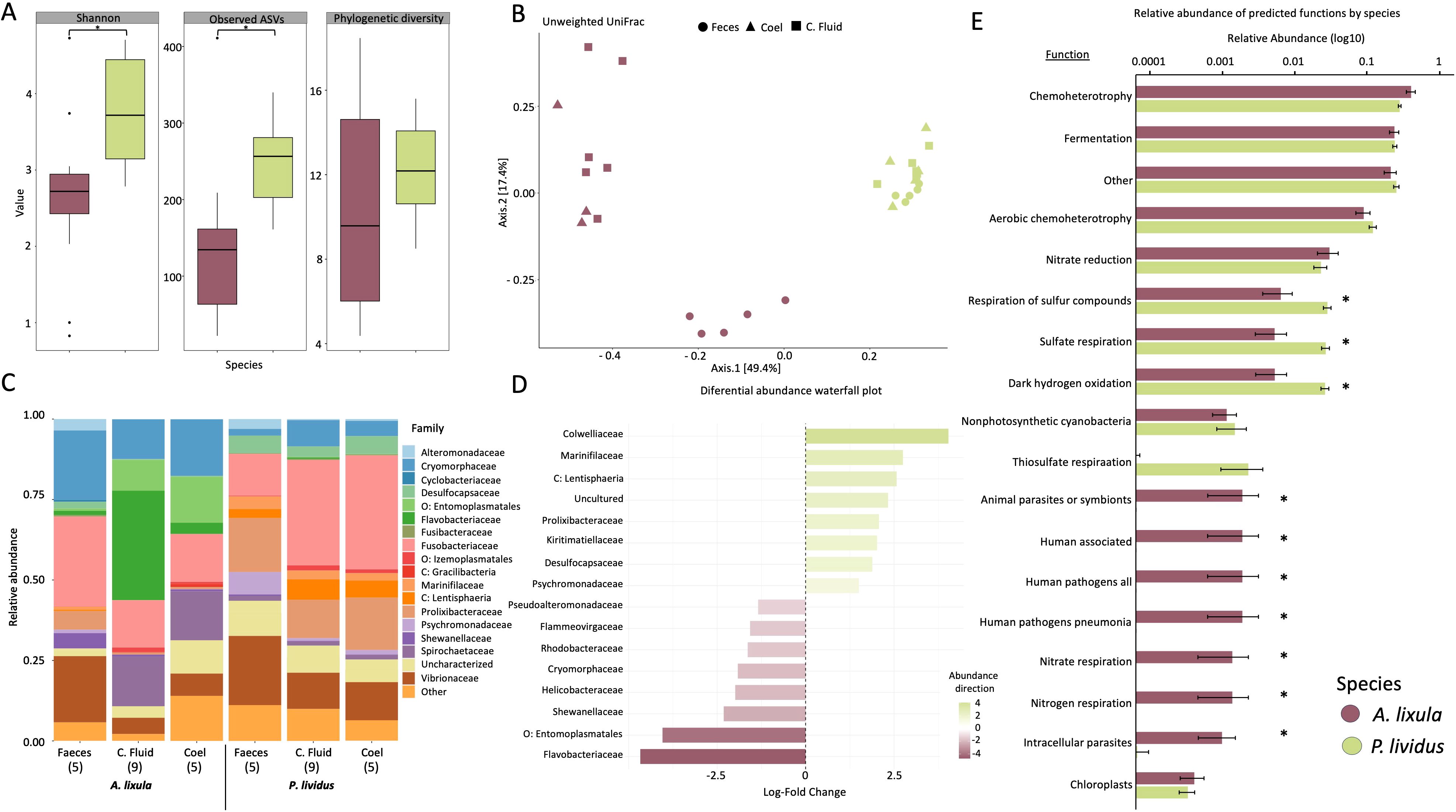
Figure 2. Overview of microbial community structure and functional potential profiles between Arbacia lixula and Paracentrotus lividus, across sample types. Color and shape coding indicates species (A. lixula in purple, P. lividus in green) and sample types (circles = feces, triangles = coelomocytes, squares = coelomic fluid). (A) Boxplots showing alpha diversity metrics: Shannon index, observed ASVs, and phylogenetic diversity between species. (B) Principal coordinates analysis (PCoA) based on unweighted UniFrac distances of microbial communities. The first two principal coordinates explain 49.4% and 17.4% of the variation, respectively. (C) Stacked bar plots of relative taxonomic abundance at the family level across species and sample types (F: feces; CF: coelomic fluid; C: coelomocytes). Sample sizes are indicated in brackets (n). Only families with abundance >0.01%) are shown; less abundant taxa are grouped under “Other.” Prefixes (e.g., p: “phylum”, o: “order”) indicate taxa not resolved at the family level. (D) Differential abundance of bacterial families between species, shown as log-fold change values. Families more abundant in P. lividus are shown in green, and those more abundant in A. lixula in purple (see Supplementary Table S7). (E) Mean relative abundances of microbial functional groups between A. lixula and P. lividus samples, with standard error bars. Asterisks indicate significant functional groups (p < 0.01) contributing to species-level differences, based on SIMPER analysis.
Beta diversity analysis based on PCoA of Unweighted and Weighted UniFrac distance matrices revealed that the first principal coordinates explained 49.4% and 45.5% of total variation, respectively, and clearly separated samples according to species (Figure 2B; Supplementary Figure S3). PERMANOVA models confirmed significant differences in microbial community composition between species (Unweighted: p = 0.001; Weighted: p = 0.001). Group dispersion analyses (PERMDISP) showed significant variation in dispersion using the Unweighted UniFrac matrix (p = 0.001), but not for the Weighted UniFrac matrix (p = 0.356) (Supplementary Tables S6). These dispersion differences were mainly driven by variation among tissue types, particularly in A. lixula, where fecal samples were more distinct compared to coelomic fluid and coelomocyte samples (Figure 2B).
The bacterial identified comprised 29 phyla across all sample types, locations, and species. The most abundant bacteria phyla were Proteobacteria, Bacteroidetes and Fusobacteriota comprising the 32.11%, 27.02% and 22.20% of relative abundance across all samples, respectively (Figure 2C; Supplementary Figure S4). The comparison of bacterial taxa between the two species revealed significant differences across several taxonomic levels and their relative abundances as identified through ANCOM-BC analysis (Supplementary Table S7, S8). Interestingly, more taxa were exclusively found in A. lixula than P. lividus (Supplementary Figure S5). The most abundant microbial phylum in P. lividus was Fusobacteria and particularly the family Fusobacteriacea (31.43%). In A. lixula the most abundant phylum was Bacteroidota (36.7%), within this phylum, the families Flavobacteriaceae and Cryomorphaceae were significantly more abundant in A. lixula (15.6% and 18.9%, respectively) than in P. lividus (0.2% and 5.9%, respectively). The phylum Proteobacteria was more predominant in P. lividus microbiota than in A. lixula (26% and 16.8%, respectively). Within this phylum, families such as Psychromonadaceae and Colwelliaceae were more abundant in P. lividus (3.4% and 2.8%) than in A. lixula (<0.5%), whereas other families such as Shewanellaceae, Pseudoalteromonadaceae, and Rhodobacteraceae were significantly more abundant in A. lixula. Five phyla, Desulfobacteriota, Verrucomicrobiota, Cyanobacteria, Planctomycetota and Campilobacterota showed higher significant abundances in P. lividus compared to A. lixula, exceeding log fold change of 2.3. Particularly, taxa within the phyla Desulfobacteriota and Verrucomicrobiota were present in P. lividus at higher abundances than 5% and in A. lixula in relative abundances lower than 1.4%. The phylum Firmicutes was more abundant in A. lixula (9.74% relative abundance) compared to P. lividus (3.80%). Notably, the genus Candidatus Hepatoplasma (LFC = 4.46), within Firmicutes, was significantly more abundant in A. lixula, reaching up to 70% relative abundance in some samples, whereas its maximum relative abundance in P. lividus samples was only 7.5%. Moreover, the phylum Spirochaeta represented 11% of average relative abundance in A. lixula microbial profiles while only 1.6% average relative abundance in P. lividus (Figure 2D; Supplementary Table S8).
Functional inference using FAPROTAX and Jaccard-based dissimilarity revealed that both host species significantly differed in the presence/absence of microbial functional traits. PERMANOVA results showed effects of species (p = 0.004) (Supplementary Table S9). SIMPER analysis identified both widespread and rare functional groups contributing to differences between A. lixula and P. lividus microbiomes. General functions such as chemoheterotrophy, fermentation, and aerobic chemoheterotrophy were shared across species. In A. lixula, microbial communities were enriched in nitrogen-cycling functions, including nitrate/nitrogen respiration (p < 0.002), as well as metabolism pathways such as methanol oxidation and methylotrophy (p < 0.001). These functions correspond to the higher relative abundance of families such as Shewanellaceae, Rhodobacteraceae, and Flavobacteriaceae, which are classified by FAPROTAX as contributors to nitrogen cycling, and in the case of Rhodobacteraceae, also to methylotrophy and methanol oxidation. Additionally, A. lixula harbored a greater abundance of bacteria associated with intracellular parasitism and potential pathogenicity (e.g., intracellular parasites, human pathogens; p < 0.01). In contrast, P. lividus microbiomes were functionally distinct due to a higher prevalence of sulfur-related metabolism, including sulfate respiration and respiration of sulfur compounds (p < 0.001). These functions correspond to the higher relative abundance of taxa such as Desulfobacterota and Campylobacteriota, which are classified by FAPROTAX as sulfate reducers and sulfur-respiring bacteria (Figure 2E; Supplementary Table S10).
3.2 Microbial profiles associated with each sample type
Among the microbial communities of the three A. lixula sample types (coelomic fluid, coelomocytes, and feces) from La Palma, fecal samples exhibited significantly higher alpha diversity. Planned contrast confirmed that feces harbored significant greater diversity than coelomic fluid (Shannon diversity: p < 0.001 and Observed ASVs: p < 0.001) and coelomocytes (Shannon: p=0.001 and Observed: p<0.001), while phylogenetic diversity did not differ significantly across tissues (p = 0.094) except between coelomocytes and feces (p=0.043). Differences between coelomic fluid and coelomocytes were not significant for any diversity metric (p > 0.15). (Figure 3A; Supplementary Table S11). Ordination of samples using PCoA based on Unweighted and Weighted UniFrac distances revealed two distinct clusters, one corresponding to fecal samples and another encompassing coelomocyte and coelomic fluid samples, indicating a separation in microbial community composition between these two groups (Figure 3B; Supplementary Figure S6). Venn diagrams also supported distinctiveness of fecal sample compared to the samples from the coelomic compartment (Supplementary Figure S7). PERMANOVA revealed significant differences in assemblage composition between feces and coelomic fluid for Unweighted UniFrac (p = 0.040), but not for Weighted UniFrac (p = 0.078). Similar trend between feces and coelomocytes for Unweighted UniFrac (p = 0.017), but not for Weighted UniFrac (p = 0.543). Differences between coelomic fluid and coelomocytes were not significant for either distance metric. Tests of homogeneity of multivariate dispersions (PERMDISP) showed no significant differences among groups (all p > 0.12; Supplementary Table S12), indicating that the observed compositional separation reflects true differences in community structure rather than unequal variability. Taxa within the phylum Proteobacteria, such as the families Vibrionaceae (20.5%), Alteromonadaceae (3.5%) and Shewanellaceae (4.7%), the phylum Fusobacteriota such as the family Fusobacteriaceae (27.9%) and the family Desulfocapsaceae (2%) within the phylum Desulfobacteriota were significantly more abundant in feces than coelomic fluid and coelomocytes, with log-fold changes exceeding 2.86 (Supplementary Table S13, S14). Conversely, the phylum Spirochaetota (family Spirochaetaceae) was primarily enriched in coelomic fluid and coelomocytes, reaching 15% of relative abundance in coelomic samples and only 0.02% in fecal samples. Within the phylum Bacteroidota, the family Flavobacteriaceae was more abundant in coelomic samples (33.9%) than in fecal samples (1.3%). Additionally, the phylum Firmicutes (order Izemoplasmatales), even at low abundances, were found in an order of magnitude higher in coelomic compartments than in feces. The heatmap visualization of the log-fold changes highlighted distinct clustering patterns, with fecal microbiota displaying stronger differential abundance patterns than the other tissue types (Figure 3C; Supplementary Tables S14).
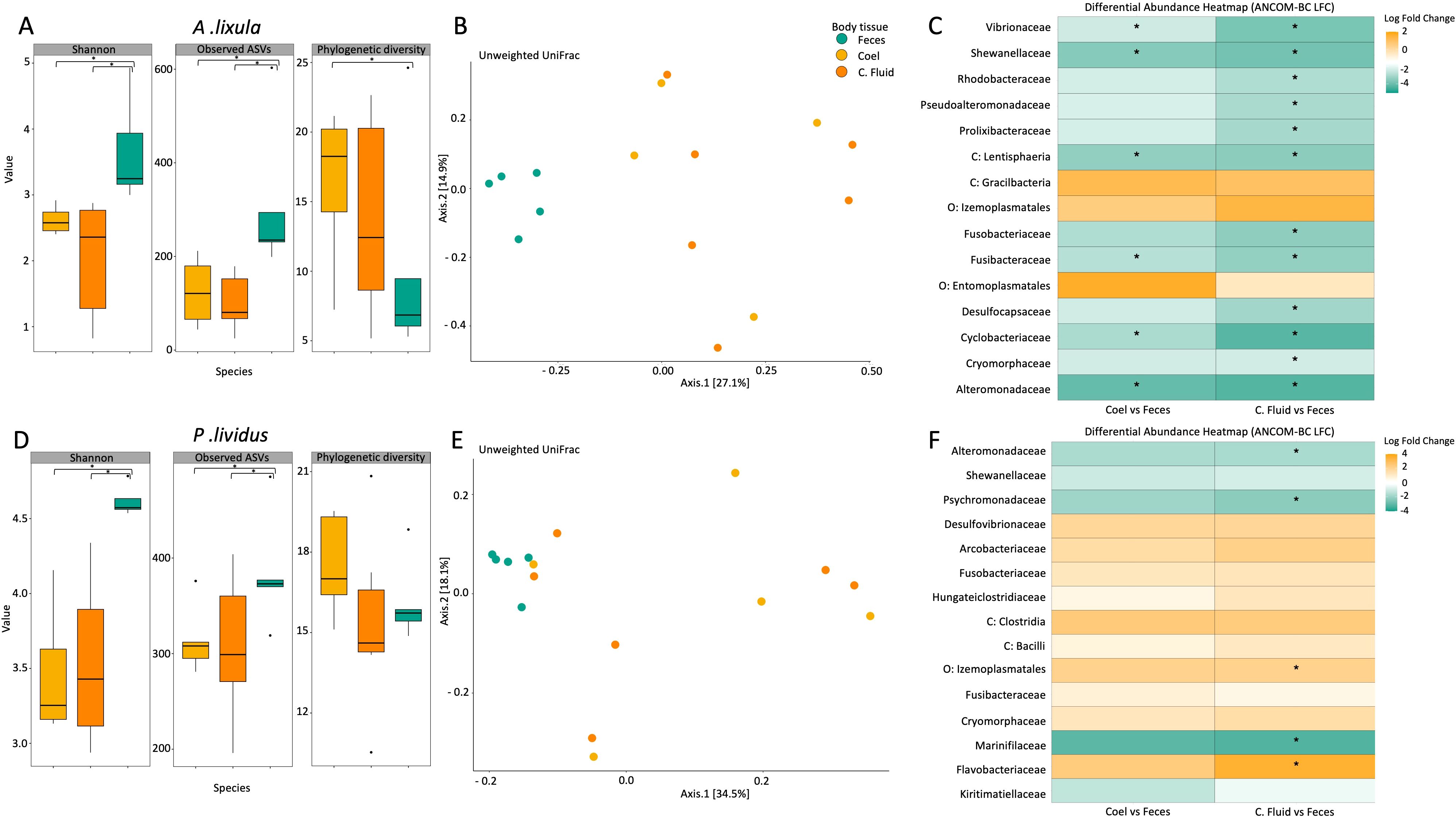
Figure 3. Microbial diversity, community structure, and differential abundance across body compartments in Arbacia lixula (A-C) and Paracentrotus lividus (D-E). Color coding indicates sample types: feces (green), coelomocytes (yellow), and coelomic fluid (orange). (A) Boxplots showing alpha diversity metrics (Shannon index, observed ASVs, and phylogenetic diversity) across sample types of A. lixula. (B) Principal coordinates analysis based on unweighted UniFrac distances of A. lixula sample types. The first two principal coordinates explain 27.1% and 14.9% of the variation, respectively. (C) Heatmap of bacterial families showing log-fold changes (ANCOM-BC results) in differential abundance between coelomocytes and feces (left column) and coelomic fluid and feces (right column) in A. lixula. Asterisks indicate significant differences (q-value < 0.05). Taxa enriched in fecal samples are highlighted in green, and those enriched in coelomic compartments are shown in orange. (D) Boxplots of alpha diversity metrics (Shannon index, observed ASVs, and phylogenetic diversity) across sample types of P. lividus. (E) Principal coordinates analysis based on Unweighted UniFrac distances of P. lividus sample types. The first two principal coordinates explain 34.5% and 18.1% of the variation, respectively. (F) Heatmap of bacterial families showing log-fold changes (ANCOM-BC results) in differential abundance between coelomocytes and feces (left column) and coelomic fluid and feces (right column) in P. lividus. Asterisks indicate significant differences. Taxa enriched in fecal samples are highlighted in green, and those enriched in coelomic compartments are shown in orange.
Among the microbial communities of the three P. lividus sample types, fecal samples of P. lividus supported higher microbial diversity than coelomic fluid and coelomocytes, whereas coelomic fluid and coelomocytes did not differ significantly. Specifically, feces exhibited significantly higher Shannon diversity and Observed ASVs, while Faith’s phylogenetic diversity did not differ between compartments (p > 0.453) (Figure 3D; Supplementary Table S15). The PCoA based on Unweighted and Weighted UniFrac distances revealed two main clusters in P. lividus, one corresponding to fecal samples and another encompassing coelomocyte and coelomic fluid samples (Figure 3E; Supplementary Figure S8). Venn diagrams also supported distinctiveness of fecal sample compared to the samples from the coelomic compartment (Supplementary Figure S9). PERMANOVA indicated no significant differences between coelomic fluid and coelomocytes across any distance metric (all p > 0.526). However, feces differed significantly from coelomic fluid in Unweighted UniFrac (p = 0.049), but not in Weighted UniFrac (p > 0.073). Tests of homogeneity of multivariate dispersions (PERMDISP) revealed no significant differences in variability between compartments across metrics (all p > 0.06), indicating that the observed differences reflect true compositional changes rather than dispersion effects (Supplementary Table S16). Significant differential abundant taxa were observed across sample types using ANCOM-BC analysis, particularly when comparing between coelomic fluid and fecal samples. However, coelomocyte samples notably presented a similar presence and abundance of microbial taxa as coelomic fluid. Microbial taxa within the phylum Proteobacteria were consistently more abundant in fecal samples (41.6% of relative abundance) than in coelomic fluid (22.05%) and coelomocytes (20.03%). Within this phylum, the families Alteromonadaceae and Psychromonadaceae were significantly more abundant in the fecal samples, with a log fold change exceeding 1.7. Additionally, the family Vibrionaceae was found at twice the abundance in fecal samples (21.5% relative abundance) compared to the other sample types (average 11.5%). In contrast, taxa within the phylum Firmicutes, particularly the order Izemoplasmatales, were significantly more abundant in the coelomic compartment. Besides that, the phylum Bacteroidota is one of the most abundant across all sample types of P. lividus, representing an average of 25% of relative abundance. The family Flavobacteriaceae presented significantly higher abundance in the coelomic and coelomic fluid samples. The relative abundance of the phylum Fusobacteriota in coelomic fluid (32.7%) and coelomocytes (35.5%) samples was twice as high compared to fecal samples (13%) (Figure 3F; Supplementary material S17, S18).
To gain deeper insight into the predicted microbial functional profiles across body compartments, ASV-assigned taxa from both A. lixula and P. lividus were annotated using the FAPROTAX database. In both species, distinct functional signatures were observed across tissue types, with fecal samples consistently enriched in diverse metabolic pathways compared to the more host-associated coelomic fluid and coelomocytes. In A. lixula, PERMANOVA did not detect statistically significant differences among sample types (p = 0.058), and the relevant pairwise comparisons were also non-significant (Supplementary Table S19). In P. lividus, however, functional profiles significantly differed among sample types (p = 0.047), with strong pairwise differences between feces and coelomocytes (p = 0.004) and coelomocytes and coelomic fluid (p = 0.027) (Supplementary Table S20). SIMPER analyses revealed consistent functional enrichments in fecal samples of both species, including aerobic chemoheterotrophy and multiple steps of the nitrogen cycles, such as nitrate reduction, nitrate/nitrite respiration, functions largely assigned by FAPROTAX to Rhodobacteraceae and Shewanellaceae. In A. lixula, fecal samples also exhibited enrichment in sulfur-related metabolic functions, including respiration of sulfur compounds, sulfate respiration, dark oxidation of sulfur compounds, and dark sulfite oxidation which correspond to the presence of Desulfobacteraceae and other sulfur-reducing lineages. In P. lividus, feces additionally featured photoheterotrophy and anoxygenic photoautotrophy, associated with members of the Rhodobacteraceae, suggesting potential light-driven metabolic processes. In contrast, the coelomic fluid and coelomocyte microbiota in both species exhibited reduced functional diversity and a higher prevalence of traits associated with animal symbionts, intracellular parasites, and putative pathogens (e.g Vibrionaceae) (Figure 4; Supplementary Table S21, S22).
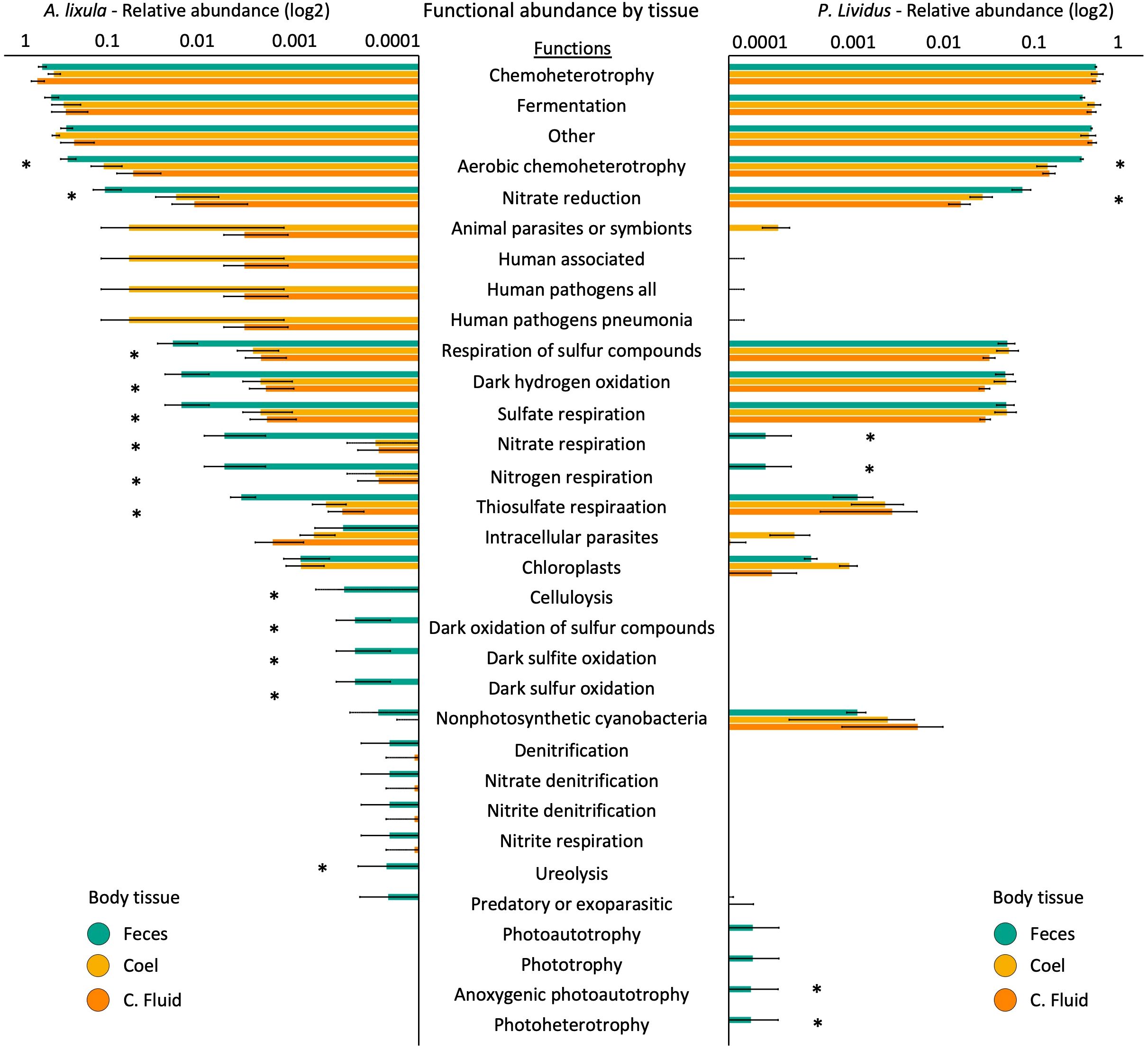
Figure 4. Predicted microbial functional profiles across tissue types in Arbacia lixula (left) and Paracentrotus lividus (right). Bar plots show the mean relative abundance of predicted microbial functions (FAPROTAX) in feces (green), coelomocytes (yellow), and coelomic fluid (orange) for each species. Only functions with a mean relative abundance >0.01% across samples are shown. Error bars represent standard error. Asterisks indicate functions that significantly differ between tissue types (p < 0.01) based on SIMPER analysis.
3.3 Microbiota variation across geographic locations
To assess geographic variation in microbiota, we focused exclusively on coelomic fluid samples, as this was the only compartment sampled for both species at both locations. Geographic comparisons showed that alpha diversity varied by location only in A. lixula (Figure 5A; Supplementary Table S23). Samples from Blanes exhibited significantly higher Shannon diversity than those from La Palma (p = 0.03), while Observed ASVs and Faith’s phylogenetic diversity did not differ significantly between locations (p > 0.428). In contrast, no significant geographic differences were detected for P. lividus across any of the alpha diversity metrics (all p > 0.48; Supplementary Table S24). However, Venn diagrams at different taxonomic levels showed that there were more unique taxa in Blanes than in La Palma in both species (Supplementary Figure S10).
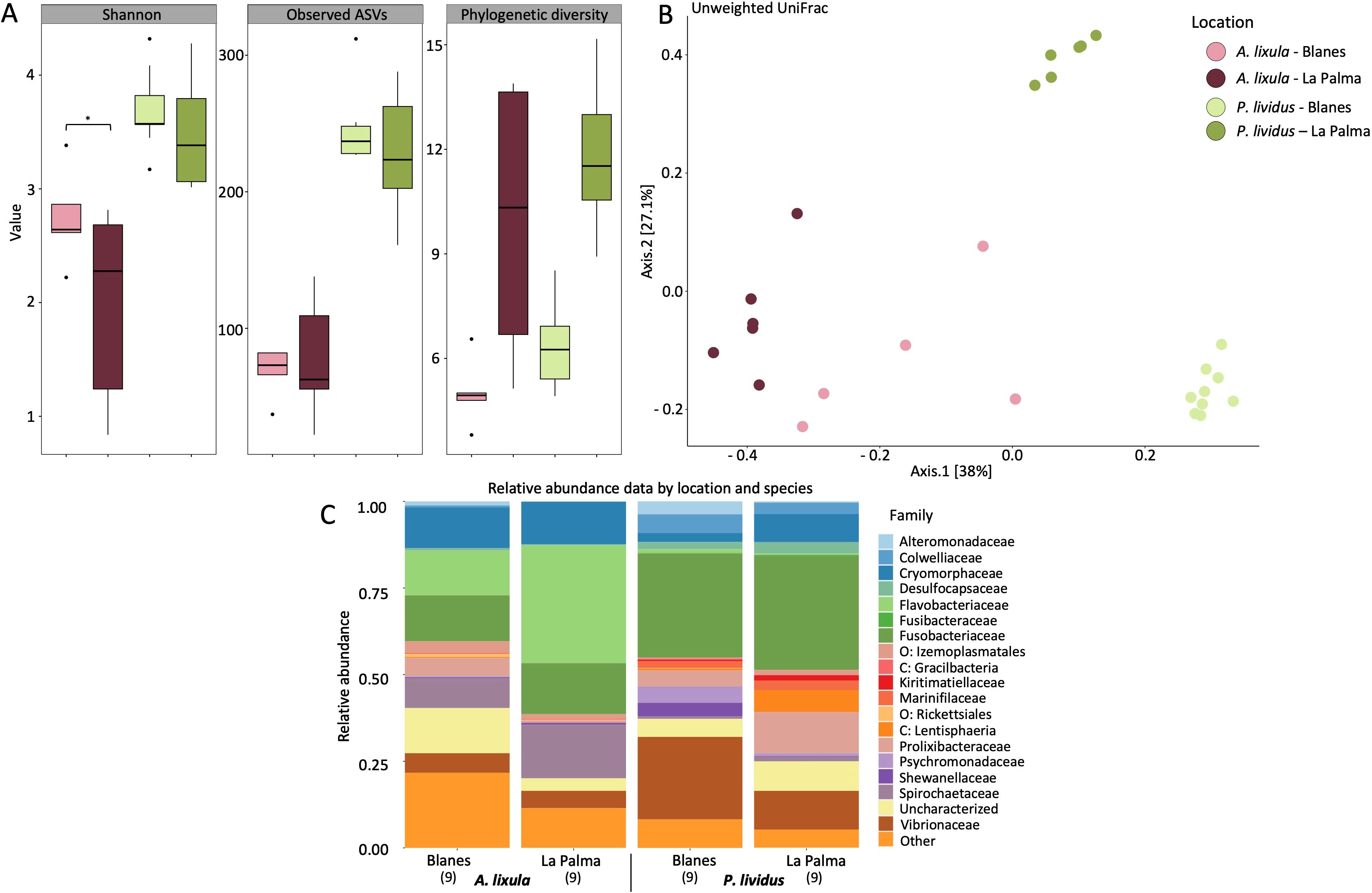
Figure 5. Microbial community diversity and composition in Arbacia lixula and Paracentrotus lividus across geographic locations, Blanes (Mediterranean Sea) and La Palma (Northeastern Atlantic). Color coding in panels A and B corresponds to species and location combinations, as indicated in the legend. (A) Boxplots showing alpha diversity metrics (Shannon index, observed ASVs, and phylogenetic diversity) by species and location. (B) Principal coordinates analysis based on unweighted UniFrac distances of microbial communities across species and locations. The first two principal coordinates explain 38% and 27.1% of the variation, respectively. (C) Stacked bar plots of relative taxonomic abundance at the family level grouped by species and location. Only families with a relative abundance >0.01% are shown; less abundant taxa are grouped under “Other”. Prefixes (e.g., p: “phylum”, o: “order”) indicate taxa not resolved at the family level. Sample sizes are indicated in brackets (n).
Geographic location also influenced microbial community composition, although patterns differed between species (Figure 5B; Supplementary Figure S11). In A. lixula, PERMANOVA indicated significant location effects across both distance metrics, Weighted UniFrac (p = 0.001), and Unweighted UniFrac (p = 0.008) (Supplementary Table S25A). By contrast, in P. lividus, location effects were significant for Unweighted UniFrac (p = 0.001), but not for Weighted UniFrac (p = 0.182; Supplementary Table S26A). In both species, PERMDISP tests showed no significant differences in within-group variability (p > 0.18), confirming that observed effects represent genuine compositional shifts rather than differences in dispersion (Supplementary Tables S25B, S26B).
The differential abundance analysis in A. lixula revealed that the phyla Proteobacteria (including the family Vibrionaceae) and Firmicutes (order Izemoplasmatales and family Mycoplasmataceae) were more abundant in Blanes (15.17% and 21.9% average relative abundance, respectively) than in La Palma (8.15% and 11.21%). In contrast, the phyla Bacteroidota and Spirochaeta were more abundant in La Palma, exceeding log fold change of 1 (Figure 5C; Supplementary Figure S12; Supplementary Table S27). However, no significant differences were found with ANCOM-BC analysis (Supplementary Table S28). In P. lividus the phylum Proteobacteria was more abundant in Blanes (47.9% average relative abundance) than in La Palma (20.23%), where the order Rickettsiales, and the families Colwelliaceae, Rhodobacteracea, Shewanellaceae and Vibrionaceae were significantly more abundant, exceeding log fold changes of 2.5. Microbial taxa within the phyla Firmicutes (families Hungateiclostridiaceae, Christensenellaceae and the order Izemoplasmatales) and Verrucomicota (Kiritimatiellaceae and the class Lentisphaeria “P.palmC41”) were found significantly more abundant in individuals from La Palma compared to Blanes. The phylum Bacteroidota was more abundant in La Palma (19.86% relative abundance) than in Blanes (11.46%), particularly the family Cryomorphaceae (Supplementary Tables S29, S30).
To explore the influence of geographic location on microbial functional potential, we used FAPROTAX annotations of ASV-assigned taxa from coelomic fluid samples of A. lixula and P. lividus collected from La Palma and Blanes (Figure 6). In A. lixula, PERMANOVA revealed no significant differences in predicted functions between locations (p = 0.303). However, SIMPER analysis identified functional groups contributing to location-based variation, with higher representation of nitrate and nitrogen respiration, ureolysis, and dark sulfur-related metabolisms (e.g., dark sulfite/sulfur oxidation) in Blanes samples, functions largely assigned to Colwelliaceae, Rhodobacteraceae and Shewanellaceae (Supplementary Table S31). In contrast, P. lividus exhibited strong functional differentiation by location, with PERMANOVA showing a significant effect of geographic origin (p = 0.002) (Supplementary Table S32). SIMPER results highlighted functional enrichments in Blanes, particularly in aerobic chemoheterotrophy, nitrate reduction, and sulfur-related metabolisms such as sulfate respiration and dark hydrogen oxidation which were associated with Desulfobacteraceae. Moreover, functions related to intracellular parasitism appeared more abundant in Blanes, largely associated with Rickettsiales.
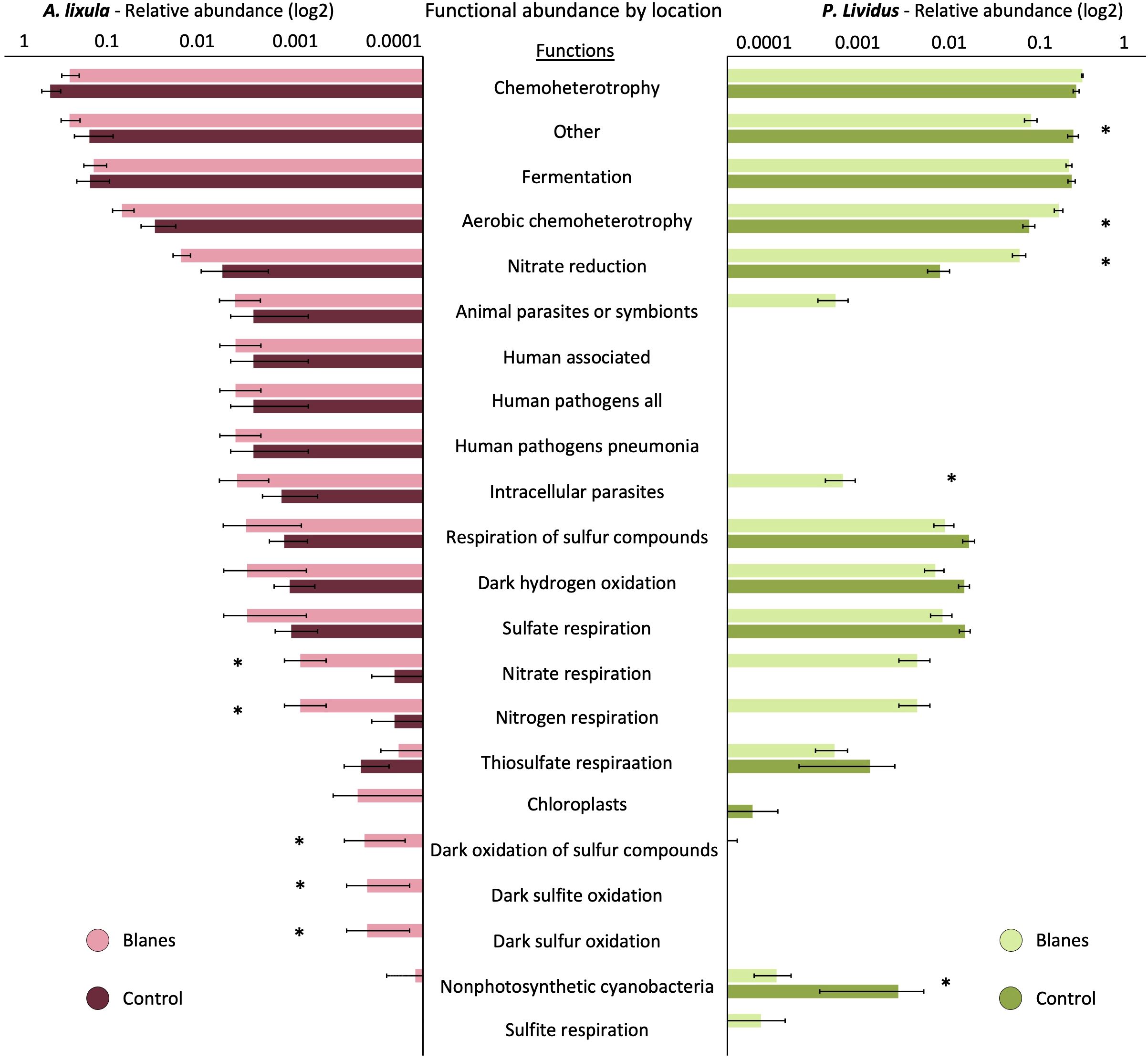
Figure 6. Predicted microbial functional profiles in Arbacia lixula (left) and Paracentrotus lividus (right) across two geographic locations, Blanes (Mediterranean Sea) and La Palma (Northeartern Atlantic). Bar plots show the mean relative abundance (log10 scale) of predicted microbial functions (FAPROTAX) by location for each species. Only functions with a mean relative abundance >0.01% across samples are shown. Error bars represent standard error. Asterisks indicate functions that significantly differ between locations (p < 0.01) based on SIMPER analysis.
4 Discussion
This study strengthens the holobiont framework by highlighting the interdependence between sea urchins and their microbiota, which performing essential roles in digestion, nutrient assimilation, and immune defense, making them integral to host physiology (Pita et al., 2018; Dittami et al., 2021). The most abundant bacterial phyla Proteobacteria, Bacteroidetes and Fusobacteria found in the studied sea urchin’s associated microbiota have been commonly reported in marine invertebrates, particularly in other sea urchins (Hakim et al., 2015; Faddetta et al., 2020; Yao et al., 2019; Rodríguez-Barreras et al., 2021). However, significant species-specific, compartmental, and geographic variation were evident, revealing dynamic microbial interactions that likely reflect sea urchin contrasting physiology, diet, and environmental adaptation.
Across both species Proteobacteria, Bacteroidota, and Fusobacteriota dominated the microbiota, consistent with patterns reported in other marine invertebrates. In P. lividus, Fusobacteriaceae and Psychromonadaceae were particularly abundant, contributing to anaerobic carbohydrate metabolism and degradation of algal polysaccharides such as cellulose, starch and alginate (Rodríguez-Barreras et al., 2021; Schwob et al., 2020; Hakim, 2019; Schram et al., 2018; Zhang et al., 2014). Enrichment of Cyanobacteria and Planctomycetota in P. lividus, also reported in other herbivorous echinoids such as Lytechinus variegatus, may support its capacity to digest macroalgae through polysaccharide degradation (Zheng et al., 2024; Meziti et al., 2007). Cyanobacteria are likely introduced via seagrass consumption, while Planctomycetes are commonly found on the surfaces of marine algae (Bondoso et al., 2017; Rodríguez-Barreras et al., 2021). The microbiota of P. lividus exhibited functional enrichment in sulfur cycling and photoautotrophy, consistent with its herbivorous diet leading to the ingestion of phototrophic microbes (Faddetta et al., 2020; Zheng et al., 2024). In contrast, A. lixula exhibited higher abundances of Bacteroidota and Firmicutes, including the families Flavobacteriaceae and Cryomorphaceae, which are known for their ability to degrade high-molecular-weight organic compounds, including proteins, lipids, and complex polysaccharides from encrusting algae and animal-derived material (Zhang et al., 2014; Yao et al., 2019; Thomas et al., 2011). Furthermore, the higher presence of Spirochaetota in A. lixula suggests microbial contributions to saccharolytic activity and nitrogen recycling, an adaptation likely related to its persistence in barren zones with fluctuating food availability (Leschine et al., 2006; Lilburn et al., 2001).
The microbial communities of A. lixula and P. lividus exhibit significant differences in composition and putative functions, which may reflect their distinct dietary niches. While both species coexist in rocky subtidal habitats, stable isotope indicate that P. lividus is primarily herbivorous, feeding on fleshy macroalgae, whereas A. lixula occupies a higher trophic level, displaying omnivorous tendencies with a notable carnivorous component (Wangensteen et al., 2011; Trenzado et al., 2012). Despite A. lixula has broader range of food items, it harboured a lower bacterial alpha diversity compared to P. lividus. This pattern aligns with previous reports that herbivorous sea urchins, such as Tripneustes gratilla and Lytechinus variegatus exhibit greater microbial alpha diversity than omnivorous echinoids (Yao et al., 2019; Rodríguez-Barreras et al., 2021). Thus, suggesting that higher bacterial alpha diversity does not necessarily correlate with broader dietary breadth. These dietary differences were also reflected in the taxonomic and functional profiles of both species associated microbiota. The microbial composition of A. lixula mirrors the microbiota observed in scavenger or detritivore echinoids, entiched in bacteria capable of degrading refractory organic matter, while P. lividus harbored microbial assemblages more specialized in algal polysaccharide degradation. Such differences parallel previous findings on omnivorous echinoids such as Echinometra lucunter and Diadema antillarum, whose host microbiota specialized in nitrogen recycling and protein metabolism (Rodríguez-Barreras et al., 2021).
Microbial profiles across body compartments in A. lixula and P. lividus revealed distinct microbial compartmentalization patterns. These findings are consistent with microbial compartmentalization observed in other echinoderms, such as holothurians, where distinct microbiota reflect ecological and physiological roles (Pagán-Jiménez et al., 2019; Schwob et al., 2020; Hakim et al., 2019). Fecal samples exhibited the highest microbial diversity and formed distinct clustering patterns, separating them from coelomic fluid and coelomocytes samples, which displayed overlapping microbial compositions. Due to the limitation of the innate gut digestive enzymes in sea urchins, microbiota likely aid in breaking down complex sugars and cellulose while contributing to essential biomolecule metabolism for protein and lipid assimilation (Miller et al., 2021). In return, these bacteria benefit from a stable, predator-free environment within the sea urchin’s digestive tract (Zheng et al., 2024). The dominance of Proteobacteria (families Vibrionaceae, Psychromonadaceae, Shewanellaceae, Alteromonadaceae), Fusobacteriota (family Fusobacteriaceae), Desulfobacteriota, and Bacteroidota in fecal samples suggest active roles in carbohydrate degradation, nitrogen fixation, and sulfate reduction, processes essential for digestion and host nutrition (Hakim et al., 2021; Meziti et al., 2007). Consistently, the fecal microbiota of A. lixula and P. lividus accordingly displayed enrichment in functions related to aerobic chemoheterotrophy, nitrate reduction, nitrogen respiration, and sulfur metabolism with Shewanellaceae, Rhodobacteraceae, and Flavobacteriaceae contributing to nitrogen cycling, and Desulfobacteraceae and related lineages associated with sulfur respiration. The presence of Vibrionaceae in both species’ fecal microbiota is notable, as Vibrio spp. are known for algal polysaccharide digestion (e.g. alginate) and nitrogen fixation (Guerinot and Patriquin, 1981; Miller et al., 2021; Hakim et al., 2015). However, Vibrionaceae taxa have also been found as opportunistic pathogens (Becker et al., 2007, 2008; Roux et al., 2015; Salazar-Forero et al., 2022), highlighting their potential to shift between mutualistic and pathogenic roles depending on host condition and environmental context. The presence of these microbial groups in both A. lixula and P. lividus suggest that despite differences in diet, similar microbial consortia play key roles in digestion and nutrient acquisition.
The coelomic fluid and coelomocytes exhibited a lower microbial diversity than fecal samples, yet their microbiota displayed slightly distinct enriched functional roles. The coelomic fluid of echinoderms contains abundant antimicrobial compounds (Dybas and Fankboner, 1986), which can influence the ability of microbes to persist in this compartment (Zhang et al., 2021). Nevertheless, a diverse microbial community was detected, consistent with previous studies on P. lividus and other echinoderms (Faddetta et al., 2020; Nakagawa et al., 2017; Zhang et al., 2021). Specifically, Firmicutes, order Izemoplasmatales, and families of Bacteroidetes, particularly Flavobacteraceae were enriched in coelomic compartments of both species. Flavobacteraceae are widely distributed in marine environments, and while their occurrence in the coelomic compartment may indicate ecological plasticity, we cannot infer transient acquisition without environmental sampling. In both species, the coelomic compartments were enriched in functional groups related to animal parasites and symbionts, aligning with previous reports that echinoderm coelomic fluid harbors unique microbial assemblages, including potentially pathogenic and symbiotic bacteria. Nakagawa et al. (2017) demonstrated that starfish coelomic fluid can be dominated by Helicobacter-related taxa and unclassified Thiotrichales, suggesting that this body compartment may serve as a reservoir for microorganisms with both pathogenic potential and beneficial roles in host physiology. Our findings support the view that the coelomic compartment may act as a reservoir for diverse microbial communities shaped by biotic and abiotic factors. However, because seawater was not sampled, we cannot directly evaluate environmental acquisition of these taxa. Moreover, recent studies have shown that A. lixula harbors a significantly higher abundance of pathogenic microbes compared to P. lividus, reinforcing the observed enrichment in intracellular parasitism-associated functions (Salazar-Forero et al., 2022).
Geographic location influenced microbiota composition in both species, with shifts more pronounced in P. lividus, while A. lixula also exhibited compositional shifts, but its microbial community appeared more conserved, suggesting a degree of ecological stability or host-driven regulation. Geographic difference is widely acknowledged as one of the primary factors influencing microbiome diversity in numerous studies on marine organisms (Hou et al., 2017) as variations in salinity, temperature, and nutrient availability can shape microbial community structure and function. In both species, the Northeastern Atlantic location, La Palma, showed higher abundances of Bacteroidota and Spirochaeta, while the phylum Proteobacteria was more abundant in the Mediterranean sea location, Blanes. The prominence of Bacteroidota in La Palma is consistent with their role in polysaccharide degradation and organic matter recycling, which has been observed in marine environments with lower nutrient availability (Kirchman, 2002), such as the waters around the Canary Islands (Bode et al., 2001; Hernández et al., 2016). The enrichment of Flavobacteriaceae in A. lixula from La Palma likely reflects their specialization in degrading algal polysaccharides (Williams et al., 2013; Teeling et al., 2016), consistent with the high macroalgal cover at La Bajita, which provides abundant organic substrates. Interestingly, Spirochaetota, often associated to low-oxygen environments and detritus-rich sediments (Lilburn et al., 2001; Dubilier et al., 2008), were also more abundant in La Palma, suggesting adaptation to local trophic conditions. In contrast, Blanes microbiota include Proteobacteria such as Pseudoalteromonas, Vibrionaceae, Shewanellaceae groups associated with bioactive secondary metabolites, nitrogen fixation and polysaccharide degradation (Bowman, 2007) (Guerinot and Patriquin, 1981). The dominance of the family Vibrionaceae in Blanes, especially in P. lividus, is particularly interesting given their possible roles in nitrogen fixation and algal polysaccharide degradation and potential pathogenicity (Becker et al., 2008; Salazar-Forero et al., 2022). The presence of bacteria associated with putative intracellular parasitism or symbionts such as these Vibrionaceae and taxa within the order Rickettsiales in higher abundance in Blanes also highlights the potential environmental stressors influencing microbial-host interactions in this region. Functional predictions reflected these taxonomic shifts, with Blanes communities enriched in nitrogen cycling pathways such as nitrate reduction and nitrogen respiration, linked to Vibrionaceae and Shewanellaceae, potentially influenced by higher nutrient availability (Galià-Camps et al., 2023). Conversely, microbial taxa enriched in La Palma were associated with metabolic pathways linked to carbon degradation and organic matter recycling, particularly through the activity of Bacteroidota and Spirochaetota, while sulfur metabolism functions were also linked to Desulfobacteraceae. Together, these results highlight that geographic location shapes both the composition and functional potential of sea urchin-associated microbiota, reflecting regional resource availability and environmental variability, though interpretations should be made cautiously given the absence of seawater samples and limited replication.
This study provides the first comparative overview of the microbiota associated with A. lixula and P. lividus across body compartments and geographic regions. We found clear differences between species as well as compartment-specific patterns in microbial assemblages, suggesting that both host identity and body compartment exert selective influences on associated bacteria. These results highlight the complexity of host–microbiota associations in echinoids and provide a foundation for future studies investigating the ecological and physiological significance of these species- and compartment-level microbial differences. However, functional predictions based on FAPROTAX should be interpreted with caution, as this approach relies on assignments from cultured representatives and only a subset of the community can be classified, potentially underrepresenting true functional diversity. Future research should therefore integrate seasonal sampling and advanced multi-omics approaches, such as metagenomics and metatranscriptomics, to further understand the dynamic interactions between environmental factors and microbiota composition and directly infer the functional potential of sea urchin microbiota. Such studies will enhance our understanding of host–microbe interactions in echinoids and provide deeper insights into the mechanisms underlying their symbiotic associations.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics statement
Ethical approval was not required for the study involving animals in accordance with the local legislation and institutional requirements because it involved non-regulated marine invertebrates (Arbacia lixula and Paracentrotus lividus), which are not subject to animal welfare legislation. All specimens were handled with care and returned alive to their original sampling locations following sample collection.
Author contributions
VA: Conceptualization, Writing – original draft, Methodology, Data curation, Supervision, Investigation, Visualization, Formal Analysis, Writing – review & editing. LS-M: Formal Analysis, Data curation, Methodology, Writing – review & editing, Investigation. RF-V: Investigation, Visualization, Data curation, Writing – review & editing, Methodology. JH: Funding acquisition, Validation, Supervision, Methodology, Writing – review & editing, Conceptualization, Resources. RP-P: Funding acquisition, Project administration, Resources, Validation, Writing – review & editing, Conceptualization, Methodology, Supervision.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was funded by the Spanish Government projects ACIDOMIC (CNS2022–135968 funded by MCIN/AEO/10.13039/501100011033, and by the European Union NextGeneration EU/PRTR), ADAPTIVE (PGC2018-100735-B-I00 funded by MICIU/AEI/10.13039/501100011033 and FEDER Una manera de hacer Europa), and ENVIOME (PID2021-128094NB-I00 funded by MCIU/AEI/10.13039/501100011033, and FEDER, UE). The project DIVERGEN from the BBVA Foundation (Ayudas Fundación BBVA a Proyectos Investigación Cientıfica 2021), and a Ramón y Cajal contract to ROCÍO PÉREZ-PORTELA (RYC2018-025070-I funded by MICIU/AEI/10.13039/501100011033 and El FSE invierte en tu futuro), and the Marie Sklodowska-Curie grant to VANESSA ARRANZ (agreement No 101105400 funded by the European Union’s Horizon Europe research and innovation program), and a predoctoral contract FPI to ROBERT FERNÁNDEZ–VILERT (PRE2022–101655 funded by MICIU/AEI/10.13039/501100011033 and FSE+). This paper is a contribution of the Consolidated Research Team: 2021 SGR 01271 Marine Biodiversity and Evolution (MBE)(AGAUR, Generalitat de Catalunya).
Acknowledgments
This research was partially performed at the OMACC (Observatorio Marino de Cambio Climático-Pta. De Fuencaliente), La Palma, Canary Islands, Spain. We greatly appreciate the support of all members of Adaptive Team and Slug Lab, Nancy Perez Negrin and Adam Santos for their help during our field trips to Fuencaliente, La Palma Island.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2025.1615711/full#supplementary-material
References
Anderson M. J. (2005). Permutational multivariate analysis of variance Vol. 26 (Auckland: Department of Statistics, University of Auckland), 32–46.
Bang C., Dagan T., Deines P., Dubilier N., Duschl W. J., Fraune S., et al. (2018). Metaorganisms in extreme environments: do microbes play a role in organismal adaptation? Zoology 127, 1–19. doi: 10.1016/j.zool.2018.02.004
Becker P. T., Egea E., and Eeckhaut I. (2008). Characterization of the bacterial communities associated with the bald sea urchin disease of the echinoid Paracentrotus lividus. J. Invertebrate Pathol. 98, 136–147. doi: 10.1016/j.jip.2007.12.002
Becker P., Gillan D. C., and Eeckhaut I. (2007). Microbiological study of the body wall lesions of the echinoid Tripneustes gratilla. Dis. Aquat. organisms 77, 73–82. doi: 10.3354/dao01821
Benjamini Y. and Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. society: Ser. B (Methodological) 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bode A., Barquero S., Varela M., Braun J. G., and de Armas D. (2001). Pelagic bacteria and phytoplankton in oceanic waters near the Canary Islands in summer. Mar. Ecol. Prog. Ser. 209, 1–17. doi: 10.3354/meps209001
Bolyen E., Rideout J. R., Dillon M. R., Bokulich N. A., Abnet C. C., Al-Ghalith G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Bonaviri C., Vega Fernández T., Fanelli G., Badalamenti F., and Gianguzza P. (2011). Leading role of the sea urchin Arbacia lixula in maintaining the barren state in southwestern Mediterranean. Mar. Biol. 158, 2505–2513. doi: 10.1007/s00227-011-1751-2
Bondoso J., Godoy-Vitorino F., Balague V., Gasol J. M., Harder J., and Lage O. M. (2017). Epiphytic Planctomycetes communities associated with three main groups of macroalgae. FEMS Microbiol. Ecol. 93, fiw255. doi: 10.1093/femsec/fiw255
Bordenstein S. R. and Theis K. R. (2015). Host biology in light of the microbiome: ten principles of holobionts and hologenomes. PloS Biol. 13, e1002226. doi: 10.1371/journal.pbio.1002226
Boudouresque C.-F., Blanfuné A., Pergent G., Pergent-Martini C., Perret-Boudouresque M., and Thibaut T. (2020). Impacts of marine and lagoon aquaculture on macrophytes in Mediterranean benthic ecosystems. Front. Mar. Sci. 7, 218. doi: 10.3389/fmars.2020.00218
Boudouresque C. F. and Verlaque M. (2001). “Ecology of paracentrotus lividus,” in Developments in aquaculture and fisheries science. (Amsterdam, The Netherlands: Elsevier).
Bowman J. P. (2007). Bioactive compound synthetic capacity and ecological significance of marine bacterial genus Pseudoalteromonas. Mar. Drugs 5, 220–241. doi: 10.3390/md504220
Brothers C. J., van der Pol W. J., Morrow C. D., Hakim J. A., Koo H., and McClintock J. B. (2018). Ocean warming alters predicted microbiome functionality in a common sea urchin. Proc. R. Soc. B 285, 20180340. doi: 10.1098/rspb.2018.0340
Bulleri F., Benedetti-Cecchi L., and Cinelli F. (1999). Grazing by the sea urchins Arbacia lixula L. and Paracentrotus lividus Lam. in the Northwest Mediterranean. J. Exp. Mar. Biol. Ecol. 241, 81–95. doi: 10.1016/S0022-0981(99)00073-8
Buschi E., Dell’Anno A., Tangherlini M., Stefanni S., Lo Martire M., Núñez-Pons L., et al. (2023). Rhodobacteraceae dominate the core microbiome of the sea star Odontaster validus (Koehler 1906) in two opposite geographical sectors of the Antarctic Ocean. Front. Microbiol. 14, 1234725. doi: 10.3389/fmicb.2023.1234725
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., and Holmes S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Carrier T. J. and Reitzel A. M. (2017). The hologenome across environments and the implications of a host-associated microbial repertoire. Front. Microbiol. 8, 802. doi: 10.3389/fmicb.2017.00802
Carrier T. J. and Reitzel A. M. (2019). Bacterial community dynamics during embryonic and larval development of three confamilial echinoids. Mar. Ecol. Prog. Ser. 611, 179–188. doi: 10.3354/meps12872
Carrier T. J. and Reitzel A. M. (2020). Symbiotic life of echinoderm larvae. Front. Ecol. Evol. 7, 509. doi: 10.3389/fevo.2019.00509
Chludil H. D., Murray A. P., Seldes A. M., and Maier M. S. (2003). Biologically active triterpene glycosides from sea cucumbers (Holothuroidea, Echinodermata). Stud. Natural products Chem. 28, 587–615. doi: 10.1016/S1572-5995(03)80150-3
Dantan L., Toulza E., Petton B., Montagnani C., Degremont L., Morga B., et al. (2024). Microbial education for marine invertebrate disease prevention in aquaculture. Rev. Aquaculture 16, 1229–1243. doi: 10.1111/raq.12893
Davis N. M., Proctor D. M., Holmes S. P., Relman D. A., and Callahan B. J. (2018). Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 1–14. doi: 10.1186/s40168-018-0605-2
Dittami S. M., Arboleda E., Auguet J.-C., Bigalke A., Briand E., Cárdenas P., et al. (2021). A community perspective on the concept of marine holobionts: current status, challenges, and future directions. PeerJ 9, e10911. doi: 10.7717/peerj.10911
Dong Y., Li Y., Ge M., Takatsu T., Wang Z., Zhang X., et al. (2023). Distinct gut microbial communities and functional predictions in divergent ophiuroid species: host differentiation, ecological niches, and adaptation to cold-water habitats. Microbiol. Spectr. 11, e02073–e02023. doi: 10.1128/spectrum.02073-23
Dubilier N., Bergin C., and Lott C. (2008). Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat. Rev. Microbiol. 6, 725–740. doi: 10.1038/nrmicro1992
Dybas L. and Fankboner P. V. (1986). Holothurian survival strategies: Mechanisms for the maintenance of a bacteriostatic environment in the coelomic cavity of the sea cucumber, Parastichopuscalifornicus. Dev. Comp. Immunol. 10, 311–330. doi: 10.1016/0145-305X(86)90022-4
Enomoto M., Nakagawa S., and Sawabe T. (2012). Microbial communities associated with holothurians: presence of unique bacteria in the coelomic fluid. Microbes Environments 27, 300–305. doi: 10.1264/jsme2.ME12020
Faddetta T., Ardizzone F., Faillaci F., Reina C., Palazzotto E., Strati F., et al. (2020). Composition and geographic variation of the bacterial microbiota associated with the coelomic fluid of the sea urchin Paracentrotus lividus. Sci. Rep. 10, 21443. doi: 10.1038/s41598-020-78534-5
Galac M. R., Bosch I., and Janies D. A. (2016). Bacterial communities of oceanic sea star (Asteroidea: Echinodermata) larvae. Mar. Biol. 163, 162. doi: 10.1007/s00227-016-2938-3
Galià-Camps C., Baños E., Pascual M., Carreras C., and Turon X. (2023). Multidimensional variability of the microbiome of an invasive ascidian species. IScience 26, 107812. doi: 10.1016/j.isci.2023.107812
Gianguzza P. and Bonaviri C. (2013). “Arbacia,” in Developments in aquaculture and fisheries science (Amsterdam, The Netherlands: Elsevier).
Guerinot M. L. and Patriquin D. (1981). The association of N2-fixing bacteria with sea urchins. Mar. Biol. 62, 197–207. doi: 10.1007/BF00388183
Hakim J. A. (2019). Genomic insight into the gut microbiome of the sea urchins Lytechinus variegatus and Strongylocentrotus purpuratus revealed distinct community compositions and their metabolic profiles. EmergentCity Press.
Hakim J. A., Green G. B., Watts S. A., Crowley M. R., Morrow C. D., and Bej A. K. (2021). Microbial composition and genes for key metabolic attributes in the gut digesta of sea urchins Lytechinus variegatus and Strongylocentrotus purpuratus using shotgun metagenomics. Curr. Issues Mol. Biol. 43, 978–995. doi: 10.3390/cimb43020070
Hakim J. A., Koo H., Dennis L. N., Kumar R., Ptacek T., Morrow C. D., et al. (2015). An abundance of Epsilonproteobacteria revealed in the gut microbiome of the laboratory cultured sea urchin, Lytechinus variegatus. Front. Microbiol. 6, 1047. doi: 10.3389/fmicb.2015.01047
Hakim J. A., Koo H., Kumar R., Lefkowitz E. J., Morrow C. D., Powell M. L., et al. (2016). The gut microbiome of the sea urchin, Lytechinus variegatus, from its natural habitat demonstrates selective attributes of microbial taxa and predictive metabolic profiles. FEMS Microbiol. Ecol. 92, fiw146. doi: 10.1093/femsec/fiw146
Hakim J. A., Schram J. B., Galloway A. W., Morrow C. D., Crowley M. R., Watts S. A., et al. (2019). The purple sea urchin Strongylocentrotus purpuratus demonstrates a compartmentalization of gut bacterial microbiota, predictive functional attributes, and taxonomic co-occurrence. Microorganisms 7, 35. doi: 10.3390/microorganisms7020035
Hereu B., Zabala M., Linares C., and Sala E. (2004). Temporal and spatial variability in settlement of the sea urchin Paracentrotus lividus in the NW Mediterranean. Mar. Biol. 144, 1011–1018. doi: 10.1007/s00227-003-1266-6
Hernández C., Sangil C., and Hernández J. (2016). A new CO2 vent for the study of ocean acidification in the Atlantic. Mar. pollut. Bull. 109, 419–426. doi: 10.1016/j.marpolbul.2016.05.040
Hou D., Huang Z., Zeng S., Liu J., Wei D., Deng X., et al. (2017). Environmental factors shape water microbial community structure and function in shrimp cultural enclosure ecosystems. Front. Microbiol. 8, 2359. doi: 10.3389/fmicb.2017.02359
Kembel S. W., Cowan P. D., Helmus M. R., Cornwell W. K., Morlon H., Ackerly D. D., et al. (2010). Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464. doi: 10.1093/bioinformatics/btq166
Kirchman D. L. (2002). The ecology of Cytophaga–Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 39, 91–100. doi: 10.1016/S0168-6496(01)00206-9
Klaoudatos D., Tziantziou L., Lolas A., Neofitou N., and Vafidis D. (2022). Population characteristics of the upper infralittoral sea urchin Arbacia lixula (Linnaeus 1758) in Eastern Mediterranean (Central Greece): An indicator species for coastal water quality. J. Mar. Sci. Eng. 10, 395. doi: 10.3390/jmse10030395
Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., Horn M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1–e1. doi: 10.1093/nar/gks808
Lahti L. and Shetty S. (2018). Introduction to the microbiome R package. Available online at: https://microbiome.github.io/tutorials (Accessed April 20, 2025).
Laport M. S., Bauwens M., Collard M., and George I. (2018). Phylogeny and antagonistic activities of culturable bacteria associated with the gut microbiota of the sea urchin (Paracentrotus lividus). Curr. Microbiol. 75, 359–367. doi: 10.1007/s00284-017-1389-5
Lawrence J. M. (2020). Developments in aquaculture and fisheries science. Ed. Lawrence J. M. (London, UK: Elsevier). doi: 10.1016/B978-0-12-819570-3.09995-9
Leschine S., Paster B. J., and Canale-Parola E. (2006). Free-living saccharolytic spirochetes: the genus Spirochaeta. Prokaryotes 7, 195–210. doi: 10.1007/0-387-30747-8_7
Li Q., Chen Y., Zhang J., Zhang S., and Li J. (2025). Specificity of benthic invertebrate gill-associated microbiome contributes to host fitness to localized heterogeneous environment in the cold seep. Sci. Total Environ. 958, 177861. doi: 10.1016/j.scitotenv.2024.177861
Lilburn T., Kim K., Ostrom N., Byzek K., Leadbetter J., and Breznak J. (2001). Nitrogen fixation by symbiotic and free-living spirochetes. Science 292, 2495–2498. doi: 10.1126/science.1060281
Lin H. and Peddada S. D. (2020). Analysis of compositions of microbiomes with bias correction. Nat. Commun. 11, 3514. doi: 10.1038/s41467-020-17041-7
Liu Z., Guo Y., Qin C., Mu X., and Zhang J. (2024). High-throughput sequencing analysis revealed a preference for animal-based food in purple sea urchins. Biology 13, 623. doi: 10.3390/biology13080623
Louca S., Parfrey L. W., and Doebeli M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277. doi: 10.1126/science.aaf4507
Marangon E., Laffy P. W., Bourne D. G., and Webster N. S. (2021). Microbiome-mediated mechanisms contributing to the environmental tolerance of reef invertebrate species. Mar. Biol. 168, 89. doi: 10.1007/s00227-021-03893-0
Marangon E., Uthicke S., Patel F., Marzinelli E. M., Bourne D. G., Webster N. S., et al. (2023). Life-stage specificity and cross-generational climate effects on the microbiome of a tropical sea urchin (Echinodermata: Echinoidea). Mol. Ecol. 32, 5645–5660. doi: 10.1111/mec.17124
Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12. doi: 10.14806/ej.17.1.200
Masasa M., Kushmaro A., Nguyen D., Chernova H., Shashar N., and Guttman L. (2023). Spatial succession underlies microbial contribution to food digestion in the gut of an algivorous sea urchin. Microbiol. Spectr. 11, e00514-23. doi: 10.1128/spectrum.00514-23
McCracken A. R., Christensen B. M., Munteanu D., Case B., Lloyd M., Herbert K. P., et al. (2023). Microbial dysbiosis precedes signs of sea star wasting disease in wild populations of Pycnopodia helianthoides. Front. Mar. Sci. 10, 1130912. doi: 10.3389/fmars.2023.1130912
McMurdie P. J. and Holmes S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8, e61217. doi: 10.1371/journal.pone.0061217
Meziti A., Kormas K. A., Pancucci-Papadopoulou M.-A., and Thessalou-Legaki M. (2007). Bacterial phylotypes associated with the digestive tract of the sea urchin Paracentrotus lividus and the ascidian Microcosmus sp. Russian J. Mar. Biol. 33, 84–91. doi: 10.1134/S1063074007020022
Miller P. M., Lamy T., Page H. M., and Miller R. J. (2021). Sea urchin microbiomes vary with habitat and resource availability. Limnology Oceanography Lett. 6, 119–126. doi: 10.1002/lol2.10189
Nakagawa S., Saito H., Tame A., Hirai M., Yamaguchi H., Sunata T., et al. (2017). Microbiota in the coelomic fluid of two common coastal starfish species and characterization of an abundant Helicobacter-related taxon. Sci. Rep. 7, 8764. doi: 10.1038/s41598-017-09355-2
Nearing J. T., Douglas G. M., Hayes M. G., MacDonald J., Desai D. K., Allward N., et al. (2022). Microbiome differential abundance methods produce different results across 38 datasets. Nat. Commun. 13, 342. doi: 10.1038/s41467-022-28034-z
Nicholson J. K., Holmes E., Kinross J., Burcelin R., Gibson G., Jia W., et al. (2012). Host-gut microbiota metabolic interactions. Science 336, 1262–1267. doi: 10.1126/science.1223813
Offret C., Jégou C., Mounier J., Fleury Y., and Le Chevalier P. (2019). New insights into the haemo-and coelo-microbiota with antimicrobial activities from Echinodermata and Mollusca. J. Appl. Microbiol. 126, 1023–1031. doi: 10.1111/jam.14184
Oksanen J., Kindt R., Legendre P., O’Hara B., Stevens M. H. H., Oksanen M. J., et al. (2007). The vegan package. Community Ecol. Package 10, 719. doi: 10.32614/CRAN.package.vegan
Pagán-Jiménez M., Ruiz-Calderón J. F., Dominguez-Bello M. G., and García-Arrarás J. E. (2019). Characterization of the intestinal microbiota of the sea cucumber Holothuria glaberrima. PloS One 14, e0208011. doi: 10.1371/journal.pone.0208011
Palacín C., Turon X., Ballesteros M., Giribet G., and López S. (1998). Stock evaluation of three littoral echinoid species on the Catalan Coast North-Western Mediterranean. Mar. Ecol. 19, 163–177. doi: 10.1111/j.1439-0485.1998.tb00460.x
Pérez-Portela R., Wangensteen O. S., Garcia-Cisneros A., Valero-Jiménez C., Palacín C., and Turon X. (2019). Spatio-temporal patterns of genetic variation in Arbacia lixula, a thermophilous sea urchin in expansion in the Mediterranean. Heredity 122, 244–259. doi: 10.1038/s41437-018-0098-6
Petersen J. M., Kemper A., Gruber-Vodicka H., Cardini U., van der Geest M., Kleiner M., et al. (2016). Chemosynthetic symbionts of marine invertebrate animals are capable of nitrogen fixation. Nat. Microbiol. 2, 1–11. doi: 10.1038/nmicrobiol.2016.195
Pita L., Rix L., Slaby B. M., Franke A., and Hentschel U. (2018). The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome 6, 46. doi: 10.1186/s40168-018-0428-1
Privitera D., Noli M., Falugi C., and Chiantore M. (2011). Benthic assemblages and temperature effects on Paracentrotus lividus and Arbacia lixula larvae and settlement. J. Exp. Mar. Biol. Ecol. 407, 6–11. doi: 10.1016/j.jembe.2011.06.030
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2024). R: A language and environment for statistical computing. R Foundation for Statistical Computing, (Vienna, Austria: R Foundation for Statistical Computing). Available online at: https://www.R-project.org/.
Rodríguez-Barreras R., Tosado-Rodríguez E. L., and Godoy-Vitorino F. (2021). Trophic niches reflect compositional differences in microbiota among Caribbean sea urchins. PeerJ 9, e12084. doi: 10.7717/peerj.12084
Roux F. L., Wegner K. M., Baker-Austin C., Vezzulli L., Osorio C. R., Amaro C., et al. (2015). The emergence of Vibrio pathogens in Europe: ecology, evolution, and pathogenesis (Paris, 11–12th March 2015). Front. Microbiol. 6, 830. doi: 10.3389/fmicb.2015.00830
Sala E., Ribes M., Hereu B., Zabala M., Alvà V., Coma R., et al. (1998). Temporal variability in abundance of the sea urchins Paracentrotus lividus and Arbacia lixula in the northwestern Mediterranean: comparison between a marine reserve and an unprotected area. Mar. Ecol. Prog. Ser. 168, 135–145. doi: 10.3354/meps168135
Salazar-Forero C. E., Reyes-Batlle M., González-Delgado S., Lorenzo-Morales J., and Hernández J. C. (2022). Influence of winter storms on the sea urchin pathogen assemblages. Front. Mar. Sci. 9, 812931. doi: 10.3389/fmars.2022.812931
Schram J. B., Kobelt J. N., Dethier M. N., and Galloway A. W. (2018). Trophic transfer of macroalgal fatty acids in two urchin species: digestion, egestion, and tissue building. Front. Ecol. Evol. 6, 83. doi: 10.3389/fevo.2018.00083
Schwob G., Cabrol L., Poulin E., and Orlando J. (2020). Characterization of the gut microbiota of the Antarctic heart urchin (Spatangoida) Abatus agassizii. Front. Microbiol. 11, 308. doi: 10.3389/fmicb.2020.00308
Simon J.-C., Marchesi J. R., Mougel C., and Selosse M.-A. (2019). Host-microbiota interactions: from holobiont theory to analysis. Microbiome 7, 1–5. doi: 10.1186/s40168-019-0619-4
Smith L. C., Arizza V., Barela Hudgell M. A., Barone G., Bodnar A. G., Buckley K. M., et al. (2018). Echinodermata: the complex immune system in echinoderms. Adv. Comp. Immunol. 7, 409–501. doi: 10.1007/978-3-319-76768-0_13
Stévenne C., Micha M., Plumier J.-C., and Roberty S. (2021). Corals and sponges under the light of the holobiont concept: how microbiomes underpin our understanding of marine ecosystems. Front. Mar. Sci. 8, 698853. doi: 10.3389/fmars.2021.698853
Teeling H., Fuchs B. M., Bennke C. M., Krüger K., Chafee M., Kappelmann L., et al. (2016). Recurring patterns in bacterioplankton dynamics during coastal spring algae blooms. elife 5, e11888. doi: 10.7554/eLife.11888
Thomas F., Hehemann J.-H., Rebuffet E., Czjzek M., and Michel G. (2011). Environmental and gut bacteroidetes: the food connection. Front. Microbiol. 2, 93. doi: 10.3389/fmicb.2011.00093
Thorsen M. S., Wieland A., Ploug H., Kragelund C., and Nielsen P. H. (2003). Distribution, identity and activity of symbiotic bacteria in anoxic aggregates from the hindgut of the sea urchin Echinocardium cordatum. Ophelia 57, 1–12. doi: 10.1080/00785236.2003.10409501
Torda G., Donelson J. M., Aranda M., Barshis D. J., Bay L., Berumen M. L., et al. (2017). Rapid adaptive responses to climate change in corals. Nat. Climate Change 7, 627–636. doi: 10.1038/nclimate3374
Trenzado C., Hidalgo F., Villanueva D., Furné M., Díaz-Casado M., Merino R., et al. (2012). Study of the enzymatic digestive profile in three species of Mediterranean sea urchins. Aquaculture 344, 174–180. doi: 10.1016/j.aquaculture.2012.03.027
Venn A. A., Loram J. E., and Douglas A. E. (2008). Photosynthetic symbioses in animals. J. Exp. Bot. 59, 1069–1080. doi: 10.1093/jxb/erm328
Wangensteen O. S., Turon X., García-Cisneros A., Recasens M., Romero J., and Palacín C. (2011). A wolf in sheep’s clothing: carnivory in dominant sea urchins in the Mediterranean. Mar. Ecol. Prog. Ser. 441, 117–128. doi: 10.3354/meps09359
Wangensteen O. S., Turon X., Perez-Portela R., and Palacin C. (2012). Natural or naturalized? Phylogeography suggests that the abundant sea urchin Arbacia lixula is a recent colonizer of the Mediterranean. Sant Francisco California, USA: PeerJ.
Wheeler B., Torchiano M., and Torchiano M. M. (2025). Package ‘lmperm’. R Package version 2:107812. doi: 10.32614/CRAN.package.lmPerm
Wickham H. (2016). ggplot2: elegant graphics for data analysis (New York, NY: Springer-Verlag New York).
Williams T. J., Wilkins D., Long E., Evans F., DeMaere M. Z., Raftery M. J., et al. (2013). The role of planktonic F lavobacteria in processing algal organic matter in coastal E ast A ntarctica revealed using metagenomics and metaproteomics. Environ. Microbiol. 15, 1302–1317. doi: 10.1111/1462-2920.12017
Yao Q., Yu K., Liang J., Wang Y., Hu B., Huang X., et al. (2019). The composition, diversity and predictive metabolic profiles of bacteria associated with the gut digesta of five sea urchins in Luhuitou fringing reef (northern South China Sea). Front. Microbiol. 10, 1168. doi: 10.3389/fmicb.2019.01168
Zhang F., Tian Y., Gao F., Chen S., Li D., and Zhou Y. (2014). Bacterial community in the intestine of the sea urchin Strongylocentrotus intermedius during digestion of Macrocystis pyrifera. Mar. Freshw. Behav. Physiol. 47, 117–127. doi: 10.1080/10236244.2014.906095
Zhang C., Yu Z., Xue Z., Li H., Zhu J., Wang L., et al. (2021). The temporal dynamics of bacteria in the coelomic fluid of sea cucumber Apostichopus japonicus after evisceration. Invertebrate Survival J. 18, 46–55. doi: 10.25431/1824-307X/isj.v18i1.46-55
Keywords: bacterial symbiosis, echinoderms, Atlantic-Mediterranean coast, high-throughput sequencing, host-microbiota interactions
Citation: Arranz V, Schmütsch-Molina L, Fernandez-Vilert R, Hernández JC and Pérez-Portela R (2025) Sea urchin holobionts: microbiome variation across species, compartments and locations in Paracentrotus lividus and Arbacia lixula. Front. Mar. Sci. 12:1615711. doi: 10.3389/fmars.2025.1615711
Received: 21 April 2025; Accepted: 30 September 2025;
Published: 16 October 2025.
Edited by:
Valerio Mazzella, Anton Dohrn Zoological Station Naples, ItalyReviewed by:
Enrico Nanetti, University of Bologna, ItalyAnna Salvatori, Anton Dohrn Zoological Station Naples, Italy
Copyright © 2025 Arranz, Schmütsch-Molina, Fernandez-Vilert, Hernández and Pérez-Portela. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vanessa Arranz, di5hcnJhbnpAdWIuZWR1